

Abstract
Liver test abnormalities are frequently observed in patients with coronavirus disease 2019 (COVID‐19) and are associated with worse prognosis. However, information is limited about pathological changes in the liver in this infection, so the mechanism of liver injury is unclear. Here we describe liver histopathology and clinical correlates of 27 patients who died of COVID‐19 in Manaus, Brazil. There was a high prevalence of liver injury (elevated alanine aminotransferase and aspartate aminotransferase in 44% and 48% of patients, respectively) in these patients. Histological analysis showed sinusoidal congestion and ischemic necrosis in more than 85% of the cases, but these appeared to be secondary to systemic rather than intrahepatic thrombotic events, as only 14% and 22% of samples were positive for CD61 (marker of platelet activation) and C4d (activated complement factor), respectively. Furthermore, the extent of these vascular findings did not correlate with the extent of transaminase elevations. Steatosis was present in 63% of patients, and portal inflammation was present in 52%. In most cases, hepatocytes expressed angiotensin‐converting enzyme 2 (ACE2), which is responsible for binding and entry of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), even though this ectoenzyme was minimally expressed on hepatocytes in normal controls. However, SARS‐CoV‐2 staining was not observed. Most hepatocytes also expressed inositol 1,4,5‐triphosphate receptor 3 (ITPR3), a calcium channel that becomes expressed in acute liver injury. Conclusion: The hepatocellular injury that commonly occurs in patients with severe COVID‐19 is not due to the vascular events that contribute to pulmonary or cardiac damage. However, new expression of ACE2 and ITPR3 with concomitant inflammation and steatosis suggests that liver injury may result from inflammation, metabolic abnormalities, and perhaps direct viral injury.
New expression of ACE2 and ITPR3 in hepatocytes with concomitant inflammation and steatosis suggests that the frequently observed liver injury in patients with COVID‐19 may result from inflammation, metabolic abnormalities, and perhaps direct viral entry.
![]()
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BMI
body mass index
- CD61
integrin subunit beta 3
- C4d
complement C4d
- C5d
complement C5d
- COVID‐19
coronavirus disease 2019
- DM
diabetes mellitus
- ITPR3
inositol 1,4,5‐triphosphate receptor 3
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
As of April 2021, there have been nearly 150 million confirmed cases of coronavirus disease 2019 (COVID‐19) worldwide, with over 3 million deaths.( 1 ) The United States and Brazil rank first and third in total number of infected individuals, with over 46 million combined cases. These two countries also account for nearly 1 million of the deaths associated with COVID‐19. Although severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the virus responsible for COVID‐19, is primarily known for its respiratory complications, extra‐pulmonary effects have been well described.( 2 ) A number of studies have found that patients with COVID‐19 have abnormal liver tests, most commonly serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) elevations, with prevalence ranging from 14.9% to 76.3% among different cohorts( 3 , 4 , 5 , 6 , 7 ) Abnormal liver tests are also associated with poorer outcomes in patients hospitalized with COVID‐19 symptoms.( 4 , 7 ) Several mechanisms of liver injury have been proposed, including inflammation related to cytokine storm, hepatic ischemia, direct viral injury, and drug‐induced liver injury, but the etiology remains unclear.( 8 )
A number of studies have examined the histopathological effects of COVID‐19 on the respiratory tract, but few have specifically examined the impact on the liver,( 9 , 10 ) in part because of limited tissue availability. Much of the existing literature is based on reports of small case series,( 11 , 12 ) although a few larger case series have been published as well.( 13 , 14 , 15 ) In those reports, histological findings included vascular damage, necrotic cell death, steatosis, and portal inflammation. Here, we report the histological findings of postmortem liver specimens for 27 patients who died of COVID‐19 in Manaus, Brazil. To gain insight about the basis for the pathological findings, these have been correlated with liver tests. We also examined immunostaining for SARS‐CoV‐2, angiotensin converting enzyme 2 (ACE2), which is thought to be the point of entry of the virus into cells,( 16 ) and the type 3 inositol trisphosphate receptor (ITPR3), which increases in acute hepatocellular injury and may relate to the nature of the response to injury.( 17 , 18 )
Methods
Study Design and Participants
Following internal review board approval (NCT04323527), liver specimens were obtained postmortem from 27 patients at a single center in Manaus, Brazil, who were hospitalized between March 23 and May 18, 2020, after testing positive for SARs‐CoV‐2 through quantitative real‐time polymerase chain reaction (PCR). Liver specimens of approximately 1 cm3 were obtained from multiple sites during necropsy and fixed in neutral buffered formalin and embedded in paraffin. Initial histopathological analysis was performed on hematoxylin and eosin or Masson’s trichrome–stained slices. Activated coagulation and platelet aggregation were evaluated by immunohistochemistry for complement C4 (C4d), complement C5 (C5d), and integrin beta 3 (CD61). Presence of viral particles in liver tissue was assessed by immunohistochemistry for SARS‐CoV‐2 spike protein as well as expression of ACE2, the putative receptor for SARS‐CoV‐2.( 16 ) Slides were reviewed by two liver pathologists and scored according to the degree of steatosis, the presence of congestion, inflammation (portal or lobular), portal vein dilatation, or fibrosis using a scale from 0 (absent) to 3 (maximum). Ischemic necrosis was identified histologically as extensive centrilobular necrosis. Normal controls were liver‐tissue samples obtained from resections of patients with colon cancer.
