

Abstract
Natural products recognized traditionally as a vital source of active constituents in pharmacotherapy. The COVID‐19 infection caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is highly transmissible, pathogenic, and considered an ongoing global health emergency. The emergence of COVID‐19 globally and the lack of adequate treatment brought attention towards herbal medicines, and scientists across the globe instigated the search for novel drugs from medicinal plants and natural products to tackle this deadly virus. The natural products rich in scaffold diversity and structural complexity are an excellent source for antiviral drug discovery. Recently the investigation of several natural products and their synthetic derivatives resulted in the identification of promising anti SARS‐CoV‐2 agents. This review article will highlight the pharmacological relevance and chemical synthesis of the recently discovered natural product and their synthetic analogs as SARS‐CoV‐2 inhibitors. The summarized information will pave the path for the natural product‐based drug discovery of safe and potent antiviral agents, particularly against SARS‐CoV‐2.
Keywords: anti-SARS-CoV-2 activity, biological activity, coronavirus, COVID-19, natural products, natural product derivatives, SARS-CoV-2
The review thoroughly discusses the current progress in natural product and their derivatives significance in SARS‐CoV‐2 treatment, predominantly focusing on pharmacological relevance and chemical synthesis. This review offers a solid basis for discovering and developing natural product‐based drugs for the SARS‐CoV‐2 treatment.

1. Introduction
The deadly SARS‐CoV‐2 virus has devoured more than four million lives as per the WHO report, dated 21st July 2021. [1] Even after eighteen months of the appearance of the first case in Wuhan, China, the number of confirmed COVID‐19 cases and deaths is still rising continuously worldwide. The COVID‐19 pandemic has caused devastating economic and social disruption in many countries around the globe and created an incredible risk to the physical and mental wellbeing of the current human era.[ 2 , 3 , 4 ] It has affected the existence of nearly every person with psychosocial consequences on a global scale. The SARS‐CoV‐2 spread rate is much faster than other viruses of the Coronavirus family, such as SARS‐CoV and MER‐CoV. SARS‐CoV‐2 similar to other viruses is constantly changing through mutation, and consequently, several variants of SARS‐CoV‐2 are emerging from various corners of the world.[ 5 , 6 , 7 ] The most common SARS‐CoV‐2 symptoms are fever, body ache, dry cough, fatigue, chills, headache, sore throat, loss of appetite, and loss of smell.[ 8 , 9 , 10 ] A few individuals with a robust immune system or who have developed herd immunity are asymptomatic. Most of the COVID‐19 infected people develop mild to moderate symptoms and recover without special treatment. In elderly patients, particularly those above 50 years or in patients with underlying medical problems, including diabetes, cardiovascular disease, respiratory disorders, and cancer, it may progress to pneumonia, acute respiratory distress syndrome, and multi‐organ failure resulting in mortality. Furthermore, it has been observed that individuals infected with SARS‐CoV‐2 can also experience gastrointestinal (GI) symptoms, neurological symptoms, or both, with or without respiratory symptoms. Besides, in few COVID‐19 patients, the symptoms last for weeks or months past the initial illness, also referred to as a post‐COVID‐19 syndrome or long COVID.[ 11 , 12 , 13 ]
With the emergence, spread, and WHO declaration of COVID‐19 as a global pandemic, scientists worldwide worked faster than ever to develop and produce a vaccine that protects against the SARS‐CoV‐2 and subsequently reduces its spread.[ 14 , 15 ] Globally, 20 vaccines have been authorized for SARS‐CoV‐2, and 86 vaccines are in development. [16] Indeed, the vaccine is playing a remarkable role in confining the pandemic, and most nations worldwide are running vaccination/immunization programs vigorously. Nevertheless, for the people who are unvaccinated, who develop a breakthrough infection, and whose body does not demonstrate a strong response to the vaccine, the SARS‐CoV‐2 drug is essential and lifesaving.[ 17 , 18 , 19 , 20 ] The drug discovery and development process are quite challenging, focusing on antiviral drugs that can reduce the virus replication by targeting the specific parts of the virus life cycle and killing the only virus, not human cells. [21]
Additionally, the virus‘s proficiency to mutate rapidly increases its adaptability and reproduction and upsurges the probabilities of developing potential resistance against the developed drugs or vaccines.[ 22 , 23 , 24 ] Researchers continuously create antiviral medications for SARS‐CoV‐2 treatment and prepare for future pandemic threats.[ 25 , 26 , 27 ] The focus is on the testing of antiviral drugs in clinical trials and on the disclosure and development of the new chemical entities as an anti‐SARS‐CoV‐2 drug.[ 28 , 29 ] To develop potent drugs for the SARS‐CoV‐2 treatment, all the enzymes and proteins responsible for viral replication and managing host cellular machinery are significant targets, including primary virus‐based and host‐based targets.[ 30 , 31 , 32 , 33 ]
Since ancient times, several natural products have been traditionally used to treat many diseases and illnesses.[ 34 , 35 , 36 ] Natural products and their synthetic derivatives have made a significant contribution to drug therapy for several diseases, particularly cancer and infectious diseases. Natural products and their analogs have been the thriving source of potential drug leads because of their therapeutic effect, chemical structure diversity, and biodiversity.[ 37 , 38 , 39 ] Moreover, due to scientific and technological advancement, the natural product‐based drug discovery program revolutionized and revealed new opportunities in recent years.
Natural products, special features including scaffold diversity, structural complexity, high molecular mass, and molecular rigidity make them valuable in a drug discovery program compared to conventional synthetic compounds. Over the years with evolution, the structure of natural products optimized to work efficiently in various biological functions, including endogenous defense mechanism regulation and interaction with other organisms. In addition, the use of natural products as a traditional medicine from ancient times gives some information regarding its efficacy and safety.[ 40 , 41 , 42 ] Indeed, the natural products are a rich source of bioactive compounds and can lead to new and promising drug leads for the treatment of various diseases by overcoming the natural product drug discovery associated challenges such as isolation, characterization, and optimization. [43] The emergence of COVID‐19 in China and the absence of conventional therapies results in traditional Chinese medicines application in 85 % of SARS‐CoV‐2 infected patients treatment. [44] Considering that natural product and natural product inspired molecules are an incredible source of drugs and drug leads to various diseases, recently the natural products and their derivatives have gained attention for SARS‐CoV‐2 treatment.[ 45 , 46 , 47 , 48 , 49 ] In the last eighteen months, researchers′ continuous efforts around the globe resulted in the identification of many natural products and their synthetic derivatives as anti‐SARS‐CoV‐2 agents with promising pharmacological relevance. This review thoroughly discusses the current progress in natural products and their derivatives significance in SARS‐CoV‐2 treatment, predominantly focusing on pharmacological relevance and chemical synthesis. It is highly recommended to perform an immense screening of the natural product and its analogs to overcome the current treatment challenges and identify potent agents for SARS‐CoV‐2 treatment and prevention. This perspective is within the context of our ongoing research theme, drug design and discovery.[ 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 ] and offers new prospects in discovering the SARS‐CoV‐2 treatment from natural products. We expect that the information presented in this review will facilitate the natural product drug discovery program associated with developing efficacious and cost‐effective SARS‐CoV‐2 treatment.
2. Structure and action mechanism of SARS‐CoV‐2
The coronavirus genome consists of 30000 nucleotides, and it encodes four structural proteins, including Spike (S) glycoprotein, Envelop (E) protein, Membrane (M) protein, Nucleocapsid (N) protein, and several non‐structural proteins (nsps) (Figure 1).[ 30 , 31 , 58 , 59 ] Inside the virus capsid (a protein shell), the N‐protein protects the viral RNA genome by coating it into a ribonucleoprotein complex. Its functioning involves viral RNA replication and transcription. The Spike (S) glycoprotein is a structural transmembrane protein positioned on the outer envelope of the virion. It enables virus entry by contacting specific receptors, particularly ACE‐2 located on the host cell‘s surface. The spike (S) glycoprotein is a homotrimer, where each monomer contains approximately 1200–1400 amino acid residues and comprises two well‐defined protein domain regions, including the S1 subunit (14–685 residues) associated with binding to host cell receptor and S2 subunit (686–1273 residues) involved in the fusion of viral and cellular membranes. Moreover, the S protein homotrimers contribute toward the virus‘s crown‐like corona; S1 and S2 subunit silhouette the bulbous head and stalk region in S protein monomers. The most abundant protein present on the virus‘s surface is M‐protein, which is involved in virus fusion, assembly, and budding function due to its membrane‐bending properties and is considered a central organizer for the coronavirus assembly. The E‐protein is a small membrane protein composed of 76–109 amino acids and plays a significant role in virus assembly and morphogenesis and virus‐host cell interaction. Hemagglutinin‐esterase dimer (HE) is present on the virus surface, and it′s crucial for virus entry but not for replication. Additionally, the SARS‐CoV‐2 structure includes highly conserved 16 nsps, liable for various functions, including the generation of replication‐transcription complex (RTC). Some of the nsps are vital for virus including chymotrypsin‐like protease (3CLpro, nsp5), papain‐like protease (PLpro, nsp3), RNA‐dependent RNA polymerase (RdRp, nsp12) in complex with cofactors nsp7, and nsp8 and helicase (nsp13), and therefore considered as promising targets for the development of SARS‐CoV‐2 treatment.
Figure 1.

Schematic representation of SARS‐CoV‐2 and its structural proteins.
The action mechanism of SARS‐CoV‐2 in a human cell involves several steps, including viral entry, replication, and RNA packing.[ 30 , 58 , 59 , 60 , 61 ] The SARS‐CoV‐2 virus enters the host cell by binding its spike (S) protein to angiotensin‐converting enzyme 2 (ACE2) receptors present on human cells′ surfaces. The spike (S) protein and various cellular proteases are highly crucial for membrane fusion, an essential phase in a virus infection cycle, and process via two pathways comprising the endosomal/clathrin‐dependent and nonendosomal/clathrin‐independent paths. The endosomal/clathrin‐dependent route involves the fusion of internalized viral particles with the host target membrane via the S2 subunit of S‐protein (low pH), followed by the S‐protein cleavage by the host protease cathepsin L. The nonendosomal/clathrin‐independent path includes the fusion with cellular membrane followed by host protease cleavage of the S protein through cell membrane‐associated proteases TMPRSS2. After membrane fusion, the viral genetic material is released into the cytoplasm, resulting in the replication/transcription complex (RTC)‐mediated replication and transcription processes. The viral RNA, after attachment with host ribosome, produce polyproteins (pp1a and pp1ab) encoding 16 nsps that go through proteolytic cleavage by the coronavirus main proteinase (3CLpro) and the papain‐like protease (PLpro). Additionally, RNA‐dependent RNA polymerase (RdRp), an essential enzyme that helps in RNA synthesis by catalyzing the RNA template‐dependent development of phosphodiester bonds, is accountable for RNA genome replication. The structural viral proteins, including E, M, and S proteins, are synthesized in the host cell‘s cytoplasm and, successively,
inserted into the endoplasmic reticulum (ER) and transported to the endoplasmic reticulum‐Golgi intermediate compartment (ERGIC). Furthermore, the nucleocapsid is shaped in the cytoplasm from the encapsidation of replicated genomes by N protein, which then merged within the ERGIC membrane, enabling the self‐ assembly and formation of new virions followed by its export from infected cells. Consequently, the new virions, surrounded by smooth‐walled vesicles, are transported to the cell membrane. Subsequently, via exocytosis, virions are exported out to infect new cells, and the infected host cell progresses towards cell death due to viral production stress in the endoplasmic reticulum.
3. Natural products and their derivatives with activity against SARS‐CoV‐2
3.1. Baicalein (1) and Baicalin (2)
Baicalein (1) (Figure 2) is a flavone bearing three hydroxy groups at C‐5, 6, and 7 positions and isolated from the roots of Scutellaria baicalensis Georgi (Huangqin) and Scutellaria lateriflora . Baicalein (1) is a traditional medicine with many pharmacological effects, including anti‐inflammation, anti‐cancer, antioxidant, antidepressant, etc.[ 62 , 63 , 64 , 65 ] It has exhibited antiviral activity against several viruses such as influenza virus A, Zika virus, dengue virus, etc.[ 66 , 67 , 68 ] Correspondingly, baicalin (2) (Figure 2), a flavone glucuronide extracted from Scutellaria baicalensis, exhibits several pharmacological activities, particularly anti‐inflammatory, antioxidant, antiapoptotic, and antiviral properties.[ 69 , 70 , 71 , 72 ]
Figure 2.

Chemical structure of Baicalein (1) and Baicalin (2) isolated from Scutellaria baicalensis roots.
3.1.1. Pharmacological relevance of baicalein (1) and baicalin (2) in SARS‐CoV‐2
In 2020, H. Su et al. identified baicalein (1) and baicalin (2) as non‐covalent, non‐peptidomimetic inhibitors of SARS‐CoV‐2 3CLpro.[ 73 , 74 ] In fluorescence resonance, energy transfer (FRET) protease assay, two fractions of S. baicalensis with major components as 1 (IC50=0.94 μM) and 2 (IC50=6.41 μM) exhibited promising SARS‐CoV‐2 3CLpro activity. Subsequently, the X‐ray protein crystallography technique was applied to discover the structural features of baicalein (1) related to binding to SARS‐CoV‐2 3CL pro [Figure 3 A–B]. [74] The crystal structure of SARS‐CoV‐2 3CLpro and 1 complex was examined (PDB 6 M2 N) to explain the potency of a small molecule baicalein (1) against the protease. Baicalein (1) was able to conceal effectively in the core of the substrate‐binding pocket. The key features for its interaction include two catalytic residues, the oxyanion loop (residues 138–145), Glu166, and the vital S1/S2 subsites. The phenolic hydroxyl groups of 1 interacted with the essential residues of S1 subsite, His 163, using water molecule. Baicalein (1) formed multiple hydrogen bonds with Leu141/Gly143/ Ser144 and secured the conformation of the flexible oxyanion loop that resulted in the stability of the tetrahedral transition state of the proteolytic reaction. The carbonyl group of 1 interacted with the main chain of Glu 166 via a hydrogen bond, and the phenyl ring formed hydrophobic interactions with multiple residues of S2 subsite.
Figure 3.

