

Abstract
Neutralizing monoclonal antibodies (mAb), novel therapeutics for the treatment of coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2), have been urgently researched from the start of the pandemic. The selection of the optimal mAb candidate and therapeutic dose were expedited using open‐access in silico models. The maximally effective therapeutic mAb dose was determined through two approaches; both expanded on innovative, open‐science initiatives. A physiologically‐based pharmacokinetic (PBPK) model, incorporating physicochemical properties predictive of mAb clearance and tissue distribution, was used to estimate mAb exposure that maintained concentrations above 90% inhibitory concentration of in vitro neutralization in lung tissue for up to 4 weeks in 90% of patients. To achieve fastest viral clearance following onset of symptoms, a longitudinal SARS‐CoV‐2 viral dynamic model was applied to estimate viral clearance as a function of drug concentration and dose. The PBPK model‐based approach suggested that a clinical dose between 175 and 500 mg of bamlanivimab would maintain target mAb concentrations in the lung tissue over 28 days in 90% of patients. The viral dynamic model suggested a 700 mg dose would achieve maximum viral elimination. Taken together, the first‐in‐human trial (NCT04411628) conservatively proceeded with a starting therapeutic dose of 700 mg and escalated to higher doses to evaluate the upper limit of safety and tolerability. Availability of open‐access codes and application of novel in silico model‐based approaches supported the selection of bamlanivimab and identified the lowest dose evaluated in this study that was expected to result in the maximum therapeutic effect before the first‐in‐human clinical trial.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Bamlanivimab is a monoclonal antibody that neutralizes severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) and underwent rapid clinical development for treatment of coronavirus disease 2019 (COVID‐19) in patients with mild or moderate disease.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This paper provides the methodology behind modeling selection of the first human dose range of bamlanivimab in a pandemic situation, and how physiologically‐based pharmacokinetic modeling was used to predict 700 mg as the lowest dose evaluated in this study that would result in maximum therapeutic effect in the absence of preclinical or clinical data.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ An open‐access in silico modeling and simulation approach to drug discovery and development facilitated an accelerated path to selecting bamlanivimab as the best drug candidate based on the projected pharmacokinetic (PK), pharmacodynamic (PD), and optimal therapeutic dose.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This work demonstrates that in silico PK/PD modeling and simulation and open‐access approaches to science can be relied upon in future drug development programs especially when speed to patient is essential.
Infections with a novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) are currently widespread and responsible for an ongoing public health emergency. As of March 9, 2020, when this work with neutralizing monoclonal antibodies (mAbs) began, there were over 114,000 cases and 3,990 deaths due to coronavirus disease 2019 (COVID‐19). That number has increased globally to more than 120 million cases and 2.6 million deaths across 192 countries or regions (as of March 16, 2021). 1 As these numbers continue to increase, there is an urgent need for safe and effective therapeutics to treat patients.
Through state‐of‐the‐art high throughput technology and the global effort to share scientific data, including genetic codes for the spike protein of SARS‐CoV‐2, 2 , 3 immunoglobulin G (IgG) mAbs, specifically engineered against the spike protein of SARS‐CoV‐2 have been developed. These mAbs have the potential to block viral attachment and entry into human cells, thus neutralizing the virus and potentially preventing and treating COVID‐19. 4 , 5 However, the process of moving from bench to patients for clinical use may take up to 10 years or more in research and development, 6 a timeframe that was not useful in the midst of a pandemic. The massive impact of COVID‐19 on both global health and economics has driven an unprecedented effort in the utilization of in silico modeling and simulation to accelerate timelines in research and development. 7 , 8 , 9 , 10
An open‐access in silico pharmacology model‐based approach was developed to project the clinical dose for a selected set of neutralizing antibodies in the preclinical screening stage to support the recommendation of the optimal neutralizing antibody. Model projected therapeutic dose is one of the many candidate selection criteria commonly used in drug discovery. 11 , 12 It is a multidimensional metric that requires inputs from multiple scientific disciplines in drug research and development. The patient population and special population characteristics, physicochemical properties of the molecules (e.g., FcRn binding and AC‐SINS score, further information is listed in the Supplementary Materials ), formulation characteristics (e.g., solution concentration, number, and volume of injections), pharmacokinetic (PK), and pharmacodynamic (PD) properties are all important criteria in the selection of the optimal neutralizing antibody and the dose. For PK properties, it is important to know where the drug is distributed and how it is cleared from the body. For PD properties, it is important to acknowledge the uncertainty of translating the in vitro potency or affinity of the antibody to bind to the virus, specifically at the S spike protein to prevent its binding to the angiotensin‐converting enzyme (ACE2) receptors on the human host cell, to in vivo or clinical potency in viral clearance. Last, it is critical to understand how the candidate mAbs and their dose‐response relationships impact the dynamics of the viral infection and elimination.
At the beginning of the pandemic in the United States, there was a paucity of data to develop the necessary PK and PD models due to tight timelines and a shortage of animal models. An extensive literature search, made possible by the open‐access practice 13 (including submissions to preprint servers and open‐access journals) of the scientific and pharmacometrics community, provided access to a physiologically‐based PK (PBPK) model for an mAb and a viral dynamic PD model of SARS‐CoV‐2. 14 This was a pivotal starting point to address the drug development questions during a pandemic.
To support the selection of anti‐SARS‐CoV‐2 neutralizing mAb candidates, and to guide the dosing recommendations for a first‐in‐human study, quantitative modeling and simulation leveraged established PK, physiologic, and statistical models and principles of pharmacology. Two modeling and simulation methods were implemented, a PBPK approach and a viral dynamic approach, to increase the confidence in dose projections.
