

Abstract
The water-soluble quencher hydrodabcyl can be activated as an N-succinimidyl ester that is readily accessible from crude hydrodabcyl and storable for a long time. With primary and secondary amines, it reacts swiftly and chemoselectively, even in the presence of other competing nucleophiles such as those typically present in natural peptides. One of the three phenolic OH groups of hydrodabcyl is amenable to selective mono-Boc protection resulting in reduced polarity, advantageous to its further use in organic synthesis. The advantages of hydrodabcyl over dabcyl in spectrometric applications are exemplified by the pH dependence of its absorbance spectra.
Introduction
In the investigation of biomolecular reactions, molecules containing a fluorophore and a neighboring dark quencher are often used for spectroscopic quantifications.1 Because of the presence of these two chromophores, the molecule is not fluorescent. If in the course of the reaction the fluorophore gets separated from the quencher, the appearing fluorescence can be measured with high sensitivity and temporal resolution. Dabcyl [4-(4′-dimethylaminophenylazo)-benzoic acid, 1] is a widely used dark quencher2−5 that exhibits no fluorescence and is appropriate for quenching many common fluorophores such as bimane, dansyl, coumarin, and fluorescein derivatives. Although its hydrophobicity is beneficial for organic synthesis, its poor water solubility hampers biological applications, which are usually carried out in aqueous solution. Moreover, random interactions with hydrophobic regions of biomacromolecules, membranes, and test tubes may lead to unwanted side effects. The use of water-soluble derivatives of dabcyl can help to overcome these issues. In a previous publication,6 we reported the synthesis of the water-soluble derivative hydrodabcyl [4-(2′,6′-dihydroxy-4′-dimethylaminophenylazo)-2-hydroxybenzoic acid, 2] and its purification by sequential cycles of precipitation and centrifugation to achieve purity, enabling its direct use in spectroscopic applications. For most purposes, however, hydrodabcyl has to be linked to target molecules. During organic synthesis, the water solubility and high polarity of hydrodabcyl turned out to be a major challenge. The encountered problems include laborious purification, low yields, or the necessity of specific protecting groups.
In this work, we describe a general strategy to activate hydrodabcyl (2) and to chemically link it to amino groups without the protection of its phenolic hydroxyl groups. This new strategy is based on the synthesis and isolation of the hydrodabcyl N-succinimidyl active ester [hydrodabcyl-ONSu (3), Scheme 1] from crude hydrodabcyl. This active ester is sufficiently reactive to afford significantly increased yields of amides when coupling it to amino groups. Moreover, active ester 3 is stable enough to allow convenient isolation, purification by column chromatography on silica gel with standard solvent systems, and long-term storage. In addition, we studied the pH-dependence of the UV/vis absorbance of hydrodabcyl (2) in order to understand the influence of the hydroxyl groups on its electrostatic properties and to characterize its spectroscopic properties in the pH range of potential applications.
Scheme 1. Strategies for the Coupling of Hydrodabcyl (2) to Amines Either via Acetyl Protection of the Phenolic OH Groups (Marked in Red) in 5 and In Situ Activation of the Carboxylic Acid to Obtain 6 [Previous Work (a–c)] or via Isolation of N-Succinimidyl (Marked in Red) Active Ester 3 to Obtain a Labeled Compound [This Work (d,e)].

Previous work: (a) 4.2 equiv. Ac2O, 6 equiv. NEt3, 0.1 equiv. DMAP, THF/DMSO; 80%. (b) Ser-Phe-EDANS, dipeptide labeled on the Phe-carboxy group as amide with 5-(2-aminoethylamino)-1-naphthalenesulfonic acid (EDANS), 1.6 equiv. propylphosphonic anhydride (T3P), 50% in THF, 3.3 equiv. NEt3, THF/DMF, 0 °C, 1.5 h. (c) piperidine/MeOH, r.t., 1 h; 30% yield over the two steps. This work: (d) 1 equiv. NHS, 1 equiv. CDI, DMF, r.t., overnight; 44%. (e) 1–2 equiv. amine (primary or secondary, HNR1R2), 2 equiv. NEt3, DMF, r.t., overnight; 70–100%. More details are provided in the Supporting Information.
Experimental Section
Chemicals and Analytical Methods
All reactions were carried out under a nitrogen or argon atmosphere using dry solvents under anhydrous conditions unless otherwise noted. DMF, carbonyldiimidazole (CDI), and all other chemicals were purchased from Sigma-Aldrich and used without further purification unless indicated otherwise. The 1H and 13C NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer. Chemical shifts are reported in parts per million (ppm) referenced with respect to residual solvent (DMSO = 2.50/39.52 ppm). The following abbreviations were used to indicate multiplicities: s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet, br s = broad singlet, and qdd = double doublet of quartet. High-resolution mass spectra (HRMS) were obtained by electrospray ionization and performed on a Q Exactive Orbitrap MS system, Thermo Fisher Scientific. For column chromatography, silica gel (40–63 μm, Merck) was used. The retention factors (Rf) were determined by thin layer chromatography on pre-coated silica plates (Merck TLC Silica gel 60 F254). The spots were visualized by UV light and stained with ceric ammonium molybdate solution, followed by treatment with a heat gun.