(A) SARS‐CoV‐2 3CL protease in complex with baicalein (1) (PDB 6 M2 N). (B) Interaction between Baicalein (1) and surrounding residues (SARS‐CoV‐2 3CL protease) (PDB 6 M2 N). [79]
In addition, the aromatic rings of 1 made S‐π and π‐π interaction with catalytic Cys145 and His41 respectively and NH2‐ π interactions with the side chain of Asn142. In contrast, the middle ring formed hydrophobic interactions with Met165. [74]
3.1.2. Synthesis of Baicalein (1)
The previous synthetic strategies to obtain Biacalein (1) from trimethoxy phenol (3), including modified conventional Baker‐Venkataraman and Wittig approach, turned out to be very low yielding.[ 75 , 76 ] In 2004, W‐H Huang reported an efficient method for the synthesis of baicalein (1) from trimethoxy phenol (3) via chalcone formation followed by demethylation (Scheme 1).[ 77 , 78 ] Two methods were applied for the synthesis of chalcone (4). The first one comprises a reaction of 3 with excess acetic acid and BF3‐Et2O followed by Claisen‐Schmidt condensation with benzaldehyde (equimolar) and KOH (catalyst) with an overall yield of 66 %. In the second approach, the trimetoxy phenol (3) was subjected to acylation with equimolar cinnamoyl chloride and BF3−Et2O, resulting in chalcone (4) with 90 % yield. Next, intramolecular oxidative cyclization of chalcone (4) with Iodine/dimethyl sulfoxide afforded molecule 5 in 87 % yield. Finally, demethylation of molecule 5 with a solution of 47 % HBr/AcOH (1: 2) under reflux conditions for 18 hours provided baicalein (1) in an 89 % yield. In 2010, D. Z. Chan et al. similarly reported the synthesis of baicalein from trimethoxy phenol (3) through chalcone (4) cyclization. However, with different ways, demethylation was performed, and the overall yield of 1 was 59 %. [79]
Scheme 1.

Synthesis of Baicalein (1) from trimethoxy phenol (3) via chalcone (4).
3.1.3. Synthesis of Baicalin (2)
An interesting molecule, baicalin (2), is a flavonoid 7‐O‐β‐D‐glucuronic glycoside, and its synthesis is associated with specific challenges, including the formation of phenolic glucuronic linkages in higher yield. Therefore, limited synthesis methodologies for baicalin (2) are available in the literature. In 1980, Mazey‐Vandor et al. reported the first total synthesis of baicalin (2) with the desired glycoside in a meager yield (44 %). [80] In 2015, Y‐F Li et al. developed an efficient multistep route for the synthesis of baicalin (2) from economical starting material in multistep with 27 % overall yield (Scheme 2). [81] For the synthesis of 2 at first, 5‐6‐diacetyl baicalein (8) was prepared from Baicalein (1) in three steps. The treatment of 1 with Ac2O and AcONa afforded fully acetylated Baicalein (6) (93 %) which then reacted with BnBr, KI, and K2CO3 for the selective substitution of reactive 7‐OAc with benzyl group and to give 7 in decent yield (91 %). Further, hydrogenolysis using Pd(OH)2/C under H2 atmosphere removed the benzyl group and provided 5‐6‐diacetyl baicalein (8) with a 90 % yield. Simultaneously, lactol 9 was prepared in three steps from D‐glucose using literature reported three conventional steps in 74 % yield. [82] Further, treatment of lactol (9) with CBr4 and PPh3 introduced the bromo group at anomeric carbon by substituting the hydroxyl group and afforded glycosyl bromide 10 in a 99 % yield. In the next step, the glycosidic linkage between donor 10 and acceptor 8 was obtained by following the classical Koenigs‐Knorr protocol, [83] with Ag2O as a promoter resulting in the synthesis of desired glycoside 11 in 92 % yield. Treatment of glycoside 11 with TBAF/HOAc deprotected TBDPS to afford compound 12 (86 %), which then subjected to Widlanski oxidation (TEMPO/BAIB), [84] to produce carboxylic acid 13 in 87 % yield. Finally, the global deprotection of all the acyl and benzyl groups was attained by reacting molecule 13 with mild base Mg(OMe)2 in methanol, resulting in the desired product baicalin (2) in 85 % yield..
Scheme 2.

Synthesis of Baicalin (2) from Baicalein (1).
3.2. Carolacton (14)
A natural product Carolacton (14) (Figure 4), is a macrolide keto‐carboxylic acid produced by the myxobacterium Sorangium cellulosum with a wide biological profile. The comprehensive evaluations have shown an association of 14 with regulatory systems, acid tolerance mechanism, biofilm formation, and folate dehydrogenase.[ 85 , 86 ] It acts as a promising inhibitor of biofilm formation in the human pathogen Streptococcus mutant, a growth inhibitor of S. pneumonia, and an inhibitor of efflux pump mutant Escherichia coli ΔtolC.[ 87 , 88 , 89 ]
Figure 4.

The chemical structure of Carolacton (14) produces by myxobacterium Sorangium cellulosum.
3.2.1. Pharmacological relevance of Carolacton (14) in SARS‐CoV‐2
In 2020, Anderson et al. revealed that methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) acts as a critical host factor for the viral RNA replication in both bat and human cells. [90] Carolacton (14) was identified as a promising MTHFD1 inhibitor that possibly worked because of its ability to bind with MTHFD1. Carolacton (14) inhibited the SARS‐CoV‐2 replication in Vero cells with IC50=0.14 μM and CC50=>0.80 μM. The in vitro results with 14 is encouraging, and therefore, carolacton (14) can be further evaluated for its effectiveness on SARS‐CoV‐2.
3.2.2. Synthesis of Carolacton (14)
To date, for the total synthesis of natural product carolacton (14), only three multistep synthesis methodologies exist in the literature. However, all the available procedure is in milligram scale and affording the carolacton with overall low yield. In 2012, Schmidt et al. reported the first total synthesis of 14 using 22 linear steps and an overall yield of 4.3 %. [91] Next, in 2014 M. S. Hallside et al. developed an efficient methodology for the synthesis of carolacton (14) in 14 linear steps and overall 7.9 % yield (Scheme 3). [92] The procedure synthesizes two major building blocks, including 19 and 26 individually, then coupling and completing the synthesis with multiple steps.
Scheme 3.

Synthesis of carolacton (14) using S. Hallside et al. methodology. [92]
Building block 19 synthesized from diol 15 in five steps. The diol 15 was subjected to oxidative cleavage using NaIO4 on silica to afford aldehyde 16, followed by Z‐selective Wittig olefination using Ph3PEtl.KHMDS in solvent THF provided 17 with an overall yield of 68 % for two steps. Further, compound 17 was subjected to the benzoate removal and Swern oxidation to afford the lactone 18 in 98 % yield. The treatment of lactone 18 with allyl magnesium bromide in the presence of Me2S⋅CuBr afforded the desired acid 19 with 60 % yield through SN2 opening of the lactone.
The synthesis of carolacton building block 26 begins by reacting Evans β‐ketoimide 20 and aldehyde 21 in the presence of (c‐Hex)2 BCl EtNMe2, Et2O to produce 22 in 81 % yield with dr of 5 : 1. Without separating diastereoisomer‘s compound, 22 subjected to methylation, reduction, and oxidation‐Witting olefination using 23 to afford enolate 24 as single diastereomer in overall 48 % yield. Next, the secondary alcohol of enolate 24 was silylated to produce TBS ether 25. The ester of compound 25 converted to an aldehyde and reacted with Leighton's (R, R)‐trans EZ‐CrotylMix to produce alcohol 26 in 61 % yield (dr 5 : 1).
The Steglich esterification reaction was applied to couple an acid 19 with an alcohol 26 to afford compound 27, then treated with 2nd generation Grubb's catalyst for ring‐closing metathesis reaction with terminal alkenes and afford lactone 28 (76 %).
Subsequently, selective hydrogenation of the olefin at Δ11,12 and PMB removal was accomplished using 5 % Pd/C in ethanol, followed by removing TBS ether with TBAF to produce diol 29 (96 % yield). The primary and secondary alcohol of 29 was oxidized using (COCl)2, DMSO, Et3N, CH2Cl2, and then NaOCl, NaH2PO4, t‐BuOH/H2O to produce compound 30 and subsequently the acetonide group removal using HF⋅pyr.H2O/MeCN provided the desired product carolacton (14).
In 2017, T. P. Kuilya et al. reported a practical synthesis of carolacton (14) on a 7.1‐mg scale and in 13 linear steps (Scheme 4). [93] The synthesis was progressed by reaction of alcohol 31 with propionic anhydride/Et3N to produce the corresponding ester that was subjected to Ireland‐Claisen rearrangement using LiHMDS/TBSCl in THF/DMPU (4 : 1) to make 32 as major isomer in 73 % yield (dr>4.7 : 1). The compound 32 was then subjected to three consecutive steps, including reaction with NH(Me)‐OMe/Et3N/DMAP for Weinreb amide formation, the addition of EtMgBr, and hydrogenation of double bond to produce alcohol 33 (67 %). Alcohol 33 was then converted to compound 34 (73 %) by oxidizing the alcohol using DMP to aldehyde, followed by Wittig olefination with PPh3CH3Br/n‐BuLi.
Scheme 4.

Synthesis of carolacton (14) using T. P. Kuilya et al. methodology. [93]
Correspondingly, compound 35 reacted with in situ generated titanium enolate from (S)‐phenylalanine‐derived N‐propionyl thiazolidinethione 36 in the presence of TiCl4 and DIPEA via Urpi diastereoselective anti acetal aldol reaction to afford compound 37 in 87 % yield (dr>15.1 : 1.0). Next, the thaizolidinethione moiety of compound 37 was reduced with DIBAL‐H to produce the corresponding aldehyde, which was reacted with chiral auxiliary 38 in the presence of TiCl4/DIPEA via Crimmins aldol reaction provided compound 39 in a decent yield (76 %) and diastereoselectivity (dr>12.5 : 1.0).The secondary alcohol of compound 39 was silylated using TBSOTf/2,6‐lutidine and then treated with DIBAL‐H to produce aldehyde 40 in overall good yield (88 %).
The compound 34 and 40 were coupled via TiCl4‐assisted syn‐aldol reaction to afford corresponding aldol adducts with 79 % yield and diastereoselectivity (dr>6 : 1). The diastereomeric mixture was then treated with MsCl/pyridine followed by DBU to produce α,β‐unsaturated ketone 41. DIBAL‐H reduced the ketone group of 41 to afford the alcohols 42 and 43 in excellent yield (93 %) and moderate diastereoselectivity (dr>2.5 : 1). Both isomers 43 and 44 reacted with acid 44 to produce ester 45 via Yamaguchi intermolecular esterification protocol and Mitsunobu esterification protocol. Next, ring‐closing metathesis reaction using Hoveyda‐Grubbs (HG‐II) catalysts converted to compound 45 into macrocyclic 46 in 74 % yield. Further, compound 46 was subjected to desilylation using TBAF, followed by oxidation of the alcohols to produce 47. Finally, the acetonide protecting group was removed using HF‐Py to provide the desired product carolacton 14 with an overall 18.8 % yield.
3.3. Chloroquine (48) and Hydroxychloroquine (49)
A molecule Chloroquine (48) (Figure 5) is an analog of the quinoline antimalarial drug quinine (50) (Figure 5), which could be extracted from the bark of the Cinchona tree ( Cinchona officinalis ). It is being used as a traditional drug to treat malaria and amoebiasis for a long time. [94] Hydroxychloroquine (49) (Figure 5) is a derivative of chloroquine (48) with an enhanced clinical safety profile during high daily dose and long‐term use. [95] Both 48 and 49 are in use to treat malaria, human immunodeficiency virus (HIV), and immune‐mediated diseases, etc.
Figure 5.