For the first method, a state‐of‐the‐art, open‐access PBPK model was critical to evaluating candidate mAbs distribution and clearance from in vitro data (Figure 1 ). 15 PBPK predictions of drug concentration‐time profiles at the target, both in plasma and lung tissue, provided dosing regimens to cover target ranges from in vitro experimental neutralization data (i.e., candidate mAb‐specific 90% inhibitory concentration (IC90) values). More specifically, this ensured the estimated doses would provide interstitial lung concentrations in excess of the 90th percentile for virus inhibition and in at least 90% of patients, based on typical interindividual PK variability seen in the patient population for typical IgG mAbs. 16 First described by Toerell in 1937, PBPK models combine physiology, patient population, and drug characteristics into a mathematical modeling framework to predict the absorption, distribution, metabolism, and excretion of drugs within specific tissues and organs. 17 , 18 These models are particularly useful during the early stages of drug discovery where information on the compounds are limited because these models can be informed by in vitro and preclinical data.
Figure 1.
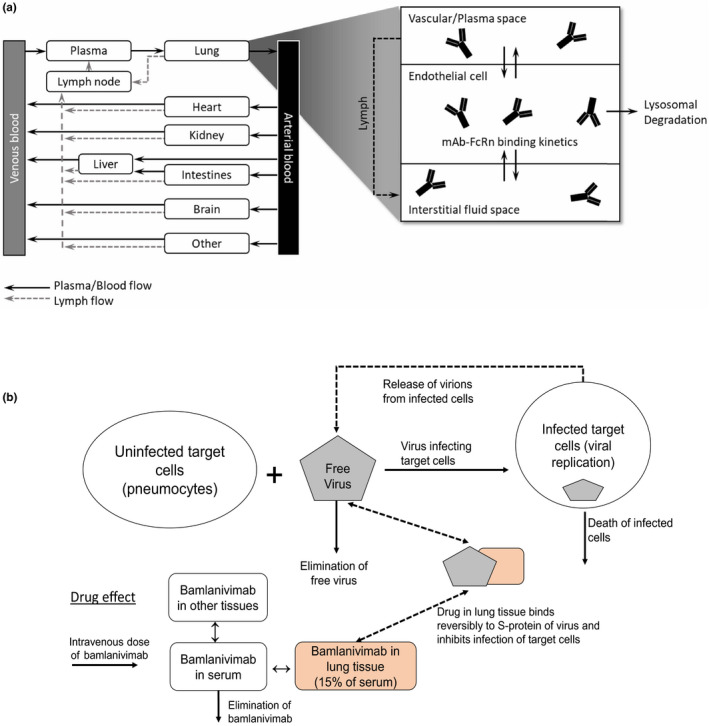
Graphical representation of modeling approaches. Diagram of physiologically‐based pharmacokinetic model for monoclonal antibodies (mAbs) with considerations for interstitial distribution through saturable endothelial, FcRN‐mediated transport mechanisms, figure concept from Jones et al. 15 (a). Diagram of viral dynamic modeling, figure concept from Cangelosi et al. 37 (b).
The aforementioned open‐access PBPK model was recently expanded to include affinity‐capture self‐interaction nanoparticle spectroscopy (AC‐SINS) as a predictor of mAb clearance and distribution. 15 Generally, mAbs with a lower AC‐SINS value, and hence a lower propensity to self‐associate, are preferable because they are more readily distributed throughout the body. AC‐SINS have also been shown to be positively correlated with in vivo mAb clearance (Spearman correlation coefficient of 0·7). 19 Qualitative knowledge on other biophysical properties were also considered.
Initially, a PBPK model using candidate mAb‐specific AC‐SINS values provided expected organ‐specific mAb exposures for all candidates. Because all candidates had AC‐SINS scores less than 3 (except one mAb, which had AC‐SINS = 8), this information alone did not allow for differentiation between the candidates. Instead, early pseudo neutralization data, and information on specific binding sites (e.g., receptor‐binding domain, N‐terminus domain, and S spike protein 1/2), were used to reduce an initial pool of candidate mAbs from 24 to the final 5 candidates. 4
Although providing mAb exposure predictions, the PBPK‐based method did not address how an mAb therapeutic may interact dynamically with the virus. For the second in silico method, a published model of SARS‐CoV‐2 viral dynamics provided unique insight into the profile of SARS‐CoV‐2 in humans early in the pandemic. 14 The model was urgently developed and published based on data from the infected patients in Wuhan, China, in December 2019. The model provided an approach to quantitatively evaluate virus‐host cell interaction and allowed incorporation of select pharmacologic mechanisms of antiviral effects, including the neutralizing mAbs.
These two quantitative modeling approaches together were used to further investigate the final five candidates using subsequently available live virus neutralization data to provide quantitative decision support to select the candidate anti‐SARS‐CoV‐2 mAbs and dose range recommendations to advance to clinical investigation.
METHODS
PBPK approach
The PBPK approach described herein focused on the primary presumption that target mAb concentrations (exposure) must be achieved and maintained at critical sites of infection in the body within a reasonable dosing range. For this development program, the goal was to ensure doses were able to achieve target concentrations in excess of the reported in vitro IC90 in the plasma and lung tissue for at least 90% of patients and for at least 14 to 28 days. Specific to this goal and to explore differentiating features among the mAb candidates, the following characteristics were considered in these analyses.