Synthesis of the Hydrodabcyl-ONSu Ester (3)
A mixture of hydrodabcyl (50 mg, 0.158 mmol) and N-hydroxysuccinimide (NHS) (18 mg, 0.158 mmol) in DMF (5 mL) was treated with CDI (26 mg, 0.158 mmol) and stirred overnight at room temperature under argon. The resulting mixture was dissolved in ethyl acetate (100 mL) and washed with aqueous citric acid (5% w/w, 2 × 50 mL) and brine (2 × 50 mL). The aqueous phases were extracted with ethyl acetate (2 × 100 mL). The organic layers were dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography on silica gel (dry package, elution with cyclohexane/ethyl acetate 1:2) to afford ester 3 (29 mg, 44%) as a dark red solid (Rf = 0.4, EtOAc); HRMS (−ESI) solution in methanol m/z: [MOMe – H]− calculated for C16H16N3O5–, 330.10954; found, 330.10951. 1H NMR (300 MHz, DMSO-d6): δ ppm 2.88 (s, 4 H), 3.09 (s, 6 H), 5.72 (s, 2 H), 7.27 (d, J = 1.7 Hz, 1 H), 7.32 (dd, J = 8.8, 1.7 Hz, 1 H), 7.89 (d, J = 8.8 Hz, 1 H), 10.62 (br s, 1 H). 13C NMR (75 MHz, DMSO-d6): δ 170.6, 170.6, 170.6, 161.3, 161.3, 160.9, 158.4, 152.9, 132.6, 125.9, 110.0, 106.8, 105.8, 91.8, 91.8, 40.1, 40.1, 25.5, 25.5. (For more details, see the Supporting Information).
General Procedure for Coupling of Hydrodabcyl to Amines (Compounds 4–11)
The amino compound (1 to 2 equiv.) was added to a stirred solution of hydrodabcyl-ONSu ester (3) (1 equiv.) in DMF (5 mL) at room temperature. The reaction mixture was stirred overnight at room temperature. Thereafter, it was diluted with ethyl acetate (100 mL) and washed with aqueous citric acid (5% w/w, 2 × 25 mL) and brine (2 × 25 mL). The aqueous phases were extracted with ethyl acetate (2 × 100 mL). The organic layers were dried over anhydrous Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography on silica gel.
Procedure for Coupling of Hydrodabcyl to a Peptide in Aqueous Solution: One-Pot Double-Functionalization of Glutathione (Compound 12)
A solution of monobromo-bimane (16.4 mg, 0.06 mmol) in acetonitrile (1.5 mL) was added to a stirred solution of glutathione (18.4 mg, 0.06 mmol) and NaHCO3 (15 mg, 0.18 mmol) in degassed water (6 mL) under argon atmosphere according to Radkowsky and Kosower.7 After 30 min, a solution of 3 (25 mg, 0.06 mmol) in DMF (2 mL) was added and the reaction was allowed to stir for another 4 h at room temperature under argon. The crude mixture was diluted with water containing 0.1% HCOOH and purified by semipreparative Amersham Äkta—900 HPLC (column Kinetex 5 μm C18 100 Å, 250 × 21.2 mm, 10 mL/min, 30% CH3CN/70% H2O + 0.1% HCOOH) to afford double-labeled glutathione (12, 5 mg, at least 20% yield after semipreparative HPLC) as a dark red solid. No other coupling products were detected. HPLC analysis showed complete consumption of 3. However, some glutathione singly labeled with bimane and free hydrodabcyl acid due to partial hydrolysis of the active ester was detected. To minimize incomplete labeling, the reaction could be optimized, for example, by adding a higher excess of 3. 1H NMR (500 MHz, DMSO-d6/D2O 9:1): δ ppm 1.64 (s, 3 H), 1.68–1.77 (m, 3 H), 1.89–2.01 (m, 1 H), 2.04–2.16 (m, 1 H), 2.26 (t, J = 6.4 Hz, 2 H), 2.30 (s, 3 H), 2.69 (dd, J = 13.5, 9.4 Hz, 1 H), 2.97 (dd, J = 13.5, 4.5 Hz, 1 H), 3.03 (s, 6 H), 3.55–3.66 (m, 2 H), 3.77 (d, J = 14.8 Hz, 1 H), 3.81 (d, J = 14.8 Hz, 1 H), 4.32 (t, J = 5.2 Hz, 1 H), 4.45 (dd, J = 9.4, 4.5 Hz, 1 H), 7.11 (dd, J = 8.5, 1.5 Hz, 1 H), 7.15 (d, J = 1.5 Hz, 1 H), 7.88 (d, J = 8.5 Hz, 1 H). 13C NMR (125.76 MHz, DMSO-d6/D2O 9:1): δ 174.5, 173.5, 172.2, 170.9, 168.1, 160.8, 160.8, 159.2, 150.1, 148.6, 147.6, 147.6, 130.8, 126.1, 113.7, 113.7, 113.5, 113.5, 111.6, 109.9, 105.3, 92.3, 92.3, 53.4, 52.6, 42.5, 40.8, 40.8, 34.4, 32.2, 27.8, 25.3, 11.8, 7.3, 7.0. HRMS (+ESI) m/z: [M + H]+ calcd for C35H41N8O12S+, 797.25592; found, 797.25302. Analytical HPLC Shimadzu (column Halo 90 Å, C18, 2.7 μm, 2.1 mm × 150 mm, 0.3 mL/min, CH3CN/H2O + 0.1% HCOOH). Method: 30% CH3CN till 7 min, from 7.5 min 40% CH3CN, tR = 6.9 min.
Titration and Absorbance Measurements
The pH was measured with a Mettler Toledo SevenMulti pH meter equipped with a microelectrode (InLab Micro, Mettler Toledo). A double beam PerkinElmer LAMBDA 750 UV/vis spectrophotometer equipped with a thermostated cuvette holder was used to record absorption spectra over a wavelength range of 250–700 nm at 20 °C in 2 mL quartz cuvettes from Hellma with a 1 cm light path. The absorbance behavior of hydrodabcyl (2) and its isopropyl amide derivative (4) has been investigated in aqueous solution over a wide range of pH values: for hydrodabcyl, from pH = 3.2 to pH = 10.5 and for its isopropyl amide derivative, from pH = 2.4 to pH = 10.5. Dabcyl (1) was investigated in aqueous solution from pH = 3.5 to pH = 10.5 and in DMSO/aqueous solution 50:50%, from pH = 2.2 to pH = 7.3.
