

Abstract
Accumulating evidence demonstrates that metabolic changes in the brain associated with neuroinflammation, oxidative stress, and mitochondrial dysfunction play an important role in the pathophysiology of mild cognitive impairment (MCI) and Alzheimer’s disease (AD). However, the neural signatures associated with these metabolic alterations and underlying molecular mechanisms are still elusive. Accordingly, we reviewed the literature on in vivo human brain 1H and 31P-MRS studies and use meta-analyses to identify patterns of brain metabolic alterations in MCI and AD. 40 and 39 studies on MCI and AD, respectively, were classified according to brain regions. Our results indicate decreased N-acetyl aspartate and creatine but increased myo-inositol levels in both MCI and AD, decreased glutathione level in MCI as well as disrupted energy metabolism in AD. In addition, the hippocampus shows the strongest alterations in most of these metabolites. This meta-analysis also illustrates progressive metabolite alterations from MCI to AD. Taken together, it suggests that 1) neuroinflammation and oxidative stress may occur in the early stages of AD, and likely precede neuron loss in its progression; 2) the hippocampus is a sensitive region of interest for early diagnosis and monitoring the response of interventions; 3) targeting bioenergetics associated with neuroinflammation/oxidative stress is a promising approach for treating AD.
Keywords: Mild cognitive impairment, Alzheimer’s disease, Neuroinflammation, Oxidative stress, Mitochondrial dysfunction, Magnetic resonance spectroscopy
1. Introduction
Alzheimer’s disease (AD) is the most common cause of age-related cognitive decline, accounting for 60–70% of total dementia cases (WHO, 2019). It is estimated that 6.2 million Americans, or 10% of the population aged 65 and older, are currently living with Alzheimer’s dementia (Alzheimer’s Association, 2021; Hebert et al., 2013). In addition, more than 11 million family members and other caregivers provided around 15.3 billion hours of care to patients with Alzheimer’s and other dementias in 2020, which is equivalent to $256.7 billion in unpaid labor (Alzheimer’s Association, 2021). Thus, AD is a substantial and growing public health and economic burden in the US.
Unfortunately, AD is often clinically diagnosed at a stage in which the underlying pathology has reached an advanced and possibly irreversible state. Therefore, one of the major challenges in AD research is to identify targets for early intervention, which in turn could substantially reduce the morbidity, mortality, and cost of care related to AD. In this vein, mild cognitive impairment (MCI) represents an earlier clinical syndrome with less functional impairment, on the path towards AD and other dementias. An estimated 15 to 20 percent of people age 65 or older have MCI (Roberts and Knopman, 2013), and conversion rates of MCI to AD have been estimated at 15% per year and almost 45% over 5 years (Gauthier et al., 2006). Therefore, MCI may be a critical stage of illness where intervention could stop or delay the progression towards dementia (Tumati et al., 2013). However, modifying the course of disease at this early stage first requires identifying treatment targets that are reliably linked to AD pathophysiology and neuronal progression.
To date, the majority of clinical trials investigating such disease-modifying therapies have sought to halt or reverse the deposition of specific proteins in the brain, namely amyloid beta (Aβ) and microtubule-associated protein tau. These trials have been anchored in the amyloid cascade hypothesis, which proposes that accumulation of Aβ plaques in the brain is a critical early driver of AD pathogenesis. Over the past two decades, FDA approved two classes of therapies, cholinesterase inhibitors and memantine, that target cholinergic and glutamatergic neurotransmission to slow decline in the symptoms of cognition and daily functioning. Only one anti-amyloid drug (aducanumab) has very recently received FDA approval, though with significant controversy. The failure rate of anti-amyloid trials has raised questions about whether the amyloid hypothesis is complete (Kuehn, 2020; Sery et al., 2013). The paucity of available disease-modifying therapies and discouraging track record of anti-amyloid trials – in conjunction with the escalating public health and economic crises associated with AD – make clear the imperative to identify alternative treatment targets.
Toward that end, accumulating evidence indicates that enhancing brain bioenergetic metabolism, with a resulting reduction in oxidative stress and/or neuroinflammation, could be a promising treatment avenue for AD. Mitochondrial dysfunction and cellular energy deficits are increasingly thought to play a critical role in aging and AD pathophysiology (Bonkowski and Sinclair, 2016; Imai and Guarente, 2014; Sonntag et al., 2017; Sorrentino et al., 2017). For example, studies have shown hypometabolism in brain regions affected by AD (Murray et al., 2014) where the mitochondrial structure is altered (Moreira et al., 2007; Hirai et al., 2001); reduced expression and activity of mitochondrial enzymes important for energy metabolism (Maurer et al., 2000); and reduced membrane potential, increased permeability, and excess production of reactive oxygen species (ROS) in mitochondria from AD brains (Onyango et al., 2016). Alterations in mitochondrial function and metabolism might also be antecedent to AD pathology, including Aβ plaques and neurofibrillary tangles (Gibson and Shi, 2010; Onyango, 2018).
Oxidative stress (OS), which occurs when there is an imbalance between oxidant and antioxidant levels in the cell resulting in increased reactive oxygen species (ROS) production, is another important metabolic facet of AD pathology. Specifically, increased levels of ROS cause damage to macromolecules within the cell, and it is this damage of lipids, proteins, and nucleic acids that give rise to pathological consequences (Bermejo et al., 2008). In the brain, ROS are eliminated by the free radical scavenger glutathione (GSH) through a chemical reaction that converts GSH to its oxidized state (GSSG) (Cabungcal et al., 2006; Dringen, 2000). As such, higher intracellular GSH levels protect cells from ROS-mediated insults. Given that neurons are particularly sensitive to oxidative damage due to the brain’s substantial metabolic requirements, alterations in GSH function can have profound effects on brain and cognitive function. Not surprisingly, research now suggests that GSH deficiencies could play a key role in the pathogenesis of various age-related neurodegenerative disorders, including AD (Bains and Shaw, 1997; Pocernich and Butterfield, 2012; Schulz et al., 2000).
As with OS, microglia-mediated neuroinflammation adversely affects brain function and is now considered a hallmark feature of various neurodegenerative diseases, including AD (Calabrese et al., 2014; Webers et al., 2020). Evidence from brain imaging supports the role of neuroinflammation in the progression of dementia. For instance, a recent meta-analysis of positron emission topography (PET) studies found evidence of increased neuroinflammation during the progression of MCI and AD (Bradburn et al., 2019). Notably, pro-inflammatory mediator expression is modulated by mitochondrial dynamics in microglial cells (Park et al., 2013). Furthermore, neuroinflammation and OS are linked closely, as neuroinflammation leads to increased OS which, in turn, causes further neuroinflammation (Fischer and Maier, 2015).
Taken together, the above findings illustrate the importance of brain bioenergetic and metabolic dysfunction in MCI and AD and, in turn, the utility of investigative methods for elucidating these pathological processes. Magnetic resonance spectroscopy (MRS) is a functional neuroimaging technique that provides one of the most direct windows into these processes. In vivo MRS is a non-invasive tool for characterizing alterations in metabolite concentration and, by extension, bioenergetic and metabolic dysfunction associated with neurodegenerative disease progression (Duarte et al., 2012). Several such metabolites in the brain are present at sufficient concentrations to be detected by 1H-MRS, including N-Acetyl Aspartate (NAA), choline-containing compounds (Cho), creatine (Cr), myo-inositol (mI), and by 31P-MRS including adenosine triphosphate (ATP) and phosphocreatine (PCr). However, unlike these metabolites which could produce well-defined peaks, GSH, γ-aminobutyric acid, glucose, lactate, etc. have small peaks with overlapping resonances resulting in more measurement variability. To date, numerous MRS studies focused on MCI and AD have shown abnormal metabolite profiles. For instance, meta-analyses of MCI (Tumati et al., 2013) and AD (Wang et al., 2015) indicated decreased NAA and increased mI levels associated with MCI and AD, respectively. However, these two meta-analyses only focused on one, either MCI or AD, and 1H-MRS studies only. These analyses lack information from both MCI and AD as well as from bioenergetics associated with OS and neuroinflammation. Therefore, it is unclear whether specific patterns of the metabolic changes exist in MCI and AD, and whether these patterns could help understand disease transition from MCI to AD or give insights on the underlying molecular mechanisms. We therefore performed a meta-analysis of in vivo 1H- and 31P-MRS studies. The central goal of this study was to investigate the alterations in most common metabolites, including antioxidant GSH and energy-related phosphates, and to further validate the model of oxidative stress and neuroinflammation associated with mitochondrial dysfunction in MCI and AD patients (Butterfield and Halliwell, 2019; Ikawa et al., 2020; Simpson and Oliver, 2020).
2. Materials and Methods
2.1. Data extraction and inclusion/exclusion criteria
We performed meta-analyses on the metabolite ratios (e.g. NAA/Cr) and absolute concentrations (quantified by the internal reference of Cr and external reference of water signal, respectively) of NAA, Cho, mI, and Cr categorized by brain location because these were the most common measures reported in MCI and AD studies. In addition, due to the small number of GSH and 31P-MRS studies, we summarized the GSH result of each study and performed meta-analyses for the averaged levels of metabolites of phosphorous compounds, e.g. PCr and ATP, regardless of brain locations. PubMed, EMBASE, Cochrane databases were searched to identify journal articles published between 1 January 1989 and 30 January 2021, using the search terms: MRS or magnetic resonance spectroscopy and (1) MCI or (2) AD or (3) brain metabolites or (4) brain phosphate metabolism. In addition, we limited the search to English language studies only. The procedure of the literature searching along with the inclusion/exclusion criteria as well as the data extraction are plotted in Fig.1 (details, see Supplementary Materials).
Figure 1.
Flowchart of studies screening, and inclusion/exclusion criteria.
2.2. Meta-analysis
The current meta-analyses were conducted using RevMan (version 5.3, Cochrane Collaboration) with a random-effects model and following the guidelines of Preferred Reporting Items for Systematic Reviews and Meta-Analyses (Moher et al., 2009). Differences in metabolite ratios and concentrations of MCI/AD relative to healthy controls (HCs) were calculated for each study. After excluding outliers (identified by the ROUT method) (Motulsky and Brown, 2006), the weighted averages of relative changes were calculated using the weight factors provided by RevMan software (version 5.3, Cochrane Collaboration). Hedges’ g was used as the effect size to analyze the differences in the means of two groups, which were divided by the pooled standard deviation (Cheung, 2015). To address potential heterogeneity, a subgroup meta-analysis was performed according to brain region, and further leave-one-out analysis (excluding the study which contributes the highest heterogeneity) was performed if the heterogeneity (I2) was larger than 50%. Meta-analyses were conducted only if the number of studies was larger than three. Otherwise, we presented summarized results from each study. Details of analyses on heterogeneity and publication biases are provided in the Supplementary Materials. Lastly, Pearson correlation was performed to determine the relationship between Hedge’s g effect sizes for metabolite concentrations and Mini-Mental State Exam (MMSE) scores in MCI/AD patients.
3. Results
3.1. Description of studies
The studies were classified according to the region of interest, as well as the reported metabolite ratios or concentrations. For in vivo 1H-MRS data, 40 MCI (1473 HCs and 1238 patients) and 39 AD (1682 HCs and 1248 patients) studies were included. Of the 40 studies that included MCI patients, 25 included data from the posterior cingulate cortex (PCC), 15 from the hippocampus or medial temporal lobe (MTL), and 7 from parietal white matter areas (PWM). Of the 39 studies that included AD patients, 23 included data from the PCC, 15 from the hippocampus or MTL, 7 from PWM, and 6 from parietal gray matter areas (PGM). Regarding 31P-MRS data, a total of 123 patients and 119 HC subjects from eight AD studies and one MCI study were included (Bottomley et al., 1992; Brown et al., 1989; Forlenza et al., 2005; Gonzalez et al., 1996; Mecheri et al., 1997; Pettegrew et al., 1994; Rijpma et al., 2018; Smith et al., 1995). Summaries of all studies included in the current analysis are provided in Table 1.
Table 1.
