

Abstract
Rationale: Primary graft dysfunction (PGD) is a severe form of acute lung injury, leading to increased early morbidity and mortality after lung transplant. Obesity is a major health problem, and recipient obesity is one of the most significant risk factors for developing PGD.
Objectives: We hypothesized that T-regulatory cells (Tregs) are able to dampen early ischemia–reperfusion events and thereby decrease the risk of PGD, whereas that action is impaired in obese recipients.
Methods: We evaluated Tregs, T cells, and inflammatory markers, plus clinical data, in 79 lung transplant recipients and 41 liver or kidney transplant recipients and studied two groups of mice on a high-fat diet (HFD), which did (“inflammatory” HFD) or did not (“healthy” HFD) develop low-grade inflammation with decreased Treg function.
Measurements and Main Results: We identified increased levels of IL-18 as a previously unrecognized mechanism that impairs Tregs’ suppressive function in obese individuals. IL-18 decreases levels of FOXP3, the key Treg transcription factor, decreases FOXP3 di- and oligomerization, and increases the ubiquitination and proteasomal degradation of FOXP3. IL-18–treated Tregs or Tregs from obese mice fail to control PGD, whereas IL-18 inhibition ameliorates lung inflammation. The IL-18–driven impairment in Tregs’ suppressive function before transplant was associated with an increased risk and severity of PGD in clinical lung transplant recipients.
Conclusions: Obesity-related IL-18 induces Treg dysfunction that may contribute to the pathogenesis of PGD. Evaluation of Tregs’ suppressive function together with evaluation of IL-18 levels may serve as a screening tool to identify obese individuals with an increased risk of PGD before transplant.
Keywords: regulatory T cells, obesity, IL-18, lung transplant, primary graft dysfunction
At a Glance Commentary
Scientific Knowledge on the Subject
Obesity is a major health problem, and transplant recipient obesity is one of the most significant risk factors for developing lung primary graft dysfunction (PGD).
What This Study Adds to the Field
By using murine models and clinical samples from lung, kidney, or liver allograft recipients, we found that obesity-related IL-18 may be the key factor that impairs the suppressive function of T-regulatory cells (Tregs) in obese individuals. Defective Treg control, in turn, may be responsible for the development of low-grade, obesity-related inflammation in affected individuals. The IL-18–driven impairment in Tregs’ suppressive function before transplant was associated with an increased risk and severity of PGD in clinical lung transplant recipients and in murine models of PGD.
Obesity is a major public health challenge that can involve low-grade chronic inflammation (1). The inflammation is believed to be associated with activation of the NLRP1 and NLRP3 inflammasomes in adipose tissues, followed by increased production of the cytokines IL-1β and IL-18 (1). IL-18 is an immunoregulatory cytokine with both pro- and antiinflammatory effects and can be produced by many cell types (2). Increased levels of IL-18 are associated with obesity, but IL-18 is currently considered as a negative regulator of inflammasome activation, fat mass accumulation, and development of obesity-induced metabolic syndrome (1, 3).
Lung primary graft dysfunction (PGD) is a severe acute lung injury, affecting 15–30% of lung recipients (4). It occurs within 72 hours of lung transplant and is associated with significant early and late post-transplant morbidity and mortality (4–7). The pathogenesis of PGD includes the initial effects of ischemia–reperfusion injury, followed by hyperactivation of innate and then adaptive immunity (5–7). PGD is associated with increased NLRP3 inflammasome activation by HMGB1 and other damage-associated molecular pattern moieties; increased levels of IL-1β and CXCL1, IL-6, IL-8, IL-17, IFNγ, and TNFα; and neutrophil and monocyte infiltration and activation with release of reactive oxygen species, catalytic proteases, and enzymes, which further promote tissue damage and inflammation (5–10). In clinical studies, a recipient’s obesity was identified as one of the most significant risk factors for developing lung PGD (6, 7, 11, 12).
In the current study, we showed that mice with obesity-related T-regulatory cell (Treg) dysfunction differed from obese mice with normal Treg function in that they had increased levels of IL-18. IL-18 impaired Tregs’ suppressive function, whereas defective Treg control, in turn, may be responsible for the development of low-grade obesity-related inflammation. In the transplant setting, IL-18–dysregulated obesity may be an important and previously unrecognized factor that significantly reduces the ability of Tregs to promote allograft survival, starting with early post-transplant events such as PGD and also affecting long-term outcomes. Some of the results of these studies have been previously reported in the form of abstracts (13–15).
Methods
Donors
Peripheral blood mononuclear cells (PBMCs) were obtained through the University of Pennsylvania Human Immunology Core. All donors signed an informed consent form.
Transplant Recipients
Lung transplant subjects were enrolled from the prospective, multicenter Lung Transplant Outcomes Group cohort study (clinicaltrials.gov identifier NCT 00457847). Patients undergoing combined organ transplant were excluded, and all other patients undergoing lung transplant aged ⩾13 years were included. PGD was defined as described in Reference 16. We analyzed 137 samples from 79 patients (mean ± SEM age, 57.24 ± 1.2 yr; 52% male).
Pediatric liver transplant recipients are described in Reference 17.
Adult Liver and Kidney Transplant Patients
Tregs were isolated from four kidney and six liver allograft recipients before transplant and at 3 months and 1 year after transplant. Patients were 52.6 ± 2.8 years old, and eight were male.
Each study center institutional review board approved corresponding studies.
More details for all patient cohorts are reported in Supplemental Methods in the online supplement.
Mice
We purchased C57BL/6 mice from The Jackson Laboratory and the Charles River Laboratory. RAG1−/− mice were purchased at The Jackson Laboratory (stock number 002216). From 4 weeks of age, control mice received normal chow (25% of kilocalories from fat; LabDiet 5015, Animal Specialties and Provisions) or a high-fat diet (HFD) (60% kilocalories from fat; D12492 formula, Research Diets, Inc.). Mice with decreased Treg function on an HFD were included in the “inflammatory” HFD (iHFD) group, whereas mice with unimpaired Treg function were included in the “healthy” HFD (hHFD) group (see Figure E1E in the online supplement).
The murine PGD model is described in Reference 18.
Murine lung transplant procedures are described in Reference 19.
All animal studies used protocols were approved by the Institutional Animal Care and Use Committees of the Children’s Hospital of Philadelphia. More details are reported in the Supplemental Methods.
Flow cytometry was performed as described in Reference 20.
Treg isolation and evaluation of Tregs’ suppressive function were performed as described in Reference 21.
ELISA and Luminex
Plasma samples of lung transplant patients were evaluated by using a custom Luminex Screening Human Assay (catalog number LXSAH-10, R&D Systems), an IL-18 Platinum ELISA (catalog number BMS267/2, eBioscience, Inc.), and an IL-18 binding peptide (IL-18BP) ELISA (catalog number EHIL18BP, Invitrogen); mouse sera were evaluated by using a Luminex assay (catalog number LX10004029301, R&D Systems), a RayBiotech Mouse Leptin ELISA Kit (catalog number ELM-Leptin-1), and a Mouse IL-18 ELISA Kit (catalog number EKC37154, Biomatik), according to manufacturer’s instructions.
qRT-PCR was performed as described in Reference 20.
A TaqMan protein assay (Thermo Fisher Scientific) was performed according to manufacturer’s protocol, except for modifications as detailed in the Supplemental Methods. Freshly isolated healthy donor Tregs were stimulated for 3 hours with CD3/28 microbeads (Dynabeads; Thermo Fisher), ±200 ng/ml IL-18 (R&D Systems), and in the presence of 3μM proteasome inhibitor PS341 (Selleck Chemicals) when indicated.
TUBE Assay
A K48 Linkage–Specific UbiTest–Magnetic TUBE Elution Kit (catalog number UM414M, LifeSensors Inc.) was used to assess ubiquitination of FOXP3. HEK-Blue IL-18 cells (catalog number hkb-hmil18, InvivoGen) were transfected with FOXP3 and HA-tagged ubiquitin ± Stub1 and analyzed as described in Reference 22.
Statistics
Treg function was calculated by using the area under the curve method as described in Reference 21 and was then adjusted for the FOXP3 purity of isolated Tregs as detailed in Reference 20. We applied parametric tests if data were normally distributed and nonparametric tests if not, and all tests are described in the figure legends. Data are shown as the mean ± SEM. A two-tailed P value of <0.05 was considered to indicate statistical significance.
For all methods, more details are reported in the Supplemental Methods.
Results
Treg Function Is Impaired in Obese Patients, but HFD Feeding in Mice Resulted in Two Different Treg Phenotypes
We found that overweight or obese patients who were listed for lung transplant (described in the Supplemental Methods and in Table E1) had impaired Treg function in comparison with normal-weight patients (Figure 1A). To ensure an absence of artifacts due to differences in the FOXP3+ purity of CD4+CD25+, MACS-isolated (Miltenyi Biotec) human Tregs, we evaluated FOXP3+ expression in isolated Tregs and then adjusted for Tregs’ suppressive function according to their FOXP3+ purity (Supplementary Methods and Figures E1A–E1D). Obese patients demonstrated significant impairment in the suppressive function of Tregs before (Figure E1D) as well as after (Figure 1B) adjustment for FOXP3+ purity. Patient obesity, evaluated as the area of subcutaneous adipose tissue (12), demonstrated an inverse correlation with Treg function (Figure 1C). We also examined Treg function in adult and pediatric liver and kidney allograft recipients and found similar results, indicating that obesity negatively impacts Treg function before (Figure 1D) but also after (Figure 1E) transplant across various solid-organ transplant populations.
Figure 1.
