

Abstract
Liver injury, characterized predominantly by elevated aspartate aminotransferase and alanine aminotransferase, is a common feature of coronavirus disease 2019 (COVID‐19) symptoms caused by severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2). Additionally, SARS‐CoV‐2 infection is associated with acute‐on‐chronic liver failure in patients with cirrhosis and has a notably elevated mortality in patients with alcohol‐related liver disease compared to other etiologies. Direct viral infection of the liver with SARS‐CoV‐2 remains controversial, and alternative pathophysiologic explanations for its hepatic effects are an area of active investigation. In this review, we discuss the effects of SARS‐CoV‐2 and the inflammatory environment it creates on endothelial cells and platelets more generally and then with a hepatic focus. In doing this, we present vascular inflammation and thrombosis as a potential mechanism of liver injury and liver‐related complications in COVID‐19.
Abbreviations
- ACE2
angiotensin converting enzyme 2
- ADAM17
a disintegrin and metalloprotease 17
- ALT
alanine aminotransferase
- Ang II
angiotensin II
- AT1R
angiotensin II receptor type 1
- COVID‐19
coronavirus disease 2019
- gp130
interleukin‐6 receptor signaling domain
- HIF
hypoxia inducible factor
- ICAM1
intracellular adhesion molecule 1
- IFN
interferon
- IL
interleukin
- IL‐6Rα
interleukin‐6 receptor alpha
- JAK
Janus kinase
- LSEC
liver sinusoidal endothelial cell
- PAI‐1
plasminogen activator inhibitor 1
- SARS‐Cov‐2
severe acute respiratory syndrome‐coronavirus 2
- sIL‐6R
soluble interleukin‐6 receptor
- STAT
signal transducer and activator of transcription
- TLR
toll‐like receptor
- TNF
tumor necrosis factor
- vWF
von Willebrand factor
Liver injury, primarily consisting of elevated transaminase levels, has been frequently observed in patients with coronavirus disease 2019 (COVID‐19) and is correlated with clinical outcomes, including mortality.( 1 , 2 , 3 ) The pathophysiologic mechanism of transaminase elevation, however, remains incompletely defined. The possibility of direct infection of hepatic cells with severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) continues to be a topic of very active research. While controversy exists on this question, overall conclusive evidence of direct viral infection causing liver injury in SARS‐CoV‐2 is lacking.( 4 ) An alternative mechanism of liver injury in SARS‐CoV‐2 is endothelial‐mediated inflammation and thrombosis. Endothelial inflammation, platelet recruitment, and thrombosis have been implicated in the pathogenesis of nonalcoholic steatohepatitis (NASH)( 5 , 6 ) and portal hypertension.( 7 ) Here, we present a discussion of endotheliopathy, altered platelet function, and inflammation in COVID‐19 and how their synergy may result in liver injury in these patients. We have also summarized clinical (Table 1) and histologic (Table 2) studies related to hepatic coagulopathy in patients with COVID‐19.
TABLE 1.
Liver and Coagulopathy in Patients with COVID‐19, Clinical Study
| Authors | Patients | Rate of | Patients With Liver Injury Had High Level of | Thrombus | ||||
|---|---|---|---|---|---|---|---|---|
| Liver Injury | ESR | Fibrinogen | D‐Dimer | FDP | Portal | Venous | ||
| McConnell et al.( 94 ) | 3,780 | 1,006 (27%) (ALT >3×) | NA | X | X | NA | NA | NA |
| Lu et al.( 127 ) | 1,361 | 125 (9%) (ALT >5× & AST >3×) | NA | NA | X | NA | NA | NA |
| Wang et al.( 128 ) | 657 | 303 (46%) (ALT >1×) | X | NA | NA | NA | NA | NA |
| Tsutsumi et al.( 129 ) | 60 | 31 (52%) (ALT >40 U/L) | NA | X | X | X | NA | NA |
| La Mura et al.( 12 ) | 1 | 1 (ALT 257 U/L) | NA | X | X | NA | X (CT) | |
| Del Hoyo et al.( 130 ) | 1 | 1 (ALT 1,065 U/L) | NA | NA | X | NA | X (CT) | |
| Franco‐Moreno et al.( 121 ) | 1 | 1 (ALT 111 U/L) | NA | X | X | NA | X (CT) | |
| Abeysekera et al.( 131 ) | 1 | 1 (ALT 55 U/L) | NA | NA | NA | NA | X (CT) | |
| Borazjani et al.( 132 ) | 1 | 1 (ALT 67 U/L) | NA | NA | NA | NA | X (CT) | |
| Randhawa et al.( 133 ) | 1 | None | NA | NA | NA | NA | X (CT) | |
| Sh Hassan et al.( 134 ) | 1 | 1 (ALT 1,325 U/L) | NA | NA | NA | NA | X (CT) | |
| Kolli and Oza( 135 ) | 1 | None | NA | NA | NA | NA | X (CT) | |
| Jeilani et al.( 136 ) | 1 | 1 (ALT 41 U/L) | NA | NA | X | NA | X (CT) | |
Abbreviations: AST, aspartate aminotransferase; CT, computed tomography; ESR, erythrocyte sedimentation rate; FDP, fibrin/fibrinogen degradation products; NA, not available; X, present.
TABLE 2.
Liver and Coagulopathy in Patients With COVID‐19, Histological Study
| Authors | Patients | Rate of | Thrombus | ||
|---|---|---|---|---|---|
| Liver Injury | Sinusoidal | Portal | Central Vein | ||
| Bugra et al.( 137 ) | 98 Postmortem liver tissues | NA | X (2 cases) | ||
| Sonzogni et al.( 56 ) | 48 Postmortem liver tissues | 25 (61%) (ALT >1×) | X (13 cases) | X (35 cases) | |
| Kondo et al.( 138 ) | 43 Postmortem liver tissues | 12 (29%) (ALT >3×) | X (23 cases) | ||
| Lagana et al.( 86 ) | 40 Postmortem liver tissues | NA | X (6 cases) | ||
| Fassan et al.( 139 ) | 25 Postmortem liver tissues | NA | X (5 cases) | X (3 cases) | |
| Schurink et al.( 140 ) | 21 Postmortem liver tissues | NA | X | ||
| Lax et al.( 141 ) | 11 Postmortem liver tissues | 4 (36%) (ALT >1×) | X (1 case) | ||
| Rapkiewicz et al.( 54 ) | 7 Postmortem liver tissues | NA | X (7 cases) | ||
| Fiel et al.( 142 ) | 2 Liver biopsy | 2 (ALT 2,074 U/L & 2,786 U/L) | X (1 case) | ||
Abbreviations: NA, not available; X, present.
Endotheliopathy in COVID‐19
While a great deal of focus has centered on COVID‐19 as a respiratory disease, it has important systemic manifestations, including on the cardiovascular and immune systems.( 8 , 9 ) In clinical practice, numerous thrombotic complications have been reported in patients, including deep vein thrombosis, strokes, myocardial infarction, aortic thrombosis, and portal vein thrombosis.( 10 , 11 , 12 , 13 ) In addition, the occurrence of a thrombotic event is associated with mortality.( 13 ) Among the features of COVID‐19 infection is a coagulopathy characterized by elevated D‐dimer and fibrinogen concentrations, with minor changes in prothrombin time and platelet count.( 11 ) Several studies have reported that increased D‐dimer is associated with severe COVID‐19 and high mortality.( 13 , 14 , 15 , 16 ) Furthermore, von Willebrand factor (vWF) activity, vWF antigen, factor VIII activity, and soluble thrombomodulin, a marker of endothelial cell activation, are considerably increased in patients with COVID‐19.( 11 , 16 ) The clinical relevance of these derangements is highlighted by the fact that mortality in patients with COVID‐19 is correlated with vWF antigen and soluble thrombomodulin levels.( 16 ) Von Willebrand factor is predominantly produced by endothelial cells, and soluble thrombomodulin is an endothelial transmembrane glycoprotein that is released following endothelial disruption or injury.( 17 ) These findings suggest that endotheliopathy is a critical component of the prothrombotic imbalance in patients with COVID‐19 and an important contributory factor to mortality in this disease. To understand the role of endotheliopathy in COVID‐19, it is important to understand normal endothelial cell physiology and thrombus formation and how this physiology is altered by the SARS‐CoV‐2 virus. The role of inflammatory signaling in creating a procoagulant endothelium will be addressed separately.