List of included MCI and AD studies.
| Literatures | Region of interest | Magnetic Field (Tesla) | Pulse sequence | Subjects number (MCI/AD/HC) | Metabolites quantification |
|---|---|---|---|---|---|
| (Jessen et al., 2000) | Medial temporal lobe, central cortical region | 1.5 | PRESS | 38 (-/20/18) | NAA/Cr, Cho/Cr |
| (Stoppe et al., 2000) | PGM, PWM | 2.0 | - | 42 (-/30/22) | NAA/Cr, Cho/Cr, mI/Cr, NAA, Cr, Cho, mI |
| (Catani et al., 2001) | PWM | 1.5 | PRESS | 36 (11/14/11) | NAA/Cr, Cho/Cr, mI/Cr |
| (Block et al., 2002) | Hippocampus, lateral temporal lobe, occipital lobe | 1.5 | - | 56 (-/34/22) | NAA/tCr, Cho/tCr |
| (Chantal et al., 2002) | MTL (Hippocampus), Frontal cortex, parietotemporal cortex | 1.5 | PRESS | 28 (-/14/14) | NAA, Cho, Cr, mI |
| (Hattori et al., 2002) | PCC, Parietooccipital WM | 3.0 | PRESS | 21 (-/9/12) | NAA/Cr, Glx/Cr, Cho/Cr, mI/Cr |
| (Schuff et al., 2002) | Bilateral hippocampus, temporal lobe, parietal lobe, frontal lobe | 1.5 | PRESS | 110 (-/56/54) | NAA |
| (Herminghaus et al., 2003) | PGM, PWM, Frontal GM, Frontal WM | 1.5 | STEAM | 43 (-/28/15) | NAA/Cr, mI/Cr, Glx/Cr, TMA/Cr |
| (Kantarci et al., 2004) | left PCC | 1.5 | PRESS | 327 (-/121/206) | NAA/Cr, Cho/Cr, mI/Cr |
| (Ackl et al., 2005) | Hippocampus, Parietal GM, Parietal WM | 1.5 | PRESS | 59 (19/18/22) | NAA/Cr, mI/Cr, mI/NAA |
| (Chao et al., 2005) | Hippocampal region, medial temporal lobe | 1.5 | PRESS | 48 (-/24/24) | NAA/Cr, NAA |
| (Metastasio et al., 2006) | PWM | 1.5 | PRESS | 54 (25/-/29) | NAA/Cr, Cho/Cr, mI/Cr |
| (Zhu et al., 2006) | PGM, PWM, Frontal GM, Frontal WM | 1.5 | - | 36 (-/14/22) | NAA/Cr, mI/Cr, NAA/mI, NAA, mI |
| (Franczak et al., 2007) | Hippocampus | 0.5 | PRESS | 10 (5/-/5) | mI/Cr, Cho/Cr, Glx/Cr, NAA/Cr, Glx/NAA, mI/NAA, NAA, mI, Cho, Cr, Glx |
| (Griffith et al., 2007) | PCC | 3.0 | PRESS | 34 (-/15/19) | NAA/Cr, Cho/Cr |
| (Kantarci et al., 2007) | PCC | 1.5 | PRESS | 194 (49/60/85) | NAA/Cr, Cho/Cr, mI/Cr |
| (Rami et al., 2007) | PCC, Temporal, Temporalparietal | 1.5 | PRESS | 89 (27/35/27) | NAA/Cr, Cho/Cr, mI/Cr, NAA, Cho, Cr, mI |
| (Kantarci et al., 2008b) | Hippocampus | 1.5 | PRESS | 243 (143/-/100) | NAA/Cr, Cho/Cr, mI/Cr |
| (Ding et al., 2008) | PCC | 1.5 | PRESS | 40 (-/20/20) | NAA/Cr, Cho/Cr, mI/Cr |
| (Olson et al., 2008) | PCC | 1.5 | STEAM | 71 (47/-/24) | NAA/Cr, NAA/Cho, NAA/mI, Cho/Cr, mI/Cr, Glx/Cr, NAA, Cho, mI, Glx, Cr |
| (Garcia Santos et al., 2008) | PCC | 1.5 | PRESS | 44 (10/-/34) | NAA/Cr, Cho/Cr, NAA/Cho, mI/Cr, NAA/mI |
| (Watanabe et al., 2008) | PCC, Hippocampus, Occipital lobe | 1.5 | PRESS | 56 (-/30/26) | NAA/Cr, mI/Cr, Cho/Cr, NAA/mI, NAA, mI, Cho, Cr |
| (Jessen et al., 2009) | MTL (Hipp) | 1.5 | PRESS | 279 (136/98/45) | NAA/Cr, mI/NAA, NAA, Cr, Cho, mI |
| (Pilatus et al., 2009) | PCC, PWM | 1.5 | PRESS | 27 (15/-/12) | NAA, Cho, mI, Cr, Glx |
| (Siger et al., 2009) | Frontal lobe G/W, PWM, PGM | 1.5 | - | 47 (14/17/16) | mI, NAA |
| (Wang et al., 2009) | PCC, Hippocampus | 3.0 | PRESS | 48 (16/16/16) | NAA/Cr, mI/Cr, Cho/Cr, mI/NAA |
| (Zhang et al., 2009) | Hippocampus, Temporalparietal | 1.5 | - | 40 (14/13/13) | NAA/Cr, mI/Cr |
| (Chao et al., 2010) | PCC | 1.5 | STEAM | 31 (13/-/18) | NAA/Cr, mI/Cr, NAA/mI |
| (Griffith et al., 2010) | PCC | 3.0 | PRESS | 71 (29/-/42) | NAA/Cr, Cho/Cr, mI/Cr |
| (Watanabe et al., 2010) | PCC, Hippocampus, Occipital, ApPoDeepWM | 1.5 | PRESS | 169 (47/70/52) | NAA, mI, Cho, Cr |
| (Foy et al., 2011) | Hippocampus | 1.5 | PRESS | 98 (21/38/39) | NAA, mI, Cho, Cr |
| (Modrego et al., 2011) | PCC | 1.5 | PRESS | 106 (71/-/35) | NAA/Cr, Cho/Cr, mI/Cr, NAA/mI, NAA |
| (Silveira de Souza et al., 2011) | PCC | 1.5 | PRESS | 68 (10/25/33) | NAA/Cr, Cho/Cr, mI/Cr, mI/NAA |
| (Zimny et al., 2011) | PCC | 1.5 | PRESS | 68 (23/30/15) | NAA/Cr, Cho/Cr, mI/Cr, mI/NAA, mI/Cho |
| (Gordon et al., 2012) | PCC | 3.0 | PRESS | 39 (-/11/28) | NAA/Cr, Cho/Cr, mI/Cr, NAA/Cho, NAA/mI, NAA, Cho, mI, Cr |
| (Lim et al., 2012) | PCC | 3.0 | STEAM | 78 (19/36/23) | NAA/Cr, mI/Cr |
| (Seo et al., 2012) | PCC, Hippocampus, ERC, Occipital WM | 3.0 | PRESS | 24 (13/-/11) | NAA/Cr, Cho/Cr |
| (Shiino et al., 2012) | PCC, bilateral hippocampus | 1.5 | PRESS | 144 (-/99/45) | NAA/Cr, Cho/Cr, mI/Cr, Glx/Cr, mI/NAA, NAA, Cr, Cho, mI, Glx |
| (Wang et al., 2012) | PCC, Hippocampus | 3.0 | PRESS | 135 (32/47/56) | NAA/Cr, Cho/Cr, mI/Cr, NAA/mI |
| (Yang et al., 2012) | PCC, PWM, Dorsal Thalamus, Lentiform nucleus | 1.5 | PRESS | 29 (14/-/15) | NAA/Cr, Cho/Cr, mI/Cr, NAA/mI, NAA, mI, Cho, Cr |
| (Targosz-Gajniak et al., 2013) | PCC, Hippocampus, Parietal lobe | 1.5 | PRESS | 76 (41/-/35) | NAA/Cr, Cho/Cr, mI/Cr, Glx/Cr, NAA/Cho |
| (Duffy et al., 2014) | Anterior and posterior cingulate | 3.0 | PRESS | 95 (54/-/41) | GSH/Cr |
| (Fayed et al., 2014) | PCC | 1.5 | PRESS | 295 (66/36/193) | Glu, Glu/Cr, Glx, Glx/Cr, mI, mI/Cr, NAA, NAA/Cr, Cho, Cho/Cr |
| (Graff-Radford et al., 2014) | PCC, frontal lobe, occipital lobe | 1.5 | PRESS | 183 (-/35/148) | NAA/Cr, Cho/Cr, mI/Cr |
| (Suriyajakryuththana et al., 2014) | Frontal and paiertal white matter | 3.0 | - | 20 (7/10/3) | NAA/Cr, Cho/Cr, mI/Cr |
| (Mandal et al., 2015) | Frontal cortex, Hippocampus | 3.0 | MEGA-PRESS | 64 (22//21/21) 66 (28/19/19) | GSH |
| (Riese et al., 2015) | PCC | 3.0 | MEGA-PRESS | 39 (15/-/21) | GABA (AU), Glx (AU), NAA (AU) |
| (Yin et al., 2015) | Hippocampus | 3.0 | PRESS | 27 (11/-/16) | NAA/Cr, mI/Cr, mI/NAA |
| (Zhu et al., 2015) | Hippocampus, Basal ganglia, Frontal lobe | 3.0 | PRESS | 86 (52/-/34) | NAA/Cr, Cho/Cr, mI/Cr |
| (Chen et al., 2016) | PCC, Hippocampus, Frontal lobe WM, PAWM | 3.0 | PRESS | 78 (38/-/40) | NAA/Cr, mI/Cr, Glu/Cr, Cho/Cr |
| (Guo et al., 2016) | ACC, PCC | 3.0 | PRESS | 44 (13/15/16) | NAA/mI, NAA/Cr, Cho/Cr, mI/Cr |
| (Waragai et al., 2017) | PCC | 1.5 | PRESS | 274 (53/21/200) | NAA/Cr, mI/Cr, NAA/mI |
| (Zeydan et al., 2017) | PCC | 3.0 | sLASER | 46 (14/-/32) | Cr, mI, Cho, Glu, NAA, Glu/mI |
| (Mullins et al., 2018) | PCC | 3.0 | PRESS | 52 (-/25/27) | Glucose, Asc, Lac, NAA, Glu, Gln, sI, PCr, mI, GSH, Ala, NAAG, GABA |
| (Marjanska et al., 2019) | PCC, OCC | 7.0 | STEAM | 49 (-/16/33) | Asc, Asp, GABA, Gln, Glu, GSH, mI, NAA, NAAG, PE, sI, Tau, Cho, Cr |
| (Oeltzschner et al., 2019) | ACC | 7.0 | STEAM | 26 (13/-/13) | GABA/tCr, Glu/tCr, GSH/tCr, NAA/tCr, NAAG/tCr, mI/tCr |
| (Shukla et al., 2020) | ACC, PCC and Cingulate | 3.0 | MEGA-PRESS | 64 (19/18/27) | GSH, NAA (AU), Cr (AU), Cho (AU) |
| (Wong et al., 2020) | Hippocampus, PCC | 7.0 | sLASER | 35 (8/9/16) | Glu, NAA |
| (Brown et al., 1989) | Frontal, Temporoparietal regions | 1.89 | - | -/17/17 | PCr/Pi, Pi, PME/PDE, PME, PCr |
| (Bottomley et al., 1992) | Whole brain | 1.5 | - | -/11/14 | PCr, NTP, PME, Pi, PDE, PCr/NTP, Pi/NTP, PCr/Pi, PDE/NTP, PME/NTP, PDE/PME |
| (Pettegrew et al., 1994) | Dorsal prefrontal cortex | 1.5 | - | -/12/21 | PME, Pi, PDE, PCr, pH |
| (Smith et al., 1995) | Frontal lobe | 1.5 | - | -/17/8 | pH, PME, Pi, PDE, PCr, Total NP, PCr/Pi, PCr/NTP, Pi/NTP, PME/PDE |
| (Gonzalez et al., 1996) | Whole brain | 1.5 | - | -/16/8 | β-NTP, PCr, PME, PDE, Pi, PCr/Pi, NTP/Pi, PME/PDE, PDE/NTP, PME/NTP |
| (Mecheri et al., 1997) | Hippocampus | 1.5 | - | -/24/11 | PME, Pi, PDE, PCr, γ-ATP, α-ATP, β-ATP |
| (Forlenza et al., 2005) | Left prefrpntal cortex | 1.5 | ISIS | -/18/16 | PME, PDE, PME/PDE, PCr, Pi, γ-ATP, α-ATP, β-ATP, total ATP |
| (Rijpma et al., 2018) | ACC, Hippocampus, retrosplenial cortex | 3 | MRSI | -/31/31 | PCr, Pi, PCr/Pi, total ATP, NAD(H), PEth, PCh, GPEth, GPCh, pH |
“-”, not available.
3.2. 1H-MRS studies in MCI and AD
The specific Hedges’ g values, heterogeneities, number of studies, and p values for each metabolite in each region of interest, as well as the overall effect are shown in Table 2. Forest plots for each metabolite were also provided in the Supplementary Materials (Fig. S1–S14). Compared to HCs, our overall analysis (Fig. 2) indicated similar alterations in metabolite concentration in both MCI and AD patients, albeit more pronounced in the latter. Specifically, we observed the following in MCI and AD patients, respectively: NAA was decreased by 9.6% and 12.9%; mI was increased by 6.3% and 7.4%; Cho was decreased by 4.3% and 5.4%; and Cr was decreased by 6.4% and 6.9%. Regarding metabolite ratios, our overall analysis showed that NAA/Cr was decreased by 6.4% and 10.8%, and mI/Cr was increased by 16.2% and 19.4%, in MCI and AD subjects, respectively. Cho/Cr did not significantly differ in either MCI or AD versus HC subjects. Regarding region-specific findings, the largest metabolite alterations were most often observed in the hippocampus in both clinical groups (Fig. 2C and Fig. 2D). Moreover, most of the observed metabolite changes were greater in AD than MCI across brain regions, such as hippocampus, PCC, and PWM, except for Cho alterations, which were only observed in the hippocampus in MCI and AD.
Table 2.
