

Abstract
De novo and inherited rare genetic disorders (RGDs) are a major cause of human morbidity, frequently involving neuropsychiatric symptoms. Recent advances in genomic technologies and data sharing have revolutionized the identification and diagnosis of RGDs, presenting an opportunity to elucidate the mechanisms underlying neuropsychiatric disorders by investigating the pathophysiology of high-penetrance genetic risk factors. Here we seek out the best path forward for achieving these goals. We think future research will require consistent approaches across multiple RGDs and developmental stages, involving both the characterization of shared neuropsychiatric dimensions in humans and the identification of neurobiological commonalities in model systems. A coordinated and concerted effort across patients, families, researchers, clinicians and institutions, including rapid and broad sharing of data, is now needed to translate these discoveries into urgently needed therapies.
RGDs are defined as genetic disorders affecting fewer than 200,000 individuals in the United States1, or 1 in 2,000 individuals; however, a few highly penetrant genetic variants that occur more frequently (such as trisomy 21, affecting 1 in 600 individuals2) are often included. RGDs can be caused by the alteration of single nucleotides in one gene, entire chromosomes with hundreds of genes, or anything in between (Fig. 1a). Though they are individually rare, the contribution of RGDs to human morbidity en masse is substantial, with 18% of protein-coding genes currently implicated in such disorders (Supplementary Table 1). RGDs affect the central nervous system (CNS) disproportionately, with CNS symptoms documented in 74%3 (Fig. 1b); neuropsychiatric manifestations account for a substantial fraction of this CNS morbidity, with RGDs diagnosed in at least 40% of cases of developmental delay4 and up to 20% of cases of autism spectrum disorder (ASD)5.
Fig. 1 |. Overview of rare genetic disorders (RGDs).
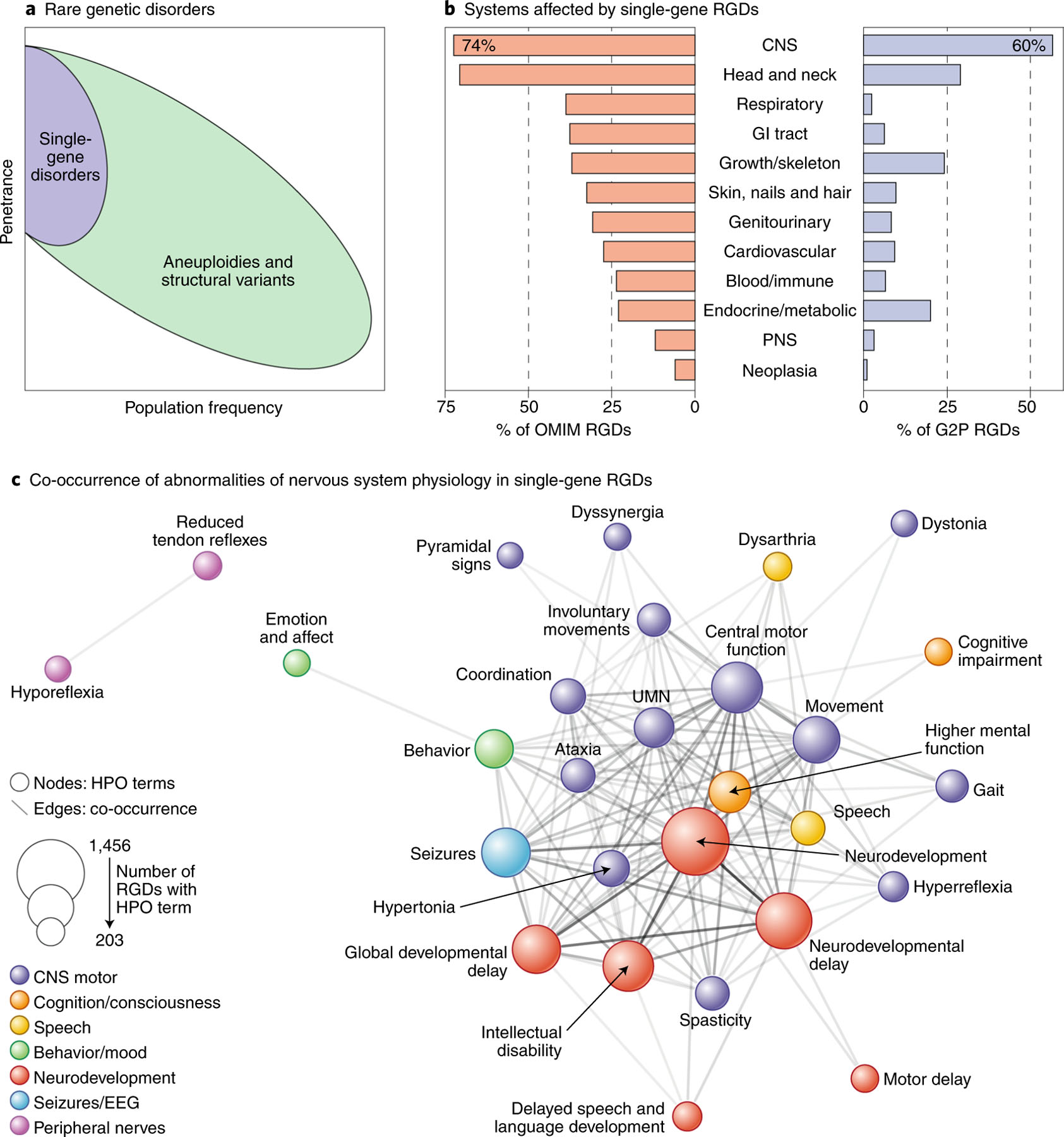
a, RGDs may be caused by variants that affect one gene (purple) or many genes (green). Many aneuploidies and structural variants arise spontaneously at higher rates than single-gene disorders, leading to comparatively high population frequencies for a given penetrance33,124. b, The CNS is involved in the majority of single-gene RGDs. c, Single-gene RGDs frequently affect multiple neuropsychiatric domains, as shown by extensive co-occurrence of Human Phenotype Ontology terms (Supplementary Table 1)125. Terms that co-occur in at least 200 RGDs are shown as nodes (colored circles, size determined by the number of RGDs), with edge weight (gray lines) determined by the degree of co-occurrence of a term between RGDs (203–1,114). Network layout is based on the Compound Spring Embedder algorithm126. OMIM, Online Mendelian Inheritance in Man; G2P, Gene2Phenotype; HPO, Human Phenotype Ontology; CNS, central nervous system; PNS, peripheral nervous system; UMN, upper motor neuron. Credit: Debbie Maizels/Springer Nature.
Some RGDs have near-complete penetrance and distinctive features that enable syndromes to be described, affected and unaffected family members to be reliably distinguished, and the underlying genetic locus identified (as for Dravet syndrome, an RGD caused by SCN1A mutations resulting in a severe form of epilepsy6,7). Newer genomic technologies, including chromosomal microarray, whole-exome sequencing (WES) and whole-genome sequencing (WGS) (see Box 1, Glossary), have identified many additional RGDs with less distinctive features, lower penetrance and/or more variable effects on phenotype. Many of these newly described RGDs have milder phenotypic impacts that may be largely brain specific (for example, 15q13.3 deletions increase risk for psychosis 18-fold8; Supplementary Table 2), in contrast to those with large effect sizes and multisystem involvement (for example, in cases of trisomy 21, intellectual disability (ID), global developmental delay and short stature are nearly universal9). Genomic technologies have also expanded the known manifestations of previously recognized RGDs (for example, many individuals with SCN2A mutations present with ASD rather than seizures10,11).
Box 1 |. Glossary.
Single-nucleotide variant (SNV)
A genetic variant in which one nucleotide (for example, C) is changed to another (for example, T).
Single-nucleotide polymorphism (SNP)
A common SNV (for example, found in ≥1% of a population).
Insertion or deletion (indel).
The gain or loss of 1–50 nucleotides, usually detected by sequencing.
Missense variant.
An SNV in a gene that changes one amino acid of the resulting protein.
Protein-truncating variant/premature termination variant (PTV).
An SNV or indel that disrupts one copy of a gene, resulting in a premature stop codon that is expected to elicit nonsense-mediated decay so that no protein is formed.
Loss-of-function (LoF) variant.
Genetic variants predicted to seriously disrupt the function of protein-coding genes, for example, a missense variant at an amino acid critical to protein function or a PTV.
pLI (probability loss-of-function intolerant) score.