Pharmacokinetic characteristics:
Distribution of candidate mAbs into target organs and the fluids interfacing with the endothelial cells of those organs i.e., lung as the organ associated with the highest viral burden and the most negative consequences of COVID‐19 disease (respiratory failure, hypoxia, and death);
mAb‐specific physicochemical properties that may affect the PKs (distribution and clearance) of the individual mAb candidates;
Patient‐to‐patient variability in mAb exposure, such that the doses recommended would expect to achieve and maintain target exposures in at least 90% of patients.
Neutralization data:
mAb‐specific neutralization half‐maximal inhibitory concentration (IC50) data, as reported by the different assay laboratories to establish the target IC90 for each candidate mAb (Table 1 ).
Table 1.
Candidate mAb dose expected to maintain concentration > IC90 over 14, 21, and 28 days for the typical patient and for 90% of patients
| mAb | Domain | ACE2 blocking | AC‐SINS score | Live virus IC50, ug/mL | Dose a , mg, expected to maintain concentration above live virus IC90 b in the lungs | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Geometric mean (min, max) | ||||||||||
| For a typical patient | For 90% of patients | |||||||||
| Day 14 | Day 21 | Day 28 | Day 14 | Day 21 | Day 28 | |||||
| 1 | S1 | No | 2·25 | 0.841 | 1,273 (918.7–2,303) | 1,664 (1,197–3,043) | 2,153 (1,542–3,988) | 3,295 (2,369–6,023) | 5,583 (3,981–10,440) | 9,236 (6,984–17,960) |
| 2 | RBD | Yes | 0 | 2.685 | 4,210 (2,607–11,580) | 5,662 (3,450–16,830) | 7,600 (4,534–25,280) | 11,190 (6,828–32,350) | 20,200 (11,890–67,670) | 37,020 (20,620–70,000) |
| 3 | NA | Yes | 0 | 1.3 |
1,977 NA |
2,599 NA |
3,387 NA |
5,145 NA |
8,838 NA |
14,990 NA |
| 4 | RBD | Yes | 0 | 1.112 | 1,684 (1,295–2,607) | 2,208 (1,692–3,450) | 2,867 (2,187–4,534) | 4,370 (3,349–6,828) | 7,446 (5,665–11,890) | 12,510 (9,370–20,620) |
| 5c | RBD | Yes | 1 | 0.04609 | 68.28 (25.16–237.5) | 88.16 (32.48–307.3) | 112.3 (41.38–392.2) | 174.3 (64.21–607.5) | 286.4 (105.4–1,002) | 454.9 (166.9–1,598) |
ACE2, angiotensin‐converting enzyme; AC‐SINS, affinity‐capture self‐interaction nanoparticle spectroscopy; IC50, half‐maximal inhibitory concentration; IC90, 90% inhibitory concentration; mAb, monoclonal antibody; NA, not applicable; Min, minimum; Max, maximum; S, spike protein; NTD, N‐terminus domain; RBD, receptor‐binding domain.
Dose for mAb 1, 2, 4, and 5 is the geometric mean of data from three assay laboratories. Data for mAb 3 was available from one assay laboratory only.
IC90 = 9 × IC50.
mAb 5 corresponds to bamlanivimab.
The PK characteristics were integrated through a PBPK model with the neutralization data to determine and compare potential starting and therapeutic doses among the candidate mAbs. The model is available online (https://github.com/metrumresearchgroup/bioPBPK) and further details of the PBPK modeling approach are available in the Supplementary Materials .
Viral dynamic modeling and simulation approach
A target‐cell limited SARS‐CoV‐2 viral dynamic model was developed based on limited data available in the literature. 14 The viral dynamic model components included the following:
a population of uninfected target cells (type II pneumocytes expressing the ACE2 receptors);
free virus available to infect the target cells and subsequent viral replication;
elimination of free virus particles (due to the host immune system);
death of infected target cells;
in vitro neutralization data, as described above.
Because no human or animal PK data were available at the time, a compartmental PK model of a typical human IgG1 mAb was used (in parallel with the development of the PBPK model) to predict the concentration profile of the selected neutralizing mAb in serum upon intravenous administration of a wide range of doses. 20 Fifteen percent drug distribution into the lungs was assumed, as expected for a typical mAb. 21 The predicted drug concentration in the lung tissue was assumed to bind to free circulating virus, thereby reducing the amount of virus available to infect target cells. A maximum effect model was incorporated that included a binding potency IC50 from live virus neutralization assays. The parameters used in the viral dynamic model are shown in Table S1 .
A range of doses was evaluated through simulations and visualization of the associated viral load profiles. In addition to the doses investigated, other simulation scenarios were evaluated, including the impact of drug administration timing relative to the onset of COVID‐19 symptoms on viral clearance. Further details of the model are found in the Supplementary Material . The model is also available online (https://github.com/metrumresearchgroup/vk‐covid19 ). The model and simulations were implemented using the R software version 3.5.0. 22
Dose selection criteria
Based on the PBPK model, candidate mAbs and ultimately the lowest bamlanivimab dose, evaluated in this study, that were expected to result in maximum effect were selected to maintain a concentration above the in vitro IC90 of viral neutralization for human host cell entry in the lung tissue for at least 28 days in 90% of the patient population. These criteria are necessarily conservative, because the risks of underdosing patients with COVID‐19 are greater than the potential safety risks of the mAb, and due to an abundance of caution in a pandemic. The efficacy of the selected dose has been investigated in ongoing clinical trials.
