

Summary
Based on epidemiology studies, Chlamydia trachomatis has been proposed as a co-factor for human papillomavirus (HPV) in the development of cervical cancer. These two intracellular pathogens have been independently reported to induce the production of extra centrosomes, or centrosome amplification, which is a hallmark of cancer cells. We developed a cell culture model to systematically measure the individual and combined effects of Chlamydia and HPV on the centrosome in the same host cell. We found that C. trachomatis caused centrosome amplification in a greater proportion of cells than HPV and that the effects of the two pathogens on the centrosome were additive. Furthermore, centrosome amplification induced by Chlamydia, but not by HPV, strongly correlated with multinucleation and required progression through mitosis. Our results suggest that C. trachomatis and HPV induce centrosome amplification through different mechanisms with the chlamydial effect being largely due to a failure in cytokinesis that also results in multinucleation. Our findings provide support for C. trachomatis as a co-factor for HPV in carcinogenesis and offer mechanistic insights into how two infectious agents may cooperate to promote cancer.
Keywords: host-pathogen interaction, co-infection, sexually-transmitted infection, multinucleation, cell cycle progression, cervical cancer
Introduction
Each year, cervical cancer causes 300,000 deaths in the world, making it the fourth most common cancer in women (Arbyn et al. 2018). Human papillomavirus (HPV) is its main etiologic agent, with 90% of cervical carcinomas linked to “high-risk” HPV types, such as HPV16 and 18 (Muñoz et al. 2003). However, not all women infected with HPV develop cervical cancer, which suggests that additional factors are involved in carcinogenesis. A number of co-factors, including smoking, long-term use of oral contraceptives, and Chlamydia trachomatis infection, have been proposed (Fonseca-Moutinho 2011; International Collaboration of Epidemiological Studies of Cervical Cancer et al. 2007; Silva, Cerqueira, and Medeiros 2014). The evidence for C. trachomatis as co-factor is based on sero-epidemiology studies showing that women with cervical cancer were more likely to have had a prior Chlamydia infection (Smith et al. 2002; Zhu et al. 2016).
HPV contributes to oncogenesis through multiple mechanisms. It causes aberrant proliferation of host cells, which supports viral DNA replication (Münger et al. 2004). It also promotes genomic instability in an infected host cell by inactivating cell cycle checkpoints, dysregulating host DNA repair pathways, and inducing centrosome abnormalities (Thomas and Laimins 1998; Banerjee et al. 2011; Spardy et al. 2009; Duensing et al. 2000). As a consequence, HPV-infected cells accumulate cellular mutations while undergoing enhanced proliferation, which together lead to malignant transformation.
The centrosome, an organelle with a key role in microtubule organization, is dysregulated in many cancer cells (Salisbury et al. 1999; Chan 2011). Normal diploid cells have a single centrosome, which duplicates in parallel to DNA in S-phase of the cell cycle. Cancer cells often contain extra, or supernumerary, centrosomes, which is a phenomenon called centrosome amplification (Lingle et al. 1998; Pihan et al. 1998). Such supernumerary centrosomes contribute to carcinogenesis by leading to chromosome missegregation, genomic instability, and enhanced cell invasiveness (Ganem, Godinho, and Pellman 2009; Godinho et al. 2014). Centrosome amplification is caused by at least three distinct mechanisms, which include cell-cell fusion, cytokinesis defects, and dysregulation of the centrosome duplication machinery (Godinho and Pellman 2014).
High risk HPV induces centrosome amplification through its oncoproteins E6 and E7. Co-expression of E6 and E7 is sufficient to increase centrosome number in normal human keratinocytes, which leads to multipolar spindles and ultimately to genomic instability (Duensing et al. 2000). Additionally, mice expressing E6 and E7 have cervical and skin lesions containing cells with multiple centrosomes (Schaeffer et al. 2004). E7 expression has been proposed to promote centrosome amplification by altering the centrosome duplication machinery, whereas the mechanism for E6-induced centrosome amplification is less understood (Duensing et al. 2006; Duensing and Münger 2002).
C. trachomatis also induces centrosome amplification (Grieshaber et al. 2006; Johnson et al. 2009). Tissue culture cells infected with this obligate intracellular bacterium formed extra centrosomes in interphase and multipolar spindles in mitosis (Grieshaber et al. 2006). Intriguingly, these phenotypes persisted after the cells were cured of the infection. C. trachomatis has been proposed to induce centrosome abnormalities by dysregulating the centrosome duplication machinery and by causing cytokinesis defects in host cells (Johnson et al. 2009; Alzhanov et al. 2009). An important caveat is that prior mechanistic investigations were mostly done in C. trachomatis-infected HeLa cells, which contain HPV-18 DNA (Schwarz et al. 1985). Thus, these studies measured the combined effects of Chlamydia and HPV on the centrosome, but not the individual contribution of C. trachomatis.
In the present study, we investigated the respective roles of HPV and Chlamydia in causing centrosome abnormalities. To accomplish this goal, we developed a cell culture system that allowed us to determine the individual and combined effects of these two sexually transmitted pathogens on the centrosome in the same host cell. Our results provide biologic plausibility for a role of Chlamydia as a co-factor for HPV in the development of cervical cancer.
Experimental procedures
Antibodies used in this study
Primary antibodies: anti-Centrin (Millipore, 04–1624), anti-γ-tubulin (Abcam, ab11321), anti-α-tubulin (Sigma, T5168), anti-Cep164 (Santa Cruz, sc-240226). Secondary antibodies for immunofluorescence microscopy: Donkey anti-Rabbit IgG Alexa Fluor 488 (Invitrogen, A21206), Donkey anti-Mouse IgG Alexa Fluor 555 (Invitrogen, A31570), Goat anti-Rat IgG Alexa Fluor 564 (Invitrogen, A11081), Donkey anti-Goat IgG Alexa Fluor 488 (Invitrogen, A21202).
Cell culture and Chlamydia infection
The parental hTERT-RPE-1 cell line was obtained from ATCC and cultured at 37°C and 5.0% CO2 in DMEM (Gibco, 11995–065) supplemented with 10% FBS (Atlanta Biologicals, S11550). A549 cells stably expressing HPV16 E6/E7 were a generous gift from Dr. Ashok Aiyar (LSU New Orleans) and were grown in DMEM supplemented with 10% FBS.
Chlamydia infection was done by infecting near-confluent cell monolayers with C. trachomatis serovar L2 (ATCC) at an MOI of 3 in SPG (200 mM sucrose, 20 mM sodium phosphate and 5 mM glutamate; pH 7.2) followed by centrifugation at 700×g for 1 hour at room temperature. As control, cells were mock infected with SPG alone. After centrifugation, the inoculum was removed and replaced with DMEM containing 10% FBS. The same infection conditions were used for RPE-1 and A549 cells.