Probability that a given gene is intolerant of PTVs based on lower-than-expected rates of PTVs in the general population, suggestive of selective pressure. Genes with high pLI scores (≥0.9) are extremely LoF intolerant, whereas genes with low pLI scores (≤0.1) may be LoF tolerant or there may be insufficient data to assess their tolerance.
Copy-number variant (CNV).
The gain (duplication) or loss (deletion) of ≥50 nucleotides (previously ≥1,000 bp), but often thousands to millions of nucleotides. Can be detected by microarray (large CNVs only) or WGS.
Structural variant (SV).
A large-scale rearrangement of DNA. Can include CNVs, but also inversions, translocations, or more complex rearrangements.
Germline de novo variant.
A new genetic variant observed in a child but not in either parent.
Somatic de novo variant.
A new genetic variant observed in some cells of an individual but not others.
Chromosomal microarray (CMA).
Technology that enables the number of copies of DNA to be assessed at thousands of locations across the genome, enabling the detection of CNVs. Many SNP genotyping microarrays detect SNPs at these locations too.
Whole-exome sequencing (WES).
Technology that assesses ~45 million individual nucleotides in regions of DNA that encode proteins (exons), enabling the detection of SNVs and indels.
Whole-genome sequencing (WGS).
Technology that assesses ~3.2 billion individual nucleotides across the genome, enabling the detection of SNVs, indels, CNVs and other SVs.
Locus (plural: loci).
A region of DNA ranging from a single nucleotide to an entire chromosome. For RGDs, it often describes a single gene or structural variant.
Mendelian disorder.
A disorder that follows a Mendelian pattern of inheritance. Changes at several loci may elicit the same disorder.
Complex disorder.
A disorder caused by multiple variants with a range of effect sizes at multiple loci, often in combination with environmental factors.
Neuropsychiatric.
Describes a behavioral or emotional disturbance due to an abnormality in the structure or function of the central nervous system.
Neuropsychiatric domain.
A psychological construct relevant to human behavior and mental disorders that can be measured along a continuum in health and disease (e.g., anxiety).
Autosomes.
Chromosomes 1–22 (in humans), in contrast to the sex chromosomes (X, Y) or mitochondria (M).
Autosomal dominant (AD).
Describes a pattern of Mendelian inheritance in which a single variant on one copy of an autosomal chromosome causes the disorder or trait.
Autosomal recessive (AR).
Describes a pattern of Mendelian inheritance in which two copies of a variant, one on each copy of an autosomal chromosome, must be present in order to cause the disorder or trait.
Compound heterozygous.
A pattern of Mendelian inheritance in which two different variants at the same locus, one on each copy of a chromosome, cause the disorder or trait.
X-linked recessive (XLR).
A pattern of Mendelian inheritance in which one variant on chromosome X causes the disorder in males only. It is not uncommon for carrier females to manifest mild or tissue-localized symptoms of disease due to normal or skewed X-inactivation of the functional copy of the gene in individual cells.
The full extent to which RGDs contribute to the manifestations of behavioral aberration, evident in several neuropsychiatric disorders, is still emerging. RGDs appear to contribute significantly to the genetic architecture of neuropsychiatric disorders characterized by high heritability, early age of onset, reduced fecundity, impaired cognition and behavioral deficits12–14. In pediatric patients referred for developmental delay, ID and/or ASD, the diagnostic yield of clinical genetic testing (microarray and/or whole-exome sequencing) is more than 30%15–19. It is important to note, however, that individuals with ASD with comorbid ID show a significantly higher rate of rare, de novo damaging variants than those with normal IQ, whereas there appears to be a greater contribution from common variants in those with higher IQ5,20,21. Therefore, RGDs contribute significantly to neurodevelopmental disorders, such as ID, ASD and epilepsy22,23; contribute modestly to child- or adolescent-onset neuropsychiatric disorders (attention deficit hyperactivity disorder (ADHD)24, schizophrenia8,25 and Tourette’s syndrome26); and contribute less to later-onset neuropsychiatric disorders (bipolar disorder and major depressive disorder8,14,27,28).
Understanding the contribution of RGDs to neuropsychiatric disorders is critical for patient care and for developing effective therapeutics. Diagnosing and understanding RGDs enables screening for specific risks, early detection, informed family planning, initiation of services and therapies, detailed prognosis and support. Notably, recent advances in genetically targeted therapies29,30 raise the possibility of treating the underlying pathology. At the same time, the importance of this research is not limited to those diagnosed with RGDs. These high-penetrance disorders also have the potential to provide key insights into the underlying biology of idiopathic neuropsychiatric disorders.
A US National Institute of Mental Health (NIMH) workshop was held in September 2017 with the goal of identifying strategies for advancing the understanding and management of neuropsychiatric disorders through a focus on RGDs (see Supplementary Table 3 for list of attendees). This Perspective outlines guidelines and recommendations that emerged from this workshop. In particular, three priorities were identified (see Box 2).
Box 2 |. Identified priorities for advancing neuropsychiatric research through a focus on RGDs.
Priority 1: Develop new strategies to consistently characterize the effects of RGDs on cognitive and behavioral traits that may result in disabilities and clinical referral, across the rapidly expanding spectrum of neuropsychiatric-associated RGDs routinely identified in the clinic.
Priority 2: Prioritize RGDs for exploration of pathophysiology through bottom-up approaches: building experimental models starting from the lower levels of the mechanistic hierarchy, such as gene expression and synaptic function, will help guide investigation and explain phenomena higher up in the hierarchy, such as macroscopic brain and behavioral alterations leading to psychiatric diagnoses.
Priority 3: Integrate the knowledge resulting from Priorities 1 and 2, above, to accelerate the development of new drugs and other therapeutic interventions in those with and without RGDs.
The overlap between rare genetic disorders and psychiatric disorders
Individuals affected by RGDs frequently have multiple neuropsychiatric symptoms, and specific RGDs frequently lead to multiple neuropsychiatric symptoms (grouped into neuropsychiatric domains) in different individuals (Fig. 1c). The co-occurrence of neuropsychiatric phenotypes within single-gene RGDs (Supplementary Table 1) reveals a highly connected network between developmental delay, behavioral abnormalities, seizures and abnormalities of the motor system (Fig. 1c).
Specific RGDs occupy different regions of this co-occurrence network, leading to widespread variation between RGDs in the domains affected (Fig. 2a,b). Notably, some RGDs appear to have strong effects on specific neuropsychiatric domains, as exemplified by the link between the RGD 22q11.2 deletion syndrome and psychosis8,31 and the putative protective effect of the reciprocal 22q11.2 duplication against psychosis32 (Fig. 2b, Extended Data Fig. 1, Supplementary Tables 2 and 4). In contrast, the association between the frequency of RGD diagnosis and IQ in ASD suggests that there is a global effect on cognition across many RGDs (Fig. 2a), and this notion is supported by large-scale screens in the general population5,13,33,34. In addition, recent evidence suggests that common genetic variation may influence the phenotypic expression of RGDs, and thus may contribute to variation in clinical presentation (Fig. 2a)35. More data are required to more precisely address the contribution of these RGD-causing mutations to neuropsychiatric traits, ideally from cohorts with consistent genotype and phenotype data ascertained without bias toward selection of individuals with particular characteristics, such as population-based birth cohorts, or with a proper strategy to control for ascertainment bias, such as assessing the same clinically presenting disorder across multiple RGDs and/or using first-degree relatives as controls34,36 to account for additional genetic and environmental factors (see Fig. 2c,d).
Fig. 2 |. Cross-domain impact of RGDs and limitations of current evidence.