In order to control the pH along the entire investigated pH range, the aqueous solution contained a mixture of buffers: N-cyclohexyl-3-aminopropanesulfonic acid (CAPS, useful pH range: 9.7–11.1), N-cyclohexyl-2-aminoethanesulfonic acid (CHES, useful pH range: 8.6–10), 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES, useful pH range: 2.5–3.5 or 6.8–8.2), 2-(N-morpholino)ethanesulfonic acid (MES, useful pH range: 5.8–6.5], and ammonium acetate (useful pH range: 3.7–5.7). The concentration of each compound in the mixture was 15 mM. In all the experiments, the pH of the solution was adjusted to 10.5 with drops of NaOH (19 M) and then gradually reduced by the dropwise addition of 5 M HCl. The concentration of dabcyl, hydrodabcyl, and hydrodabcyl-isopropyl amide was 12.7 μM in all experiments.
Results and Discussion
Shortcomings of the One-Pot Coupling of Hydrodabcyl (2) to Amines in the Presence of Transacylation Reagents
Hydrodabcyl (2) is to be linked to its target via an amide bond between its carboxyl group and an amino group. In the standard one-pot amidation protocol for amino acids, the carboxylic acid is activated by dicyclohexylcarbodiimide (DCC),8 followed by the formation of an active ester with hydroxybenzotriazole or NHS and eventually acylation of the amine. However, such a formation of an active ester in situ, that is, in the presence of amino derivatives, failed with hydrodabcyl (2) because of the low reactivity of its carboxyl group, which is attached to an electron-rich phenyl ring. Reactions with the stronger acylation catalyst N,N-dimethylaminopyridine (DMAP) worked but gave a high yield of 85% only in the case of the isopropyl amide of 2. The one-pot amidation of hydrodabcyl with functionalized primary amines proceeded with poor yields throughout, for example, with only 10% for the primary amide of the NαBoc-lysine ethyl ester. The coupling to peptides containing unprotected competing O-nucleophiles such as serine esters failed completely. Although we had previously reported the use of protected tris(acetyl)hydrodabcyl (5) for the labeling of a serine-containing peptide (6) in ca. 30% yield (Scheme 1), this strategy is not generally applicable due to the electrophilic nature and migration tendency of the acetyl groups. For instance, the reaction of tris(acetyl)hydrodabcyl (5) with the primary amine of the NαBoc-lysine ethyl ester exclusively afforded the primary acetamide of the Nα(Boc)-Nε(acetyl)-lysine ethyl ester. These findings led us to the conclusion that the isolation of the active ester intermediate might be required to achieve efficient coupling between hydrodabcyl (2) and structurally diverse amines.
Separation of Activation and Coupling Steps by Isolation of the Active Ester of Hydrodabcyl
For efficient labeling of amino derivatives with hydrodabcyl (2), we decided to disconnect activation and coupling by isolating the hydrodabcyl-ONSu active ester, (3 in Scheme 1). We tried out three condensation reagents for the synthesis of 3: DCC, ethyl-dimethylaminopropyl carbodiimide (EDC), and CDI. With DCC, ester 3 was obtained in less than 30% yield. Moreover, the byproduct dicyclohexylurea led to problems during purification because it is of similar polarity as 3 and cannot be removed completely from the product. The water-soluble EDC failed to produce substantial amounts of condensation products and led to the formation of emulsions upon extraction. With CDI, the desired active ester 3 was obtained in a pure form and 44% yield.
The active ester 3 is surprisingly stable and allows easy handling during aqueous workup, standard column chromatography, and long-term storage in the dark. As a solid, 3 is completely stable for at least one week at room temperature and for at least eight months in the fridge at 4 °C or freezer at −20 °C (see NMR spectra in Figure S1 in the Supporting Information). Solutions of 3 in DMSO were found to be stable for at least three months at room temperature.
Coupling of the Hydrodabcyl Active Ester to Amino Derivatives
In order to evaluate the applicability of 3 in peptide chemistry and biology, we assessed its reactivity toward different commercially available amino acid derivatives in five exemplary test reactions A–E (Scheme 2).
Scheme 2. Test Reactions of Hydrodabcyl Active Ester 3 with Primary and Secondary Amines (in Blue).
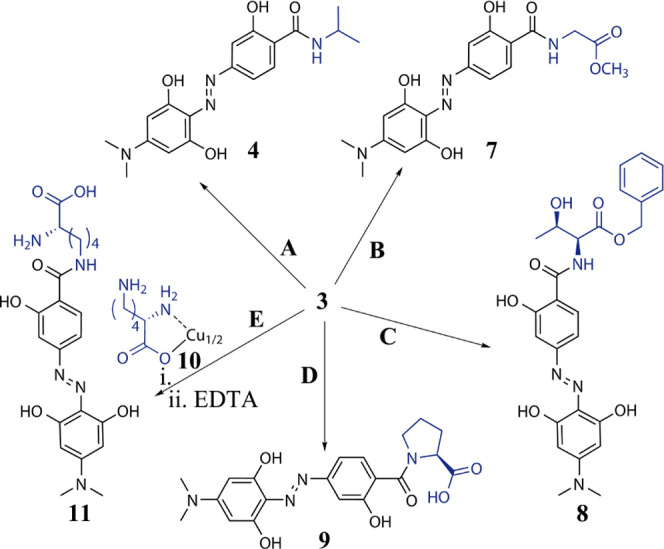
(A) 1.2 equiv. Isopropylamine, DMF; 99%; (B) 1.1 equiv. Glycine Methyl Ester Hydrochloride, 1.1 equiv. NEt3, DMF, r.t., Overnight; 99%; (C) l-threonine Benzylester Oxalate, 2 equiv. NEt3, DMF, r.t., Overnight; 70%; (D) 2 equiv. l-proline, 2 equiv. NEt3, DMF, r.t., Overnight; 92%; and (E) Two-Step Reaction: i. 2 equiv. l-lysine Copper(II) Complex 10, Water/DMF and ii. 25 mL of 0.1% Na2EDTA Solution; 30%. More details are provided in the Supporting Information.