Demographics and clinical data were obtained from chart review and included age at admission, sex, body mass index (BMI), oxygen saturation at hospital admission, diabetes status, use of mechanical ventilation, prior and current medication history, serum tests of liver function (ALT, AST, and total bilirubin), hemoglobin, and white blood cell (WBC) count with differential, platelets, and creatinine.
Statistical Methods
Summary statistics were calculated for pathological findings and clinical laboratory findings. Mean, SD, median, and range were reported for quantitative variables; counts and percentages were reported for categorical variables. Spearman’s rank correlation test was used to evaluate correlations between pathological variables and clinical/lab findings. Pathological variables (original scale 0‐3) were dichotomized as 0 versus 1+, and associations with clinical variables were explored using Student t test and Wilcoxon rank‐sum test.
Results
Demographics and clinical characteristics, including laboratory findings of all patients, are summarized in Tables 1 and 2, respectively. Of the 27 patients, the mean age was 59 years, 7 (25.9%) were female, the mean BMI was 27.3 (range 21.5‐37.7), and 22.2% had diabetes mellitus (DM). The mean pulse oximetry was 94% (range 69‐100) following admission, and all patients required supplemental oxygen within the first day of hospitalization. Of those, 87% received mechanical ventilation. In terms of medications administered starting at day 1 of hospitalization, 48.4% and 19.3% of patients received vasopressors and ACE inhibitors, respectively. Most hospitalized individuals received antibiotics (65.4%) and antivirals (71%).
TABLE 1.
Clinical Characteristics of All Patients
| Characteristic | All Patients |
|---|---|
| n = 27 (100%) | |
| Baseline and demographics | |
| Age (years), mean (range) | 59 (20‐80) |
| Sex | 7 females |
| 20 males | |
| BMI, mean (range) | 27.3 (21.5‐37.7) |
| Comorbidities | |
| Obesity (%) | 7 (25.9) |
| DM (%) | 6 (22.2) |
| Symptoms and hospital course | |
| O2 saturation at admission | 94 (69‐100) |
| Supplemental O2 (%) | 27 (100) |
| Invasive mechanical ventilation | 23 (85.2) |
| Previous medications received | |
| ACE inhibitor | 6 (22.2) |
| Captopril | 5 (18.5) |
| Enalopril/hydrochlorothiazide | 1 (3.7) |
| Antiarrhythmic | |
| Amiodarone | 1 (3.7) |
| Antibiotics | |
| Amoxicillin | 2 (7.4) |
| Azithromycin | 15 (55.5) |
| Ceftriaxone | 20 (74.1) |
| Cephalexin | 1 (3.7) |
| Cephepime | 1 (3.7) |
| Clarithromycin | 6 (22.2) |
| Clindamycin | 1 (3.7) |
| Piperacillin/tazobactam | 1 (3.7) |
| Vancomycin | 1 (3.7) |
| Antiretrovirals | 0 (0) |
| Immunobiologics | 0 (0) |
| Bronchodilators (fenoterol, ipratropium, aminophylline) | 1 (3.7) |
| Ibuprofen | 1 (3.7) |
| Statin (simvastatin) | 1 (3.7) |
| Steroids (hydrocortisone) | 1 (3.7) |
| Medications received during day 1 | |
| ACE inhibitor (captopril) | 2 (7.4) |
| Antibiotics | 27 (100) |
| Amoxicillin | 0 (0) |
| Azithromycin | 22 (81.5) |
| Ceftriaxone | 0 (0) |
| Cephalexin | 0 (0) |
| Cephepime | 0 (0) |
| Clarithromycin | 4 (14.8) |
| Clindamycin | 1 (3.7) |
| Piperacillin/tazobactam | 1 (3.7) |
| Vancomycin | 0 (0) |
| Anticoagulation | 18 (66.7) |
| Enoxaparin | 15 (55.5) |
| Heparin | 4 (14.8) |
| Antivirals (Tamiflu) | 22 (81.5) |
| Bronchodilators | 0 (0) |
| Insulin | 3 (11.1) |
| Steroids | 4 (14.8) |
| Hydrocortisone | 2 (7.4) |
| Prednisone | 1 (3.7) |
| Methylprednisone | 1 (3.7) |
| Vasopressors | 14 (51.8) |
| Dobutamine | 0 (0) |
| Dopamine | 0 (0) |
| Norepinephrine | 14 (51.8) |
| Laboratory values, mean (range)/number | |
| AST (units/L) | 92.2 (29.3‐343)/13 |
| ALT (IU/L) | 100.61 (22.1‐533.3)/15 |
| Total bilirubin (mg/dL) | 1.13 (0.15‐3.08)/14 |
| Hemoglobin (g/dL) | 11.0 (6.2‐14.5)/27 |
| Platelets (103/μL) | 217.8 (25‐437)/27 |
| Leukocytes (103/μL) | 12.4 (5.2‐32.0)/27 |
| Creatinine (mg/dL) | 3.7 (0.63‐16.3)/27 |
TABLE 2.