Based on the viral dynamic model, at the time of this analysis, the lowest dose evaluated in this study expected to result in maximum effect was selected based on the fastest time from the onset of symptoms to achieve maximum viral clearance. This approach was independent of the IC90 approach of the PBPK‐based approach. The PBPK model predictions were subsequently translated to a population PK model for a priori optimization of the PK sample collection times used in the clinical investigation. Simulations of the expected range of plasma mAb concentration‐time profile included estimates of expected interindividual variability of clearance and volume parameters for IgG mAbs 16 and were later compared with the actual observed PK data from the first‐in‐human study (NCT04411628). As a benchmark, it is typical to evaluate the predictive performance of these models by determining whether the predicted concentration‐time profiles were within two‐fold of the observed data. 23 , 24
RESULTS
Prospective PBPK modeling
Predictions of the expected PK profiles, differentiated over a range of AC‐SINS (0 to 8), indicated that the expected plasma concentration profile would vary across the AC‐SINS range for the final selected mAb candidates (Figure 2 ). The PK profiles show plasma clearance increases, and hence exposure decreases, with higher AC‐SINS values (Figure 2 ). All five drug candidates, had AC‐SIN values within the favorable range of 0 to 3 and shared similar expected PK profiles. Although this factor did not allow for differentiation between the candidates, it did provide reassurance regarding the expected overall PK and an understanding of the expected exposure profiles in the various tissues and organs throughout the body. Notably, the interstitial concentrations in the lung tended to be the same or lower than in other organs of interest (e.g., heart, gastrointestinal, and kidneys; data not shown). This further supported the lung tissue concentrations as the bellwether site for evaluating dosing regimen recommendations. Notably, the lung tissue concentrations were ~ 17‐fold lower than (6‐6·5% of) the corresponding plasma prediction (Figure 3 ).
Figure 2.
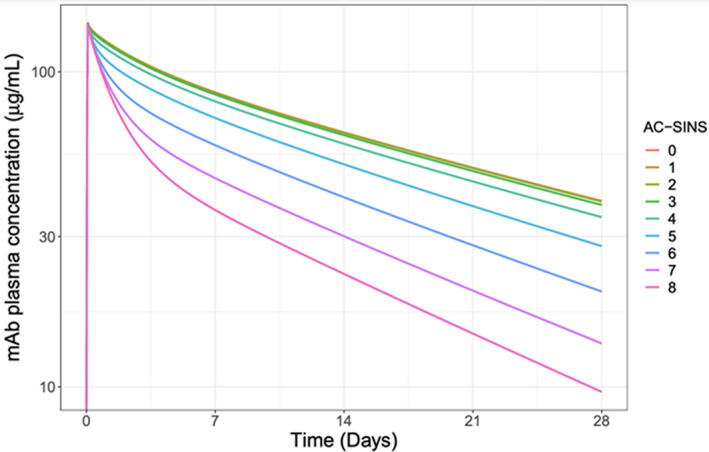
Simulated candidate mAb plasma pharmacokinetic profiles. Simulation of the expected plasma mAb concentration‐time profiles for mAbs with a range of AC‐SINS scores over 28 days. The mAbs with AC‐SINS scores from 0–8 are color coded as per the legend. Simulation assumes a 71 kg individual infused with 700 mg of antibody over 2 hours. AC‐SINS, affinity‐capture self‐interaction nanoparticle spectroscopy; mAb, monoclonal antibody.
Figure 3.
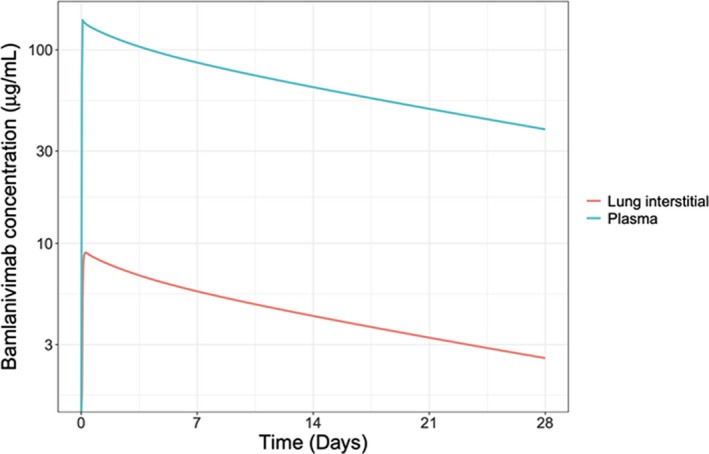
Representative simulated plasma and lung mAb concentration‐time profile. Physiologically‐based pharmacokinetic model output depicting the differential mAb concentration‐time profile expected in plasma and lung tissue over 28 days. Data are simulated for a mAb with an AC‐SINS score of 1. Simulation assumes a 71 kg individual infused with 700 mg of antibody over 2 hours. AC‐SINS, affinity‐capture self‐interaction nanoparticle spectroscopy; mAb, monoclonal antibody.
The simulated patient population PK profiles showed the model predicted variability between subjects was consistent with the variability observed following mAb administration in humans (e.g., the % coefficient of variation for clearance was ~ 30–50%; Figure 4 ). 7 An overlay of predicted bamlanivimab drug concentrations with observed PK data from the first human study (NCT04411628) demonstrated that the PBPK modeling approach predicted human PKs very well, and was therefore accurate with respect to selecting doses (Figure 4 ). The first human study of bamlanivimab was a randomized, double‐blind, sponsor unblinded, placebo‐controlled, single ascending dose first‐in‐human trial in hospitalized patients with COVID‐19. 25
Figure 4.