Hedges’g for MCI and AD studies in each region of interest before leave-one-out analysis.
| Metabolite | Region of interest | MCI |
AD |
||||||
|---|---|---|---|---|---|---|---|---|---|
| k | Hedges’g [95% CI] | I2 | p value | k | Hedges’g [95% CI] | I2 | p value | ||
| NAA/Cr | PCC | 19 | −0.53 [−0.78, −0.28] | 80% | <0.001 | 19 | −0.96 [−1.22, −0.69] | 82% | <0.001 |
| Hippocampus | 12 | −0.37 [−0.59, −0.14] | 61% | 0.001 | 9 | −0.85 [−1.20, −0.50] | 84% | <0.001 | |
| PWM | 5 | −0.34 [−0.85, 0.17] | 71% | 0.19 | 6 | −0.85 [−1.43, −0.26] | 84% | 0.004 | |
| PGM | - | - | - | - | 4 | −0.78 [−1.71, 0.14] | 92% | 0.1 | |
| overall | 36 | −0.46 [−0.62, −0.30] | 76% | <0.001 | 38 | −0.90 [−1.10, −0.71] | 84% | <0.001 | |
| mI/Cr | PCC | 18 | 0.36 [0.12, 0.65] | 83% | 0.003 | 19 | 0.85 [0.52, 1.18] | 89% | <0.001 |
| Hippocampus | 10 | 0.70 [0.37, 1.03] | 79% | <0.001 | 6 | 0.90 [0.60, 1.19] | 62% | <0.001 | |
| PWM | 5 | 0.69 [0.10, 1.29] | 78% | 0.02 | 6 | 0.72 [0.23, 1.21] | 77% | 0.004 | |
| PGM | - | - | - | - | 4 | 0.93 [0.54, 1.32] | 57% | <0.001 | |
| overall | 33 | 0.52 [0.33, 0.71] | 80% | <0.001 | 35 | 0.85 [0.65, 1.06] | 85% | <0.001 | |
| Cho/Cr | PCC | 16 | 0.14 [−0.08, 0.35] | 70% | 0.15 | 15 | 0.39 [0.20, 0.58] | 62% | <0.001 |
| Hippocampus | 8 | −0.02 [−0.38, 0.34] | 80% | 0.92 | 6 | −0.24 [−0.45, −0.03] | 41% | 0.02 | |
| PWM | 4 | −0.04 [−0.31, 0.23] | 0% | 0.76 | 3 | −0.26 [−0.51, −0.02] | 0% | 0.04 | |
| overall | 28 | 0.08 [−0.08, 0.24] | 69% | 0.35 | 24 | 0.15 [−0.04, 0.34] | 78% | 0.13 | |
| GSH/Cr | overall | 2 | 0.16 [−0.76, 1.08] | 89% | 0.73 | 1 | 0.49 [−0.06, 1.04] | - | - |
| NAA | PCC | 10 | −0.77 [−1.07, −0.48] | 70% | <0.001 | 8 | −1.13 [−1.38, −0.88] | 44% | <0.001 |
| Hippocampus | 5 | −0.79 [−1.22, −0.37] | 71% | <0.001 | 8 | −0.96 [−1.27, −0.65] | 81% | <0.001 | |
| PWM | - | - | - | - | 3 | −0.28 [−0.51, −0.06] | 0% | 0.01 | |
| PGM | - | - | - | - | 4 | −0.74 [−1.17, −0.31] | 72% | <0.001 | |
| overall | 15 | −0.78 [−1.01, −0.55] | 68% | <0.001 | 23 | −0.90 [−1.09, −0.70] | 78% | <0.001 | |
| mI | PCC | 7 | 0.26 [−0.01, 0.53] | 56% | 0.06 | 6 | 1.00 [0.29, 1.72] | 93% | 0.006 |
| Hippocampus | 4 | 0.16 [−0.25, 0.58] | 68% | 0.44 | 5 | 0.35 [−0.10, 0.79] | 87% | 0.13 | |
| PWM | 3 | 0.68 [0.36, 1.01] | 0% | <0.001 | 3 | 0.45 [−0.01, 0.91] | 66% | 0.06 | |
| PGM | - | - | - | - | 3 | 0.25 [0.01, 0.49] | 0% | 0.04 | |
| overall | 14 | 0.31 [0.10, 0.52] | 62% | 0.004 | 17 | 0.51 [0.25, 0.78] | 86% | <0.001 | |
| Cho | PCC | 8 | −0.06 [−0.33, 0.20] | 56% | 0.64 | 7 | 0.60 [−0.06, 1.26] | 92% | 0.08 |
| Hippocampus | 4 | −0.53 [−0.84, −0.22] | 49% | <0.001 | 5 | −0.60 [−0.75, −0.45] | 0% | <0.001 | |
| overall | 12 | −0.24 [−0.47, −0.01] | 67% | 0.04 | 12 | 0.02 [−0.39, 0.42] | 92% | 0.93 | |
| Cr | PCC | 7 | −0.57 [−1.02, −0.12] | 79% | 0.01 | 6 | −0.37 [−0.85, 0.11] | 82% | 0.13 |
| Hippocampus | 4 | −0.46 [−0.66, −0.27] | 0% | <0.001 | 5 | −0.41 [−0.58, −0.24] | 22% | <0.001 | |
| overall | 11 | −0.50 [−0.76, −0.24] | 68% | <0.001 | 11 | −0.40 [−0.63, −0.18] | 69% | <0.001 | |
| GSH | overall | 2 | −1.01 [−1.27, −0.76] | 0% | <0.001 | 3 | −0.40 [−2.37, 1.58] | 98% | 0.69 |
k, number of studies; I2, heterogeneity. “-”, not available. The threshold for significance is p < 0.05 showed as bold.
Figure 2. Semi-quantifications of metabolite ratios and concentrations.
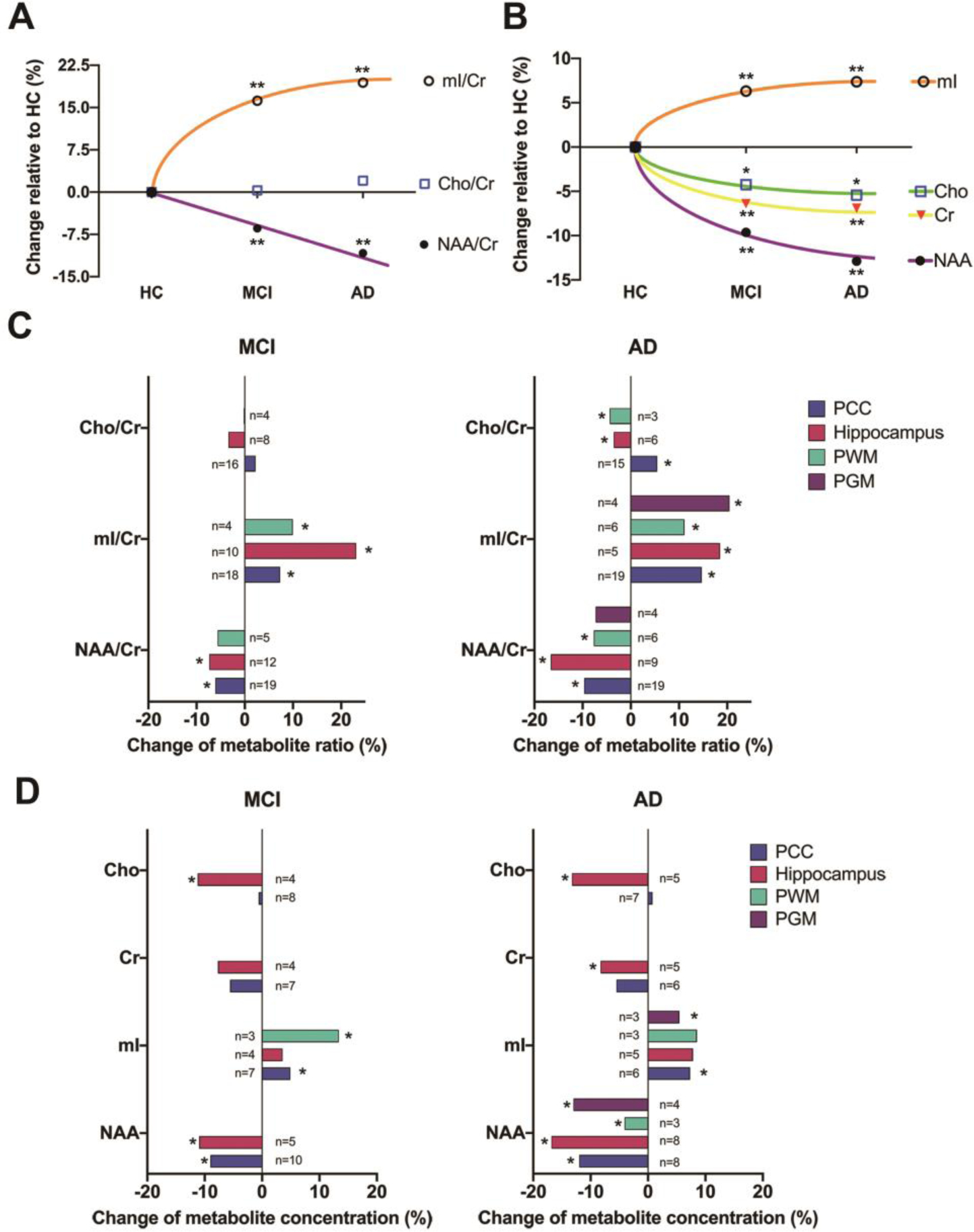
Panel (A) and Panel (B) respectively depicts the overall metabolite ratios and concentrations alterations in mild cognitive impairment (MCI) and Alzheimer’s disease (AD) compared to healthy control (HC). Panel (C) and Panel (D) depicts the metabolite ratios and concentrations, respectively, with the focus on the alterations in specific brain regions in MCI and AD compared to HC. *, p < 0.05; **, p < 0.01 (compared to HC) after leave-one-out analysis; The other abbreviations have the same meanings as noted in the main manuscript.
After excluding studies that contributed to the highest heterogeneity (see forest plots in Supplementary Materials), the results were not changed significantly apart from the following: Increased mI concentration in the PCC in MCI patients (p value changed from 0.06 to 0.002 after excluding Pilatus et al., 2009), decreased overall Cho concentrations in AD (p value changed from 0.93 to 0.04 after excluding Marjanska et al., 2019), and decreased Cr concentrations in the PCC of AD patients (p value changed to from 0.13 to 0.002 after excluding Marjanska et al., 2019).
To date, four studies have measured GSH levels in MCI and AD subjects respectively, and this precluded region-specific meta-analysis of those data. Instead, we performed an overall meta-analysis. Relative to HC subjects, lower GSH concentrations were reported in MCI and AD patients by (Mandal et al., 2015) and (Shukla et al., 2020), respectively, while a separate study reported a non-significant difference in AD (Marjanska et al., 2019). The GSH/Cr ratio was reported as either increased or no significant difference in MCI and AD patients (Duffy et al., 2014; Mullins et al., 2018; Oeltzschner et al., 2019). The overall effect sizes of GSH/Cr ratio and GSH concentrations are shown in Table 2.
Pearson correlation was performed to determine the association of alterations in metabolite concentrations and the decreases in MMSE scores in MCI/AD patients compared to HCs. A strong negative correlation (r = −0.86, p = 0.006) was observed between Hedge’s g effect size for MMSE scores and mI concentrations in the hippocampus of AD patients. A positively trending correlation (r = 0.62, p = 0.08) was also observed between Hedge’s g for NAA and MMSE scores in the hippocampus (Fig. 3). No significant correlation of metabolite concentrations with MMSE scores was observed in PCC and PWM. All of the Hedge’s g for metabolite and MMSE scores were extracted from the forest plots in the Supplementary Materials.
Figure 3. Correlations between Hedge’s g effect sizes for metabolite concentrations and Mini-Mental State Exam (MMSE) scores in the hippocampus of MCI/AD patients.
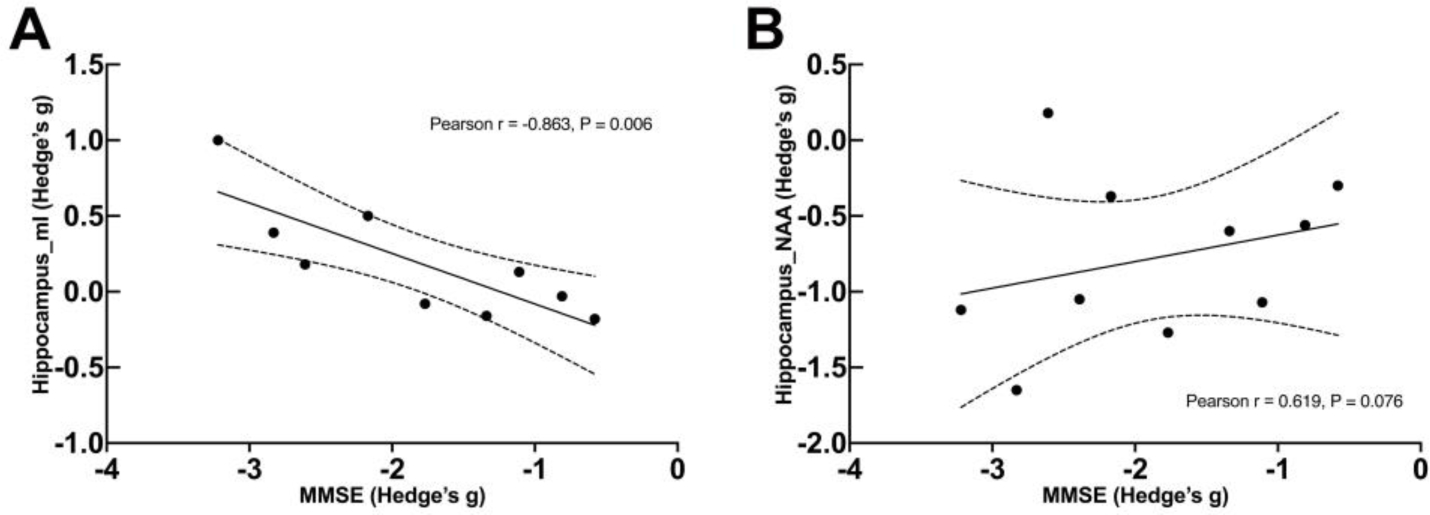
Panel (A) depicts the correlations between Hedge’s g for myo-inositol (mI) and MMSE scores. Panel (B) depicts the correlations between Hedge’s g for N-acetyl aspartate (NAA) and MMSE scores.