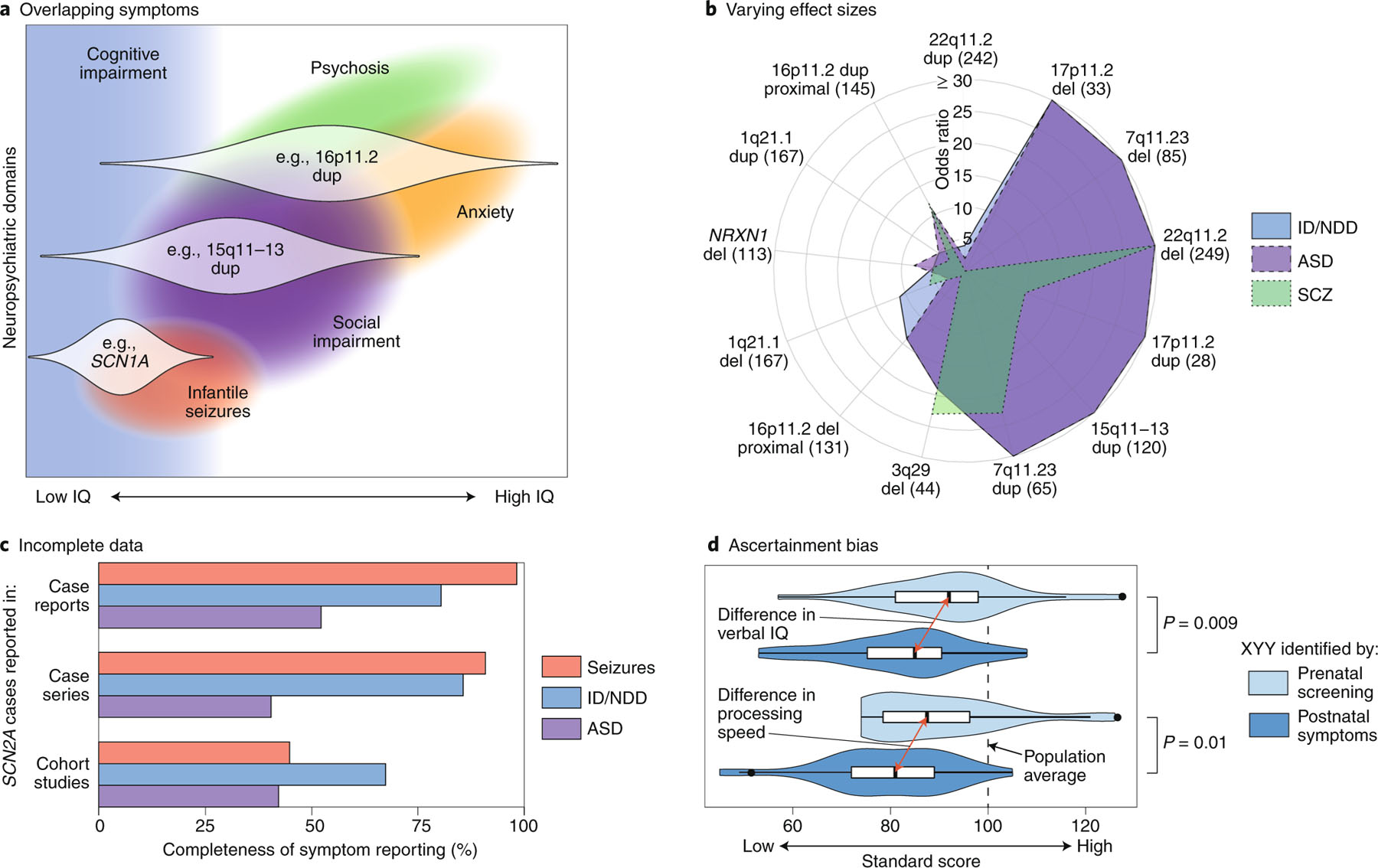
a, A theoretical model of how three RGDs (16p11.2 dup, 15q11–13 dup and SCN1A) impact multiple neuropsychiatric domains across different individuals (distribution across affected individuals shown as white violin plots)34,127,128. b, A polar plot showing the varying effect sizes (odds ratios) of different CNVs on the diagnosis of ID/NDD, ASD and SCZ (Supplementary Table 2, Extended Data Fig. 1). The number of CNV cases are shown in parentheses5,8,32,129–131; the UK Biobank was used for controls33. c, The completeness of symptom reporting for SCN2A mutations varies widely between publications132. Case reports describe a single SCN2A mutation in one case or family; case series describe multiple SCN2A cases; cohort studies describe hundreds of cases with the same disorder (for example, ID/NDD), some of which are found to have SCN2A mutations. This reporting bias, which is likely to be present for most RGDs, complicates comparisons across neuropsychiatric domains and between RGDs. d, The severity of symptoms in XYY aneuploidy varies between cases ascertained by prenatal screening (light blue) and those ascertained on the basis of clinical symptoms (dark blue)133. This ascertainment bias, which is also likely to be present for most RGDs, also complicates cross-disorder comparisons and potentially inflates estimates of effect size and penetrance. ID, intellectual disability; NDD, neurodevelopmental delay; ASD, autism spectrum disorder; SCZ, schizophrenia; IE: infantile epilepsy; CNV, copy-number variant. Credit: Debbie Maizels/Springer Nature.
Further confounding our understanding is that the phenotypic severity within each RGD varies widely (Fig. 2a). Presently, our knowledge of individuals with RGDs is biased toward those who are clinically identified and thus likely more severely affected, with milder cases (Fig. 2d) and atypical presentations under-represented12,13. At the same time, studies of brain, cognition and behavior in RGDs (i.e., phenotyping studies) often have the opposite bias, assessing only high-functioning individuals. This ascertainment bias complicates genetic counseling, as it is not yet possible to provide an accurate representation of prognosis.
To address Priority 1 (Box 2), we think that proper characterization of RGD effects on brain-related phenotypes requires moving away from categorical diagnoses (such as ASD) to neuropsychiatric domains assessed as quantitative traits (such as social impairment)37, as advocated for by the NIMH Research Domain Criteria (RDoC) initiative38,39, and assessing traits consistently across carriers who were referred to the clinic (probands) and carriers who were not (family members, individuals from unselected populations) regardless of presenting diagnosis (Fig. 2d)36,40,41. Capturing the full spectrum of phenotypic severity and controlling for ascertainment bias that often leads to inflated estimates of severity (Fig. 2d)42 may require conducting population-level analyses of hundreds of thousands of individuals, for example, using birth cohorts or health system registries13,33,34,43, and applying this experimental design across generations to capture early- and late-onset manifestations34,36,44,45.
Prioritizing RGDs for research
We think that the allocation of resources for research into an RGD should be commensurate with the level of evidence for genetic association to a specific neuropsychiatric domain. In order for resources to be provided for research, genome-wide statistical association should generally be demonstrated for a neuropsychiatric domain4,46,47 in a defined cohort4,5,8,23,48–50; however, for very rare neuropsychiatric disorders (such as childhood-onset schizophrenia), a lower level of evidence may be appropriate51. Further, in order to identify supporting mechanistic insights, we assert that it is important that functional data are used to enhance these statistical models (for example, protein-truncating variant (PTV) as opposed to missense mutation data, pLI score; see Box 1, Glossary)52. For genes for which clear functional assays are available, systematic evaluation of observed variants in both cases and controls11,53 may also contribute to statistical association. In the absence of clear biomarkers (see below) indicating a role for a genetic variant in the etiology of disease, observing downstream, nonspecific effects for individual variants, such as altered synaptic function or behavioral assays, is not a substitute for identifying a statistical association between the genetic variant and the human condition. For genetic loci containing multiple genes (such as CNVs), association evidence for a locus does not automatically imply that a single gene is driving the effect. For many recurrent CNVs, multiple genes with smaller individual effects appear to contribute to the overall risk (Supplementary Fig. 1)5,54. The additive contribution of polygenic, common risk should also be considered5,21,54,55.
For the majority of RGDs associated with neuropsychiatric domains, the evidence for statistical association comes from the observation of multiple de novo mutations at the same gene or locus in independent cases4,5,8,23,48–50. Statistical association is calculated from three metrics: (1) the number of independent cases (affected individuals) with de novo mutations at the gene or locus; (2) the denominator, i.e., the total number of cases assessed to find the mutations; and (3) the mutation rate, i.e. the chance of observing a similar de novo mutation in an unaffected individual. Mutation rate is highly dependent on gene size, with larger genes having higher mutation rates (Fig. 3a) and therefore requiring more de novo mutations in cases to demonstrate association (Fig. 3b)10,46,47. Defining these three metrics is straightforward in research cohorts4,52; however, when considering multiple clinical reports56, the denominator becomes all neuropsychiatric cases that have undergone genetic testing worldwide. Estimating this worldwide denominator as 100,000 at present, at least 5–10 confirmed and independent de novo PTV mutations are required to achieve genome-wide association, depending on gene size (Fig. 3b, Extended Data Fig. 2).
Fig. 3 |. Thresholds for genome-wide significant association.