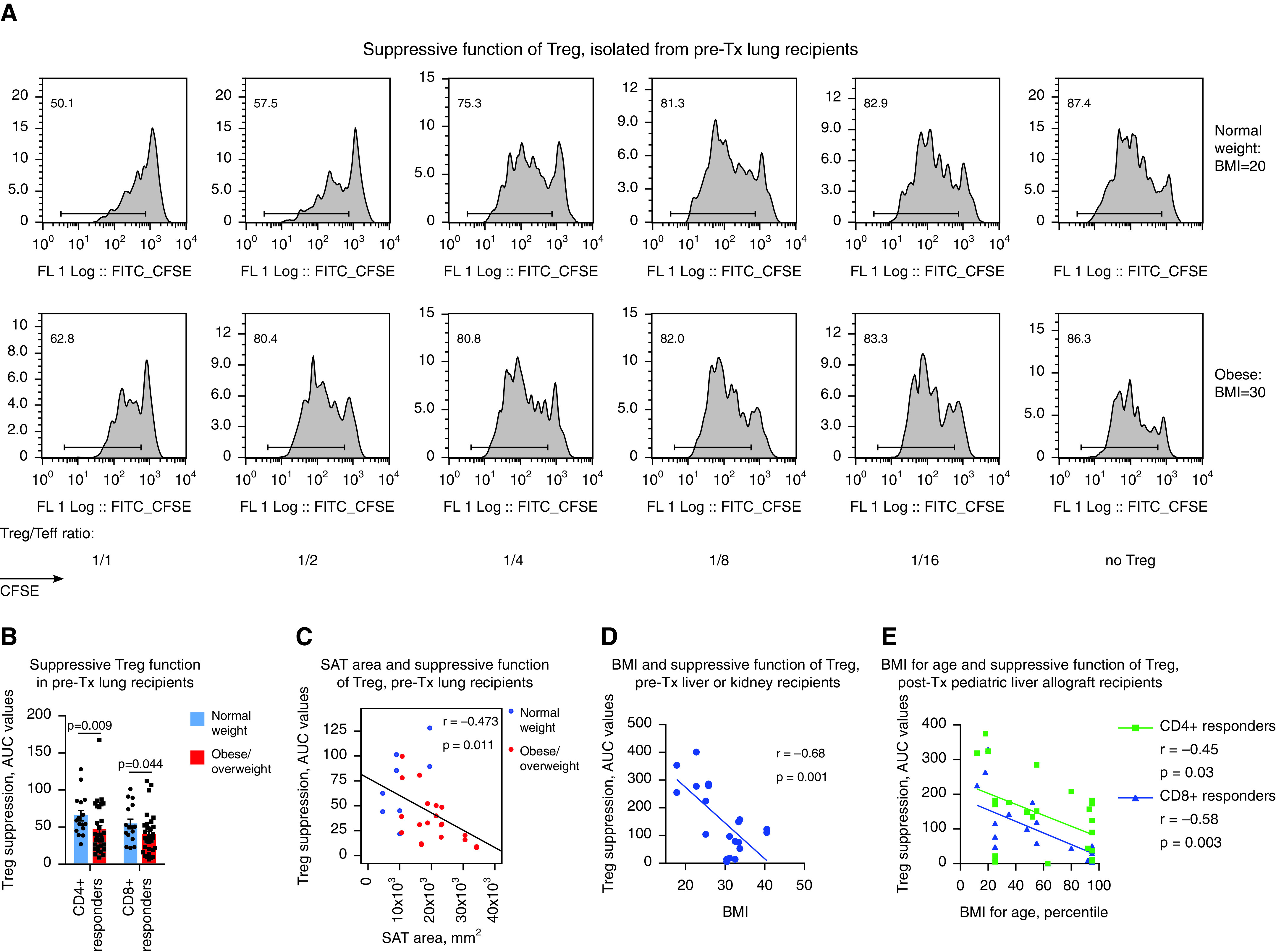
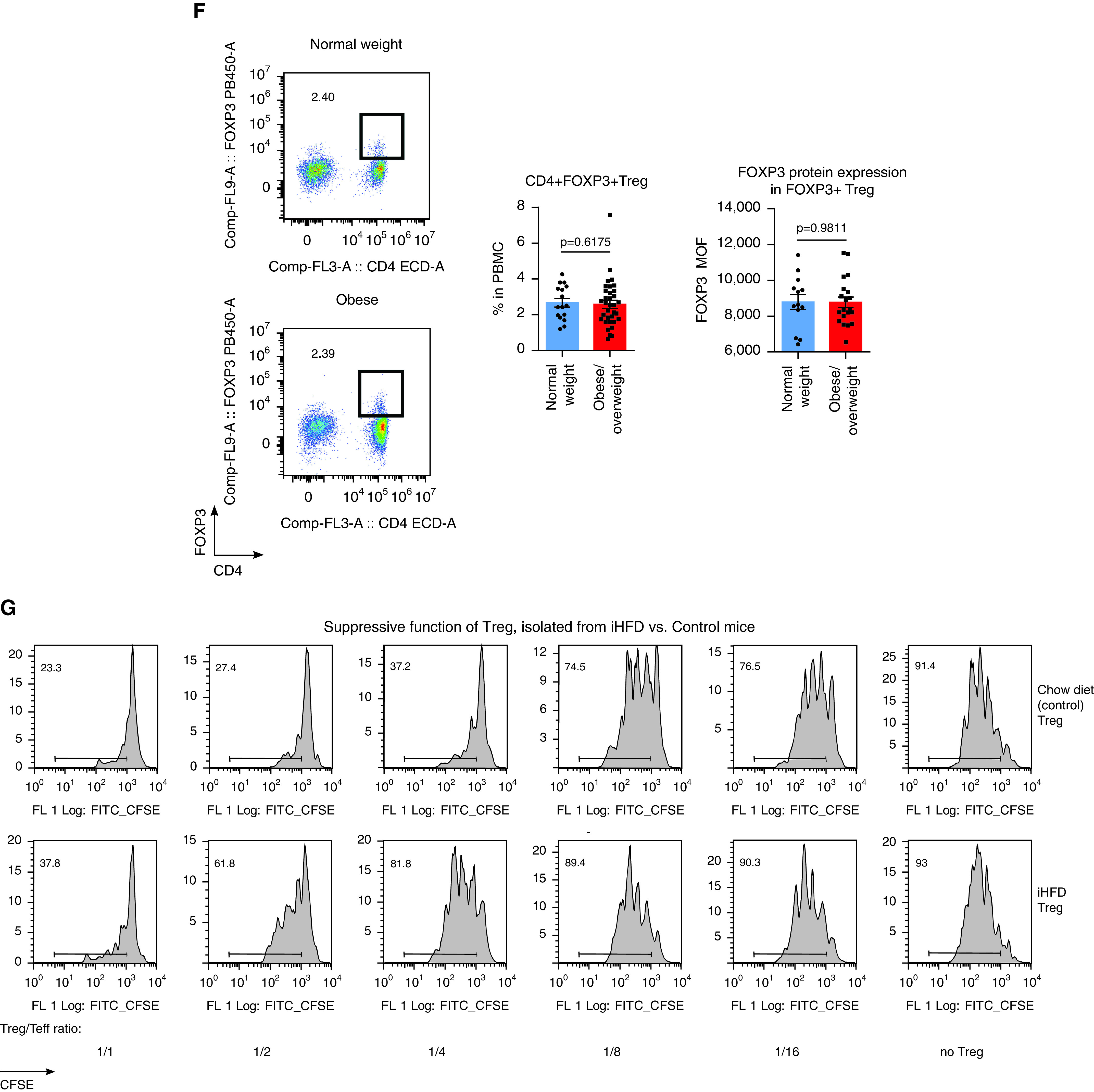
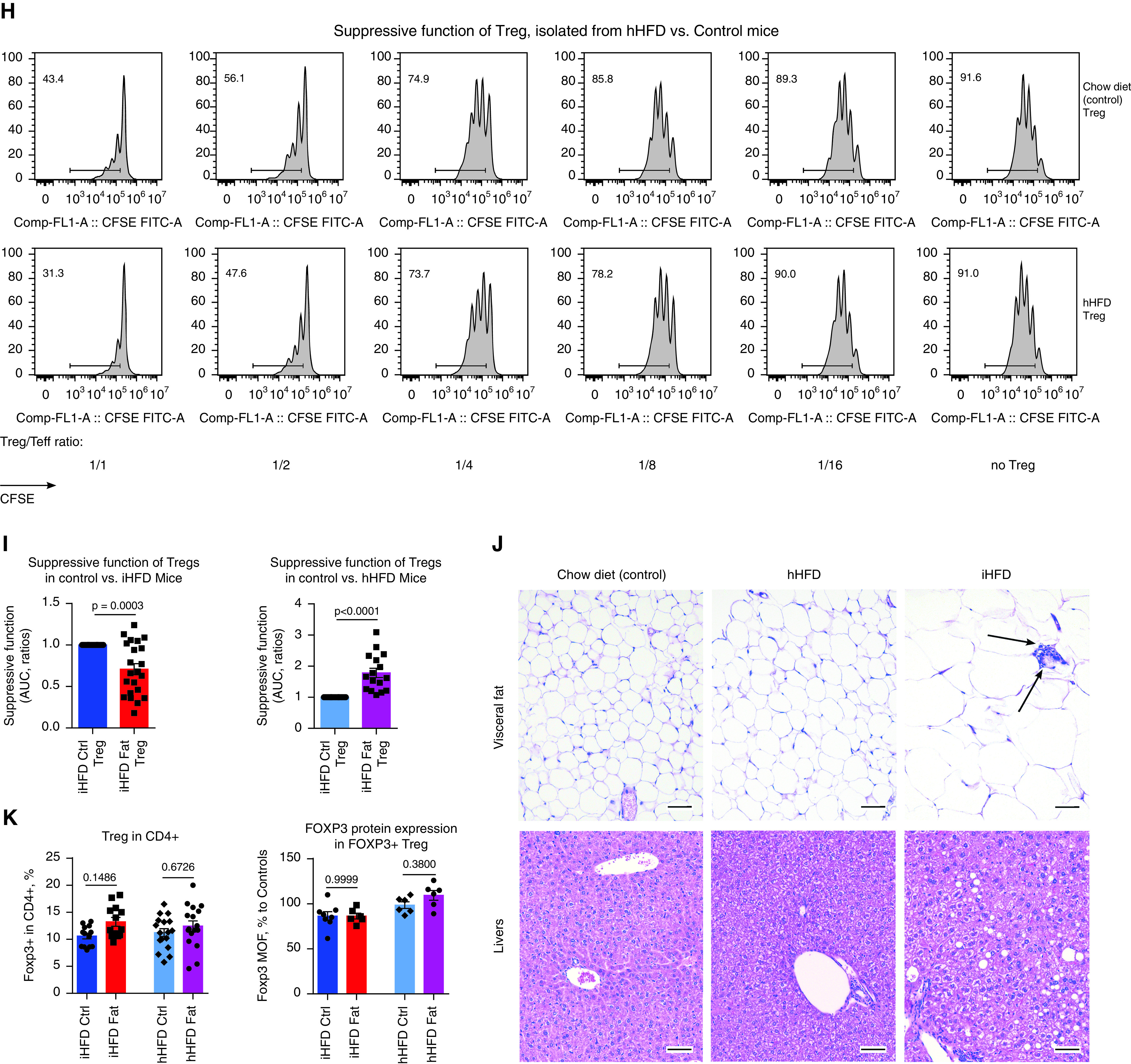
T-regulatory cell (Treg) function is impaired in obese patients, but high-fat diet (HFD) feeding in mice resulted in two different Treg phenotypes. (A and B) The pretransplant (pre-Tx) suppressive function of Tregs is impaired in overweight or obese patients. (A) CFSE plots for CD4+ responders and (B) statistics of Treg function adjusted for FOXP3+ Treg purity after isolation (n = 57). The percentages of FOXP3+ expression in Tregs in A were 50.6% (normal weight) and 52.6% (obese). Data on Treg function before adjustment for FOXP3+ purity are shown at Figure E1D and Table E10 in the online supplement, and details on the evaluation and standardization of suppression assays are provided in the Supplementary Methods. (C) Tregs’ suppressive function, combined for CD4+ and CD8+ responders, was inversely correlated with the area of SAT as measured before Tx in lung Tx recipients (n = 14). (D) The pre-Tx suppressive function of Tregs negatively correlates with patient body mass index (BMI) values in adult liver (n = 7) and kidney (n = 3) recipients; data for Treg controlling divisions of CD4+ and CD8+ responders are combined together (n = 20). (E) The post-Tx suppressive function of Tregs negatively correlates with patient BMI-for-age values in pediatric liver allograft recipients with stable allograft function (n = 24). (F) Representative plots (left) and statistics (middle, n = 50) showing the pre-Tx number of FOXP3+ Tregs in PBMCs and the levels of FOXP3 in Tregs (right, n = 33) in lung Tx recipients are shown. Clinical and demographic characteristics, inflammatory markers in the plasma, and the flow cytometry characteristics of T cells and Tregs of lung Tx recipients are presented in detail in Tables E3–E5. (G–I) The suppressive function of Tregs isolated from control (Ctrl), “inflammatory” HFD (iHFD), and “healthy” HFD (hHFD) mice were compared by using in vitro suppressive assays. (G and H) Representative and (I) quantitative data are shown (iHFD mice vs. Ctrl mice, n = 22; hHFD mice vs. ctrl mice, n = 16). (J) Histology of visceral fat and livers (hematoxylin and eosin staining; scale bars, 100 μm); arrows indicate an accumulation of inflammatory cells that is also known as a “crown,” which was observed at least once per slide in iHFD fat samples. Sixty samples in total were evaluated and included iHFD Ctrl (n = 12), iHFD fat (n = 29), hHFD Ctrl (n = 9), and hHFD fat (n = 10) samples. (K) Results from flow cytometry of the spleens and lymph nodes of Ctrl, iHFD, and hHFD mice; the percentage of Foxp3+ Tregs among CD4+ cells (left, n = 36); and the levels of Foxp3 in Tregs (right, n = 26; as determined by using a Dako CyAn flow cytometer) are shown. More data are presented in Figures E1I–E1L. Because male and female mice in both the iHFD group and the hHFD group only differed in terms of weight, all presented data show both sexes combined, unless otherwise specified. ANOVA with Sidak’s multiple comparisons test was used in B, Spearman’s test was used in C, Pearson’s test was used in D and E; a Mann-Whitney U test (middle) and an unpaired t test (right) were used in F, a one-sample t test with n = 1 (Ctrl) hypothetical value was used in I, and ANOVA with Tukey’s multiple comparison test (left) and a Wilcoxon signed rank test (right) were used in K. AUC = area under the curve; CFSE = carboxyfluorescein succinimidyl ester; Comp = compensatory; ECD = extracellular domain; FITC = fluorescein isothiocyanate; FL = fluorescence; MOF = median of FL; PBMC = peripheral blood mononuclear cell; SAT = subcutaneous adipose tissue; Teff = T-effector cell.
Overweight or obese and normal-weight patients showed no differences in clinical or demographic data, the levels of inflammatory markers in plasma, or the activation or maturation characteristics of Tregs and T cells as assessed by flow cytometry, with the exception of increased leptin levels being noted in overweight or obese patients (Tables E3–E5). Their numbers of Tregs and amounts of FOXP3 protein per cell were also comparable (Figure 1F). Therefore, significant impairment in Tregs’ suppressive function as determined in vitro does not correspond with evident inflammatory abnormalities in vivo, at least for the tested parameters.