3.3. 31P-MRS studies in MCI and AD
Due to the small number of 31P-MRS studies, we were unable to analyze differential effects as a function of specific brain region. Moreover, only one 31P-MRS study to date has examined MCI subjects (Mandal et al., 2012), and it did not show significant differences in phosphodiester (PDE), inorganic phosphate (Pi), PCr, ATP, or phosphomonoester (PME) in the hippocampus compared to HC subjects. Regarding AD studies, a significant decrease was observed in PDE level (Effect size in hedge’s g (ES) = −1.15 [−2.23, −0.06], p = 0.04, I2 = 88%), whereas differences in PME, Pi, PCr, PCr/Pi, ATP, and pH were not significant compared to HC subjects. After accounting for heterogeneity via leave-one-out analyses, we observed significantly increased PME (ES = 0.52 [0.14, 0.90], p = 0.007, I2 =16%) and Pi (ES = 0.34 [0.01, 0.67], p = 0.050, I2 = 28%), and significantly decreased PCr/Pi (ES = −0.82 [−1.31, −0.32], p = 0.001, I2 = 0%) in AD patients. However, PDE was not significantly different (ES = −0.66 [−1.51, 0.19], p = 0.13, I2 = 80%). Forest plots were shown in the Supplementary Materials (Fig. S15).
4. Discussion
The current study aimed to characterize and quantify oxidative stress and neuroinflammation associated with bioenergetic and metabolic abnormalities in MCI and AD via meta-analysis of prior research that has utilized in vivo 1H-MRS and 31P-MRS. The results from our meta-analyses revealed several noteworthy patterns, which we interpret in greater detail below: (1) Concentrations of NAA and Cr were significantly lower in MCI and AD patients compared to HC subjects, while mI was significantly higher, and these patterns were preserved across different brain regions; (2) alterations in Cho were observed only in the hippocampus in both clinical groups; (3) significantly reduced overall GSH concentration was found in MCI but not AD patients; (4) compared to other brain regions, the hippocampus showed the most significant alterations for most of these metabolites in both MCI and AD patients (Fig. 2C and Fig. 2D), which aligns with extensive research highlighting the centrality of this region in AD disease pathophysiology and progression; and (5) after performing the leave-one-out analyses to account for heterogeneity, we noted significantly increased PME and Pi and decreased PCr/Pi in AD, which could be associated with mitochondrial dysfunction and oxidative stress.
4.1. Significance of metabolic alterations in MCI and AD
Our meta-analyses showed consistently decreased NAA/Cr and NAA concentrations in both patient groups in almost all regions assessed, which broadly aligns with previous research in MCI/AD (Wong et al., 2020) and other neuropsychiatric disorders, such as schizophrenia (Whitehurst et al., 2020). This is noteworthy given that NAA exists exclusively within neurons and plays crucial and diverse roles in the nervous system. The generation of NAA is correlated with mitochondrial function and is thought to play a role in neuroenergetics (Moffett et al., 2007). Therefore, the observed reductions in NAA and NAA/Cr in MCI/AD may reflect neuronal loss in addition to nonstructural and physiological changes associated with impaired mitochondrial activity (Bornstein et al., 2020).
The observed increase in mI concentration in MCI/AD is important given that mI has been proposed as a glial marker, is a constituent of the lipid component of biomembranes, and plays an important role in the phosphatidylinositol second messenger system (Brand et al., 1993; Kim et al., 2005; Rae, 2014). Altered cerebral mI concentrations are implicated in many neuropsychiatric disorders (Sekar et al., 2019). For example, elevated mI levels have been observed in AD, gliomatosis cerebri, diabetes melitus, systemic lupus erythematosus, multiple sclerosis, etc (Chhetri, 2019). As an indicator of the glial activation, mI is considered the most likely candidate MRS-marker of inflammation in AD (Chaney et al., 2019). Decreased NAA and increased mI have been generally reported and associated with each other in the same regions (for instance, PCC, hippocampus, and PWM, as illustrated in Fig.2) in AD, which suggests a link between increased neuroinflammation and decreased neuron viability (Chaney et al., 2019). It has been reported that reduced NAA and increased mI are associated with increased cerebrospinal fluid tau, and mI is negatively correlated with cerebrospinal fluid Aβ−42, reduction of which indicates brain Aβ amyloidosis (Piersson et al., 2020). In addition, mI levels have been shown to predict the progression to AD with a 70% sensitivity and 85% specificity (Targosz-Gajniak et al., 2013). Thus, accumulating evidence suggests that increases in mI concentration may precede decreases in NAA in AD, with increased mI potentially reflecting neuroinflammation in AD (Chaney et al., 2019; Kantarci et al., 2008a), though further investigation is needed to confirm this dynamic.
Interestingly, the current study observed variable correlations between NAA and mI levels and cognitive performance on the MMSE. This finding is not only consistent with previous research in MCI and AD (Ackl et al., 2005; Foy et al., 2011), but also in stroke, hereditary ataxias, and alcohol dependence (Krahe et al., 2020; Morley et al., 2020; Wang et al., 2017). Changes in NAA concentration are closely related to MMSE and the cognitive part of the AD Assessment Scale scores (Jessen et al., 2000). Moreover, NAA and mI are also relevant to performance in verbal memory testing (Auditory Verbal Learning Test) and general cognition (Dementia Rating Scale) (Kantarci et al., 2002). Thus, NAA and mI may be the most sensitive MRS markers to monitor AD progression and treatment response, as indicated by Fig. 2.
As with NAA, we observed reductions in Cr overall and in almost all regions in both MCI and AD patients. Cr (including PCr) is a well-known energy shuttle which also serves as an intracellular buffer for ATP by providing a ready supply of high energy phosphate through the creatine kinase reaction. Findings from rodent research suggested that Cr exerts a neuroprotective effect by buffering ATP levels (Beal, 2011). Similarly, beneficial effects of Cr supplementation have been shown in neurodegenerative and neurological diseases linked with mitochondrial dysfunction (Adhihetty and Beal, 2008; Andres et al., 2008; Marques and Wyse, 2019; Smith et al., 2014). Interestingly, the observed alterations in mI/Cr in the current study were faster/larger than that of NAA/Cr (Fig. 2A). This is noteworthy given that Cr is often used as an internal reference for in vivo MRS quantification based on the assumption that Cr is constant and stable in the altered pathophysiological conditions (Foy et al., 2011; Shukla et al., 2020). However, this notion needs should be taken with caution in light of the current meta-analysis showing decreases in Cr concentrations in both MCI and AD patients relative to healthy controls. As a consequence, the metabolite ratio values involving Cr may be differentially affected across clinical and healthy comparison groups, potentially complicating the interpretation of these ratios. Therefore, we further analyzed the absolute concentrations such as NAA, mI, and Cho (Fig. 2B) and found that NAA concentrations were largely altered more than that of mI, which is the opposite of what we observed in the NAA/Cr and mI/Cr metabolite ratios. The absolute Cho concentration was also shown to be decreased, while the Cho/Cr ratio remained unchanged (Fig. 2A, 2B). These differential results call into question the practice of normalizing metabolite levels by using Cr as an internal reference, and this should be carefully considered in future research.
The current analysis found reductions in Cho concentration in MCI/AD, though only in the hippocampus. Cho is considered as a cell membrane marker which indicates cellular proliferation (Duarte et al., 2012). Changes in Cho reflect non-steady-state alterations in membrane turnover in pathological states, such as AD, tumors, inflammation, infections, and schizophrenia (Rae, 2014). Cortical acetylcholine (ACh) is critical for the cognitive processes and ACh deficit has been reported to be a primary event in the early stage of AD and related to the cognitive symptoms of dementia (Muir, 1997). Previous research has also found that increasing ACh can enhance cholinergic functioning and subsequently improve AD patient outcomes (Akincioğlu and Gülçin, 2020; Saxena and Dubey, 2019), partially through the suppression of inflammation by administration of acetylcholinesterase inhibitors (Pollak et al., 2005). In addition, being a byproduct of ACh hydrolysis, free choline is an endogenous and selective α7 agonist for nicotinic ACh receptors (nAChR) (Alkondon et al., 1997). Targeting an α7 nAChR has been suggested as a possible strategy to reduce microglial activation (and neuroinflammation), thereby enhancing the clearance of misfolded and aggregated proteins that typify AD (Hernandez et al., 2010; Medeiros et al., 2014). High Cho dietary intake has also been associated with reduced pro-inflammatory markers in serum (Detopoulou et al., 2008). Regretfully, ACh level cannot be directly quantified by in vivo 1H-MRS, although one recent study suggested that Cho MRS had the potential to serve as a proxy of ACh function in human brains (Lindner et al., 2017). 1H-MRS detectable Cho signal are mostly from glycerophosphocholine and phosphocholine, which provide free choline for synthesizing ACh (Rae, 2014; Wang et al., 2008). Thus, our finding of decreased Cho levels in both MCI and AD may indicate increased inflammation and cell membrane changes, particularly in hippocampus. However, it is worth noting that increased Cho levels have also been reported as a non-specific marker of neuroinflammation in patients with HIV (Zahr et al., 2014). Therefore, the exact interpretation of changes in Cho signal is complicated due to the multiple metabolic pathways involved.
GSH is an important antioxidant that displays remarkable metabolic and regulatory versatility (Dwivedi et al., 2020). In the context of relatively few GSH studies in MCI/AD, the current meta-analysis observed overall reductions in GSH levels in MCI but not AD, raising the possibility that GSH reductions may play an important role in the early stages of AD before later increasing as the disease progresses. This seemingly paradoxical trajectory may have some indirect empirical support from rodent models of other neurodegenerative diseases. For example, higher brain GSH levels were found in R6/2 Huntington’s Disease mice due to GSH overproduction induced by oxidative stress (Tkac et al., 2007). Similarly, McLaughlin et al. found frontal cortex GSH increases in GT-tg bi-genic mice after short-term Tat protein exposure and induced oxidative stress (McLaughlin et al., 2017). Thus, GSH may remain near normal, or even increase slightly as indicated in the early stages of some illnesses, but then is reduced as the disease progresses. To test this hypothesis, more evidence is needed. Of note, studies measuring GSH in the brain by in vivo MRS have been controversial, with no consistent pattern reported in the limited publications that exist. These discrepancies may reflect the challenge of measuring GSH by in vivo MRS due to the limited sensitivity and spectral resolution of MRI scanners, low concentration of GSH (~ 1mM), and partially overlapped resonances with other metabolites.
For 31P studies, we couldn’t meta-analyze relevant MCI data due to the paucity of prior research but decreased PDE and PCr/Pi levels and increased PME and Pi levels were observed in the AD studies. The increased Pi and decreased PCr/Pi are consistent with the notion of abnormal energy metabolism associated with mitochondrial dysfunction and oxidative stress in AD. However, most of these studies involved small sample sizes, were performed at 1.5T, and were conducted decades ago (Forlenza et al., 2005; Gonzalez et al., 1996; Mecheri et al., 1997; Pettegrew et al., 1994; Smith et al., 1995). Thus, further research with larger samples and using more advanced techniques are needed to better characterize and understand potential alterations in 31P metabolite concentration in AD.
4.2. Hippocampus-specific metabolic alterations
The current meta-analysis found the largest and most consistent alterations in metabolite concentration in the hippocampus (Fig. 2C and Fig. 2D). This is not surprising given that the hippocampus is both critical for learning/memory and anatomically central to AD neuropathology progression. Longitudinal fMRI studies reveal that early hippocampal hyperactivation is a predictor of cognitive decline in patients with MCI and early AD (O’Brien et al., 2010). Different hippocampal sub-regions, namely the CA1 and dentate gyrus, appear particularly vulnerable to early neuritic plaque aggregation associated with altered synaptic density within the perforant pathway (Hyman et al., 1986), possibly reflecting the sensitivity of CA1 to microglial mediated neuroinflammation in response to neuritic plaque pathology (Moodley and Chan, 2014). A previous study also demonstrated hippocampal atrophy is the most robust structural MRI biomarker of AD at the prodromal stage (Risacher et al., 2009).
Of note, the current study also found a significant correlation between mI concentrations and MMSE scores in AD patients, as well as a positively trending correlation between NAA concentrations and MMSE scores, both of which were observed in the hippocampus but not in the other brain regions (Fig. 3). This finding suggests that increases in mI concentration and decreases in NAA concentration in the hippocampus are associated with declines in cognitive function. Taken together, it seems that hippocampus would be the most sensitive among all the brain regions during the progression of AD and treatment response.
4.3. Molecular mechanisms of AD suggested by metabolites’ changes
AD is a complex and multi-factorial disorder involving mitochondrial dysfunction, OS, neuroinflammation, Aβ and tau burden, etc (Fig. 4). Over 90% of all cases are first diagnosed after age 65. Earlier ages of onset are rare and are usually associated with a dominant genetic mutation of amyloid precursor protein and presenilin (PS1 and PS2), which represents only 1–5% of AD cases. However, most of the molecular AD research in cells and mice has used these mutations in the search for the cause of AD (Behl, 2017), thus, the models based on Aβ might be not adequately modeling human AD. In addition, Aβ alone appears to be insufficient for the development of AD, and changes in tau are nonspecific to AD and are seen in other neurodegenerative disorders as well as in nondemented elderly individuals (Behl, 2017). Even though Aβ proves to be correlated with AD, the results of amyloid-targeting therapies for AD have been controversial in clinical trials (Herrup, 2015; Kuehn, 2020). Aducanumab becomes the first FDA-approved anti-amyloid drug in June 2021. Aβ burden as well as tau burden relevant to the consequences of other pathological mechanisms of AD need further studies. For instance, it has been found that both OS and mitochondrial dysfunction induce aggregation of misfolded proteins, such as Aβ and tau (Lévy et al., 2019). Aβ plaques could be detected and engulfed by microglia through TAM (Tyro3, Axl, and Mer) receptors (Huang et al., 2021). Besides, microglia are capable of binding to soluble Aβ oligomers via cell surface receptors including cluster of differentiation CD36, CD14, and Toll-like receptors (TLR4, 6 and 9), etc. The binding of Aβ to CD36, TLR4 or TLR6 results in activation of microglia and further leads to the production of proinflammatory cytokines and chemokines such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α which may directly impair neuronal function (El Khoury et al., 2003; Ye et al., 2013). These findings suggest a close connection between amyloidogenesis and neuroinflammation (Webers et al., 2020).