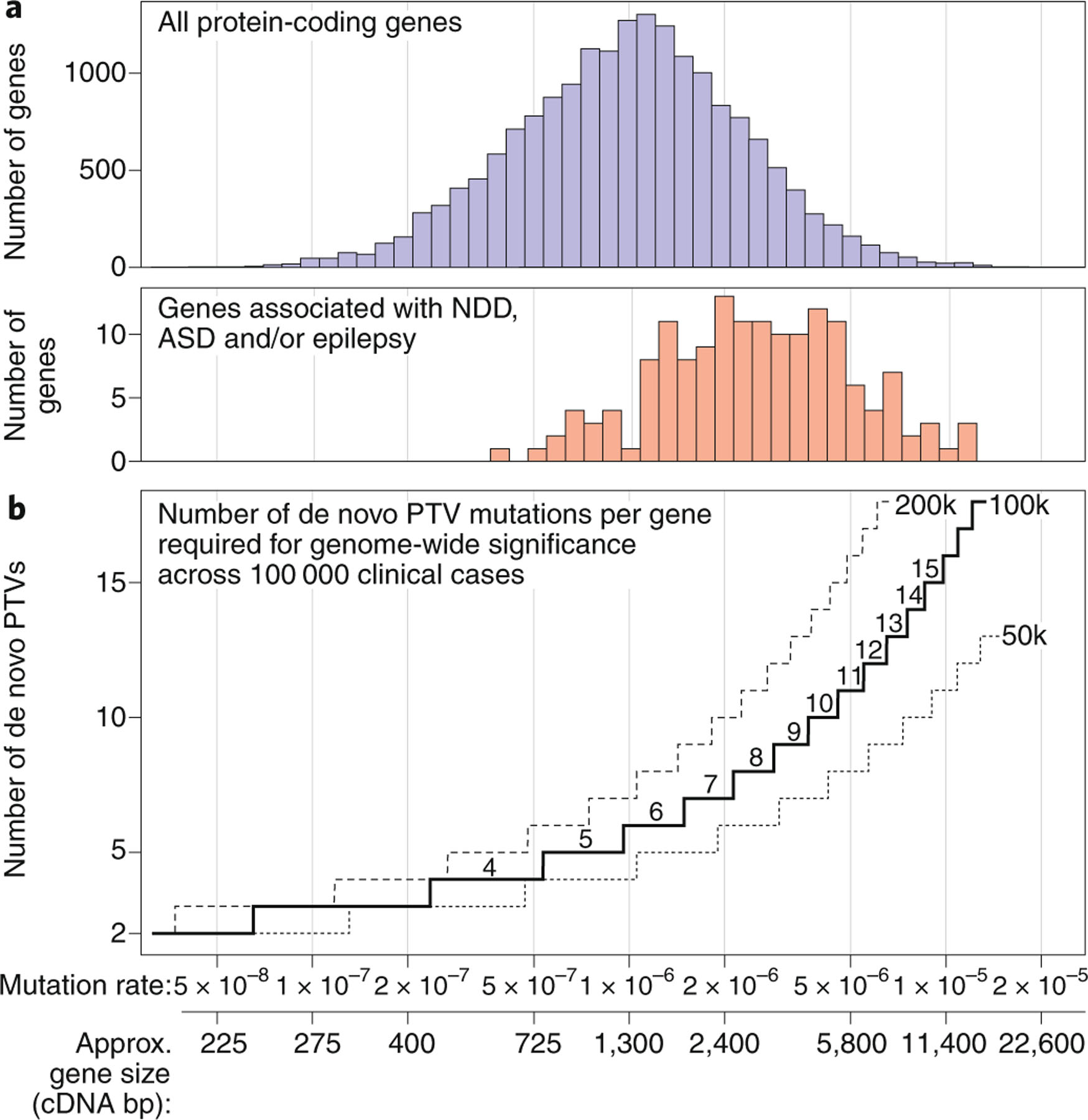
a, Mutation rates vary across genes based primarily on gene size (cDNA) but also on sequence (for example, GC content)10,47. Genes associated with neurodevelopmental delay, ASD and/or epilepsy to date tend to be large, with higher mutation rates4,23,52. b, Tens of thousands of individuals with neuropsychiatric disorders have been sequenced to date. To estimate the number of independent de novo protein-truncating variants (PTVs) across multiple case reports that are required for reliable association of a gene with a neuropsychiatric disorder, we used a Poisson distribution. Expected mutation rates in 50,000, 100,000 and 200,000 individuals are estimated from controls across the range of gene sizes (see Supplementary Methods and Extended Data Fig. 2)5,47. ASD, autism spectrum disorder; NDD, neurodevelopmental delay; GC, guanine-cytosine134. Credit: Debbie Maizels/Springer Nature.
Once a genetic locus meets the threshold for genome-wide association, other factors should be considered in selecting which RGDs to study, including the effect size on behavioral domains, level of risk for a categorical disease manifestation, population frequency, tractability for experimentation and therapeutic potential. Given the complexity of neurobiology, there are important roles for both detailed assessment of individual genes and consistent assessments across multiple genes.
Progressing from genotype to mechanisms
The second priority we identified is the design of bottom-up approaches in order to progress from genetic etiology to mechanism. Because RGDs are genetically defined, they provide an unparalleled opportunity to understand the pathophysiology of psychiatric domains and conditions. However, making the link between a genetic variant and the underlying mechanism (Fig. 4a) for a psychiatric domain or disorder presents a significant challenge57,58. Conceptually, the problem can be divided into two parts: first, to link a genetic variant to a biochemical and/or biological consequence in a specific type of neuron or glial cell, and second, to connect a functional change in a set of cells to a defect in a circuit in a specific anatomical region of the brain at a particular time in development. Although linking these diverse levels seems daunting, the prospects for success are greatly improved by the ongoing development of experimental and conceptual tools, from wet-bench to bioinformatic approaches58,59.
Fig. 4 |. Functional assays across disorders and models.
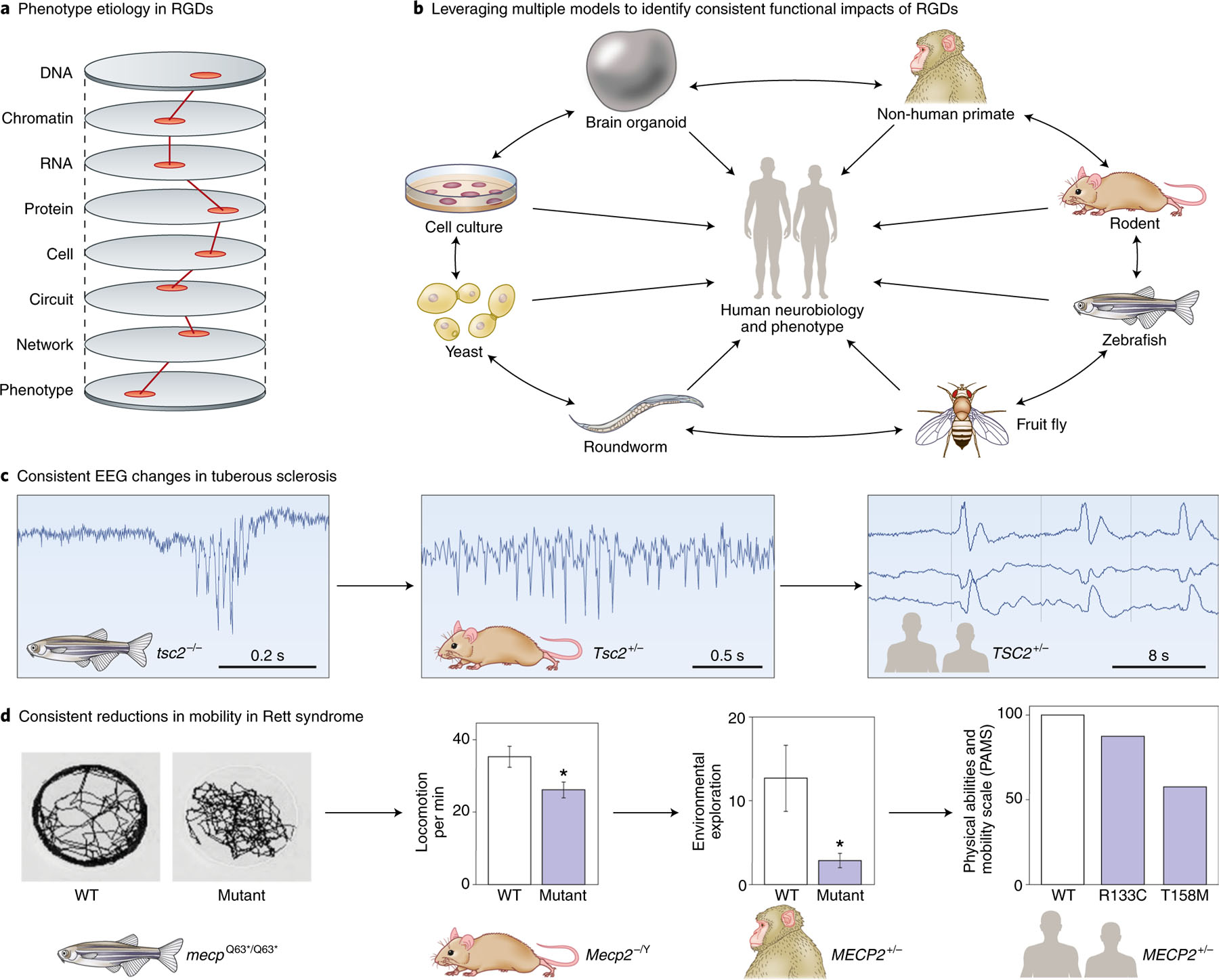
a, To understand the etiology of neuropsychiatric disorders, we need to identify the minimal ‘causal path’ by which the effects of the RGD lead to the phenotype, as shown by the hypothetical red line. Future therapeutics or biomarkers would be expected to interact with this causal path. b, No model experimental system perfectly recapitulates the human brain. By performing similar assays across multiple models, we can identify consistent consequences of RGDs, while leveraging the strengths of each model. These need to be related back to humans through similar assays or testing model predictions. c, Seizure activity is consistently observed in models of tuberous sclerosis (TSC2), though a homozygote model is used in zebrafish135–137. d, Mobility is consistently reduced in models of Rett syndrome (MECP2); as with TSC2, the specific genetic lesion assessed varies between models138–141. Credit: Debbie Maizels/Springer Nature.