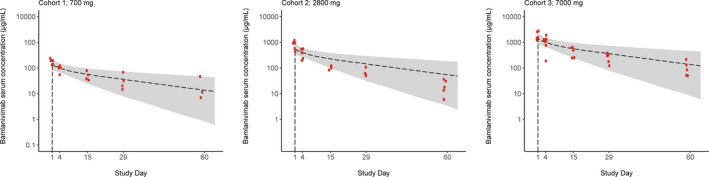
Overlay of pharmacokinetic (PK) profiles as predicted a priori using the physiologically‐based pharmacokinetic (PBPK) model with observed data from the first‐in‐human trial. Bamlanivimab serum concentrations from cohorts of patients receiving 700, 2,800, or 7,000 mg of bamlanivimab. Red data points are the observed clinical data from each of the respective three cohorts. The grey shaded area represents the 90% prediction interval from PBPK modeling with the black dotted line representing the median.
Efficacy of projected dose ranges was primarily assessed in terms of candidate mAbs’ ability to maintain lung tissue concentrations at or above the in vitro IC90 for periods of 14‐, 21‐, and 28‐days post‐administration. The required dose was estimated for both a typical patient, and also to provide coverage for 90% of patients (Table 1 ), as determined from the simulated distribution of patient‐to‐patient variability in mAb exposure. The simulation predicted a 3‐ to 4‐fold higher dose would be required to achieve the target lung tissue concentrations for 90% of patients compared with the typical patient.
Prospective viral dynamic modeling and simulation results
Simulations demonstrated that early drug administration (less than 3 days from symptom onset) would result in greater reduction in lung viral load compared with placebo (Figure 5 ), with late drug administration (greater than 7 days) having a smaller benefit. Further simulations using the viral dynamic model indicated that a range of doses (100 mg to 700 mg) would be effective in reducing the viral load in patients. However, the intention was to determine the lowest dose that would result in the most rapid clearance of virus. This dose was determined to be 700 mg, with higher doses resulting in overlapping viral load profiles, and thus a flat dose‐response between the 700 and 7,000 mg dose (Figure 6 ). 26 Therefore, at the time of this analysis, 700 mg was predicted to be the lowest dose evaluated in this study, that would result in maximum efficacy.
Figure 5.
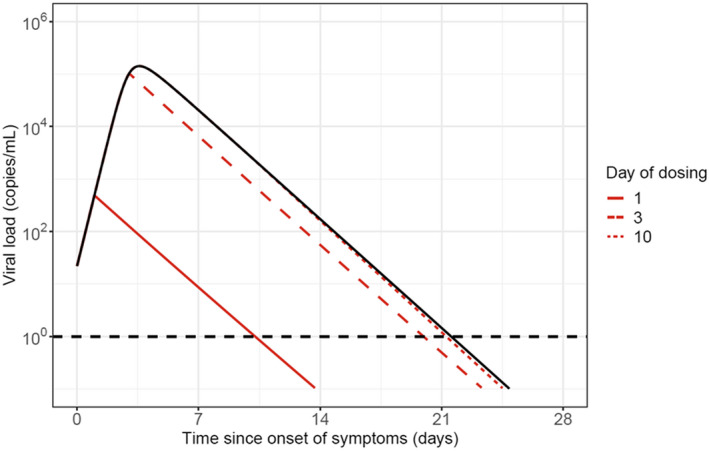
Deterministic simulation of lung viral load profiles over time. Example simulations demonstrating that earlier drug administration is associated with greater reduction in viral load relative to placebo. Colored lines represent a simulated viral load profile for a typical patient infused with bamlanivimab (700 mg) on day 1, 3, or 10 and the black line represents simulated placebo.
Figure 6.
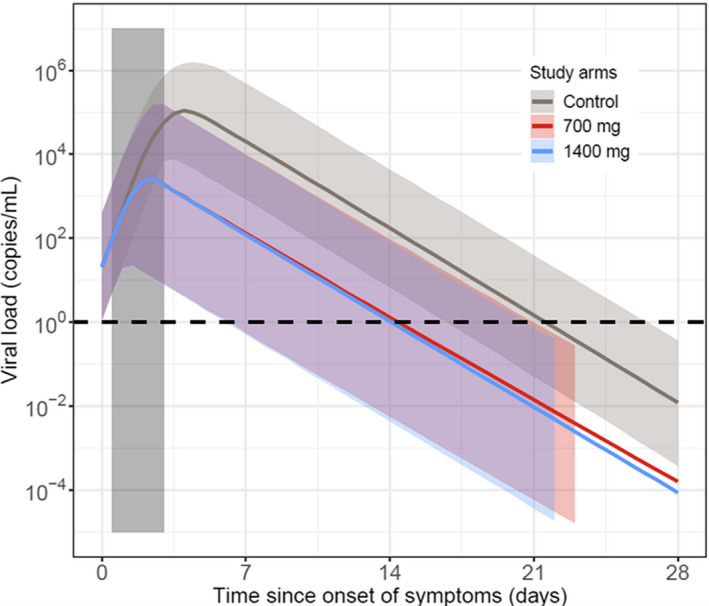
The 95% prediction interval of simulated lung viral load profiles from the severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) viral dynamic model. Monte Carlo simulation including variability in pharmacokinetic (PK) and pharmacodynamic (PD) parameters, as well as variability in the time of dosing relative to the onset of symptoms. All study arms received dosing on day 0. Colored shaded bars represent the 95% prediction interval of each treatment arm and the grey shaded bar represents uniform distribution of time of dosing from symptom onset.