Generation of RPE-1 cell lines expressing HPV16 E6/E7 oncoproteins
The pLXSN-HPV16 E6/E7 retroviral vector was obtained from Addgene (Plasmid #52394). From this vector, the pLXSN-Empty control vector was generated using Gibson assembly with forward primer 5′-TCCTCTAGAGTCCTGTAATCCTACCATGGCTGATCCTGCAG-3′ and reverse primer 5′-GATTACAGGACTCTAGAGGATC-3′. The pLXSN retroviral vectors and helper plasmid were co-transfected in 293T cells with calcium phosphate (293T cells and helper plasmid were generous gifts from Dr. Aimee Edinger, UC Irvine). Viral particles were collected 48 hours post transfection and used to infect RPE-1 cell monolayers with 10ug/mL polybrene (Sigma). Colonies were pooled after 10 days of selection with 600ug/mL G418 (Fisher, BP-918). HPV16 E6/E7 expressions were confirmed via RT-PCR. Forward primer 5′-GCAAGCAACAGTTACTGCG-3′ and reverse primer 5′-GGTTTCTCTACGTGTTCTTG-3′ were used to detect HPV16 E6 expression and primer pair 5′-CAGCTCAGAGGAGGAGGATG-3′ and 5′-GCCCATTAACAGGTCTTCCA-3′ were used to detect HPV16 E7 expression.
Immunofluorescence microscopy
Cells, grown and infected on glass coverslips, were fixed in 100% ice-cold methanol for 10 minutes. Cells were permeabilized and incubated in blocking buffer (2% FBS, 0.1% Triton) for 30 minutes at room temperature. C. trachomatis and host cell DNA was stained with NucBlue (Invitrogen, P36985). Centrosomes were detected with antibodies to Centrin to observe centrioles, Cep164 to mark mother centrioles and γ-tubulin to observe pericentriolar material. Mitotic spindles and microtubule regrowth were visualized with anti-α-tubulin antibody. Coverslips were mounted with ProLong Glass Antifade containing NucBlue (Invitrogen, P36985). Immunofluorescence microscopy images were acquired on a Zeiss Axiovert 200M microscope.
Microtubule regrowth assay
Cells grown on coverslips in growth medium supplemented with 25mM HEPES, were first incubated on ice for 40 minutes to depolymerize microtubules and then shifted to room temperature for 4 minutes to allow microtubule regrowth. Cells were rinsed for 40 seconds with microtubule buffer (60mM PIPES, 25mM HEPES, 10mM EGTA, 2mM MgCl2, 0.25nM Nocodazole, 0.25nM Paclitaxel, pH 6.9) and fixed with ice-cold methanol for 7 minutes prior to immunofluorescence microscopy analysis with antibodies to α-tubulin and γ-tubulin.
Pharmacological inhibition of cell cycle progression and centrosome duplication
Mock or Chlamydia-infected RPE-1 cells were arrested in S-phase by incubating them with 2mM thymidine (ACROS Organics, 226740050) in standard growth medium for 24 hours, starting at 12 hpi. Cells were arrested in G2 by first incubating them with 2mM thymidine for 18 hours, followed by release from the thymidine block for 6 hours and incubation with 10μM RO-3306 (TOCRIS, cat # 4181) in standard medium for 12 hours. Both the S-phase and the G2 arrest experiments were analyzed at 36 hpi using immunofluorescence microscopy.
To inhibit centrosome duplication, Chlamydia, HPV and HPV+Chlamydia cells were incubated with 125nM of the Plk4 inhibitor centrinone (MedChemExpress HY-18682) in standard growth medium starting at 1 hpi. The experiment was evaluated by immunofluorescence microscopy at 36 hpi.
EdU labeling
Cells undergoing S-phase were identified using the Click-iT EdU Cell Proliferation kit (Invitrogen, C10337). Control or HPV cells were grown on coverslips and infected with Chlamydia at an MOI of 3. At 36 hpi, cells were incubated with 10μM EdU for 30 minutes and fixed with 4% PFA. EdU labeled cells were detected following the manufacturer’s protocol.
Statistical analyses
For each experiment, 3 independent biological replicates were performed, and the results are presented as mean ± SD. Data were analyzed by unpaired, two-tailed t-tests with Welch’s correction on Graph Pad PRISM software version 8.
Results
An experimental system for studying the effects of Chlamydia and HPV on the centrosome
We developed a cell culture model that allowed us to separate the effects of HPV and Chlamydia trachomatis (referred to as Chlamydia hereafter) on the centrosome in the same human cell (Fig. 1A). We used retinal pigment epithelial cells (RPE-1) as the host cell because these diploid epithelial cells are neither cancerous nor transformed by HPV, unlike HeLa or A2EN cells that are commonly utilized for Chlamydia infection (Buckner et al. 2016). To mimic the effects of HPV on the centrosome, we generated an RPE-1 cell line that stably expresses the viral oncoproteins HPV16 E6 and E7 (referred to as “HPV cells”) (Fig. S1). Prior studies showed that ectopic co-expression of the HPV oncoproteins E6 and E7 was sufficient to induce centrosome amplification (Duensing et al. 2000; Duensing and Münger 2002). Ectopic E6/E7 expression is proposed to mimic the effect of HPV on the centrosome, with the advantage that this approach does not require a stratified epithelium typically used in an HPV infection model (Bienkowska-Haba et al. 2018). However, because we are not performing actual HPV infections, we cannot exclude the possibility that other HPV factors may contribute to the phenotypes measured in this study. RPE-1 cells transduced with an empty vector lacking these viral oncogenes served as a negative control (“control cells”).
Figure 1. Chlamydia and HPV have additive effects on the host cell centrosome.