Histological Characteristics of All Patients
| Pathology | |
|---|---|
| Steatosis | 27 of 27 |
| Grade 0 | 10 of 27 |
| Grade 1 | 10 of 27 |
| Grade 2 | 6 of 27 |
| Grade 3 | 1 of 27 |
| Portal inflammation | 27 of 27 |
| Grade 0 | 13 of 27 |
| Grade 1 | 12 of 27 |
| Grade 2 | 2 of 27 |
| Lobular inflammation | 27 of 27 |
| Grade 0 | 25 of 27 |
| Grade 1 | 2 of 27 |
| Congestion | 27 of 27 |
| Grade 0 | 4 of 27 |
| Grade 1 | 12 of 27 |
| Grade 2 | 5 of 27 |
| Grade 3 | 6 of 27 |
| Ischemic necrosis | 27 of 27 |
| Grade 0 | 1 of 27 |
| Grade 1 | 9 of 27 |
| Grade 2 | 12 of 27 |
| Grade 3 | 5 of 27 |
| Fibrosis | 27 of 27 |
| Grade 0 | 18 of 27 |
| Grade 1 | 6 of 27 |
| Grade 2 | 3 of 27 |
Median liver test values were 59.1 (range 29.3‐343.0) for AST, 50.8 (range 22.1‐533.3) for ALT, and 1.13 (range 0.28‐3.08) for bilirubin. Median WBC was 11.07 (range 5.19‐31.95), and median platelet count was 180.5 (range 25.0‐437.0). Most patients (80.7%) experienced kidney injury with median creatinine of 2.58 mg/dL (range 0.64‐16.10) (Table 2).
Most liver specimens had evidence of congestion (n = 23; 85.2%) and ischemic necrosis (n = 26; 96.3%), whereas 17 (63%) had steatosis, of which 5 demonstrated moderate or severe steatosis (Fig. 1). Significant fibrosis was less common and was only observed in 8 (31%) of the specimens. Portal vein dilatation was seen in 19 (70%) patients. Fourteen patients (52%) had portal inflammation, whereas only 2 (7.4%) had lobular inflammation, suggesting that the COVID‐19 infection is associated with hepatic inflammation in over half of these patients (Fig. 1).
FIG. 1.
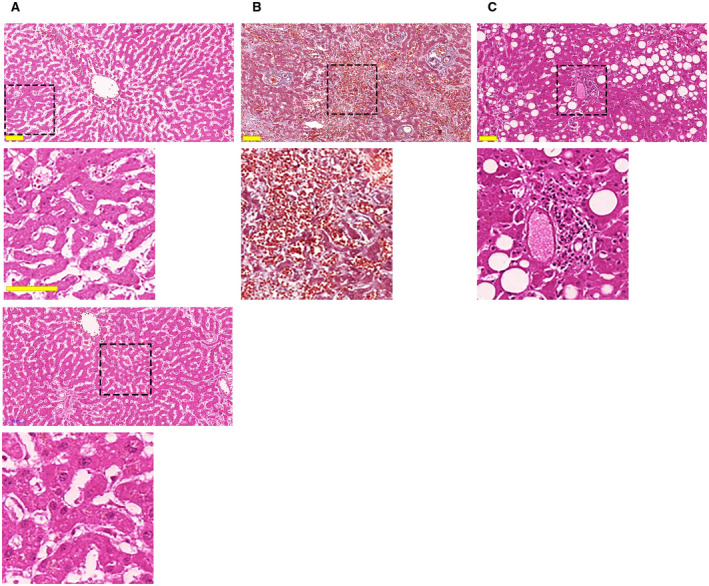
COVID‐19 infection is associated with ischemic necrosis, congestion, steatosis, and portal inflammation in most patients. (A) Representative hematoxylin and eosin staining of a liver section demonstrates areas of necrosis as evidenced by the uniform cytosolic staining and absence of nuclei. This pattern was observed in 26 of 27 patients. A magnified view of the dotted square area is shown in the inset below. The bottom panel and inset show an area not affected by necrosis, as demonstrated by the light eosin staining and presence of normal‐sized nuclei. (B) Masson’s trichrome staining shows diffuse blood retention and congestion in liver sinusoids (inset). This was observed in 23 of 27 patients. (C) Areas of macrovesicular steatosis, as highlighted by the typical round negative images, in the liver of a representative patient with COVID‐19. This was observed in 17 of 27 patients. Representative area of portal inflammation, as shown by the presence of inflammatory infiltrate surrounding portal tracts, were similarly observed (inset) in 14 of 27 patients. Scale bar, 100 μm.
Next, we investigated potential relationships between selected clinical and histological findings in this group of patients (Table 3). Neither ischemic necrosis, congestion, steatosis, nor inflammation were correlated with transaminase elevations, despite the fact that each of these histological findings were present in over half of the patients. Similarly, these histologic findings did not correlate with neutrophil count, lymphocyte count, or BMI, even though each of those clinical parameters has been related to outcomes in patients with COVID‐19.( 4 , 19 ) In fact, Spearman’s rank correlation analysis revealed that the only laboratory values significantly associated with histological findings (highlighted in blue in Table 3) were ALT (correlation = 0.77, P = 0.0008) and AST (correlation = 0.63, P = 0.0166), which were positively correlated with the presence of fibrosis, seen in only one‐third of specimens.