Endothelial Cell Function and Thrombus Formation in General
In noninflamed tissues, vascular endothelial cells maintain blood fluidity, regulate blood flow, control vessel wall permeability, and maintain quiescence of circulating leukocytes.( 18 ) Thrombosis is the pathological process of blood clot (or thrombus) formation on the inner surface of the endothelium. Blood clots consist of accumulated platelets (platelet plug) and a mesh of cross‐linked fibrin.( 19 ) The formation of blood clots involves interplay among various cell types and cascades of coagulation factors.( 20 ) Platelet adhesion to the injured endothelial cells is an early step in thrombosis. Attached platelets are activated by the action of thrombin (also known as activated factor II), which facilitates additional recruitment of circulating platelets to the injury site to form a platelet plug. Thrombin is generated from prothrombin by a series of well‐described cascades of coagulation factors. Damaged endothelial cells and hepatocytes produce a procoagulant molecule, tissue factor, that binds and activates circulating procoagulant molecule factor VII. Activated factor VII proteolytically cleaves factor X to form active factor X, which then converts prothrombin to its active form thrombin. In addition to platelet activation and aggregation, thrombin facilitates a cascade of coagulation events to generate fibrin and cross‐links fibrin chains to form a large fibrin mesh.( 19 , 21 , 22 )
In normal hemostatic conditions, unnecessary blood clotting is inhibited by multiple antithrombotic factors produced by healthy endothelial cells. First, endothelial cells express tissue factor pathway inhibitor (TFPI), which inhibits the actions of the factor‐VIIa–tissue‐factor complex. Second, endothelial cells synthesize and display heparan sulfate proteoglycans on their cell surface; these activate antithrombin III to inhibit thrombin generation. Third, endothelial cells also express thrombomodulin, which diverts the activity of thrombin toward activation of protein C, an inhibitor of coagulation. Fourth, endothelial cells suppress platelet activation.( 23 )
Endothelial Effects of SARS‐CoV‐2 Infection
SARS‐CoV‐2 uses the angiotensin converting enzyme 2 (ACE2) receptor for internalization, aided by transmembrane protease serine 2 protease.( 24 , 25 ) The ACE2 receptor is expressed in several organs, including the lung, heart, liver, kidney, and intestine.( 26 , 27 ) ACE2 receptors are also expressed by endothelial cells.( 28 ) A high level of transcriptomic changes has been reported in both the lymphatic and vascular endothelial cell population in COVID‐19 human tissue biopsies from lung, kidney, liver, and heart, using a single‐cell and single‐nucleus RNA sequencing approach.( 29 ) In this study, however, almost no liver endothelial cells are recognized to express ACE2. Thus, expression of ACE2 in endothelial cells may be tissue specific.
ACE converts angiotensin I to angiotensin II (AngII), while ACE2 converts AngII to angiotensin 1‐7 (Ang1‐7). AngII binds AngII receptor type 1 (AT1R) and exerts proinflammatory, pro‐oxidant, and vasoconstrictive effects. In opposition, Ang1‐7 binds to the Mas receptor and mediates anti‐inflammatory, antioxidant, and vasodilatory effects.( 30 , 31 , 32 ) The AngII–AT1R axis also activates a disintegrin and metalloprotease 17 (ADAM17). ADAM17 cleaves the membrane‐bound interleukin (IL)‐6 receptor α (IL‐6Rα) (Fig. 1) and membrane‐bound tumor necrosis factor alpha (TNFα) on the cell membrane, generating soluble IL‐6Rα and TNFα.( 32 , 33 ) Additionally, AngII induces expression of plasminogen activator inhibitor 1 (PAI‐1) in endothelial cells by AT1R. PAI‐1 suppresses tissue‐type and urokinase‐type plasminogen activator function, resulting in suppression of fibrinolysis. The resulting PAI‐1/plasminogen activator imbalance creates a hypercoagulable state.( 31 , 34 , 35 ) SARS‐CoV‐2 binding to ACE2 attenuates ACE2 activity through internalization. Attenuation of ACE2 can lead to a dominant AngII effect, including enhanced inflammation and a hypercoagulable state.
FIG. 1.
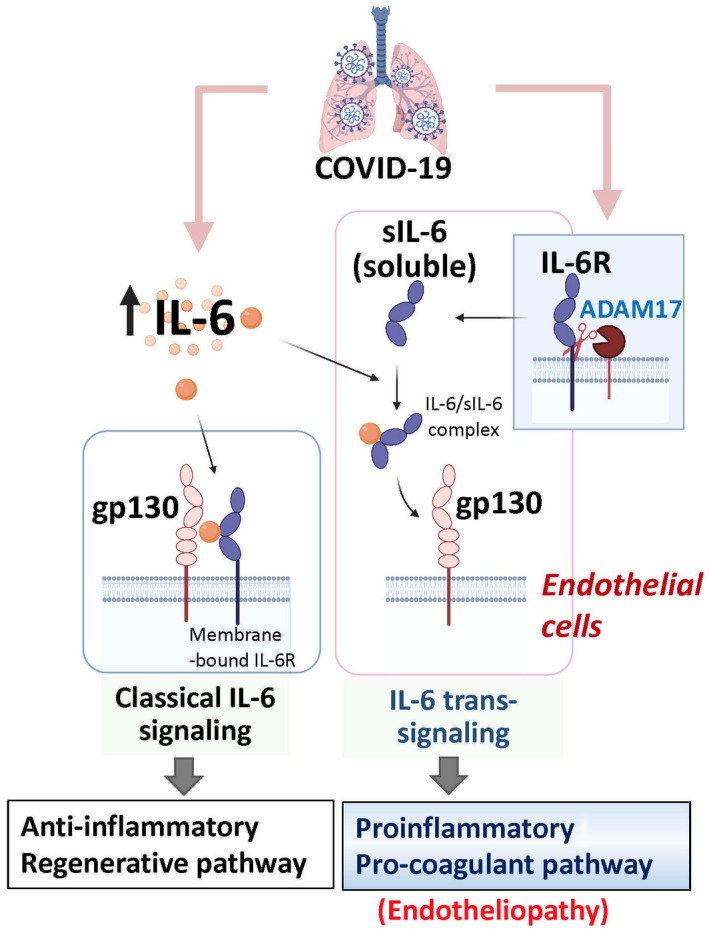
IL‐6 signaling (classical vs. trans‐signaling) in COVID‐19. Excessive inflammatory cytokine signaling, particularly through IL‐6, is thought to be an important factor in the pathogenesis of COVID‐19. IL‐6 induces downstream signaling through JAK/STAT activation through two pathways. One is classical IL‐6 signaling through IL‐6 binding to the ligand‐binding alpha subunit of its receptor (gp80/IL‐6Rα) and subsequently recruiting the signaling beta subunit (gp130) to produce downstream signaling. This pathway is thought to be anti‐inflammatory and to promote liver regeneration. The other is trans‐signaling that occurs with IL‐6 binding to a soluble form of the receptor alpha subunit. ADAM17, which is typically increased in inflammatory conditions, cleaves the membrane‐bound IL‐6Rα, increasing sIL‐6R and creating opportunity for formation of the IL‐6/sIL‐6R complex, which then interacts with gp130 on target cells that may not express membrane‐bound IL‐6Ra. IL‐6 trans‐signaling is thought to be the major route of IL‐6 signaling to LSECs and has been implicated in endotheliopathy (proinflammatory and procoagulant state) in COVID‐19. The sIL‐6R levels are increased in COVID‐19, 143 with a likely result of increased trans‐signaling. Abbreviation: gp80, interleukin‐6 receptor ligand‐binding domain.
Other Effects of COVID‐19 on the Endothelium
SARS‐CoV‐2 endotheliopathy may be further enhanced by other clinical factors, including hypoxemia (secondary to acute lung injury/acute respiratory distress syndrome [ALI/ARDS]) and hyperfibrinogenemia. Hypoxia attenuates the anticoagulant function of endothelial cells through suppression of thrombomodulin.( 36 ) Further, hypoxia inducible factor (HIF)‐1α and HIF‐2α may also produce a prothrombotic endothelium. HIF‐2α represses TFPI expression.( 37 , 38 ) Furthermore, expression of PAI‐1 is regulated by HIF‐1α and HIF‐2α, and hypoxia may up‐regulate PAI‐1.( 37 ) In addition, hypoxia activates the nuclear factor kappa B signaling pathway in macrophages and neutrophils,( 39 , 40 ) and HIF‐1α up‐regulates ADAM17, IL‐6, and TNFα.( 41 , 42 ) Through these multiple pathways, hypoxia may promote further hypercoagulation and inflammation.
During hyperfibrinogenemia, fibrinogen may contribute to endothelial dysfunction.( 43 ) Fibrinogen binds to endothelial cells through the interactions with cell adhesion molecules, such as intercellular adhesion molecule 1 (ICAM1), integrin α5β1, and integrin α5β3.( 44 , 45 ) ICAM1 is constitutively present on endothelial cells, but its expression is increased by proinflammatory cytokines. ICAM1 plays an important role in both innate and adaptive immune responses and is involved in the transendothelial migration of leukocytes to sites of inflammation.( 46 ) Hyperfibrinogenemia may promote further inflammation.( 47 )
Functional assessment of the systemic microvasculature in COVID‐19 has produced mixed results. The most commonly used technique for this assessment in patients has been sublingual videomicroscopy. Both microcirculatory impairment( 48 , 49 ) and intact microcirculatory function( 50 ) have been described by this method in patients with COVID‐19. An additional report defined radiographically diagnosed perfusion deficits in the lungs and kidneys of patients with COVID‐19, potentially indicative of a systemic microangiopathy.( 51 ) The important role of systemic endotheliopathy beyond the liver in COVID‐19 has been reviewed,( 52 ) highlighting the critical interplay between inflammatory factors, endothelial cells, and platelets in disease pathogenesis throughout the body.