To pinpoint the subcellular location of a specific gene implicated in a disorder, it is possible to use genetic tagging with fluorescent proteins and imaging using ultra-resolution microscopy in human neurons created from stem cells in vitro. The function of the gene can be determined by using CRISPR-Cas9 genome engineering tools to knock out or overexpress the gene and observing the cellular consequences using a combination of high-content microscopy, calcium and voltage imaging, high-throughput electrophysiology and RNA sequencing. Although these tools have traditionally not been amenable to the study of multiple variants, the development of automated microscopes and patch clamps that are used for high-throughput drug screening is rapidly changing this. To establish the time in development and the anatomical location that is affected by a particular genetic variant, a set of gene atlases of the developing human brain have been generated by the Allen Institute and other groups that allow gene expression to be ascribed to specific types of cells in specific regions of the brain at particular times in development (http://portal.brain-map.org). Moreover, recent progress in recapitulating neural development in vitro using pluripotent stem cells offers a powerful platform to manipulate and investigate the roles of specific disease genes in human neurons and glial cells60. This recent work demonstrates the high reproducibility and validity of these systems for studying many human brain developmental processes.
By looking for cell types that are enriched in the expression of specific genes, it has been possible to determine that the phenotypes associated with particular RGDs are caused by dysfunction in a specific type of cell. For example, phenotypes associated with mutations in the gene SCN1A that cause Dravet syndrome are due to defects in fast-spiking interneurons61, and glutamatergic neurons in the cerebral cortex are implicated in ASD52,62–64. A similar strategy can be used to identify the developmental window during which a particular genetic variant leads to a disorder by narrowing down the expression of the gene to particular periods during development, as is the case for mutations in FOXG1, which likely lead to neurodevelopmental disorders by affecting the differentiation of cortical glutamatergic neurons during brain development65. Similar approaches have been applied to structural and functional neuroimaging data, testing multiple brain regions and circuits to identify those that are distinct versus shared across disorders66–68, as well as neuroanatomic patterns that predict subsequent disease69–71. Advances in data sharing have enabled the analysis of large-scale, multisite cohorts72–74. Large-scale harmonized phenotypic data across RGDs could be analyzed in a similar manner and integrated with imaging and genomic data.
Identifying the circuits
The development of new, effective treatments is hindered by our current very limited understanding of the circuit abnormalities that underlie different neuropsychiatric disorders. RGDs offer a unique roadmap to the identification of these circuits because distinct underlying biochemical defects have now been shown to manifest with common phenotypes. For example, genes with roles in glutamatergic receptors (such as GRIN2B), sodium channel activity (such as SCN2A) and chromatin remodeling (such as CHD2) can all manifest both in the behaviors of ASD and in seizures5,23,52. In all likelihood, these mechanistically distinct genetic causes of disorders have common phenotypic manifestations through convergent effects on key circuits. The convergent effects of the mutations responsible for these circuit abnormalities therefore offer a unique set of probes to identify those circuits.
Although in vitro tools are essential for identifying the biochemical and cell biological consequences of genetic variants, identifying the neuronal circuits that lead to changes in behavior requires a more intact system. Well-defined and replicable genetic loci associated with RGDs provide a unique and indispensable opportunity to generate experimental animal models. These experimental models do not replicate the complexity of human neuropsychiatric disorders, but are essential as scientific tools to interrogate the effects of disorder-associated genes and variants on circuit properties75. In fact, it is at this juncture of genetics and neurobiology that RGDs present an unmatched advantage to investigate pathophysiological mechanisms across multiple levels (Fig. 4a). Using a combination of recently developed technologies such as optogenetics, chemogenetics, trans-synaptic tracers and single-cell sequencing in both animal models and human cells76–79, we now have the ability to determine the effect of genetic variants on specific cells and networks with unprecedented detail.
Genetically engineered mouse lines can offer important insights into gene function that provide a basis for probing pathological mechanisms in RGDs. Although behavioral phenotypes of mouse models may not resemble human disorders, the molecular, cellular and circuit phenotypes are more likely to be informative. Cross-species validation of animal models provides evidence for evolutionarily conserved electrophysiological signatures and cellular phenotypes that may enhance confidence in the relevance of the genetic perturbation (Fig. 4b–d)80–82. Regardless of the approach, replicable and rigorously collected data are beneficial, including replication of key findings in a second cohort83.
Nonhuman primate (NHP) experimental systems, such as are used in studying depression84 and schizophrenia85–88, offer several advantages, including structural and functional similarity of the prefrontal cortex to humans, presence of similar cortical–subcortical circuits and complex behavior repertoires89. These benefits come at the expense of lower throughput and substantially greater costs and ethical concerns. As with other models, validation of resulting findings in humans will be essential.
Alongside animal models, advances in cell reprogramming and differentiation, as well as CRISPR-Cas9 methods, enable the study of human neurons and glial cells in specific RGDs90,91. In particular, three-dimensional (3D) cultures, also known as organoids, resembling specific brain regions and in-vitro-assembled 3D cultures to model inter-regional cell-cell interactions92 hold promise in capturing previously inaccessible aspects of human development. Transplantation of human-derived neural cells into animals could reveal defects associated with circuit-wide integration in RGDs. Moving forward, further improvements (for example, cellular maturation, spatiotemporal control, scalability) and validation of human cellular models will be needed90.
The discovery of biomarkers for patient selection for clinical trials, confirmation of target engagement (i.e., determining that the intervention has the predicted effect on its hypothesized mechanism of action) or prediction of treatment response that translates between species, including humans, would transform the utility of such experimental systems81,82,93,94. Without such biomarkers, our best hope is to look for convergence across multiple RGDs in multiple experimental systems. Rather than advocating for one animal or cellular experimental system, we should encourage the development of multiple avenues of investigation80 for the RGDs that rank highest in the prioritization criteria (above) and for assays that can be compared across multiple systems (Fig. 4b–d)95. In all animal or cellular experimental systems, genetic background can affect the results profoundly96, and experiments that replicate results across multiple strains or clones should be encouraged97.