TABLE 3.
Spearman Correlation Analysis Between Laboratory and Histopathological Findings
| Steatosis (0‐3) | Portal II (0‐3) | Lobular II (0‐3) | Congestion (0‐3) | Ischemic Necrosis (0‐3) | Portal Vein Dilatation (0‐3) | Fibrosis (0‐3) | BMI | ALT | AST | Leukocyte Count | Lymphocyte Count | Neutrophil Count | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Steatosis (0‐3) | 1 | ||||||||||||
| Portal II (0‐3) | 0.53051 | 1 | |||||||||||
| 0.0044 | |||||||||||||
| Lobular II (0‐3) | 0.36597 | 0.36511 | 1 | ||||||||||
| 0.0605 | 0.0611 | ||||||||||||
| Congestion (0‐3) | 0.04095 | −0.07579 | −0.13451 | 1 | |||||||||
| 0.8393 | 0.7071 | 0.5036 | |||||||||||
| Ischemic necrosis (0‐3) | −0.01805 | 0.07824 | −0.10707 | −0.10131 | 1 | ||||||||
| 0.9288 | 0.6981 | 0.595 | 0.6151 | ||||||||||
| Portal vein dilatation (0‐3) | −0.04093 | −0.13179 | −0.00944 | 0.33943 | −0.21092 | 1 | |||||||
| 0.8394 | 0.5123 | 0.9627 | 0.0833 | 0.291 | |||||||||
| Fibrosis (0‐3) | 0.0626 | 0.26797 | −0.19003 | 0.27808 | 0.49679 | −0.08063 | 1 | ||||||
| 0.7613 | 0.1857 | 0.3525 | 0.169 | 0.0098 | 0.6954 | ||||||||
| BMI | −0.02899 | 0.15606 | −0.07265 | −0.10567 | 0.06596 | −0.29558 | −0.25665 | 1 | |||||
| 0.8859 | 0.437 | 0.7188 | 0.5999 | 0.7437 | 0.1344 | 0.2056 | |||||||
| ALT | 0.02307 | −0.11525 | −0.18558 | 0.22267 | 0.26971 | −0.02454 | 0.76765 | −0.06434 | 1 | ||||
| 0.9349 | 0.6825 | 0.5079 | 0.4251 | 0.331 | 0.9308 | 0.0008 | 0.8198 | ||||||
| AST | −0.03131 | 0.07957 | 0.24081 | 0.1439 | 0.09423 | 0.04886 | 0.62616 | −0.16502 | 0.78022 | 1 | |||
| 0.9154 | 0.7869 | 0.4069 | 0.6236 | 0.7486 | 0.8683 | 0.0166 | 0.5729 | 0.001 | |||||
| Leukocyte count | 0.06776 | −0.09099 | 0 | 0.25721 | −0.12488 | −0.14159 | 0.17336 | 0.13686 | 0.34066 | 0.2967 | 1 | ||
| 0.7422 | 0.6585 | 1 | 0.2046 | 0.5433 | 0.4902 | 0.4073 | 0.4708 | 0.2333 | 0.3249 | ||||
| Lymphocyte count | −0.09747 | 0.20797 | −0.17323 | −0.07458 | −0.18824 | 0.08213 | 0.07741 | 0.06443 | −0.11001 | −0.25034 | −0.55591 | 1 | |
| 0.6357 | 0.308 | 0.3974 | 0.7173 | 0.3571 | 0.69 | 0.713 | 0.7352 | 0.7081 | 0.4094 | 0.0014 | |||
| Neutrophil count | 0.08479 | −0.23741 | 0.36572 | 0.02619 | 0.13921 | 0.0707 | −0.17078 | −0.12229 | 0.03297 | 0.22527 | 0.48565 | −0.89941 | 1 |
| 0.6805 | 0.2429 | 0.0662 | 0.8989 | 0.4976 | 0.7314 | 0.4144 | 0.5197 | 0.9109 | 0.4593 | 0.0065 | <0.0001 | ||
| Creatinine | −0.10508 | 0.23337 | −0.05774 | −0.03779 | 0.13583 | 0.03396 | −0.15506 | 0.15272 | −0.0989 | 0.04945 | 0.01454 | 0.00813 | 0.04757 |
| 0.6095 | 0.2512 | 0.7793 | 0.8546 | 0.5082 | 0.8692 | 0.4592 | 0.4204 | 0.7366 | 0.8725 | 0.9403 | 0.9666 | 0.8064 |
Blue color denotes statistically significant correlations.
Because most of the specimens had histological evidence of congestion and portal vein dilatation, but not other vascular alterations such as sinusoidal edema, hemorrhage, or endothelial lifting/denudation, we next investigated whether COVID‐19 infection was associated with coagulation activation in the liver. Expression of C4d and C5d were assessed by immunohistochemistry as markers of activation of coagulation within the liver vasculature. CD61 expression was used as an indicator of local platelet activation. C4d staining, a marker of complement activation, was present in 6 (22%) samples (Fig. 2). A second marker of coagulation activation, C5d, was not detected in any of the specimens (not shown). Expression of both coagulation markers was present in positive control specimens from a kidney transplant rejection allograft (Fig. 2). Expression of CD61 was observed in only 4 (14.8%) of the liver‐tissue samples (Fig. 2). Together, these results demonstrate that SARS‐CoV‐2 infection does not routinely induce local activation of coagulation cascades and thrombotic events in the liver. This in turn suggests that the sinusoidal congestion observed in most of the samples is likely due to extrahepatic vascular events typical of serious SARS‐CoV‐2 infection.