DISCUSSION
To combat this pandemic at the onset and to get treatment to patients as soon as possible, unprecedented pandemic‐ready efforts were taken chronologically for a novel therapeutic agent for the treatment of COVID‐19:
Team formation: assembly of expert scientists across functions, including pharmacometric expertise and therapeutic area consultants.
Establishment of criteria for dose selection (rapid reduction in viral load) including mitigation strategy for translational uncertainty for safety and efficacy.
Developing PK/PD models and conducting simulations to determine efficacious doses and doses that would result in maximum reduction in viral load.
A starting dose of 700 mg was used in the first in human study in patients based on the PK/PD modeling presented in this paper. Higher doses were included as additional study arms for further evaluation of safety and to account for translational uncertainty.
Learn and confirm approach throughout the remainder of drug development between 2020 and 2021. Clinical trial data was subsequently obtained (and is still being collected) to further inform and develop the PK/PD model to inform future dose selection to build on the current models that we built with no clinical data of bamlanivimab.
Apply learning to next generation antibody development.
Bamlanivimab is a potent neutralizing mAb with a long half‐life, similar to other antibodies, 27 and was predicted to have a low efficacious dose compared with other candidate mAbs. This prediction was based on its ability to maintain a concentration above the in vitro IC90 of viral cell entry neutralization in the lung tissue for 14, 21, and 28 days, and to rapidly decrease SARS‐CoV‐2 viral load following early administration relative to the onset of COVID‐19 symptoms. This was determined for both a typical patient and for a simulated virtual patient population. The latter provided a projected dose that would achieve the target concentration in at least 90% of patients and generally provided a more conservative estimate of the efficacious dose level that was 3‐ to 4‐fold higher than those in the typical patient (at the 50th percentile). Because the PBPK‐based population predictions indicated that doses as low as 175 to 500 mg of bamlanivimab were potentially efficacious (defined as reduction in viral load), and the viral dynamic modeling approach suggested 700 mg would result in the most rapid viral clearance, a 700 mg starting dose was recommended given that it encompasses the effects of the lower dose range. Given the importance of minimizing underdosing patients, out of an abundance of caution in a pandemic situation and accounting for model uncertainty, this dose protected against potential uncertainty of in vitro to in vivo translation. However, it remains a possibility that lower doses could also achieve maximal effect. The initial first‐in‐human trial proceeded with the recommended dose of 700 mg as a starting dose and escalated to 2,800 mg, and 7,000 mg to investigate the limit of safety and tolerability. This was afforded by the speed brought by the model‐supported early clinical program used to obtain the emergency use authorization for bamlanivimab.
With regard to the use of the PBPK model along with the mAb‐specific AC‐SINS values, there were several factors that led to the confidence in using this approach. These included an understanding of the dispositional properties of mAbs and the reported validation work across dozens of existing mAbs, that demonstrated a remarkable predictive performance when translating from in vivo to predict human mAb PKs.
Model validation indicated that the PBPK model was able to accurately predict in vivo PK for antibodies a priori using in vitro data. 6 The core of the PBPK model, including organs, basic topology, and physiological parameters, was originally described by Shah and Betts. 21 Jones et al. expanded this to include a mechanistic description of FcRn‐mAb dynamics within a tissue compartment and a clearance mechanism to describe nonspecific interactions within each organ compartment using AC‐SINS data. 15 The inclusion of the FcRn recycling system for IgG‐based mAbs was important in this modeling application as it allowed for the differentiation of drug exposures specifically at the site of action (i.e., the lung interstitial space where the SARS‐CoV‐2 virus would attempt ACE2‐mediated cell entry for replication). The importance of quantifying the in vivo response directly at the site of action when using experimental in vitro data was recently highlighted by Fan et al. 7 The mAb concentrations in lung tissues have been reported to be ~ 15% of corresponding serum concentrations. 21 , 28 In addition, mAb concentrations in the tissue, the fluid in between the outer endothelial lining and the plasma membranes of cells, have been estimated to be ~ 2‐fold to 4‐fold lower than corresponding total tissue concentrations. 29 Model predictions were in line with these expectations: lung tissue concentrations were ~ 17‐fold lower than (6–6.5% of) the corresponding plasma/serum prediction (Figure 3 ). These results align with recent recommendations to use model‐based support, 30 and more specifically PBPK modeling, 7 to quantitatively characterize the target site concentrations to inform dosing recommendations.
The initial clinical trial results were in good agreement with the presented PBPK and viral kinetic modeling and simulation predictions from several angles. First, the observed serum concentration‐time (PK) data from all three bamlanivimab doses were in remarkable agreement with the a priori predictions from the PBPK model (Figure 4 ). This was a notable external confirmation of the open‐access work reported by Jones et al. and demonstrated the importance of these shared contributions to advancing public health. 15 Second, interim analyses in patients with recently diagnosed COVID‐19 infections showed similar viral load reduction, prevention of hospitalization, and symptom relief across the studied dose range of 700–7,000 mg. 26 This suggested that 700 mg was at the plateau of drug effect and, as such, studies of a lower dose range are underway. Second, the viral dynamic modeling suggested that early drug treatment would result in more significant reduction in viral load. This prediction was confirmed by the inability of bamlanivimab to help hospitalized patients who were at an advanced stage of their disease. 31 Implemented in the first quarter of 2020, our viral dynamic modeling approach was conducted with paucity of data. Currently, much clinical data have been gathered that can be used to develop more robust viral dynamic models and improve accuracy of predictions. Future modeling work should incorporate all these data, including data about the incubation period and the time between infection and the onset of symptoms. The approach presented in this paper can be built upon using updated models as they are developed and published, and thus lead to more robust predictions.
