

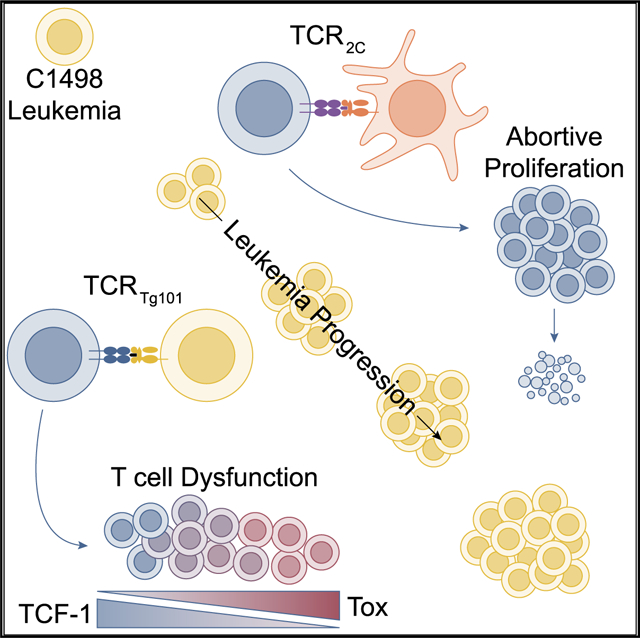
SUMMARY
The existence of a dysfunctional CD8+ T cell state in cancer is well established. However, the degree to which CD8+ T cell fates are influenced by the context in which they encounter cognate tumor antigen is less clear. We previously demonstrated that CD8+ T cells reactive to a model leukemia antigen were deleted by antigen cross-presenting type 1 conventional dendritic cells (cDC1s). Here, through a study of T cell receptor (TCR) transgenic CD8+ T cells (TCRTg101) reactive to a native C1498 leukemia cell antigen, we uncover a different mode of T cell tolerance in which TCRTg101 undergo progressive expansion and differentiation into an exhausted state. Antigen encounter by TCRTg101 requires leukemia cell major histocompatibility complex (MHC)-I expression and is independent of DCs, implying that leukemia cells directly mediate the exhausted TCRTg101 phenotype. Collectively, our data reveal that leukemia antigens are presented to CD8+ T cells via discrete pathways, leading to distinct tolerant states.
In brief
In order to track antigen-specific CD8+ T cell fates in leukemia-bearing hosts, Chen et al. generate a leukemia-specific TCR transgenic mouse (Tg101). They find that leukemia-specific CD8+ T cells expand and acquire a profoundly dysfunctional phenotype, which is mediated through direct antigen presentation by leukemia cells.
Graphical Abstract
INTRODUCTION
CD8+ T cells are key effectors of anti-tumor immune responses. In order to evade immune recognition and elimination, cancers exploit pathways that undermine CD8+ T cell responses, thereby facilitating cancer progression and metastasis (Gajewski et al., 2006). Although the existence of a dysfunctional CD8+ T cell phenotype has been demonstrated across many cancers, the underlying mechanisms and manifestations of T cell dysfunction are varied. Moreover, the degree to which tumor-specific CD8+ T cell fates are shaped by unique interactions with antigen-presenting cells (APCs) has not been well-defined.
Our group has been focused on characterizing immune evasion mechanisms in hosts with hematologic malignancies. We previously demonstrated in leukemia-bearing mice that cross-presentation of a leukemia-specific antigen by splenic CD8α+ type 1 conventional dendritic cells (cDC1s) induced the deletion of a CD8+ T cell population expressing a high-affinity T cell receptor (TCR; referred to as TCR2C) (Kline et al., 2018; Zhang et al., 2013). Interestingly, presentation of the antigen by cDC1s in draining lymph nodes (dLNs) of mice with locally implanted tumors derived from the same leukemia cells resulted in the robust activation of the identical CD8+ T cell population (Kline et al., 2018; Zhang et al., 2013), revealing that the environmental context in which a tumor antigen is encountered can confer drastically disparate CD8+ T cell functional states.
In order to elucidate the extent to which deletion was a fate shared by other leukemia-specific CD8+ T cells, we generated a TCR transgenic mouse strain (referred to as Tg101) harboring a clonal CD8+ T cell population specific for a naturally expressed, major histocompatibility complex (MHC) class I-restricted antigen on murine C1498 leukemia cells. In striking contrast to our previous observations (Zhang et al., 2013), CD8+ T cells from Tg101 mice (TCRTg101) expanded to relatively large numbers in leukemia-bearing animals rather than being deleted. However, TCRTg101 expansion in this context was accompanied by the progressive acquisition of an exhausted phenotype that could not be effectively reversed. Interestingly, the in vivo differentiation of TCRTg101 into an exhausted state occurred independently of DCs but rather required direct presentation of cognate antigen by leukemia cells. These results reveal that multiple mechanisms mediate CD8+ T cell tolerance in leukemia and further highlight that the context in which a CD8+ T cell encounters its cognate leukemia antigen is a critical factor in determining the subsequent tolerance phenotype that ensues.
RESULTS
TCRTg101 develop along the CD8 lineage and are leukemia specific
Tg101 transgenic mice were generated from TCR-α and -β chains of a CD8+ T cell clone (T15) specific for an undefined antigen expressed by C1498 leukemia cells (Boyer et al., 1997). Tg101 mice were born in Mendelian ratios, developed normally, and showed no gross symptoms or signs of poor health. Tg101 mice were crossed onto a Rag2−/− background to prevent rearrangement of endogenous TCR-α and -β loci. Thymocytes from Rag2−/− Tg101 mice developed into CD8 single-positive cells (Figures S1A–S1C), and TCRTg101 were uniformly CD8+ in secondary lymphoid organs (SLOs) (Figures S1D–S1F). TCRTg101 exhibited no evidence of self-reactivity and maintained a naive phenotype that persisted in SLOs of older mice (Figures S1G–S1K).
To confirm their specificity for syngeneic C1498 leukemia cells, CellTrace Violet (CTV)-labeled TCRTg101 were cultured with C1498 cells or with other syngeneic cancer cell lines, including B16.F10 melanoma and EL4 thymoma, or with splenocytes from C57BL/6 mice. CTV dilution of TCRTg101 occurred only upon co-culture with C1498 cells but not with B16.F10 cells, EL4 cells, or C57BL/6 splenocytes (Figure 1A), indicating that TCRTg101 specifically recognize an antigen expressed on C1498 leukemia cells. Because TCRTg101 failed to proliferate when co-cultured with syngeneic splenocytes and maintained a naive phenotype in Tg101 transgenic mice, it is unlikely that their cognate antigen is derived from a normal self-protein. Together, these results suggest that TCRTg101 recognize a leukemia-specific antigen.
Figure 1. TCRTg101 recognize an H2-Kb-restricted antigen on C1498 cells.
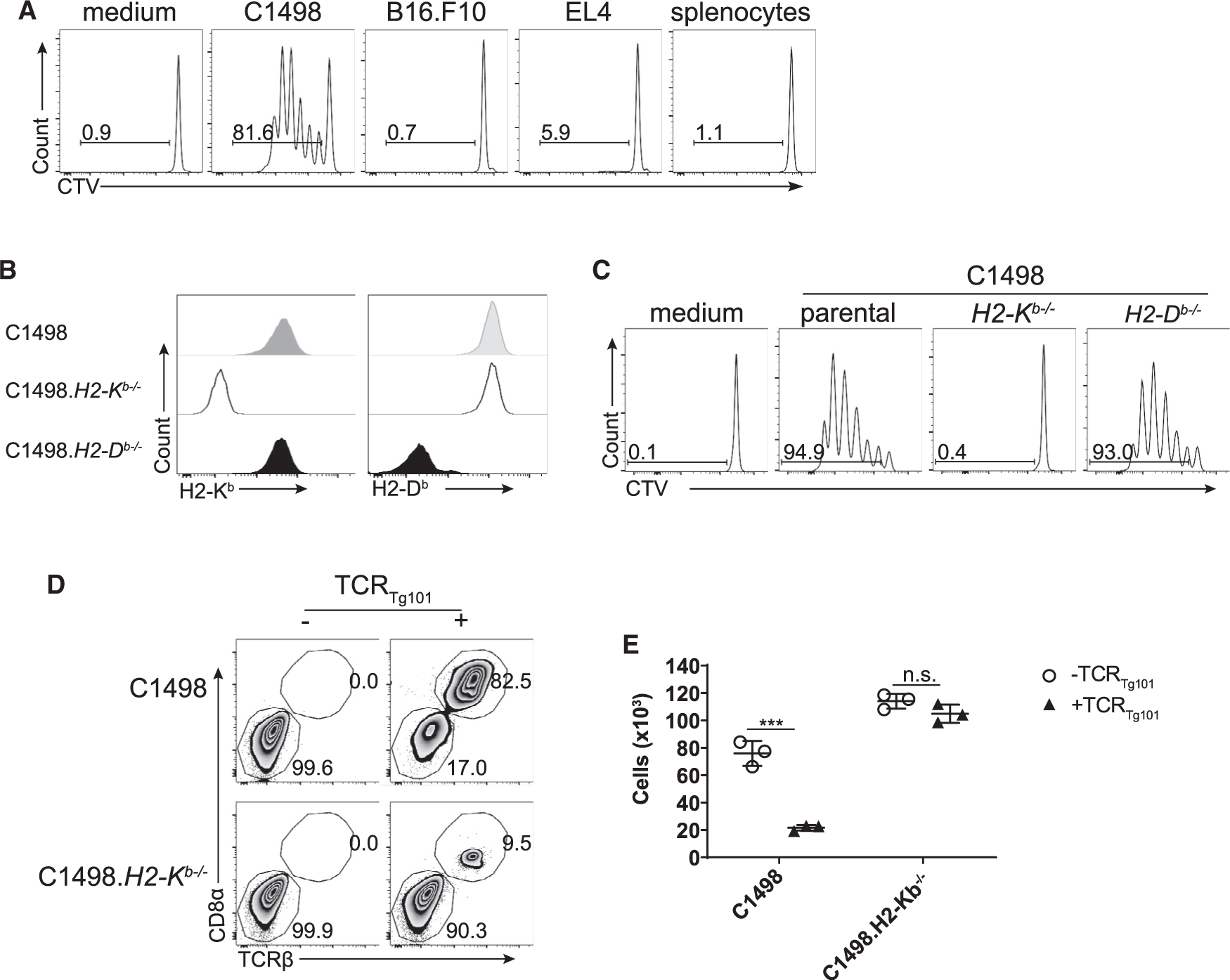
(A) CTV-labeled TCRTg101 were cultured for 72 h with the indicated tumor cell lines or with splenocytes from C57BL/6 mice. Proliferation of TCRTg101, as measured by CTV dilution, was analyzed by flow cytometry. Representative FACS plots are shown.
(B) Expression of H2-Kb and H2-Db on parental C1498 cells, C1498.H2-Kb−/− cells, and C1498.H2-Db−/− cells. Representative FACS plots are displayed.
(C) Proliferation of CTV-labeled TCRTg101 cultured for 72 h with C1498 cells, C1498.H2-Kb−/− cells, or C1498.H2-Db−/− cells. Representative FACS plots are shown.
(D and E) Live C1498 or C1498.H2-Kb−/− cells (2 × 104) were cultured for 72 h alone or with 1 × 105 TCRTg101. Subsequently, numbers of viable C1498 or C1498.H2-Kb−/− cells were enumerated. Representative FACS plots are shown in (D). Summary data are presented in (E) as mean ± SD. Data are representative of 2 to 3 independent experiments with 3 wells/group. ***p < 0.001; n.s., not significant.
C1498 cells deficient in the MHC class I molecules H2-Kb (Kb) or H2-Db (Db) were generated in order to define the restricting MHC class I molecule for the Tg101 antigen (Figure 1B). Only Kb−/−, but not parental or Db−/− C1498, cells were incapable of stimulating proliferation of TCRTg101 in vitro, demonstrating that TCRTg101 recognize a Kb-restricted leukemia antigen (Figure 1C). Importantly, parental C1498 cells, but not C1498 Kb−/− cells, were susceptible to TCRTg101-mediated killing in vitro (Figures 1D and 1E). Thus, TCRTg101 recognize a leukemia-specific antigen directly presented by C1498 leukemia cells and, in doing so, are capable of eliminating C1498 cells in vitro.