To further study the effects of obesity on Treg function, we induced obesity by feeding an HFD to C57BL/6 mice. Surprisingly, although some mice reproduced the phenotype with dysfunctional Tregs while receiving an HFD (Figures 1G and 1I), as in our clinical samples, others had no decline in or even had enhancement of the suppressive function of Tregs while receiving an HFD (Figures 1H and 1I). We categorized mice into iHFD and hHFD groups, corresponding to the effects of the HFD on their Treg function (Figure E1E). iHFD and hHFD mice gained weight equally, had no differences in glucose tolerance, had upregulated leptin levels in sera, and had no differences in Treg or conventional T-cell numbers or in FOXP3 protein expression in Tregs (Figures 1K and E1F–E1L). Histologic evaluation of visceral fat showed adipocyte enlargement, which was more pronounced in iHFD mice, and was accompanied by crown-like structures (Figure 1J). The livers of hHFD and iHFD mice showed steatosis in comparison with those of control mice, which was more marked in the iHFD mice than in the hHFD mice (Figure 1J). Therefore, iHFD mice, but not hHFD mice, have impaired Treg function and more pronounced histologic abnormalities in fat and liver tissues.
Murine Models of Obesity Indicate that IL-18 Contributes to Impaired Treg Function
We evaluated the sera of mice for the set of 19 cytokines and chemokines (Figures 2A and E2A). Half of them tended to be increased in overweight or obese mice, with more pronounced differences being shown in the iHFD group than in the hHFD group, and 37% of cytokines and chemokines showed no clear changes and were present at levels within a normal range. However, most tested markers had expression levels much lower than the levels present in acute LPS-induced inflammation, suggesting that iHFD mice had low-grade, nonapparent systemic inflammation.
Figure 2.
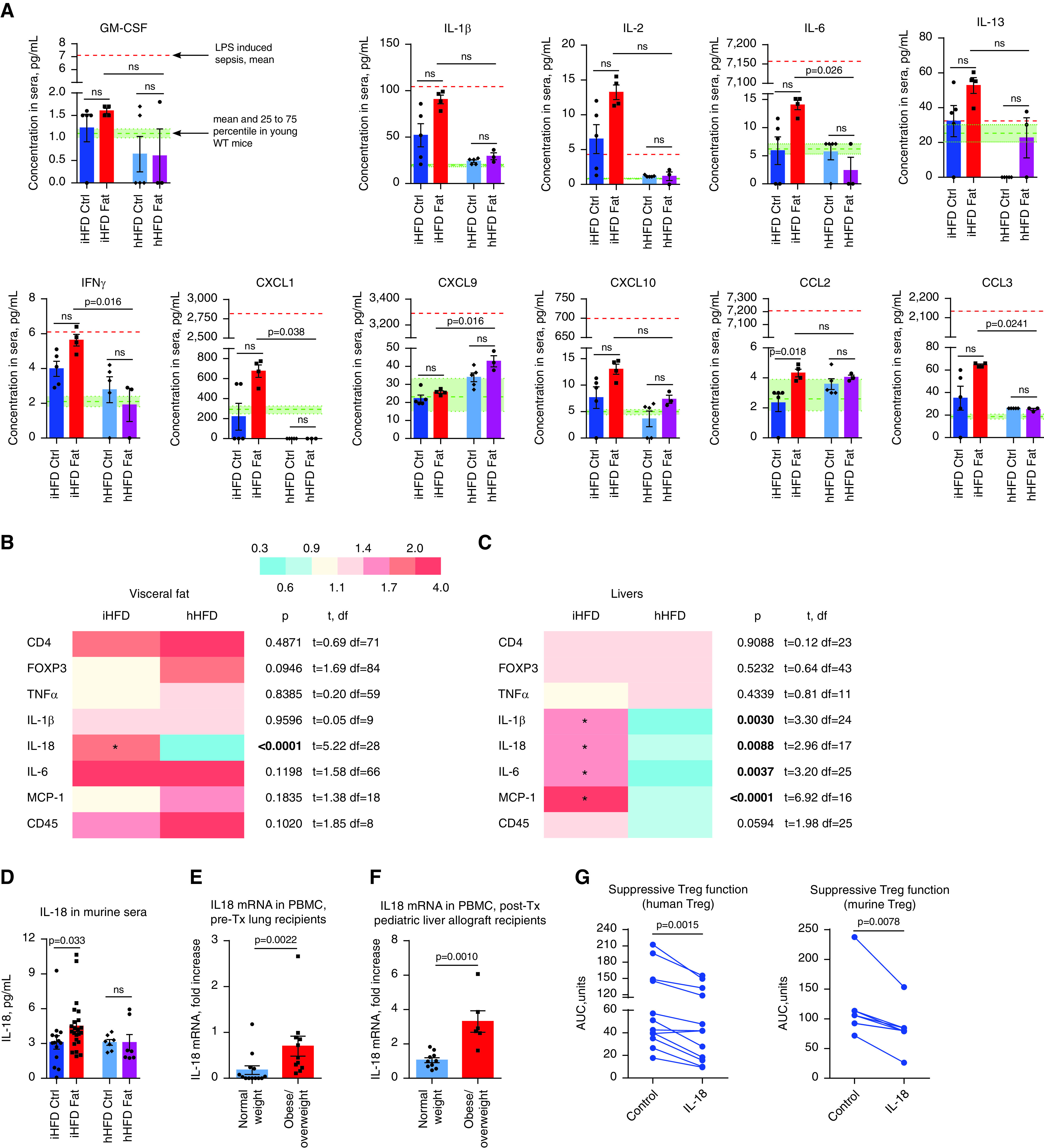
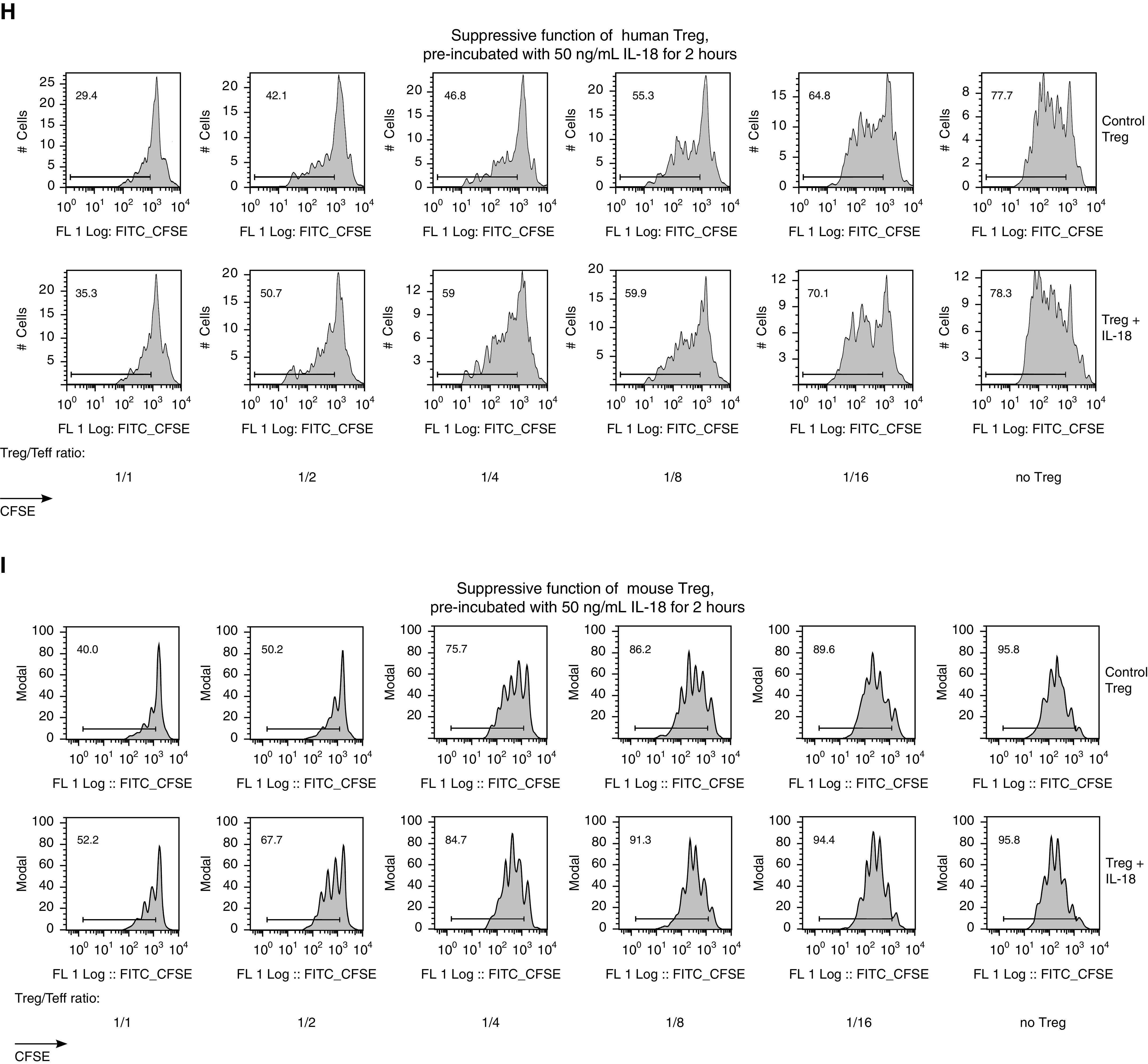
Identification of IL-18 related to obesity and effects of IL-18 on T-regulatory cells (Tregs). (A) Eighteen sera samples pooled from 21 mice of both sexes were evaluated for 19 cytokines and chemokines by using a Luminex assay. Six sera samples were pooled from eight young, 6- to 9-week-old WT mice of both sexes and served to provide the mean and interquartile range for the “normal” range of tested markers. Normal-range data are shown in the graphs as shaded green areas with green dotted lines. One sample of pooled sera from three female mice at 2 hours after LPS injection (i.p., 1 mg/kg LPS) served as the level indicating a high-inflammation condition (model of sepsis), which is shown in the graphs as orange dashed lines. More data are presented in Figure E2A in the online supplement. (B and C) Quantitative PCR (qPCR) of the visceral fat (B) and livers (C) of “inflammatory” high-fat diet (iHFD) and “healthy” high-fat diet (hHFD) mice, presented as heatmaps of fold differences of the corresponding mRNA expression over the control (Ctrl) diet samples in each group; notable differences are highlighted with an asterisk. Then the fold differences for iHFD and hHFD mice were compared with each other by using multiple t tests with Holm-Sidak correction, and the resulting test values are shown to the right of the heatmaps. Significant differences are marked by bold type. The number of samples evaluated for each marker (median [interquartile range]) are as follows: 18 (6.25–21.5) iHFD fat samples, 28.5 (5.5–52.75) hHFD fat samples, 15.5 (7–16) iHFD liver samples, and 11 (9.5–11.75) hHFD liver samples. (D) IL-18 levels in 51 pooled sera samples from 54 mice were evaluated by using an ELISA. (E) Pre-Tx IL-18 was measured by using qPCR analysis of peripheral blood mononuclear cells (PBMCs) from lung Tx recipients (n = 24). (F) IL-18 mRNA was evaluated by using qPCR analysis of the CD4-depleted PBMCs of children with stable liver allografts (n = 16). (G–I) Effects of IL-18 on Tregs’ suppressive function in humans (in G [left] and H) and mice (in G [right] and I). Tregs were treated with 50 ng/ml IL-18 for 2 hours, washed two times, and used for the suppression assay. Representative examples in H and I with corresponding statistics in G are shown. In G, n = 12 in six experiments, with data for CD4+ and CD8+ responders combined (left), and n = 8 in five experiments with n = 5 Ctrls (right). Kruskal-Wallis and Dunn’s tests were used in A and D, multiple t tests with Holm-Sidak correction were used in B and C, a Mann-Whitney U test was used in E and F, and a Wilcoxon matched-pair test was used in G. AUC = area under the curve; CFSE = carboxyfluorescein succinimidyl ester; FITC = fluorescein isothiocyanate; FL = fluorescence; ns = not significant; Teff = T-effector cell; Tx = transplant; WT = wild type.