In November of 2020, bamlanivimab received emergency use authorization in patients recently diagnosed with COVID‐19 (with mild to moderate symptoms) and was not indicated for use in hospitalized patients. 32 This emergency use authorization was later revoked to complete transition to bamlanivimab together with etesevimab in the United States. 33 These recent clinical trial findings, which are in agreement with the model‐based predictions, lend confidence to the two modeling and simulation approaches presented here and suggests that such methods should be relied upon more in future drug development programs.
In an effort to combat the current COVID‐19 pandemic, an unparalleled international scientific response has been launched with the aim to understand all aspects of the SARS‐CoV‐2 virus and, in particular, to develop therapeutic strategies that are able to treat or prevent COVID‐19. This pandemic has highlighted the importance of open science, data‐sharing, and new means of communication among members of the scientific community. 34 , 35 Underscoring this statement, transparent and fully reproducible science, as provided by Jones et al. 2019 15 and again here in our Supplementary Materials (model also available on github), were paramount to successfully supporting and planning clinical trials; in this example, model‐informed decision support supplanted potentially years of clinical efforts to identify this effective dose range. Relying on advanced technologies, such as modeling and simulation to contract the need for otherwise lengthy dose‐ranging clinical investigations, has allowed the realization of condensed development timelines, which went from years to months.
As a direct consequence of the modeling work described here, the initial first‐in‐human trials included a dose that was 10 times lower than a priori experience (developing other antiviral therapeutics 36 ) had suggested. Given the enormous financial and logistical costs associated with producing and distributing drugs to such a vast patient population, the implications of this drastically reduced efficacious dose from a public health perspective are highly significant.
This work also supports further dosing regimen optimization studies. These results, when available, could lead to considerable improvements in the scale with which this treatment may be delivered, thereby further influencing the global efforts to control COVID‐19 infections. At a minimum, results from these further clinical investigations, in conjunction with continued model‐inform decision support, will lend to the learn‐confirm paradigm that continues to improve all therapeutics development; an all‐too‐important need as researchers continue to confront diseases affecting the world’s population.
FUNDING
Eli Lilly and Company was responsible for study design, data collection, data analysis, data interpretation, manuscript preparation, and the decision to submit the paper for publication. The corresponding authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
CONFLICT OF INTEREST
J.Y.C., E.C., and A.N. are employees and stockholders of Eli Lilly and Company. A.E., E.J., T.K., and M.R. are employees of Metrum Research Group, a vendor of Eli Lilly and Company.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. E.C., A.N., and J.Y.C. designed the research. E.C., A.E., E.J., M.R., and T.K. performed the research. E.C., A.E., E.J., M.R., T.K., and J.Y.C. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
Bamlanivimab emerged from the collaboration between Eli Lilly and Company and AbCellera Biologics Inc. to create antibody therapies for the prevention and treatment of COVID‐19. Bamlanivimab was developed by Eli Lilly and Company following its discovery by AbCellera Biologics Inc. and scientists at the National Institute of Allergy and Infectious Diseases Vaccine Research Center. The authors wish to thank Holly Green, PhD (Eli Lilly and Company) and Katherine Kay, PhD (Metrum Research Group), for writing assistance provided during the preparation of this manuscript. The authors also thank Lan Ni, PhD, Carl McMillian, PhD, Charles Benson, MD (Eli Lilly and Company), Kyle Baron, PhD, and Tim Waterhouse, PhD (Metrum Research Group) for scientific review and input and finally the authors greatly appreciate the support from Daniel Skovronsky, MD, PhD (Eli Lilly and Company), for overall strategic support.
DATA AVAILABILITY STATEMENT
The PBPK model and viral dynamic model used in this paper are available online (https://github.com/metrumresearchgroup/bioPBPK, https://github.com/metrumresearchgroup/vk‐covid19).