Chemical structure of chloroquine (48) and hydroxychloroquine (49), analogs of antimalarial drug quinine (50) extracted from the bark of Cinchona tree.
3.3.1. Pharmacological relevance of Chloroquine (48) and Hydroxychloroquine (49) in SARS‐CoV‐2
Chloroquine (48) exhibited the efficiency to block the interaction of RBD of SARS‐CoV to ACE‐2 with an ED50 of 4.4 μM under cell culture conditions.[ 96 , 97 ] Recently, Wang et al. reported that 48 inhibits the SARS‐CoV‐2 infection with an EC50 of 1.13 μM, CC50 > 100 μM, and SI > 88.50 in Vero E6 cells. Moreover, in in vivo studies, it showed immune‐modulating activity and increased antiviral effect. [98] Furthermore, hydroxychloroquine (49) is discovered as an effective molecule for SARS‐CoV‐2 treatment with good in vitro anti‐SARS‐CoV‐2 activities (EC50=0.72 μM) better than 48 (EC50=5.47 μM) and with the capacity to effectively reduce the viral copy number of SARS‐CoV‐2.[ 99 , 100 ] These two‐reported molecules 48 and 49 inhibit the SARS‐CoV/SARS‐CoV‐2 possibly because of their ability to block virus infection by increasing the endosomal pH essential for membrane fusion between virus and host cell and interferes with the glycosylation of cellular receptor ACE‐2. The reported studies with chloroquine phosphate displayed its potency to promote a negative virus conversion, inhibit the exacerbation of pneumonia, and shorten the disease course. [101] Therefore, it was expected that 48 might improve the treatment success rate, shorten hospital stay time, and improve patient outcomes. [102] The initial clinical trial studies, including 49 and azithromycin in combination (EUCTR2020‐000890‐25) and with only 49, exhibited promising results with a significant reduction in viral load, however; both the trials expressed the requirement for larger sample size for further validating the results.[ 103 , 104 ] Hydroxychloroquine (49) treatment on 62 patients in China displayed promising results and significantly reduced the patients′ clinical recovery time, and encouraged the resolution of pneumonia (ChiCTR2000029559). [105] The clinical trials conducted with large‐scale COVID‐19 patients (368 and 1376) in the USA were unfavorable in terms of 49 efficacy and safety. In 368 COVID‐19 patients, 49 by itself or in combination with azithromycin was ineffective in decreasing the risk of death or mechanical ventilation over supportive care. Moreover, treatment with only 49 increases the mortality rate. [106] Similarly, 1376 patient‘s clinical studies, including moderate to severe COVID‐19 infection, showed 49 ineffectiveness in COVID‐19 treatment. Patients receiving 49 (811) and patients with no given drug (placebo group) showed equivalent risk from intubation or death. [107] Indeed, the low cost of 48 and 49 make it an approachable drug for COVID‐19 treatment in middle and low‐income countries; however, the associated side effects, including ventricular arrhythmias, QT prolongation, retinopathy, and other cardiac‐related toxicity, make it life‐threatening, especially for critically ill patients and with cardiovascular diseases.[ 108 , 109 , 110 ] In June 2020, the FDA revoked the emergency authorization for hydroxychloroquine (49) and chloroquine (48) as a treatment for COVID‐19, and the WHO decided to suspend the SOLIDARITY trial of hydroxychloroquine (49).[ 111 , 112 ]
3.3.2. Synthesis of Chloroquine (48)
For the synthesis of chloroquine (48) [7‐chloro‐4‐(4‐diethylamino‐1‐methylbutylamino)‐quinoline], several procedures are available, and the majority of them include coupling between 4,7‐dichloroquinoline (55) with 4‐diethylamino‐1‐methylbutylamine (59) as a final step (Scheme 5).[ 113 , 114 ]
Scheme 5.

Synthesis of chloroquine (48) from 4,7‐dichloroquinoline (55) and 4‐diethylamino‐1‐methylbutylamine (59).
Component 4, 7‐dichloroquinoline (55) can be efficiently prepared in various ways starting from 3‐chloroaniline (51).[ 115 , 116 , 117 , 118 ] In 2007, Margolis et al. reported an efficient procedure for synthesizing 4,7‐ dichloroaniline, beginning with the reaction of 51 with 52 in the presence of triethylorthoformate under refluxing conditions to afford 53 in 82 % yield. [119] Next, compound 53 was subjected to ring closure by refluxing in Ph2O to afford 7‐chloro‐4‐hydroxyquinoline (54), which was subsequently treated with POCl3 to produce 4,7‐ dichloroaniline (55). Further, the reaction of 55 with 4‐diethylamino‐1‐methylbutylamine (59) under Suzuki coupling condition afforded the desired final product quinolone in 74 % yield. In addition, a considerable number of methods are reported for the synthesis of 59 as well.[ 120 , 121 ] The treatment of 3‐acetylbutyrolactone (56) prepared from acetoacetic acid ester and ethylene oxide) with acid, HBr leads to ester hydrolysis followed by decarboxylation to produce 1‐bromo‐4‐pentanone (57). The reaction of 57 with diethylamine resulted in a substitution reaction to afford 1‐diethylamino‐4‐pentanone (58), which was then subjected to reductive amination using H2, NH3, and Raney nickel as a catalyst to produce the desired compound 4‐diethyl‐1‐methylbutylamine (59) in decent yield.
3.3.3. Synthesis of Hydroxychloroquine (49)
Correspondingly, several effective synthetic routes are reported to prepare hydroxychloroquine (49).[ 122 , 123 , 124 , 125 , 126 , 127 ] One of the procedures reported by Ashok et al. in 2005 started with the carbonyl group protection of 5‐chloro‐2‐pentanone (60) using ethylene glycol in the presence of PTSA to produce compound 61 (Scheme 6). [128] Next, reaction of 61 with 2‐(ethylamino)ethanol (62) in toluene leads to nucleophilic substitution reaction to afford 63, which on treatment with conc. HCl provided the deprotected compound 64. Further, compound 64 was subjected to ammoniation and hydrogenation using Raney Nickel in high pressure to afford 2‐((4‐aminopentyl)(ethyl)amino)ethanol (65), which subsequently reacted with 4,7‐dichloro quinoline (55) in the presence of NaOH and KI to produce the hydroxychloroquine (49) (CLQ‐OH) in 76 % yield. In the end, the hydroxychloroquine sulfate (CLQ‐OH sulfate) was prepared from CLQ‐OH (49) by treating it with H2SO4/MeOH in good yield (77 %).
Scheme 6.

Synthesis of hydroxychloroquine (49) from 4,7‐dichloroquinoline (55) with 2‐((4‐aminopentyl)(ethyl)amino)ethanol (65).
3.4. Emetine (66)
Emetine (66) (Figure 6) is a tetrahydroisoquinoline alkaloid isolated from the root of Carapichea ipecacuanha, the common name ipecacuanha. Emetine (66) is a protein synthesis inhibitor with a wide range of pharmacological relevance, including its application in the treatment of intestinal and extra‐intestinal amoebiasis, inducing vomiting, inhibiting malaria, and reducing inflammation and cough.[ 129 , 130 , 131 , 132 ] In addition, emetine (66) is a safe and effective antiviral agent at a low dose. It possesses antiviral activity against many DNA and RNA viruses comprising Ebolavirus, Zika virus, rabies virus, HIV‐1 influenza, and coronavirus.[ 133 , 134 ] Particularly in coronavirus, emetine (66) displayed effectiveness in micromolar range against Human Coronavirus OC43 (HCoV‐OC43), Human Coronavirus NL63 (HCoV‐NL43), Coronavirus mouse hepatitis virus A59 (MHV‐A59), SARS‐CoV and MERS‐CoV.[ 135 , 136 ]
Figure 6.

Chemical structure of emetine (66) isolated from the root of Carapichea ipecacuanha.
3.4.1. Pharmacological relevance of emetine (66) in SARS‐CoV‐2
In 2020, Choy et al. evaluated the antiviral effect of 66 against SARS‐CoV‐2 virus in Vero E6 cells with observed EC50=0.46 μM. Emetine (66) (0.195 μM) in combination with remdesivir (6.25 μM) exhibited a synergistic inhibitory effect (64.9 %) against SARS‐CoV‐2 replication. [137] In the same year, N. Kumar and coworkers evaluated the in vitro antiviral efficacy of 66 against SARS‐CoV‐2 with cytotoxicity concentration 50 (CC50)=1603.8 nM, effective concentration 50 (EC50)=0.147 nM and selective index (CC50/EC50)=10910.4. [138] Furthermore, the 66 demonstrated the ability to protect the chicken embryo against lethal challenge with infectious bronchitis virus (IBV), reduce SARS‐CoV‐2 RNA and protein synthesis, inhibit the interaction of SARS‐CoV‐2 RNA and eIF4E (eukaryotic translation initiator factor 4E) and reduce the SARS‐CoV‐2 replication in the target cell via RK/MNK1/eIF4E signaling pathway. Further, Wang et al. in 2020 performed several in vitro and in vivo studies with 66 to explore its relevance in SARS‐CoV‐2 treatment. [139] Emetine (66) exhibited promising results in the in vitro studies using Vero cells, including antiviral replication effect with EC50 = 0.007 μM, selectivity index (SI) = 280, and CC50 = 1.96 μM in the cytotoxicity assay and dose‐dependent viral entry blocking effect with an EC50 = 0.019 μM. Next, in vitro anti‐inflammatory effects of 66 in the M1‐polarized THP‐1 macrophages were examined. Results revealed the effectiveness of emetine in reducing the lipopolysaccharide (LPS) induced interleukin‐6 (IL‐6) protein level and cytokine TNF‐α expression at 0.01 and 0.1 μM, respectively. In vivo pharmacokinetic study suggested that 66 was enriched in the SARS‐CoV‐2 targeted tissues, including lung, kidney, and liver. Mainly, in the lungs, it can reach quickly and retain its high concentration over 12 hours. At the single oral dose of 1 mg/kg, the effective concentration of emetine in the lung at 12 h was up to 1.8 μM in mice and 1.6 μM in rats (200 fold higher than the EC50 of the drug). Initial studies suggest the effectiveness of 66 in viral infection treatment; however, further in vivo studies with 66 are required to prove its significance in SARS‐CoV‐2 treatment.
3.4.2. Synthesis of Emetine (66)
Several efficient synthetic methodologies are available in the literature to synthesize emetine (66).[ 140 , 141 , 142 , 143 , 144 ] Most of the synthetic procedures for the synthesis of 66 utilize homoveratrylamine [2‐(3,4‐dimethoxyphenyl)ethylamine] (67) as a starting material (Scheme 7). [145] The reaction of homoveratrylamine (67) with β‐(α ′‐cyano)propylglutaric acid (68) under catalytic hydrogenation conditions in the presence of H2/PtO leads to the formation of an intermediate amine (69) through reductive amination and release of ammonia. Under the same reaction condition intramolecular cyclization of amine 69 afforded 1‐[2‐(3,4‐dimethoxyphenyl)‐ethyl]‐4‐carbethoxymethyl‐6‐ethylpiperidone‐2 (70). Next, the reaction of lactam 70 with POCl3 afforded 1‐[2‐(3,4‐dimethoxyphenyl)‐ethyl]‐4‐carbethoxymethyl‐6‐ethylpiperidone‐2 (71) via heterocyclization. Further treatment of 71 with homoveratrylamine (67) afforded amide 72, which on reaction with POCl3 undergoes cyclization to afford isoquinoline derivative 73. Finally, the pyridine ring of 73 was hydrogenated using H2/PtO to provide a racemic mixture of products (74), and from this racemic mixture, the desired emetine (66) can be isolated.
Scheme 7.

Synthesis of emetine (66) from homoveratrylamine (67).
3.5. Glycyrrhizin (GLR) (75)
Glycyrrhizin (75) (Figure 7) is also known as glycyrrhizic acid (GLR), a triterpenoid sapononin, sweet and isolated from the roots of the plant‘s Glycyrrhiza glabra (licorice), G. uralensis Fisch and G. inflata Bat and G. triphylla, etc. [146] Glycyrrhiza glabra (licorice) root is used as food and as a medicinal plant. GLR mainly exist as two epimers that is 18α‐glycyrrhizic acid (18α‐GLR) and 18β‐glycyrrhizic acid (18β‐GLR), and its structure includes a glycosidic linkage between glycyrrhetinic acid (76) (Figure 7) (GA, the sapogenin moiety) with two residues of glucuronic acid. It is used in foodstuffs, beverages, and cosmetics as a natural emulsifier and gel‐forming agent.
Figure 7.

Chemical structure of glycyrrhizin (75) isolated from the roots of the plants′ Glycyrrhiza glabra and glycyrrhetinic acid (76).
Since 1985, it is approved in the USA as an additive in foods and has received Generally Recognized As Safe (GRAS) status. Additionally, glycyrrhizin (75) has a comprehensive pharmacological profile, including antioxidant, anti‐inflammatory, anti‐allergic, anti‐microbial, antiviral, anti‐parasite, and anticancer.[ 147 , 148 , 149 , 150 ] Glycyrrhizin (75) also has broad‐spectrum antiviral activity against many RNA and DNA viruses such as SARS coronavirus, herpes virus, HIV, hepatitis virus, influenza viruses, cytomegaloviruses, and respiratory syncytial virus..[ 151 , 152 , 153 , 154 , 155 , 156 ]
3.5.1. Pharmacological relevance of glycyrrhizin (75) in SARS‐CoV‐2
Recent reports by Luo et al. revealed that glycyrrhizin (75) binds to ACE‐2 and inhibits SARS‐CoV‐2 binding to ACE‐2 and virus adsorption into the cell. [157] Yu et al. reported that 75 disrupts the interaction between receptor‐binding domain (RBD) of SARS‐CoV‐2 and ACE‐2 and therefore exhibits anti‐coronavirus activity. [158] In 2020, A Krawczyk and coworkers evaluated the aqueous licorice root extract for in vitro neutralization activity against SARS‐CoV‐2, identified 75 as an active compound, and discovered respective viral neutralization mechanisms. [159] The results revealed that 75, a primary active ingredient of the licorice root, effectively neutralizes SARS‐CoV‐2 by inhibiting 3CL protease (Mpro) in vitro. Glycyrrhizin (75) at a concentration of 2000 μM (1.6 mg/ml) inhibited Mpro activity and at a concentration of 30 μM (0.024 mg/ml) reduced Mpro activity by 70.3 %. A greener approach is followed for the bioproduction of GLR (75) from engineered Saccharomyces cerevisiae obtained from yeast cell factories. [160] Additionally, GLR (75) can be bio‐transformed into GA (76) by using the enzyme β‐glucuronidase. [161]
3.6. Homoharringtonine (77)
Homoharringtonine (77) (Figure 8) is an alkaloid isolated from the plant Cephalotoxus fortune [162] In 2012, the US Food and Drug Administration (FDA) approved Omacetaxine, a semi‐synthetic form of 77, as an anticancer agent for treating chronic myeloid leukemia. [163] Homoharringtonine (77) displayed antiviral activity against a wide range of viruses, including varicella‐zoster virus, herpes simplex virus‐1, pseudorabies virus, porcine epidemic diarrhea virus, murine hepatitis virus, rabies virus, hepatitis B virus, Newcastle disease virus, and echovirus 1.[ 164 , 165 , 166 ] Additionally, 77 displayed binding the 80S ribosome and inhibiting viral protein synthesis by interfering with the chain elongation. [167]
Figure 8.
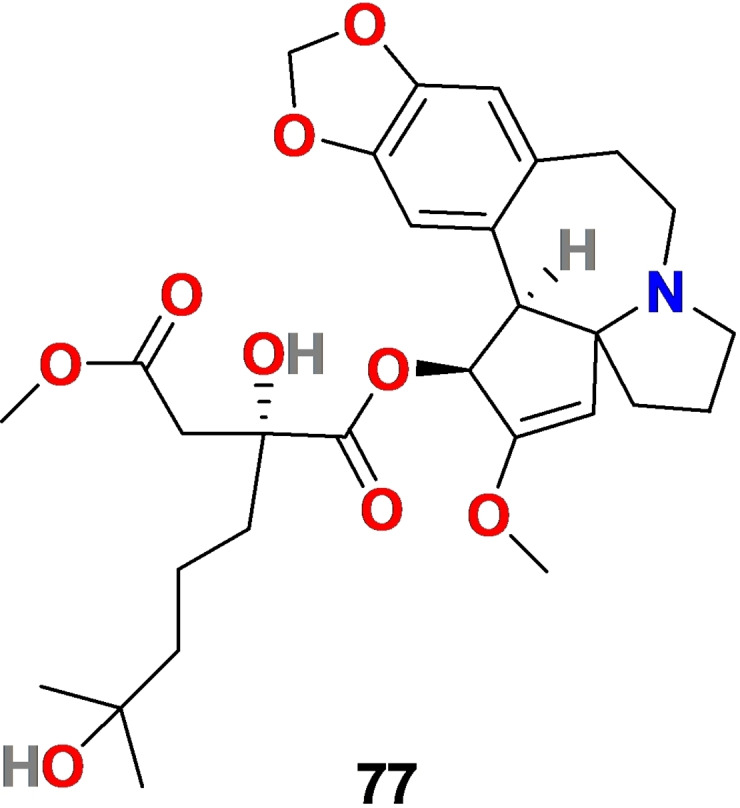
Chemical structure of Homoharringtonine (77) isolated from the plant Cephalotoxus fortun
3.6.1. Pharmacological relevance of Homoharringtonine (77) in SARS‐CoV‐2
In 2020, Choy et al. examined the in vitro (Vero E6 cells) inhibition of SARS‐CoV‐2 with Homoharringtonine (77) and results exhibited encouraging results with EC50 = 2.14 μM (reduction in viral RNA copy), EC50 = 2.55 μM (reduction in infectious virus) and CC50 = 59.75 μM. [168]
3.6.2. Synthesis of Homoharringtonin (77)
Homoharringtonin (77) is a Cephalotaxus alkaloid and exclusively two synthetic strategies reported for its synthesis in the literature. In 2008, Eckelbarger et al. reported the total synthesis of 77 in milligram scale (8.5 mg) by using 1,3‐dipolar cycloaddition, aziridine rearrangement, reductive; Neber rearrangements, and strain‐accelerated acylation reactions.. [169] In 2019, X. Ju et al. reported the 12 linear step synthesis of 77 on a 2.4 mg scale and with good enantioselectivity (Scheme 8). [170]
Scheme 8.