FIG. 2.
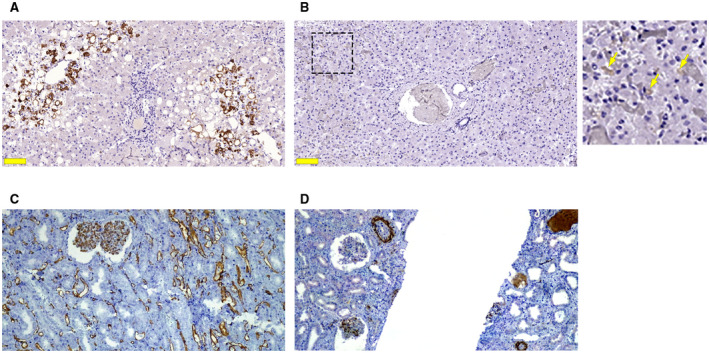
Expression of activated coagulation factors is uncommon in COVID‐19 liver specimens. (A) Positive C4d immunohistochemical staining (brown), a marker of activated complement cascade, was observed in 22% of the liver samples from patients with COVID‐19. (B) Positive staining for CD61 was detected in 14% of the 27 COVID‐19 liver samples. Light brown staining (arrows in the inset) is seen along the sinusoidal walls. Scale bar, 100 μm. Positive CD61 was present in control samples of kidney tissue (data not shown). (C) Positive control staining for C4d in kidney tissue from a transplant rejection allograft. (D) Cd5 positive staining was similarly found in kidney tissue from the same patient.
Evidence from both genomic analysis of viral particles and structural data suggests that the ectoenzyme ACE2 mediates internalization of SARS‐CoV‐2 into human cells.( 16 , 20 ) Thus, we analyzed the expression of ACE2 in liver specimens of patients with COVID‐19. A previous study suggested that ACE2 is present in cholangiocytes but absent in hepatocytes of healthy individuals.( 21 ) However, our results indicate that ACE2 was minimally expressed in both hepatocytes and cholangiocytes in control livers (Fig. 3). In contrast, ACE2 expression was significantly (P < 0.0001) increased in both of these epithelial cell types in our cohort of patients with COVID‐19 (Fig. 3). To determine whether SARS‐CoV‐2 actually enters hepatocytes or cholangiocytes, liver samples were stained with serum against SARS‐CoV‐2 spike protein, and a lung specimen of an infected patient was used as a positive control. Although the alveolar cells in the lung were labeled by this, none of the liver specimens demonstrate specific staining for the spike protein (Fig. 3). Thus, although ACE2 expression is increased in both hepatocytes and cholangiocytes in this cohort of patients with COVID‐19, the findings do not provide evidence for viral entry into either of these liver cell types.
FIG. 3.
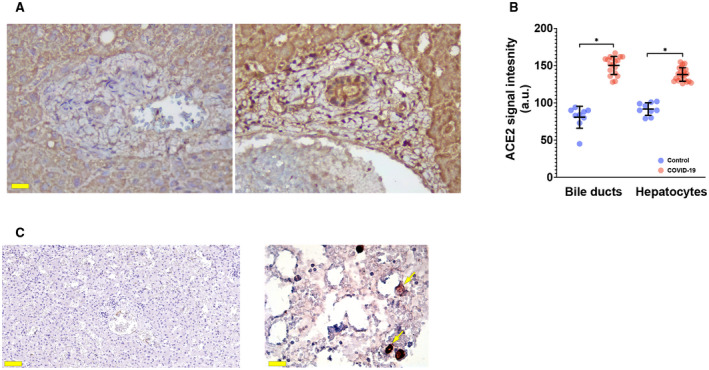
ACE2 is present but SARS‐CoV‐2 Spike protein is not detected in livers of patients with COVID‐19. (A) Immunohistochemistry from representative liver specimens shows that expression of ACE2, the receptor for SARS‐CoV‐2, is increased in our cohort of COVID‐19 liver samples (left), relative to a histologically normal control (right). Scale bar, 20 μm. (B) Quantitative analysis shows that ACE2 expression is increased in both hepatocytes and cholangiocytes; *P < 0.0001. Measurements were made in a blinded fashion in 18‐21 microscopic fields from each of 5 separate patients with COVID‐19 and in nine fields from each of 3 separate controls. (C) Immunohistochemistry shows absence of SARS‐CoV‐2 spike protein in a representative liver sample of a patient with COVID‐19 patient. Scale bar, 100 μm. In contrast, staining was detected in alveolar cells (arrows) in a lung sample from one of the patients, which was used as a positive control (right). Scale bar, 20 μm.