In test reaction A, isopropylamide 4 of hydrodabcyl could be obtained quantitatively when employing active ester 3 instead of hydrodabcyl. In test reaction B, glycine methyl ester hydrochloride reacted quantitatively with 3 to give the desired hydrodabcyl amide 7. In test reaction C, the threonine benzyl ester with the unprotected hydroxy group afforded amide 8 in 70% yield. In test reaction D, the secondary amino group of proline in the presence of the unprotected carboxy group was N-acylated to give amide 9 in over 90% yield. Test reaction E showed that lysine can be regioselectively functionalized on the ε-amino group by employing its copper chelate complex910, which is soluble only in water. This reaction highlights the advantage of active ester 3 being applicable in aqueous solutions. Direct decomplexation with EDTA in a nonoptimized one-pot procedure gave ε-(hydrodabcylamido)lysine 11 in 30% yield.
The selectivity for primary and secondary amino groups that were observed in the presence of oxygen nucleophiles such as alcohol and carboxyl groups (test reactions C and D, respectively, in Scheme 2) was also confirmed in the presence of aromatic azacycles such as indole and imidazole (data not shown). To verify the possibility of selectively functionalizing the N-terminus of an unprotected peptide in aqueous solution, we carried out one-pot double-functionalization of glutathione with hydrodabcyl and bimane. Bimane is a fluorescent azacycle that is widely used for labeling glutathione due to the straightforward linking procedure. Moreover, the emission spectrum of bimane overlaps with the absorption spectrum of hydrodabcyl. Because a dark quencher is often used in combination with a fluorophore to create FRET-based fluorogenic probes,1 this test proves the real-world applicability of the new hydrodabcyl-ONSu active ester (3) in aqueous media. The reaction conditions were based on the standard procedure for functionalizing glutathione with bimane,7 followed by simply adding 3 to the same mixture (details in the Experimental Section). Double-labeled glutathione (12, Scheme 3) and no other coupling products were detected by HPLC, indicating that the reaction proceeded with high selectivity.
Scheme 3. Test Reaction of Hydrodabcyl Active Ester 3 with an Unprotected Peptide; Glutathione in Black Doubly Labeled with Bimane in Green and Hydrodabcyl in Red (12) is Obtained by a One-Pot Synthesis in Aqueous Solution (See Details in the Experimental Section).

Importance of Intramolecular Hydrogen Bonds for the Reactivity of the Phenol Groups
Hydrodabcyl (2) contains three phenolic hydroxyl groups, which do not require protection for activation and coupling under the mildly basic conditions described above. We explain their reduced nucleophilicity by their engagement in intramolecular H bonds. As shown in Scheme 4, two of these hydroxyl groups can be involved in pericyclic keto–enol tautomerism.10 Even though the enol-form 2a is probably dominant, the molecule can also exist in another tautomer (structure 2b), resulting from proton transfer within hydrogen bonds.
Scheme 4. Regioselective Mono-Boc-Protection of Hydrodabcyl (2) due to Engagement of Two of the Three Hydroxyl Groups in Intramolecular H Bonds Leading to Stable Six-Membered Rings.

The reaction sequence 2a–2d is the suggested mechanism for the observed proton–deuteron exchange in ring B. It is drawn only once to avoid overfill; however, it concerns both aromatic protons of ring B. The exchange of the second aromatic proton occurs via the same mechanism after rotation of the ring in 2d.
Moreover, the intramolecular hydrogen bonds between the diazo-ortho-phenol groups and the neighboring nitrogen atoms differ in the size of the rings that they generate, a five- versus a six-membered ring (Scheme 4). Quantum chemical calculations show that the H–N distance is 1.7 Å in the six-membered ring and 2.3 Å in the five-membered ring, suggesting that the six-membered ring is more stable due to steric restraints.
The reduced stability of the hydrogen bond in the five-membered ring may lead to a slightly higher acidity of the involved hydroxyl group compared to the two other hydroxyl groups of hydrodabcyl (2). This effect becomes obvious in the reaction of hydrodabcyl (2) with an excess of Boc2O, resulting in the selective monoprotection of one OH group of the B ring, most likely because of its engagement in the less stable five-membered ring of 2 (Scheme 4). A similar selectivity has also been described for flavonoids, where the β-ketone protects the adjacent phenol from alkylation under basic conditions.11,12 Surprisingly, proton–deuteron exchange in DMSO-d6-D2O (9:1) solution showed the complete disappearance of the signal corresponding to the aromatic protons in the B ring after one night at room temperature. Because the exchange of aromatic protons is unlikely in aqueous solutions, the transition between 2b and 2d can be explained by postulating the presence of the nonaromatic tautomer 2c (NMR spectra in the Supporting Information).
Originally, we synthesized the mono-Boc derivative 13 in order to protect 2 before its activation as the active ester 3. However, we found that the activation eventually proceeded well also without protection. Nevertheless, the Boc protection can be used when 2 needs to be purified because the precipitation of crude 2 is time consuming and does not always lead to high purity. Even though in the present work, we did not use 13 for further applications, its selective synthesis highlights the importance of intramolecular H bonds for the decreased reactivity of the phenolic hydroxyl groups in 2. Derivative 13 may also be used when hydrodabcyl needs to be introduced at an early stage of a longer synthesis, and it facilitates isolation and purification steps due to its reduced polarity. In contrast to unprotected hydrodabcyl (2), derivative 13 can be extracted from the water phase with ethyl acetate and purified by normal phase column chromatography. It is also soluble in methanol, whereas 2 is only soluble in DMSO or water. From these observations, we conclude that the intramolecular hydrogen bonds in the six-membered rings reduce the acidity and, therefore, also the nucleophilicity of the involved hydroxyl groups enabling isolation and coupling of the hydrodabcyl-ONSu ester without the need of protective groups.