Alterations of Platelet Function in COVID‐19
Platelet function in COVID‐19 is an area of intense interest and very active research. The high rate of thrombotic complications in this disease( 53 ) raised suspicion for a hypercoagulable platelet phenotype, and recent histologic series have confirmed platelet‐rich microthrombi in multiple organs, including the lungs, liver, renal, and cardiac microcirculation.( 54 , 55 ) These studies are concordant with another recently published autopsy series describing thrombosis in the liver sinusoids, suggesting that platelet hyperactivity in COVID‐19 may be an important mediator of the frequent liver injury seen in these patients.( 56 )
The phenotype of platelets in COVID‐19 continues to be defined. Recent primate studies of SARS‐CoV‐2 infection have demonstrated up‐regulation of platelet activation in response to infection.( 57 ) Studies in patients have demonstrated increased platelet aggregation on blood smears in severe COVID‐19( 58 ) and elevated serum markers of platelet activation.( 16 ) Details of platelet function in COVID‐19, however, remain controversial. Several reports have demonstrated through RNA sequencing and flow cytometry that platelets from patients with COVID‐19 exhibited mitochondrial dysfunction and hyperactivation, as measured by increased P‐selectin positivity and aggregation.( 59 , 60 ) Interestingly, the latter study showed defective generation of “procoagulant” platelets, as measured by annexin V positivity after stimulation, in COVID‐19 compared to healthy controls but suggested that platelets unable to form a procoagulant phenotype may hyperaggregate to cause vascular pathology.( 60 ) It has been additionally reported that platelets in COVID‐19 demonstrated increased responsiveness to vWF due to increased aggregation in response to ristocetin (a proaggregatory factor( 61 )) but impaired aggregation in response to agonists, such as adenosine diphosphate, collagen, and arachidonic acid.( 62 , 63 )
One area of more broad agreement concerning platelet function in COVID‐19 is platelet–leukocyte interactions. In particular, platelet–neutrophil aggregates have been shown to be increased in COVID‐19 in several reports( 59 , 64 , 65 ) and are likely mediated by up‐regulation of platelet P‐selectin. Platelet–neutrophil interactions have been shown to mediate neutrophil extracellular trap formation, which is associated with thrombosis, in influenza( 66 ) infection as well as in COVID‐19.( 65 )
Another aspect of platelet phenotype under investigation in COVID‐19 is its role in cytokine signaling. In contrast to platelets from healthy donors, platelets from patients with COVID‐19 released higher levels of IL‐1β, monocyte chemoattractant protein‐1/CC motif chemokine ligand 2, and IL‐18, among other cytokines, implicating platelets directly in the dysregulated inflammation of the disease.( 64 )
While platelet dysfunction is becoming increasingly defined as a feature of COVID‐19, the specific mechanisms mediating it remain largely unknown, with three main possibilities emerging as candidates: 1) Inflammatory cytokine signaling (e.g., IL‐6), 2) toll‐like receptor (TLR) signaling, 3) direct infection by platelets with SARS‐CoV‐2.
Inflammatory Cytokine Signaling: The Role of IL‐6
Excessive inflammatory cytokine signaling, particularly through IL‐6, is thought to be an important factor in the pathogenesis of COVID‐19. In addition to its other effects, IL‐6 may be mediating COVID‐induced coagulopathy by interacting with platelets and contributing to the activated platelet phenotype seen in this disease.( 16 ) In patients receiving IL‐6 cytokine therapy for cancer, platelets showed enhanced aggregation and degranulation,( 67 ) and in vitro studies also demonstrated increased platelet activation with IL‐6.( 68 ) In addition, plasma IL‐6 has been shown to correlate with the abundance of platelet‐derived microparticles, which are generated as a consequence of platelet activation.( 69 ) Interestingly, platelets do not possess a membrane‐bound form of the IL‐6 receptor but do contain the soluble form (sIL‐6R), which is released following platelet activation and may provide the manner in which platelets are able to respond to IL‐6 through trans‐signaling.( 70 ) In a colitis model of experimental inflammation, IL‐6 was found to be the key mediator of thrombosis and platelet hyper‐reactivity, suggesting a role for IL‐6‐mediated thrombosis in other disease states as well.( 71 ) The specifics of IL‐6 signaling and its effect on platelets in COVID‐19 is an area of ongoing research.
TLR Signaling
Platelets possess functional TLRs, including TLR7, which recognizes single‐stranded RNA and so could provide a possible mechanism for platelet response to SARS‐CoV‐2. A recent study examining the platelet transcriptomic response to SARS‐CoV‐2 interestingly found messenger RNA from the virus inside a small number of platelets.( 59 ) This was corroborated in an additional study showing the association of SARS‐CoV‐2 RNA with platelets.( 72 ) Because platelet expression of ACE2 remains controversial, although it has been described,( 73 ) TLR7 provides a possible alternative mechanism by which viral RNA may enter platelets, and this finding bolsters the suggestion that TLR7‐mediated signaling may be playing a role in platelet activation in COVID‐19.( 59 )
Direct Infection of Platelets
Infection of platelets by SARS‐CoV‐2 remains an area of controversy and active investigation. Reports have found both that platelets do( 73 , 74 ) and do not( 59 ) express ACE2, the receptor necessary for SARS‐CoV‐2 infection. Additional studies are awaited to further define whether direct infection causes platelet abnormalities in COVID‐19.
Inflammatory Signaling in COVID‐19
Accumulating evidence suggests that the severity of COVID‐19 is associated with increased levels of cytokines, such as IL‐1β, IL‐2, IL‐6, IL‐8, interferon‐γ (IFN‐γ)‐induced protein 10 (IP‐10), TNF‐α, IFN‐γ, macrophage inflammatory protein 1α and 1β, and vascular endothelial growth factor.( 75 , 76 , 77 ) In previous outbreaks of Middle East respiratory syndrome‐coronavirus, a coronavirus related to SARS‐CoV‐2, proinflammatory cytokines, such as IL‐2, IL‐6, and IL‐8, have been identified( 78 , 79 ) as key players during infection and possible triggers of liver injury. Del Valle et al.( 77 ) reported that high serum IL‐6, IL‐8, and TNF‐α levels at the time of hospitalization are strong and independent predictors of patient survival in COVID‐19 (n = 1,484) and suggested that serum IL‐6 and TNF‐α levels should be considered in the management and treatment of patients with COVID‐19. An additional report identified a triad of IL‐6, IP‐10, and IL‐10 as excellent predictors of a severe disease course in COVID‐19.( 80 ) On the other hand, it was reported that the cytokine profile in patients with severe COVID‐19 does not differ from moderate COVID‐19, ARDS, and sepsis.( 81 ) However, several important therapeutic differences between COVID‐19‐associated cytokine storms and many other cytokine storm disorders have been reported.( 82 ) Notably, although blood clotting disorders can occur throughout a cytokine storm, thromboembolic events seem to occur more frequently in cytokine storms associated with COVID‐19.( 83 )
Liver Injury and COVID‐19
Liver Injury as a Manifestation of Endotheliopathy, Platelet Dysfunction, and Inflammation in COVID‐19
The mechanism of liver injury in COVID‐19 infection is likely multifactorial and associated with immune dysregulation and cytokine storm, hypoxic/ischemic injury, and drug‐induced hepatotoxicity.( 84 , 85 ) Understanding the hepatic manifestations of the coagulopathy of COVID‐19 will give additional insight into liver injury in this disease (Fig. 2). One recent series of postmortem liver biopsies from patients with COVID‐19 reported portal or sinusoidal vascular thrombosis in at least 50% of patients.( 56 ) High D‐dimer values were also almost universally present in the cohort of patients (96%), and elevated alanine aminotransferase (ALT) was also highly prevalent (62%), suggesting a relationship between liver vascular thrombosis, coagulopathy, and liver injury.( 56 ) Additional pathology of COVID‐19 livers at autopsy has also demonstrated platelet‐fibrin microthrombi in the hepatic sinusoids along with some portal vein platelet aggregates. In some cases, these findings were associated with ischemic‐type damage in the liver.( 54 ) Another series of liver pathology at autopsy demonstrated what were termed “thrombotic bodies.” These were inclusions in the sinusoids that were positive for a platelet marker, clusters of differentiation 61 (CD61).( 86 )
FIG. 2.