TCRTg101 accumulate and acquire an exhausted phenotype in leukemia-bearing hosts
After confirming TCRTg101 specificity for C1498 leukemia cells, their behavior in leukemia-bearing mice was characterized. Adoptive transfer of TCRTg101 activated in vitro with anti-CD3 and anti-CD28 antibodies modestly extended the survival of C1498-challenged mice by approximately 8 to 9 days (Figure 2A). Conversely, adoptive transfer of large numbers of naive TCRTg101 had no impact on the survival of leukemia-bearing mice (Figure 2B), which was suggestive of an acquired tolerant state among TCRTg101 in vivo. For comparison, adoptive transfer of naive or in vitro-activated TCR2C failed to extend the survival of leukemia-bearing mice (Figures S2A and S2B). To further investigate the hypothesis that TCRTg101 acquired a dysfunctional phenotype in leukemia-bearing hosts, the fate of naive CTV-labeled TCRTg101 was assessed following adoptive transfer into C57BL/6 hosts that received an intravenous (i.v.) challenge with C1498 cells the following day. 7 days later, TCRTg101 remained largely undivided and had not upregulated CD44 expression in the spleen, where we have previously demonstrated leukemia antigen cross-presentation occurs (Kline et al., 2018). In the liver, which is a primary site of leukemia progression in the C1498 model (Zhang et al., 2009), very few TCRTg101 had proliferated, although a subset began to express CD44 (Figures 2C–2G), indicating that most TCRTg101 had not yet encountered cognate antigens. Expression of co-inhibitory receptors (PD-1, LAG-3, TIM3, TIGIT) on TCRTg101 was negligible at this time point (Figure 2I). By day 13, a fraction of TCRTg101 had begun to proliferate and upregulate CD44 expression (Figures 2C–2G), and a subset expressed PD-1 and LAG-3 (Figures 2H and 2I). Finally, at days 17 to 18, TCRTg101 had proliferated extensively, particularly in livers of leukemia-bearing animals (Figures 2C–2G). At this late time point, the majority of TCRTg101 were CD44+, PD-1+, and LAG-3+, and many co-expressed TIM3 and TIGIT (Figure 2I). Between days 7 and 18, TCRTg101 expanded up to 16-fold in livers of leukemia-bearing mice (Figure 2G). The expansion and upregulation of co-inhibitory receptors by TCRTg101 is in contrast to previous observations with TCR2C, which were rapidly and efficiently deleted in mice with leukemia (Kline et al., 2018; Zhang et al., 2013), and is consistent with the acquisition of a dysfunctional phenotype.
Figure 2. TCRTg101 expand and acquire a dysfunctional phenotype in leukemia-bearing animals.
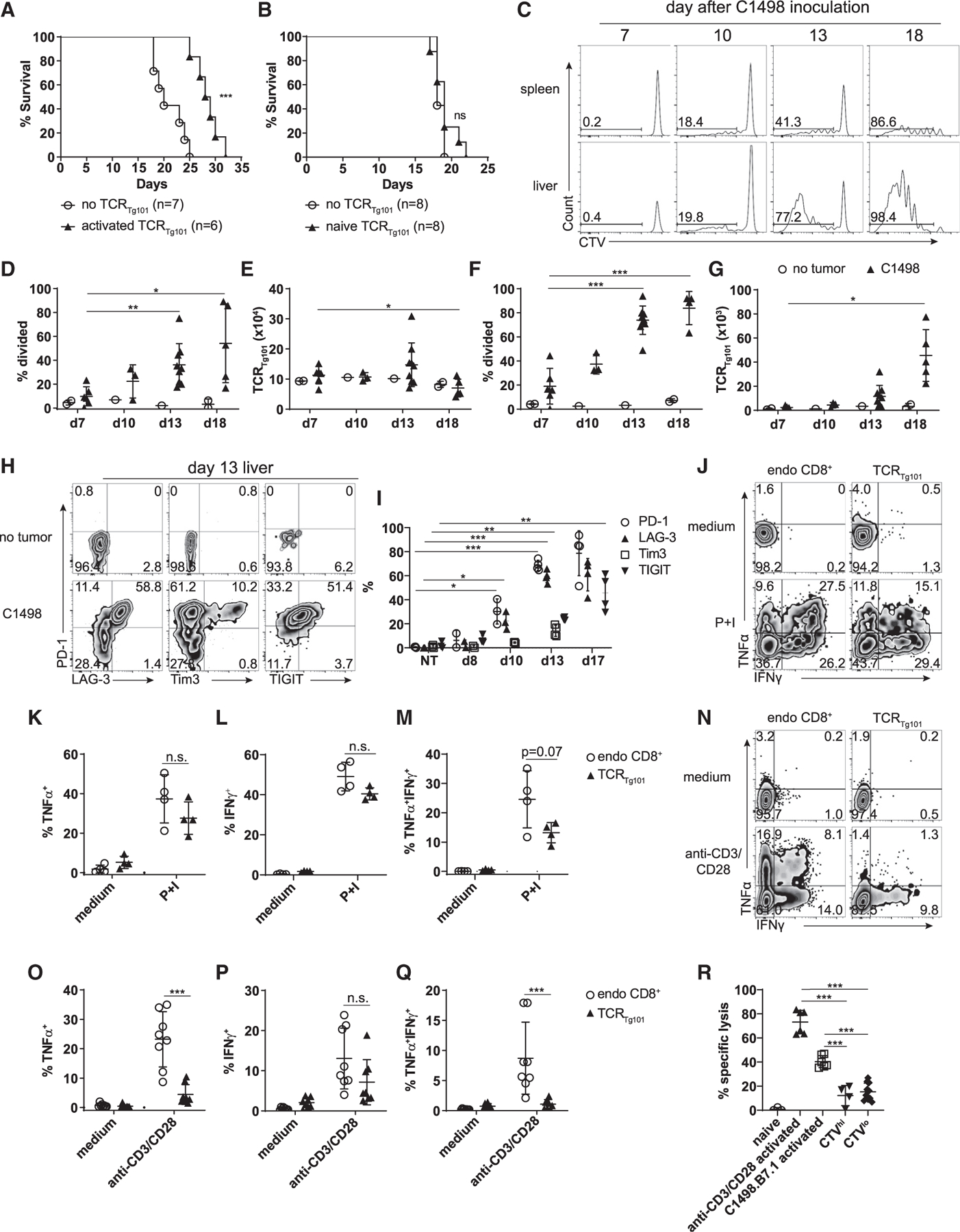
(A and B) Survival of C57BL/6 mice challenged i.v. with C1498 cells (106) and transferred or not with in vitro activated (A) or naive (B) TCRTg101 (4 × 106) 3 days later. Data are pooled from 2 independent experiments with 3–4 mice/group.
(C–G) 2 × 106 CTV-labeled TCRTg101 (CD45.2) were transferred into B6.SJL mice (CD45.1) followed by an i.v. challenge with 106 C1498 leukemia cells 1 day later.
(C) Representative FACS plots showing CTV dilution of adoptively transferred TCRTg101 in spleens and livers of leukemia-bearing mice at the indicated time points.
(D–G) Quantitative data showing the percentage of divided TCRTg101 (D and F) and TCRTg101 number (E and G) in spleens (D and E) or livers (F and G) of leukemia-bearing mice over time. Data are pooled from 2 to 3 independent experiments with 2 to 4 mice/group including data from leukemia-free control mice.
(H) Representative FACS plots showing expression of PD-1, LAG-3, TIM3, and TIGIT on TCRTg101 in livers of tumor-free or leukemia-bearing mice.
(I) Quantitative data from (H) as mean ± SD.
(J–Q) Mononuclear cells isolated from livers of mice 13 to 14 days (J–M) or 17 to 18 days (N–Q) after C1498 cell inoculation were restimulated with PMA + ionomycin (P+I) (J–M) or anti-CD3 and anti-CD28 antibodies (N–Q) for 4 h. Cytokine production was analyzed by flow cytometry. Gating was performed on TCRβ+CD8+CD45.1 cells (endogenous CD8+ T cells) or TCRβ+CD8+CD45.2 cells (TCRTg101).
(J and N) Representative FACS plots showing TNFα and IFNγ production by endogenous CD8+ T cells or TCRTg101.
Quantitative data are shown in (K)–(M) and (O)–(Q) as mean ± SD.
(R) TCRTg101 killing assay. CTVhi and CTVlo TCRTg101 (CD45.1.2) were FACS-purified from livers of mice 17 to 18 days after C1498 inoculation and were co-cultured for 20 h with equal numbers of C1498 cells and EL4 cells (both CD45.2) labeled with different CTV concentrations. Naive TCRTg101 purified from control Tg101 mice, or those activated in vitro with anti-CD3 and anti-CD28 antibodies or with C1498.B7.1 cells served as negative and positive controls for C1498 cell lysis, respectively. Specific lysis was analyzed by flow cytometry. Gating was performed on live CD8−CD45.2 cells.
Data are representative (K–M) of or pooled (I and O–R) from 2 independent experiments with 2 to 4 mice/group and shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant; NT, no tumor.
To directly assess the functional capacity of TCRTg101 in mice with advanced leukemia, mononuclear cells were isolated from livers of leukemia-bearing mice 14 and 18 days following TCRTg101 adoptive transfer (13 to 17 days following C1498 cell challenge) and were restimulated ex vivo with anti-CD3 and anti-CD28 antibodies or phorbol 12-myristate 13-acetate (PMA) and ionomycin. TCRTg101 produced non-significantly lower levels of tumor necrosis factor (TNF)α and interferon (IFN)γ (or both) than endogenous CD8+ T cells at day 14 (Figure 2J–2M). At day 18, effector cytokine production by TCRTg101 had further declined, and when compared with endogenous CD8+ T cells, TCRTg101 expressed significantly lower levels of TNFα and tended to also be poorer producers of IFNγ (Figures 2N–2P). Importantly, a significantly smaller proportion of TCRTg101 produced both effector cytokines at this time point (Figure 2Q). Lack of polyfunctional cytokine production has been recurrently observed in dysfunctional tumor-infiltrating CD8+ T cells (Ahmadzadeh et al., 2009; Sakuishi et al., 2010; Thommen and Schumacher, 2018). Granzyme B (GzmB) expression induced by ex vivo restimulation with anti-CD3 and anti-CD28 antibodies was similar in liver-infiltrating TCRTg101 and endogenous CD8+ T cells (Figures S3A–S3C). Interestingly, ex vivo restimulation of TCRTg101 with C1498 cells failed to activate effector cytokine production or GzmB expression. However, restimulation of TCRTg101 with C1498 cells engineered to express the T cell co-stimulatory molecule B7.1 (C1498.B7.1) induced cytokine production and GzmB expression, albeit to a lesser degree than anti-CD3 and anti-CD28 antibodies (Figures S3A–S3C).
To determine the extent to which extrinsic inhibitory signals delivered by C1498 cells or other immune cells present after Ficoll-based enrichment of liver-resident mononuclear cells were extrinsically mediating the poor effector function of ex vivo restimulated TCRTg101, liver-resident CD8+ T cells (including TCRTg101) were purified via positive selection at day 18 following C1498 challenge. The function of purified or bulk endogenous CD8+ T cells and TCRTg101 was examined following ex vivo restimulation with anti-CD3 and anti-CD28 antibodies. TNFα production by purified endogenous CD8+ T cells and TCRTg101 was slightly higher than the same T cell populations restimulated in the presence of contaminating immune cells and C1498 cells (bulk) while IFNγ production was unaffected (Figures S3D–S3F), suggesting that the acquired dysfunction of TCRTg101 was largely cell intrinsic.
To directly examine the cytolytic capability of TCRTg101 in leukemia-bearing animals, CTVhi (naive/undivided) and CTVlo (dysfunctional) TCRTg101 (CD45.1.2) were separately fluorescence-activated cell sorting (FACS)-purified from livers of leukemia-bearing mice 18 days after adoptive transfer (see Figure 4A) and cultured with equal numbers of C1498 cells and EL4 cells (both CD45.2) labeled with 2 different CTV concentrations. As a positive control for C1498 cell cytolysis, TCRTg101 isolated from naive Tg101 mice were restimulated in vitro with anti-CD3 and anti-CD28 antibodies or with gamma-irradiated C1498.B7.1 cells for 3 days prior to co-culture with CTV-labeled C1498 cells and EL4 cells. As a negative control for C1498 cell cytolysis, unstimulated TCRTg101 freshly isolated from naive Tg101 mice were utilized. Specific lysis was measured by comparing relative proportions of viable C1498 and EL4 cells after a 20-hour co-culture with the indicated TCRTg101 populations, compared with those present in culture wells lacking TCRTg101. As expected, unstimulated TCRTg101 from control Tg101 mice failed to eliminate C1498 cells, while in vitro anti-CD3 and anti-CD28 antibody-stimulated and C1498.B7.1-stimulated TCRTg101 from control Tg101 mice effectively and specifically killed C1498 cells (Figure 2R). CTVhi TCRTg101 from livers of leukemia-bearing mice poorly lysed C1498 cells, which was not surprising given that they were likely of a naive phenotype. Finally, CTVlo TCRTg101 from livers of leukemia-bearing animals were also unable to effectively kill C1498 cells directly ex vivo (Figure 2R), despite their inducible expression of GzmB (Figures S3A and S3C). Collectively, these observations indicated that as TCRTg101 expanded in leukemia-bearing animals, they concomitantly acquired a dysfunctional phenotype.
Figure 4. The exhausted TCRTg101 phenotype is profound.