Figure 4. A schematic view of mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic response in the progression of AD.
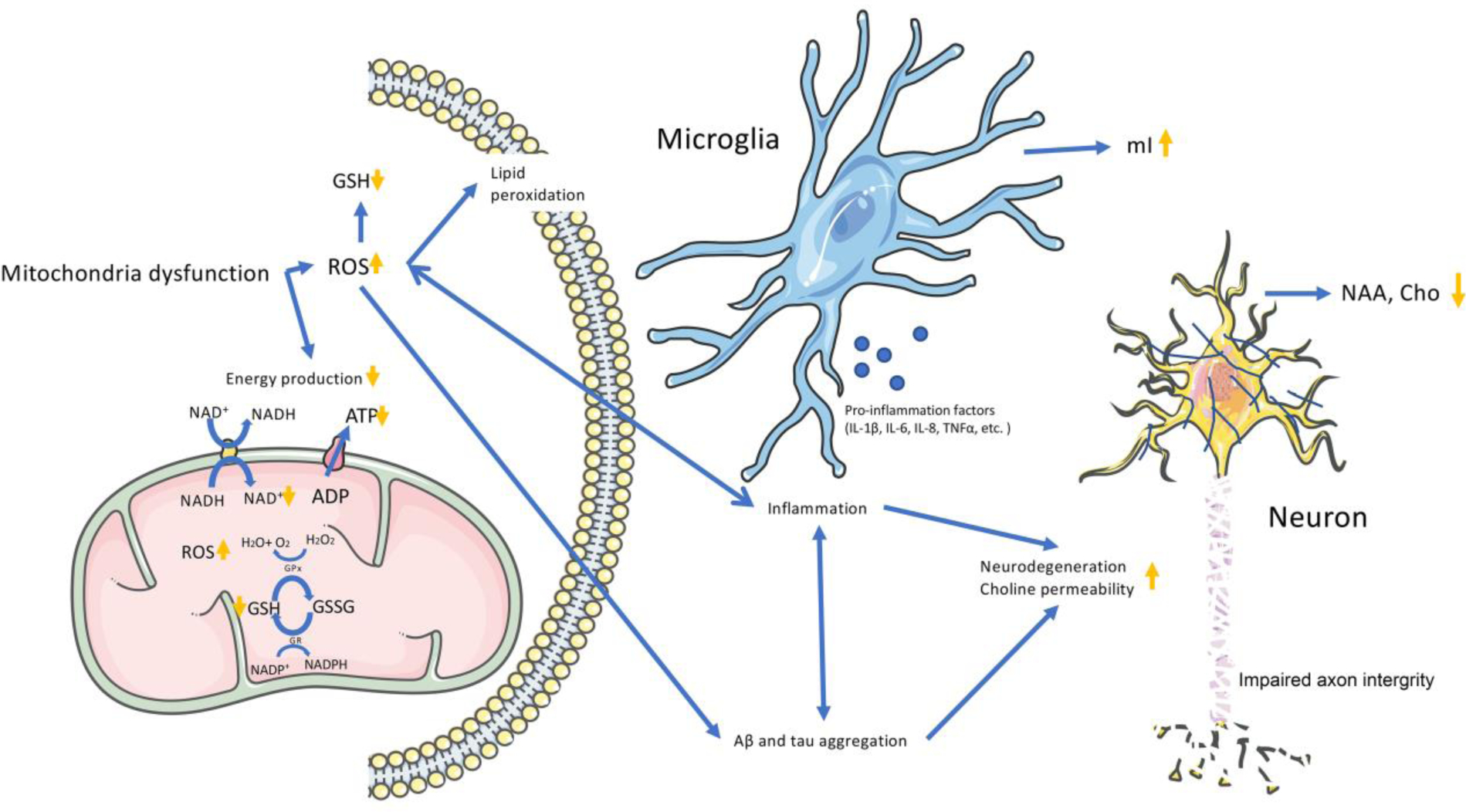
Mitochondrial dysfunction results in decreased phosphocreatine (PCr) and adenosine triphosphate (ATP), overproduction of reactive oxygen species (ROS), and occurrence of oxidative stress manifested in decreased nicotinamide adenine dinucleotide (NAD+) and glutathione (GSH) levels (Bhat et al., 2015). The overproduction of ROS would further lead to lipid peroxidation, neuroinflammation and Aβ/tau aggregation (Ikawa et al., 2020). Neuroinflammation is induced by microglia and astrocytes activation, accompanying with the increased level of mI. The neuroinflammation and/or Aβ/tau aggregation ultimately result in neural dysfunction with increased choline (Cho) permeability, then lead to decreased NAA and Cho levels, and finally aggravate the progression to AD.
Although the nature of AD pathogenesis is complex, it is known that OS plays a key role starting in early AD stages (Chang et al., 2014). Under physiological conditions, there is a balance between oxidant and antioxidant species. OS occurs when the level of oxidants present exceeds the levels of antioxidants present (Dalle-Donne et al., 2006). States of intensive energy metabolism and inflammation lead to the production of ROS, which can have toxic effects that damage proteins, lipids, and DNA, as well as result in neuron death (Bermejo et al., 2008; Kuka et al., 2013). ROS are eliminated by the free radical scavenger GSH in the brain (Cabungcal et al., 2006; Dringen, 2000), thus alteration in GSH metabolism may have profound effects on neurons.
OS, neuroinflammation, and mitochondrial dysfunction are intertwined in the pathology of AD and could act as a consequence of each other (Fig. 4). Thus, it’s hard to tell which is the primary upstream cause in the progression of AD. The mitochondrial cascade hypothesis indicates that mitochondrial dysfunction is considered a primary and early event in the pathological cascade of AD (Swerdlow, 2020; Swerdlow et al., 2010). Mitochondrial dysfunction has also been observed prior to amyloid plaque deposition (Gillardon et al., 2007). Evidence suggests that mitochondrial dysfunction could result in overproduction of ROS/reactive nitrogen species leading to OS (Bhat et al., 2015; Calabrese et al., 2005). In AD, mitochondrial abnormalities are considered the main source of OS (Perry et al., 2002). Besides mitochondrial dysfunction, there are other possible causes of oxidative stress in the brain, e.g., neuroinflammation, protein aggregation, and decreased antioxidant defenses (Simpson and Oliver, 2020). Previous PET studies also indicated that mitochondrial dysfunction has shown in the early stage of AD, and this mitochondrial-related energy failure may precede glycolysis-related hypometabolism in regions with pathologically confirmed early neurodegeneration in AD, e.g. hippocampus (Terada et al., 2020). On the other hand, mitochondrial dysfunction in microglial cells has been found to inhibit portions of the IL-4-induced alternative response, which is associated with a reduction of inflammation (Ferger et al., 2010). Notably, besides the Aβ pathway, microglia activated by OS also releases proinflammatory cytokines such as IL-1β, IL-6, IL-8, and TNFα, and up-regulates the expression of chemokines such as C-C motif ligand (CCL)-2, chemokine receptors CCR3 and CCR5, resulting in local inflammatory responses, causing the death of neural cells (Heneka et al., 2015; Xia et al., 1998). Taken together, a schematic framework of hypothesized mechanism of mitochondrial dysfunction, oxidative stress, and neuroinflammation with the altered metabolites in AD was proposed in Fig. 4, which illustrated that the ROS formation, enhanced activation of microglia and other immune cells, and expression of cytokines are associated with neuroinflammation in AD (Kinney et al., 2018). OS and neuroinflammation are inextricably linked, neuroinflammation leads to increased OS which in turn causes subsequent inflammation (Fischer and Maier, 2015), and antioxidants protect neurons by reducing OS and chronic inflammation in AD (Prasad, 2017). In that sense, early intervention of AD could target mitochondrial dysfunction, OS, and neuroinflammation. And enhancing brain bioenergetics and antioxidant supplementation could be potential ways to treat AD at an early stage (Perez Ortiz and Swerdlow, 2019; Petrovic et al., 2020; Viña et al., 2011). For instance, raising brain nicotinamide adenine dinucleotide (NAD) levels has recently aroused intense scholarly attention as a potential treatment intervention (Hou et al., 2018; Hikosaka et al., 2021; Wang et al., 2021; Rajman et al., 2018; Trammell et al., 2016), which can also reduce neuroinflammation in a mouse model of AD (Hou et al., 2021).
5. Limitations
Several limitations of our meta-analysis should be mentioned. Firstly, the heterogeneity we observed between studies may relate to factors such as age, illness duration, diagnosis, symptom severity, and medication exposure, which may vary between cohorts. Secondly, data quality is another concern. Cramér–Rao Lower Bounds values, which are used to estimate metabolite goodness of fit, were not provided for most of the studies. Therefore, we cannot fully evaluate how MRS data quality affected the results of our meta-analyses. Thirdly, the number of GSH and 31P studies were too small to permit region-specific analyses. Fourthly, the etiology of MCI patients and the disease stage amongst AD patients were also not considered in this review due to insufficient information provided in the published MRS studies with MCI and AD patients. Lastly, MRI platforms, MRS sequences, and quantification methods (e.g., correcting partial volume effect of gray/white matter and cerebrospinal fluid) differed between studies and could not be corrected due to insufficient details provided.
6. Conclusion
In conclusion, the present meta-analysis indicates a similar pattern of metabolic alterations in MCI and AD, albeit with increased severity in AD. These results (particularly in Fig. 2) support the hypothesis (Fig.3) that neuroinflammation and oxidative stress associated with mitochondrial dysfunction play a critical role in AD etiopathogenesis and pathophysiology (Chaney et al., 2019; Gadhave et al., 2020; Tobore, 2019), and eventually lead to decreased neuronal function, which is implied by the changes in in vivo MRS markers of increased mI and decreased NAA, GSH, and PCr/Pi. Among all the MRS markers, NAA and mI seem most sensitive in detecting the progression of AD. The hippocampus is the most sensitive during AD progression, suggesting it could be an important brain region for early diagnosis and prevention. Lastly, targeting abnormal bioenergetic processes associated with neuroinflammation/oxidative stress could be a promising approach for earlier intervention in AD.
Supplementary Material
Highlights:
Neuroinflammation and oxidative stress (OS) may occur in the early stages of AD;
Neuroinflammation and OS likely precede neuronal loss in AD;
The hippocampus is the most vulnerable brain region in the progression of AD.
Funding
This work was supported by the National Institute of Health [grant numbers R01MH114982, R01AG066670, R01MH095809, P50MH115846, K24MH104449].