In visceral fat, IL-18 was the only cytokine whose expression significantly differed in the visceral fat of iHFD versus hHFD mice, being upregulated only in iHFD fat samples (Figure 2B). IL-18 was also upregulated in the livers and sera of iHFD mice but was not upregulated in hHFD mice (Figures 2C and 2D). In addition, iHFD mice had upregulation of other inflammatory cytokines in their livers (Figure 2C). Therefore, IL-18 was identified as potential candidate factor to explain the differences between obesity that affected Treg function (iHFD) and obesity with preserved Treg function (hHFD). IL-18 levels were increased in overweight or obese transplant recipients before and after transplant (Figures 2E, 2F, and E2B). Preincubation of human (Figures 2G and 2H) or murine (Figures 2G and 2I) Tregs with IL-18 significantly impaired their suppressive function.
Mechanism of IL-18–mediated Effects
The levels of IL-18Rα were significantly higher in Tregs than in other lymphocytes (Figures 3A–3C). Incubation of Tregs with IL-18 gradually increased phosphorylation of IRAK4, one of the central elements in the signal transduction of IL-1/IL-18 receptors (Figures 3C and 3D), suggesting that Tregs have an active IL-18R–MyD88 pathway that is activated by IL-18.
Figure 3.
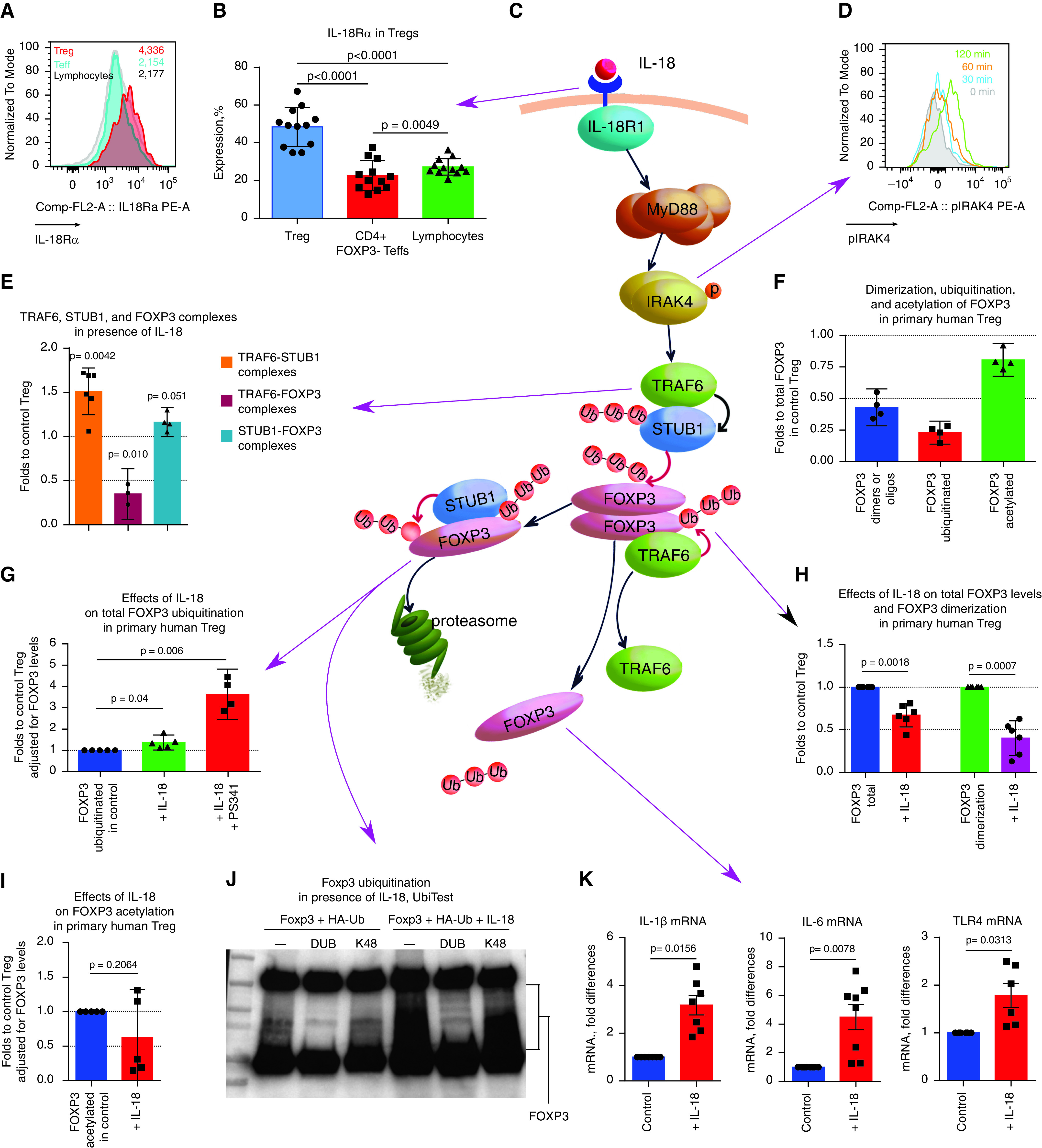
Mechanism of IL-18 effects on T-regulatory cells (Tregs). (A and B) Representative example (A) and corresponding statistics (B) for flow cytometry evaluation of IL-18Rα (CD218α) expression in human CD4+FOXP3+ Tregs (red), CD4+FOXP3− Teffs (blue), and CD4− lymphocytes (gray). (n = 12). (C) The proposed mechanism of IL-18 effects, which is explained in detail at the end of the figure legend. (D) Donor peripheral blood mononuclear cells were incubated for 0–120 minutes with 200 ng/ml IL-18, and pIRAK4 expression was evaluated by using flow cytometry in CD4+FOXP3+ Tregs. Representative data of three experiments are shown. (E) Tregs were stimulated for 3 hours with CD3/28 microbeads ± 200 ng/ml IL-18 and were evaluated by using a TaqMan protein assay; data are shown for three donors in four experiments. (F) TaqMan protein assay evaluation of freshly isolated healthy donor Tregs. The FOXP3 di- or oligomer forms, ubiquitinated FOXP3, and acetylated FOXP3 calculated as the percentage of total FOXP3 protein levels in the same Tregs are shown. (G–I) Tregs were stimulated for 3 hours with CD3/28 microbeads ± 200 ng/ml IL-18 (in G–I) and ±3 μM proteasome inhibitor PS341 (in G). (G) FOXP3 ubiquitination in presence of IL-18. (H) Total level of FOXP3 protein and FOXP3 di- and oligomer forms in the presence of IL-18. (I) FOXP3 acetylation in the presence of IL-18. In F–I, five donors were evaluated in 10 experiments. (J) 293T cells were transfected with Foxp3 and HA-tagged ubiquitin, treated with 200 ng/ml IL-18 for 2 hours plus the proteasome inhibitor bortezomib, and subjected to TUBB pull-downs; ubiquitinated substrates were eluted and treated with nonspecific DUB (deubiquitinase) or K48 linkage–specific DUB (K48); and proteins were analyzed by using Western blotting. One experiment out of three is shown. (K) Quantitative PCR analysis of healthy donor Tregs, stimulated as shown in E; more data are presented in Figure E3 in the online supplement (donor Treg experiments, n = 4; quantitative PCR experiments, n = 17). (C) Proposed mechanism of IL-18 effects. Purple arrows connect our experimental data to corresponding events in the mechanism. Upon binding of IL-18 to IL-18R (event in A), a signaling cascade is initiated. IL-18R recruits MyD88, followed by IRAK4 phosphorylation (shown as p in the diagram) and activation (event in D) (2). This Myddosome complex recruits and activates TRAF6. We suggest that TRAF6 forms a complex with STUB1 and activates its ligase E3 function toward FOXP3 protein (event in E). STUB1 increases K48-linked polyubiquitination of FOXP3, which is known to facilitate proteasomal degradation of FOXP3 (event in G) (26), which then leads to a decline of the total FOXP3 protein level (event in H). As a result of K48-linked polyubiquitination, the dimerization and oligomerization of FOXP3 protein significantly decreases (event in H). In turn, TRAF6, which was associated with FOXP3 complex and provided stabilizing K-63–linked polyubiquitination (50), releases from complexes with FOXP3 (event in E). ANOVA with Tukey’s multiple comparison test was used in B, a one-sample t test with n = 1 (control) hypothetical value was used in E and G–I, and a Wilcoxon test was used in K. Comp = compensatory; FL = fluorescence; Teff = T-effector cell.