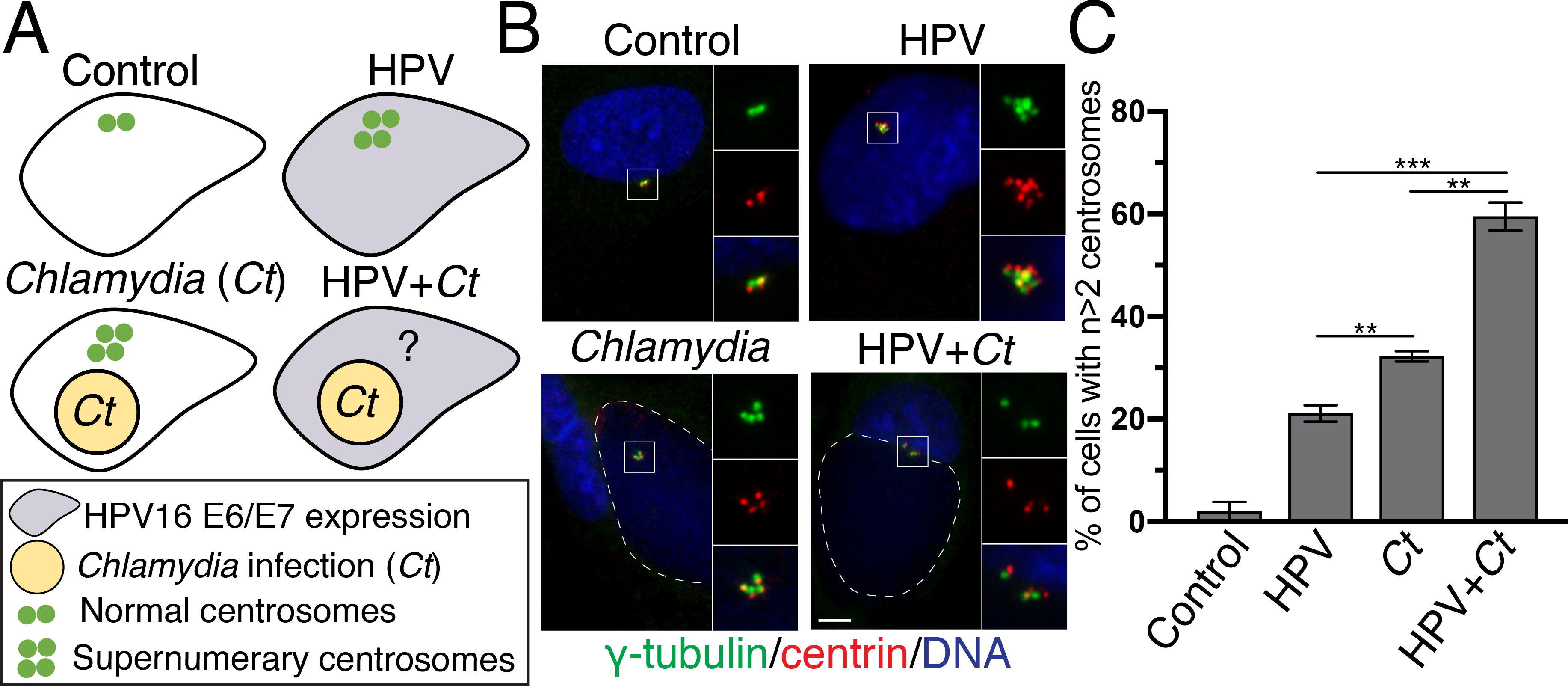
(A) Schematic representation of the experimental design to study the separate and combined effect of Chlamydia and HPV on the centrosome. Control cells are RPE-1 cells transfected with an empty plasmid. “HPV cells” are RPE-1 cells that stably express HPV16 oncoproteins E6 and E7 (gray cell). “Chlamydia cells” are RPE-1 cells infected with Chlamydia trachomatis (yellow circle), while “HPV+Chlamydia” cells are RPE-1 cells that stably express HPV16 oncoproteins and that are infected with C. trachomatis. Individual centrosomes are presented as green dots.
(B) Control RPE-1 cells or HPV cells, grown on coverslips, were either mock-infected or infected with C. trachomatis L2 at an MOI of 3. Samples were fixed at 36 hours post infection (hpi). Centrosomes were visualized with antibodies to γ-tubulin (green) and centrin (red), host and chlamydial DNA was detected with DAPI (blue). Chlamydial inclusions are outlined with white dashed lines. Scale bar: 5 μm.
(C) The percentage of host cells with supernumerary centrosomes (n>2 centrosomes) from the four different conditions is shown. 100 cells were analyzed for each condition. For the samples with a Chlamydia infection, only infected cells were examined and quantified. Data are represented as mean ± SD (n=3); **P≤0.01 and ***P<0.001.
We infected either control cells with C. trachomatis to produce “Chlamydia cells”, or HPV cells to generate “HPV+Chlamydia cells”. For these infections, we used C. trachomatis serovar L2 because this strain has been used as an experimental model to study C. trachomatis-induced centrosome amplification (Grieshaber et al. 2006; Johnson et al. 2009). This strain is representative of other C. trachomatis strains, such as the genital serovars D and G, that produce comparable levels of centrosome amplification (Grieshaber et al. 2006).
We then compared the percentage of control, HPV, Chlamydia and HPV+Chlamydia cells with amplified centrosomes. We detected centrosomes by immunofluorescence microscopy with antibodies to the centrosomal marker proteins γ-tubulin and centrin2, which stain the pericentriolar material (PCM) and centrioles, respectively. Centrosome amplification was defined as cells harboring more than 2 centrosomes (n>2 γ-tubulin dots) (Fig. 1B).
Chlamydia and HPV dysregulated centrosome number in an additive manner
This systematic approach revealed different effects of these two pathogens on the centrosome. Control cells had only few supernumerary centrosomes (Prevalence of 1.9%). Chlamydia cells showed a higher prevalence of amplified centrosomes than HPV cells (Prevalence of 32.2% vs 21.1%) (Fig. 1C), but the highest percentage of extra centrosomes (59.5%) was seen in HPV+Chlamydia cells. These data demonstrated that Chlamydia had a greater effect on the centrosome than HPV, and that together, they caused more centrosome amplification than either infectious agent alone (Fig. 1C). Thus, HPV and Chlamydia cause centrosome amplification in a host cell through additive mechanisms.
We also examined effects of HPV and Chlamydia on the centrosome of A549 cells, which are HPV-negative lung carcinoma cells. Similar to our results with RPE-1 cells, there was a greater prevalence of centrosome amplification in Chlamydia cells when compared to HPV cells. However, because E6/E7 expression did not cause a statistically significant increase in the percentage of A549 cells with amplified centrosomes, we were unable to test if the effects of HPV and Chlamydia on centrosome amplification are additive (Fig. S2). We conclude from these experiments that unlike for HPV, the prominent effects of Chlamydia in dysregulating centrosome number is independent of the cell type used as the host cell.
Chlamydia- and HPV-induced centrosome amplification did not disrupt the function of the centrosome in organizing microtubules
We next tested if Chlamydia and HPV altered the function of the centrosome, which organizes microtubules by controlling their nucleation and anchoring. HPV, Chlamydia and HPV+Chlamydia cells all formed a radial array of interphase microtubules that was indistinguishable from that of control cells (Fig. 2A). There was also no difference in the growth kinetics of microtubules as measured in regrowth assays (Fig. 2B).
Figure 2. Centrosome amplification does not disrupt centrosome function.