Therapeutic development
Aside from some metabolic disorders98, very few RGDs currently have treatments guided by genetic findings. Recent advances in genetic therapy, including the landmark improvement of infant mortality and neurological function in patients with spinal muscular atrophy (SMA) treated with the antisense oligonucleotide nusinersen29 or Zolgensma gene replacement30, offer grounds for cautious optimism. However, potential therapies have yet to modify cognitive or behavioral symptoms in humans in fragile X syndrome99, tuberous sclerosis100 or Rett syndrome101, despite promising results in mice102–104. Gene dosage is important in many such disorders, with too much or too little causing symptoms11,105, presenting challenges in titrating therapy. Consequently, recessive loss-of-function disorders, such as SMA, may be easier to treat than dominant loss-of-function disorders. Delivery of a therapeutic to the brain presents an additional challenge, further complicated if the therapy needs to be targeted to a specific cell type or brain region.
The extent to which neuropsychiatric symptoms are modifiable in humans varies by domain, age and RGD. Although some domains can be modified years after onset (for example, seizures, psychosis, attention), others may require early treatment, as suggested by the critical period of visual cortex development106 or the relationship between early management of phenylalanine hydroxylase deficiency (PKU) and adult IQ98. With this in mind, therapies that have failed in adults and adolescents are being tested in 2–6-year-olds with fragile X syndrome107 and infants with tuberous sclerosis108,109. Prenatal screening for many RGDs is feasible with current technology, and there is precedent for neonatal treatment for highly penetrant RGDs29.
Clinical trials require predefined outcome measures. In neuropsychiatric disorders, this usually entails caregiver questionnaires regarding symptom severity and functioning. There is considerable need for more reliable, sensitive and objective measures. Measures of brain function that can be compared to model systems, such as EEG or functional MRI, should be included in trial protocols to help demonstrate target engagement by the therapy and refine dosing for future trials94. Establishing multisite databases of individuals with RGDs, alongside genotype and longitudinal phenotype data, for example, by extracting knowledge from patient’s clinical notes110,111, would reduce the costs of trials and help identify subgroups with the greatest potential to respond. Finally, clinical trials need to be of sufficient size to yield clear positive or null results99,112,113.
Phenotyping and harmonization
To date, most phenotypic descriptions have focused on individual RGDs, ranging from case series to quantitative assessments of large patient registries. Such studies provide vital, clinically relevant insights that can have important therapeutic implications114,115. At the same time, the overlap among neuropsychiatric domains, within and between RGDs, provides the opportunity for cross-domain and cross-disorder analyses44,116, which may identify biologically defined subcategories of neuropsychiatric domains117.
Achieving this vision will require a community-wide initiative to collate consistent, harmonized genetic data with quantitative measures of cognition and behavior across multiple RGDs and multiple neuropsychiatric domains. No single set of clinical data elements adequately describes all RGDs. The variables to collect in a natural history study should be broad and based on features of the disorder, including morbidities that are most important to patients, those that are most likely to be life limiting, potential prognostic characteristics and those that may help formulate sensitive clinical endpoints118. The resulting data repository should reflect existing National Institutes of Health (NIH) data-sharing policies and standards, and become part of the NIH data sharing ecosystem to secure access for researchers, clinicians and institutions, enabling a wide range of hypothesis-testing with no restrictions on data use and appropriate human subjects and privacy safeguards119,120. This effort will also require standardized phenotypic ontologies, which can be achieved by adapting existing ontologies to ensure that adequate and comprehensive terminology exists for neuropsychiatric symptom manifestations across the lifespan.
Other stakeholder actions
The challenges and opportunities presented above require coordinated efforts among the community of stakeholders to fund, execute, coordinate and communicate research (see Supplementary Table 5). Patient- and family-based organizations fulfill an essential role of providing support and decreasing isolation experienced by patients and caregivers; these organizations also have a key role in forming interactive networks that connect them with researchers and health care systems (for example, to keep families informed about the latest clinical trials and research findings). Although some examples of interactive research and family-based networks for RGDs already exist, they are not all focused on or inclusive of neuropsychiatric disorders, and efforts need to be made to use these networks for such purposes. Such networks will contribute not only to larger sample sizes, which are critical for research progress, but also to organizing healthcare systems to collect consistent phenotypes that include neuropsychiatric measures to enhance data harmonization, as well as implementing standards for genotyping and phenotyping. Academic health systems, in partnership with other health care systems, can play a pivotal role in these networks by expanding the reach of their initiatives and imparting knowledge gained from the research. Some registries and databases will use a federated approach, and some will utilize a centralized repository approach. Both will be useful for specific purposes, with federated approaches allowing for more flexibility in individualization and ‘real-time’ accrual of data, and centralized repositories providing more digested, pre-harmonized data that require less work by the user before analysis, but also restricts usage. Both types of networks will clearly be useful, and will continue to be expanded upon with respect to data types included, with electronic health records (EHRs) quickly becoming an additional data source.
Recently, clinical notes from patient EHRs have been identified as important sources of clinical and demographic information. This has led to the development of a variety of knowledge-extraction tools that transform the raw text content of clinical notes into structured associations between patients and phenotypes defined in terminologies such as the Systematized Nomenclature of Medicine–Clinical Terms and the Human Phenotype Ontology. Knowledge extracted from clinical notes is of increased value to research into RGDs given the scarcity of patient data, necessitating the use of natural language processing engines to extract structured knowledge from clinical notes110. In addition, biobanks are increasingly being developed to support organized collections of biological specimens, genomic data and associated clinical information—including EHR data—from broadly consented, diverse patient populations contributing to RGD research121–123.
Partnerships among patient and family organizations, academic sites and other health systems will provide an important platform for working with funding agencies to emphasize the need for the implementation of core, cross-diagnostic phenotypic measures and the use of broad neuropsychiatric measures to facilitate data harmonization. This approach will culminate in early collaboration and coordination across groups working in this space, resulting in the design of maximally effective initiatives, data sharing and dissemination of information and knowledge gained. Furthermore, partnerships will create a platform for collaboration with pharmaceutical companies, encouraging alliances in preclinical and translational research toward drug development. However, data harmonization for collaboration necessarily requires data sharing. The community should follow NIH and established standards in the field for phenomic, genomic and biosample sharing. Although this can be done retrospectively, optimal sharing will begin before data collection, and include sharing of variables and even study methodology, to ensure replication and maximal generalizability of findings.
Drug development programs require a strong scientific foundation, including detailed understanding of the natural history of a disorder. Because of the small numbers of patients affected, and because clinical experience is dispersed among clinical referral centers, the natural history of RGDs is rarely well characterized. The US Food and Drug Administration advises sponsors to evaluate the depth and quality of existing natural history knowledge early in drug development118. Close collaboration with patient and family groups and all stakeholders is essential to design and conduct impactful natural history studies for future potential drug design and approval.
Several examples (Supplementary Table 5) illustrate the potential contributions of such convergent efforts. The benefits of coordination across stakeholders are immense, and could create resources enabling investigators to merge and integrate available databases, examine existing data across disorders, and define the best data formats for prospective studies that use consistent broad and deep phenotyping to facilitate harmonization efforts. Furthermore, it fosters complementary approaches for building resources and establishing cohorts (for example, mining of electronic medical records, clinical recruitment, patient advocacy) with synergism that propels the field.
Conclusions
We think that RGDs hold important biological clues into the mechanisms underlying complex neuropsychiatric disorders, such as ASD and schizophrenia, that can be leveraged in model experimental systems, with the view to understanding the etiology underlying the full spectrum of these neuropsychiatric conditions. Although the individual features of each RGD are important, it is the commonalities across RGDs that hold the greatest promise to transform our understanding and management of neuropsychiatric disorders. Identifying these commonalities in phenotype and neurobiology will require coordination and sharing of methods, data and resources across individuals and institutions, following the lead of the NIH and genomics community. Although RGDs and the neuropsychiatric domains they impact pose substantial challenges, the opportunity these rare disorders offer for illuminating mechanisms is likely to be transformative for scientists, clinicians and, ultimately, patients.
Extended Data
Extended Data Fig. 1 |. Impact of RGDs on neuropsychiatric domains.