pH Dependence of the UV/Vis Absorption Spectra as an Example of the Superiority of Hydrodabcyl (2) over Dabcyl (1)
As hydrodabcyl (2) was designed to be employed in spectroscopic investigations, a meticulous characterization of its spectroscopic properties is appropriate. One major aspect for applications in aqueous solution is the pH dependence of its absorbance. In addition to the protonation sites shared with dabcyl (carboxyl and diazo groups), hydrodabcyl bears three hydroxyl groups, the influence of which on its absorbance properties needs to be explored (Figures 1 and 2). Both hydrodabcyl 2 and dabcyl 1 comprise push–pull systems linking an electron-donating dimethylamino group and an electron-withdrawing acyl group. This push–pull system generates an intramolecular charge transfer, which is responsible for the main absorption band around 450 nm. We first analyzed the pH dependence of the absorbance of dabcyl in 50% DMSO where it is well soluble. The acidification of the solution (gray arrow in Figure 1a) induced a bathochromic shift of the major absorption band (blue arrow) accompanied by a hyperchromic effect. The dashed lines indicate the pH range (Figure 1a) where the carboxylate (red line) and the diazo groups (blue line) are expected to get protonated. In aqueous solution above pH = 6, the absorbance spectrum of 1a showed a similar maximum at 450 nm. Upon acidification (gray arrow in Figure 1b), a decrease of the major band was observed in the pH range where the carboxylate starts to get protonated.13−15 However, the resulting uncharged molecule 1b is poorly soluble and prone to precipitation in aqueous media.16 Therefore, the significant drop in absorbance at 450 nm below pH = 6 (red arrow in Figure 1b) was mainly due to the precipitation of 1b. The formation of visible aggregates8 due to precipitation results in spectral deformation. Not only is the absorbance at 450 nm drastically diminished, but the gain in absorbance at 580 nm is attenuated as well, due to fewer absorbing molecules in the illuminated volume (blue arrow).
Figure 1.
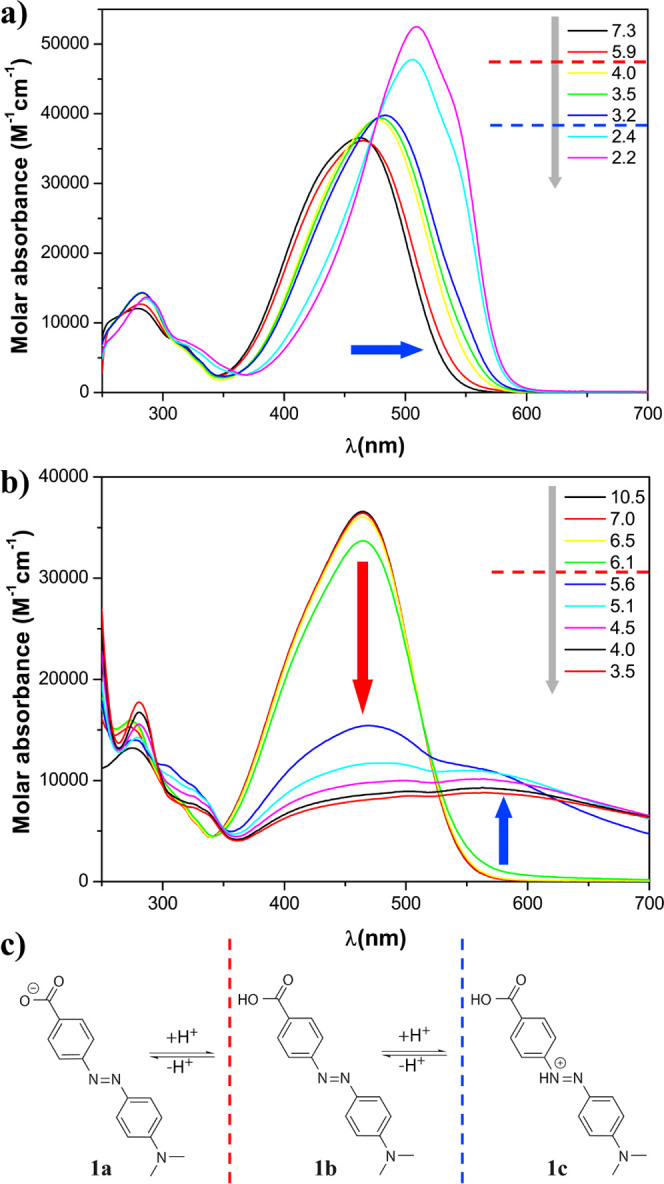
pH dependence of the molar absorbance of dabcyl (1) at 20 °C. The pH was controlled with an aqueous mixture of buffers CAPS, CHES, HEPES, MES, and ammonium acetate, each present at 15 mM. (a) 50% DMSO and 50% buffer mix; (b) 100% aqueous mixture of buffers; and (c) protonation states of 1. The dashed lines indicate the qualitative estimation of the transition between the protonation states. The gray arrows represent the experimental acidification of the solution.
Figure 2.