To study the biological effects of IL-18 on primary unmanipulated human Tregs, we employed a recently developed TaqMan protein assay that enables assessment of protein while using very small cell counts (23). We have observed increased TRAF6–STUB1 complexes, decreased TRAF6–FOXP3 complexes, and a trend toward increased STUB1–FOXP3 complexes in Tregs after exposure to IL18 (Figure 3E). We evaluated FOXP3 post-translational modifications in freshly isolated human Tregs to establish a normal range, as there are no published data in this regard. In healthy donor Tregs, ∼50% of FOXP3 protein was present as dimers and/or oligomers, ∼25% of total FOXP3 was ubiquitinated, and ∼80% of total FOXP3 was acetylated (Figure 3F). Exposure of Tregs to IL-18 led to a significant decrease in the total FOXP3 protein level (to a median of 69% of initial levels) and a twofold reduction of FOXP3 dimerization and/or oligomerization (Figure 3G). Such assembly of FOXP3 into higher-order structures is critical for Treg function (24). Regarding post-translational modifications, we observed a substantial increase in FOXP3 ubiquitination but did not observe a substantial increase in FOXP3 acetylation (Figures 3H and 3I). The UbiTest assay (25) confirmed that in the presence of IL-18, FOXP3 ubiquitination was significantly increased, with substantial increase of K48-linked ubiquitination being shown, and showed that STUB1 may be involved in that process (Figures 3J and E3A).
IL-18 led to increased mRNA expression of the proinflammatory cytokines IL-1β and IL-6 and upregulated expression of TLR4 (Figure 3K) in Tregs. TLR4 may increase Treg sensitivity to proinflammatory TLR4 ligand signals, which further declines Treg function and stability (26). Conversely, there were no significant changes in the Treg mRNA expression of other cytokines (IL-2, IL-10, IL-17a, IFN-β, TNF-α), in Treg-associated markers (CTLA-4, TGFβ, GARP, GITR), or in “Treg-locking” transcription factors (IRF4, SATB1, EOS, LEF1) that we previously shown to be associated with the enhanced suppressive capability of human intratumoral Tregs (20) (Figures E3B–E3F). On the basis of our results, we suggest a mechanism of IL-18 effects on Tregs (Figure 3C).
In conjunction with increased FOXP3 ubiquitination and disrupted di- and oligomerization of FOXP3, the stability of FOXP3 expression under stimulatory conditions was significantly altered. Thus, Tregs from iHFD mice lost Foxp3 expression during the 3 days of in vitro stimulation, whereas Tregs from control and hHFD mice maintained their Foxp3 expression better (Figures 4A–4C). Correspondingly, human CD4+ cells from overweight or obese individuals were unable to upregulate FOXP3 expression with CD3/CD28 stimulation, whereas human CD4+ cells from normal-weight individuals significantly upregulated FOXP3 (Figures 4D and 4E). The addition of an IL-18 inhibitor (IL-18BP) (1) restored the ability of CD4+ cells to upregulate FOXP3 (Figure 4F).
Figure 4.
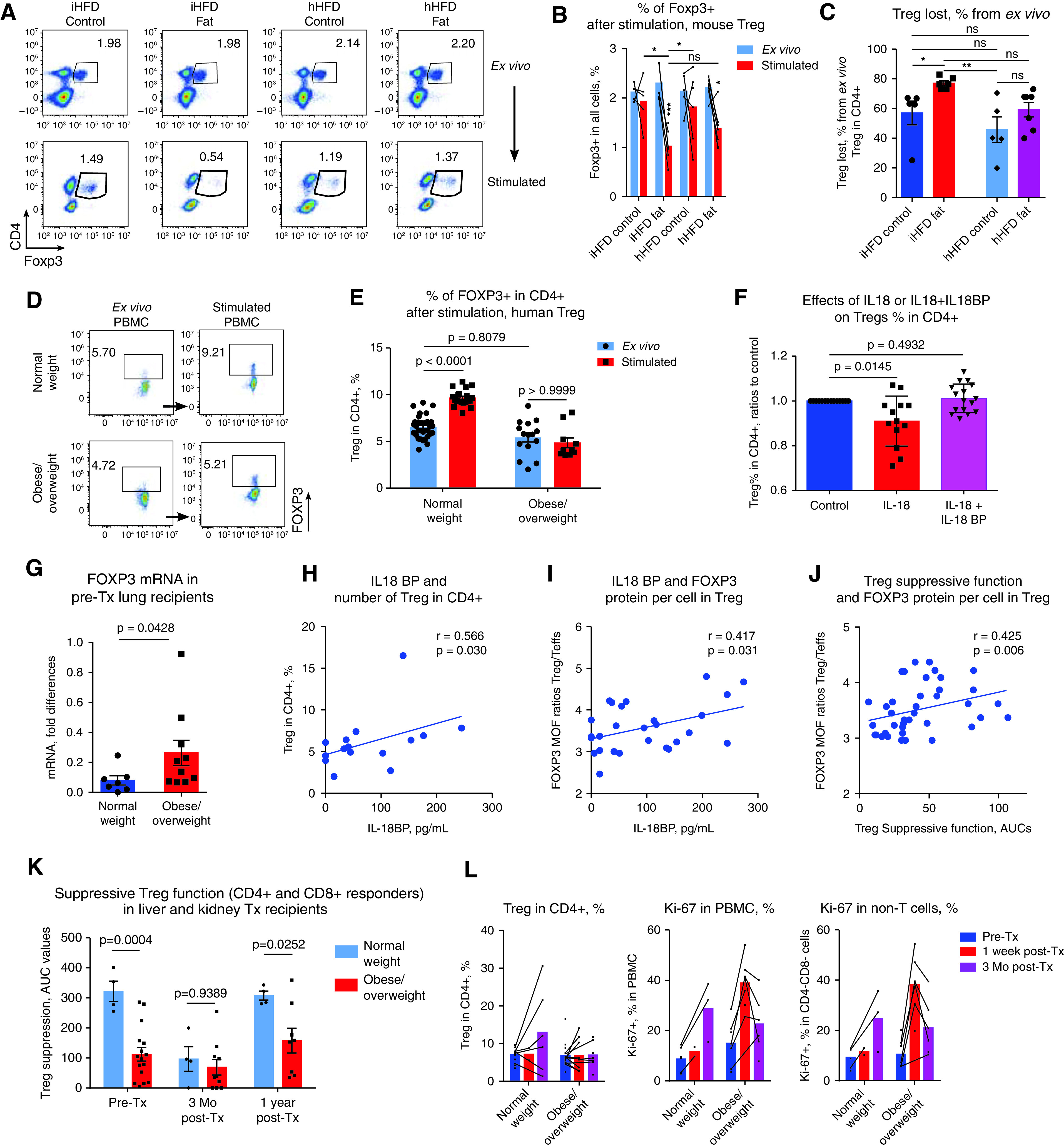
T-regulatory cell (Treg) stability, post-transplant (post-Tx) events, and primary graft dysfunction (PGD) in clinical settings. (A–C) Twenty-four samples from 56 mice were stimulated for 3 days with soluble CD3 antibodies (1 μg/ml), and Foxp3 expression was evaluated by using flow cytometry before and after stimulation. (A) Representative plots and (B and C) corresponding statistics are shown. (D and E) Peripheral blood mononuclear cells (PBMCs) from healthy donors (n = 5) and pre-Tx PBMCs from lung Tx recipients (n = 17) were stimulated overnight with CD3 microbeads, and FOXP3 expression was evaluated by using flow cytometry. (D) Representative plots and (E) corresponding statistics are shown. (F) Donor PBMCs were incubated for 2 hours with 200 ng/ml IL-18 ± IL-18 binding peptide (IL-18BP), washed, and stimulated overnight with CD3 microbeads (3.6 beads/cell), and FOXP3 expression was evaluated on the next day by using flow cytometry (n = 12 donors and n = 7 experiments). (G) FOXP3 mRNA expression was evaluated in pre-Tx PBMCs from lung Tx recipients (n = 17).(H–J) Pre-Tx plasma from obese/overweight lung Tx recipients was evaluated by using an ELISA for levels of IL-18BP. FOXP3 protein expression levels in Tregs (CD4+FOXP3+) and T-effector cells (Teffs) (CD4+FOXP3−) were evaluated by using flow cytometry. Then the FOXP3 median of fluorescence (MOF) ratios in Tregs were divided into FOXP3 MOF ratios in Teffs to eliminate the effects of absolute MOF units; MOF was evaluated in different experiments by using different flow cytometers. The resulting FOXP3 MOF ratios reflect the differences in FOXP3 protein levels in Tregs over those of the corresponding negative controls (Teffs). (H) Correlations between levels of IL-18BP and Treg numbers among
Overweight or obese individuals maintain FOXP3 mRNA level upregulation (Figure 4G). In conjunction with the unaffected levels of FOXP3+ protein per Treg cell and the equal numbers of Tregs (Figure 1F), the upregulation of FOXP3 mRNA in the PBMCs of overweight or obese individuals suggests a need for constant enhanced production of FOXP3 protein in Tregs due to its decreased stability. Correspondingly, in overweight or obese patients, blood levels of IL-18BP positively correlated with the number of Tregs among CD4+ cells and with FOXP3 protein levels per Treg (Figures 4H and 4I), whereas Tregs’ suppressive function, in turn, correlates with FOXP3 protein levels (Figure 4J).