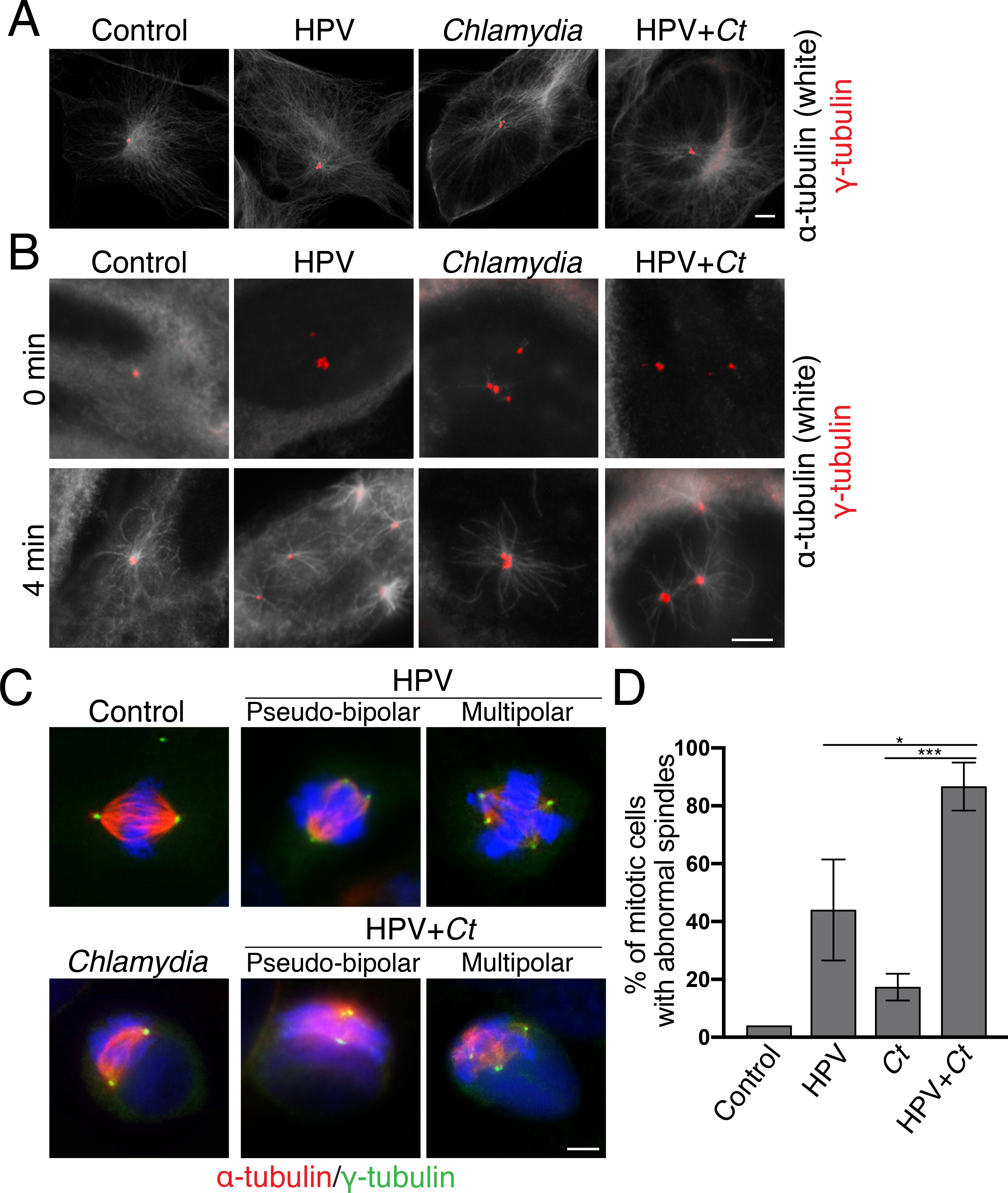
(A) Immunofluorescence images of mock or Chlamydia-infected RPE-1 or HPV cells fixed at 36hpi in interphase. Microtubules were visualized with α-tubulin antibodies (white) and centrosomes were detected with γ-tubulin antibodies (red). Scale bar: 5μm.
(B) Microtubule regrowth assays are shown for cells of each of the four experimental conditions. Cells were incubated on ice to depolymerize microtubules (0 min) and then shifted to 37°C for 4 minutes to allow microtubule nucleation and polymerization (4 min). Cells were fixed at the indicated time points and analyzed by immunofluorescence microscopy, detecting microtubules and centrosomes as described in (A). Scale bar: 5μm.
(C) Immunofluorescence images of mitotic cells in the samples in (A). Mitotic spindles were identified with antibodies to α-tubulin antibodies (red) and centrosomes were visualized with γ-tubulin antibodies (green). DNA was visualized with DAPI (blue). Scale bar: 5μm.
(D) The percentage of mitotic cells with abnormal spindles in each of the four conditions is shown. Both pseudo-bipolar (centrosome clustering) and multipolar spindles were considered abnormal. 50 mitotic cells were analyzed for each condition. For the samples with a Chlamydia infection, only infected cells were examined and quantified. Data are represented as mean ± SD (n=3); *P≤0.05 and ***P<0.001.
We further analyzed spindle formation to assess centrosome function in mitosis (Fig. 2C). HPV and HPV+Chlamydia cells showed a higher prevalence of abnormal spindles, including multipolar and pseudo-bipolar spindles, than control or Chlamydia cells (Figs. 2C and 2D). This finding suggests that the abnormal interphase centrosomes in HPV and HPV+Chlamydia cells may promote the formation of abnormal mitotic spindles. In contrast, while a high percentage of Chlamydia cells had supernumerary centrosomes in interphase (Fig. 1C), only few of these cells actually progressed into mitosis and formed abnormal spindles. We conclude that all cells with abnormal centrosomes were able to form mitotic spindles and that cell cycle progression to reach mitosis may be disrupted in Chlamydia cells with supernumerary centrosomes.
Chlamydia cells with amplified centrosomes are multinucleated and defective in cell cycle progression
We observed that Chlamydia was more likely to cause host cell multinucleation than HPV. 32.9% of Chlamydia cells and 32.1% of HPV+Chlamydia cells were multinucleated, compared to only 3.9% of HPV cells and 1.0% of control cells (Fig. 3A). In Chlamydia cells, multinucleation and centrosome amplification were often present in the same cell. Consistent with this observation, we determined a ϕ coefficient of 0.94 between the two phenotypes, which indicates a strong correlation (Fig. 3B). In contrast, HPV cells had a low ϕ coefficient of 0.23. HPV+Chlamydia cells displayed an intermediate correlation (ϕ coefficient = 0.49), consistent with a mixed effect (Fig. 3B).
Figure 3. Multinucleation is strongly correlated with centrosome amplification in Chlamydia cells but not in HPV cells.