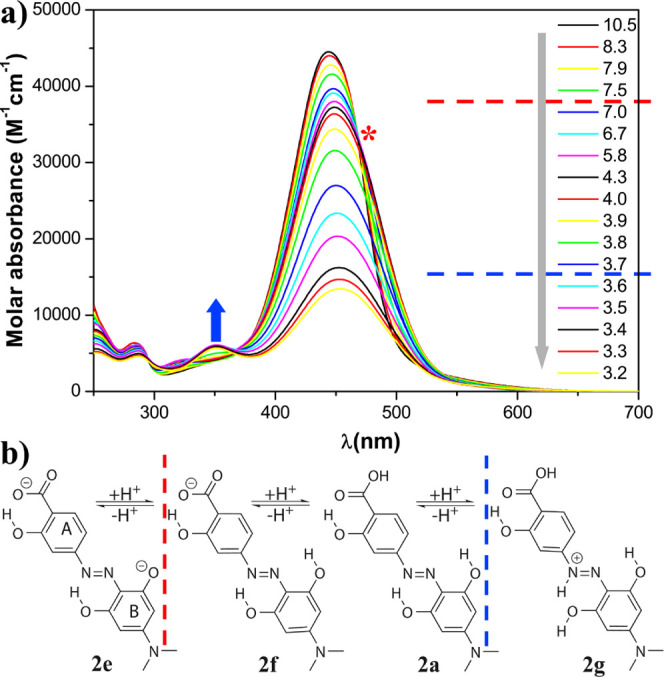
(a) pH dependence of the molar absorbance at 20 °C of hydrodabcyl (2) in an aqueous mixture of buffers CAPS, CHES, HEPES, MES, and ammonium acetate, each one present at 15 mM and (b) protonation states. The gray arrow represents the experimental acidification of the solution. The red dashed line indicates the putative pH range of the protonation of the phenolate, and the red asterisk indicates the corresponding spectral effect, that is, the isosbestic point. The blue dashed line indicates the putative pH range of the protonation of the diazo group, and the blue arrow indicates the corresponding spectral effect, that is, the appearance of the local maximum at 350 nm.
In contrast to dabcyl (1), the acidification of an aqueous solution of hydrodabcyl (2) (Figure 2a) neither led to a sudden decrease of the major band of the spectrum nor to the appearance of an additional band at a large wavelength. Consequently, the quenching ability of hydrodabcyl remained constant over a broader pH range, which is a significant advantage when it is used in dual probes for quantitative fluorimetric assays.
In the applied pH range, hydrodabcyl (2) can exist at least in four different protonation states, namely dianion 2e, monoanion 2f, neutral 2a, and cation 2g (Figure 2b). The isosbestic point at 467 nm in the pH range 10–6 is due to a small red shift of the main band, which could be attributed to the protonation of one phenolate of ring B in 2e (red asterisk in Figure 2a). At pH lower than 6, we observe only a decrease without a shift of the main absorption band. The formation of neutral 2a and cation 2g occurs in a similar pH range and cannot be distinguished by qualitative inspection of the spectra. The presence of cation 2g below pH 3.7 can be associated with the appearance of a small band at about 350 nm (blue arrow).
In most practical applications, hydrodabcyl (2) will be linked to a biomolecule through its carboxyl group. To mimic the spectroscopic behavior of hydrodabcyl in its function as an amide label, we used its isopropylamide 4 as a surrogate for the chromophore resulting from the functionalization of a peptide. The protonation states of 4 are different from those of 2, namely the monophenolate 4a, the neutral compound 4b, and the mono cation 4c (Figure 3b). However, similar arguments to rationalize the pH dependence of their UV/vis absorbance can be raised. The main absorption maximum shows similar pH dependence as in hydrodabcyl with an isosbestic point at 463 nm between pH 8 and 3.3 (red asterisk in Figure 3a), showing the equilibrium between 4a and 4b. Additionally, below pH = 2.6, we observe the appearance of a small band at 350 nm and of a shoulder at about 500 nm associated with the protonation of the diazo group in 4c (blue arrow and asterisk in Figure 3a). In contrast to hydrodabcyl (2), the latter changes in the absorbance of 4 occur at significantly lower pH values due to the absence of the negatively charged carboxylate. Additionally, in order to compare the spectroscopic behavior of dabcyl and hydrodabcyl as amide labels, which is the relevant form in most of their applications, we synthetized dabcyl-isopropylamide (14) (protocol in the Supporting Information), a nonhydroxylated congener of 4. However, 14 was completely insoluble in aqueous solution, and its absorption spectra could not be recorded. This result indicates that dabcyl may reduce the solubility of the peptide to which it is linked, underlining once more its inferiority to hydrodabcyl in aqueous solutions. A detailed quantitative analysis of the pH behavior of the compounds presented in this work is ongoing and will be the object of a future publication.
Figure 3.
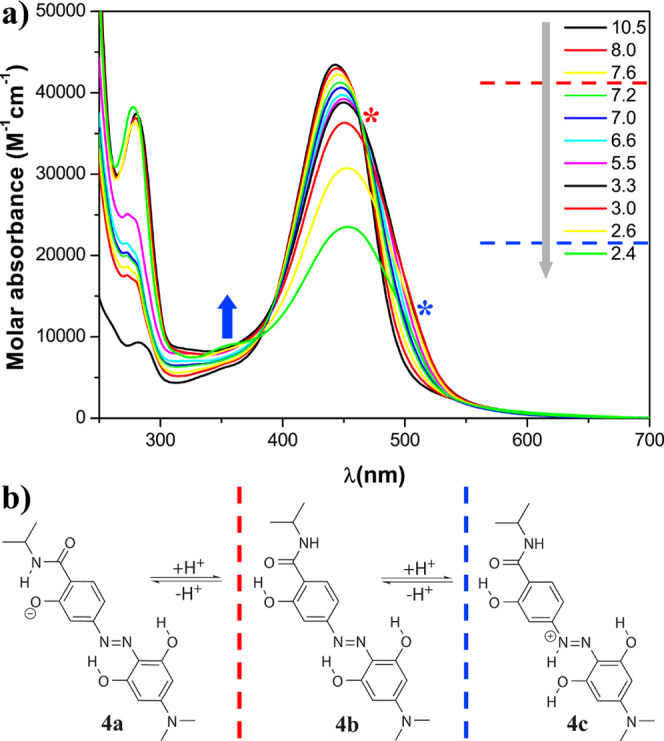
(a) pH dependence of the molar absorbance at 20 °C of hydrodabcyl isopropylamide (4) in an aqueous mixture of buffers CAPS, CHES, HEPES, MES, and ammonium acetate, each present at 15 mM and (b) protonation states. The gray arrow represents the experimental acidification of the solution. The red dashed line indicates the putative pH range of the protonation of the phenolate, and the red asterisk indicates the corresponding spectral effect, that is, the isosbestic point. The blue dashed line indicates the putative pH range of the protonation of the diazo group, and the corresponding spectral effects are indicated by the blue arrow showing the appearance of the local maximum at 350 nm and the blue asterisk showing the shoulder at around 500 nm visible below pH 2.6.