Synthesis of Homoharringtonin (77) using X Ju et al. methodology. [170]
The TBS‐protected hydroxyl pentanone 78 was subjected to Claisen‐type condensation and cyclization to afford furanone 79. Methylation of 79 using NAH, HMPA, and Me2SO4 leads to the formation of methoxyfuran 80, which is then coupled to known benzyl chloride 81 in the presence of 2,6 lutidine CF3CH2OH to produce compound 82. Next, the deprotection of the alcohol group followed by its activation by converting to the corresponding mesylate provided 83. The compound 83 on treatment with LiOH causes removal of TFA group and cyclization to produce compound 84 in good yield (62–85 %). Subsequently, the treatment of compound 84 with DDQ in solvent trifluoroethanol provided (±) cephalotaxinone (85) in 60 % yield via transannular Mannich cyclization and single‐electron oxidation; transannular reaction. A resolution‐based strategy using Noyori's condition was applied for the conversion of 85 to (−)‐cephalotaxine (86) with very high enantioselectivity (97 % ee) at 50 % conversion (krel=278). Finally, the (−)‐Cephalotaxine (86) was converted to desired (−)‐homoharringtonine (HHT) (77) in excellent enantioselectivity (>99 %) following the three‐step procedure, including reaction with compound 87 in strain accelerated acylation.
3.7. Ivermectin (88)
Ivermectin 88 (Figure 9) is derivative of the natural product avermectin, a macrocyclic lactone isolated from the bacterium Streptomyces avermitilis. It is approved by the FDA as an antiparasitic drug and has a good safety profile.[ 171 , 172 ]
Figure 9.

Chemical structure of ivermectin (88), a derivative of natural product avermectin isolated from the bacterium Streptomyces avermitilis.
3.7.1. Pharmacological relevance of ivermectin (88) in SARS‐CoV‐2
Recently, ivermectin (88) exhibited activity against SARS‐CoV‐2 and viral RNA replication.[ 173 , 174 ] In in vitro studies, 88 at 5 μM causes approximately 5000 fold reduction in viral RNA at 48 h. The studies showed that ivermectin (88) inhibited SARS‐CoV‐2 replication with IC50 of 2.5 μM in Vero/hSLAM cells at an MOI of 0.1. [175] Additionally, it was observed by Schmith et al. that ivermectin (88) couldn′t reach up to IC50 (2.5 μM) in the lung at a single oral dose; therefore, combination therapy and inhaled treatment were suggested. [176] The safety profile of molecule ivermectin (88) in humans has been documented; [177] however, so far, there are no clinical reports related to SARS‐CoV‐2 available in the literature. Further, ivermectin (88) is registered for different clinical trials in the USA and UK to its effect on treating SARS‐CoV‐2. [178]
3.7.2. Synthesis of Ivermectin [B1a (91) & B1b (92)]
Selective, catalytic hydrogenation of the cis‐22,23‐ double bond of the avermectin [B1a (89) & B1b (90)], a microbial natural product isolated from Streptomyces avermitilis, by using Wilkinson's catalyst [chlorotris(triphenylphosphine)rhodium(I) {RhCl(PPh3)3] to afford Ivermectin [B1a (91) & B1b (92)] (Scheme 9). [179]
Scheme 9.

Synthesis of Ivermectin [B1a (91) & B1b (92)] by catalytic hydrogenation of the cis‐22,23‐ double bond of the avermectin [B1a (89) & B1b (90)].
3.8. Resveratrol (93)
Resveratrol (93) (Figure 10) is a natural polyphenol compound (3, 5, 4′‐trihydroxy‐trans‐stilbene) that belongs to the phytoalexin family of phytochemicals. It is found abundantly in many plants and fruits, including grapes, mulberry, blueberries, cranberries, peanuts, etc. [180] Resveratrol (93) exhibits anti‐inflammatory, anticoagulant, antithrombotic, and antiviral properties against a range of viruses, mainly MERS‐CoV, pseudorabies virus, Zika virus, influenza virus, etc.[ 181 , 182 , 183 , 184 , 185 , 186 , 187 ]
Figure 10.
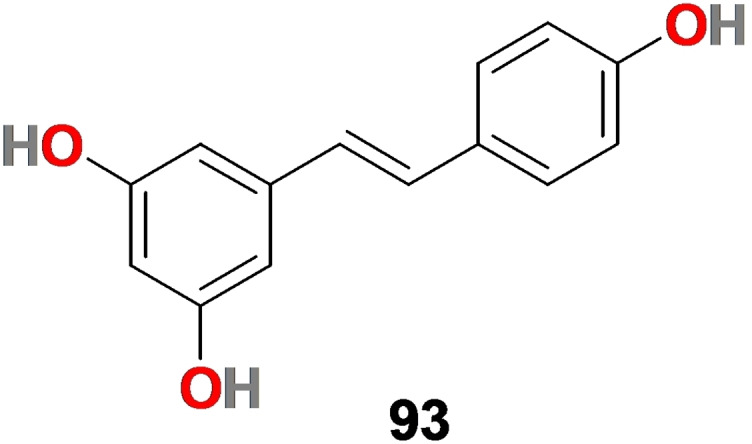
Chemical structure of resveratrol (93).
3.8.1. Pharmacological relevance of resveratrol (93) in SARS‐CoV‐2
The mortality associated with COVID‐19 infection is related to an upsurge in thrombosis and inflammation. Therefore resveratrol with antithrombotic and anti‐inflammatory properties would be a suitable candidate for treating SARS‐CoV‐2 disease.[ 188 , 189 ] Taking into consideration these facts, Y. Liu et al. studied the effect of resveratrol (93) in inhibiting the replication of SARS‐CoV‐2 using quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) and immunofluorescence assay. [190] The results disclosed that molecule 93 considerably inhibited the SARS‐CoV‐2 replication in the in vitro cell culture with an EC50 = 4.48 μM and could block viral entry into the cell. In addition, 93 endured a good safety tracking record and were used as a food supplement. Its large dose of up to 600 mg per day is used in chronic illness and immunomodulatory disorders with no profound adverse effect. [191]
3.8.2. Synthesis of Resveratrol (93)
Resveratrol (93) has wide‐ranging pharmacological significance. Therefore several synthetic methodologies are reported in the literature, mainly involving Horner Emmons, Pd catalyzed Heck or Suzuki, Perkin, Wittig type reactions and Ru catalyzed cross‐metathesis, etc.[ 192 , 193 , 194 , 195 , 196 , 197 , 198 ] In the year 2015, F. Lara‐Ochoa et al. reported an interesting four‐step synthesis methodology for resveratrol (93) from commercially available starting material using Sonogashira type reaction (Scheme 10). [199] The 3,5‐dimethoxy‐1‐ethynyl‐benzene (94) and 4‐iodoanisole (95) were subjected to Sonogashira coupling reaction using PPh3, Pd(OAc)2, and pyrrolidine to afford molecule 96 in 98 % yield. Following, the stereospecific reduction molecule 96 with LiAlH4 produced the E/Z mixture 97 in ratio 96 : 4. The crude E/Z mixture of molecule 96 was treated with a catalytic amount of diphenyl disulfide in THF to produce only trans isomer 98 through double bond isomerization. Consequently, the methyl group in molecule 98 was de‐protected using BBr3 and dichloromethane solvent to produce the desired product resveratrol 93 with an overall 63 % yield.
Scheme 10.

Synthesis of Resveratrol (93) from 3,5‐dimethoxy‐1‐ethynyl‐benzene (94) and 4‐iodoanisole (95).
3.9. Remdesivir (99)
Remdesivir (GS‐5734) (99) (Figure 11) is a C‐nucleoside analog, particularly an adenosine derivative that mimics naturally occurring nucleosides. Remdesivir (99) is a prodrug, and its synthesis is solely based on the structural modification of tubercidin (100) (Figure 11), an antibiotic and adenosine analog, isolated from Streptomyces tubercidicus. [200] The presence of cyano group (−CN) at C‐1 position was inspired by natural cyanide toyocamycin (101) (Figure 11), isolated from Streptomyces toyocmnsis. [201] Remdesivir (99) developed by Gilead Science with broad‐spectrum antiviral activity against wide range of RNA viruses including Ebola virus, SARS‐CoV (EC50 = 69 nM) and MERS‐CoV (EC50=20 nM).[ 202 , 203 , 204 ]
Figure 11.

Chemical structure of Remdesivir (99) synthesized by structural modification of tubercidin (100) isolated from Streptomyces tubercidicus. The CN group in 99 was inspired by natural cyanide toyocamycin (101) isolated from Streptomyces toyocmnsis.
3.9.1. Pharmacological relevance of Remdesivir (GS‐5734) (99) in SARS‐CoV‐2
Recently, Wang et al. revealed that Remdesivir (99) exhibited promising activity in reducing SARS‐CoV‐2 replication in Vero E6 cells with EC50 = 0.77 μM and selectivity index=129.87. [204] In another study, Choy et al. in 2020 reported anti‐SARS‐CoV‐2 activity of remdesivir (99) with EC50 = 23.15 μM when viral load fitted in logarithmic scale. [205]
Remdesivir (99) showed effectiveness in the early stage of virus entry and inhibited virus infection in human liver cell line Huh‐7 cells. The action mechanism of nucleoside prodrug 99 in CoVs is proceeded by inhibiting the RdRp and evading the action of the exoribonuclease (nsp14). [206] Further studies revealed that molecule 99 binds to the RNA‐binding channel of SARS‐CoV‐2 RNA‐dependent RNA polymerase and cyano group (−CN) substitution in molecule 99 plays a significant role in inhibiting the viral RNA replication SARS‐CoV‐2.[ 207 , 208 ] Yin et al. reported a cryo‐electron microscopy structure of the SARS‐CoV‐2 RdRp in complex with a 50‐base template‐primer RNA and remdesivir (99) at 2.5‐angstrom resolution (PDB 7BV2) (Figure 12A–B). [209] The complex structure indicated that the partial double‐stranded RNA template is placed into the central channel of the RdRp, where a molecule 99 is covalently assimilated into the primer strand at the first replicated base pair and terminates chain elongation. Later on, Bravo et al. reported 3.9‐Å‐resolution cryo‐EM reconstruction of a remdesivir‐stalled RNA‐dependent RNA polymerase complex (PDB 7 L1F) (Figure 12C). [210] The structure reveals the full incorporation of three copies of remdesivir monophosphate (RMP) and a partially incorporated fourth RMP in the active site suggesting the structural basis for remdesivir‐induced stalling SARS‐CoV‐2 RNA‐dependent RNA polymerase. Further, to examine the therapeutic potential of molecule 99 in patients, several clinical trials (NCT04323761 and NCT04292899) were conducted.[ 211 , 212 , 213 , 214 , 215 ] On May 1, 2020, the United States Food and Drug Administration (US FDA) issued emergency use authorization of remdesivir (99) for COVID‐19 treatment. [216] However, the results from remdesivir (99) clinical trials and usage were disseminated and broadly discussed. [217] In Nov 2020, the World Health Organization (WHO) commended against the use of remdesivir (99) in hospitalized patients, irrespective of disease severity, due to lack of evidence supporting its effectiveness in improving survival and other outcomes in COVID‐19 infected patients. [218]
Figure 12.