a, Many RGDs impact cognition, measured by IQ. For CNVs, the decrease in IQ (x axis) can be predicted by considering the pLI score of the genes within the CNV. CNVs that are predicted to markedly reduce IQ are more likely to be de novo (y axis), based on logistic regression (blue line) of 2,743 CNVs detected in patients with neurodevelopmental disorders and the general population (gray distributions at top and bottom). Updated analysis from ref. 54. b, In Fig. 2, we show the odds ratio for ID/NDD, ASD and SCZ across different CNV loci. Here, we show an equivalent plot for single-gene RGDs. Insufficient control data exist to estimate odds ratio, and therefore we show the percentage of cases with ID/NDD, ASD, and IE based on curated publication review applied equally across genes (https://dbd.geisingeradmi.org) with the number of cases are shown in parentheses (see Supplementary Table 2 for numbers). Abbreviations: ID, intellectual disability; NDD, neurodevelopmental delay; ASD, autism spectrum disorder; SCZ, schizophrenia; IE, infantile epilepsy; pLI, probability loss-of-function intolerant.
Extended Data Fig. 2 |. Thresholds for genome-wide significant association with de novo PTVs.

a, Gene mutability is a function of gene length (cDNA) and sequence context (particularly GC content). b, RGD gene discovery from exome sequencing has been driven by de novo mutations, leading to a bias towards larger genes with higher mutability. c, Thresholds of statistical association (colored lines) are estimated for a given number of de novo PTV mutations (3, 5, 10, and 20) as cohort size (x axis) and gene mutability/size (y axis) varies. P values are estimated based on the rate of de novo PTV mutations in controls4 and a Poisson distribution (see Methods for details). Abbreviations: pLI, probability of loss-of-function intolerance; ASD, autism spectrum disorder; DDD: Deciphering Developmental Disorders; GC content, guanine-cytosine content.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This paper offers a synthesis of the ideas generated at the NIMH-sponsored workshop “Rare Genetic Disease Workshop: Window into Genomic Risk and Resilience of Mental Disorders,” held in September 2017, with the goal of discussing research and clinical opportunities presented by recent discoveries of RGDs with high risk for developmental neuropsychiatric disorders. Analyses utilize data generated by the Saguenay Youth Study, OMIM (https://www.omim.org), ExAC (http://exac.broadinstitute.org/) and the DECIPHER Consortium, including the Developmental Disorders Genotype-Phenotype Database (DDG2P, https://decipher.sanger.ac.uk/info/ddg2p). A full list of centers that contributed to the generation of the DECIPHER data is available at http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. We also thank G. Senthil for helpful feedback on the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41591-019-0581-5.
Peer review information Hannah Stower was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.US Food and Drug Administration. Orphan Drug Act. (1983).
- 2.Loane M. et al. Twenty-year trends in the prevalence of Down syndrome and other trisomies in Europe: impact of maternal age and prenatal screening. Eur. J. Hum. Genet. 21, 27–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKusick-Nathans Institute of Genetic Medicine. Online Mendelian Inheritance in Man, OMIM® (Johns Hopkins University, Baltimore, MD, USA: ) https://omim.org/ (accessed 28 April 2018). [Google Scholar]
- 4.McRae JF et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders SJ et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catterall WA, Kalume F & Oakley JC NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escayg A. et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat. Genet. 24, 343–345 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Marshall CR et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukowski AF, Milojevich HM & Eales L. Cognitive functioning in children with down syndrome: current knowledge and future directions. Adv. Child Dev. Behav. 56, 257–289 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Sanders SJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Shalom R. et al. Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82, 224–232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Power RA et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry 70, 22–30 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Stefansson H. et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Sanders SJ et al. Whole genome sequencing in psychiatric disorders: the WGSPD consortium. Nat. Neurosci. 20, 1661–1668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reuter MS et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 74, 293–299 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Schaefer GB et al. Array comparative genomic hybridization findings in a cohort referred for an autism evaluation. J. Child Neurol. 25, 1498–1503 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Retterer K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Sawyer SL et al. Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 89, 275–284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tammimies K. et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314, 895–903 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Kosmicki JA et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 49, 504–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiner DJ et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49, 978–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.EuroEPINOMICS-RES Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heyne HO et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat. Genet. 50, 1048–1053 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Ganna A. et al. Ultra-rare disruptive and damaging mutations influence educational attainment in the general population. Nat. Neurosci. 19, 1563–1565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh T. et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 49, 1167–1173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willsey AJ et al. De Novo coding variants are strongly associated with Tourette disorder. Neuron 94, 486–499.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fromer M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purcell SM et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finkel RS et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Schneider M. et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 171, 627–639 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rees E. et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol. Psychiatry 19, 37–40 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kendall KM et al. Archival report cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK Biobank subjects. Biol. Psychiatry 83, 103–110 (2016). [DOI] [PubMed] [Google Scholar]
- 34.D’Angelo D. et al. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 73, 20–30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niemi MEK et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 562, 268–271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moreno-De-Luca A. et al. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry 72, 119–126 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Insel TR The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am. J. Psychiatry 171, 395–397 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Cuthbert BN & Insel TR Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med. 11, 126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuthbert BN Research Domain Criteria: toward future psychiatric nosologies. Dialog-. Clin. Neurosci. 17, 89–97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Constantino JN et al. Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview-revised. J. Autism Dev. Disord. 33, 427–433 (2003). [DOI] [PubMed] [Google Scholar]
- 41.van Os J & Reininghaus U. Psychosis as a transdiagnostic and extended phenotype in the general population. World Psychiatry 15, 118–124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen L. et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry 5, 573–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Männik K. et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA. 313, 2044–2054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simons Vip C, Spiro JE & Chung WK Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron 73, 1063–1067 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Stessman HA, Bernier R & Eichler EE A genotype-first approach to defining the subtypes of a complex disease. Cell 156, 872–877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He X. et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 9, e1003671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Rubeis S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iossifov I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh T. et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn K. et al. High rate of disease-related copy number variations in childhood onset schizophrenia. Mol. Psychiatry 19, 568–572 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satterstrom FK et al. Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. Preprint at 10.1101/484113v3 (2018). [DOI]
- 53.Mighell TL, Evans-Dutson S & O’Roak BJ A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotypes relationships. Am. J. Hum. Genet. 102, 943–955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huguet G. et al. Measuring and estimating the effect sizes of copy number variants on general intelligence in community-based samples. JAMA Psychiatry 75, 447–457 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geschwind DH Autism: many genes, common pathways? Cell 135, 391–395 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng H. et al. Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 28, 2900–2919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chakravarti A, Clark AG & Mootha VK Distilling pathophysiology from complex disease genetics. Cell 155, 21–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gandal MJ, Leppa V, Won H, Parikshak NN & Geschwind DH The road to precision psychiatry: translating genetics into disease mechanisms. Nat. Neurosci. 19, 1397–1407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parikshak NN, Gandal MJ & Geschwind DH Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 16, 441–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amin ND & Paşca SP Building models of brain disorders with three-dimensional organoids. Neuron 100, 389–405 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Sun Y. et al. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. eLife 5, e13073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willsey AJ et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parikshak NN et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Velmeshev D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cargnin F. et al. FOXG1 orchestrates neocortical organization and cortico-cortical connections. Neuron 100, 1083–1096.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guloksuz S, Pries LK & van Os J. Application of network methods for understanding mental disorders: pitfalls and promise. Psychol. Med. 47, 2743–2752 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Sheffield JM et al. Transdiagnostic associations between functional brain network integrity and cognition. JAMA Psychiatry 74, 605–613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cao H. et al. Toward leveraging human connectomic data in large consortia. Generalizability of fMRI-based brain graphs across sites, sessions, and paradigms. Cereb. Cortex (2018). [DOI] [PMC free article] [PubMed]