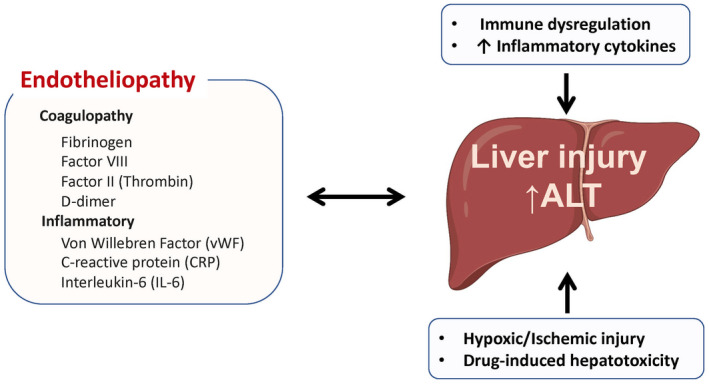
Potential mechanisms of liver injury in patients with COVID‐19. Abbreviation: CRP, C‐reactive protein.
Interestingly, a common feature of liver pathology in COVID‐19 seems to be hepatic steatosis. As demonstrated in models of nonalcoholic fatty liver disease (NAFLD), activation of coagulation is capable of driving hepatic steatosis, and this may be a novel mechanism linking the thrombosis and steatosis that are prevalent in the livers of patients with COVID‐19.( 87 )
The role of PAI‐1 in COVID‐19 liver injury is potentially interesting as elevated PAI‐1 has been associated with NAFLD and NASH.( 88 , 89 ) PAI‐1 is known as a marker of endothelial cell injury,( 90 ) and overproduced PAI‐1 binds to TLR4 on macrophages, inducing the secretion of proinflammatory cytokines and chemokines.( 91 ) Recently, PAI‐1 has been demonstrated to be elevated due to IL‐6 signaling to vascular cells in COVID‐19,( 92 ) and this occurrence in the liver may contribute to microvascular thrombosis, inflammation, and steatosis.
IL‐6 Signaling (Classical vs. Trans‐Signaling) and Endotheliopathy
Beyond its effect on PAI‐1, IL‐6 has been reported to be a key factor in acute liver injury in COVID‐19.( 79 ) IL‐6 signals to cells either directly through the membrane‐bound IL‐6 ligand‐binding domain (gp80) and the signal‐transducing component of the receptor (gp130) on the same cell or by trans‐signaling, in which a soluble form of the ligand‐binding receptor domain binds IL‐6 and complexes with gp130 alone on the cell membrane( 92 , 93 ) (Fig. 1). Liver sinusoidal endothelial cells are not thought to express the membrane‐bound ligand‐binding domain for IL‐6. We reported that IL‐6 trans‐signaling induced liver sinusoidal endotheliopathy with neutrophil infiltration and a hypercoagulable liver sinusoidal endothelial cell (LSEC) phenotype( 94 ) (Fig. 3). Patients with COVID‐19 with liver injury demonstrated elevated levels of IL‐6, serum markers of hypercoagulability, increased LSEC dysfunction on histology as evidenced by increased vWF, and increased platelet accumulation and neutrophil infiltration in the liver. Because of the association of liver injury with both elevated IL‐6 and a hypercoagulable and inflammatory LSEC phenotype, further experiments were done exploring the role of IL‐6 trans‐signaling in LSECs. These studies demonstrated that IL‐6 trans‐signaling produces a hypercoagulable and inflammatory LSEC phenotype through Janus kinase‐signal transducer and activator of transcription (JAK/STAT) activation, blockable with JAK inhibitors. This suggests that LSEC endotheliopathy may be an important mechanism by which liver injury occurs in COVID‐19. More broadly, elevated plasma ICAM1, indicative of endothelial damage, has been recently defined as a predictor of mortality in patients with both cirrhosis and COVID‐19 or sepsis.( 95 ) IL‐6 trans‐signaling induced ICAM1 up‐regulation by LSECs in our study, indicating that this pathway of inflammatory endotheliopathy may be broadly important for key outcomes in our patients with infection and inflammation.
FIG. 3.
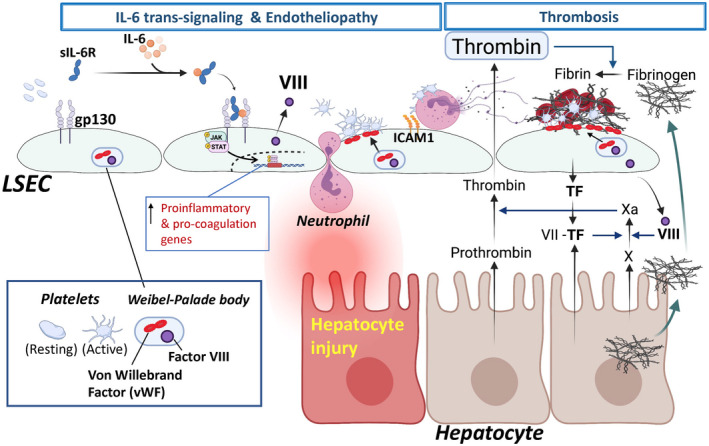
Liver sinusoidal endotheliopathy and liver injury in COVID‐19. IL‐6 trans‐signaling and endotheliopathy: IL‐6 is highly elevated in patients with COVID‐19 and is related to liver injury. LSECs are not thought to express the membrane‐bound ligand‐binding domain for IL‐6. However, the complex of IL‐6 and sIL‐6R binds to gp130 on the cell membrane, inducing IL‐6/JAK/STAT signaling, known as IL‐6 trans‐signaling. The IL‐6 trans‐signaling induces liver sinusoidal endotheliopathy with neutrophil infiltration and a hypercoagulable LSEC phenotype. IL‐6 trans‐signaling increases expression of proinflammatory (ICAM1, CXCL1, CXCL2, P‐selectin, and E‐selectin) and procoagulation genes (factor VIII and vWF). Weibel‐Palade body includes vWF and factor VIII in LSECs. IL‐6 trans‐signaling promotes movement of vWF to the LSEC surface, facilitating platelet attachment, an initial step of the thrombus formation. Endotheliopathy is associated with neutrophil recruitment, potentially leading to hepatocyte injury. Platelet–neutrophil interactions mediate NET formation. Microvascular thrombosis: Thrombus (blood clots) consists of accumulated platelets (platelet plug) and a mesh of cross‐linked fibrin. Thrombosis involves interplay among various cell types and cascades of coagulation factors. Platelet adhesion to the injured LSECs is an early step in thrombosis. Attached platelets are activated by the action of thrombin (also known as activated factor II), which facilitates additional recruitment of circulating platelets to the injury site to form a platelet plug. Thrombin is generated from prothrombin by a series of well‐described cascades of coagulation factors. Damaged endothelial cells and hepatocytes produce a procoagulant molecule, TF, which binds and activates circulating procoagulant molecule factor VII. Activated factor VII proteolytically cleaves factor X to form Xa, which then converts prothrombin to its active form, thrombin. In addition to platelet activation and aggregation, thrombin facilitates a cascade of coagulation events to generate fibrin and cross‐links fibrin chains to form a large fibrin mesh. Patients with COVID‐19 show an elevated fibrinogen level. Abbreviations: CXCL, chemokine (C‐X‐C motif) ligand 1; NET, neutrophil extracellular trap; TF, tissue factor; Xa, active factor X.
IL‐6 may also provide an interesting link between poor outcomes observed in alcohol‐related liver disease and COVID‐19. IL‐6 has been shown to be up‐regulated by ethanol consumption and in alcohol‐related liver disease.( 96 , 97 ) While IL‐6 is a potent factor for inducing liver regeneration( 98 ) and may in some cases play a protective role in the liver,( 99 ) it has also been associated with poor outcomes in acute‐on‐chronic liver failure (ACLF),( 100 , 101 ) suggesting a complex physiology. Given the proinflammatory effects of IL‐6 trans‐signaling in the liver vasculature and the frequent observation of hepatic vascular pathology in COVID‐19, up‐regulated inflammatory IL‐6 signaling may be contributing to poor outcomes in alcohol‐related liver disease.
Type I IFNs
Interesting results have also been obtained in liver organoids exposed to inflammatory cytokines typical of COVID‐19. Type I IFNs have been shown in this setting to up‐regulate ACE2 expression on hepatocytes and facilitate SARS‐CoV‐2 infection. Further studies are awaited to define this process in vivo in patients infected with SARS‐CoV‐2.( 102 )
Biliary Injury and Cholestasis in COVID‐19
In addition to a hepatocellular pattern of enzyme elevation, liver injury in COVID‐19 can manifest with a cholestatic pattern as well.( 103 , 104 ) This has been hypothesized to be due to ACE2 expression on biliary epithelial cells leading to direct viral infection. Interestingly, however, two reports of cholestasis following COVID‐19 infection suggest a possible role for endotheliopathy, noting hepatic artery branches in the portal tract with endothelial swelling with luminal narrowing, portal vein endophlebitis (inflammation of the intima area of a vein), and endothelialitis (leukocyte attachment to the vascular wall) and thrombotic material in portal vein branches.( 103 , 105 ) COVID‐19 cholestasis has been reported as a long‐term complication of infection,( 105 ) leading to the need for liver transplantation( 106 ) and highlighting its importance for further mechanistic study.