(A): The nsp12‐nsp7‐nsp8 complex bound to the template‐primer RNA and triphosphate form of Remdesivir (RTP) (PDB 7BV2). (B) Interaction between Remdesivir triphosphate and surrounding residues (RNA dependent RNA polymerase (PDB 7BV2)[209] (C): SARS‐CoV‐2 RdRp in complex with 4 Remdesivir monophosphates (PDB 7 L1F). [210]
3.9.2. Synthesis of Remdesivir (99)
Considering the pharmacological relevance of Remdesivir (99), researchers have developed an efficient methodology for its synthesis, including first‐generation, second‐generation, and gram‐scale synthesis of 99.[ 219 , 220 ] Recently, in the year 2019, Wang et al. developed an efficient method for the synthesis of 99 by reacting a molecule 103 and D‐ribose‐derived 5‐alcohol (109) in high purity and yield under asymmetric catalysis conditions and gram scale (Scheme 11). [221] For the synthesis of 103, the treatment of compound 102 with OP(OPh)Cl2, Et3N in dichloromethane solvent produces 103.[ 222 , 223 ]
Scheme 11.

Synthesis of Remdesivir (99) by coupling blocks 103 and 108 in gram scale using Wang et al. methodology. [221]
Conversely, D‐ribose‐derived 5‐alcohol (7) can be synthesized from reported methodology, including the coupling of 104 and 105 in the presence of TMSCl, PhMgCl, iPrMgCl‐LiCl in solvent THF to produce 106 in 40 % yield. Following, the treatment of 106 with TMSCN, TfOH, TMSOTf in dichloromethane solvent at very low temperature (−78 °C) afford cyano compound 107 in 85 % yield followed by deprotection of benzyl group using BCl3 in dichloromethane solvent produced 108 in 80 % yield. The vicinal diol of 108 was protected using 2,2‐dimethoxy propane, H2SO4, and acetone to produce compound 109 (90 %). [224] The reaction of compounds 103 and 109 in the presence of 2,6‐lutidine, and 4 Å MS catalyzed by chiral catalyst 110 at lower temperature gave 111 through Phosphoamidation in decent yield (89 %) and high chiral purity (99 %). Finally, compound 111 was treated with 37 % hydrochloric acid to produce the final desired product remdesivir (99) via deprotection in a 73 % yield. In 2021, the group reported a 10‐gram scale one‐pot synthesis of a molecule 99 in 70 % yield and 99.3/0.7 d.r. by combining the phosphoramidation using chiral catalyst 112 and protecting group removal steps followed by one‐step recrystallization. [225]
3.10. EIDD‐1931 (113) and EIDD‐2801 (115)
EIDD‐1931 (113) (Figure 13), also known as β‐D‐N4‐hydroxycytidine (NHC), is a ribonucleoside analog, especially a cytidine analog derived from the essential natural product uridine (114) (Figure 13) found in human plasma and displayed antiviral activity against a broad range of RNA viruses including Ebola and SARS‐CoV..[226–228]
Figure 13.

Chemical structure of EIDD‐1931 (113) and EIDD‐2801 (115) derived from natural product uridine (114).
3.10.1. Pharmacological relevance of EIDD‐1931 (113) and EIDD‐2801(115)
Recent studies revealed the efficiency of EIDD‐1931 (113) at controlling SARS‐CoV‐2 replication. It exhibited anti‐SARS‐CoV‐2 activity in Calu‐3‐cells with IC50=0.08 μM and in Vero cells with IC50=0.30 μM and with low cytotoxicity (CC50>10 μM). In addition, EIDD‐1931 (113), in a concentration‐dependent manner (0.01–0.10 μM), effectively inhibited the SARS‐CoV‐2 proliferation in primary human airway epithelial (HAE) cells. [229] The structural modification of the broad‐spectrum antiviral agent EIDD‐1931 (113) leads to the discovery of the N‐nucleoside analog EIDD‐2801 (115) (Figure 13). EIDD‐2801 (115) is an orally bioavailable antiviral drug discovered by Plemper's group at Emory University with the aptitude to effectively hydrolyze in vivo and with good selectivity (therapeutic window>1713).[ 230 , 231 , 232 ] The EIDD‐2801 (115) worked efficiently in mice infected with MERS‐CoV or SARS‐CoV by significantly improving pulmonary function.
The EIDD‐2801 (115) related human data are lacking; however, its potential effectiveness in vitro and in vivo makes it an interesting pharmacophore for further studies.
3.10.2. Synthesis of EIDD‐2801 (115)
Recently, Emory University researchers reported an efficient five‐step synthetic route using uridine as starting material for the synthesis of EIDD‐2801 (115) in gram scale (Scheme 12). [233] The synthesis started with one pot acetonide protection of 3,4‐diols using acetone, H2SO4, and then NEt3 afforded an intermediate 116, followed by selective esterification of 5‐hydroxy group of uridine afforded the compound 117 in 99 % yield. Next, hydroamination was performed by activating 117 using 1,2,4‐triazole moiety to produce 118, which was then treated with hydroxylamine, iPrOH to afford 119 by replacing the triazole group with hydroxylamine with an overall 17 % yield. Finally, the deprotection of the acetonide group using formic acid afforded 115 after crystallization; however, the yield of the final deprotection step is not disclosed in the patent.
Scheme 12.

Synthesis of EIDD‐2801 (115) in gram scale from uridine 114 using the patented methodology. [233]
In the year 2021, A Steiner et al. reported an improved methodology for the synthesis of EIDD‐2801 (115) starting from uridine (114) in better yield (Scheme 13). [234] The procedure was initiated by performing triazolation of 114 via one‐pot TMS protection, POCl3 promoted traizole coupling using Et3N, and deportation from MeOH/AcOH mixture to produce 120 in 88 % yield. Subsequently, 120 was subjected to one pot acetonide protection and esterification reaction to produce compound 122 via intermediate 121 in quantitative yield. Finally, the hydroxyamination (NH2OH x H2O/MeOH) and continuous flow acetonide deprotection (H2SO4/MeOH) lead to the formation of desired product, EIDD‐2801 (115) (69 %) via intermediate 119 with an overall 61 % yield.
Scheme 13.

Synthesis of EIDD‐2801 (115) from uridine 114 using A Steiner et al. methodology. [234]
4. Conclusion and perspective
The world is struggling to overcome the COVID‐19 pandemic. The war against COVID‐19 can prevail by developing new efficacious and innocuous therapeutic agents to prevent or treat this highly infectious virus. Natural products are a rich source of new therapeutics against various infectious diseases and cancer for a long time. With the emergence of COVID‐19, one approach followed in China and developing countries to tackle this contagious virus is by using herbal remedies for its treatment. The traditional herbal medicines are a rich reservoir of molecules with antiviral activity against a wide range of virus strains and work proficiently by interfering with the virus life cycle, including viral entry, replication, assembly, and release, and also the virus‐host‐specific interactions.
Several molecules from the natural source have been discovered in the last few months with promising SARS‐CoV‐2 inhibition activity at the molecular and cellular levels. This review offers an insightful view of recently discovered natural product and their synthetic analogs with favorable pharmacological profiles against the SARS‐CoV‐2 activity. In addition, the chemical synthesis methodologies of some of the natural products are discussed in this article. Even though several natural products as active compounds or extracts exhibited anti‐SARS‐CoV‐2 properties, in any case, advanced testing in animals, randomized placebo clinical trials, and human cytotoxicity and dosage evaluation is still required. As apparent from the chemical structures of the natural products included in this review, most of the natural products with anti‐SARS‐CoV‐2 activity bear high molecular weight with vast scaffold diversity and structural complexity. The natural products and synthetic analogs, including carolacton, emetine, glycyrrhizin, homoharringtonine, ivermectin, and remdesivir, are huge skeletons with a less aromatic ring system. The presence of many tetrahedral carbon atoms, chiral carbon, hydrogen bond donors and acceptors, and oxygen atom in proportion to nitrogen and halogen atoms are standard structural features of these natural products. In addition, most of them are sterically more complex structures with cyclization and greater molecular rigidity. The baicalein and resveratrol are small molecules with benzene rings and many alcohol (−OH) groups. In contrast, chloroquine, hydroxyl chloroquine, EIDD‐1931, and EIDD‐2801 are small structures with heteroaromatic rings and many oxygens (O) and nitrogen (N) atoms. For example, small natural products, baicalein, and resveratrol with no chiral carbon can be synthesized efficiently in minimal steps. In contrast, large natural products and analogs with multifunctional groups and stereocenter, including carolacton, homoharringtonine, remdesivir, etc., bearing stereocenters are synthetically challenging, mainly when synthetic methodologies are difficult to apply in academia and industry. At present, due to the lack of adequate and safe therapeutics for COVID‐19 treatment, it's necessary to identify promising hits and leads that can be developed into successful therapeutics for SARS‐CoV‐2 treatment. One approach to achieving this objective probably involves rigorous screening of the natural products for anti‐SARS‐CoV‐2 bioactivity. Natural products are exceptionally valued lead compounds; however, structural modifications are desired for clinical applications. Structural modification is a multidimensional process and vital to upsurge potency and selectivity, lessen side effects and structural complexity (reducing the number of stereo centers) and improve the lead compound‘s physicochemical, biochemical, and pharmacokinetic properties. The identification of the natural product as a hit compound followed by its modification for commercial use through drug research and development is a time‐consuming and challenging process; therefore, we believe that the need of the hour to tackle COVID‐19 is global research cooperation between academia and pharmaceutical companies particularly between organic and medicinal chemists. The collaboration will optimize natural product leads and enrich the anti‐SARS‐CoV‐2 molecule library with natural product analogs, natural product hybrids, and natural product‐inspired molecules.
Natural products are successful in drug discovery because of their vast complex structural diversity. Recent advancement techniques have facilitated the human understanding of natural product conformation and structural features essential for activity. The natural product metabolites′ biological and pharmacological activities are usually better than randomly synthesized compounds. The plants and herbs are natural laboratories continuously synthesizing simple to highly complex molecules, and indeed, it will continue in the future. Nature is a rich, reliable source of undiscovered natural products that can provide defense against various developing infections. In the past, natural products have been a precious source of pharmaceutical molecules, and correspondingly, in the future, possibly the cure for COVID‐19 will emerge from nature. We believe that the natural product;s pharmacological relevance and chemical synthesis route discussed in this review provides a solid basis for discovering and developing natural product‐based drugs for the SARS‐CoV‐2 treatment.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr. Shagufta is an Associate Professor of Chemistry at American University of Ras Al Khaimah, UAE. She attended the Department of Chemistry and Biochemistry, Stephenson Life Science Research Center, University of Oklahoma, USA and Leiden Academic Centre for Drug Research, Leiden University, Leiden, The Netherlands as a Postdoctoral research associate. She received her Ph.D. in Chemistry (2008) from Central Drug Research Institute/ Lucknow University, India. Her research focused on organic and medicinal chemistry. She is involved in the development of novel chemical entities as anticancer and anti SARS‐CoV‐2 agents. Her research area include medicinal chemistry, homogeneous and heterogeneous catalysis for organic transformations and nanotechnology.

Biographical Information
Dr. Irshad Ahmad is an Associate Professor of Chemistry at American University of Ras Al Khaimah, UAE. He received his PhD in Chemistry (2006) from Central Salt and Marine Chemicals Research Institute/ Bhavnagar University, India. He attended the Korea Research Institute of Chemical Technology, S. Korea as an invited scientist and subsequently, pursued research at the Vant Hoff Institute for Molecular Sciences, University of Amsterdam, The Netherlands; Leibniz Institute for Surface Modification, Leipzig, Germany; Department of Chemistry and Biochemistry, Stephenson Life Science Research Center, University of Oklahoma, USA. He was a Visiting Professor (2019) at University of Toronto, St. George Campus,Toronto, Canada. His research area is homogeneous and heterogeneous asymmetric catalysis for organic transformation, supramolecular chemistry, nanotechnology and pharmaceutical chemistry.