- 69.Anticevic A. et al. Association of Thalamic dysconnectivity and conversion to psychosis in youth and young adults at elevated clinical risk. JAMA Psychiatry 72, 882–891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bruno JL et al. Longitudinal identification of clinically distinct neurophenotypes in young children with fragile X syndrome. Proc. Natl. Acad. Sci. USA 114, 10767–10772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hazlett HC et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bearden CE & Thompson PM Emerging global initiatives in neurogenetics: the Enhancing Neuroimaging Genetics through Meta-analysis (ENIGMA) consortium. Neuron 94, 232–236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thompson PM et al. ENIGMA and the individual: predicting factors that affect the brain in 35 countries worldwide. Neuroimage 145 Pt B, 389–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson PM et al. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 8, 153–182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.National Advisory Mental Health Council Workgroup on Genomics. Opportunities and Challenges of Psychiatric Genetics (NAHMC, 2018). [Google Scholar]
- 76.Skene NG et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 50, 825–833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deisseroth K, Etkin A & Malenka RC Optogenetics and the circuit dynamics of psychiatric disease. J. Am. Med. Assoc. 313, 2019–2020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stoodley CJ et al. Author Correction: Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat. Neurosci. 21, 1016 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Anthony TE et al. Control of stress-induced persistent anxiety by an extra-amygdala septohypothalamic circuit. Cell 156, 522–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stewart AM & Kalueff AV Developing better and more valid animal models of brain disorders. Behav. Brain Res. 276, 28–31 (2015). [DOI] [PubMed] [Google Scholar]
- 81.LeBlanc JJ et al. Visual evoked potentials detect cortical processing deficits in Rett syndrome. Ann. Neurol. 78, 775–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lovelace JW, Ethell IM, Binder DK & Razak KA Translation-relevant EEG phenotypes in a mouse model of Fragile X Syndrome. Neurobiol. Dis. 115, 39–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chadman KK, Yang M & Crawley JN Criteria for validating mouse models of psychiatric diseases. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 150B, 1–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Galvão-Coelho NL, Galvão ACM, da Silva FS & de Sousa MBC Common marmosets: a potential translational animal model of juvenile depression. Front. Psychiatry 8, 175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oikonomidis L. et al. A dimensional approach to modeling symptoms of neuropsychiatric disorders in the marmoset monkey. Dev. Neurobiol. 77, 328–353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mao P, Cui D, Zhao X-D & Ma Y-Y Prefrontal dysfunction and a monkey model of schizophrenia. Neurosci. Bull. 31, 235–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kotani M. et al. The atypical antipsychotic blonanserin reverses (+)-PD128907- and ketamine-induced deficit in executive function in common marmosets. Behav. Brain Res. 305, 212–217 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Clarke HF et al. Orbitofrontal dopamine depletion upregulates caudate dopamine and alters behavior via changes in reinforcement sensitivity. J. Neurosci. 34, 7663–7676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou Y. et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 570, 326–331 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Pașca SP The rise of three-dimensional human brain cultures. Nature 553, 437–445 (2018). [DOI] [PubMed] [Google Scholar]
- 91.Bredenoord AL, Clevers H & Knoblich JA Human tissues in a dish: The research and ethical implications of organoid technology. Science 355, eaaf9414 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Birey F. et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang J. et al. A resting EEG study of neocortical hyperexcitability and altered functional connectivity in fragile X syndrome. J. Neurodev. Disord. 9, 11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sahin M. et al. Discovering translational biomarkers in neurodevelopmental disorders. Nat. Rev. Drug Discov. 18, 235–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Donaldson ZR & Hen R. From psychiatric disorders to animal models: a bidirectional and dimensional approach. Biol. Psychiatry 77, 15–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spencer CM et al. Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res. 4, 40–56 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aylor DL et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 21, 1213–1222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vockley J. et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet. Med. 16, 188–200 (2014). [DOI] [PubMed] [Google Scholar]
- 99.Berry-Kravis EM et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 17, 280–299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Krueger DA et al. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 4, 877–887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.O’Leary HM et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann. Clin. Transl. Neurol. 5, 323–332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guy J, Gan J, Selfridge J, Cobb S & Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Henderson C. et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med. 4, 152ra128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dolan BM et al. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 110, 5671–5676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jacquemont S. et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 478, 97–102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wiesel TN & Hubel DH Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017 (1963). [DOI] [PubMed] [Google Scholar]
- 107.Berry-Kravis E AFQ056 for language learning in children with FXS. https://clinicaltrials.gov/ct2/show/NCT02920892.
- 108.Bebin M Preventing epilepsy using vigabatrin in infants with tuberous sclerosis complex. https://clinicaltrials.gov/ct2/show/NCT02849457.
- 109.Jozwiak S Long-term, prospective study evaluating clinical and molecular biomarkers of epileptogenesis in a genetic model of epilepsy—Tuberous Sclerosis Complex (EPISTOP). https://clinicaltrials.gov/ct2/show/NCT02098759.
- 110.Kothari C. et al. Phelan-McDermid syndrome data network: integrating patient reported outcomes with clinical notes and curated genetic reports. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 177, 613–624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kohane IS Using electronic health records to drive discovery in disease genomics. Nat. Rev. Genet. 12, 417–428 (2011). [DOI] [PubMed] [Google Scholar]
- 112.Berry-Kravis E. et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 8, 321ra5 (2016). [DOI] [PubMed] [Google Scholar]
- 113.van der Vaart T, Overwater IE, Oostenbrink R, Moll HA & Elgersma Y. Treatment of cognitive deficits in genetic disorders: a systematic review of clinical trials of diet and drug treatments. JAMA Neurol. 72, 1052–1060 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Wolff M. et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Guerrini R & Falchi M. Dravet syndrome and SCN1A gene mutation related-epilepsies: cognitive impairment and its determinants. Dev. Med. Child Neurol. 53 Suppl 2, 11–15 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Moreno-De-Luca D, Moreno-De-Luca A, Cubells JF & Sanders SJ Cross-disorder comparison of four neuropsychiatric CNV loci. Curr. Genet. Med. Rep. 2, 151–161 (2014). [Google Scholar]
- 117.Demkow U & Wolańczyk T. Genetic tests in major psychiatric disorders-integrating molecular medicine with clinical psychiatry—why is it so difficult? Transl. Psychiatry 7, e1151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER) & Center for Biologics Evaluation and Research (CBER). Rare diseases: common issues in drug development guidance for industry. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM458485.pdf (2019).
- 119.Collins R. What makes UK Biobank special? Lancet 379, 1173–1174 (2012). [DOI] [PubMed] [Google Scholar]
- 120.Senthil G, Dutka T, Bingaman L & Lehner T. Genomic resources for the study of neuropsychiatric disorders. Mol. Psychiatry 22, 1659–1663 (2017). [DOI] [PubMed] [Google Scholar]
- 121.Bastarache L. et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science 359, 1233–1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pedersen CB et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol. Psychiatry 23, 6–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rusk N. The UK Biobank. Nat. Methods 15, 1001 (2018). [DOI] [PubMed] [Google Scholar]
- 124.An J-Y & Sanders SJ Appreciating the population-wide impact of copy number variants on cognition. Biol. Psychiatry 82, 78–80 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Köhler S. et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 45 D1, D865–D876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Finucane BM et al. 15q duplication syndrome and related disorders. in Gene Reviews (eds. Pagon RA et al.) (University of Washington, Seattle: ) https://www.ncbi.nlm.nih.gov/books/NBK367946/ (2016). [Google Scholar]
- 128.Miller IO & Sotero de Menezes MA SCN1A seizure disorders. in Gene Reviews (eds. Pagon RA et al. ) (University of Washington, Seattle: ) https://www.ncbi.nlm.nih.gov/books/NBK1318/ (2007). [PubMed] [Google Scholar]
- 129.Kirov G. et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry 75, 378–385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moreno-De-Luca D. et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 18, 1090–1095 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Malhotra D & Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sanders SJ et al. Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci. 41, 442–456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Joseph L. et al. Characterization of autism spectrum disorder and neurodevelopmental profiles in youth with XYY syndrome. J. Neurodev. Disord. 10, 30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 107, 961–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Scheldeman C. et al. mTOR-related neuropathology in mutant tsc2 zebrafish: Phenotypic, transcriptomic and pharmacological analysis. Neurobiol. Dis. 108, 225–237 (2017). [DOI] [PubMed] [Google Scholar]
- 136.Kelly E. et al. mGluR5 modulation of behavioral and epileptic phenotypes in a mouse model of tuberous sclerosis complex. Neuropsychopharmacology 43, 1457–1465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shukla G. et al. Magnetoencephalographic identification of epileptic focus in children with generalized electroencephalographic (EEG) Features but focal imaging abnormalities. J. Child Neurol. 32, 981–995 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Pietri T. et al. The first mecp2-null zebrafish model shows altered motor behaviors. Front. Neural Circuits 7, 118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu Y. et al. Characterization of Rett Syndrome-like phenotypes in Mecp2-knockout rats. J. Neurodev. Disord. 8, 23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen Y. et al. Modeling Rett syndrome using TALEN-edited MECP2 mutant cynomolgus monkeys. Cell 169, 945–955.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pidcock FS et al. Functional outcomes in Rett syndrome. Brain Dev. 38, 76–81 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.