Finally, we examined whether hepatocytes expressed ITPR3 in this cohort of patients with COVID‐19. This intracellular calcium release channel is not expressed in hepatocytes under normal conditions, as previously shown by immunohistochemistry studies of histologically normal liver adjacent to resected colorectal cancer metastasis.( 22 ) However, it becomes expressed in response to hepatocellular damage, both during acute types of liver injury such as ischemia‐reperfusion injury( 18 ) and yellow fever infection,( 17 ) and during chronic types of liver injury such as chronic viral hepatitis, alcohol‐associated liver disease, and nonalcoholic steatohepatitis.( 22 ) Here, we found that each of 10 patients with COVID‐19 in whom this was examined had mild but consistent ITPR3 staining in their hepatocytes (Fig. 4). This provides supportive evidence that hepatocellular injury routinely occurs in such patients.
FIG. 4.
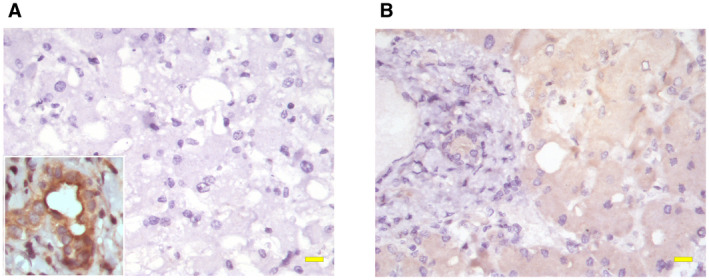
Hepatocytes express ITPR3 in COVID‐19 liver specimens. (A) ITPR3 staining was not observed in hepatocytes in histologically normal controls. Note that ITPR3 staining is present in normal bile ducts (inset), which serves as a positive control for the stain. (B) In contrast, positive ITPR3 staining (brown) in hepatocytes was observed in each of 10 liver samples from patients with COVID‐19. Scale bar, 20 μm.
Discussion
Liver‐test abnormalities are a frequent occurrence in patients with COVID‐19. In cohorts ranging from 12 to 1,827 patients, liver tests at admission were reported to be elevated in 40%‐66.9% (AST) and 41.6%‐67.5% (ALT) of patients, respectively.( 4 ) Moreover, elevated AST and ALT are associated with a poorer prognosis in hospitalized patients with COVID‐19. Postmortem liver histology of SARS‐CoV‐2‐infected patients suggested that lobular inflammation, vascular alterations, and steatosis are the main histological findings.( 13 , 14 ) Here, we described clinical and histological correlates of liver pathology in 27 patients who died with confirmed COVID‐19 disease. Our results corroborate the finding that vascular congestion in the liver is seen in most cases,( 13 ) although two separate studies found this to be less common.( 14 , 23 ) The current work extends previous observations by noting that CD61 and C4d labeling usually are absent, indicating a lack of locally activated coagulation factors and platelet aggregation within the liver. Although a local CD61 positivity of about 40%( 23 ) has been recently reported, our results reinforce the idea that vascular events observed in the liver of infected patients are likely due to the well‐documented systemic coagulopathy of COVID‐19 disease, which is characterized by thrombocytopenia and increased D‐dimer levels as well as different degrees of thrombosis, affecting vessels ranging from small to large capacity in multiple organs.( 24 ) Alternatively, our results suggest that liver vascular alterations are secondary to the ischemia caused by the cardio‐respiratory dysfunction characteristic of severe COVID‐19 cases.( 25 )
Our data failed to identify SARS‐CoV‐2 in liver cells of infected individuals. This is in contrast to findings that SARS‐CoV‐2 is present in most liver specimens, which was examined by in situ hybridization( 13 ) as well as by PCR.( 14 ) It is possible that those molecular methods may have been more sensitive than the immunochemistry used in this study. Alternatively, methods such as PCR do not provide spatial information, so it is possible that the virus detected by that approach was not localized to hepatocytes. One key factor related to the presence of the virus in parenchymal cells of the liver is the expression of the receptor for the virus, the transmembrane protein ACE2. We detected ACE2 in both hepatocytes and cholangiocytes in infected SARS‐Cov‐2 specimens, but minimal to no expression in normal tissues. This is in partial agreement with data from normal liver tissues that reported no ACE2 expression in hepatocytes.( 21 ) However, contrary to our findings, that report also found significant expression of ACE2 in cholangiocytes and endothelial cells. This discrepancy might be attributed to differences in ACE2 expression between healthy liver tissues and liver samples from patient with COVID‐19. The finding that ACE2 becomes expressed in the liver of patients with COVID‐19 also may explain why SARS‐CoV‐2 can become detectable in the liver in these patients,( 13 , 14 ) albeit at levels below what can be appreciated by immunochemistry. Another potential factor affecting ACE2 expression is the use of ACE blockers. For example, use of ACE blockers has been shown to increase ACE2 expression in epithelial cells of the small intestine.( 26 ) In the liver, the use of ACE blockers appears to potentiate the fibrosis‐associated increase in ACE2 expression in stellate cells. Nonetheless, the samples used here to evaluate ACE2 expression were all from patients who were not taking any ACE blocker medication.