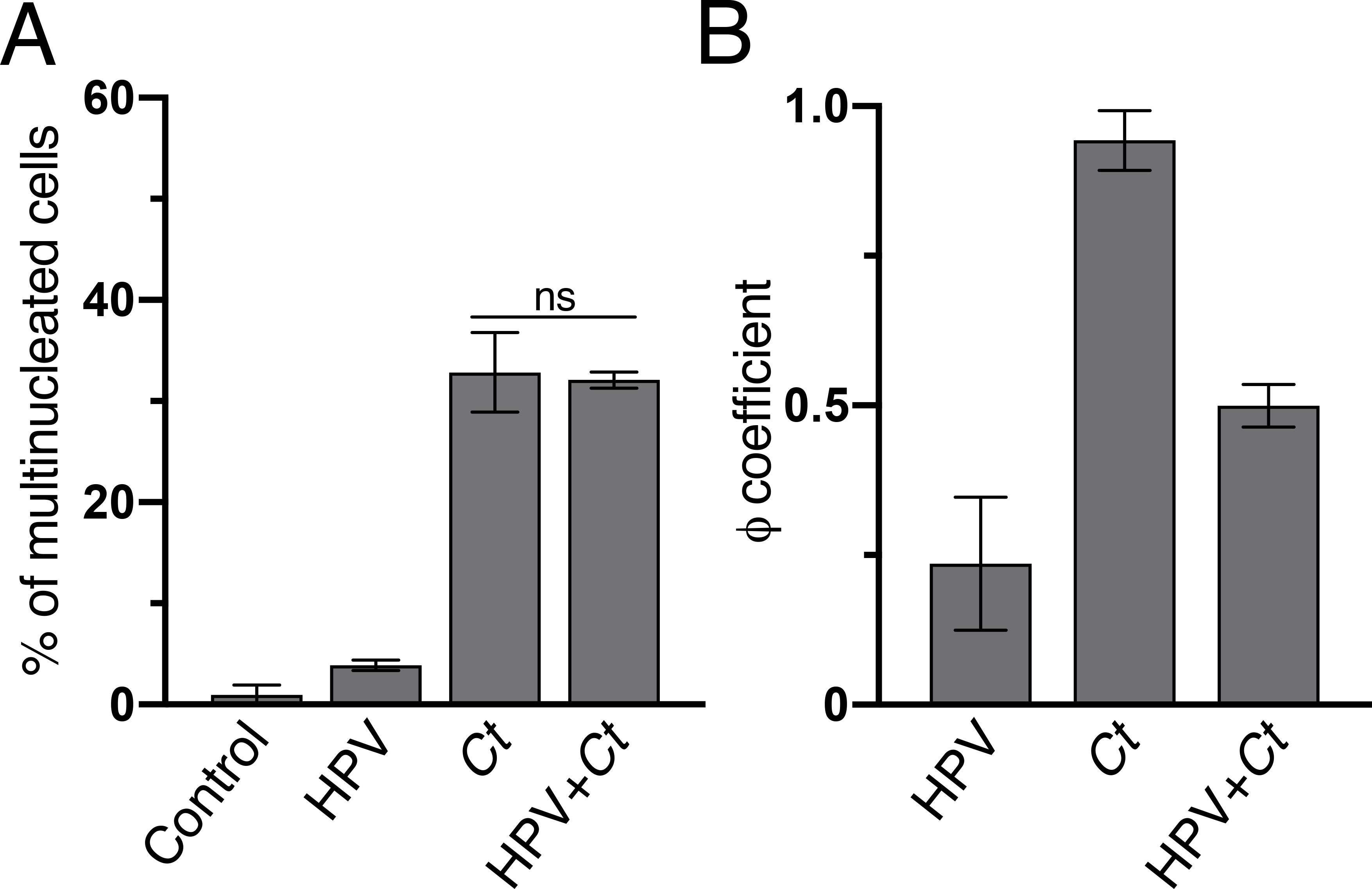
(A) The percentage of multinucleated host cells described in Figure 1C is shown. 100 cells were examined for each condition. For Chlamydia cells, only infected cells were counted. Data are shown as mean ± SD (n=3); ns: not statistically significant.
(B) ϕ coefficients between multinucleation and centrosome amplification were calculated for the HPV, Chlamydia, and HPV+Chlamydia cells of Figure 1C. ϕ coefficients range from −1 to 1, with −1 or +1 indicating perfect negative or positive relationships, respectively, while 0 shows no relationship between the two phenotypes. The control sample was not included due to low level of centrosome amplification in these cells. Data are shown as mean ± SD (n=3).
As multinucleation can cause cells to arrest in the G1 phase of the cell cycle (Ganem et al. 2009; Hart, Adams, and Draviam 2021), we compared cell cycle progression in mono- and multinucleated Chlamydia and HPV+Chlamydia cells. Mononucleated Chlamydia and HPV+Chlamydia cells, as well as multi-nucleated HPV+Chlamydia, incorporated EdU, a marker for S-phase entry, to similar extents (Fig. S3). In contrast, the percentage of EdU-positive multinucleated Chlamydia cells was reduced (Fig. S3), suggesting that these cells become arrested in a pre-S-phase stage of the cell cycle, likely in G1.
Chlamydia-induced centrosome amplification and multinucleation result from a cytokinesis defect
The strong correlation between centrosome amplification and multinucleation in Chlamydia cells is indicative of a cytokinesis defect in the host cell, a reported consequence of Chlamydia infection (Alzhanov et al. 2009; Sun et al. 2016). To test this hypothesis, we counted the number of Cep164-positive foci in HPV, Chlamydia, and HPV+Chlamydia cells, focusing on cells with supernumerary centrosomes (Fig. S4). In a normal mitotic cell, the two mature Cep164-positive centrioles are passed on to the two daughter cell after cytokinesis (Schmidt et al. 2012). The presence of two mature centrioles in the same cell is therefore a strong indicator of a cytokinesis defect. Greater than 60% of Chlamydia cells had two Cep164-positive foci, whereas HPV cells contained predominantly a single Cep164 focus. HPV+Chlamydia cells showed an intermediate phenotype (Fig. S4).
As a complementary approach, we prevented Chlamydia, HPV and HPV+Chlamydia cells from reaching cytokinesis by blocking cell cycle progression either in S-phase or G2 and then measuring the prevalence of amplified centrosomes and multinucleation in each cell population. We induced a cell cycle arrest in S-phase by treating cells with thymidine. We also arrested cells in G2 by first synchronizing cells in S-phase, with thymidine treatment and washout, and then incubating them with the G2 inhibitor RO-3306 (Ma and Poon 2017). Each of these cell cycle manipulations decreased the prevalence of multinucleation as well as centrosome amplification in Chlamydia cells, but not in HPV cells (Figs. 4A and 4B). Once again, HPV+Chlamydia cells showed an intermediate phenotype. Thus, multinucleation and centrosome amplification are closely linked in Chlamydia cells, with both phenotypes depending on progression through the cell cycle. Together, these results indicate that centrosome amplification in Chlamydia, but not in HPV cells, may be the consequence of a cytokinesis defect. They also provide further support that these pathogens contribute to centrosome abnormalities through different mechanisms in the same host cell.
Figure 4. Multinucleation and centrosome amplification in Chlamydia-infected cells require mitotic progression.