In order to provide a quick overview of the pH dependence of the absorption band that is most relevant for the potential applications, we plotted the absorbance at the maximum wavelength as a function of pH for the three compounds under investigation. For all of them, the decreasing pH induced a moderate bathochromic shift (6–8 nm). Hypochromism, however, was by far the most pronounced effect. Therefore, we monitored the absorbance at the wavelength for which we had initially measured the maximum at basic pH and which we deemed most meaningful for a description of the overall tendency. The molar absorbance (Figure 4a) showed a steep drop at pH = 5.5 for the parent compound dabcyl (1, green curve) but only at pH = 3.5 for hydrodabcyl (2, red curve) and at an even lower pH value for its isopropylamide (4, black curve). This effect has already been discussed in detail above (Figures 1–3). Nonetheless, Figure 4a shows a clear and concise roundup of the cardinal features. In the case of hydrodabcyl (2), the decrease in absorbance is attributed to the protonation in a similar pH range of the carboxylate and of the diazo group. In the case of hydrodabcyl isopropylamide (4), the decrease in absorbance at low pH can only be attributed to the protonation of the diazo group because it does not contain a carboxyl group. The abrupt decrease in absorption observed for dabcyl (1) is mainly due to aggregation, followed by precipitation in concomitance with the protonation of the carboxylate. In contrast, the precipitation of hydrodabcyl (2) is negligible and occurs at a much lower pH value due to the higher solubility of the compound. Likewise, no precipitation of 4 was detected in the investigated pH range. The fact that the protonation of the carboxylate of 2 occurs at a much lower pH value than that of 1 is due to two different effects. On the one hand, the intramolecular H bond at the carboxylate group in 2 (Figure 4b) allows for the delocalization of its negative charge to the phenolate. This delocalization over three oxygen atoms results in the stabilization of the negative charge and thus in a decrease of the acidity when compared to 1. On the other hand, the precipitation of 1 withdraws the protonated form constantly from the equilibrium in solution and leads to an apparent shift of the protonation equilibrium to higher pH values.
Figure 4.
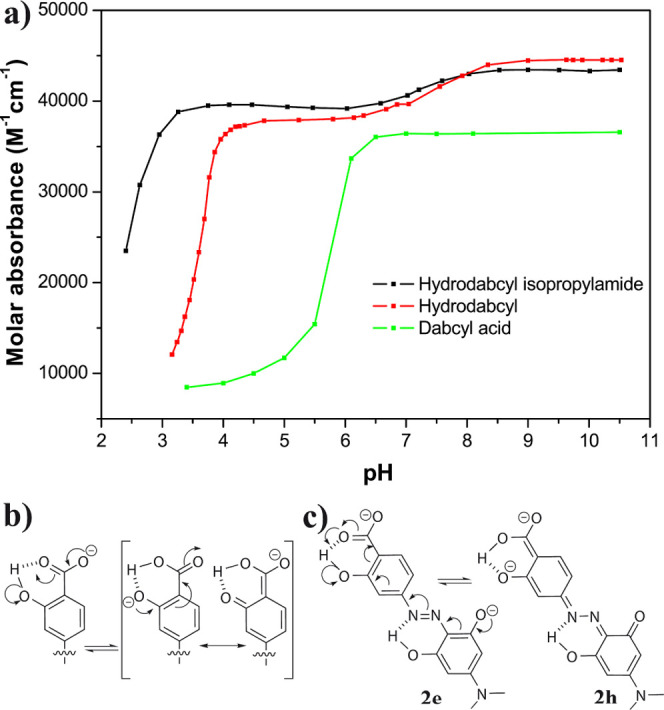
pH dependence of the molar absorbance of hydrodabcyl (2, red), hydrodabcyl isopropylamide (4, black), and dabcyl (1, green). (a) Evolution of the signal detected at the wavelength of maximal absorbance (445 nm for hydrodabcyl and its isopropylamide and 463 nm for dabcyl). Each compound was dissolved in a buffer mix at 20 °C (15 mM each of CAPS, CHES, HEPES, MES, and acetate buffer). The line connecting the experimental points is only a visual guide. (b) Three-center delocalization of the carboxylate anion leading to higher acidity of the carboxyl group in the hydroxylated compound. (c) Delocalization of the phenolates leading to comparably high acidity of the phenolic OH groups.
The absorbance change of small amplitude for both hydroxylated compounds 2 and 4 (red and black curves in Figure 4a) in the pH range between 6 and 9 can be attributed to the deprotonation of a phenol group. This deprotonation occurs at a lower pH value than expected for a standard phenol group and can be explained with the mesomeric stabilization by the carbonyl group forming a vinylogous carboxylate 2e (Figure 4c). In addition, the two negative charges can be delocalized over tautomer 2h.
The obvious advantage of hydrodabcyl (2) and its isopropylamide (4) in comparison with dabcyl (1) is their ability to retain the absorption properties over a broader pH range because of their enhanced solubility and the charge delocalization mediated by intramolecular H bonds.