We also were not able to appreciate any significant bile duct injury in this study, as demonstrated by the absence of epithelial degeneration around biliary structures, further suggesting that cholangiocytes are not directly targeted by SARS‐CoV‐2. However, this topic requires more investigation, as bile duct organoids are susceptible to infection by SARS‐CoV‐2 in vitro.( 27 ) Moreover, a recent report in 3 patients has suggested that prolonged cholangiopathy may occur following recovery from COVID‐19.( 28 )
Another frequent histological finding was the presence of steatosis, observed in 63% of the 27 cases in this study. This finding was observed in most patients in two additional case series( 13 , 14 ); however, it was less frequent (30%) in a third autopsy report.( 23 ) Notably, in our study, the presence of steatosis did not correlate with the presence of obesity or DM, two well‐established co‐morbidities in fatty liver disease. Moreover, this incidence of steatosis is appreciably higher than that reported in larger cohorts in Latin America and Brazil.( 29 , 30 ) This raises the possibility that fat accumulation could be due to other factors operating either before or during the infection by SARS‐CoV‐2. Alcohol consumption could explain the presence of steatosis in some of the livers. However, no specimens had any additional histological evidence of alcohol‐associated steatohepatitis, and previous history of alcohol consumption was not available from the charts. Medications such as steroids could also contribute to fat accumulation in the liver, but only a minority of the patients (n = 4; 14.8%) were administered these drugs either before or during hospitalization. History of viral hepatitis infection (hepatitis B or hepatitis C) could also account for the steatosis in our cohort, but these data were not available. A direct effect of SARS‐CoV‐2 in inducing lipid accumulation in hepatocytes is a final possibility. Hepatic lipid accumulation occurs acutely in yellow fever infection involving the liver,( 17 ) and both SARS‐CoV‐2 and yellow fever are associated with new expression of ITPR3 in hepatocytes, which alters mitochondrial function in a way that may contribute to the development of steatosis.( 17 ) Further studies will be needed to understand whether this is the pathological mechanism linking COVID‐19 infection to liver steatosis.
Over 80% of patients hospitalized for COVID‐19 may have elevated transaminases,( 4 ) yet the reason for this very frequently observed form of hepatocellular injury remains unclear. Patients with COVID‐19 may receive a variety of medications, but in most cases transaminase elevations are not related to any one specific medication.( 4 ) The current work furthermore provides evidence that the extent of transaminase elevations is not correlated with hepatic congestion, steatosis, or inflammation, which are the most common histologic abnormalities found on autopsy.( 13 , 14 ) On the other hand, this and other( 13 , 14 ) autopsy studies provide complementary pieces of evidence to suggest that hepatocytes may become directly infected with SARS‐CoV‐2, so it is conceivable that transaminase elevations reflect a direct cytopathic effect of the virus. Another intriguing observation by us and others( 14 ) is the frequency of steatosis in patients with COVID‐19, especially because we found that this histological finding was not correlated with either elevated BMI or the presence of DM. It is well known that each of these two conditions increases the risk of nonalcoholic fatty liver disease,( 31 ) which in turn contributes to an inflammatory state.( 32 ) Our findings raise the question of whether SARS‐CoV‐2 independently leads to steatosis, which then may contribute to the cytokine storm, which is thought to lead to worse outcomes.( 33 ) Consistent with this hypothesis is the observation that increased transaminase is associated with worst outcomes.( 4 ) Further work, likely in cell cultures and organoids, animal models, or using liver biopsy specimens, would be needed to understand whether SARS‐CoV‐2 has a cytopathic effect on hepatocytes, whether and how it induces steatosis, and whether either of these effects lead to release of cytokines or other factors from hepatocytes that have systemic effects that contribute to morbidity and mortality.
Acknowledgment
The authors thank the support of the Liver Center UFMG and the CloroCovid‐19 team. The authors would also like to thank the support of Fundação de Amparo à Pesquisa de Minas Gerais (FAPEMIG, Brazil); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil).
Supported by the National Institutes of Health (P30 DK34989, P01 DK57751, R01 DK114041, and R01 DK112797) and the Gladys Phillips Crofoot Professorship.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. WHO Coronavirus Disease (COVID‐19) Dashboard. In: World Health Organization; 2021. https://covid19.who.int. Accessed April 9, 2021.