COVID‐19 Therapeutics and Liver Injury
Current therapeutics for COVID‐19 that may impact endotheliopathy and liver injury include those targeting the virus itself (remdesivir), IL‐6 and its downstream signaling pathways (tocilizumab and baricitinib), and systemic inflammation (dexamethasone). One report suggests that dexamethasone may ameliorate endothelial injury in COVID‐19.( 107 ) The nuclear receptor subfamily 3 group C member 1 (NR3C1) receptor, a key target of dexamethasone, has also been shown by single‐cell RNA sequencing to be coexpressed with IL‐6 in endothelial cells, suggesting dampening of endothelial IL‐6 production as a key action of corticosteroids in COVID‐19.( 108 ) Administration of the IL‐6 receptor antagonist tocilizumab has been suggested to improve coagulation parameters in COVID‐19, which may indicate improved endothelial function as well.( 109 ) The combination of remdesivir with the JAK inhibitor baricitinib was recently shown to improve outcomes in COVID‐19 compared to remdesivir alone,( 110 ) and baricitinib was also shown to improve COVID‐19 outcomes compared with standard of care, including dexamethasone and remdesivir.( 111 ) Treatment with a JAK inhibitor would be expected to ameliorate IL‐6‐mediated endotheliopathy in the liver. A major limitation in applying these therapeutic approaches to liver injury, however, is the frequent exclusion of patients with aspartate aminotransferase or ALT above 5 times the upper limit of normal from the studies and the association of remdesivir, tocilizumab, and baricitinib with liver injury.( 112 , 113 , 114 ) We speculate that application of therapy to prevent endothelial‐mediated liver injury would need to occur early in the course of hospitalization, but this should be clarified in future studies. Given the lack of data showing a benefit of directing therapy toward liver injury in COVID‐19 and the risks of the therapies themselves to the liver and the patient overall, more data are needed to determine which, if any, patients with liver injury require specific treatment.
Conclusion: Consequences of Liver Injury in COVID‐19
COVID‐19 disease in patients with preexisting liver disease has been a topic of active research, and vascular pathology in the liver may be particularly important to the outcomes of these patients. In patients with cirrhosis, SARS‐CoV‐2 infection has been associated with both decompensation and ACLF.( 115 , 116 ) While the specific mechanisms of decompensation remain under investigation, vascular dysfunction due to the endotheliopathy and platelet activation described above could be playing a role in worsening portal hypertension in these patients.
In addition to the acute setting, the long‐term consequences of liver injury in COVID‐19 remain unknown. The question of long‐term effects has been given additional importance by evidence that COVID‐19 viral antigens may be present in liver tissue up to 6 months following recovery.( 117 ) In the lungs, fibrosis seems to be a consequence of COVID‐19 disease,( 118 ) and basic models of pulmonary fibrosis have implicated activation of coagulation in this process.( 119 ) Whether this occurs in the liver remains unknown. Multiorgan proteomics recently completed in patients with COVID suggests that profibrotic pathways are up‐regulated in the liver due to COVID‐19, and follow‐up data are needed for evaluation of any long‐term consequences.( 120 ) In one case, portal vein thrombosis occurred in a patient without cirrhosis after resolution of SARS‐CoV‐2 infection with negative real‐time polymerase chain reaction,( 121 ) suggesting ongoing endotheliopathy and coagulopathy. A study of survivors of acute SARS‐CoV‐2 infection found that patients had an elevated risk of liver test abnormalities when followed for months after their infection, suggesting some possible long‐term sequelae for the liver.( 122 ) Additional reports have raised the possibility of persistent liver inflammation and liver fat accumulation as detected by magnetic resonance imaging following COVID‐19( 123 ) and the potential for increasing liver stiffness over time following COVID‐19,( 124 ) which must be confirmed in future studies. Metabolomic profiling of patients recovered from COVID‐19 also demonstrates elevated taurine concentrations, which are potentially indicative of liver injury, 3 months after recovery, and further study in this area will be of interest.( 125 ) The mechanism of any long‐term liver injury remains speculative, but endotheliopathy has been reported to be sustained following COVID‐19,( 126 ) suggesting endothelial‐mediated inflammation as a possible mechanism. Long‐term consequences of liver injury in COVID‐19 will require ongoing study and observation of patients.
Supported by the National Institutes of Health (grants 1R01 DK130362‐01, 1R01DK117597, 1R01AA025342 to Y.I.), American Association for the Study of Liver Diseases Clinical, Translational, and Outcomes Research Award (to M.M.), Yale Center for Clinical Investigation Scholar Award (to M.M.); and the International Research Fund for Subsidy of Kyushu University School of Medicine Alumni (to R.K.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship
- 1. Saviano A, Wrensch F, Ghany MG, Baumert TF. Liver disease and corona virus disease 2019: from pathogenesis to clinical care. Hepatology 2021;74:1088‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phipps MM, Barraza LH, LaSota ED, Sobieszczyk ME, Pereira MR, Zheng EX, et al. Acute liver injury in COVID‐19: prevalence and association with clinical outcomes in a large U.S cohort. Hepatology 2020;72:807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hundt MA, Deng Y, Ciarleglio MM, Nathanson MH, Lim JK. Abnormal liver tests in COVID‐19: a retrospective observational cohort study of 1,827 patients in a major U.S. hospital network. Hepatology 2020;72:1169‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marjot T, Webb GJ, Barritt AS, Moon AM, Stamataki Z, Wong VW, et al. COVID‐19 and liver disease: mechanistic and clinical perspectives. Nat Rev Gastroenterol Hepatol 2021;18:348‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Furuta K, Guo Q, Pavelko KD, Lee J‐H, Robertson KD, Nakao Y, et al. Lipid‐induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest 2021;131:e143690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641‐655. [DOI] [PubMed] [Google Scholar]
- 7. Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, et al. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology 2019;157:193‐209 e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu PP, Blet A, Smyth D, Li H. The science underlying COVID‐19: implications for the cardiovascular system. Circulation 2020;142:68‐78. [DOI] [PubMed] [Google Scholar]
- 9. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID‐19. Nature 2020;584:463‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, et al. Venous and arterial thromboembolic complications in COVID‐19 patients admitted to an academic hospital in Milan, Italy. Thromb Res 2020;191:9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Helms J, Tacquard C, Severac F, Leonard‐Lorant I, Ohana M, Delabranche X, et al.; CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis) . High risk of thrombosis in patients with severe SARS‐CoV‐2 infection: a multicenter prospective cohort study. Intensive Care Med 2020;46:1089‐1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. La Mura V, Artoni A, Martinelli I, Rossio R, Gualtierotti R, Ghigliazza G, et al. Acute portal vein thrombosis in SARS‐CoV‐2 infection: a case report. Am J Gastroenterol 2020;115:1140‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, Berger JS. Thrombosis in hospitalized patients with COVID‐19 in a New York City health system. JAMA 2020;324:799‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054‐1062. Erratum in: Lancet 2020;395:1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liao D, Zhou F, Luo L, Xu M, Wang H, Xia J, et al. Haematological characteristics and risk factors in the classification and prognosis evaluation of COVID‐19: a retrospective cohort study. Lancet Haematol 2020;7:e671‐e678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goshua G, Pine AB, Meizlish ML, Chang C‐H, Zhang H, Bahel P, et al. Endotheliopathy in COVID‐19‐associated coagulopathy: evidence from a single‐centre, cross‐sectional study. Lancet Haematol 2020;7:e575‐e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. López‐Aguirre Y, Páramo JA. Endothelial cell and hemostatic activation in relation to cytokines in patients with sepsis. Thromb Res 1999;94:95‐101. [DOI] [PubMed] [Google Scholar]
- 18. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation 2007;115:1285‐1295. [DOI] [PubMed] [Google Scholar]
- 19. Violi F, Basili S, Raparelli V, Chowdary P, Gatt A, Burroughs AK. Patients with liver cirrhosis suffer from primary haemostatic defects? Fact or fiction? J Hepatol 2011;55:1415‐1427. [DOI] [PubMed] [Google Scholar]