Abbreviations:
- Aβ
amyloid beta
- ACC
anterior cingulate cortex
- ACh
acetylcholine
- AD
Alzheimer’s disease
- ATP
adenosine triphosphate
- Cho
choline-containing compounds
- Cr
creatine
- ETC
electron transport chain
- fMRI
function magnetic resonance imaging
- GSH
glutathione
- GSSG
oxidized glutathione
- HC
healthy control
- IL
interleukin
- MCI
mild cognitive impairment
- mI
myo-inositol
- MMSE
Mini-Mental State Exam
- MRS
magnetic resonance spectroscopy
- MTL
medial temporal lobe
- NAA
N-acetyl aspartate
- nAChR
nicotinic ACh receptors
- NAD
nicotinamide adenine dinucleotide
- PCC
posterior cingulate cortex
- PCr
phosphocreatine
- PDE
phosphodiester
- PET
positron emission topography
- PGM
parietal gray matter
- Pi
inorganic phosphate
- PME
phosphomonoester
- PWM
parietal white matter
- OS
oxidative stress
- ROS
reactive oxygen species
- TCA
tricarboxylic acid
- TLR
Toll-like receptors
- TNF
tumor necrosis factor
Footnotes
Declaration of interest
Declarations of interests: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackl N, Ising M, Schreiber YA, Atiya M, Sonntag A, Auer DP, 2005. Hippocampal metabolic abnormalities in mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 384, 23–28. 10.1016/j.neulet.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Adhihetty PJ, Beal MF, 2008. Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromol. Med. 10, 275–290. 10.1007/s12017-008-8053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akincioğlu H, Gülçin İ, 2020. Potent acetylcholinesterase inhibitors: potential drugs for Alzheimer’s disease. Mini Rev. Med. Chem. 20, 703–715. 10.2174/1389557520666200103100521. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX, 1997. Choline is a selective agonist of α7 nicotinic acetylcholine receptors in the rat brain neurons. Eur. J. Neurosci. 9, 2734–2742. 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association, 2021. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 10.1002/alz.12328. [DOI] [PubMed] [Google Scholar]
- Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR, 2008. Functions and effects of creatine in the central nervous system. Brain Res. Bull. 76, 329–343. 10.1016/j.brainresbull.2008.02.035. [DOI] [PubMed] [Google Scholar]
- Bains JS, Shaw CA, 1997. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Rev. 25, 335–358. 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- Beal MF, 2011. Neuroprotective effects of creatine. Amino Acids 40, 1305–1313. 10.1007/s00726-011-0851-0. [DOI] [PubMed] [Google Scholar]
- Behl C, 2017. Amyloid in Alzheimer’s disease: guilty beyond reasonable doubt? Trends Pharmacol. Sci. 38, 849–851. 10.1016/j.tips.2017.07.002. [DOI] [PubMed] [Google Scholar]
- Bermejo P, Martin-Aragon S, Benedi J, Susin C, Felici E, Gil P, Ribera JM, Villar AM, 2008. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 42, 162–170. 10.1080/10715760701861373. [DOI] [PubMed] [Google Scholar]
- Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, Ganie SA, 2015. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 74, 101–110. 10.1016/j.biopha.2015.07.025. [DOI] [PubMed] [Google Scholar]
- Block W, Jessen F, Träber F, Flacke S, Manka C, Lamerichs R, Keller E, Heun R, et al. , 2002. Regional N-acetylaspartate reduction in the hippocampus detected with fast proton magnetic resonance spectroscopic imaging in patients with Alzheimer disease. Arch. Neurol. 59, 828–834. 10.1001/archneur.59.5.828. [DOI] [PubMed] [Google Scholar]
- Bonkowski MS, Sinclair DA, 2016. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 17, 679–690. 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein R, Gonzalez B, Johnson SC, 2020. Mitochondrial pathways in human health and aging. Mitochondrion 54, 72–84. 10.1016/j.mito.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley PA, Cousins JP, Pendrey DL, Wagle WA, Hardy CJ, Eames FA, McCaffrey RJ, Thompson DA, 1992. Alzheimer dementia: quantification of energy metabolism and mobile phosphoesters with P-31 NMR spectroscopy. Radiology 183, 695–699. 10.1148/radiology.183.3.1584923. [DOI] [PubMed] [Google Scholar]
- Bradburn S, Murgatroyd C, Ray N, 2019. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a meta-analysis. Ageing Res. Rev. 50, 1–8. 10.1016/j.arr.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Brand A, Richter-Landsberg C, Leibfritz D, 1993. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev. Neurosci. 15, 289–298. 10.1159/000111347. [DOI] [PubMed] [Google Scholar]
- Brown GG, Levine SR, Gorell JM, Pettegrew JW, Gdowski JW, Bueri JA, Helpern JA, Welch KMA, 1989. In vivo 31P NMR profiles of Alzheimer’s disease and multiple subcortical infarct dementia. Neurology 39, 1423–1427. 10.1212/wnl.39.11.1423. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Halliwell B, 2019. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabungcal JH, Nicolas D, Kraftsik R, Cuenod M, Do KQ, Hornung JP, 2006. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: relevance to schizophrenia. Neurobiol. Dis. 22, 624–637. 10.1016/j.nbd.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R, 2014. Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front. Cell. Neurosci. 8, 430. 10.3389/fncel.2014.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, et al. , 2005. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 233, 145–162. 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Catani M, Cherubini A, Howard R, Tarducci R, Pelliccioli GP, Piccirilli M, Gobbi G, Senin U, et al. , 2001. 1H-MR spectroscopy differentiates mild cognitive impairment from normal brain aging. Neuroreport 12, 2315–2317. 10.1097/00001756-200108080-00007. [DOI] [PubMed] [Google Scholar]
- Chaney A, Williams SR, Boutin H, 2019. In vivo molecular imaging of neuroinflammation in Alzheimer’s disease. J. Neurochem. 149, 438–451. 10.1111/jnc.14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YT, Chang WN, Tsai NW, Huang CC, Kung CT, Su YJ, Lin WC, Cheng BC, et al. , 2014. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer’s disease: a systematic review. Biomed. Res. Int. 2014, 182303. h 10.1155/2014/182303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantal S, Labelle M, Bouchard RW, Braun CM, Boulanger Y, 2002. Correlation of regional proton magnetic resonance spectroscopic metabolic changes with cognitive deficits in mild Alzheimer disease. Arch. Neurol. 59, 955–962. 10.1001/archneur.59.6.955. [DOI] [PubMed] [Google Scholar]
- Chao LL, Mueller SG, Buckley ST, Peek K, Raptentsetseng S, Elman J, Yaffe K, Miller BL, et al. , 2010. Evidence of neurodegeneration in brains of older adults who do not yet fulfill MCI criteria. Neurobiol. Aging 31, 368–377. 10.1016/j.neurobiolaging.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LL, Schuff N, Kramer JH, Du AT, Capizzano AA, O’Neill J, Wolkowitz OM, Jagust WJ, et al. , 2005. Reduced medial temporal lobe N-acetylaspartate in cognitively impaired but nondemented patients. Neurology 64, 282–289. 10.1212/01.Wnl.0000149638.45635.Ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SQ, Cai Q, Shen YY, Xu CX, Zhou H, Zhao Z, 2016. Hydrogen proton magnetic resonance spectroscopy in multidomain amnestic mild cognitive impairment and vascular cognitive impairment without dementia. Am. J. Alzheimers Dis. Other Dement. 31, 422–429. 10.1177/1533317515628052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung MW-L, 2015. Computing effect sizes for meta-analysis, in: Meta-Analysis. John Wiley & Sons, Ltd, Chichester, UK, pp. 48–80. [Google Scholar]
- Chhetri DR, 2019. Myo-inositol and its derivatives: their emerging role in the treatment of human diseases. Front. Pharmacol. 10, 1172. 10.3389/fphar.2019.01172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane Collaboration, 2014. Review Manager (RevMan) (Version 5.3). Copenhagen: The Nordic Cochrane Centre. [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A, 2006. Biomarkers of oxidative damage in human disease. Clin. Chem. 52, 601–623. 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- Detopoulou P, Panagiotakos DB, Antonopoulou S, Pitsavos C, Stefanadis C, 2008. Dietary choline and betaine intakes in relation to concentrations of inflammatory markers in healthy adults: the ATTICA study. Am. J. Clin. Nutr. 87, 424–430. 10.1093/ajcn/87.2.424. [DOI] [PubMed] [Google Scholar]
- Ding B, Chen KM, Ling HW, Zhang H, Chai WM, Li X, Wang T, 2008. Diffusion tensor imaging correlates with proton magnetic resonance spectroscopy in posterior cingulate region of patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 25, 218–225. 10.1159/000113948. [DOI] [PubMed] [Google Scholar]
- Dringen R, 2000. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 62, 649–671. 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Duarte JM, Lei H, Mlynárik V, Gruetter R, 2012. The neurochemical profile quantified by in vivo 1H NMR spectroscopy. NeuroImage 61, 342–362. 10.1016/j.neuroimage.2011.12.038. [DOI] [PubMed] [Google Scholar]
- Duffy SL, Lagopoulos J, Hickie IB, Diamond K, Graeber MB, Lewis SJ, Naismith SL, 2014. Glutathione relates to neuropsychological functioning in mild cognitive impairment. Alzheimers Dement. 10, 67–75. 10.1016/j.jalz.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Dwivedi D, Megha K, Mishra R, Mandal PK, 2020. Glutathione in brain: overview of its conformations, functions, biochemical characteristics, quantitation and potential therapeutic role in brain disorders. Neurochem. Res. 45, 1461–1480. 10.1007/s11064-020-03030-1. [DOI] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD, 2003. CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 197, 1657–1666. 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayed N, Andres E, Viguera L, Modrego PJ, Garcia-Campayo J, 2014. Higher glutamate+glutamine and reduction of N-acetylaspartate in posterior cingulate according to age range in patients with cognitive impairment and/or pain. Acad. Radiol. 21, 1211–1217. 10.1016/j.acra.2014.04.009. [DOI] [PubMed] [Google Scholar]
- Ferger AI, Campanelli L, Reimer V, Muth KN, Merdian I, Ludolph AC, Witting A, 2010. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 7, 45. 10.1186/1742-2094-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R, Maier O, 2015. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid. Med. Cell. Longev. 2015, 610813. 10.1155/2015/610813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlenza OV, Wacker P, Nunes PV, Yacubian J, Castro CC, Otaduy MC, Gattaz WF, 2005. Reduced phospholipid breakdown in Alzheimer’s brains: a 31P spectroscopy study. Psychopharmacology 180, 359–365. 10.1007/s00213-005-2168-8. [DOI] [PubMed] [Google Scholar]
- Foy CM, Daly EM, Glover A, O’Gorman R, Simmons A, Murphy DG, Lovestone S, 2011. Hippocampal proton MR spectroscopy in early Alzheimer’s disease and mild cognitive impairment. Brain Topogr. 24, 316–322. 10.1007/s10548-011-0170-5. [DOI] [PubMed] [Google Scholar]
- Franczak M, Prost RW, Antuono PG, Mark LP, Jones JL, Ulmer JL, 2007. Proton magnetic resonance spectroscopy of the hippocampus in patients with mild cognitive impairment: a pilot study. J. Comput. Assist. Tomogr. 31, 666–670. 10.1097/RCT.0b013e318031bc31. [DOI] [PubMed] [Google Scholar]
- Gadhave K, Kumar D, Uversky VN, Giri R, 2020. A multitude of signaling pathways associated with Alzheimer’s disease and their roles in AD pathogenesis and therapy. Med. Res. Rev. 10.1002/med.21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Santos JM, Gavrila D, Antunez C, Tormo MJ, Salmeron D, Carles R, Jimenez Veiga J, Parrilla G, et al. , 2008. Magnetic resonance spectroscopy performance for detection of dementia, Alzheimer’s disease and mild cognitive impairment in a community-based survey. Dement. Geriatr. Cogn. Disord 26, 15–25. 10.1159/000140624. [DOI] [PubMed] [Google Scholar]
- Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, Belleville S, Brodaty H, et al. , 2006. Mild cognitive impairment. Lancet 367, 1262–1270. 10.1016/s0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Shi Q, 2010. A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J. Alzheimers Dis. 20 Suppl 2, S591–607. 10.3233/jad-2010-100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K, Kraut N, Hengerer B, 2007. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 7, 605–616. 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- Gonzalez RG, Guimaraes AR, Moore GJ, Crawley A, Cupples LA, Growdon JH, 1996. Quantitative in vivo 31P magnetic resonance spectroscopy of Alzheimer disease. Alzheimer Dis. Assoc. Disord 10, 46–52. [PubMed] [Google Scholar]
- Gordon ML, Kingsley PB, Goldberg TE, Koppel J, Christen E, Keehlisen L, Kohn N, Davies P, 2012. An open-label exploratory study with memantine: correlation between proton magnetic resonance spectroscopy and cognition in patients with mild to moderate Alzheimer’s disease. Dement. Geriatr. Cogn. Dis. Extra. 2, 312–320. 10.1159/000341604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford J, Boeve BF, Murray ME, Ferman TJ, Tosakulwong N, Lesnick TG, Maroney-Smith M, Senjem ML, et al. , 2014. Regional proton magnetic resonance spectroscopy patterns in dementia with Lewy bodies. Neurobiol. Aging 35, 1483–1490. 10.1016/j.neurobiolaging.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith HR, den Hollander JA, Stewart CC, Evanochko WT, Buchthal SD, Harrell LE, Zamrini EY, Brockington JC, et al. , 2007. Elevated brain scyllo-inositol concentrations in patients with Alzheimer’s disease. NMR Biomed. 20, 709–716. 10.1002/nbm.1132. [DOI] [PubMed] [Google Scholar]