Hence, in the presence of IL-18, levels of FOXP3 protein and FOXP3 di- and oligomerization declined, whereas FOXP3 ubiquitination (total and K48-linked) rose. These events increase proteasomal degradation of FOXP3, allow derepression of genes normally suppressed in Tregs, and compromise FOXP3 stability in stimulatory or inflammatory conditions to the detriment of Tregs’ suppressive function.
Postoperative Events and PGD
We evaluated Tregs’ suppressive function 3 months after transplant in available samples and found that it tended to decrease at 3 months after transplant in normal-weight patients but that it did not decrease in obese patients (Figure E4A). Similar trends were observed in liver and kidney recipients. Thus, it seems that in the first weeks after transplant, when maximal doses of immunosuppressive therapy are employed, the effects of obesity on Tregs were masked by the still more inhibitory effects of high-calcineurin inhibitors (17) and by acute early post-transplant events (Figure 4K). However, at 1 year after transplant, as soon as Treg function was restored, overweight or obese patients demonstrated the same differences in Treg function as they did before transplant (Figure 4K), suggesting that the negative effects of obesity on Treg function were not ameliorated by post-transplant immunosuppressive therapy.
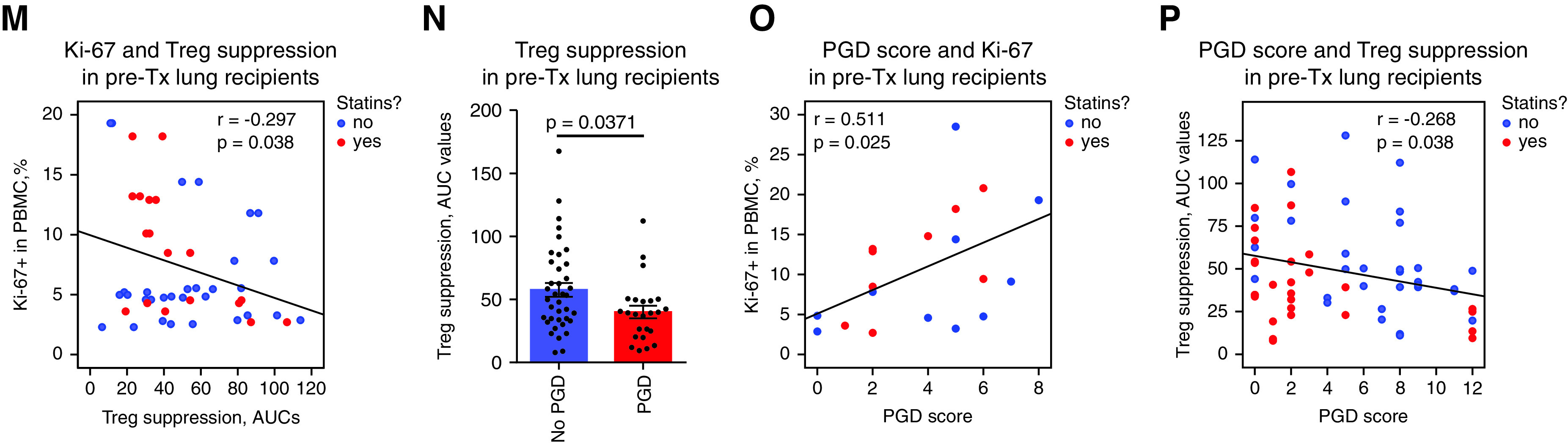
By using flow cytometry evaluation, we observed a trend toward increased Treg numbers at 3 months after transplant, which may reflect attempts of Tregs to control early alloreactive responses. Of note, this trend existed only in normal-weight patients (not in obese/overweight patients) (Figure 4I, left). We also found that overweight or obese lung recipients (but not normal-weight lung recipients) had substantial peaks of Ki-67 expression early after transplant (Figures 4I, E4B, and E4C). Those peaks may reflect the inability of their Tregs to properly control post-transplant activation of immune cells. Because Tregs’ suppressive function in vitro demonstrated a strong negative correlation with ex vivo Ki-67 expression (Figure 4M), we suggest that Ki-67 may serve as a marker that can be used to indirectly evaluate the in vivo suppressive function of Tregs.
In our cohort of lung allograft recipients, 42.9% of patients developed grade 3 PGD. Patients who did (PGD present) or did not (PGD absent) develop PGD had no significant differences in their clinical or demographic data and had no differences in medications for their comorbidities, with the exception of statin therapy (Tables E6 and E7). Within statin users, only 21.1% (4 patients) developed PGD, whereas 50% (14 patients) developed PGD in the statin-free group (Pearson chi-square P = 0.045). Use of statins is an important factor for decreasing the risk of PGD (27), and statin therapy has an impact on metabolic and immune processes (28). However, in our cohort, there were only seven patients with normal weight who received statin therapy, and only one of them developed PGD, so we were unable to perform further stratification of patients receiving statins.
We found that patients with PGD had significant impairment to Tregs’ suppressive function before transplant (Figure 4N). No other tested parameters differed before transplant between patients with PGD and patients without PGD (Tables E8 and E9). To combine the severity and longevity data of PGD into one score, we summed patients’ daily PGD scores from Day 0 to Day 3 after transplant to generate a total PGD score for each transplant recipient. Ki-67 expression in PBMCs positively correlated with PGD scores (Figure 4O), whereas Tregs’ suppressive function negatively correlated with PGD scores (Figure 4P). Notably, the inverse correlations of Tregs’ suppressive function with PGD scores were much stronger when patients were evaluated separately according to their statin status (r = −0.401, P = 0.034 for statins users; r = −0.373, P = 0.03 for statin-free patients; Pearson test). Collectively, our data suggest that the impaired suppressive function of Tregs, evaluated ex vivo (Treg suppression assay) and in vivo (Ki-67 expression), may be related to a higher risk and more pronounced severity of PGD, whereas pretransplant use of statins was associated with a reduced risk of PGD.
IL-18 Impairs the Protective Function of Tregs in a Murine Model of PGD
Tregs are well established as suppressors of immune and inflammatory responses, with most attention being given to their control of adaptive immunity. However, the contribution of Tregs to the prevention of PGD has not been explored experimentally. We developed a lung ischemia–reperfusion injury model in Rag1−/− mice. These mice lack mature T cells or B cells, such that any Treg effects must involve direct interactions with innate immune cells and/or nonimmune components such as the lung epithelium and endothelium. We found that just 0.5 million Tregs, injected the night before operation, were able to dramatically decrease inflammation associated with ischemia–reperfusion, as shown by the significantly reduced mRNA levels of cytokines in ischemic lungs (Figures 5A, 5B, E4D, and E4E) and by the markedly reduced histologic injury (Figures 5E, 5F, and E4J). In contrast, IL-18–pretreated Tregs lost their ability to control lung ischemia–reperfusion injury in RAG1−/− mice (Figures 5C–5F, E4F, E4G, and E4J). Therefore, Tregs can directly limit innate immunity–driven inflammation in an in vivo PGD model and do not require mediation by T or B cells, but this protective capacity is lost upon Treg exposure to IL-18. To maximize the translational value of our study, we performed a series of experiments with lungs that underwent 24 hours of cold ischemia and were then transplanted into three types of recipients: iHFD normal-weight control mice, iHFD obese mice, and iHFD obese mice treated with IL-18BP (Figure 5G). Obese mice that did not receive IL-18BP treatment showed significantly enhanced inflammation, whereas obese mice that received IL-18BP treatment showed alleviation of the most negative effects of their obesity (Figures 5H–5J and E4H–E4J).
Figure 5.