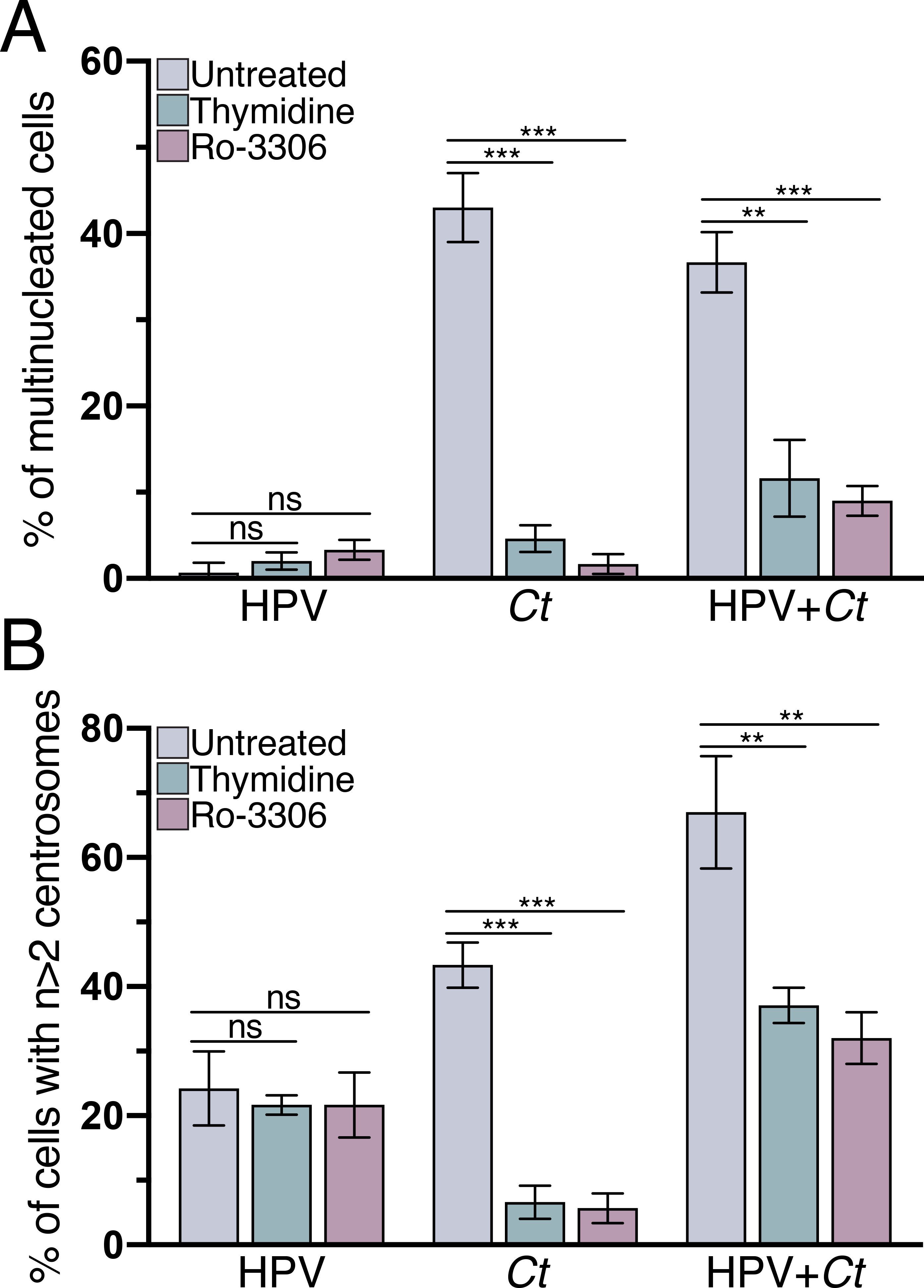
The cells of our four experimental conditions were arrested in S-phase with thymidine, or in G2 by the addition of RO-3306. Untreated samples were incubated in equivalent volume of DMSO. The percentage of cells with (A) multiple nuclei and (B) supernumerary centrosomes is shown. 100 cells were analyzed for each condition at 36hpi. Data are represented as mean ± SD (n=3); **P≤0.01, ***P<0.001, ns: not statistically significant.
Our experimental set-up allowed us to examine an alternative model in which centrosome amplification in Chlamydia cells was proposed to cause multinucleation (Brown et al. 2014). To test this order of events, we prevented centrosome duplication through the use of the Plk4 inhibitor centrinone, which has been shown to block new centriole assembly in RPE-1 cells (Wong et al. 2015). Centrinone treatment significantly reduced centrosome amplification in HPV, Chlamydia and HPV+Chlamydia cells (Fig. 5A), which is consistent with published results on Plk4 inhibition (Johnson et al. 2009; Korzeniewski, Treat, and Duensing 2011). However, centrinone treatment did not prevent multinucleation in Chlamydia cells (Fig. 5B), suggesting that centrosome amplification is not necessary for Chlamydia-induced multinucleation.
Figure 5. Multinucleation in Chlamydia-infected cells does not depend on centrosome amplification.
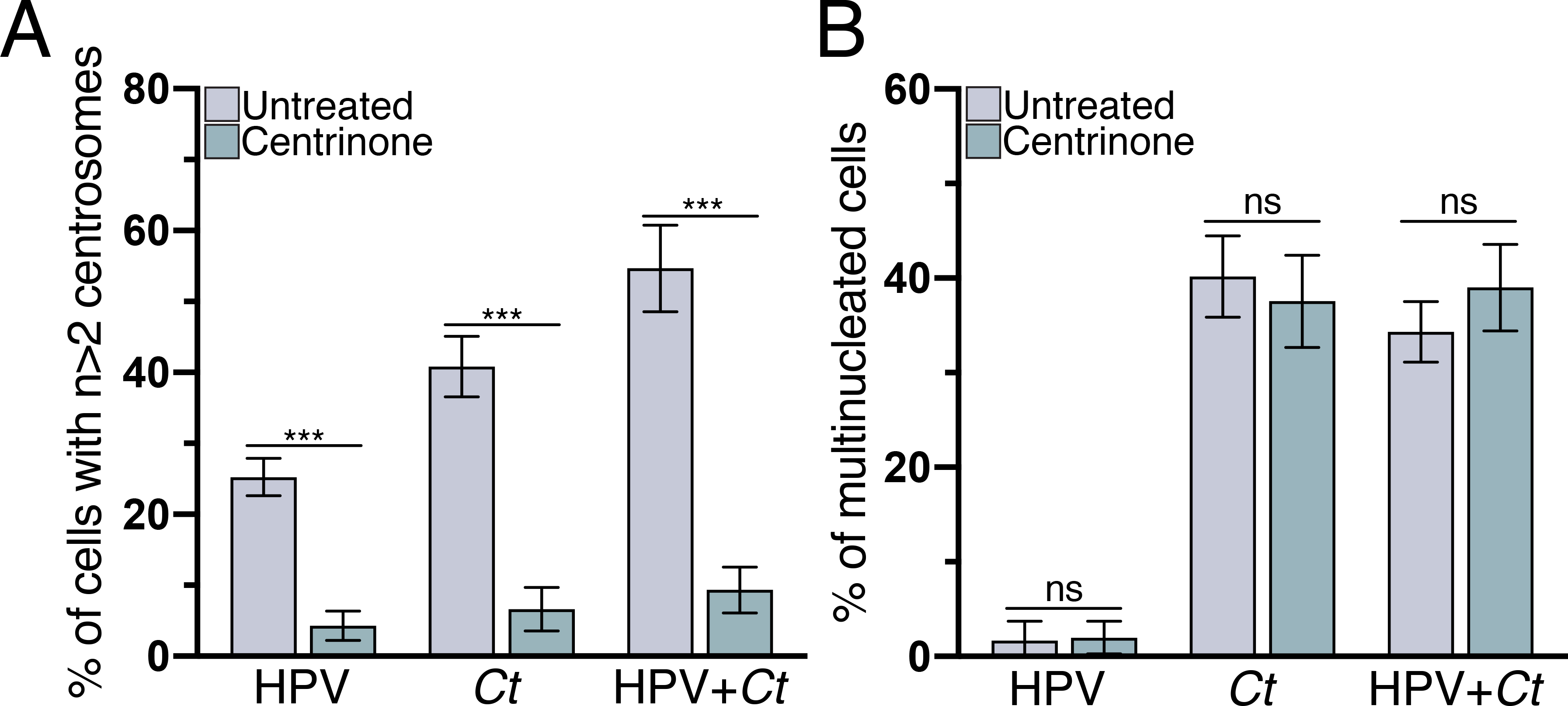
The percentage of HPV, Chlamydia, and HPV+Chlamydia cells with (A) supernumerary centrosomes or (B) multiple nuclei after treatment with the Plk4 inhibitor centrinone is shown. Untreated samples were incubated in equivalent volume of DMSO. 100 cells were analyzed for each condition at 36hpi. Data are represented as mean ± SD (n=3); ***P<0.001.