- 20. McConnell M, Iwakiri Y. Biology of portal hypertension. Hepatol Int 2018;12(Suppl. 1):11‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Page EM, Ariens RAS. Mechanisms of thrombosis and cardiovascular complications in COVID‐19. Thromb Res 2021;200:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luyendyk JP, Schoenecker JG, Flick MJ. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood 2019;133:511‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007;7:803‐815. [DOI] [PubMed] [Google Scholar]
- 24. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 2020;181:281‐292.e286. Erratum in: Cell 2020;183:1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271‐280 e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qi F, Qian S, Zhang S, Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun 2020;526:135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single‐cell RNA‐seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019‐nCoV infection. Front Med 2020;14:185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1‐9. Circ Res 2000;87:E1‐E9. [DOI] [PubMed] [Google Scholar]
- 29. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe A, et al. COVID‐19 tissue atlases reveal SARS‐CoV‐2 pathology and cellular targets. Nature 2021;595:107‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang K, Gheblawi M, Oudit GY. Angiotensin converting enzyme 2: a double‐edged sword. Circulation 2020;142:426‐428. [DOI] [PubMed] [Google Scholar]
- 31. Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin‐angiotensin‐aldosterone system in COVID‐19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta 2020;507:167‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension 2018;71:804‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riethmueller S, Somasundaram P, Ehlers JC, Hung C‐W, Flynn CM, Lokau J, et al. Proteolytic origin of the soluble human IL‐6R in vivo and a decisive role of N‐glycosylation. PLoS Biol 2017;15:e2000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor‐1 in cultured endothelial cells. A potential link between the renin‐angiotensin system and thrombosis. J Clin Invest 1995;95:995‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura S, Nakamura I, Ma L, Vaughan DE, Fogo AB. Plasminogen activator inhibitor‐1 expression is regulated by the angiotensin type 1 receptor in vivo. Kidney Int 2000;58:251‐259. [DOI] [PubMed] [Google Scholar]
- 36. Ogawa S, Gerlach H, Esposito C, Pasagian‐Macaulay A, Brett J, Stern D. Hypoxia modulates the barrier and coagulant function of cultured bovine endothelium. Increased monolayer permeability and induction of procoagulant properties. J Clin Invest 1990;85:1090‐1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res 2019;181:77‐83. [DOI] [PubMed] [Google Scholar]
- 38. Cui XY, Skretting G, Tinholt M, Stavik B, Dahm AEA, Sahlberg KK, et al. A novel hypoxia response element regulates oxygen‐related repression of tissue factor pathway inhibitor in the breast cancer cell line MCF‐7. Thromb Res 2017;157:111‐116. [DOI] [PubMed] [Google Scholar]
- 39. Fang H‐Y, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia‐inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009;114:844‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia‐induced neutrophil survival is mediated by HIF‐1alpha‐dependent NF‐kappaB activity. J Exp Med 2005;201:105‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia‐inducible factor mediates hypoxic and tumor necrosis factor alpha‐induced increases in tumor necrosis factor‐alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem 2007;282:33714‐33724. [DOI] [PubMed] [Google Scholar]
- 42. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 2014;41:518‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grobler C, Maphumulo SC, Grobbelaar LM, Bredenkamp JC, Laubscher GJ, Lourens PJ, et al. Covid‐19: the rollercoaster of fibrin(ogen), D‐dimer, von Willebrand factor, P‐selectin and their interactions with endothelial cells, platelets and erythrocytes. Int J Mol Sci 2020;21:5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D. Fibrinogen induces endothelial cell permeability. Mol Cell Biochem 2008;307:13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suehiro K, Gailit J, Plow EF. Fibrinogen is a ligand for integrin alpha5beta1 on endothelial cells. J Biol Chem 1997;272:5360‐5366. [DOI] [PubMed] [Google Scholar]
- 46. Lawson C, Wolf S. ICAM‐1 signaling in endothelial cells. Pharmacol Rep 2009;61:22‐32. [DOI] [PubMed] [Google Scholar]
- 47. Rubel C, Fernández GC, Rosa FA, Gómez S, Bompadre MB, Coso OA, et al. Soluble fibrinogen modulates neutrophil functionality through the activation of an extracellular signal‐regulated kinase‐dependent pathway. J Immunol 2002;168:3527‐3535. [DOI] [PubMed] [Google Scholar]
- 48. Kanoore Edul VS, Caminos Eguillor JF, Ferrara G, Estenssoro E, Siles DSP, Cesio CE, et al. Microcirculation alterations in severe COVID‐19 pneumonia. J Crit Care 2021;61:73‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rovas A, Osiaevi I, Buscher K, Sackarnd J, Tepasse P‐R, Fobker M, et al. Microvascular dysfunction in COVID‐19: the MYSTIC study. Angiogenesis 2021;24:145‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hutchings SD, Watchorn J, Trovato F, Napoli S, Mujib SF, Hopkins P, et al. Microcirculatory, endothelial, and inflammatory responses in critically ill patients with COVID‐19 are distinct from those seen in septic shock: a case control study. Shock 2021;55:752‐758. [DOI] [PubMed] [Google Scholar]
- 51. Idilman IS, Telli Dizman G, Ardali Duzgun S, Irmak I, Karcaaltincaba M, Inkaya AC, et al. Lung and kidney perfusion deficits diagnosed by dual‐energy computed tomography in patients with COVID‐19‐related systemic microangiopathy. Eur Radiol 2021;31:1090‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gu SX, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID‐19 thromboinflammation. Nat Rev Cardiol 2021;18:194‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Malas MB, Naazie IN, Elsayed N, Mathlouthi A, Marmor R, Clary B. Thromboembolism risk of COVID‐19 is high and associated with a higher risk of mortality: a systematic review and meta‐analysis. EClinicalMedicine 2020;29:100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rapkiewicz AV, Mai X, Carsons SE, Pittaluga S, Kleiner DE, Berger JS, et al. Megakaryocytes and platelet‐fibrin thrombi characterize multi‐organ thrombosis at autopsy in COVID‐19: a case series. EClinicalMedicine 2020;24:100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao CL, Rapkiewicz A, Maghsoodi‐Deerwester M, Gupta M, Cao W, Palaia T, et al. Pathological findings in the postmortem liver of patients with coronavirus disease 2019 (COVID‐19). Hum Pathol 2021;109:59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sonzogni A, Previtali G, Seghezzi M, Grazia Alessio M, Gianatti A, Licini L, et al. Liver histopathology in severe COVID 19 respiratory failure is suggestive of vascular alterations. Liver Int 2020;40:2110‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aid M, Busman‐Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, et al. Vascular disease and thrombosis in SARS‐CoV‐2‐infected rhesus macaques. Cell 2020;183:1354‐1366.e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rampotas A, Pavord S. Platelet aggregates, a marker of severe COVID‐19 disease. J Clin Pathol 2020. 10.1136/jclinpath-2020-206933. [DOI] [PubMed] [Google Scholar]
- 59. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID‐19. Blood 2020;136:1317‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Denorme F, Manne BK, Portier I, Petrey AC, Middleton EA, Kile BT, et al. COVID‐19 patients exhibit reduced procoagulant platelet responses. J Thromb Haemost 2020;18:3067‐3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kornblith LZ, Robles AJ, Conroy AS, Hendrickson CM, Calfee CS, Fields AT, et al. Perhaps it's not the platelet: ristocetin uncovers the potential role of von Willebrand factor in impaired platelet aggregation following traumatic brain injury. J Trauma Acute Care Surg 2018;85:873‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ruberto F, Chistolini A, Curreli M, Frati G, Marullo AGM, Biondi‐Zoccai G, et al.; COVID‐19 Group . Von Willebrand factor with increased binding capacity is associated with reduced platelet aggregation but enhanced agglutination in COVID‐19 patients: another COVID‐19 paradox? J Thromb Thrombolysis 2021;52:105‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Heinz C, Miesbach W, Herrmann E, Sonntagbauer M, Raimann FJ, Zacharowski K, et al. Greater Fibrinolysis Resistance but No Greater Platelet Aggregation in Critically Ill COVID‐19 Patients. Anesthesiology 2021;134:457‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Taus F, Salvagno G, Canè S, Fava C, Mazzaferri F, Carrara E, et al. Platelets promote thromboinflammation in SARS‐CoV‐2 pneumonia. Arterioscler Thromb Vasc Biol 2020;40:2975‐2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Petito E, Falcinelli E, Paliani U, Cesari E, Vaudo G, Sebastiano M, et al.; COVIR study investigators . Association of neutrophil activation, more than platelet activation, with thrombotic complications in coronavirus disease 2019. J Infect Dis 2021;223:933‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun 2019;10:1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oleksowicz L, Puszkin E, Mrowiec Z, Isaacs R, Dutcher JP. Alterations in platelet function in patients receiving interleukin‐6 as cytokine therapy. Cancer Invest 1996;14:307‐316. [DOI] [PubMed] [Google Scholar]