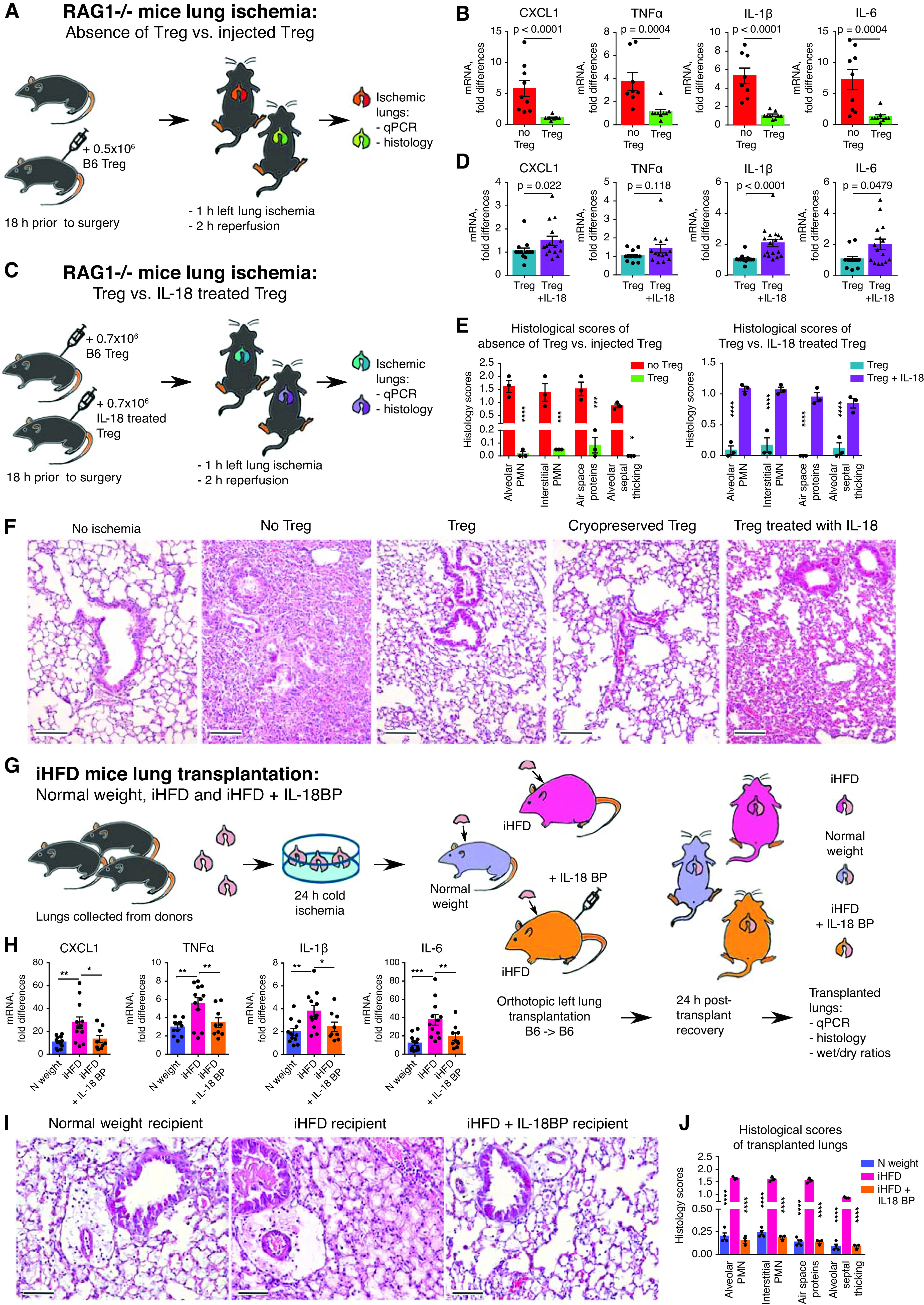
Two murine models of primary graft dysfunction. (A, B, E, and F) RAG1−/− mice (n = 12) received PBS or 0.5 × 106 T-regulatory cells (Tregs) intravenously; 18 hours later, lung ischemia–reperfusion was performed, which consisted of 1 hour of left lung ischemia and 2 hours of reperfusion; and then lungs were collected for quantitative PCR (qPCR) analysis (B) and histologic evaluation (E [left] and F). (C–F) RAG1−/− mice (n = 17) received intravenous 0.7 × 106 Tregs, which were preliminarily treated with 200 ng/ml IL-18 or PBS for 3 hours and then washed and cryopreserved. Eighteen hours later, lung ischemia–reperfusion was performed, which consisted of 1 hour of left lung ischemia and 2 hours of reperfusion; then both lungs were collected for (C) qPCR analysis and (H [right] and F) histologic evaluation. The median number (interquartile range) of samples evaluated for each marker is as follows: 7 (7–8) in B; 13.5 (12–14.75) in D. (F) Representative examples and (E) scoring from histology of left lungs (hematoxylin and eosin staining; scale bars, 100 μm; n = 12). (G–I) Wild-type B6 donor lungs were kept for 24 hours at 4°C to induce prolonged cold ischemia. On the next day, those lungs were transplanted into three different recipients: normal (N)-weight male mice receiving a control “inflammatory” high-fat diet (iHFD), obese male mice receiving an iHFD, and obese mice receiving an iHFD and IL-18 binding peptide (IL-18BP) treatment. For the last group, we injected 7 mg/kg IL-18BP intraperitoneally at 1 hour before operation. On the next day, both lungs were collected for dry/wet evaluation (see Figure E4H in the online supplement) before undergoing (H) qPCR and (I and J) histologic evaluation (scale bars, 100 μm). Ten mice in total were evaluated for qPCR analysis and histologic scoring: four with N weight, three untreated iHFD mice, and three IL-18BP–treated iHFD mice. More data are presented in Figures E4D–E4J. A Mann-Whitney U test was used in B and D, and two-way ANOVA with a Sidak’s test was used in E, H, and J. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. PMN = polymorphonuclear neutrophils.
Discussion
This study identified IL-18 as the most promising target to explain the differences between obesity affecting Treg function (iHFD) and obesity with preserved Treg function (hHFD). Although the vast majority of HFD models and clinical data describe low-grade local and systemic inflammation caused by obesity (29–31), none of them identified defects in Tregs due to increased levels of IL-18. There are reports of decreased (32, 33), increased (34), or unaltered (35) Treg numbers, and one study (36) reported that there were detrimental effects of hyperlipidemia on Treg function in HFD mice.
Prior studies have suggested contradictory effects of IL-18 on Tregs. Thus, IL-18 was reported to enhance Treg function (37, 38). Conversely, another study reported a significant decrease in the number of human engrafted Tregs and reported aggravated graft-versus-host disease in mice treated with human IL-18 in vivo but found no effects of IL-18 on human Tregs’ suppressive function in vitro (39). However, in the latter study, the authors studied Tregs expanded in vitro but did not study freshly isolated Tregs. Lastly, IL-18– and IL-18R–deficient mice, infected with Helicobacter pylori, were reported to have defects in the number and function of Tregs (40). This report contrasted with another study showing that IL-18–deficient mice had normal Treg numbers and function (41). Hence, to our knowledge, we are the first to evaluate the direct effects of IL-18 on freshly isolated human and murine Tregs and to extend those data in vivo by showing that although IL-18 impaired the ability of Tregs to control lung inflammation in a murine model of PGD, the use of an IL-18 inhibitor, IL-18BP, ameliorated inflammation in the transplanted lungs of obese recipients.
The basis for the systemic increase of IL-18 levels in iHFD mice and in obese transplant patients is currently unknown but may be related to activation of inflammasome pathways. Transplant patients demonstrating “IL-18–accompanied” inflammatory obesity in the current study had end-stage disease of the kidney, liver, or lung, all of which are known to result in ongoing inflammasome activation (42). Therefore, we can suggest that those individuals are prone to developing systemic increased IL-18 levels, which may be further provoked by obesity, and as a result, their Tregs may be much less able to control naive immune activation early after transplant.
In line with our data, elevated serum IL-18 levels correlated with hepatocyte injury and systemic inflammation in obese children (43). Another recent study found that tetracycline inhibits inflammasome–caspase-1 signaling in patients with acute respiratory distress syndrome, which led to decreased production of IL-1β and IL-18 and significantly diminished lung injury and inflammation (44). The long-term effects of obesity on allograft survival with regard to IL-18 and Treg function deserve further studies, but current data report inferior patient and graft survival as being linked to obesity (45, 46), and our own data from pediatric and adult recipients confirms that.
Inflammasome activation, IL-18, and activation of innate immunity are important for ischemia–reperfusion injury and early post-transplant outcomes (8–10, 47). The major role of adaptive immunity, regulated by Tregs, in long-term allograft acceptance and function is well known (48). Conversely, the role of Tregs in controlling activation of innate immunity and lung injury is only beginning to be recognized (49). The current study introduces two previously unrecognized but important players: the role of IL-18 in obesity, with detrimental effects of IL-18 on Tregs being shown, and an important role of Tregs in the direct control of early inflammation driven by lung ischemia–reperfusion in a PGD model. These findings may have relevance to the pretransplant evaluation of patients listed as candidates for lung transplant and to identifying those at increased risk of developing PGD.
Footnotes
Supported by NIH grants R01HL114468 (W.W.H.), R01HL154241-01A1 (W.W.H.), R01HL114626 (D.J.L.), and U01HL145435, K24HL115354, R01087115, R01HL113252, and R01HL114626 (J.D.C.).