- 2. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA 2020;323:1239‐1242. [DOI] [PubMed] [Google Scholar]
- 3. Cai Q, Huang D, Yu H, Zhu Z, Xia Z, Su Y, et al. COVID‐19: abnormal liver function tests. J Hepatol 2020;73:566‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hundt MA, Deng Y, Ciarleglio MM, Nathanson MH, Lim JK. Abnormal liver tests in COVID‐19: a retrospective observational cohort study of 1,827 patients in a major U.S. hospital network. Hepatology 2020;72:1169‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Zheng L, Liu L, Zhao M, Xiao J, Zhao Q. Liver impairment in COVID‐19 patients: a retrospective analysis of 115 cases from a single centre in Wuhan city, China. Liver Int 2020;40:2095‐2103. [DOI] [PubMed] [Google Scholar]
- 6. Bloom PP, Meyerowitz EA, Reinus Z, Daidone M, Gustafson J, Kim AY, et al. Liver biochemistries in hospitalized patients with COVID‐19. Hepatology 2021;73:890‐900. [DOI] [PubMed] [Google Scholar]
- 7. Phipps MM, Barraza LH, LaSota ED, Sobieszczyk ME, Pereira MR, Zheng EX, et al. Acute liver injury in COVID‐19: prevalence and association with clinical outcomes in a large U.S. cohort. Hepatology 2020;72:807‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang C, Shi L, Wang FS. Liver injury in COVID‐19: management and challenges. Lancet Gastroenterol Hepatol 2020;5:428‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vasquez‐Bonilla WO, Orozco R, Argueta V, Sierra M, Zambrano LI, Muñoz‐Lara F, et al. A review of the main histopathological findings in coronavirus disease 2019. Hum Pathol 2020;105:74‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chornenkyy Y, Mejia‐Bautista M, Brucal M, Blanke T, Dittmann D, Yeldandi A, et al. Liver pathology and SARS‐CoV‐2 detection in formalin‐fixed tissue of patients with COVID‐19. Am J Clin Pathol 2021;155:802‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fiel MI, El Jamal SM, Paniz‐Mondolfi A, Gordon RE, Reidy J, Bandovic J, et al. Findings of hepatic severe acute respiratory syndrome coronavirus‐2 infection. Cell Mol Gastroenterol Hepatol 2021;11:763‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID‐19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020;8:420‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sonzogni A, Previtali G, Seghezzi M, Grazia Alessio M, Gianatti A, Licini L, et al. Liver histopathology in severe COVID 19 respiratory failure is suggestive of vascular alterations. Liver Int 2020;40:2110‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lagana SM, Kudose S, Iuga AC, Lee MJ, Fazlollahi L, Remotti HE, et al. Hepatic pathology in patients dying of COVID‐19: a series of 40 cases including clinical, histologic, and virologic data. Mod Pathol 2020;33:2147‐2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hooper JE, Padera RF, Dolhnikoff M, da Silva LFF, Duarte‐Neto AN, Kapp ME, et al. A postmortem portrait of the coronavirus disease 2019 (COVID‐19) pandemic: a large multi‐institutional autopsy survey study. Arch Pathol Lab Med 2021;145:529‐535. [DOI] [PubMed] [Google Scholar]
- 16. Shang J, Ye G, Shi KE, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS‐CoV‐2. Nature 2020;581:221‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lemos FDO, França A, Lima Filho ACM, Florentino RM, Santos ML, Missiaggia DG, et al. Molecular mechanism for protection against liver failure in human yellow fever infection. Hepatol Commun 2020;4:657‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lima Filho ACM, França A, Florentino RM, dos Santos ML, de Oliveira Lemos F, Missiaggia DG, et al. Inositol 1,4,5‐trisphosphate receptor type 3 plays a protective role in hepatocytes during hepatic ischemia‐reperfusion injury. Cell Calcium 2020;91:102264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simadibrata DM, Calvin J, Wijaya AD, Ibrahim NAA. Neutrophil‐to‐lymphocyte ratio on admission to predict the severity and mortality of COVID‐19 patients: a meta‐analysis. Am J Emerg Med 2021;42:60‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu R, Zhao X, Li J, Niu P, Yang BO, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. a first step in understanding SARS pathogenesis. J Pathol 2004;203:631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guerra MT, Florentino RM, Franca A, Lima Filho AC, Dos Santos ML, Fonseca RC, et al. Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 2019;68:1676‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bryce C, Grimes Z, Pujadas E, Ahuja S, Beasley MB, Albrecht R, et al. Pathophysiology of SARS‐CoV‐2: the Mount Sinai COVID‐19 autopsy experience. Mod Pathol 2021;34:1456‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lo MW, Kemper C, Woodruff TM. COVID‐19: complement, coagulation, and collateral damage. J Immunol 2020;205:1488‐1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berlin DA, Gulick RM, Martinez FJ. Severe Covid‐19. N Engl J Med 2020;383:2451‐2460. [DOI] [PubMed] [Google Scholar]
- 26. Vuille‐dit‐Bille RN, Camargo SM, Emmenegger L, Sasse T, Kummer E, Jando J, et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE‐inhibitors. Amino Acids 2015;47:693‐705. [DOI] [PubMed] [Google Scholar]
- 27. Zhao B, Ni C, Gao R, Wang Y, Yang LI, Wei J, et al. Recapitulation of SARS‐CoV‐2 infection and cholangiocyte damage with human liver ductal organoids. Protein Cell 2020;11:771‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roth NC, Kim A, Vitkovski T, Xia J, Ramirez G, Bernstein D, et al. Post‐COVID‐19 cholangiopathy: a novel entity. Am J Gastroenterol 2021;116:1077‐1082. [DOI] [PubMed] [Google Scholar]
- 29. Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2019;69:2672‐2682. [DOI] [PubMed] [Google Scholar]
- 30. Cotrim HP, Parise ER, Oliveira CPMS, Leite N, Martinelli A, Galizzi J, et al. Nonalcoholic fatty liver disease in Brazil. Clinical and histological profile. Ann Hepatol 2011;10:33‐37. [PubMed] [Google Scholar]
- 31. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 32. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349‐364. [DOI] [PubMed] [Google Scholar]
- 33. Moore JB, June CH. Cytokine release syndrome in severe COVID‐19. Science 2020;368:473‐474. [DOI] [PubMed] [Google Scholar]