- 68. Oleksowicz L, Mrowiec Z, Isaacs R, Dutcher JP, Puszkin E. Morphologic and ultrastructural evidence of interleukin‐6 induced platelet activation. Am J Hematol 1995;48:92‐99. [DOI] [PubMed] [Google Scholar]
- 69. Ueba T, Nomura S, Inami N, Nishikawa T, Kajiwara M, Iwata R, et al. Correlation and association of plasma interleukin‐6 and plasma platelet‐derived microparticles, markers of activated platelets, in healthy individuals. Thromb Res 2010;125:e329‐e334. [DOI] [PubMed] [Google Scholar]
- 70. Marta RF, Goette NP, Lev PR, Chazarreta CD, Pirola CJ, Molinas FC. Normal platelets possess the soluble form of IL‐6 receptor. Cytokine 2005;29:13‐17. [DOI] [PubMed] [Google Scholar]
- 71. Senchenkova EY, Komoto S, Russell J, Almeida‐Paula LD, Yan L‐S, Zhang S, et al. Interleukin‐6 mediates the platelet abnormalities and thrombogenesis associated with experimental colitis. Am J Pathol 2013;183:173‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, et al. Platelets can associate with SARS‐Cov‐2 RNA and are hyperactivated in COVID‐19. Circ Res 2020;127:1404‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang SI, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS‐CoV‐2 binds platelet ACE2 to enhance thrombosis in COVID‐19. J Hematol Oncol 2020;13:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sahai A, Bhandari R, Koupenova M, Freedman J, Godwin M, McIntyre T, et al. SARS‐CoV‐2 receptors are expressed on human platelets and the effect of aspirin on clinical outcomes in COVID‐19 patients. Res Sq 2020; 10.21203/rs.3.rs-119031/v1. [DOI] [Google Scholar]
- 75. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497‐506. Erratum in: Lancet 2020;395:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al.; HLH Across Speciality Collaboration, UK . COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 2020;395:1033‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Del Valle DM, Kim‐Schulze S, Huang H‐H, Beckmann ND, Nirenberg S, Wang BO, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med 2020;26:1636‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fehr AR, Channappanavar R, Perlman S. Middle East respiratory syndrome: emergence of a pathogenic human coronavirus. Annu Rev Med 2017;68:387‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Effenberger M, Grander C, Grabherr F, Griesmacher A, Ploner T, Hartig F, et al. Systemic inflammation as fuel for acute liver injury in COVID‐19. Dig Liver Dis 2021;53:158‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Laing AG, Lorenc A, del Molino del Barrio I, Das A, Fish M, Monin L, et al. A dynamic COVID‐19 immune signature includes associations with poor prognosis. Nat Med 2020;26:1623‐1635. Erratum in: Nat Med 2020;26:1663; Nat Med 2020;26:1951. [DOI] [PubMed] [Google Scholar]
- 81. Wilson JG, Simpson LJ, Ferreira AM, Rustagi A, Roque J, Asuni A, et al. Cytokine profile in plasma of severe COVID‐19 does not differ from ARDS and sepsis. JCI Insight 2020;5:e140289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med 2020;383:2255‐2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Klok FA, Kruip M, van der Meer N, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID‐19: an updated analysis. Thromb Res 2020;191:148‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Garrido I, Liberal R, Macedo G. Review article: COVID‐19 and liver disease‐what we know on 1st May 2020. Aliment Pharmacol Ther 2020;52:267‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chew M, Tang Z, Radcliffe C, Caruana D, Doilicho N, Ciarleglio MM, et al. Significant liver injury during hospitalization for COVID‐19 is not associated with liver insufficiency or death. Clin Gastroenterol Hepatol 2021;19:2182‐2191.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lagana SM, Kudose S, Iuga AC, Lee MJ, Fazlollahi L, Remotti HE, et al. Hepatic pathology in patients dying of COVID‐19: a series of 40 cases including clinical, histologic, and virologic data. Mod Pathol 2020;33:2147‐2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kopec AK, Abrahams SR, Thornton S, Palumbo JS, Mullins ES, Divanovic S, et al. Thrombin promotes diet‐induced obesity through fibrin‐driven inflammation. J Clin Invest 2017;127:3152‐3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Targher G, Bertolini L, Scala L, Zenari L, Lippi G, Franchini M, et al. Plasma PAI‐1 levels are increased in patients with nonalcoholic steatohepatitis. Diabetes Care 2007;30:e31‐e32. [DOI] [PubMed] [Google Scholar]
- 89. Chang M‐L, Hsu C‐M, Tseng J‐H, Tsou Y‐K, Chen S‐C, Shiau S‐S, et al. Plasminogen activator inhibitor‐1 is independently associated with non‐alcoholic fatty liver disease whereas leptin and adiponectin vary between genders. J Gastroenterol Hepatol 2015;30:329‐336. [DOI] [PubMed] [Google Scholar]
- 90. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL‐6 trans‐signaling induces plasminogen activator inhibitor‐1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A 2020;117:22351‐22356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Matsuyama T, Kubli SP, Yoshinaga SK, Pfeffer K, Mak TW. An aberrant STAT pathway is central to COVID‐19. Cell Death Differ 2020;27:3209‐3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Garbers C, Heink S, Korn T, Rose‐John S. Interleukin‐6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov 2018;17:395‐412. [DOI] [PubMed] [Google Scholar]
- 93. Rose‐John S. IL‐6 trans‐signaling via the soluble IL‐6 receptor: importance for the pro‐inflammatory activities of IL‐6. Int J Biol Sci 2012;8:1237‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. McConnell MJ, Kawaguchi N, Kondo R, Sonzogni A, Licini L, Valle C, et al. Liver injury in COVID‐19 and IL‐6 trans‐signaling‐induced endotheliopathy. J Hepatol 2021;75:647‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kaur S, Hussain S, Kolhe K, Kumar G, Tripathi DM, Tomar A, et al. Elevated plasma ICAM1 levels predict 28‐day mortality in cirrhotic patients with COVID‐19 or bacterial sepsis. JHEP Rep 2021;3:100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hong F, Kim W‐H, Tian Z, Jaruga B, Ishac E, Shen X, et al. Elevated interleukin‐6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol‐induced apoptosis in the liver: involvement of induction of Bcl‐2 and Bcl‐x(L) proteins. Oncogene 2002;21:32‐43. [DOI] [PubMed] [Google Scholar]
- 97. Deviere J, Content J, Denys C, Vandenbussche P, Schandene L, Wybran J, et al. High interleukin‐6 serum levels and increased production by leucocytes in alcoholic liver cirrhosis. Correlation with IgA serum levels and lymphokines production. Clin Exp Immunol 1989;77:221‐225. [PMC free article] [PubMed] [Google Scholar]
- 98. Schmidt‐Arras D, Rose‐John S. IL‐6 pathway in the liver: from physiopathology to therapy. J Hepatol 2016;64:1403‐1415. [DOI] [PubMed] [Google Scholar]
- 99. Hou X, Yin S, Ren R, Liu S, Yong L, Liu Y, et al. Myeloid‐cell‐specific IL‐6 signaling promotes microRNA‐223‐enriched exosome production to attenuate NAFLD‐associated fibrosis. Hepatology 2021;74:116‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Clària J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al.; CANONIC Study Investigators of the EASL‐CLIF Consortium and the European Foundation for the Study of Chronic Liver Failure (EF‐CLIF) . Systemic inflammation in decompensated cirrhosis: characterization and role in acute‐on‐chronic liver failure. Hepatology 2016;64:1249‐1264. [DOI] [PubMed] [Google Scholar]
- 101. Fischer J, Silva TE, Soares e Silva PE, Colombo BS, Silva MC, Wildner LM, et al. From stable disease to acute‐on‐chronic liver failure: circulating cytokines are related to prognosis in different stages of cirrhosis. Cytokine 2017;91:162‐169. [DOI] [PubMed] [Google Scholar]
- 102. Stebbing J, Sánchez Nievas G, Falcone M, Youhanna S, Richardson P, Ottaviani S, et al. JAK inhibition reduces SARS‐CoV‐2 liver infectivity and modulates inflammatory responses to reduce morbidity and mortality. Sci Adv 2021;7:eabe4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Bütikofer S, Lenggenhager D, Wendel Garcia PD, Maggio EM, Haberecker M, Reiner CS, et al. Secondary sclerosing cholangitis as cause of persistent jaundice in patients with severe COVID‐19. Liver Int 2021;41:2404‐2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Da BL, Suchman K, Roth N, Rizvi A, Vincent M, Trindade AJ, et al.; Northwell COVID‐19 Research Consortium . Cholestatic liver injury in COVID‐19 is a rare and distinct entity and is associated with increased mortality. J Intern Med 2021;290:470‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Roth NC, Kim A, Vitkovski T, Xia J, Ramirez G, Bernstein D, et al. Post‐COVID‐19 cholangiopathy: a novel entity. Am J Gastroenterol 2021;116:1077‐1082. [DOI] [PubMed] [Google Scholar]