Acknowledgements
The authors, Dr. Shagufta and Dr. Irshad Ahmad are thankful to the School of Graduate Studies and Research, American University of Ras Al Khaimah for their support. We would like to give special acknowledgement to Ms. Sumayya Mohammad Adam Mohammad Rahma, Ms. Taif Ahmed Al Shabi and Ms. Bdour Haitham Alhourani, undergraduate students of Biotechnology department for their contribution to literature search.
Shagufta, I. Ahmad, ChemistrySelect 2021, 6, 11502.
References
- 1.“WHO Coronavirus (COVID-19) Dashboard” to be found under https://covid19.who.int/, 2021.
- 2. Haleem A., Javaid M., Vaishya R., Curr. Med. Res. Pract. 2020, 10, 78–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.“ Everyone Included: Social Impact of COVID-19′′ to be found under https://www.un.org/development/desa/dspd/everyone-included-covid-19.html, 2021.
- 4. Ibn-Mohammed T., Mustapha K. B., Godsell J., Adamu Z., Babatunde K. A., Akintade D. D., Acquaye A., Fuji H., Nadiaye M. M., Yamoah F. A., Koh S. C. L., Resour. Conserv. Recycl. 2021, 164, 105169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hu B., Guo H., Zhou P., Shi Z.-L., Nat. Rev. Microbiol. 2021, 19, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.“What You Need to Know about Variants” to be found under https://www.cdc.gov/coronavirus/2019-ncov/variants/variant.html, 2021.
- 7.“SARS-CoV-2 Variant Classifications and Definitions” to be found under https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html, 2021.
- 8.“ COVID-19: Clinical features” to be found under https://www.uptodate.com/contents/covid-19-clinical-features, 2021.
- 9. Wang C., Wang Z., Wang G., Lau J. Y.-N., Zhang K., Li W., C Sig. Transduct. Target Ther. 2021, 6, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peng Z., Lei B., Wei Y., Jian-Jiang W., Medicine 2020, 99, e21618.32872018 [Google Scholar]
- 11. Yong S. J., Infect. Dis. (Lond). 2021, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nalbandian A., Sehgal K., Gupta A., Madhavan M. V., McGroder C., Stevens J. S., Cook J. R., Nordvig A. S., Shalev D., Sehrawat T. S., Ahluwalia N., Bikdeli B., Dietz D., Der-Nigoghossian C., Liyanage-Don N., Rosner G. F., Bernstein E. J., Mohan S., Beckley A. A., Seres D. S., Choueiri T. K., Ureil N., Ausiello J. C., Accili D., Freedberg D. E., Baldwin M., Schwartz A., Brodie D., Garcia C. K., Alkind M. S. V., Connors J. M., Bilezikien J. P., Landry D. W., Yan E. Y., Nat. Med. 2021, 27, 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raveendran A. V., Jayadevan R., Sashidharan S., Diabetes Metab. Syndr. Obes. 2021, 15, 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krammer F., Nature 2020, 586, 516–527. [DOI] [PubMed] [Google Scholar]
- 15. Abdulla Z. A., Al-Bashir S. M., Al-Salih N. S., Aldamen A. A., Abdulazeez M. Z., Pathogens 2021, 10, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.“COVID-19 vaccine tracker” to be found under https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-vaccine-tracker, 2021.
- 17. Richman D. D., Glob. Health Med. 2021, 3, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Awadasseid A., Wu Y., Tanaka Y., Zhang W., Biomed. Pharmacother. 2021, 137, 111330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shyr Z. A., Gorshkov K., Chen C. Z., Zheng W., J. Pharmacol. Exp. Ther. 2020, 375, 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abd El-Aziz T. M., Stockand J. D., Infect. Genet. Evol. 2020, 83, 104327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poduri R., Joshi G., Jagadeesh G., Cell. Signalling 2020, 74, 109721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J., Lu H., Biosci. Trends. 2021, 15, 126–128. [DOI] [PubMed] [Google Scholar]
- 23.W. T. Harvey, A. M. Carabelli, B. Jackson, R. K. Gupta, E. C. Thomson, E. M. Harrison, C. Ludden, R. Reeve, A. Rambaut, COVID-19 Genomics UK (COG-UK) Consortium, S. J. Peacock, D. L. Robertson. Nat. Rev. Microbiol. 2021, 19, 409–424. [DOI] [PMC free article] [PubMed]
- 24. Kennedy D. A., Read A. F., PLoS Biol. 2020, 18, e3001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghanbari R, Teimoori A., Sadeghi A., Mohamadkhani A., Rezasoltani S., Asadi E., Jouyban A., Sumner S. C. J., Future Microbiol. 2020, 15, 1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng L., Shen L., Xu J., Tian X., Liu F., Wang J., Tian G., Yang J., Zhou L., Sci. Rep. 2021, 11, 6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahamad S., Branch S., Harrelson S., Hussain M. K., Saquib M., Khan S., Eur. J. Med. Chem. 2021, 209, 112862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Riva L., Yuan S., Yin X., Martin-Sancho L., Matsunaga N., Pache L., Brugstaller-Muehlbacher S., De Jesus P. D., Teriete P., Hull M. V., Chang M. W., Chan J. F.-W., Cao J., Poon V. K.-M., Herbert K. M., Cheng K., Nguyen T.-T. H., Rubanov A., Pu Y., Nguyen C., Choi A., Rathnasinghe R., Schotsaert M., Miorin L., Dejosez M., Zwaka T. P., Sit K.-Y., Martinez-Sobrido L., Liu W. C., White K. M., Chapman M. E., Lendy E. K., Glynne R. J., Albrecht R., Ruppin E., Mesecar A. D., Johnson J. R., Benner C., Sun R., Schultz P. G., Su A. I., Garcia-Sastre A., Chatterjee A. K., Yuen K.-Y., Chanda S. K., Nature 2020, 586, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shagufta, Ahmad I., Eur. J. Med. Chem. 2021, 213, 113157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gil C., Ginex T., Maestro I., Nozal V., Gil L. B., Geijo M. A. C., Urquiza J., Ramírez D., Alonso C., Campillo N. E., Martinez A., J. Med. Chem. 2020, 63, 12359–12386. [DOI] [PubMed] [Google Scholar]
- 31. Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X., J. Med. Chem. 2020, 63, 12256–12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frecer V., Miertus S., RSC Adv. 2020, 10, 40244–40263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghosh A. K., Brindisi M., Shahabi D., Chapman M. E., Mesecar A. D., ChemMedChem 2020, 15, 907–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, International Natural Product Sciences Taskforce, C. T. Supuran, Nat. Rev. Drug Discov. 2021, 20, 200–216. [DOI] [PMC free article] [PubMed]
- 35. Harvey A. L., Drug Discovery Today 2008, 13, 894–901. [DOI] [PubMed] [Google Scholar]
- 36. Dias D. A., Urban S., Roessner U., Metabolites 2012, 2, 303–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barnes E. C., Kumar R., Davis R. A., Nat. Prod. Rep. 2016, 33, 372–381. [DOI] [PubMed] [Google Scholar]
- 38. Li J. W.-H., Vederas J. C., Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- 39. Lawson A. D. G., MacCoss M., Heer J. P., J. Med. Chem. 2018, 61, 4283–4289. [DOI] [PubMed] [Google Scholar]
- 40. Yuan H., Ma Q., Ye L., Piao G., Molecules 2016, 21, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wangkheirakpam S., Natural Products and Drug Discovery, An integrated Approach. Elsevier, Amsterdam, 2018, 29–56. [Google Scholar]
- 42. Corson T. W., Crews C. M., Cell 2007, 130, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harrison C., Nat. Biotechnol. 2014, 32, 403–404. [DOI] [PubMed] [Google Scholar]
- 44. Yang Y., Islam M. S., Wang J., Li Y., Chen X., Int. J. Biol. Sci. 2020, 16, 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fielding B. C., da Silva C., Bezerra Filho M., Ismail N. S. M., Sousa D. P., Molecules 2020, 25, 5496. [Google Scholar]
- 46. Chinsembu K. C., Rev. Bras. Farmacogn. 2020, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Z., Yang L., Front. Pharmacol. 2020, 11,1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li D., Hu J., Li D., Yang W., Yin S. F., Qiu R., Top. Curr. Chem. (Cham). 2021, 379, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Christy M. P., Uekusa Y., Gerwick L., Gerwick W. H., J. Nat. Prod. 2021, 84, 161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shagufta, Ahmad I., Mathew S., Rehman S., RSC Med. Chem. 2020, 11, 438–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shagufta, Ahmad I., Inorganica. Chimica. Acta. 2020, 506, 119521. [Google Scholar]
- 52. Shagufta, Ahmad I., Eur. J. Med. Chem. 2018, 143, 515–531. [DOI] [PubMed] [Google Scholar]
- 53. Shagufta, Ahmad I., MedChemComm 2017, 8, 871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shagufta, Ahmad I., Panda G., Eur. J. Med. Chem. 2017, 133, 139–151. [DOI] [PubMed] [Google Scholar]
- 55. Shagufta, Ahmad I., Eur. J. Med. Chem. 2016, 116, 267–280. [DOI] [PubMed] [Google Scholar]
- 56. Ahmad I., Shagufta, Eur. J. Med. Chem. 2015, 102, 375–386. [DOI] [PubMed] [Google Scholar]
- 57. Ahmad I., Shagufta, Int. J. Pharm. Pharm. Sci. 2015, 7, 19–27. [Google Scholar]
- 58. Boopathi S., Poma A. B., Kolandaivel P., J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar]
- 59. Huang Y., Yang C., Xu X. F., Xu W., Liu S. W., Acta. Pharmacologica Sinica. 2020, 41, 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Duan Y., Yao Y., Kumar S. A., Zhu H. L., Chang J., Drug Discovery Today 2020, 25, 1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zheng L., Zhang L., Huang J., Nandakumar K. S., Liu S., Cheng K., Eur. J. Med. Chem. 2020, 205, 112687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim Y. J., Kim H. J., Lee J. Y., Kim D. H., Kang M. S., Park W., Viruses 2018, 10, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gao Y., Snyder S. A., Smith J. N., Chen Y. C., Med. Chem. Res. 2016, 25, 1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shieh D. E., Liu L. T., Lin C. C., Anticancer Res. 2000, 20, 2861–2865. [PubMed] [Google Scholar]
- 65. Xiong Z., Jiang B., Wu P.-F., Tian J., Shi L.-L., Gu J., Hu Z.-L., Fu H., Wang F., Chen J.-G., Biol. Pharm. Bull. 2011, 34, 253–259. [DOI] [PubMed] [Google Scholar]
- 66. Ding Y., Dou J., Teng Z., Yu J., Wang T., Lu N., Wang H., Zhou C., Arch. Virol. 2014, 159, 3269–3278. [DOI] [PubMed] [Google Scholar]
- 67. Oo A., Teoh B. T., Sam S. S., Bakar S. A., Zandi K., Arch. Virol. 2019, 164 585–593. [DOI] [PubMed] [Google Scholar]
- 68. Zandi K., Teoh B.-T., Sam S.-S., Wong P.-F., Mustafa M. R., Abubakar S., BMC Complementary Altern. Med. 2012, 12, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lee W., Ku S.-K., Bae J.-S., Inflammation 2015, 38, 110–125. [DOI] [PubMed] [Google Scholar]
- 70. Peng-Fei L., Fu-Gen H., Bin-Bin D., Tian-Sheng D., Xiang-Lin H., Ming-Qin Z., J. Food Sci. Technol. 2013, 50, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Takahashi H., Chen M. C., Pham H., Matsuo Y., Ishiguro H., Reber H. A., Takeyama H., Hines O. J., Eibl G., Biochim. Biophys. Acta. 2011, 1813, 1465–1474. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moghaddam E., Teoh B. T., Sam S. S., Lani R., Hassandarvish P., Chik Z., Yueh A., Abubakar S., Zandi K., Sci. Rep. 2014, 4, 5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Su H., Yao S., Zhao W., Li M., Liu J., Shang W., Xie H., Ke C., Gao M., Yu K., Liu H., Shen J., Tang W., Zhang L., Zuo J., Jiang H., Bai F., Wu Y., Ye Y., Xu Y., bioRxiv 2020, doi: 10.1101/2020.04.13.038687. [DOI] [Google Scholar]
- 74. Su H., Yao S., Zhao W., Li M., Liu J., Shang W., Xie H., Ke C., Gao M., Yu K., Liu H., Shen J., Tang W., Zhang L., Zuo J., Jiang H., Bai F., Wu Y., Ye Y., Xu Y., Acta. Pharmacologica Sinica. 2020, 41, 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ares J. J., Outt P. E., Kakodkar S. V., Buss R. C., Geiger J. C., J. Org. Chem. 1993, 58, 7903–7905. [Google Scholar]
- 76. Hercouet A., LeCorre M., LeFloc'h Y., Synthesis 1982, 7, 597–598. [Google Scholar]
- 77. Huang W. H., Chien P. Y., Yang C. H., Lee A. R., Chem. Pharm. Bull. 2003, 51, 339–340. [DOI] [PubMed] [Google Scholar]
- 78.J. Shaw, A.-R. Lee, W.-H. Huang. US 2004/0242907 A1, 2004.
- 79. Chen D.-Z., Yang J., Yang B., Wu Y.-S., Wu T., J. Asian Nat. Prod. Res. 2010, 12, 124–128. [DOI] [PubMed] [Google Scholar]
- 80. Mazey-Vandor G., Farkas L., Kanzel I., Nogradi M., Chem. Ber. 1945, 1980, 113. [Google Scholar]
- 81. Li Y.-F., Yu B., Sun J.-S., Wang R.-X., Tetrahedron Lett. 2015, 56, 3816–3819. [Google Scholar]
- 82. Panchadhayee R., Misra A. K., J. Carbohydr. Chem. 2010, 29, 76–83. [Google Scholar]
- 83. Koenigs W., Knorr E., Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. [Google Scholar]
- 84. Epp J. B., Widlanski T. S., J. Org. Chem. 1999, 64, 293–295. [DOI] [PubMed] [Google Scholar]