- Griffith HR, Okonkwo OC, den Hollander JA, Belue K, Copeland J, Harrell LE, Brockington JC, Clark DG, et al. , 2010. Brain metabolic correlates of decision making in amnestic mild cognitive impairment. Aging Neuropsychol. Cogn. 17, 492–504. 10.1080/13825581003646135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Liu X, Hou H, Wei F, Chen X, Shen Y, Chen W, 2016. 1H-MRS asymmetry changes in the anterior and posterior cingulate gyrus in patients with mild cognitive impairment and mild Alzheimer’s disease. Compr. Psychiatry 69, 179–185. 10.1016/j.comppsych.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Hattori N, Abe K, Sakoda S, Sawada T, 2002. Proton MR spectroscopic study at 3 Tesla on glutamate/glutamine in Alzheimer’s disease. Neuroreport 13, 183–186. 10.1097/00001756-200201210-00041. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, Evans DA, 2013. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80, 1778–1783. 10.1212/wnl.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, et al. , 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. 10.1016/s1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herminghaus S, Frölich L, Gorriz C, Pilatus U, Dierks T, Wittsack H-J, Lanfermann H, Maurer K, et al. , 2003. Brain metabolism in Alzheimer disease and vascular dementia assessed by in vivo proton magnetic resonance spectroscopy. Psychiat. Res. Neuroimaging 123, 183–190. 10.1016/s0925-4927(03)00071-4. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT, 2010. Loss of α7 nicotinic receptors enhances β-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 30, 2442–2453. 10.1523/jneurosci.5038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K, 2015. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 18, 794–799. 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- Hikosaka K, Yaku K, Okabe K, Nakagawa T, 2021. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr. Neurosci. 24, 371–383. 10.1080/1028415x.2019.1637504. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, et al. , 2001. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 21, 3017–3023. 10.1523/jneurosci.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Lautrup S, Cordonnier S, Wang Y, Croteau DL, Zavala E, Zhang Y, Moritoh K, et al. , 2018. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U. S. A. 115, E1876–e1885. 10.1073/pnas.1718819115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, Mattson MP, Croteau DL, et al. , 2021. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. U. S. A. 118, e2011226118. 10.1073/pnas.2011226118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Happonen KE, Burrola PG, O’Connor C, Hah N, Huang L, Nimmerjahn A, Lemke G, 2021. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 22, 586–594. 10.1038/s41590-021-00913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR, 1986. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann. Neurol. 20, 472–481. 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- Ikawa M, Okazawa H, Nakamoto Y, Yoneda M, 2020. PET imaging for oxidative stress in neurodegenerative disorders associated with mitochondrial dysfunction. Antioxidants 9. 10.3390/antiox9090861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L, 2014. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471. 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen F, Block W, Träber F, Keller E, Flacke S, Papassotiropoulos A, Lamerichs R, Heun R, et al. , 2000. Proton MR spectroscopy detects a relative decrease of N-acetylaspartate in the medial temporal lobe of patients with AD. Neurology 55, 684–688. 10.1212/wnl.55.5.684. [DOI] [PubMed] [Google Scholar]
- Jessen F, Gür O, Block W, Ende G, Frölich L, Hammen T, Wiltfang J, Kucinski T, et al. , 2009. A multicenter 1H-MRS study of the medial temporal lobe in AD and MCI. Neurology 72, 1735–1740. 10.1212/WNL.0b013e3181a60a20. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Knopman DS, Dickson DW, Parisi JE, Whitwell JL, Weigand SD, Josephs KA, Boeve BF, et al. , 2008a. Alzheimer disease: postmortem neuropathologic correlates of antemortem 1H MR spectroscopy metabolite measurements. Radiology 248, 210–220. 10.1148/radiol.2481071590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Petersen RC, Boeve BF, Knopman DS, Tang-Wai DF, O’Brien PC, Weigand SD, Edland SD, et al. , 2004. 1H MR spectroscopy in common dementias. Neurology 63, 1393–1398. 10.1212/01.wnl.0000141849.21256.ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Petersen RC, Przybelski SA, Weigand SD, Shiung MM, Whitwell JL, Negash S, Ivnik RJ, et al. , 2008b. Hippocampal volumes, proton magnetic resonance spectroscopy metabolites, and cerebrovascular disease in mild cognitive impairment subtypes. Arch. Neurol. 65, 1621–1628. 10.1001/archneur.65.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Smith GE, Ivnik RJ, Petersen RC, Boeve BF, Knopman DS, Tangalos EG, Jack CR Jr., 2002. 1H magnetic resonance spectroscopy, cognitive function, and apolipoprotein E genotype in normal aging, mild cognitive impairment and Alzheimer’s disease. J. Int. Neuropsychol. Soc 8, 934–942. 10.1017/s1355617702870084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Weigand SD, Petersen RC, Boeve BF, Knopman DS, Gunter J, Reyes D, Shiung M, et al. , 2007. Longitudinal 1H MRS changes in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 28, 1330–1339. 10.1016/j.neurobiolaging.2006.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, McGrath BM, Silverstone PH, 2005. A review of the possible relevance of inositol and the phosphatidylinositol second messenger system (PI-cycle) to psychiatric disorders—focus on magnetic resonance spectroscopy (MRS) studies. Hum. Psychopharmacol 20, 309–326. 10.1002/hup.693. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT, 2018. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590. 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahe J, Binkofski F, Schulz JB, Reetz K, Romanzetti S, 2020. Neurochemical profiles in hereditary ataxias: a meta-analysis of magnetic resonance spectroscopy studies. Neurosci. Biobehav. Rev. 108, 854–865. 10.1016/j.neubiorev.2019.12.019. [DOI] [PubMed] [Google Scholar]
- Kuehn BM, 2020. In Alzheimer research, glucose metabolism moves to center stage. JAMA 323, 297–299. 10.1001/jama.2019.20939. [DOI] [PubMed] [Google Scholar]
- Kuka S, Tatarkova Z, Racay P, Lehotsky J, Dobrota D, Kaplan P, 2013. Effect of aging on formation of reactive oxygen species by mitochondria of rat heart. Gen. Physiol. Biophys. 32, 415–420. 10.4149/gpb_2013049. [DOI] [PubMed] [Google Scholar]
- Lévy E, El Banna N, Baïlle D, Heneman-Masurel A, Truchet S, Rezaei H, Huang ME, Béringue V, et al. , 2019. Causative links between protein aggregation and oxidative stress: a review. Int. J. Mol. Sci 20. 10.3390/ijms20163896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim TS, Hong YH, Lee HY, Choi JY, Kim HS, Moon SY, 2012. Metabolite investigation in both anterior and posterior cingulate gyri in Alzheimer’s disease spectrum using 3-tesla MR spectroscopy. Dement. Geriatr. Cogn. Disord. 33, 149–155. 10.1159/000338177. [DOI] [PubMed] [Google Scholar]
- Lindner M, Bell T, Iqbal S, Mullins PG, Christakou A, 2017. In vivo functional neurochemistry of human cortical cholinergic function during visuospatial attention. PLoS One. 12(2):e0171338. 10.1371/journal.pone.0171338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Akolkar H, Tripathi M, 2012. Mapping of hippocampal pH and neurochemicals from in vivo multi-voxel 31P study in healthy normal young male/female, mild cognitive impairment, and Alzheimer’s disease. J. Alzheimers Dis. 31 Suppl 3, S75–86. 10.3233/JAD-2012-120166. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Saharan S, Tripathi M, Murari G, 2015. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 78, 702–710. 10.1016/j.biopsych.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Marjanska M, McCarten JR, Hodges JS, Hemmy LS, Terpstra M, 2019. Distinctive neurochemistry in Alzheimer’s disease via 7 T in vivo magnetic resonance spectroscopy. J. Alzheimers Dis. 68, 559–569. 10.3233/JAD-180861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques EP, Wyse ATS, 2019. Creatine as a neuroprotector: an actor that can play many parts. Neurotox. Res. 36, 411–423. 10.1007/s12640-019-00053-7. [DOI] [PubMed] [Google Scholar]
- Maurer I, Zierz S, Möller HJ, 2000. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462. 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Paris JJ, Mintzopoulos D, Hymel KA, Kim JK, Cirino TJ, Gillis TE, Eans SO, et al. , 2017. Conditional human immunodeficiency virus transactivator of transcription protein expression induces depression-like effects and oxidative stress. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2, 599–609. 10.1016/j.bpsc.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecheri G, Marie-Cardine M, Sappey-Marinier D, Bonmartin H, Albrand G, Ferry G, Coppard-Meyer N, Courpron P, 1997. In vivo hippocampal 31P NMR metabolites in Alzheimer’s disease and ageing. Eur. Psychiatry 12, 140–148. 10.1016/s0924-9338(97)80203-9. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Castello NA, Cheng D, Kitazawa M, Baglietto-Vargas D, Green KN, Esbenshade TA, Bitner RS, et al. , 2014. α7 Nicotinic receptor agonist enhances cognition in aged 3xTg-AD mice with robust plaques and tangles. Am. J. Pathol. 184, 520–529. 10.1016/j.ajpath.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Metastasio A, Rinaldi P, Tarducci R, Mariani E, Feliziani FT, Cherubini A, Pelliccioli GP, Gobbi G, et al. , 2006. Conversion of MCI to dementia: role of proton magnetic resonance spectroscopy. Neurobiol. Aging 27, 926–932. 10.1016/j.neurobiolaging.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Modrego PJ, Fayed N, Sarasa M, 2011. Magnetic resonance spectroscopy in the prediction of early conversion from amnestic mild cognitive impairment to dementia: a prospective cohort study. BMJ Open 1, e000007. 10.1136/bmjopen-2010-000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM, 2007. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog. Neurobiol. 81, 89–131. 10.1016/j.pneurobio.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, 2009. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Br. Med. J. 339, 332–336. 10.1136/bmj.b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley KK, Chan D, 2014. The hippocampus in neurodegenerative disease. Front. Neurol. Neurosci. 34, 95–108. 10.1159/000356430. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, et al. , 2007. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 3, 614–615. 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- Morley KC, Lagopoulos J, Logge W, Chitty K, Moustafa AA, Haber PS, 2020. Brain N-acetyl aspartate and associations with cognitive impairment in alcohol dependent patients. J. Clin. Exp. Neuropsychol. 42, 111–117. 10.1080/13803395.2019.1685078. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Brown RE, 2006. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123. 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir JL, 1997. Acetylcholine, aging, and Alzheimer’s disease. Pharmacol. Biochem. Behav. 56, 687–696. 10.1016/s0091-3057(96)00431-5. [DOI] [PubMed] [Google Scholar]
- Mullins R, Reiter D, Kapogiannis D, 2018. Magnetic resonance spectroscopy reveals abnormalities of glucose metabolism in the Alzheimer’s brain. Ann. Clin. Transl. Neurol. 5, 262–272. 10.1002/acn3.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J, Tsui WH, Li Y, McHugh P, Williams S, Cummings M, Pirraglia E, Solnes L, et al. , 2014. FDG and amyloid PET in cognitively normal individuals at risk for late-onset Alzheimer’s disease. Adv. J. Mol. Imaging 4, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC, Sperling RA, 2010. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology 74, 1969–1976. 10.1212/WNL.0b013e3181e3966e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeltzschner G, Wijtenburg SA, Mikkelsen M, Edden RAE, Barker PB, Joo JH, Leoutsakos JS, Rowland LM, et al. , 2019. Neurometabolites and associations with cognitive deficits in mild cognitive impairment: a magnetic resonance spectroscopy study at 7 Tesla. Neurobiol. Aging 73, 211–218. 10.1016/j.neurobiolaging.2018.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson BL, Holshouser BA, Britt W 3rd, Mueller C, Baqai W, Patra S, Petersen F, Kirsch WM, 2008. Longitudinal metabolic and cognitive changes in mild cognitive impairment patients. Alzheimer Dis. Assoc. Disord. 22, 269–277. 10.1097/WAD.0b013e3181750a65. [DOI] [PubMed] [Google Scholar]
- Onyango IG, 2018. Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease. Neural Regen. Res. 13, 19–25. 10.4103/1673-5374.224362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango IG, Dennis J, Khan SM, 2016. Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies. Aging Dis. 7, 201–214. 10.14336/ad.2015.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Choi H, Min JS, Park SJ, Kim JH, Park HJ, Kim B, Chae JI, et al. , 2013. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 127, 221–232. 10.1111/jnc.12361. [DOI] [PubMed] [Google Scholar]
- Perez Ortiz JM, Swerdlow RH, 2019. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 176, 3489–3507. 10.1111/bph.14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Cash AD, Smith MA, 2002. Alzheimer disease and oxidative stress. J. Biomed. Biotechnol. 2, 120–123. 10.1155/s1110724302203010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic S, Arsic A, Ristic-Medic D, Cvetkovic Z, Vucic V, 2020. Lipid peroxidation and antioxidant supplementation in neurodegenerative diseases: A review of human studies. Antioxidants 9. 10.3390/antiox9111128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR, 1994. Alterations of cerebral metabolism in probable Alzheimer’s disease: a preliminary study. Neurobiol. Aging 15, 117–132. 10.1016/0197-4580(94)90152-x. [DOI] [PubMed] [Google Scholar]
- Piersson AD, Mohamad M, Rajab F, Suppiah S, 2020. Cerebrospinal fluid amyloid beta, Tau levels, apolipoprotein, and 1H-MRS brain metabolites in Alzheimer’s disease: a systematic review. Acad. Radiol. 10.1016/j.acra.2020.06.006. [DOI] [PubMed] [Google Scholar]
- Pilatus U, Lais C, Rochmont Adu M, Kratzsch T, Frolich L, Maurer K, Zanella FE, Lanfermann H, et al. , 2009. Conversion to dementia in mild cognitive impairment is associated with decline of N-actylaspartate and creatine as revealed by magnetic resonance spectroscopy. Psychiatry Res. 173, 1–7. 10.1016/j.pscychresns.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Pocernich CB, Butterfield DA, 2012. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta. 1822, 625–630. 10.1016/j.bbadis.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak Y, Gilboa A, Ben-Menachem O, Ben-Hur T, Soreq H, Yirmiya R, 2005. Acetylcholinesterase inhibitors reduce brain and blood interleukin-1β production. Ann. Neurol. 57, 741–745. 10.1002/ana.20454. [DOI] [PubMed] [Google Scholar]
- Prasad KN, 2017. Oxidative stress and pro-inflammatory cytokines may act as one of the signals for regulating microRNAs expression in Alzheimer’s disease. Mech. Ageing Dev. 162, 63–71. 10.1016/j.mad.2016.12.003. [DOI] [PubMed] [Google Scholar]