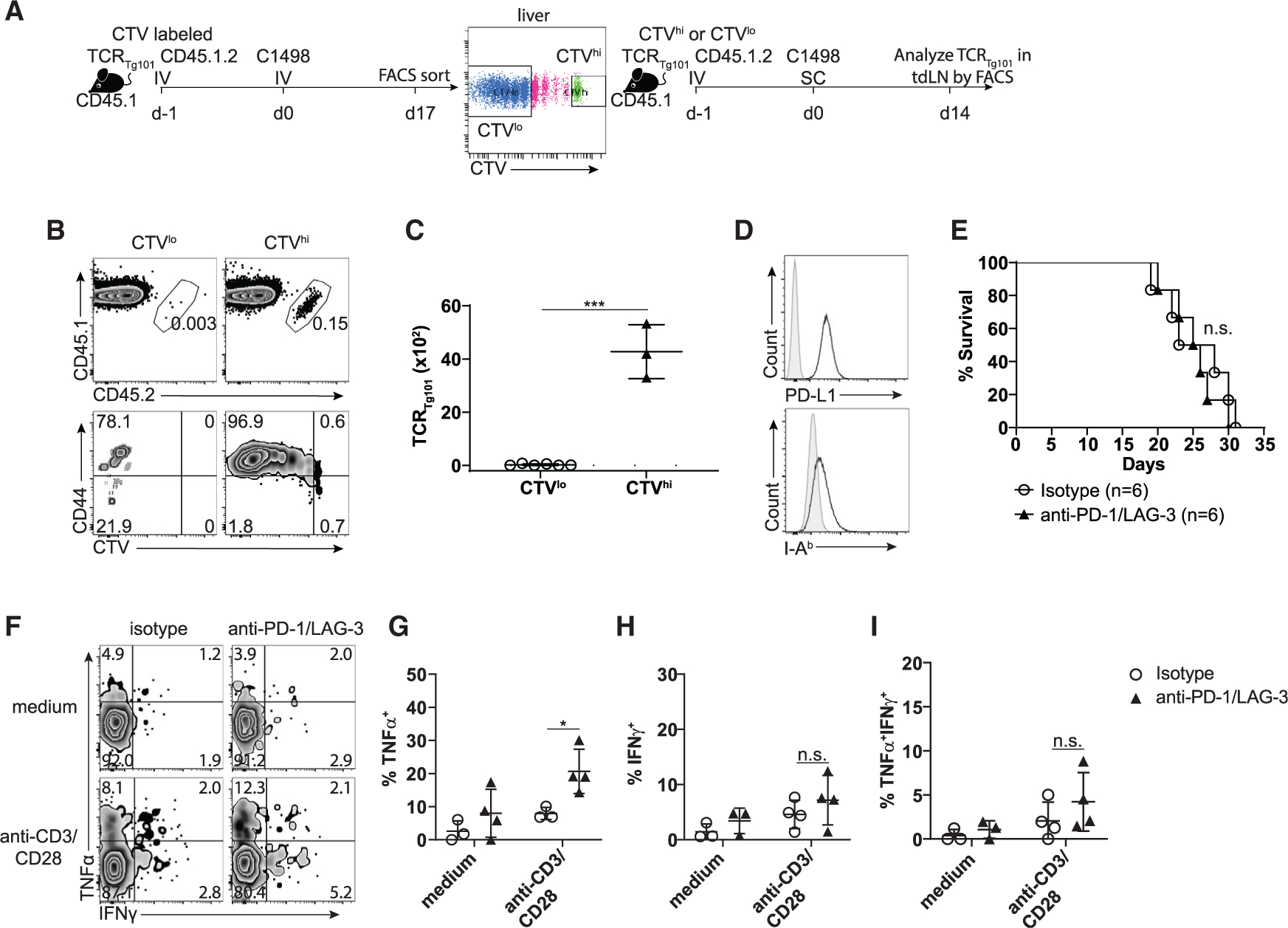
(A) Experimental design: 8 × 106 CTV-labeled TCRTg101 (CD45.1.2) were transferred into B6.SJL (CD45.1) mice followed by i.v. challenge with C1498 leukemia cells 1 day later. Naive (CTVhi) or exhausted (CTVlo) TCRTg101 were isolated from livers of day-17 leukemia-bearing mice by FACS and were separately transferred into secondary B6.SJL hosts. These B6.SJL mice received a s.c. C1498 cell (106) challenge the following day. On day 14, numbers and CD44 expression of TCRTg101 in tdLNs were analyzed by flow cytometry.
(B) Representative FACS plots showing CTV dilution and CD44 expression of TCRTg101 in tdLNs of secondary B6.SJL mice with s.c. C1498 tumors.
(C) Quantified data showing numbers of TCRTg101 in tdLNs of secondary B6.SJL mice with s.c. C1498 tumors.
(D) Expression of PD-L1 and I-Ab on C1498 cells when analyzed directly ex vivo. Shaded histograms represent staining with appropriate isotype control antibodies.
(E) TCRTg101 (8 × 106) were adoptively transferred into C57BL/6 mice. 1 day later, mice were challenged i.v. with C1498 cells (106) and were treated with anti-PD-1 and anti-LAG-3 or isotype control antibodies. Survival was assessed.
(F–I) Cytokine production by TCRTg101 in leukemia-bearing mice treated with 4 doses of isotype control or anti-PD-1/anti-LAG-3 antibody therapy. Mononuclear cells isolated from livers on day 16 were restimulated ex vivo with anti-CD3 and anti-CD28 antibodies and cytokine production was assessed. Gating was performed on TCRβ+CD8+CD45.1.2 cells (TCRTg101).
(F) Representative FACS plots showing TNFα and IFNγ production by TCRTg101.
Quantitative data are shown in (G)–(I) as mean ± SD. Data are pooled from 2 independent experiments with 2 to 3 mice/group. *p < 0.05; ***p < 0.001; n.s., not significant.
Although TCRTg101 exhibited clear evidence of functional impairment, particularly at late stages of the disease, they largely retained proliferative capacity. Only TCRTg101, having undergone greater than 6 to 7 cell divisions, showed decreased BrdU incorporation (Figures S3G and S3H). Furthermore, while a small subset of TCRTg101 expressed activated caspase 3, most were viable, suggesting that the acquisition of effector dysfunction was not necessarily associated with the induction of apoptosis (Figures S3I and S3J). These results demonstrate that proliferative capacity and effector function were largely uncoupled in TCRTg101 until the point at which the dysfunctional program was fully established—an observation previously described among antigen-specific CD8+ T cells in tumors (Schietinger et al., 2016).
Dysfunctional TCRTg101 acquire a transcriptional program canonically associated with T cell exhaustion
To identify molecular programs associated with the observed dysfunctional TCRTg101 phenotype, RNA sequencing (RNA-seq) was performed on TCRTg101 isolated from livers of leukemia-bearing mice at several time points following adoptive transfer (days 6 to 7, 13 to 14, and 16 to 18) and on TCRTg101 from control (leukemia-free) mice (Figure 3A). Principal-component analysis (PCA) indicated that TCRTg101 from control animals, and those from leukemia-bearing animals at mid (days 13 to 14) to late (day 16 to 18) time points, exhibited very different transcriptional programs (Figure 3B). Complete clustering of the samples based on Euclidean distance revealed 2 distinct TCRTg101 clusters, including one containing TCRTg101 from control mice, and the other containing TCRTg101 isolated from leukemia-bearing mice at mid and late time points (Figure 3C). Interestingly, one early (days 6 to 7) time point TCRTg101 sample clustered with TCRTg101 from control mice, while the other day-6-to-7 TCRTg101 sample clustered with day-13-to-14 TCRTg101 from leukemia-bearing mice (Figures 3B and 3C), likely indicating that TCRTg101 had already encountered leukemia antigen in one sample but not the other. In order to identify differentially expressed genes by TCRTg101 at specific time points during the course of leukemia progression, we performed pairwise comparisons of gene expression profiles at the time points indicated above (Figure S4). In total, 4,075 genes were significantly differentially expressed across all pairwise comparisons (Table S1).
Figure 3. TCRTg101 acquire a transcriptional program enriched for genes associated with T cell anergy and exhaustion.
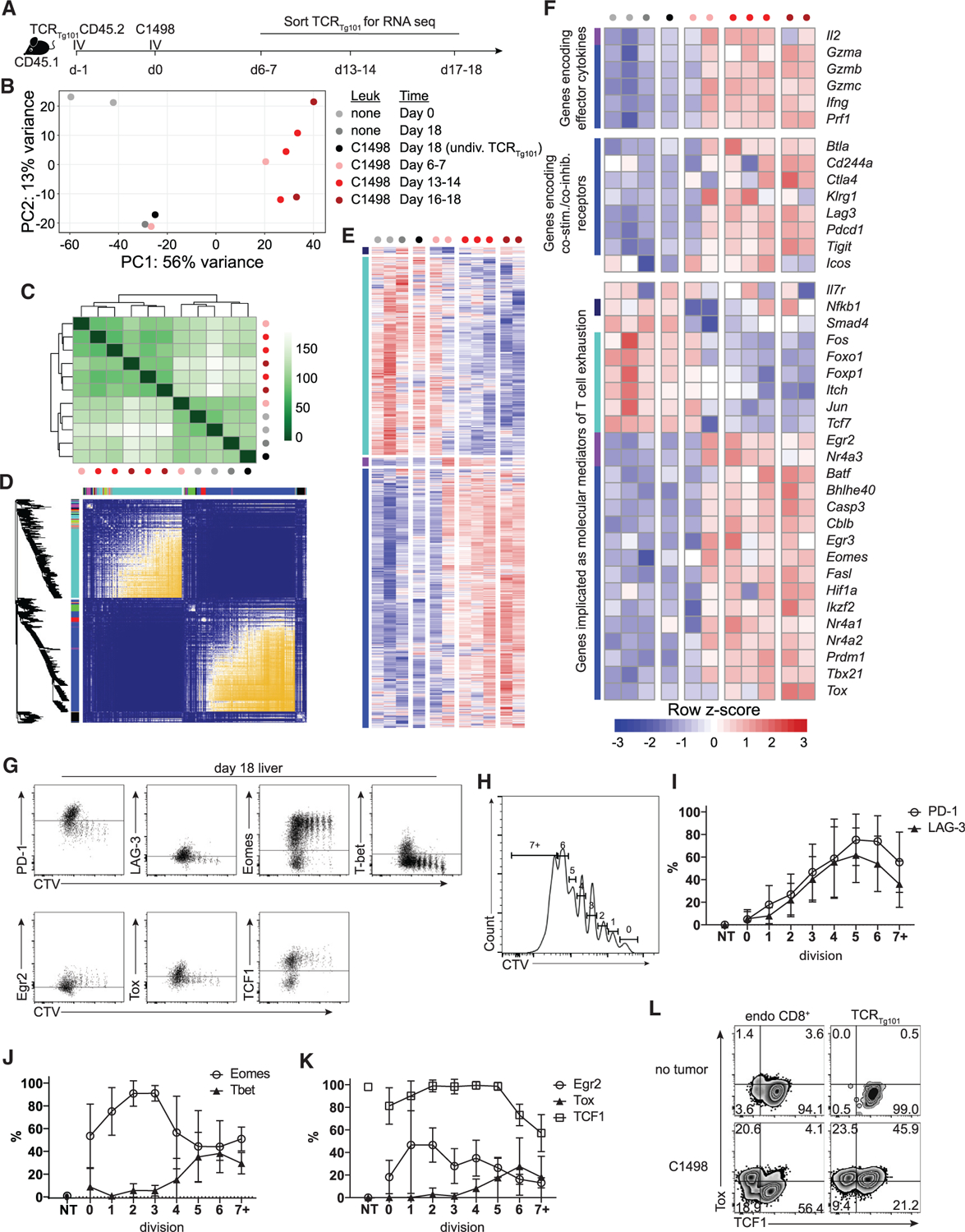
(A) Experimental design.
(B) Principal-component analysis (PCA).
(C) Complete clustering of TCRTg101 samples at the indicated time points based on their Euclidean distance.
(D) Heatmap of topological overlap based on weighted gene correlation network analysis of TCRTg101 transcriptomes.
(E) Heatmap showing up/downregulation or transient up/downregulation of genes from 2 major and 2 minor nodules, respectively.
(F) Heatmap showing up/downregulation of selected genes of interest (co-inhibitory receptors, transcription factors, effector cytokines, cell-cycle genes, survival genes) in TCRTg101 isolated from leukemia-bearing mice at indicated time points.
(G–L) PD-1, LAG-3, Eomes, T-bet, Egr2, Tox, and TCF1 expression in CTV-labeled TCRTg101 from livers of leukemia-bearing mice at day 18.
(G) Representative FACS plots showing expression of the indicated proteins by CTV dilution (cell division) in TCRTg101.
(H) Gating strategy for (I)–(K).
(I–K) Quantitative data showing expression of PD-1 and LAG-3 (I), Eomes and T-bet (J), and Egr2, Tox, and TCF1 (K) in TCRTg101 according to cell division number. Data are presented as mean ± SD
(L) Representative FACS plots showing expression of Tox and TCF1 in liver-resident endogenous CD8+ T cells TCRTg101.
To identify gene modules potentially involved in the regulation of TCRTg101 dysfunction, we performed a weighted gene correlation network analysis (Langfelder and Horvath, 2008), an unbiased analysis of gene co-regulation previously utilized to define transcriptional networks associated with CD8+ T cell exhaustion in the context of chronic viral infection (Doering et al., 2012). Here, 2 major gene modules were observed—one defined by gene transcripts that were progressively upregulated (indicated in blue in Figures 3D and 3E) and another characterized by gene transcripts that were progressively downregulated over the course of the experiment (indicated in turquoise). 2 minor gene clusters containing genes that were transiently up- or downregulated (purple and navy, respectively) in TCRTg101 early in leukemia-bearing mice were also identified (Figures 3D and 3E). Within the module that contained significantly upregulated genes were several encoding co-inhibitory receptors, including Pdcd1, Tigit, Lag3, and Ctla4 (Figure 3F). Somewhat surprisingly, genes also contained within this cluster were those encoding effector cytokines (Ifng) and cytolytic molecules (Gzma, Gzmb, Gzmc, Prf1). Finally, within this cluster were genes encoding transcription factors previously associated with T cell exhaustion or anergy, including Tox, Nr4a family members, Egr3, and Cblb (Chen et al., 2019a; Doering et al., 2012; Jadhav et al., 2019; McLane et al., 2019; Philip et al., 2017; Seo et al., 2019; Sowell and Kaech, 2016). Conversely, within the module containing significantly downregulated genes were those encoding for transcription factors such as Tcf7 and Foxo1, which antagonize terminal differentiation into a dysfunctional state (Figure 3F) (Chen et al., 2019b; Delpoux et al., 2018; Sade-Feldman et al., 2018; Siddiqui et al., 2019). Although these gene expression patterns suggest that TCRTg101 acquire an exhausted phenotype in leukemia-bearing mice, they could also be consistent with the induction of CD8+ T cell anergy, as significant overlap exists among gene sets associated with various dysfunctional T cell states (Waugh et al., 2016). Together, these results implied that the hyporesponsive TCRTg101 phenotype was coupled with transcriptional changes previously associated with T cell exhaustion/dysfunction.