- 85. Solinski A. E., Scharnow A. M., Fraboni A. J., Wuest W. M., ACS Infect. Dis. 2019, 5, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Fu C., Sikandar A., Donner J., Zaburannyi N., Herrmann J., Reck M., Wgner-Dobler I., Koehnke J., Muller R., Nat. Commun. 2017, 8, 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jansen R., Irschik H., Huch V., Schummer D., Steinmetz H., Bock M., Schmidt T., Kirschning A., Muller R., Eur. J. Org. Chem. 2010, 7, 1284–1289. [Google Scholar]
- 88. Donner J., Reck M., Bergmann S., Kirschning A., Muller R., Wagner-Dobler I., Sci. Rep. 2016, 6, 29677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kunze B., Reck M., Dötsch A., Lemme A., Schummer D., Irschik H., Steinmetz H., Wganer-Dobler I., BMC Microbiol. 2010, 10, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Anderson D. E., Cui J., Ye Q., Huang B. Y., Zu W. H., Gong J., et al, bioRxiv. 2020. doi: 10.1101/2020.03.29.014209. [Google Scholar]
- 91. Schmidt T., Kirschning A., Angew. Chem. Int. Ed. 2012, 51, 1063–1066; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1087–1091. [Google Scholar]
- 92. Hallside M. S., Brzozowski R. S., Wuest W. M., Phillips A. J., Org. Lett. 2014, 16, 1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kuilya T. K., Goswami R. K., Org. Lett. 2017, 19, 2366–2369. [DOI] [PubMed] [Google Scholar]
- 94. Plantone D., Koudriavtseva T., Clin. Drug Invest. 2018, 38, 653–671. [DOI] [PubMed] [Google Scholar]
- 95. Marmor M. F., Kellner U., Lai T. Y., Melles R. B., Mieler W. F., Ophthalmology 2016, 123, 1386–1394. [DOI] [PubMed] [Google Scholar]
- 96. Gao J., Tian Z., Yang X., BioSci. Trends 2020, 14, 72–73. [DOI] [PubMed] [Google Scholar]
- 97. Vincent M. J., Bergeron E., Benjannet S., Erickson B. R., Rollin P. E., Ksiazek T. G., Seidah N. G., Nichol S. T., Virol. J. 2005, 2, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell Res. 2020, 30, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C., Zhan S., Lu R., Li H., Tan W., Liu D., Clin. Infect. Dis. 2020, 71, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lan L., Xu D., Ye G., Xia C., Wang S., Li Y., Xu H., Jama 2020, 323, 1502–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gao J., Tian Z., Yang X., Biosci. Trends. 2020, 14, 72–73. [DOI] [PubMed] [Google Scholar]
- 102. Jie Z., He H., Xi H., Zhi Z., Zhonghua Jiehe He Huxixi Jibing Zazhi 2020, 43, 185–188. [DOI] [PubMed] [Google Scholar]
- 103. Gautret P., Lagier J.-C., Parola P., Hoang V. T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V. E., Dupont H. T., Honore S., Colson P., Chabri_ere E., La Scola B., Rolain J.-M., Brouqui P., Raoult D., Int. J. Antimicrob. Agents. 2020, 56, 105949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chen J., Liu D., Liu L., Liu P., Xu Q., Xia L., Ling Y., Huang D., Song S., Zhang D., Qian Z., Li T., Shen Y., Lu H., J. Zhejiang Univ. 2020, 49, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen Z., Hu J., Zhang Z., Jiang S., Han S., Yan D., Zhuang R., Hu B., Zhang Z., MedRxiv. 2020, 10.1101/2020.03.22.20040758. [DOI] [Google Scholar]
- 106. Magagnoli J., Narendran S., Pereira F., Cummings T. H., Hardin J. W., Sutton S. S., Ambati J., Med (N Y). 2020, 1, 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Geleris J., Sun Y., Platt J., Zucker J., Baldwin M., Hripcsak G., Labella A., Manson D. K., Kubin C., Barr R. G., Sobieszczyk M. E., Schluger N. W., N. Engl. J. Med. 2020, 382, 2411–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen C., Wang F., Lin C., Clin. Toxicol. 2006, 44, 173–175. [DOI] [PubMed] [Google Scholar]
- 109. Stas P., Faes D., Noyens P., Int. J. Cardiol. 2008, 127, e80–e82. [DOI] [PubMed] [Google Scholar]
- 110. Yaylali S. A., Sadigov F., Erbil H., Ekinci A., Akcakaya A. A., Int. Ophthalmol. 2013, 33, 627–634. [DOI] [PubMed] [Google Scholar]
- 111.′′Coronavirus (COVID-19) Update: FDA Revokes Emergency Use Authorization for Chloroquine and Hydroxychloroquine” to be found under https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and, 2020.
- 112.′′Q&A : Hydroxychloroquine and COVID-19” to be found under https://www.who.int/news-room/q-a-detail/q-a-hydroxychloroquine-and-covid-19, 2020.
- 113. Vardanyan R. S., Hruby V. J., Chapter 37 - Drugs for Treating Protozoan Infections, Synthesis of Essential Drugs, Elsevier, Amsterdam, 2006, 559–582. [Google Scholar]
- 114. Li D., Hu J., Li D., Yang W., Yin S. F., Qiu R., Top. Curr. Chem. (Cham.). 2021, 379, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Price Ch. C., Roberts R. M., J. Am. Chem. Soc. 1946, 68, 1204–1208. [DOI] [PubMed] [Google Scholar]
- 116.Ch. F. Gerschickter, L. M. Rice, U. S. Pat. 2.947.664, 1960.
- 117. Surrey A. R., Hammer H. F., J. Am. Chem. Soc. 1946, 68, 113–116. [DOI] [PubMed] [Google Scholar]
- 118.E. H. Northey, P. F. Dreisbach, U. S. Pat. 2.478.125, 1949.
- 119. Margolis B. J., Long K. A., Laird D. L., Ruble J. C., Pulley S. R., J. Org. Chem. 2007, 72, 2232–2235. [DOI] [PubMed] [Google Scholar]
- 120.W. Schuelemann, F. Schoenhoefer, A. Wingler. Ger. Pat. 486.079, 1924.
- 121. Elderfield R. C., Gensler W. J., Brody F., Head J. D., Dickerman S. C., Wiederhold L., Cramer Ch. B., Hageman H. A., Kreyse F. J., Griffing J. M., Kupchan S. M., Newman B., Maynard J. T., J. Am. Chem. Soc. 1946, 68, 1579–1584.20994986 [Google Scholar]
- 122.A. R. Surrey. U. S. Pat. 2.546.658, 1951.
- 123. Surrey A. R., Hammer H. F., J. Am. Chem. Soc. 1950, 72, 1814–1815. [Google Scholar]
- 124.R. Alexander. US2546658, 1951.
- 125.Y. S. Min, H. S. Cho, K. W. Mo. WO201002715, 2010.
- 126. Yu E., Mangunuru H. P. R., Telang N. S., Kong C. J., Verghese J., Gilliland S. E., Ahmad S., Dominey R. N., Gupton B. F., Beilstein J. Org. Chem. 2018, 14, 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.G. B. Frank, A. Saeed, H. P. R Mangunure, N. S. Telang. WO2019165337, 2019.
- 128.K. Ashok, V. K. Dhansukhlal, S. Dharmendra, N. Sanjay, B. Sanjay, J. Atul. WO2005062723, 2005.
- 129. Shrapnel B. C., Johnson C. M., Sandground J. H., Am. J. Trop. Med. Hyg. 1946, 26, 293–310. [DOI] [PubMed] [Google Scholar]
- 130. Wong W., Bai X. C., Brown A., Fernandez I. S., Hansses E., Condron M., Tan Y. H., Baum J., Scheres S. H. W., eLife 2014, 3, e03080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Akinboye E. S., Rosen M. D., Denmeade S. R., Kwabi-Addo B., Bakare O., J. Med. Chem. 2012, 55, 7450–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Bleasel M. D., Peterson G. M., Pharmaceuticals. 2020, 13, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Yang S., Xu M., Lee E. M., Gorshkov K., Shiryaev S. A., He S., Sun W., Cheng Y.-S., Hu X., Tharappel A. M., Lu B., Pinto A., Farhy C., Huang C.-T., Zhang Z., Zhu W., Wu Y., Zhou Y., Song G., Zhu H., Shamim K., Martinez-Romero C., Garcia-Sastre A., Preston R. A., Jayaweera D. T., Huang R., Huang W., Xia M., Simeonov A., Ming G., Qiu X., Terskikh A. V., Tang H., Song H., Zheng W., Cell Discov. 2018, 4, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. MacGibeny M. A., Koyuncu O. O., Wirblich C., Schnell M. J., Enquist L. W., PLoS Pathog. 2018, 14, e1007188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Chaves Valadão A. L., Abreu C. M., Dias J. Z., Arantes P., Verli H., Tanuri A., de Aguiar R. S., Molecules 2015, 20, 11474–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dyall J., Coleman C. M., Hart B. J., Venkataraman T., Holbrook M. R., Kindrachuk J., et al., Antimicrob. Agents Chemother. 2014, 58, 4885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Shen L., Niu J., Wang C., Huang B., Wang W., Zhu N., Deng Y., Wang H., Ye F., Cen S., Tan W., J. Virol. 2019, 93, e00023–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Choy K. T., Wong A. Y., Kaewpreedee P., Sai S. F., Chen D., Hui K. P. Y., Chu D. K. W., Chan M. C. W., Cheung P. P.-H., Huang X., Peiris M., Yen H. L., Antiviral Res. 2020, 178, 104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kumar R., Khandelwal N., Chander Y., Riyesh T., Gulati B. R., Pal Y., Tripathi B. N., Barua S., Kumar N., bioRxiv 2020. doi: 10.1101/2020.11.29.401984. [DOI] [Google Scholar]
- 140. Wang A., Sun Y., Liu Q., Wu H., Liu J., He J., Yu J., Chen Q. Q., Ge Y., Zhang Z., Hu C., Chen C., Qi Z., Zou F., Liu F., Hu J., Zhao M., Huang T., Wang B., Wang L., Wang W., Wang W., Ren T., Liu J., Sun Y., Fan S., Wu Q., Liang C., Sun L., Su B., Wei W., Liu Q., Mol. Biomed. 2020, 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Clark D. E., Holton P. G., Meredith R. F. K., Ritchie A. C., Walker T., Whiting K. D. E., J. Chem. Soc. 1962, 2479–2490. [Google Scholar]
- 142. Van Tamelen E. E., Placeway C., Schiemenz G. P., Wrigh I. G., J. Am. Chem. Soc. 1969, 91, 7359–7371. [Google Scholar]
- 143. Kametani T., Suzuki Y., Terasava H., Ihara M., J. Chem. Soc. Perkin Trans. 1 1979, 1211–1217. [Google Scholar]
- 144. Takano S., Sasaki M., Kanno H., Shishido K., Ogasawar K., J. Org. Chem. 1978, 43, 4169–4172. [Google Scholar]
- 145. Fujii T., Yoshifuji S., Quinolizidines V., Tetrahedron 1980, 36, 1539–1545. [Google Scholar]
- 146. Sharma S., Anand N., Chapter 14 - Natural Products. Approaches to Design and Synthesis of Antiparasitic Drugs, Volume 25, 1st Edition. Elsevier, Amsterdam, 1997, 347–383. [Google Scholar]
- 147. Bailly C., Vergoten G, Pharmacol. Ther. 2020, 214, 107618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Li X. L., Zhou A. G., Zhang L., W. J. Chen. Int. J. Mol. Sci. 2011, 12, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Akamatsu H., Komura J., Asada Y., Niwa Y., Planta Med. 1991, 57, 119–21. [DOI] [PubMed] [Google Scholar]
- 150. Han S., Sun L., He F., Che H., Sci. Rep. 2017, 7, 7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wang L., Yang R., Yuan B., Liu Y., Liu C., Acta Pharm. Sin. B 2015, 5, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Roohbakhsh A., Iranshahy M., Iranshahi M., Curr. Med. Chem. 2016, 23, 498–517. [DOI] [PubMed] [Google Scholar]
- 153. Gomaa A. A., Abdel-Wadood Y. A., Phytomedicine Plus 2021, 1, 100043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sun Z. G., Zhao T. T., Lu N., Yang Y. A., Zhu H. L., Mini-Rev. Med. Chem. 2019, 19, 826–832. [DOI] [PubMed] [Google Scholar]
- 155. Al-Kamel H., Grundmann O., Mini-Rev. Med. Chem. 2021. doi: 10.2174/1389557521666210210160237. [DOI] [PubMed] [Google Scholar]
- 156. Murck H., Front. Immunol. 2020, 11, 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chrzanowski J., Chrzanowska A., Graboń W., Phytother. Res. 2021, 35, 629–636. [DOI] [PubMed] [Google Scholar]
- 158. Luo Y., Liu D., Li J., Int. J. Antimicrob. Agents. Int. J. Antimicrob. Agents. 2020, 55, 105995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Yu S., Zhu Y., Xu J., Yao G., Zhang P., Wang M., Zhao Y., Lin G., Chen H., Chen L., Zhang J., Phytomedicine 2021, 85,153364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. van de Sand L., Bormann M., Alt M., Schipper L., Heilingloh C. S., Todt D., Dittmer U., Elsner C., Witzke O., Krawczyk A., bioRxiv 2020. doi: 10.1101/2020.12.18.423104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang C., Su X., Sun M., Zhang M., Wu J., Xing J., Wang Y., Xue J., Liu X., Sun W., Chen S., Microb. Cell Fact. 2019, 18, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Li Q., Jiang T, Liu R., Feng X., Li C., Appl. Microbiol. Biotechnol. 2019, 103, 4813–4823. [DOI] [PubMed] [Google Scholar]
- 163. Powell R. G., Weisleder D., Smith C. R., Rohwedder W. K., Tetrahedron Lett. 1970, 11, 815–818. [DOI] [PubMed] [Google Scholar]
- 164. Mullard A., Nat. Rev. Drug Discovery 2013, 12, 87–90. [DOI] [PubMed] [Google Scholar]
- 165. Andersen P. I., Krpina K., Ianevski A., Shtaida N., Jo E., Yang J., Koit S., Tenson T., Hukkanen V., Anthonsen M. W., Bjoras M., Evander M., Windisch M. P., Zusinaite E., Kainov D. E., Viruses 2019, 11, 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cao J., Forrest J. C., Zhang X., Antiviral Res. 2015, 114, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Romero M. R., Serrano M. A., Earth T., Alvarez M., Marin J. J., Planta Med. 2007, 73, 552–558. [DOI] [PubMed] [Google Scholar]