- Rae CD, 2014. A guide to the metabolic pathways and function of metabolites observed in human brain 1H magnetic resonance spectra. Neurochem. Res. 39, 1–36. 10.1007/s11064-013-1199-5. [DOI] [PubMed] [Google Scholar]
- Rajman L, Chwalek K, Sinclair DA, 2018. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 27, 529–547. 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami L, Gomez-Anson B, Bosch B, Sanchez-Valle R, Monte GC, Villar A, Molinuevo JL, 2007. Cortical brain metabolism as measured by proton spectroscopy is related to memory performance in patients with amnestic mild cognitive impairment and Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 24, 274–279. 10.1159/000107487. [DOI] [PubMed] [Google Scholar]
- Riese F, Gietl A, Zolch N, Henning A, O’Gorman R, Kalin AM, Leh SE, Buck A, et al. , 2015. Posterior cingulate gamma-aminobutyric acid and glutamate/glutamine are reduced in amnestic mild cognitive impairment and are unrelated to amyloid deposition and apolipoprotein E genotype. Neurobiol. Aging 36, 53–59. 10.1016/j.neurobiolaging.2014.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijpma A, van der Graaf M, Meulenbroek O, Olde Rikkert MGM, Heerschap A, 2018. Altered brain high-energy phosphate metabolism in mild Alzheimer’s disease: A 3-dimensional 31P MR spectroscopic imaging study. NeuroImage Clin. 18, 254–261. 10.1016/j.nicl.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC, 2009. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr. Alzheimer Res. 6, 347–361. 10.2174/156720509788929273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R, Knopman DS, 2013. Classification and epidemiology of MCI. Clin. Geriatr. Med. 29, 753–772. 10.1016/j.cger.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena M, Dubey R, 2019. Target enzyme in Alzheimer’s disease: Acetylcholinesterase inhibitors. Curr. Top. Med. Chem. 19, 264–275. 10.2174/1568026619666190128125912. [DOI] [PubMed] [Google Scholar]
- Schuff N, Capizzano AA, Du AT, Amend DL, O’Neill J, Norman D, Kramer J, Jagust W, et al. , 2002. Selective reduction of N-acetylaspartate in medial temporal and parietal lobes in AD. Neurology 58, 928–935. 10.1212/wnl.58.6.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J, 2000. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 267, 4904–4911. 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Sekar S, Grandjean J, Garnell JF, Willems R, Duytschaever H, Seramani S, Su H, Ver Donck L, et al. , 2019. Neuro-metabolite profiles of rodent models of psychiatric dysfunctions characterised by MR spectroscopy. Neuropharmacology 146, 109–116. 10.1016/j.neuropharm.2018.11.021. [DOI] [PubMed] [Google Scholar]
- Seo SW, Lee JH, Jang SM, Kim ST, Chin J, Kim GH, Kim JH, Roh JH, et al. , 2012. Neurochemical alterations of the entorhinal cortex in amnestic mild cognitive impairment (aMCI): a three-year follow-up study. Arch. Gerontol. Geriatr. 54, 192–196. 10.1016/j.archger.2011.04.002. [DOI] [PubMed] [Google Scholar]
- Sery O, Povova J, Misek I, Pesak L, Janout V, 2013. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: a review. Folia Neuropathol. 51, 1–9. 10.5114/fn.2013.34190. [DOI] [PubMed] [Google Scholar]
- Shiino A, Watanabe T, Shirakashi Y, Kotani E, Yoshimura M, Morikawa S, Inubushi T, Akiguchi I, 2012. The profile of hippocampal metabolites differs between Alzheimer’s disease and subcortical ischemic vascular dementia, as measured by proton magnetic resonance spectroscopy. J. Cereb. Blood Flow Metab. 32, 805–815. 10.1038/jcbfm.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, Mandal PK, Tripathi M, Vishwakarma G, Mishra R, Sandal K, 2020. Quantitation of in vivo brain glutathione conformers in cingulate cortex among age-matched control, MCI, and AD patients using MEGA-PRESS. Hum. Brain Mapp. 41, 194–217. 10.1002/hbm.24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siger M, Schuff N, Zhu X, Miller BL, Weiner MW, 2009. Regional myo-inositol concentration in mild cognitive impairment using 1H magnetic resonance spectroscopic imaging. Alzheimer Dis. Assoc. Disord. 23, 57–62. 10.1097/WAD.0b013e3181875434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira de Souza A, de Oliveira-Souza R, Moll J, Tovar-Moll F, Andreiuolo PA, Bottino CM, 2011. Contribution of 1H spectroscopy to a brief cognitive-functional test battery for the diagnosis of mild Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 32, 351–361. 10.1159/000334656. [DOI] [PubMed] [Google Scholar]
- Simpson DSA, Oliver PL, 2020. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 9, 743. 10.3390/antiox9080743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CD, Pettigrew LC, Avison MJ, Kirsch JE, Tinkhtman AJ, Schmitt FA, Wermeling DP, Wekstein DR, et al. , 1995. Frontal lobe phosphorus metabolism and neuropsychological function in aging and in Alzheimer’s disease. Ann. Neurol. 38, 194–201. 10.1002/ana.410380211. [DOI] [PubMed] [Google Scholar]
- Smith RN, Agharkar AS, Gonzales EB, 2014. A review of creatine supplementation in age-related diseases: more than a supplement for athletes. F1000Res. 3, 222. 10.12688/f1000research.5218.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag KC, Ryu WI, Amirault KM, Healy RA, Siegel AJ, McPhie DL, Forester B, Cohen BM, 2017. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 7, 14038. 10.1038/s41598-017-14420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, et al. , 2017. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 552, 187–193. 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppe G, Bruhn H, Pouwels PJ, Hänicke W, Frahm J, 2000. Alzheimer disease: absolute quantification of cerebral metabolites in vivo using localized proton magnetic resonance spectroscopy. Alzheimer Dis. Assoc. Disord. 14, 112–119. 10.1097/00002093-200004000-00009. [DOI] [PubMed] [Google Scholar]
- Suriyajakryuththana W, Tuntiyatorn L, Teepprasarn N, Sukying C, 2014. Proton magnetic resonance spectroscopy in mild cognitive impairment and Alzheimer’s disease: a preliminary study. J. Med. Assoc. Thai. 97, 407–414. [PubMed] [Google Scholar]
- Swerdlow RH, 2020. The mitochondrial hypothesis: dysfunction, bioenergetic defects, and the metabolic link to Alzheimer’s disease. Int. Rev. Neurobiol. 154, 207–233. 10.1016/bs.irn.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, Burns JM, Khan SM, 2010. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 20 Suppl 2, S265–279. 10.3233/jad-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targosz-Gajniak MG, Siuda JS, Wicher MM, Banasik TJ, Bujak MA, Augusciak-Duma AM, Opala G, 2013. Magnetic resonance spectroscopy as a predictor of conversion of mild cognitive impairment to dementia. J. Neurol. Sci. 335, 58–63. 10.1016/j.jns.2013.08.023. [DOI] [PubMed] [Google Scholar]
- Terada T, Obi T, Bunai T, Matsudaira T, Yoshikawa E, Ando I, Futatsubashi M, Tsukada H, et al. , 2020. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 94, e1592–e1604. 10.1212/wnl.0000000000009249. [DOI] [PubMed] [Google Scholar]
- Tkac I, Dubinsky JM, Keene CD, Gruetter R, Low WC, 2007. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J. Neurochem 100, 1397–1406. 10.1111/j.1471-4159.2006.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobore TO, 2019. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer’s disease. Neurol. Sci. 40, 1527–1540. 10.1007/s10072-019-03863-x. [DOI] [PubMed] [Google Scholar]
- Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, et al. , 2016. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 7, 12948. 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumati S, Martens S, Aleman A, 2013. Magnetic resonance spectroscopy in mild cognitive impairment: systematic review and meta-analysis. Neurosci. Biobehav. Rev. 37, 2571–2586. 10.1016/j.neubiorev.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Viña J, Lloret A, Giraldo E, Badia MC, Alonso MD, 2011. Antioxidant pathways in Alzheimer’s disease: possibilities of intervention. Curr. Pharm. Des. 17, 3861–3864. 10.2174/138161211798357755. [DOI] [PubMed] [Google Scholar]
- Wang H, Tan L, Wang HF, Liu Y, Yin RH, Wang WY, Chang XL, Jiang T, et al. , 2015. Magnetic resonance spectroscopy in Alzheimer’s disease: systematic review and meta-analysis. J. Alzheimers Dis. 46, 1049–1070. 10.3233/jad-143225. [DOI] [PubMed] [Google Scholar]
- Wang SY, Wang M, Wang XX, Chen W, Sheng C, Gong ZK, 2017. Study on the clinical application of the MRS in the cognitive assessment after stroke. Eur. Rev. Med. Pharmacol. Sci. 21, 2437–2442. [PubMed] [Google Scholar]
- Wang T, Xiao S, Li X, Ding B, Ling H, Chen K, Fang Y, 2012. Using proton magnetic resonance spectroscopy to identify mild cognitive impairment. Int. Psychogeriatr. 24, 19–27. 10.1017/S1041610211000962. [DOI] [PubMed] [Google Scholar]
- Wang X, He HJ, Xiong X, Zhou S, Wang WW, Feng L, Han R, Xie CL, 2021. NAD+ in Alzheimer’s disease: Molecular mechanisms and systematic therapeutic evidence obtained in vivo. Front. Cell Dev. Biol. 9, 668491. 10.3389/fcell.2021.668491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XC, Du XX, Tian Q, Wang JZ, 2008. Correlation between choline signal intensity and acetylcholine level in different brain regions of rat. Neurochem. Res. 33, 814–819. 10.1007/s11064-007-9509-4. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhao C, Yu L, Zhou W, Li K, 2009. Regional metabolic changes in the hippocampus and posterior cingulate area detected with 3-Tesla magnetic resonance spectroscopy in patients with mild cognitive impairment and Alzheimer disease. Acta. Radiol. 50, 312–319. 10.1080/02841850802709219. [DOI] [PubMed] [Google Scholar]
- Waragai M, Moriya M, Nojo T, 2017. Decreased N-acetyl aspartate/myo-inositol ratio in the posterior cingulate cortex shown by magnetic resonance spectroscopy may be one of the risk markers of preclinical Alzheimer’s disease: A 7-year follow-up study. J. Alzheimers Dis. 60, 1411–1427. 10.3233/JAD-170450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Shiino A, Akiguchi I, 2008. Absolute quantification in proton magnetic resonance spectroscopy is superior to relative ratio to discriminate Alzheimer’s disease from Binswanger’s disease. Dement. Geriatr. Cogn. Disord. 26, 89–100. 10.1159/000144044. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Shiino A, Akiguchi I, 2010. Absolute quantification in proton magnetic resonance spectroscopy is useful to differentiate amnesic mild cognitive impairment from Alzheimer’s disease and healthy aging. Dement. Geriatr. Cogn. Disord. 30, 71–77. 10.1159/000318750. [DOI] [PubMed] [Google Scholar]
- Webers A, Heneka MT, Gleeson PA, 2020. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 98, 28–41. 10.1111/imcb.12301. [DOI] [PubMed] [Google Scholar]
- Whitehurst TS, Osugo M, Townsend L, Shatalina E, Vava R, Onwordi EC, Howes O, 2020. Proton magnetic resonance spectroscopy of N-acetyl aspartate in chronic schizophrenia, first episode of psychosis and high-risk of psychosis: a systematic review and meta-Analysis. Neurosci. Biobehav. Rev 119, 255–267. 10.1016/j.neubiorev.2020.10.001. [DOI] [PubMed] [Google Scholar]
- Wong D, Atiya S, Fogarty J, Montero-Odasso M, Pasternak SH, Brymer C, Borrie MJ, Bartha R, 2020. Reduced hippocampal glutamate and posterior cingulate N-acetyl aspartate in mild cognitive impairment and Alzheimer’s disease is associated with episodic memory performance and white matter integrity in the cingulum: a pilot study. J. Alzheimers Dis. 73, 1385–1405. 10.3233/JAD-190773. [DOI] [PubMed] [Google Scholar]
- Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT, 1998. Immunohistochemical study of the β-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am. J. Pathol. 153, 31–37. 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZX, Huo SS, Cheng XF, Xu ZF, Cao Z, Zeng JX, Xiao YY, You KZ, et al. , 2012. Quantitative multivoxel proton MR spectroscopy study of brain metabolites in patients with amnestic mild cognitive impairment: a pilot study. Neuroradiology 54, 451–458. 10.1007/s00234-011-0900-0. [DOI] [PubMed] [Google Scholar]
- Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, Qian G, Zheng JC, 2013. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem. 125, 897–908. 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Wu W, Liu R, Liang X, Yu T, Chen X, Feng J, Guo A, et al. , 2015. APOE genotype and age modifies the correlation between cognitive status and metabolites from hippocampus by a 2D 1H-MRS in non-demented elders. PeerJ 3, e1202. 10.7717/peerj.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Mayer D, Rohlfing T, Sullivan EV, Pfefferbaum A, 2014. Imaging neuroinflammation? A perspective from MR spectroscopy. Brain Pathol. 24, 654–664. 10.1111/bpa.12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeydan B, Deelchand DK, Tosakulwong N, Lesnick TG, Kantarci OH, Machulda MM, Knopman DS, Lowe VJ, et al. , 2017. Decreased glutamate levels in patients with amnestic mild cognitive impairment: an sLASER proton MR spectroscopy and PiB-PET study. J. Neuroimaging 27, 630–636. 10.1111/jon.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Li M, Sun ZZ, Zhu B, Yuan L, Wang Y, Xu Y, 2009. Evaluation of functional MRI markers in mild cognitive impairment. J. Clin. Neurosci. 16, 635–641. 10.1016/j.jocn.2008.07.080. [DOI] [PubMed] [Google Scholar]
- Zhu X, Cao L, Hu X, Dong Y, Wang H, Liu F, Sun Z, 2015. Brain metabolism assessed via proton magnetic resonance spectroscopy in patients with amnestic or vascular mild cognitive impairment. Clin. Neurol. Neurosurg. 130, 80–85. 10.1016/j.clineuro.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Zhu X, Schuff N, Kornak J, Soher B, Yaffe K, Kramer JH, Ezekiel F, Miller BL, et al. , 2006. Effects of Alzheimer disease on fronto-parietal brain N-acetyl aspartate and myo-inositol using magnetic resonance spectroscopic imaging. Alzheimer Dis. Assoc. Disord. 20, 77–85. 10.1097/01.wad.0000213809.12553.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimny A, Szewczyk P, Trypka E, Wojtynska R, Noga L, Leszek J, Sasiadek M, 2011. Multimodal imaging in diagnosis of Alzheimer’s disease and amnestic mild cognitive impairment: value of magnetic resonance spectroscopy, perfusion, and diffusion tensor imaging of the posterior cingulate region. J. Alzheimers Dis. 27, 591–601. 10.3233/JAD-2011-110254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.