Direct analysis of co-inhibitory receptor and transcription factor protein expression in TCRTg101 from livers of late-stage, leukemia-bearing mice was next performed to validate key RNA-seq findings. Consistent with previous observations (Figures 2H and 2I) and RNA-seq analysis, PD-1 and LAG-3 were upregulated on TCRTg101 in a manner that directly correlated with in vivo proliferation (Figures 3G–3I). Furthermore, Eomes was highly upregulated in TCRTg101 upon antigen encounter and during early rounds of proliferation but was subsequently downregulated with continued TCRTg101 expansion (Figures 3G and 3J). A similar but delayed pattern was observed for TCF1, a transcription factor associated with “stem-like” properties and responsiveness to PD-1 blockade therapy among CD8+ tumor-infiltrating lymphocytes (Sade-Feldman et al., 2018; Siddiqui et al., 2019). TCF1 expression was high in naive TCRTg101 and those undergoing early proliferation but decreased significantly in TCRTg101 that had proliferated extensively (Figures 3G and 3K). Conversely, transcription factors associated with CD8+ T cell dysfunction/exhaustion, including Egr2 and Tox (Scott et al., 2019; Zheng et al., 2012), were upregulated in TCRTg101 following antigen encounter. Egr2 expression showed an earlier peak, whereas Tox expression was upregulated at a point where most TCRTg101 had undergone 4 or more divisions (Figures 3G and 3K). Endogenous CD8+ T cells in livers of leukemia-bearing mice showed 2 clear populations of Tox and TCF1 expression (TCF1+Tox− and TCF1−Tox+), suggesting that TCF1 downregulation was associated with Tox expression, as might be expected (Figure 3L). Conversely, among liver-resident TCRTg101, downregulation of TCF1 was not required for Tox upregulation (Figure 3L), which has previously been reported among dysfunctional, antigen-specific CD8+ T cells (Sekine et al., 2020). Thus, TCRTg101 progressively downregulated expression of transcription factors associated with plasticity and differentiation into effector and memory subsets and upregulated expression of transcription factors known to drive an exhaustion program in dysfunctional CD8+ T cells, both in tumors and chronic viral infections.
The dysfunctional TCRTg101 phenotype is T cell intrinsic and irreversible
Having clearly established that TCRTg101 exhibit a dysfunctional phenotype in leukemia-bearing animals, we sought to determine the extent to which TCRTg101 function could be rescued by removing them from the leukemia environment and introducing them into secondary recipients that subsequently received a subcutaneous (s.c.) C1498 cell challenge (Figure 4A). Thus, CTV-labeled TCRTg101 were isolated from livers of day-17 leukemia-bearing mice. Non-proliferating TCRTg101 (naive; CTVhi) and TCRTg101, having undergone >4 rounds of cell division (exhausted; CTVlo), were separately isolated and adoptively transferred into secondary recipients. 1 day later, secondary hosts were challenged with C1498 cells s.c. On day 14, numbers and proliferation of TCRTg101 were assessed in tumor-dLNs (tdLNs) (Figure 4A). Naive (CTVhi) TCRTg101 expanded vigorously and upregulated CD44 in tdLNs of secondary recipients, as expected (Figures 4B and 4C). In contrast, exhausted (CTVlo) TCRTg101 were almost completely undetectable in tdLNs or within C1498 tumors, spleens, or other selected organs of secondary hosts (Figures 4B, 4C, and S5A). To determine if resting dysfunctional (CTVlo) TCRTg101 in vitro with cytokine support could restore their ability to survive and expand, CTVhi and CTVlo TCRTg101 isolated from livers of leukemia-bearing animals were cultured with interleukin (IL)-2, IL-7, and IL-15 for 5 days, at which point, CTVhi TCRTg101 had expanded 1.5-fold; conversely, very few CTVlo TCRTg101 survived (Figures S5B and S5C). Finally, to determine whether optimal ex vivo stimulation through the TCR, CD28, and provision of IL-12 could rescue the survival and function of dysfunctional TCRTg101, CTVhi and CTVlo TCRTg101 were purified from livers of day-17 leukemia-bearing mice and restimulated ex vivo with anti-CD3 and anti-CD28 antibodies or with C1498.B7.1 cells along with IL-2 and IL-12 for 5 days. As shown in Figure S5D, CTVhi TCRTg101 expanded markedly upon restimulation with anti-CD3 and anti-CD28 antibodies or with C1498.B7.1 cells plus IL-2 and IL-12, while again, very few CTVlo TCRTg101 survived. Furthermore, anti-CD3 and anti-CD28 antibodies plus IL-2/IL-12 restimulation of CTVhi TCRTg101 induced robust production of TNFα and IFNγ and expression of GzmB. Restimulation of CTVhi TCRTg101 with C1498.B7.1 cells plus IL-2/IL-12 induced uniform GzmB expression, while production of IFNγ and TNFα was less impressive (Figures S5E–S5L). With regard to CTVlo TCRTg101, restimulation with anti-CD3 and anti-CD28 antibodies plus IL-2/IL-12 induced partial GzmB expression and IFNγ/TNFα production among the few surviving cells, whereas absolute numbers of effector cytokine-producing and GzmB-expressing CTVlo TCRTg101 were strikingly decreased when compared to their CTVhi TCRTg101 counterparts (Figures S5E–S5L). Taken together, these results suggest that TCRTg101 exhaustion was intrinsic and not readily reversible upon removal from the leukemia environment.
The dysfunctional TCRTg101 phenotype was temporally associated with upregulation of multiple co-inhibitory receptors, primarily PD-1 and LAG-3 (Figures 2H and 2I). To determine whether interruption of PD-1/PD-L1 and LAG-3/MHC class II interactions could rescue the function of exhausted TCRTg101, mice transferred with TCRTg101 and challenged i.v. with C1498 cells were treated with anti-PD-1 and anti-LAG-3 antibodies. C1498 cells expressed both PD-L1 and low levels of MHC class II molecules (I-Ab), the ligands for PD-1 and LAG-3, respectively, when analyzed ex vivo (Figure 4D). Despite these observations, treatment with anti-PD-1 and anti-LAG-3 antibodies failed to enhance the survival of leukemia-bearing mice transferred with TCRTg101 (Figure 4E) and did little to reverse the dysfunctional TCRTg101 phenotype (Figures 4F–4I). These results indicated that either co-inhibitory receptor expression was dispensable in promoting the exhausted TCRTg101 phenotype, or, more likely, that the exhaustion program evolved to become independent of key T cell checkpoint receptors.
In vivo antigen recognition by TCRTg101 requires direct presentation by leukemia cells
We previously showed that leukemia antigen recognition by and deletion of TCR2C was mediated by antigen cross-presenting splenic CD8α+ cDC1s (Kline et al., 2018). Moreover, in leukemia-bearing Batf3−/− mice largely devoid of cDC1s (Hildner et al., 2008), TCR2C maintained a naive phenotype and failed to proliferate (Kline et al., 2018). Despite their near-complete inability to engage cognate antigen following C1498.SIY leukemia cell challenge in the absence of cross-presenting cDC1s, TCR2C clearly recognized antigens and proliferated following direct presentation by C1498.SIY cells in vitro, possibly with higher sensitivity than TCRTg101 (Figure 5A). However, without knowing the actual affinity of TCRTg101 for cognate antigens or the relative extent to which the antigens recognized by TCR2C versus TCRTg101 are expressed on C1498 cells, this conclusion is speculative. To determine if cross-presentation by cDC1s was similarly required for in vivo recognition of the Tg101 antigen, TCRTg101 were transferred into Batf3−/− or Batf3+/+ mice that were challenged i.v. with C1498 cells the following day. Numbers and proliferation of TCRTg101 were similar in leukemia-bearing Batf3−/− and Batf3+/+ mice, indicating that cDC1s—the major subset of cross-presenting DCs—were not required for antigen recognition and expansion of TCRTg101 in vivo (Figures S6A–S6F).
Figure 5. Direct antigen presentation by leukemia cells is required for in vivo antigen recognition by TCRTg101.
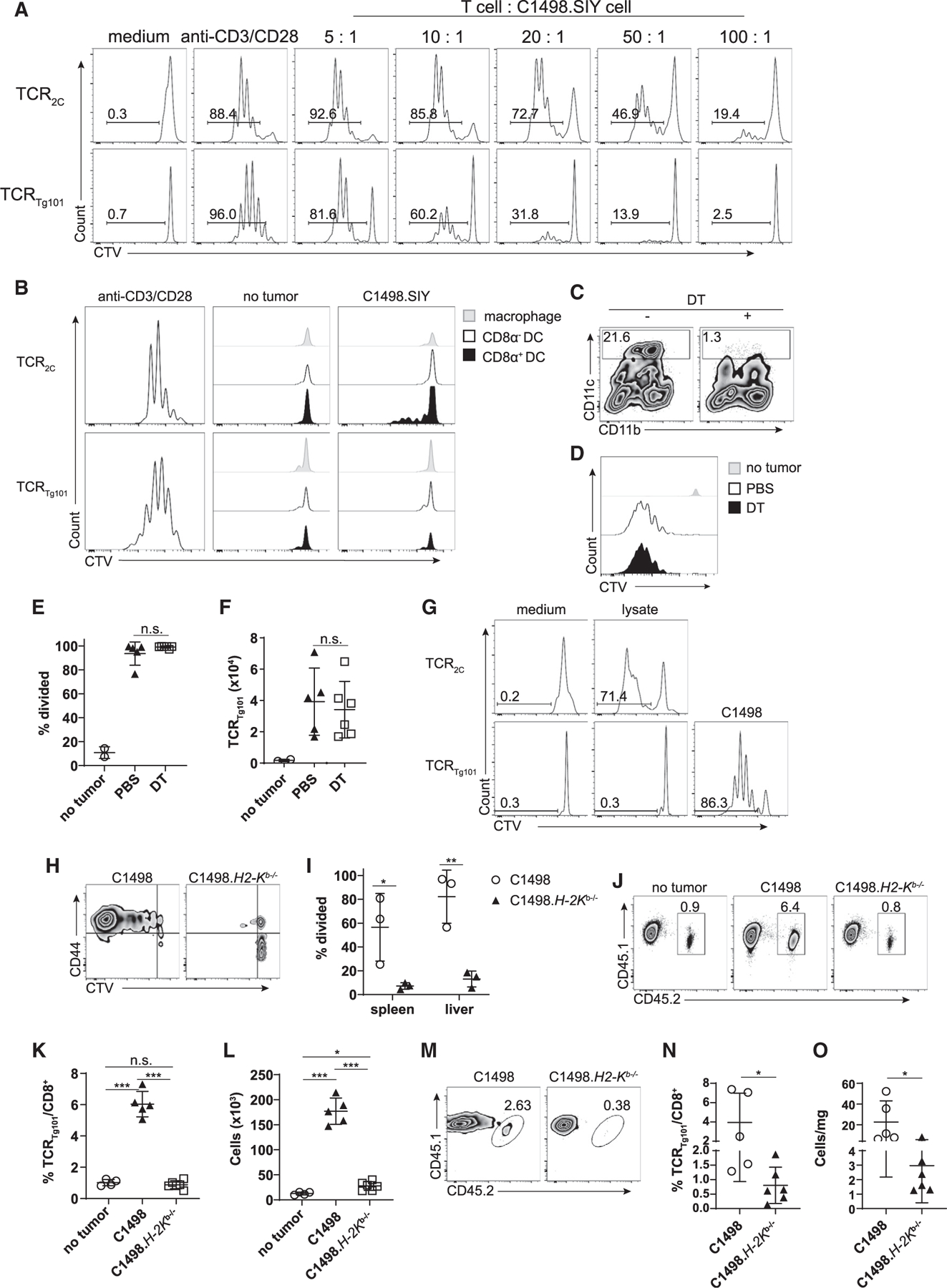
(A) TCR2C or TCRTg101 (1 × 105) were CTV-labeled and cultured in vitro with C1498.SIY cells at various ratios for 72 h, at which point TCR2C and TCRTg101 proliferation, as measured by CTV dilution, was assessed by flow cytometry. TCR2C and TCRTg101 stimulated with anti-CD3 and anti-CD28 antibodies or left unstimulated served as positive and negative controls for proliferation, respectively.
(B) CTV dilution profiles of TCR2C or TCRTg101 cultured for 72 h with CD8α+ DCs, CD8 α− DCs, or macrophages isolated from spleens of naive C57BL/6 mice or C57BL/6 mice challenged 3 h earlier with C1498.SIY cells i.v.
(C–F) CTV-labeled TCRTg101 (CD45.1.2) were adoptively transferred into CD11c-DTR bone marrow chimeric mice (CD45.2) that received DT or PBS prior to and following i.v. C1498 cell challenge.
(C) Representative FACS plot showing frequencies of CD11c+ cells in spleens of CD11c-DTR bone marrow chimeras after DT or PBS treatment. Gating was performed on live CD3−CD19−CD11c+ cells.