- 106. Lee A, Wein AN, Doyle MBM, Chapman WC. Liver transplantation for post‐COVID‐19 sclerosing cholangitis. BMJ Case Rep 2021;14.e244168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kim WY, Kweon OJ, Cha MJ, Baek MS, Choi SH. Dexamethasone may improve severe COVID‐19 via ameliorating endothelial injury and inflammation: a preliminary pilot study. PLoS One 2021;16:e0254167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Awasthi S, Wagner T, Venkatakrishnan AJ, Puranik A, Hurchik M, Agarwal V, et al. Plasma IL‐6 levels following corticosteroid therapy as an indicator of ICU length of stay in critically ill COVID‐19 patients. Cell Death Discov 2021;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Di Nisio M, Potere N, Candeloro M, Spacone A, Pieramati L, Ferrandu G, et al. Interleukin‐6 receptor blockade with subcutaneous tocilizumab improves coagulation activity in patients with COVID‐19. Eur J Intern Med 2021;83:34‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al.; ACTT‐2 Study Group Members . Baricitinib plus remdesivir for hospitalized adults with Covid‐19. N Engl J Med 2021;384:795‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Marconi VC, Ramanan AV, de Bono S, Kartman CE, Krishnan V, Liao R, et al.; COV‐BARRIER Study Group . Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID‐19 (COV‐BARRIER): a randomised, double‐blind, parallel‐group, placebo‐controlled phase 3 trial. Lancet Respir Med 2021; 10.1016/S2213-2600(21)00331-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Takahashi T, Luzum JA, Nicol MR, Jacobson PA. Pharmacogenomics of COVID‐19 therapies. NPJ Genom Med 2020;5:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Aleem A, Mahadevaiah G, Shariff N, Kothadia JP. Hepatic manifestations of COVID‐19 and effect of remdesivir on liver function in patients with COVID‐19 illness. Proc (Bayl Univ Med Cent) 2021;34:473‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kim MS, Jung SY, Lee SW, Li H, Koyanagi AI, Kronbichler A, et al. Hepatobiliary adverse drug reactions associated with remdesivir: the WHO International Pharmacovigilance Study. Clin Gastroenterol Hepatol 2021;19:1970‐1972.e1973. [DOI] [PubMed] [Google Scholar]
- 115. Marjot T, Moon AM, Cook JA, Abd‐Elsalam S, Aloman C, Armstrong MJ, et al. Outcomes following SARS‐CoV‐2 infection in patients with chronic liver disease: an international registry study. J Hepatol 2021;74:567‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sarin SK, Choudhury A, Lau GK, Zheng M‐H, Ji D, Abd‐Elsalam S, et al.; APASL COVID Task Force, APASL COVID Liver Injury Spectrum Study (APCOLIS Study‐NCT 04345640) . Pre‐existing liver disease is associated with poor outcome in patients with SARS CoV2 infection; the APCOLIS Study (APASL COVID‐19 Liver Injury Spectrum Study). Hepatol Int 2020;14:690‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cheung CCL, Goh D, Lim X, Tien TZ, Lim JCT, Lee JN, et al. Residual SARS‐CoV‐2 viral antigens detected in GI and hepatic tissues from five recovered patients with COVID‐19. Gut 2021; 10.1136/gutjnl-2021-324280. [DOI] [PubMed] [Google Scholar]
- 118. Zou J‐N, Sun L, Wang B‐R, Zou Y, Xu S, Ding Y‐J, et al. The characteristics and evolution of pulmonary fibrosis in COVID‐19 patients as assessed by AI‐assisted chest HRCT. PLoS One 2021;16:e0248957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shea BS, Probst CK, Brazee PL, Rotile NJ, Blasi F, Weinreb PH, et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2017;2:e86608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nie X, Qian L, Sun R, Huang BO, Dong X, Xiao QI, et al. Multi‐organ proteomic landscape of COVID‐19 autopsies. Cell 2021;184:775‐791.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Franco‐Moreno A, Piniella‐Ruiz E, Montoya‐Adarraga J, Ballano‐Franco C, Alvarez‐Miguel F, Peinado‐Martinez C, et al. Portal vein thrombosis in a patient with COVID‐19. Thromb Res 2020;194:150‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Daugherty SE, Guo Y, Heath K, Dasmariñas MC, Jubilo KG, Samranvedhya J, et al. Risk of clinical sequelae after the acute phase of SARS‐CoV‐2 infection: retrospective cohort study. BMJ 2021;373:n1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dennis A, Wamil M, Alberts J, Oben J, Cuthbertson DJ, Wootton D, et al.; COVERSCAN study investigators . Multiorgan impairment in low‐risk individuals with post‐COVID‐19 syndrome: a prospective, community‐based study. BMJ Open 2021;11:e048391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bende F, Tudoran C, Sporea I, Fofiu R, Bâldea V, Cotrău R, et al. A multidisciplinary approach to evaluate the presence of hepatic and cardiac abnormalities in patients with post‐acute COVID‐19 syndrome‐a pilot study. J Clin Med 2021;10:2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Holmes E, Wist J, Masuda R, Lodge S, Nitschke P, Kimhofer T, et al. Incomplete systemic recovery and metabolic phenoreversion in post‐acute‐phase nonhospitalized COVID‐19 patients: implications for assessment of post‐acute COVID‐19 syndrome. J Proteome Res 2021;20:3315‐3329. [DOI] [PubMed] [Google Scholar]
- 126. Fogarty H, Townsend L, Morrin H, Ahmad A, Comerford C, Karampini E, et al.; Irish COVID‐19 Vasculopathy Study (iCVS) investigators . Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J Thromb Haemost 2021;19:2546‐2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lu JY, Anand H, Frager SZ, Hou W, Duong TQ. Longitudinal progression of clinical variables associated with graded liver injury in COVID‐19 patients. Hepatol Int 2021;15:1018‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wang M, Yan W, Qi W, Wu D, Zhu L, Li W, et al. Clinical characteristics and risk factors of liver injury in COVID‐19: a retrospective cohort study from Wuhan, China. Hepatol Int 2020;14:723‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tsutsumi T, Saito M, Nagai H, Yamamoto S, Ikeuchi K, Lim LA, et al. Association of coagulopathy with liver dysfunction in patients with COVID‐19. Hepatol Res 2021;51:227‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Del Hoyo J, López‐Muñoz P, Fernández‐de la Varga M, Garrido‐Marín A, Valero‐Pérez E, Prieto M, et al. Hepatobiliary and pancreatic: a fatal case of extensive splanchnic vein thrombosis in a patient with Covid‐19. J Gastroenterol Hepatol 2020;35:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Abeysekera KW, Karteszi H, Clark A, Gordon FH. Spontaneous portomesenteric thrombosis in a non‐cirrhotic patient with SARS‐CoV‐2 infection. BMJ Case Rep 2020;13:e238906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Borazjani R, Seraj SR, Fallahi MJ, Rahmanian Z. Acute portal vein thrombosis secondary to COVID‐19: a case report. BMC Gastroenterol 2020;20:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Randhawa J, Kaur J, Randhawa HS, Kaur S, Singh H. Thrombosis of the portal vein and superior mesenteric vein in a patient with subclinical COVID‐19 infection. Cureus 2021;13:e14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sh Hassan AA, Alsaleh ME, Alsaleh ME, Al Zaher FA, Almajed FA, Alkhudhair AM, et al. Budd‐Chiari syndrome: a case report of a rare presentation of COVID‐19. Cureus 2021;13:e12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kolli S, Oza VM. SARS‐CoV‐2 and portal vein thrombosis: a rare gastrointestinal manifestation of COVID‐19. Cureus 2021;13:e14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jeilani M, Hill R, Riad M, Abdulaal Y. Superior mesenteric vein and portal vein thrombosis in a patient with COVID‐19: a rare case. BMJ Case Rep 2021;14:e244049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bugra A, Das T, Arslan MN, Ziyade N, Buyuk Y. Postmortem pathological changes in extrapulmonary organs in SARS‐CoV‐2 rt‐PCR‐positive cases: a single‐center experience. Ir J Med Sci 2021;May 7:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kondo R, Kawaguchi N, McConnell MJ, Sonzogni A, Licini L, Valle C, et al. Pathological characteristics of liver sinusoidal thrombosis in COVID‐19 patients: a series of 43 cases. Hepatol Res 2021;51:1000‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Fassan M, Mescoli C, Sbaraglia M, Guzzardo V, Russo FP, Fabris R, et al. Liver histopathology in COVID‐19 patients: a mono‐institutional series of liver biopsies and autopsy specimens. Pathol Res Pract 2021;221:153451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID‐19: a prospective autopsy cohort study. Lancet Microbe 2020;1:e290‐e299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, et al. Pulmonary arterial thrombosis in COVID‐19 with fatal outcome: results from a prospective, single‐center, clinicopathologic case series. Ann Intern Med 2020;173:350‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Fiel MI, El Jamal SM, Paniz‐Mondolfi A, Gordon RE, Reidy J, Bandovic J, et al. Findings of severe hepatic severe acute respiratory syndrome coronavirus‐2 infection. Cell Mol Gastroenterol Hepatol 2021;11:763‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Patra T, Mayer K, Geerling L, Isbell TS, Hoft DF, Brien J, Pinto AK. SARS‐CoV‐2 spike protein promotes IL‐6 trans‐signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathogens 2020;16:e1009128. [DOI] [PMC free article] [PubMed] [Google Scholar]