Discussion
To investigate how C. trachomatis could contribute to HPV-mediated carcinogenesis, we measured the respective effects of these intracellular pathogens on the centrosome in the same host cell. We found that Chlamydia induced more cells to have amplified centrosomes than HPV and that these pathogens together caused an even higher percentage of cells with supernumerary centrosomes. These additive effects, together with our mechanistic analyses, suggest that Chlamydia and HPV induce centrosome amplification through different mechanisms. This study thus provides evidence that C. trachomatis, as a co-factor for HPV, may contribute to the development of cervical cancer by enhancing centrosome defects.
Chlamydia has been reported to cause centrosome amplification (Grieshaber et al. 2006; Johnson et al. 2009). These prior cell culture studies have predominantly used transformed cell lines that have an HPV background, such as HeLa cells (Grieshaber et al. 2006; Johnson et al. 2009; Knowlton et al. 2013). Although Chlamydia was found to also induce centrosome amplification in HPV-negative cell lines, such as NIH3T3 or HFF, these studies did not separate or compare the effects on the centrosome caused by either Chlamydia, HPV, or both pathogens together (Grieshaber et al. 2006; Johnson et al. 2009; Knowlton et al. 2013). Furthermore, comparing centrosome amplification between different Chlamydia-infected HPV-negative and positive cell lines can be difficult. We avoided these issues by developing a cell culture model that uses the same cellular background to measure the respective effects of Chlamydia and HPV on the centrosome.
HPV has been shown to induce centrosome amplification through its oncoproteins E6 and E7. E7 is proposed to be the primary driver of centrosome amplification in an HPV infection. This conclusion is based on data showing that E7 dysregulates the centrosome duplication machinery in a Cdk2-dependent manner (Duensing et al. 2006) and that transient E7 expression is sufficient to cause centrosome amplification (Duensing et al. 2001; Duensing and Münger 2002). In contrast, E6 has been proposed to play a lesser role in centrosome amplification by disabling the p53-dependent checkpoint (Duensing et al. 2001; Duensing and Münger 2002). The loss of this checkpoint could lead to a cytokinesis defect (Bunz et al. 1998; Duensing et al. 2001), in which the nucleus and the two centrosomes duplicate normally, but the cell fails to divide, resulting in a multinucleated cell with extra centrosomes (Cosenza and Krämer 2016).
This present study compared the mechanisms through which Chlamydia and HPV produce supernumerary centrosomes. We propose that Chlamydia-induced centrosome amplification is the result of a cytokinesis defect. This idea is supported by the observation that centrosome amplification in Chlamydia cells strongly correlated with host cell multinucleation and required progression through mitosis. Additionally, most Chlamydia cells with supernumerary centrosomes had two Cep164-positive foci. In contrast, centrosome amplification in HPV cells did not correlate with multinucleation and was independent of cell cycle progression. Furthermore, most HPV cells with amplified centrosomes only contained one mature Cep164-positive mother centriole.
Together, these data suggest that HPV and Chlamydia induce centrosome defects through different mechanisms in the same host cell. While HPV primarily dysregulate the centrosome duplication machinery through E7 (Duensing et al. 2001; Duensing and Münger 2002), Chlamydia appears to cause centrosome dysregulation by disrupting cytokinesis. Consistent with this model, blocking cell cycle progression in HPV+Chlamydia cells partially reduced the prevalence of amplified centrosomes because it eliminated the contribution of Chlamydia, but not HPV, to this phenotype. Overall, our results suggest that these two pathogens activate two distinct pathways to induce centrosome dysregulation, although the respective contribution of each pathway to centrosome amplification appears to be cell type specific (Fig. S2).
Chlamydia is proposed to block cytokinesis through multiple mechanisms, including the physical presence of the inclusion and expression of the chlamydial proteins, CT223 (IPAM) or the protease CPAF (Greene 2001, Alzanov, 2009, Sun 2011, Brown 2014). The latter two studies both described evidence for a link between multinucleation and centrosome amplification, but Brown and colleagues suggested that multinucleation is the consequence of CPAF-induced centrosome amplification in Chlamydia-infected cells (Brown et al., 2014). Our data, however, suggests that the cytokinesis defect is upstream of the other two phenotypes because centrinone treatment blocked centrosome amplification in Chlamydia cells, but did not prevent multinucleation. Currently, it is not clear if centrosome amplification and multinucleation are functionally linked or if they are two independent consequences of the cytokinesis defect.
The presence of Chlamydia and HPV in the same cells produced a high prevalence of abnormal spindles but did not to affect the function of the centrosome in organizing microtubules in interphase or mitosis. As supernumerary centrosomes can lead to spindle defects, we propose that Chlamydia and HPV together produce abnormal spindles by altering centrosome number rather than function. These abnormal spindles may lead to chromosome mis-segregation and aneuploidy (Zhou et al. 1998), thereby providing a mechanism by which Chlamydia may contribute to HPV-induced carcinogenesis.
Our study provides evidence for Chlamydia-induced cell cycle dysregulation in the host cell. We observed that fewer multinucleated Chlamydia cells progressed through the cell cycle than either mononucleated Chlamydia or multinucleated HPV+Chlamydia cells (Fig. S3). Thus, in addition to the known effect on cytokinesis, which leads to centrosome amplification and multinucleation, Chlamydia appears to disrupt progression through interphase. This Chlamydia-induced cell cycle arrest is likely in G1 and may be the consequence of either centrosome amplification or multinucleation. Centrosome amplification and multinucleation have been reported to lead to a G1 arrest through p53-dependent or p53-independent mechanisms, respectively (Hart et al. 2021; Mikule et al. 2007). However, as Chlamydia infection is known to induce p53 degradation, the presence of multiple nuclei in an infected cells is more likely to induce this G1 arrest in a p53-independent manner (González et al. 2014:2; Hart et al. 2021; Siegl et al. 2014). In HPV+Chlamydia cells, the presence of E6 and E7 may release the G1 arrest, possibly by degrading retinoblastoma protein (pRB) (Boyer, Wazer, and Band 1996; Giacinti and Giordano 2006), resulting in cell cycle progression and the formation of aberrant mitotic spindles.