(D) Representative FACS plots showing CTV dilution of TCRTg101 (TCRb+CD8+CD45.1.2 cells) in livers of naive or C1498 cell-challenged CD11c-DTR bone marrow chimeric mice (treated with DT or PBS) at day 12.
(E and F) Quantitative data from (D) are shown.
(G) CTV dilution profiles of TCR2C and TCRTg101 cultured with BMDCs pulsed with C1498.SIY or C1498 cell lysates, respectively. CTV dilution profile of TCRTg101 cultured with live C1498 cells was included as a positive control (lower right). Representative FACS plots are shown.
(H and I) 2 × 106 CTV-labeled TCRTg101 (CD45.2) were transferred i.v. into B6.SJL (CD45.1) mice followed by an i.v. challenge with 106 C1498 or C1498.H2-Kb−/− cells the following day. On day 18, TCRTg101 proliferation was assessed.
(H) Representative FACS plots showing CTV dilution and CD44 expression on TCRTg101 in livers of mice previously challenged with C1498 or C1498.H2-Kb−/− cells.
(I) Quantitative data from (H) are shown.
(J–L) 2 × 106 CTV-labeled TCRTg101 (CD45.1.2) were transferred i.v. into B6.SJL (CD45.1) mice followed by a s.c. challenge with 106 C1498 or C1498.H2-Kb−/− cells the following day. 14 days later, TCRTg101 frequencies and numbers were assessed in tdLNs and tumors.
(J) Representative FACS plots showing TCRTg101 frequencies in tdLNs of mice with s.c. C1498 or Kb−/− C1498 tumors.
(K and L) Quantitative data showing frequencies (K) and numbers (L) of TCRTg101 in tdLNs of mice with s.c C1498 or Kb−/− C1498 tumors.
(M) Representative FACS plots showing TCRTg101 frequencies among total CD8+ T cell populations in s.c. C1498 or Kb−/− C1498 tumors.
(N and O) Quantitative data showing TCRTg101 frequencies (N) and numbers/mg of tumor tissue (O).
Data are representative (I) of or pooled (E–F, K–L, and M–O) from 2 independent experiments with 3 mice/group and presented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Cross-presentation is a major pathway by which tumor antigens are displayed to CD8+ T cells (Hildner et al., 2008; Theisen et al., 2018). The observation that cDC1s were dispensable for TCRTg101 expansion in leukemia-bearing animals suggested either that the Tg101 antigen was cross-presented by a separate APC population or that a different antigen-presentation pathway was involved altogether. To address the first possibility, CD8α+ cDC1, CD11b+ cDC2, and CD11b+CD11clo/− cells (monocytes/macrophages) were isolated from spleens of mice challenged i.v. 3 h earlier with C1498 cells expressing the Kb-restricted SIYRYYGL peptide antigen (C1498.SIY) recognized by TCR2C. We have demonstrated that a fraction of splenic CD8α+ cDC1s engulf C1498 cell-derived material and cross-present the SIY antigen to TCR2C at this time point (Kline et al., 2018). Consistent with previous results, CD8 α+ cDC1s, but not CD11b+ cDC2s or macrophages/monocytes, induced proliferation of TCR2C directly ex vivo, consistent with their ability to cross-present the SIY antigen. However, none of these APC populations supported TCRTg101 activation ex vivo (Figure 5B), arguing that the Tg101 antigen was not effectively cross-presented, at least at the time point assessed in this experiment.
To determine whether DCs were at all capable of presenting the Tg101 antigen in vivo, ItgaxDTR-EGFP (CD11c-DTR) bone marrow chimeric mice were generated. 8 weeks later, CD11c-DTR bone marrow chimeric animals received adoptive transfer of TCRTg101 followed 1 day later by i.v. C1498 cell challenge. CD11c-DTR chimeras also received DT or PBS 1 day prior to TCRTg101 transfer and every 2 days thereafter for a total of 7 doses. As shown in Figure 5C, DT effectively depleted CD11c+ cells from CD11c-DTR bone marrow chimeric animals. Surprisingly, TCRTg101 numbers and proliferation profiles were identical in the presence or absence of CD11c+ cells (Figures 5D–5F), indicating that DCs are dispensable for in vivo antigen presentation to TCRTg101. Lastly, bone-marrow-derived DCs (BMDCs) pulsed with C1498 cell lysates were also incapable of inducing TCRTg101 proliferation in vitro (Figure 5G), consistent with our in vivo findings. Collectively, these observations revealed that the Tg101 antigen was not efficiently displayed through cross-presentation and suggested that an alternative presentation pathway mediated antigen recognition by TCRTg101 in vivo.
To determine whether direct Tg101 antigen presentation by C1498 leukemia cells was required for in vivo antigen encounter by TCRTg101, their expansion was compared in mice challenged i.v. with parental C1498 or C1498 Kb−/− cells. TCRTg101 expanded in mice challenged with parental C1498 cells as expected. However, no TCRTg101 expansion occurred in mice inoculated with C1498 Kb−/− cells (Figures 5H and 5I). A caveat of this experiment was that the survival of mice challenged with C1498 Kb−/− cells was significantly prolonged compared to mice challenged with parental C1498 cells. This difference persisted in C57BL/6 mice, in which natural killer (NK) cells were depleted, and in Rag−/−γc−/− mice, which lacked T cells, B cells, and NK cells. These results suggested that engraftment and/or progression of C1498 Kb−/− leukemia was impaired, potentially through enhanced phagocytosis of leukemia cells with diminished MHC class I expression. Fortunately, parental C1498 and C1498 Kb−/− tumors grew similarly following s.c. inoculation in C57BL/6 mice. TCRTg101 expanded vigorously in tdLNs of mice with localized tumors derived from parental C1498 cells but were significantly reduced in frequency and number within tdLNs of mice with localized C1498 Kb−/− tumors (Figures 5J–5L). Furthermore, TCRTg101 frequencies and numbers per mg of tumor tissue were significantly higher in mice with parental C1498 versus C1498 Kb−/− tumors (Figures 5M–5O). These results strongly suggested that the Tg101 antigen was directly presented to TCRTg101 by C1498 cells in vivo.
Disparate fates of two leukemia-specific CD8+ T cell clones
Finally, the in vivo behavior of TCRTg101 and TCR2C was directly compared in leukemia-bearing animals. Because the site and context of initial antigen encounter may be important factors in determining subsequent leukemia-specific CD8+ T cell fates, it was of interest to determine the site at which TCR2C and TCRTg101 first encountered antigens in vivo. Because our previous work indicated that TCR2C encountered cognate antigen in the spleen very early after leukemia challenge (Kline et al., 2018), TCR2C expansion and CD44 upregulation were assessed 2 days following i.v. C1498.SIY cell challenge (3 days following TCR2C adoptive transfer) in various organs, including the spleen, liver, liver-dLNs (celiac and portal), skin-dLNs, bone marrow, and blood. As predicted, at this early time point, TCR2C had already begun to expand and upregulate CD44 expression almost exclusively in the spleens of leukemia-challenged animals (Figures S6G and S6H). Experiments outlined in Figures 2C–2G suggested that TCRTg101 encountered cognate antigen later than TCR2C, likely around 7 to 10 days following C1498 cell challenge. In fact, TCRTg101 proliferation and CD44 upregulation was not detectable in leukemia-challenged mice prior to day 7. However, 7 days following C1498 challenge, prior to proliferation, TCRTg101 began to upregulate CD44 expression to a higher degree in the liver compared to the spleen, bone marrow, LNs, or blood (Figures S6G and S6I). This result suggested that TCRTg101 initially encountered cognate antigen in the liver via direct presentation by C1498 cells (Figure 5).
As expected, TCR2C expanded rapidly in spleens of mice challenged i.v. with C1498.SIY cells, decreased in number by day 7, and by day 14 were similar in number to TCR2C in leukemia-naive mice (Figures 6A–6C). Similarly, TCR2C expanded minimally by day 7 in livers of leukemia-bearing animals but subsequently decreased to nearly undetectable numbers by day 14 (Figures 6A, 6B, and 6D). These results are consistent with our previous reports showing that TCR2C undergo abortive proliferation and subsequent deletion in mice with leukemia (Kline et al., 2018; Zhang et al., 2013). In stark contrast, TCRTg101 expanded modestly in spleens of mice challenged with parental C1498 cells over time and accumulated significantly in livers of leukemia-bearing mice, particularly when compared with TCR2C (Figures 6A–6D). Thus, although tolerance is effectively established in TCR2C and TCRTg101 in mice with leukemia, the underlying mechanisms are entirely different. Whereas tolerance of TCR2C occurs primarily through deletion requiring antigen cross-presenting cDC1s, TCRTg101 tolerance is mediated by the progressive acquisition of a dysfunctional phenotype requiring direct presentation of the Tg101 antigen by leukemia cells.
Figure 6. Disparate fates of leukemia-specific CD8+ T cell clones.
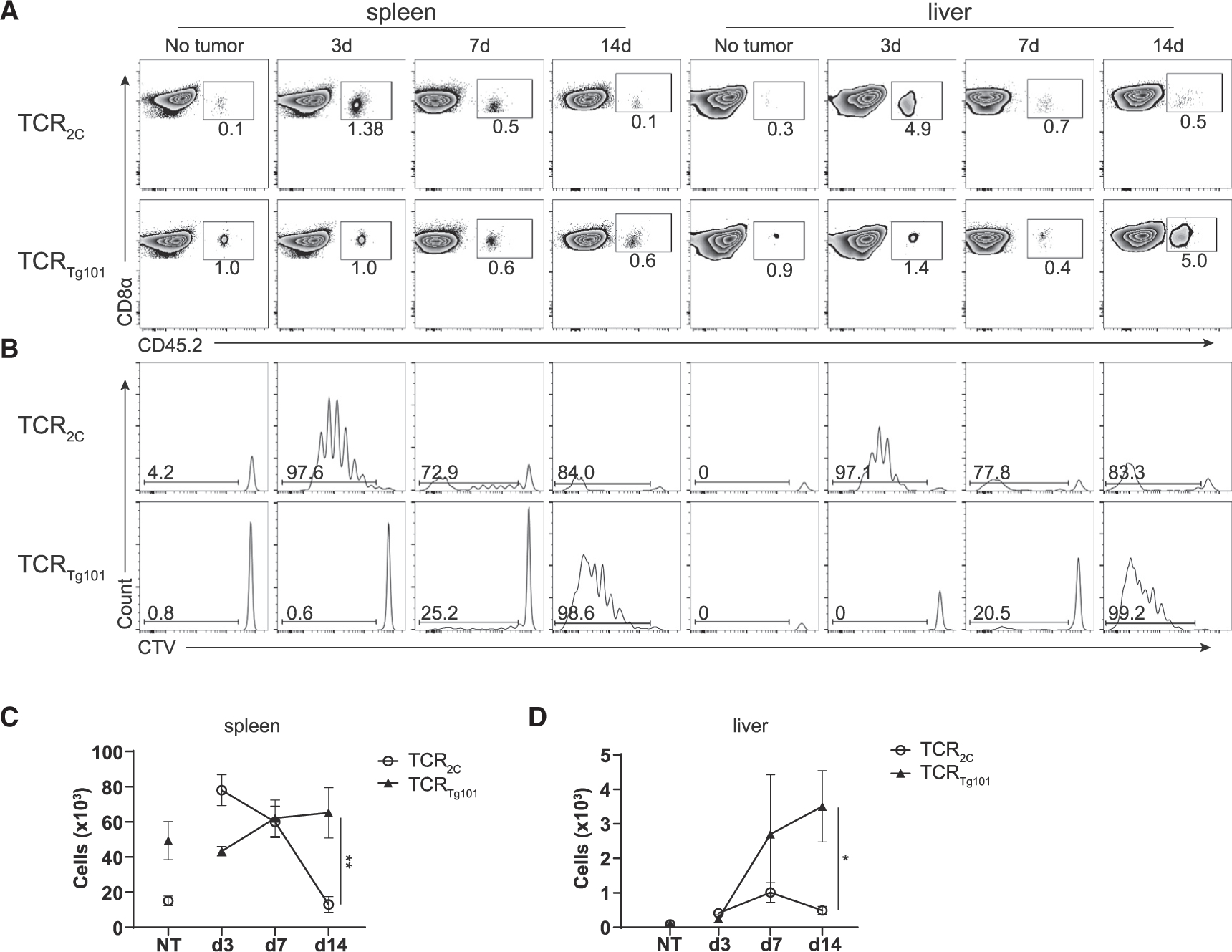
CTV-labeled TCRTg101 (CD45.2) or TCR2C (CD45.1.2) (106 each) were adoptively transferred into individual B6.SJL (CD45.1) mice 1 day prior to i.v. challenge with C1498.SIY cells.
(A and B) Representative FACS plots depicting the frequencies (A) and CTV dilution profiles (B) of TCRTg101 and TCR2C in spleens or livers of leukemia-bearing mice at the indicated time points.
(C and D) Numbers of TCRTg101 and TCR2C in spleens (C) and livers (D) of leukemia-bearing mice at the indicated time points.
Data are pooled from 2 independent experiments with 2 to 4 mice/group and presented as mean ± SD. *p < 0.05; **p < 0.01.