- 168. Dong H. J., Wang Z. H., Meng W., Li C. C., Hu Y. X., Zhou L., Wang X. J., Viruses 2018, 10, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Choy K. T., Wong A. Y., Kaewpreedee P., Sia S. F., Chen D., Hui K. P. Y., Chu D. K. W., Chan M. C. W., Cheung P. P. H., Huang X., Peiris M., Yen H. L., Antiviral Res. 2020, 178, 104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Eckelbarger J. D., Wilmot J. T., Epperson M. T., Thakur C. S., Shum D., Antczak C., Tarassishin L., Djaballah H., Gin D. Y., Chem. Eur. J. 2008, 14, 4293–4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ju X., Beaudry C. M., Angew. Chem. Int. Ed. 2019, 58, 6752–6755; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6824–6827. [Google Scholar]
- 172. Campbell W. C., Fisher M. H., Stapley E. O., Albers-Schonberg G., Jacob T. A., Science 1983, 221, 823–828. [DOI] [PubMed] [Google Scholar]
- 173. Campbell W. C., Angew. Chem. Int. Ed. 2016, 55, 10184–10189; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10338–10343. [Google Scholar]
- 174. Chaccour C., Hammann F., Ramon-Garcia S., Rabinovich N. R., Am. J. Trop. Med. Hyg. 2020, 102, 1156–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Chaccour C., Hammann F., Rabinovich N. R., Malar. J. 2017, 16, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Caly L., Druce J. D., Catton M. G., Jans D. A., Wagstaf K. M., Antiviral Res. 2020, 178, 104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schmith V. D., Zhou J., Lohmer L. R. L., Clin. Pharmacol. Ther. 2020, 108, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Buonfrate D., Salas-Coronas J., Muñoz J., Maruri B. T., Rodari P., Castelli F., Zammarchi L., Bianchi L., Gobbi F., Cabezas-Fernandez T., Requena-Mendez a., Godbole G., Silva R., Romero M., Chiodini P. L., Bisoff Z., Lancet Infect. Dis. 2019, 19, 1181–1190. [DOI] [PubMed] [Google Scholar]
- 179. ′′Oxford University adds ivermectin to PRINCIPLE trial for Covid-19′′ to be found under https://www.clinicaltrialsarena.com/news/ivermectin-principle-trial-covid/. 2021.
- 180. Campbell W. C., Fisher M. H., Stapley E. O., Albers-Schonberg G., Jacob T. A., Science 1983, 221, 823–828. [DOI] [PubMed] [Google Scholar]
- 181. Burns J., Yokota T., Ashihara H., Lean M. E., Crozier A., J. Agric. Food Chem. 2002, 50, 3337–3340. [DOI] [PubMed] [Google Scholar]
- 182. Baur J. A., Sinclair D. A., Nat. Rev. Drug Discovery 2006, 5, 493–506. [DOI] [PubMed] [Google Scholar]
- 183. Liu F.-C., Tsai Y.-F., Tsai H.-I., Yu H.-P., Mediators Inflammation 2015, 643763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Olas B., Wachowicz B., Platelets 2005, 16, 251–260. [DOI] [PubMed] [Google Scholar]
- 185. Campagna M., Rivas C., Biochem. Soc. Trans. 2010, 38, 50–3. [DOI] [PubMed] [Google Scholar]
- 186. Abba Y., Hassim H., Hamzah H., Noordin M. M., Adv. Virol. 2015, 184241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Zhao X., Tong W., Song X., Jia R., Li L., Zou Y., He C., Liang X., Lv C., Jing B., Lin J., Yin L., Ye G., Yue G., Wang Y., Yin Z., Viruses 2018, 10, 457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Lin S. C., Ho C. T., Chuo W. H., Li S., Wang T. T., Lin C.-C., BMC Infect. Dis. 2017, 17, 144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Marinella M. A., Int. J. Clin. Pract. 2020, 74, e13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Giordo R., Zinellu A., Eid A. H., Pintus G., Molecules 2021, 26, 856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Yang M., Wei J., Huang T., Lei L., Shen C., Lai J., Yang M., Liu L., Yang Y., Liu G., Liu Y., Phytother. Res. 2021, 35, 1127–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Brown V. A., Patel K. R., Viskaduraki M., Crowell J. A., Perloff M., Booth T. D., Piccirilli G., Cancer Res. 2010, 70, 9003–9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Yu J., Gaunt M. J., Spencer J. B., J. Org. Chem. 2002, 67, 4627–4629. [DOI] [PubMed] [Google Scholar]
- 194. Lebel H., Ladjel C., Bréthous L., J. Am. Chem. Soc. 2007, 129, 13321–13326. [DOI] [PubMed] [Google Scholar]
- 195. Bazin M.-A., El Kihel L., Lancelot J.-C., Rault S., Tetrahedron Lett. 2007, 48, 4347–4351. [Google Scholar]
- 196. Solladie G., Pasturel-Jacope'a Y., Maignanb J., Tetrahedron 2003, 59, 3315–3321. [Google Scholar]
- 197. Alonso F., Riente P., Yus M., Tetrahedron Lett. 2009, 50, 3070–3073. [Google Scholar]
- 198. Ferré-Filmon K., Delaude L., Demonceau A., Noels A. F., Eur. J. Org. Chem. 2005, 15, 3319–3325. [Google Scholar]
- 199. Hilt G., Hengst C., J. Org. Chem. 2007, 72, 7337–42. [DOI] [PubMed] [Google Scholar]
- 200. Lara-Ochoa F., Sandoval-Minero L. C., Espinosa-Pérez G., Tetrahedron Lett. 2015, 56, 5977–5979. [Google Scholar]
- 201. Patil S. A., Otter B. A., Klein S., Tetrahedron Lett. 1994, 35, 5339–5342. [Google Scholar]
- 202. Nishimura H., Katagiri K., Sato K., Mayama M., Shimaoka N., J. Antibiot. 1956, 9, 60–62. [PubMed] [Google Scholar]
- 203. Sheahan T. P., Sims A. C., Graham R. L., Menachery V. D., Gralinski L. E., Case J. B., Leist S. R., Pyrc K., Feng J. Y., Trantcheva I., Bannister R., Park Y., Babusis D., Clarke M. O., Mackman R. L., Spahn J. E., Palmiotti C. A., Siegel D., Ray A. S., Cihlar T., Jordan R., Denison M. R., Baric R. S., Sci. Transl. Med. 2017, 9, eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Mulangu S., Dodd L. E., R. T. Davey Jr. , Mbaya O. T., Proschan M., Mukadi D., Manzo M. L., Nzolo D., Oloma A. T., Ibanda A., Ali R., Coulibaly S., Levine A. C., Grais R., Diaz J., Lane H. C., Muyembe-Tamfum J.-J., PALM Writing Group for the PALM Consortium Study Team, N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar]
- 205. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell Res. 2020, 30 (3), 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Choy K. T., Wong A. Y., Kaewpreedee P., Sia S. F., Chen D., Hui K. P. Y., Chu D. K. W., Chan M. C. W., Cheung P. P., Huang X., Peiris M., Yen H.-l., Antiviral Res. 2020, 178, 104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Agostini M. L., Andres E. L., Sims A. C., Graham R. L., Sheahan T. P., Lu X., Smith E. C., Case J. B., Feng J. Y., Jordan R., Ray A. S., Cihlar T., Siegel D., Mackman R. L., Clarke M. O., Baric R. S., Denison M. R., mBio. 2018, 9, e00221–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wu C. R., Liu Y., Yang Y. Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H., Acta Pharm. Sin. B. 2020, 10, 766–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Zhang L., Zhang D., Yuan C., Wang X. W., Li Y. F., Jia X. L., Cheung P. P. H., Huang X., bioRxiv 2020. doi: 10.1101/2020.04.27.063859. [DOI] [Google Scholar]
- 210. Yin W., Mao C., Luan X., Shen D. D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M., Chang S., Xie Y. C., Tian G., Jiang H. W., Tao S. C., Shen J., Jiang Y., Jiang H., Xu Y., Zhang S., Zhang Y., Xu H. E., Science. 2020, 368, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Bravo J. P. K., Dangerfield T. L., Taylor D. W., Johnson K. A., Mol. Cell 2021, 81, 1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Beigel J. H., Tomashek K. M., Dodd L. E., Mehta A. K., Zingman B. S., Kalil A. C., Hohmann E., Chu H. Y., Luetkemeyer A., Kline S., Castilla D. L., Finberg R. W., Dierberg K., Tapson V., Hsieh L., Patterson T. F., Paredes R., Sweeney D. A., Short W. R., Touloumi G., Lye D. C., Ohmagari N., Oh M., Ruiz-Palacious G. M., Benfield T., Fatkenheuer G., Kortepeter M. G., Atmar R. L., Creech C. B., Lundgren J., Babiker A. G., Pett S., Neaton J. D., Burgess T. H., Bonnett T., Green M., Makowski M., Osinusi A., Nayak S., Lane H. C., N. Engl. J. Med. 2020, 383, 1813–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.′′Gilead Announces Results from Phase 3 Trial of Investigational Antiviral Remdesivir in Patients with Severe COVID-19” to be found under https://www.gilead.com/news-and-press/press-room/press-releases/2020/4/gilead-announcesresults-from-phase-3-trial-of-investigational-antiviral-remdesivir-in-patients-with-severe-covid-19, 2020.
- 214.′′Gilead's Suspension of Covid-19 Trials in China Should Serve as a Bellwether for Studies in Other Countries, Clin. Trials Arena”to be found under https://www.clinicaltrialsarena.com/comment/gilead-remdesivir-covid-19-china-trials/, 2020.
- 215.′′Montefiore and Einstein Assess Remdesivir and Baricitinib for Covid-19, Clin. Trials Arena” to be found under https://www.clinicaltrialsarena.com/news/remdesivirplus-baricitinib-covid-19/, 2020.
- 216.′′Global Coronavirus COVID-19 Clinical Trial Tracker′′ to be found under https://www.covid-trials.org/ 2020.
- 217. ′′Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment” to be found under https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment. 2020.
- 218. Singh A. K., Singh A., Singh R., Misra A., Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.′′WHO recommends against the use of remdesivir in COVID-19 patients” to be found under. https://www.who.int/news-room/feature-stories/detail/who-recommends-against-the-use-of-remdesivir-in-covid-19-patients. 2020.
- 220. Siegel D., Hui H. C., Doerffler E., Clarke M. O., Chun K., Zhang L., Neville S., Carra E., Lew W., Ross B., Wang Q., Wolfe L., Jordan R., Soloveva V., Knox J., Perry J., Perron M., Stray K. M., Barauskas O., Feng J. Y., Xu Y., Lee G., Rheingold A. L., Ray A. S., Bannister R., Streckley R., Swaminathan S., Lee W. A., Bavaria S., Cihlar T., Lo M. K., Warren T. K., Mackman R. L., J. Med. Chem. 2017, 60, 1648–1661. [DOI] [PubMed] [Google Scholar]
- 221. Li D., Hu J., Li D., Yang W., Yin S. F., Qiu R., Top. Curr. Chem. (Cham). 2021, 379, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Wang M., Zhang L., Huo X. H., Zhang Z. F., Yuan Q. J., Li P. P., Chen J. Z., Zou Y. S., Wu Z. X., Zhang W. B., Angew. Chem. Int. Ed. 2020, 59, 20814–20819; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21000–21005. [Google Scholar]
- 223.T. Butler, A. Cho, C. Kim, O. Saunders, L. J. Zhang, J. Parrish. EP2268642. 2011.
- 224. Metobo S. E., Xu J., Saunders O. L., Butler T., Aktoudianakis E., Cho A., Kim C. U., Tetrahedron Lett. 2012, 53, 484–486. [Google Scholar]
- 225. Wit E. D., Feldmannb F., Cronin J., Jordanc R., Okumurad A., Thomas T., Scott D., Cihlar T., Feldmann H., Proc. Natl. Acad. Sci. USA. 2020, 117, 6771–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Gannedi V., Villuri B. K., Reddy S. N., Ku C.-C., Wong C.-H., Hung S.-C., J. Org. Chem. 2021, 8, 4977–4985. [DOI] [PubMed] [Google Scholar]
- 227. Agostini M. L., Pruijssers A. J., Chappell J. D., Gribble J., Lu X. T., Andres E. L., Bluemling G. R., Lockwood M. A., Sheahan T. P., Sims A. C., Natchus M. G., Saindane M., Kolykhalov A. A., Painter G. R., Baric R. S., Denison M. R., J. Virol. 2019, 93, e01348–e01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Yamamoto T., Koyama H., Kurajoh M., Shoji T., Tsutsumi Z., Moriwaki Y., Clin. Chim. Acta. 2011, 412, 1712–1724. [DOI] [PubMed] [Google Scholar]
- 229. Cho A., Saunders O. L., Butler T., Zhang L. J., Xu J., Vela J. E., Feng J. Y., Ray A. S., Kim C. U., Bioorg. Med. Chem. Lett. 2012, 22, 2705–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Rothan H. A., Byrareddy S. N., J. Autoimmun. 2020, 109, 102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Toots M., Yoon J. J., Cox R. M., Hart M., Sticher Z. M., Makhsous N., Plesker R., Barrena A. H., Reddy P. G., Mitchell D. G., Shean R. C., Bluemling G. R., Kolykhalov A. A., Greninger A. L., Natchus M. G., Painter G. R., Plemper R. K., Sci. Transl. Med. 2019, 11, eaax5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Toots M., Yoon J. J., Hart M., Natchus M. G., Painter G. R., Plemper R. K., Transl. Res. 2020, 218, 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Sheahan T. P., Sims A. C., Zhou S. T., Graham R. L., Pruijssers A. J., Agostini M. L., Leist S. R., Schafer A., Dinnon K. H., Stevens L. J., Chappell J. D., Lu X., Hughes T. M., George A. S., Hill C. S., Montgomery S. A., Brown A. J., Bluemling G. R., Natchus M. G., Saindane M., Kolykhalov A. A., Painter G., Harcourt J., Tamin A., Thornburg N. J., Swanstrom R., Denison M. R., Baric R. S., Sci. Transl. Med. 2020, 12, eabb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.G. R. Painter, D. B. Guthrie, G. R. Bluemling, M. R. Natchus. WO 2019/113462 A1, 2019.
- 235. Steiner A., Znidar D., Ötvös S. B., Snead D. R., Dallinger D., Kappe C. O., Eur. J. Org. Chem. 2020, 43, 6736–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]