Conclusions
The dark quencher hydrodabcyl (2) can be conveniently activated for amide bond formation in the form of its N-succinimidyl ester (3), which is stable in the solid state and may be stored for months in the fridge or as DMSO solution at room temperature. Hydrodabcyl N-succinimidyl ester (3) reacts selectively and swiftly at room temperature with primary and secondary amines in good to quantitative yields. This selective amide bond formation also takes place in the presence of other protein-typical nucleophilic functional groups such as hydroxyl, carboxyl, and the aromatic imino groups of imidazole and indole. Hydrodabcyl N-succinimidyl ester (3) can even be used in water. In fact, we showed that an unprotected peptide (glutathione) can be successfully labeled with bimane and hydrodabcyl N-succinimidyl ester (3) by a one-pot synthesis in aqueous solution. Furthermore, mono-Boc protection of hydrodabcyl (2) proceeds regioselectively affording a derivative with properties convenient for organic synthesis of other derivatives. The observed chemical and physical behavior of hydrodabcyl can be rationalized by the presence of intramolecular hydrogen bonds. Two of the three hydroxyl groups are engaged in pericyclic keto–enol tautomerism, which explains their decreased nucleophilicity. Due to the charge delocalization mediated by intramolecular H bonds and their increased solubility, hydrodabcyl (2) and its isopropylamide 4 retained their UV/vis absorption properties over a wider pH range in comparison to the parent compound dabcyl (1). The pH dependence characteristics presented in this paper emphasize the superiority of hydrodabcyl over dabcyl for applications in aqueous solution.
Acknowledgments
We are indebted to Prof. Stéphane Quideau who gave us the opportunity to use the facilities of his lab at the University of Bordeaux. We are very thankful to Dr. Philippe Peixoto for fruitful discussions and very helpful ideas. The present work was supported by the DFG grant BO 3578/1 and by the SFB 1357 (project C03). This publication was funded by the University of Bayreuth Open Access Publishing Fund.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04891.
Synthetic protocols and NMR spectra of new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Johansson M. K.; Cook R. M. Intramolecular Dimers: A New Design Strategy for Fluorescence-Quenched Probes. Chem.—Eur. J. 2003, 9, 3466–3471. 10.1002/chem.200304941. [DOI] [PubMed] [Google Scholar]
- Bernacchi S.; Mély Y. Exciton Interaction in Molecular Beacons: A Sensitive Sensor for Short Range Modifications of the Nucleic Acid Structure. Nucleic Acids Res. 2001, 29, e62 10.1093/nar/29.13.e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi E. D.; Wang G. T.; Krafft G. A.; Erickson J. Novel Fluorogenic Substrates for Assaying Retroviral Proteases by Resonance Energy Transfer. Science 1990, 247, 954–958. 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- Tyagi S. Taking DNA Probes into a Protein World. Nat. Biotechnol. 1996, 14, 947–948. 10.1038/nbt0896-947b. [DOI] [PubMed] [Google Scholar]
- Tyagi S.; Marras S. A. E.; Kramer F. R. Wavelength-Shifting Molecular Beacons. Nat. Biotechnol. 2000, 18, 1191–1196. 10.1038/81192. [DOI] [PubMed] [Google Scholar]
- Kempf O.; Kempf K.; Schobert R.; Bombarda E. Hydrodabcyl: A Superior Hydrophilic Alternative to the Dark Fluorescence Quencher Dabcyl. Anal. Chem. 2017, 89, 11893–11897. 10.1021/acs.analchem.7b03488. [DOI] [PubMed] [Google Scholar]
- Radkowsky A. E.; Kosower E. M. Bimanes. 17. (Haloalky1)- 1,5-Diazabicyclo[ 3.3.0loctadienediones (Halo-9,LO-Dioxabimanes): Reactivity toward the Tripeptide Thiol, Glutathione. J. Am. Chem. Soc. 1986, 108, 4527–4531. 10.1021/ja00275a045. [DOI] [Google Scholar]
- Hayne J. D.; White J. M.; McLean C. A.; Villemagne V. L.; Barnham K. J.; Donnelly P. S. Synthesis of Oxorhenium(V) and Oxotechnetium(V) Complexes That Bind to Amyloid-β Plaques. Inorg. Chem. 2016, 55, 7944–7953. 10.1021/acs.inorgchem.6b00972. [DOI] [PubMed] [Google Scholar]
- Wiejak S.; Masiukiewicz E.; Rzeszotarska B. A Large Scale Synthesis of Mono- and Di-Urethane Derivatives of Lysine. Chem. Pharm. Bull. 1999, 47, 1489–1490. 10.1248/cpb.47.1489. [DOI] [PubMed] [Google Scholar]
- Sıdır İ.; Gülseven Sıdır Y.; Berber H.; Taşal E. A Study on Solvatochromism of Some Monoazo Dye Derivatives. J. Mol. Liq. 2013, 178, 127–136. 10.1016/j.molliq.2012.11.011. [DOI] [Google Scholar]
- Karimova E. R.; Spirikhin L. V.; Baltina L. A.; Abdullin M. I. Synthesis and Identification of Quercetin Benzyl Ethers. Russ. J. Gen. Chem. 2014, 84, 1711–1715. 10.1134/s1070363214090126. [DOI] [Google Scholar]
- Lu K.; Chu J.; Wang H.; Fu X.; Quan D.; Ding H.; Yao Q.; Yu P. Regioselective Iodination of Flavonoids by N-Iodosuccinimide under Neutral Conditions. Tetrahedron Lett. 2013, 54, 6345–6348. 10.1016/j.tetlet.2013.09.051. [DOI] [Google Scholar]
- Tobey S. W. The Acid Dissociation Constant of Methyl Red. A Spectrophotometric Measurement. J. Chem. Educ. 1958, 35, 514–515. 10.1021/ed035p514. [DOI] [Google Scholar]
- Daniels F.; Alberty R. A.; Williams J. W.; Cornwell C. D.; Bender P.; Harriman J. E.. Experimental Physical Chemistry; McGraw-Hill, 1970. [Google Scholar]
- Khouri S. J.; Abdel-Rahim I. A.; Alshamaileh E. M.; Altwaiq A. M. Equilibrium and Structural Study of M-Methyl Red in Aqueous Solutions: Distribution Diagram Construction. J. Solut. Chem. 2013, 42, 1844–1853. 10.1007/s10953-013-0068-9. [DOI] [Google Scholar]
- Tawarah K. M.; Khouri S. J. The Tautomeric and Acid-Base Equilibria of p-Methyl Red in Aqueous Solutions. Dyes Pigments 1992, 20, 261–270. 10.1016/0143-7208(92)87025-v. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.