DISCUSSION
The acquisition of a dysfunctional phenotype among antigen-specific CD8+ T cells in the solid tumor environment is a well-recognized phenomenon (Gajewski et al., 2006; Thommen and Schumacher, 2018), which in many cases results from chronic TCR stimulation by antigen-expressing cancer cells capable of avoiding immune-mediated elimination (Pauken and Wherry, 2015). This repeated and suboptimal CD8+ T cell activation is accompanied by co-inhibitory receptor upregulation, which, along with transcriptional, epigenetic, and metabolic re-programming, leads to a progressive and eventually irreversible exhausted state (Pauken and Wherry, 2015; Philip et al., 2017; Schietinger et al., 2016; Scott et al., 2019; Williams et al., 2017). Because immune checkpoint blockade therapy can restore (at least partially) the function of exhausted CD8+ T cells in a subset of patients with cancer (Ribas and Wolchok, 2018; Sharma and Allison, 2015), defining the mechanisms that regulate tumor-specific CD8+ T cell tolerance has garnered much attention in the past decade.
In contrast to solid tumors, mechanisms that promote CD8+ T cell tolerance to hematologic cancers have not been as well-elucidated. However, we previously reported a unique T cell tolerance mechanism in which antigen cross-presenting cDC1s induced the deletion of high-affinity, leukemia-specific CD8+ T cells (TCR2C) (Kline et al., 2018; Zhang et al., 2013). Here, we have characterized the behavior of a CD8+ T cell clone specific for a naturally expressed leukemia antigen, TCRTg101, which was not deleted but rather accumulated in leukemia-bearing hosts. TCRTg101 expansion, however, was coupled with the acquisition of an exhausted phenotype, characterized by co-inhibitory receptor upregulation, transcriptional reprogramming, and diminished effector function. The markedly disparate fates of TCR2C and TCRTg101 in leukemia-bearing animals might be explained by several factors. First, in vivo antigen encounter by TCR2C and TCRTg101 was mediated by different APCs. Leukemia antigen cross-presenting cDC1 were required for antigen encounter by and subsequent deletion of TCR2C (Kline et al., 2018). Conversely, TCRTg101 encountered antigen via direct presentation by C1498 leukemia cells that lack expression of classical T cell co-stimulatory receptors. Second, unique properties associated with the leukemia antigens recognized by TCR2C and TCRTg101 could have conferred the divergent outcomes observed. For example, high-affinity interactions occurring between TCR2C and SIY antigen cross-presenting cDC1s preferentially induced initial proliferation but subsequent deletion of TCR2C. On the other hand, presumably lower affinity interactions between TCRTg101 and C1498 leukemia cells, or reduced Tg101 antigen presentation on the leukemia cell surface associated with decreased TCRTg101 avidity, may have promoted their differentiation into an exhausted state (Figure 5A) (Dougan et al., 2013). Finally, the local environments in which these unique CD8+ T cell clones encountered their cognate antigens may also have impacted their ensuing fates. TCR2C initially recognized cognate antigen in the spleen (Figures S6G and S6H and Kline et al., 2018), while antigen recognition by TCRTg101 occurred later in the disease course and primarily within the liver, an organ associated with immune-suppressive properties (Kubes and Jenne, 2018). Clearly, further investigation will be required to formally address these important questions independently. Regardless, our results imply that numerous mechanisms underlie antigen-specific CD8+ T tolerance in the leukemia-bearing host and suggest that by the point at which a patient is diagnosed with acute leukemia, high-affinity leukemia-specific CD8+ T cells may be largely absent, having already been deleted, leaving behind a pool of exhausted, lower-affinity CD8+ T cells, the function of which may or may not be restorable with immunotherapeutic intervention.
Another key finding of our work was the inability to restore functional competence of exhausted TCRTg101, which indicated that once fully established, TCRTg101 dysfunction was profound and possibly irreversible. The upregulation of transcription factors in TCRTg101, such as Tox, Nr4a1, Nr4a2, and Nr4a3, previously shown to mediate epigenetic re-programming of terminally exhausted CD8+ T cells in solid cancers (Chen et al., 2019a; Liu et al., 2019; Scott et al., 2019), as well as those known to promote an anergic T cell phenotype, including Egr2 and Egr3 (Safford et al., 2005; Zheng et al., 2012), supports the notion that the functionally unresponsive TCRTg101 phenotype at some point becomes fully imprinted as the leukemia progresses in the host. Interestingly, however, even in mice with advanced leukemia, exhausted TCRTg101 largely retained proliferative capability, indicating that the regulation of cell-cycle progression and effector function are uncoupled until very late stages of TCRTg101 dysfunction, which has been previously demonstrated in exhausted CD8+ T cells in settings of cancer and chronic viral infection (Schietinger et al., 2016; Wherry, 2011). Furthermore, while upregulation of PD-1 and LAG-3 was observed on the majority of dysfunctional TCRTg101, combined PD-1 and LAG-3 blockade therapy did little to enhance effector cytokine production or control disease progression, which was somewhat surprising given the maintained expression of TCF1 in a significant proportion of TCRTg101 in the leukemia environment (Figures 3G, 3K, and 3L). Regardless, this result indicated that the mechanisms driving the dysfunctional TCRTg101 program were (or at some point became) independent of key T cell checkpoint receptors (Scott et al., 2019; Singer et al., 2016). Finally, despite their functional incompetence, TCRTg101 continued to express mRNAs for effector cytokines (Ifng) and cytolytic proteins (Gzmb, Prf1) at high levels. Presumably, post-transcriptional mechanisms were negatively regulating translation of these effector molecules, leading to the observed decrease in IFNγ production and cytolytic capacity of dysfunctional TCRTg101 (Salerno et al., 2018). Regardless, this finding supports the notion that the dysfunctional state acquired by TCRTg101 is associated with partially maintained, albeit ineffectual, effector programs. Overall, these results may be important to consider when therapeutic strategies aimed at reversing CD8+ T cell dysfunction in the leukemia context are being developed. However, a potential caveat with regard to transplantable murine leukemias is their tendency to progress primarily in the liver compared to bone marrow, as is the case in the human disease. This is a long-standing limitation of such models, and our data should be interpreted with that knowledge in mind.
Surprisingly, classical cross-presentation was dispensable for in vivo antigen encounter by TCRTg101. In vitro experiments also failed to detect cross-presentation of the Tg101 antigen by professional APCs. On the contrary, direct antigen presentation by leukemia cells was necessary for inducing TCRTg101 expansion and eventual dysfunction in vivo. The reasons for the inability of the Tg101 antigen to be displayed through cross-presentation are unknown, but low abundance, or failure of the DC immunoproteasome to efficiently generate the antigenic peptide recognized by TCRTg101, are possible explanations. This question can be directly addressed once the peptide antigen recognized by TCRTg101 has been identified, which is an area of active investigation in our laboratory. Regardless, results outlined in Figures 1 and S1 indicate that the Tg101 antigen is likely leukemia-specific. Once defined, MHC multimers can also be utilized to study the behavior of endogenous CD8+ T cells specific for the Tg101 antigen, and the true affinity of TCRTg101 for cognate antigen can be established.
In conclusion, our findings advance the understanding of the mechanisms associated with CD8+ T cell tolerance in hematological cancers such as leukemia. We show that the fates of leukemia-specific CD8+ T cell clones are highly divergent and are governed, at least in part, by the context in which their cognate antigen is recognized. Regardless of the underlying CD8+ tolerance mechanism (deletion versus exhaustion), neither is reversible through immune checkpoint blockade therapy, which may help to explain the disappointing efficacy of anti-PD-1 monotherapy in patients with acute leukemia (Berger et al., 2008). In the context of deletional CD8+ T cell tolerance mediated by cDC1s, we have shown that targeting cDC1 activation with toll-like receptor (TLR), CD40, or simulator of interferon genes (STING) agonists can be effective in restoring functional anti-leukemia CD8+ T cell responses (Curran et al., 2016; Kline et al., 2018; Zhang et al., 2013). Reinvigorating the function of exhausted, leukemia-specific CD8+ T cells may be possible through epigenetic reprogramming (Peng et al., 2015), although this is speculative and will require further experimentation. Finally, we expect that Tg101 mice will be useful to other investigators interested in studying tumor-specific CD8+ T cell exhaustion.
Study limitations
A transplantable murine leukemia model served as the basis for the studies and related conclusions presented above. The extent to which the C1498 leukemia model recapitulates the biology of human leukemia is debatable. Furthermore, although we have thoroughly characterized the behavior of TCRTg101 in leukemia-bearing animals and have identified the cells that present antigen to TCRTg101, without knowing the nature of the Tg101 antigen, we are unable to draw conclusions regarding the natural affinity of this CD8+ T cell clone for cognate antigen and the impact on its behavior in vivo, nor are we able to identify and investigate the biology of endogenous CD8+ T cells reactive to the antigen.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Justin Kline, jkline@medicine.bsd.uchicago.edu.
Material availability
Tg101 mice generated in the study this study is available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
RNA sequencing data has been deposited at GEO and is publicly available as of the date of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD45.2 | Biolegend | Clone: 104; Cat# 109820; 109806; 109814 |
| CD45.1 | Biolegend | Clone: A20; Cat#110722; 110706; 110708; 110714 |
| CD3e | Biolegend, BioxCell | Clone: CD3e (145–2C11); Cat#100204; BE0001–1 |
| TCRβ | Biolegend | Clone: H57–597;Cat#109208 |
| CD8α | Biolegend; eBioscience | Clone: 53–6.7; Cat# 100744; 100734; Cat #45–0081-82 |
| CD4 | Biolegend | Clone:RM4.5; Cat#100528 |
| Thy1.2 | Biolegend | Clone: 30-H12; 105326 |
| CD11b | Biolegend | Clone:M1/70; Cat#101263 |
| H2-Db | Biolegend | Clone:KH95; Cat# 111507 |
| H2-Kb | Biolegend | Clone: AF6–88.5; Cat#116518 |
| I-Ab/I-Eb | eBioscience | Clone: (M5/114.15.2); Cat#17–5958-82 |
| PD-1 | eBioscience | Clone: RMP1–30; Cat#25–5982-82 |
| TIM3 | Biolegend | Clone: RMT3–23; Cat#119704 |
| LAG-3 | Biolegend; BioXcell | Clone: C9B7W; Cat#125210; BE0174 |
| TIGIT | eBioscience | Clone:GIGD7;Cat# 12–9501-82 |
| TNFα | eBioscience | Clone:Mp6-XT22; Cat#12–7321-82 |
| IFNγ | BD | Clone: XMG1.2; Cat#554413 |
| Granzyme B | Biolegend | Clone: QA16A02; Cat#372211 |
| B220 | Biolegend | Clone:RA3–6B2; Cat#103206 |
| CD44 | Biolegend | Clone: IM7; Cat#103030 |
| CD62L | Biolegend | Clone: Mel-14; |
| CD69 | Biolegend | Clone: H1.2F3; |
| CD11c | Biolegend | Clone: HL3; |
| Eomes | Biolegend | Clone: Dan11mag; |
| T-bet | Biolegend | Clone: 4B10; |
| Egr2 | Biolegend | Clone: erongr2; |
| TOX | Miltenyi | Clone: REA473; Cat#130–118-474 |
| CD28 | BioXCell | Clone: PV-1; Cat#BE0015–5 |
| CD16/32 | Bio X Cell | 2.4g2;Cat# BE0307 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PMA | Sigma | Cat# P1585 |
| ionomycin | Sigma | Cat# I0634 |
| CellTrace Violet Cell Proliferation Kit, for flow cytometry | Invitrogen | Cat# C34557 |
| APC BrDU Kit | BD | Cat# 552598 |
| Foxp3 staining kit | eBioscience | Cat#00–5523–00 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat#L10119 |
| 1X DPBS GIBCO 14190–250 | GIBCO 14190–250 | Cat#14190–250 |
| RPMI 1640 | GIBCO | Cat#11875–119 |
| DMEM | GIBCO | Cat#11965–118 |
| Fetal Bovine Serum | Gemini | Cat#100–106 |
| Penicillin/Streptimycin | GIBCO | Cat#15140–122 |
| 0.05% Trypsin EDTA | Cat#25300062 | |
| Dnase I | Roche | Cat#10104159001 |
| Collgenase IV | Sigma | Cat#C5138 |
| IL-2 | Biolegend | Cat#575402 |
| IL-7 | Peprotech | Cat#217–17 |
| IL-15 | Biolegend | Cat#566301 |
| GM-CSF | Biolegend | Cat#576304 |
| CD8a Microbeads (mouse) | Miltenyi | Cat#130–117–044 |
| Venor GeM Mycoplasma Detection Kit, PCR-based | Sigma | Cat#MP0025–1KT |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| C1498 | ATCC | Cat#TIB-49 |
| C1498.SIY | This lab | Zhang et al., 2009 |
| C1498.B7.1 | This paper | N/A |
| B16.F10 | A gift from Thomas Gajewski, University of Chicago | N/A |
| EL4 | A gift from Thomas Gajewski, University of Chicago | N/A |
| C1498.H2-Kb−/− | This lab | Kline et al., 2018 |
| C1498.H2-Db−/− | This paper | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 | Taconic | Cat# B6 |
| B6.SJL-Ptprca/BoyAiTac | Taconic | Cat# 4007 |
| Rag2−/− | Taconic | Cat# RAGN12 |
| 2C TCR transgenic mice | Thomas Gajewski lab | Sha et al., 1988 |
| CD11cDTR/GFP B6.FVB-Tg (ItgaxDTR-EGFP) | Jackson Laboratory | Cat# 004509 |
| Batf3−/− | Jackson Laboratory | Cat# 013755 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer XmaI-intron-Va10 forward | IDT | 5′CATCTCCCGGGGCCACACAAG CACCATGAAGAGGCTGCTGTGCTCTCTGC3′ |
| Primer SacII-intron-TRAJ9 reverse | IDT | 5′CACCGCGGTAATTTAAATCAAGT TTCTCATTGCACTCACTTGGA TCAACCAACAAGCTTGTTCCTG3′ |
| Primer XhoI-intron-TRBV5 forward | IDT | 5′AGCCACCTCGAGCCTGATTCCACCATG AGCTGCAGGCTTCTCCTCTATGTTTC3′ |
| Primer SacII-intron-TRBJ1–6 reverse | IDT | 5′CTGCAACCGCGGTCAGAAATGGAGCCCC CATACCTGTCACAGTGAGCCGGGTGCCTG3′ |
|
| ||
| Recombinant DNA | ||
|
| ||
| pTα and pTβ cassettes | Diane Mathis lab | Kouskoff et al., 1995 |
| pLEGFP-N1 | Clontech | 6059–1 |
|
| ||
| Software and algorithms | ||
|
| ||
| Flowjo_V10 | Treestar | Version 10; https://www.flowjo.com/ |
| Prism | GraphPad | Version 8; https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator | Adobe | Version 25.3.1, https://www.adobe.com/ |
|
| ||
| Deposited data | ||
|
| ||
| RNA-Seq | This paper | GEO # GSE186268 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 mice (H-2b; CD45.2) and B6.SJL-Ptprca/BoyAiTac mice (H-2b; CD45.1) were purchased from Taconic Biosciences and breed in our facility. Rag2−/− and 2C TCR transgenic mice (Sha et al., 1988) were bred in our facility. CD11cDTR/GFP B6.FVB-Tg (ItgaxDTR-EGFP) mice and Batf3−/− mice were purchased from Jackson Labs and bred in our facility. Mice were maintained in a specific pathogen-free environment. 6–12 week-old mice, age and sex matched whenever possible, were used for experiments. Animal experimentation was carried out under a protocol approved by an Institutional Animal Use and Care Committee at The University of Chicago.