References
- 1. Dong, E. , Du, H. & Gardner, L. An interactive web‐based dashboard to track COVID‐19 in real time. Lancet Infect. Dis. 20, 533–534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones, B.E. et al. The neutralizing antibody, LY‐CoV555, protects against SARS‐CoV‐2 infection in non‐human primates. Sci. Transl. Med. (2021). 10.1126/scitranslmed.abf1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walls, A.C. , Park, Y.‐J. , Tortorici, M.A. , Wall, A. , McGuire, A.T. & Veesler, D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 181, 281–292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corr, P. & Williams, D. The pathway from idea to regulatory approval: examples for drug development. In Conflict of interest in medical research education and practice (eds. Lo, B. & Field, M.J.) (Nat. Acad. Press, Washington DC, 2009). [PubMed] [Google Scholar]
- 7. Fan, J. et al. Connecting hydroxychloroquine in vitro antiviral activity to in vivo concentration for prediction of antiviral effect: a critical step in treating COVID‐19 patients. Clin. Infect. Dis. 2020. 10.1093/cid/ciaa623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallo, J.M. Hybrid physiologically‐based pharmacokinetic model for remdesivir: application to SARS‐CoV‐2. Clin. Transl. Sci 14, 1082–1091 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu, Q. , Ahadpour, M. , Rocca, M. & Huang, S.‐M. Clinical pharmacology regulatory sciences in drug development and precision medicine: current status and emerging trends. AAPS J. 23, 1–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jang, S. , Rhee, J.‐Y. , Wi, Y.M. & Jung, B.K. Viral kinetics of SARS‐CoV‐2 over the preclinical, clinical, and postclinical period. Int. J. Infect. Dis. 102, 561–565 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nayak, S. et al. Getting innovative therapies faster to patients at the right dose: impact of quantitative pharmacology towards first registration and expanding therapeutic use. Clin. Pharmacol. Ther. 103, 378–383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galluppi, G.R. et al. Industrial perspective on the benefits realized from the FDA's model‐informed drug development paired meeting pilot program. Clin. Pharmacol. Ther. 110, 1172–1175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arrizabalaga, O. , Otaegui, D. , Vergara, I. , Arrizabalaga, J. & Méndez, E. Open access of COVID‐19‐related publications in the first quarter of 2020: a preliminary study based in PubMed. F1000Res. 9, 649 (2020). 10.12688/f1000research.24136.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim, K.S. et al. Modelling SARS‐CoV‐2 dynamics: implications for therapy. medRxiv (2020). 10.1101/2020.03.23.20040493. [DOI] [Google Scholar]
- 15. Jones, H.M. et al. A physiologically‐based pharmacokinetic model for the prediction of monoclonal antibody pharmacokinetics from in vitro data. CPT Pharmacometrics Syst. Pharmacol. 8, 738–747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dirks, N.L. & Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 633–659 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Teorell, T. Kinetics of distribution of substances administered to the body, I: the extravascular modes of administration. J. Archives Internationales de Pharmacodynamie et de Therapie 57, 226–240 (1937). [Google Scholar]
- 18. Zhuang, X. & Lu, C. PBPK modeling and simulation in drug research and development. Acta Pharm. Sin. B 6, 430–440 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Avery, L.B. et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. Mabs 10, 244–255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chakraborty, A. et al. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti‐interleukin‐1β monoclonal antibody. Clin. Pharmacokinet. 51, e1–e18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah, D.K. & Betts, A.M. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. Mabs 5, 297–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. R Core Team . R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2020) <https://www.R‐project.org/> [Google Scholar]
- 23. Sager, J.E. , Yu, J. , Ragueneau‐Majlessi, I. & Isoherranen, N. Physiologically Based Pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab. Dispos. 43, 1823–1837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malik, P.R.V. & Edginton, A.N. Physiologically‐based pharmacokinetic modeling vs. allometric scaling for the prediction of infliximab pharmacokinetics in pediatric patients. CPT Pharmacometrics Syst. Pharmacol. 8, 835–844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen, P. et al. First in human study of bamlanivimab in a randomized trial of hospitalized patients with COVID‐19. Clin. Pharmacol. Ther. (2021). 10.1002/cpt.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen, P. et al. SARS‐CoV‐2 neutralizing antibody LY‐CoV555 in outpatients with Covid‐19. N. Engl. J. Med. 384, 229–237 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keizer, R.J. , Huitema, A.D. , Schellens, J.H. & Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 493–507 (2010). [DOI] [PubMed] [Google Scholar]
- 28. Magyarics, Z. et al. Randomized, double‐blind, placebo‐controlled, single‐ascending‐dose study of the penetration of a monoclonal Antibody Combination (ASN100) targeting staphylococcus aureus cytotoxins in the lung epithelial lining fluid of healthy volunteers. Antimicrob. Agents Chemother. 2019, e00350–e00419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jadhav, S.B. , Khaowroongrueng, V. , Fueth, M. , Otteneder, M.B. , Richter, W. & Derendorf, H. Tissue distribution of a therapeutic monoclonal antibody determined by large pore microdialysis. J. Pharm. Sci. 106, 2853–2859 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Prescription Drug User Fee Act Reauthorization: Testimony by Janet Woodcock, M.D., Peter Marks, M.D., PhD., and Jeffrey E. Shuren, M.D. before the Committee on Health, Education, Labor and Pensions, United States Senate, 115th Congress (2017).
- 31. Eli Lilly and Company . Lilly Statement Regarding NIH’s ACTIV‐3 Clinical Trial <https://www.lilly.com/news/stories/statement‐activ3‐clinical‐trial‐nih‐covid19>
- 32. US Food and Drug Administration . Fact sheet for health care providers emergency use authorization (EUA) of bamlanivimab <https://wwwfdagov/media/143603/download> (2020).
- 33. Eli Lilly and Company . Lilly requests revocation of emergency use authorization for bamlanivimab alone to complete transition to bamlanivimab and etesevimab together for treatment of COVID‐19 in the U.S. <https://investorlillycom/news‐releases/news‐release‐details/lilly‐requests‐revocation‐emergency‐use‐authorization> (2021).
- 34. Hiscott, J. et al. The global impact of the coronavirus pandemic. Cytokine Growth Factor Rev. 53, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zastrow, M. Open Science takes on COVID‐19. Nature 581, 109–111 (2020). [DOI] [PubMed] [Google Scholar]
- 36. Mulangu, S. et al. A randomized, controlled trial of Ebola virus disease therapeutics. N. Engl. J. Med. 381, 2293–2303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cangelosi, R.A. , Schwartz, E.J. & Wollkind, D. A quasi‐steady‐state approximation to the basic target‐cell‐limited viral dynamics model with a non‐cytopathic effect. Front. Microbiol. 9, 54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The PBPK model and viral dynamic model used in this paper are available online (https://github.com/metrumresearchgroup/bioPBPK, https://github.com/metrumresearchgroup/vk‐covid19).