Author Contributions: T.A. designed and performed most of the experiments (except surgery), evaluated data, performed statistical analyses, and wrote the manuscript. T.Z., L.M.C., D.N., L.W., and U.H.B. performed flow cytometry, cell isolation, and cell culture experiments. Z.W. performed lung ischemia–reperfusion experiments, L.W. performed lung transplants, and T.Z. and L.M.C. collected and prepared tissue samples. R.H., T.G., I.E.S., and J.J. managed mice colonies as well as the corresponding diets and housing requirements and provided mice for studies. R.H. prepared microscopy sections with hematoxylin and eosin staining. A.S. performed TUBE assays. T.R.B. analyzed hematoxylin and eosin–stained sections. M.A. assisted with collection of clinical blood samples. M.H.L., J.M.D., E.C., D.S.W., D.J.L., and J.D.C. conceived and managed corresponding clinical studies and provided reagents, patient samples, and clinical data. T.A., R.A.S., J.M.D., J.D.C., and W.W.H. designed experiments and analyzed data. W.W.H., U.H.B., M.H.L., J.M.D., R.A.S., E.C., D.S.W., D.J.L., and J.D.C. edited the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202012-4306OC on August 4, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Lee MK, Yvan-Charvet L, Masters SL, Murphy AJ. The modern interleukin-1 superfamily: divergent roles in obesity. Semin Immunol . 2016;28:441–449. doi: 10.1016/j.smim.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 2. Rex DAB, Agarwal N, Prasad TSK, Kandasamy RK, Subbannayya Y, Pinto SM. A comprehensive pathway map of IL-18-mediated signalling. J Cell Commun Signal . 2020;14:257–266. doi: 10.1007/s12079-019-00544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murphy AJ, Kraakman MJ, Kammoun HL, Dragoljevic D, Lee MK, Lawlor KE, et al. IL-18 production from the NLRP1 inflammasome prevents obesity and metabolic syndrome. Cell Metab . 2016;23:155–164. doi: 10.1016/j.cmet.2015.09.024. [DOI] [PubMed] [Google Scholar]
- 4. Diamond JM, Lee JC, Kawut SM, Shah RJ, Localio AR, Bellamy SL, et al. Lung Transplant Outcomes Group. Clinical risk factors for primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med . 2013;187:527–534. doi: 10.1164/rccm.201210-1865OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer KC. Recent advances in lung transplantation. F1000Res. 2018;7 doi: 10.12688/f1000research.15393.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenheck J, Pietras C, Cantu E. Early graft dysfunction after lung transplantation. Curr Pulmonol Rep . 2018;7:176–187. doi: 10.1007/s13665-018-0213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Porteous MK, Diamond JM, Christie JD. Primary graft dysfunction: lessons learned about the first 72 h after lung transplantation. Curr Opin Organ Transplant . 2015;20:506–514. doi: 10.1097/MOT.0000000000000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weigt SS, Palchevskiy V, Belperio JA. Inflammasomes and IL-1 biology in the pathogenesis of allograft dysfunction. J Clin Invest . 2017;127:2022–2029. doi: 10.1172/JCI93537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cantu E, Lederer DJ, Meyer K, Milewski K, Suzuki Y, Shah RJ, et al. CTOT Investigators. Gene set enrichment analysis identifies key innate immune pathways in primary graft dysfunction after lung transplantation. Am J Transplant . 2013;13:1898–1904. doi: 10.1111/ajt.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kreisel D, Goldstein DR. Innate immunity and organ transplantation: focus on lung transplantation. Transpl Int . 2013;26:2–10. doi: 10.1111/j.1432-2277.2012.01549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lederer DJ, Kawut SM, Wickersham N, Winterbottom C, Bhorade S, Palmer SM, et al. Lung Transplant Outcomes Group. Obesity and primary graft dysfunction after lung transplantation: the Lung Transplant Outcomes Group Obesity Study. Am J Respir Crit Care Med . 2011;184:1055–1061. doi: 10.1164/rccm.201104-0728OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anderson MR, Udupa JK, Edwin E, Diamond JM, Singer JP, Kukreja J, et al. Adipose tissue quantification and primary graft dysfunction after lung transplantation: The Lung Transplant Body Composition Study. J Heart Lung Transplant . 2019;38:1246–1256. doi: 10.1016/j.healun.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hancock WW, Zhang T, Beier UH, Samanta A, Han R, Wang L, et al. Obesity induced IL-18 production impairs FOXP3+ Treg function and increases risk of primary graft dysfunction in lung transplant recipients [abstract] Am J Transplant . 2019;19:1008–1009. [Google Scholar]
- 14.Akimova T, Zhang T, Beier UH, Samanta A, Han R, Wang L, et al. Obesity leads to increased IL-18, Treg impairment and increased risk of PGD [abstract] J Heart Lung Transplant 201938S329. [Google Scholar]
- 15. Akimova T, Zhang T, Beier UH, Jiao J, Diamond JM, Christie JD, et al. Obesity-driven IL-18 inhibits the suppressive function of FOXP3+ T-regulatory (Treg) cells [abstract] J Immunol. 2018;200:166.152. [Google Scholar]
- 16. Christie JD, Carby M, Bag R, Corris P, Hertz M, Weill D. ISHLT Working Group on Primary Lung Graft Dysfunction. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction part II: definition. A consensus statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant . 2005;24:1454–1459. doi: 10.1016/j.healun.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 17. Akimova T, Kamath BM, Goebel JW, Meyers KE, Rand EB, Hawkins A, et al. Differing effects of rapamycin or calcineurin inhibitor on T-regulatory cells in pediatric liver and kidney transplant recipients. Am J Transplant . 2012;12:3449–3461. doi: 10.1111/j.1600-6143.2012.04269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T lymphocytes mediate acute pulmonary ischemia-reperfusion injury. J Thorac Cardiovasc Surg . 2009;137:695–702, discussion 702. doi: 10.1016/j.jtcvs.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Outtz Reed H, Wang L, Kahn ML, Hancock WW. Donor-host lymphatic anastomosis after murine lung transplantation. Transplantation . 2020;104:511–515. doi: 10.1097/TP.0000000000003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akimova T, Zhang T, Negorev D, Singhal S, Stadanlick J, Rao A, et al. Human lung tumor FOXP3+ Tregs upregulate four “Treg-locking” transcription factors. JCI Insight . 2017;2:e94075. doi: 10.1172/jci.insight.94075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Akimova T, Levine MH, Beier UH, Hancock WW. Standardization, evaluation, and area-under-curve analysis of human and murine Treg suppressive function. Methods Mol Biol . 2016;1371:43–78. doi: 10.1007/978-1-4939-3139-2_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao Y, Nagai Y, Deng G, Ohtani T, Zhu Z, Zhou Z, et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep . 2014;7:1471–1480. doi: 10.1016/j.celrep.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenwood C, Ruff D, Kirvell S, Johnson G, Dhillon HS, Bustin SA. Proximity assays for sensitive quantification of proteins. Biomol Detect Quantif . 2015;4:10–16. doi: 10.1016/j.bdq.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chae WJ, Henegariu O, Lee SK, Bothwell AL. The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. Proc Natl Acad Sci USA . 2006;103:9631–9636. doi: 10.1073/pnas.0600225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang L, Kumar S, Dahiya S, Wang F, Wu J, Newick K, et al. Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory cell function and promotes antitumor immunity. EBioMedicine . 2016;13:99–112. doi: 10.1016/j.ebiom.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity . 2013;39:272–285. doi: 10.1016/j.immuni.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raphael J, Collins SR, Wang XQ, Scalzo DC, Singla P, Lau CL, et al. Perioperative statin use is associated with decreased incidence of primary graft dysfunction after lung transplantation. J Heart Lung Transplant . 2017;36:948–956. doi: 10.1016/j.healun.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 28. Liberale L, Carbone F, Montecucco F, Sahebkar A. Statins reduce vascular inflammation in atherogenesis: a review of underlying molecular mechanisms. Int J Biochem Cell Biol . 2020;122:105735. doi: 10.1016/j.biocel.2020.105735. [DOI] [PubMed] [Google Scholar]
- 29. Lee YS, Wollam J, Olefsky JM. An integrated view of immunometabolism. Cell . 2018;172:22–40. doi: 10.1016/j.cell.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McLaughlin T, Ackerman SE, Shen L, Engleman E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest . 2017;127:5–13. doi: 10.1172/JCI88876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao L, Zhong S, Qu H, Xie Y, Cao Z, Li Q, et al. Chronic inflammation aggravates metabolic disorders of hepatic fatty acids in high-fat diet-induced obese mice. Sci Rep. 2015;5:10222. doi: 10.1038/srep10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wagner NM, Brandhorst G, Czepluch F, Lankeit M, Eberle C, Herzberg S, et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity (Silver Spring) . 2013;21:461–468. doi: 10.1002/oby.20087. [DOI] [PubMed] [Google Scholar]
- 33. Luczyński W, Wawrusiewicz-Kurylonek N, Iłendo E, Bossowski A, Głowińska-Olszewska B, Krętowski A, et al. Generation of functional T-regulatory cells in children with metabolic syndrome. Arch Immunol Ther Exp (Warsz) . 2012;60:487–495. doi: 10.1007/s00005-012-0198-6. [DOI] [PubMed] [Google Scholar]
- 34.van der Weerd K, Dik WA, Schrijver B, Schweitzer DH, Langerak AW, Drexhage HA, et al. Morbidly obese human subjects have increased peripheral blood CD4+ T cells with skewing toward a Treg- and Th2-dominated phenotype. Diabetes. 2012;61:401–408. doi: 10.2337/db11-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Donninelli G, Del Cornò M, Pierdominici M, Scazzocchio B, Varì R, Varano B, et al. Distinct blood and visceral adipose tissue regulatory T cell and innate lymphocyte profiles characterize obesity and colorectal cancer. Front Immunol . 2017;8:643. doi: 10.3389/fimmu.2017.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bagley J, Yuan J, Iacomini J. Impact of hyperlipidemia on alloimmunity. Curr Opin Organ Transplant . 2017;22:14–21. doi: 10.1097/MOT.0000000000000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol . 2015;8:1226–1236. doi: 10.1038/mi.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell . 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carroll RG, Carpenito C, Shan X, Danet-Desnoyers G, Liu R, Jiang S, et al. Distinct effects of IL-18 on the engraftment and function of human effector CD8 T cells and regulatory T cells. PLoS One . 2008;3:e3289. doi: 10.1371/journal.pone.0003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest . 2012;122:1082–1096. doi: 10.1172/JCI61029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zeiser R, Zambricki EA, Leveson-Gower D, Kambham N, Beilhack A, Negrin RS. Host-derived interleukin-18 differentially impacts regulatory and conventional T cell expansion during acute graft- versus-host disease. Biol Blood Marrow Transplant . 2007;13:1427–1438. doi: 10.1016/j.bbmt.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol . 2019;15:501–520. doi: 10.1038/s41581-019-0158-z. [DOI] [PubMed] [Google Scholar]
- 43. Flisiak-Jackiewicz M, Bobrus-Chociej A, Tarasów E, Wojtkowska M, Białokoz-Kalinowska I, Lebensztejn DM. Predictive role of interleukin-18 in liver steatosis in obese children. Can J Gastroenterol Hepatol . 2018;2018:3870454. doi: 10.1155/2018/3870454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peukert K, Fox M, Schulz S, Feuerborn C, Frede S, Putensen C, et al. Inhibition of caspase-1 with tetracycline ameliorates acute lung injury. Am J Respir Crit Care Med . 2021;204:53–63. doi: 10.1164/rccm.202005-1916OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heinbokel T, Floerchinger B, Schmiderer A, Edtinger K, Liu G, Elkhal A, et al. Obesity and its impact on transplantation and alloimmunity. Transplantation . 2013;96:10–16. doi: 10.1097/TP.0b013e3182869d2f. [DOI] [PubMed] [Google Scholar]
- 46. Dick AA, Perkins JD, Spitzer AL, Lao OB, Healey PJ, Reyes JD. Impact of obesity on children undergoing liver transplantation. Liver Transpl . 2010;16:1296–1302. doi: 10.1002/lt.22162. [DOI] [PubMed] [Google Scholar]
- 47. Liu C, Chen J, Liu B, Yuan S, Shou D, Wen L, et al. Role of IL-18 in transplant biology. Eur Cytokine Netw . 2018;29:48–51. doi: 10.1684/ecn.2018.0410. [DOI] [PubMed] [Google Scholar]
- 48. Burrell BE, Nakayama Y, Xu J, Brinkman CC, Bromberg JS. Regulatory T cell induction, migration, and function in transplantation. J Immunol . 2012;189:4705–4711. doi: 10.4049/jimmunol.1202027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang L, Wang X, Tong L, Wang J, Dou M, Ji S, et al. Recovery from acute lung injury can be regulated via modulation of regulatory T cells and Th17 cells. Scand J Immunol . 2018;88:e12715. doi: 10.1111/sji.12715. [DOI] [PubMed] [Google Scholar]
- 50. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 directs FOXP3 localization and facilitates regulatory T-cell function through K63-linked ubiquitination. EMBO J . 2019;38:e99766. doi: 10.15252/embj.201899766. [DOI] [PMC free article] [PubMed] [Google Scholar]