Generation of Tg101 TCR transgenic mice
Tg101 T cell receptor (TCR) transgenic mice were generated in our laboratory in collaboration with the Transgenic/ES cell technology mouse core facility at the University of Chicago. The Tg101 TCR was derived from a C1498 specific CD8+ T cell clone (T15) (Boyer et al., 1997). TCR Vα10 (TRAV13) and Vβ1 (TRBV5) genes of the T15 TCR were cloned into pTα and pTβ cassettes (Kouskoff et al., 1995). Tg101 TCR DNA was amplified by PCR using primers for the Vα chain and the Vβ chain (below). The pTα10 and pTβ1 plasmids were linearized using SalI (pTα) and KnpI (pTβ), respectively. Via pronuclear injection, linearized plasmids were separately introduced into fertilized eggs of F1 (C57BL/6J) mice. TCR Vα10 and TCR Vβ1 transgenic founder mice were identified by Vα10 and Vβ1 CDR3 spectratyping. TCR Vα10 and TCR Vβ1 transgenic founder mice were crossed to obtain TCR Vα10/Vβ1 (Tg101) mice. Tg101 founder mice were then crossed onto a Rag2−/− background to prevent rearrangement of endogenous TCR loci.
XmaI-intron-Va10 forward:
5’CATCTCCCGGGGCCACACAAGCACCATGAAGAGGCTGCTGTGCTCTCTGC3’
SacII-intron-TRAJ9 reverse:
5’CACCGCGGTAATTTAAATCAAGTTTCTCATTGCACTCACTTGGATCAACCAACAAGCTTGTTCCTG3’
XhoI-intron-TRBV5 forward:
5’AGCCACCTCGAGCCTGATTCCACCATGAGCTGCAGGCTTCTCCTCTATGTTTC3’
SacII-intron-TRBJ1–6 reverse:
5’CTGCAACCGCGGTCAGAAATGGAGCCCCCATACCTGTCACAGTGAGCCGGGTGCCTG3’
Cell lines
The C1498 leukemia cell line (H-2b) was originally purchased from ATCC. C1498 cells expressing the Kb-restricted model SIY (SIYRYYGL) peptide antigen were previously generated in our laboratory (Zhang et al., 2009). C1498.B7.1 cells were engineered by retroviral transduction using the pLEGFP-B7.1 plasmid. B16.F10 and EL4 cell lines were provided by Dr. Thomas Gajewski. Cell lines were routinely tested for mycoplasma contamination using a VenorTM GEM mycoplasma detection kit (Sigma). The H2-Kb-deficient (Kb−/−) C1498 cell line was generated as previously described (Kline et al., 2018). The H2-Db-deficient (Db−/−) C1498 cell line was generated via CRISPR/Cas9 targeting using the following guide sequences: forward - CACCGACCCGCGCGGGTCTGAGTCG; reverse - AAACCGACTCAGACCCGCGCGGGTC.
All the cell lines described in this manuscript are of a H2b haplotype. C1498 derivative cell lines and B16.F10 cells were cultured in DMEM, and EL4 cells were cultured in RPMI-1640 (Invitrogen) both supplemented with 10% FBS, 2-mercaptoethanol, essential amino acids, penicillin and streptomycin at 37°C.
METHOD DETAILS
Flow cytometry
Spleens, lymph nodes, livers, peripheral blood, and bone marrow were harvested from mice, mechanically dissociated with a 3 mL syringe plunger, and pushed through 70 um mesh filter to generate single cell suspensions (spleens, livers, lymph nodes). Liver mononuclear cells were enriched over a Ficoll gradient (BD Biosciences). Following red blood cell lysis and Fc receptor blockade using anti-CD16/32 antibodies, cells were stained with the following directly conjugated antibodies (clones): CD45.2 (104), CD45.1 (A20), CD3ε (145–2C11), TCRβ (H57–597), CD8α (53–6.7), CD4 (RM4.5), Thy1.2 (30-H12), CD11b (M1/70), H2-Db (KH95), H2-Kb (AF6–88.5), I-Ab/I-Eb (M5/114.15.2), PD-1 (RMP1–30), TIM3 (RMT3–23), LAG-3 (C9B7W), TIGIT (GIGD7), TNFα (Mp6-XT22), IFNγ (XMG1.2), Granzyme B (QA16A02), B220 (RA3–6B2), CD44 (IM7), CD62L (Mel-14), CD69 (H1.2F3), CD11c (HL3), Eomes (Dan11mag), T-bet (4B10), Egr2 (erongr2) and TOX (REA473). Fixable viability dyes (Invitrogen) were used to exclude dead cells. Flow cytometry was performed on LSRII or LSRFortessa cytometers (BD Biosciences). Analysis was performed using FlowJo software (Treestar). Fluorescence-activated cell sorting (FACS) was performed using a FACSAria (BD Biosciences). Intracellular antibody staining was performed using a Foxp3 staining kit (eBioscience).
T cell labeling with CellTrace Violet
TCRTg101 were enriched from spleens of Tg101 mice using a mouse CD8 microbead kit (Miltenyi). Isolated TCRTg101 were washed once with PBS and labeled with CellTrace Violet (CTV) (Invitrogen) for 20 minutes at 37°C and quenched with 10% FBS in RPMI-10. Prior to adoptive transfer, CTV-labeled TCRTg101 were washed twice more with PBS.
In vitro TCRTg101 proliferation assay
105 CTV-labeled TCRTg101 were plated in 96 well, flat-bottom tissue culture plates with (or without) 2×104 γ-irradiated tumor cells (12,000 rads), or with freshly-harvested spleen cells from C57BL/6 mice, and cultured at 37°C for 3 days. Cells were then harvested, and TCRTg101 proliferation, measured by CTV dilution, was analyzed by flow cytometry.
In vivo TCRTg101 proliferation assay
1–2×106 CTV-labeled, congenically-marked TCRTg101 or TCR2C were adoptively transferred i.v. through the lateral tail vein into B6.SJL mice (CD45.1) or into C57BL/6 mice (CD45.2). One day later, 106 C1498 cells (or derivative C1498 cell lines) were inoculated i.v. At subsequent time points indicated, TCRTg101 number, proliferation, co-inhibitor receptor expression, and effector function was analyzed in spleens and livers by flow cytometry. TCR2C numbers were assessed similarly.
In vitro antigen cross-presentation assay
Bone marrow cells isolated from femurs of B6.SJL mice (CD45.1) were cultured with 40 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Biolegend). One-half of the medium was removed on day 2 and replaced with fresh GM-CSF-supplemented medium warmed to 37°C. On day 3, the culture medium was discarded and again replaced with fresh, warmed, GM-CSF-supplemented medium (20 ng/ml). This process was repeated until day 8. At this point, non- and loosely adherent cells (BMDCs) were harvested by gentle washing with PBS and plated at a concentration of 1×105 cells/well in round-bottom 96-well plates. BMDCs were then pretreated with 20 ng/ml LPS for 2 hours and pulsed with C1498 or C1498.SIY cell lysates (3 tumor cell lysates to 1 BMDC). Next, CTV-labeled, congenically-marked TCR2C or TCRTg101 were added and cultured at 37°C for 3 days. Cells in each well were then harvested, and CTV dilution of TCR2C or TCRTg101 was analyzed by flow cytometry as a read out for in vitro antigen cross-presentation.
Ex vivo antigen cross-presentation assay
8×106 C1498.SIY cells were injected i.v. into B6.SJL mice. Three hours later, spleens were harvested and injected with 1 mg/ml collagenase IV (Sigma),20 mg/ml DNase I (Roche) in RPMI with 2% FBS and were incubated at 37°C for 15–20 minutes. Spleens were then dissociated with a 3 mL plunger and passed through a 70 um mesh filter to generate single cell suspensions. CD3+ and CD19+ cells were depleted by positive selection. Remaining cells were stained with fluorescently-labeled antibodies directed against CD3ε (145–2C11), CD19(eBio1D3), CD11c (HL3), CD8α (53–6.7), and CD11b (M1/70). After excluding CD3+ and CD19+ cells, CD8α+ DCs (cDC1) CD11b+ DCs (cDC2), and CD11b+CD11clo/− (monocytes/macrophages) cells were separately isolated by FACS. Sorted APC populations were cultured 1:1 with purified, congenically-marked, CTV-labeled TCR2C or TCRTg101 for 65–72 hours in 96 well, round bottom tissue culture plates. Subsequently, CTV dilution of TCR2C or TCRTg101 was assessed by flow cytometry as a read out for ex vivo antigen cross-presentation.
Ex vivo TCRTg101 restimulation and intracellular cytokine staining
Mononuclear cells isolated from livers of mice at the indicated time points following C1498 cell inoculation were restimulated with plate-bound anti-CD3 antibody (clone: 145–2C11; 1 μg/ml) plus soluble anti-CD28 antibody (clone PV-1, 1 ug/ml), or with PMA plus ionomycin, for 1 hour, followed by 4 hours in the presence of 1 mg/ml GolgiPlug (BD Biosciences). Intracellular staining of effective cytokines was performed using FoxP3 intracellular staining kit (eBiosciences). In some experiments, irradiated (12,000 rads) C1498.B7.1 cells were utilized to restimulate TCRTg101 ex vivo overnight prior to intracellular cytokine staining and flow cytometric analysis.
Ex vivo TCRTg101 killing assay
CTVhi or CTVlo TCRTg101 were separately FACS-purified from livers of mice 17–18 days after i.v. C1498 cell inoculation and 1×104 cells were cultured in vitro with equal numbers of C1498 cells and EL4 cells (1×104 each) labeled with two different concentrations of CTV, along with IL-2 (50 U/ml) and IL-12 (20 ng/ml) in 384 well round bottom tissue culture plates. In negative control wells, C1498 cells and EL4 cells were plated in the absence of TCRTg101. Approximately 20 hours later, proportions of surviving C1498 and EL4 cells in each well were analyzed by flow cytometry. Naive TCRTg101 isolated from spleens of Tg101 mice were activated in vitro for 72 hours with anti-CD3 and anti-CD28 antibodies or irradiated C1498.B7.1 cells prior to co-culture with C1498 cells and EL4 cells to serve as positive controls for C1498 cell lysis. Naive, unstimulated TCRTg101 isolated from spleens of Tg101 mice were also co-cultured with C1498 cells and EL4 cells to serve as a negative control for C1498 cell lysis. Specific lysis was calculated as follows (Noto et al., 2013):
Generation of bone marrow chimeric (BMC) mice
C57BL/6 mice received total body irradiation (900 rads) and were reconstituted 16 hours later with 2.5×106 bone marrow cells isolated from CD11c-DTR mice. Eight weeks later, these bone marrow chimeric mice received TCRTg101 adoptive transfer, followed 1 day later by an i.v. C1498 cell challenge. To deplete CD11c+ cells from leukemia-bearing CD11c-DTR bone marrow chimeric mice, diphtheria toxin (DT) (500 ng) was administered i.p. 2 days prior to C1498 cell inoculation and continued every 48 hours for a total of 7 doses.
Survival experiments
106 C1498 cells were inoculated i.v. into C57BL/6 mice three days before adoptive transfer of 4×106 naive or anti-CD3 and anti-CD28 activated TCRTg101 or TCR2C. Control groups of C57BL/6 mice received an i.v. C1498 cell challenge but did not receive TCRTg101 or TCR2C adoptive cell transfer. Survival was monitored. In experiments involving immune checkpoint blockade therapy, mice transferred with TCRTg101 and challenged with C1498 cells were subsequently administered anti-PD-1 (RMP1–14) and anti-LAG-3 (C9B7W) antibodies (BioXcell) intra-peritoneal, 200 μg each, beginning on day 6, and continued every other day for 2 weeks. Control mice received isotype control antibodies at the same dose and schedule. Survival was monitored.
RNA sequencing
TCRTg101 in livers and spleens of leukemia-bearing mice were isolated at various time points following adoptive transfer (day 0, day 6–7, day 13–14, day 16–18) by FACS and were re-suspended in Trizol (Life Technologies). TCRTg101 RNA was isolated via chloroform extraction. Low input RNA sequencing was performed in the University of Chicago Genomic Core Facility on the Illumina HiSeq 2500 platform in two batches. Reads were mapped onto the University of California Santa Cruz mouse genome using kallisto (Bray et al., 2016). Genes with fewer than 10 reads in at least 6 samples were filtered out, resulting in a dataset of 11,164 genes. Differential gene expression analysis was performed on raw aligned read counts using DESeq2 (Love et al., 2014), with batch effects accounted for in the design formula. Genes were considered to be differentially expressed if they had an adjusted p value < 0.05 using a Benjamini-Hochberg test (FDR). Counts per gene were regular log (rlog)-transformed, and batch effects were removed using the removeBatch-Effect function from limma (Ritchie et al., 2015) for principal component analysis (PCA), sample clustering based on Euclidean distance, and heatmaps depicting z-scores of gene expression. rlog-transformed, batch-corrected counts were also used for weighted gene correlation network analysis (WGCNA) (Langfelder and Horvath, 2008) while additionally filtering out the 50% of genes with the lowest variance to reduce noise, resulting in a set of 5,582 genes. Adjacency was determined using a signed analysis and a soft thresholding power of 14, which was determined by scale-free fit index (Zhang and Horvath, 2005). 12 clusters of genes were initially identified, 2 of which contained a combined 4,269 genes (76.5% of the dataset), and roughly corresponded to genes whose expression increased or decreased over the experimental time course. Of the other 10 clusters, 2 were identified as containing genes which were transiently upregulated or downregulated. These two clusters contained 448 genes, combined. The remaining 865 genes were assigned to eight different clusters, which appeared to be the result of high variance within sample groups, either due to low overall gene expression or low outlier values. No conclusions were drawn regarding these clusters due to uncertainty in the data.
QUANTIFICATION AND STATISTICAL ANALYSIS
Grouped data were analyzed via two-way ANOVA with Bonferroni post-tests. Survival differences were analyzed with the Log-rank test. Statistics were performed using GraphPrism software. Data are presented as mean ± SD unless otherwise indicated. A p value of < 0.05 was considered statistically significant. Additional description of statistical methods for individual experiments can be found in the figure legends.
Supplementary Material
Highlights.
A native leukemia antigen-specific TCR transgenic mouse strain (Tg101) is generated
Leukemia-specific T cells (TCR2C) are deleted by CD8α+ DCs
TCRTg101 became dysfunctional via direct interactions with leukemia cells
Distinct modes of leukemia antigen display confer unique CD8+ T cell tolerant states
ACKNOWLEDGMENTS
The authors would like to acknowledge the University of Chicago Transgenics/ES Cell Technology Mouse Core Facility and, in particular, Linda Degenstein for assistance in generating Tg101 mice. We are also grateful to Marlieke Jongsma, PhD, for generating Kb−/− and Db−/− C1498 cell lines. This work was funded by the Janet Rowley Discovery Fund to J.K. and grants R01 HL56067 and R37 AI 34495 to B.R.B.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109991.
REFERENCES
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, and Rosenberg SA (2009). Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, Koren-Michowitz M, Shimoni A, and Nagler A (2008). Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 14, 3044–3051. [DOI] [PubMed] [Google Scholar]
- Boyer MW, Vallera DA, Taylor PA, Gray GS, Katsanis E, Gorden K, Orchard PJ, and Blazar BR (1997). The role of B7 costimulation by murine acute myeloid leukemia in the generation and function of a CD8+ T-cell line with potent in vivo graft-versus-leukemia properties. Blood 89, 3477–3485. [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Chen J, López-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, and Rao A (2019a). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. (2019b). TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51, 840–855.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran E, Chen X, Corrales L, Kline DE, Dubensky TW Jr., Duttagupta P, Kortylewski M, and Kline J (2016). STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep. 15, 2357–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpoux A, Michelini RH, Verma S, Lai CY, Omilusik KD, Utzschneider DT, Redwood AJ, Goldrath AW, Benedict CA, and Hedrick SM (2018). Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8+ T cells. J. Exp. Med 215, 575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ (2012). Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho HI, Jaenisch R, Celis E, and Ploegh HL (2013). Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunol. Res 1, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, and Harlin H (2006). Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev 213, 131–145. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, Leonard WJ, Greenleaf WJ, Ahmed R, and Goronzy JJ (2019). Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl. Acad. Sci. USA 116, 14113–14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DE, MacNabb BW, Chen X, Chan WC, Fosco D, and Kline J (2018). CD8α+ Dendritic Cells Dictate Leukemia-Specific CD8+ T Cell Fates. J. Immunol 201, 3759–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Signorelli K, Benoist C, and Mathis D (1995). Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods 180, 273–280. [DOI] [PubMed] [Google Scholar]
- Kubes P, and Jenne C (2018). Immune Responses in the Liver. Annu. Rev. Immunol 36, 247–277. [DOI] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, Hao J, Alekseev A, Khong H, Chen T, et al. (2019). Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Noto A, Ngauv P, and Trautmann L (2013). Cell-based flow cytometry assay to measure cytotoxic activity. J. Vis. Exp 82, e51105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, and Wherry EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, and Powell JD (2005). Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol 6, 472–480. [DOI] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno F, Guislain A, Freen-Van Heeren JJ, Nicolet BP, Young HA, and Wolkers MC (2018). Critical role of post-transcriptional regulation for IFN-γ in tumor-infiltrating T cells. OncoImmunology 8, e1532762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 45, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine T, Perez-Potti A, Nguyen S, Gorin JB, Wu VH, Gostick E, Llewellyn-Lacey S, Hammer Q, Falck-Jones S, Vangeti S, et al. (2020). TOX is expressed by exhausted and polyfunctional human effector memory CD8+ T cells. Sci. Immunol 5 (49), eaba7918. [DOI] [PubMed] [Google Scholar]
- Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, et al. (2019). TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 116, 12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha WC, Nelson CA, Newberry RD, Kranzt DM, Russell JH, and Loh DY (1988). Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature 335 (6187), 271–274. 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10. [DOI] [PubMed] [Google Scholar]
- Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. (2016). A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 166, 1500–1511 e1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell RT, and Kaech SM (2016). Probing the Diversity of T Cell Dysfunction in Cancer. Cell 166, 1362–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen DJ, Davidson JT 4th, Briseñ o CG, Gargaro M, Lauron EJ, Wang Q, Desai P, Durai V, Bagadia P, Brickner JR, et al. (2018). WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen DS, and Schumacher TN (2018). T Cell Dysfunction in Cancer. Cancer Cell 33, 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh KA, Leach SM, Moore BL, Bruno TC, Buhrman JD, and Slansky JE (2016). Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model. J. Immunol 197, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ (2011). T cell exhaustion. Nat. Immunol 12, 492–499. [DOI] [PubMed] [Google Scholar]
- Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, and Gajewski TF (2017). The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med 214, 381–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- Zhang L, Gajewski TF, and Kline J (2009). PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood 114, 1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen X, Liu X, Kline DE, Teague RM, Gajewski TF, and Kline J (2013). CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J. Clin. Invest 123, 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zha Y, Driessens G, Locke F, and Gajewski TF (2012). Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med 209, 2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data has been deposited at GEO and is publicly available as of the date of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD45.2 | Biolegend | Clone: 104; Cat# 109820; 109806; 109814 |
| CD45.1 | Biolegend | Clone: A20; Cat#110722; 110706; 110708; 110714 |
| CD3e | Biolegend, BioxCell | Clone: CD3e (145–2C11); Cat#100204; BE0001–1 |
| TCRβ | Biolegend | Clone: H57–597;Cat#109208 |
| CD8α | Biolegend; eBioscience | Clone: 53–6.7; Cat# 100744; 100734; Cat #45–0081-82 |
| CD4 | Biolegend | Clone:RM4.5; Cat#100528 |
| Thy1.2 | Biolegend | Clone: 30-H12; 105326 |
| CD11b | Biolegend | Clone:M1/70; Cat#101263 |
| H2-Db | Biolegend | Clone:KH95; Cat# 111507 |
| H2-Kb | Biolegend | Clone: AF6–88.5; Cat#116518 |
| I-Ab/I-Eb | eBioscience | Clone: (M5/114.15.2); Cat#17–5958-82 |
| PD-1 | eBioscience | Clone: RMP1–30; Cat#25–5982-82 |
| TIM3 | Biolegend | Clone: RMT3–23; Cat#119704 |
| LAG-3 | Biolegend; BioXcell | Clone: C9B7W; Cat#125210; BE0174 |
| TIGIT | eBioscience | Clone:GIGD7;Cat# 12–9501-82 |
| TNFα | eBioscience | Clone:Mp6-XT22; Cat#12–7321-82 |
| IFNγ | BD | Clone: XMG1.2; Cat#554413 |
| Granzyme B | Biolegend | Clone: QA16A02; Cat#372211 |
| B220 | Biolegend | Clone:RA3–6B2; Cat#103206 |
| CD44 | Biolegend | Clone: IM7; Cat#103030 |
| CD62L | Biolegend | Clone: Mel-14; |
| CD69 | Biolegend | Clone: H1.2F3; |
| CD11c | Biolegend | Clone: HL3; |
| Eomes | Biolegend | Clone: Dan11mag; |
| T-bet | Biolegend | Clone: 4B10; |
| Egr2 | Biolegend | Clone: erongr2; |
| TOX | Miltenyi | Clone: REA473; Cat#130–118-474 |
| CD28 | BioXCell | Clone: PV-1; Cat#BE0015–5 |
| CD16/32 | Bio X Cell | 2.4g2;Cat# BE0307 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PMA | Sigma | Cat# P1585 |
| ionomycin | Sigma | Cat# I0634 |
| CellTrace Violet Cell Proliferation Kit, for flow cytometry | Invitrogen | Cat# C34557 |
| APC BrDU Kit | BD | Cat# 552598 |
| Foxp3 staining kit | eBioscience | Cat#00–5523–00 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat#L10119 |
| 1X DPBS GIBCO 14190–250 | GIBCO 14190–250 | Cat#14190–250 |
| RPMI 1640 | GIBCO | Cat#11875–119 |
| DMEM | GIBCO | Cat#11965–118 |
| Fetal Bovine Serum | Gemini | Cat#100–106 |
| Penicillin/Streptimycin | GIBCO | Cat#15140–122 |
| 0.05% Trypsin EDTA | Cat#25300062 | |
| Dnase I | Roche | Cat#10104159001 |
| Collgenase IV | Sigma | Cat#C5138 |
| IL-2 | Biolegend | Cat#575402 |
| IL-7 | Peprotech | Cat#217–17 |
| IL-15 | Biolegend | Cat#566301 |
| GM-CSF | Biolegend | Cat#576304 |
| CD8a Microbeads (mouse) | Miltenyi | Cat#130–117–044 |
| Venor GeM Mycoplasma Detection Kit, PCR-based | Sigma | Cat#MP0025–1KT |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| C1498 | ATCC | Cat#TIB-49 |
| C1498.SIY | This lab | Zhang et al., 2009 |
| C1498.B7.1 | This paper | N/A |
| B16.F10 | A gift from Thomas Gajewski, University of Chicago | N/A |
| EL4 | A gift from Thomas Gajewski, University of Chicago | N/A |
| C1498.H2-Kb−/− | This lab | Kline et al., 2018 |
| C1498.H2-Db−/− | This paper | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 | Taconic | Cat# B6 |
| B6.SJL-Ptprca/BoyAiTac | Taconic | Cat# 4007 |
| Rag2−/− | Taconic | Cat# RAGN12 |
| 2C TCR transgenic mice | Thomas Gajewski lab | Sha et al., 1988 |
| CD11cDTR/GFP B6.FVB-Tg (ItgaxDTR-EGFP) | Jackson Laboratory | Cat# 004509 |
| Batf3−/− | Jackson Laboratory | Cat# 013755 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer XmaI-intron-Va10 forward | IDT | 5′CATCTCCCGGGGCCACACAAG CACCATGAAGAGGCTGCTGTGCTCTCTGC3′ |
| Primer SacII-intron-TRAJ9 reverse | IDT | 5′CACCGCGGTAATTTAAATCAAGT TTCTCATTGCACTCACTTGGA TCAACCAACAAGCTTGTTCCTG3′ |
| Primer XhoI-intron-TRBV5 forward | IDT | 5′AGCCACCTCGAGCCTGATTCCACCATG AGCTGCAGGCTTCTCCTCTATGTTTC3′ |
| Primer SacII-intron-TRBJ1–6 reverse | IDT | 5′CTGCAACCGCGGTCAGAAATGGAGCCCC CATACCTGTCACAGTGAGCCGGGTGCCTG3′ |
|
| ||
| Recombinant DNA | ||
|
| ||
| pTα and pTβ cassettes | Diane Mathis lab | Kouskoff et al., 1995 |
| pLEGFP-N1 | Clontech | 6059–1 |
|
| ||
| Software and algorithms | ||
|
| ||
| Flowjo_V10 | Treestar | Version 10; https://www.flowjo.com/ |
| Prism | GraphPad | Version 8; https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator | Adobe | Version 25.3.1, https://www.adobe.com/ |
|
| ||
| Deposited data | ||
|
| ||
| RNA-Seq | This paper | GEO # GSE186268 |