We hypothesize that these combined effects on the centrosome occur through co-infection, with HPV infection preceding the Chlamydia infection. Both sexually transmitted agents are highly prevalent, making co-infection likely. These pathogens each infect the stratified epithelia of the cervix, but they do so at different locations, with HPV infecting basal cells (Spurgeon and Lambert 2017), while Chlamydia infects the superficial cell layer (Murall et al. 2019). It is known, however, that HPV-infected basal cells divide, differentiate and migrate to the epithelial surface (Pinidis et al. 2016), thus providing a population of HPV-infected cells that can be infected acutely by Chlamydia. Our HPV/Chlamydia cell culture model mimics this sequence of events by taking cells expressing HPV E6 and E7 and then infecting them with C. trachomatis.
Our data is consistent with a ‘hit-and-run’ model, in which C. trachomatis infects a cervical cell that has an on-going HPV infection and contributes to HPV-induced carcinogenesis by augmenting centrosomal defects in these cells. Chlamydia typically causes a lytic infection, but we propose that some co-infected cells survive the Chlamydia infection and have enhanced centrosomal defects that promote their progression into cancer cells. This model is supported by data showing that cervical cancer cells do not have evidence of active Chlamydia infection (Wallin et al. 2002). In addition, cervical cancer has been associated with serological evidence of past, rather than current, Chlamydia infection (Wallin et al. 2002). There is also evidence that cells can be cleared of a C. trachomatis infection with antibiotics, while retaining amplified centrosome number (Grieshaber et al. 2006) or can survive through a non-lytic process called extrusion (Hybiske and Stephens 2007).
Our findings provide biologic plausibility for C. trachomatis as a co-factor for HPV in carcinogenesis and have implications for the management of HPV and C. trachomatis infections. Based on this study, HPV-infected women who have had a prior C. trachomatis infection may be at a higher risk for the development of cervical cancer. Our findings suggest that current screening for cervical cancer, which is based on Pap smear identification of premalignant cells and HPV test may not be adequate (Fontham et al. 2020). Enhanced screening for past C. trachomatis infection could be performed with an antibody blood test, but not with the standard C. trachomatis test, which is a nucleic acid amplification test (NAAT) that only detects an active or resolving infection (Meyer 2016). If Chlamydia does contribute to cervical cancer, there will also be a greater need to identify and treat active infections and to develop a vaccine.
Supplementary Material
Take Away:
Chlamydia and HPV induce centrosome amplification in an additive manner
Chlamydia-induced centrosome amplification is linked to host cell multinucleation
Chlamydia-induced centrosome amplification requires cell cycle progression
Chlamydia and HPV cause centrosome amplification through different mechanisms
This study supports Chlamydia as a co-factor for HPV in carcinogenesis
Acknowledgments
We would like to thank Dr. Karl Munger for his input and guidance on generating our HPV16 E6 and E7-expressing RPE-1 cell line, and Dr. Aimee Edinger and Brendan Finicle for their assistance with lentiviral transductions. We also thank Dr. Ashok Aiyar for generously providing us with the A549 cell lines. This research was supported by the California Cancer Research Consortium (CCRC grant # 582146 to C.S.) and the National Institutes of Health (R21 AI128723 and R01 AI153410 to M.T. and C.S.). K.W. was supported by a National Institutes of Health T32 training grant (AI141346). K.J.M was supported by a National Science Foundation graduate research fellowship (ID 2018262334).
Footnotes
Conflict of interest:
None
References
- Alzhanov Damir T., Weeks Sara K., Burnett Jeffrey R., and Rockey Daniel D.. 2009. “Cytokinesis Is Blocked in Mammalian Cells Transfected with Chlamydia Trachomatis Gene CT223.” BMC Microbiology 9:2. doi: 10.1186/1471-2180-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbyn Marc, Smith Sara B., Temin Sarah, Sultana Farhana, Castle Philip, and Collaboration on Self-Sampling and HPV Testing. 2018. “Detecting Cervical Precancer and Reaching Underscreened Women by Using HPV Testing on Self Samples: Updated Meta-Analyses.” BMJ (Clinical Research Ed.) 363:k4823. doi: 10.1136/bmj.k4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N. Sanjib, Wang Hsu-Kun, Broker Thomas R., and Chow Louise T.. 2011. “Human Papillomavirus (HPV) E7 Induces Prolonged G2 Following S Phase Reentry in Differentiated Human Keratinocytes.” The Journal of Biological Chemistry 286(17):15473–82. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienkowska-Haba Malgorzata, Luszczek Wioleta, Myers Julia E., Keiffer Timothy R., Stephen DiGiuseppe Paula Polk, Bodily Jason M., Scott Rona S., and Sapp Martin. 2018. “A New Cell Culture Model to Genetically Dissect the Complete Human Papillomavirus Life Cycle.” PLoS Pathogens 14(3):e1006846. doi: 10.1371/journal.ppat.1006846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, and Band V. 1996. “E7 Protein of Human Papilloma Virus-16 Induces Degradation of Retinoblastoma Protein through the Ubiquitin-Proteasome Pathway.” Cancer Research 56(20):4620–24. [PubMed] [Google Scholar]
- Brown Heather M., Knowlton Andrea E., Snavely Emily, Nguyen Bidong D., Richards Theresa S., and Grieshaber Scott S.. 2014. “Multinucleation during C. Trachomatis Infections Is Caused by the Contribution of Two Effector Pathways.” PloS One 9(6):e100763. doi: 10.1371/journal.pone.0100763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner Lyndsey R., Amedee Angela M., Albritton Hannah L., Kozlowski Pamela A., Lacour Nedra, McGowin Chris L., Schust Danny J., and Quayle Alison J.. 2016. “Chlamydia Trachomatis Infection of Endocervical Epithelial Cells Enhances Early HIV Transmission Events.” PloS One 11(1):e0146663. doi: 10.1371/journal.pone.0146663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, and Vogelstein B. 1998. “Requirement for P53 and P21 to Sustain G2 Arrest after DNA Damage.” Science (New York, N.Y.) 282(5393):1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Chan Jason Yongsheng. 2011. “A Clinical Overview of Centrosome Amplification in Human Cancers.” International Journal of Biological Sciences 7(8):1122–44. doi: 10.7150/ijbs.7.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosenza Marco Raffaele, and Krämer Alwin. 2016. “Centrosome Amplification, Chromosomal Instability and Cancer: Mechanistic, Clinical and Therapeutic Issues.” Chromosome Research: An International Journal on the Molecular, Supramolecular and Evolutionary Aspects of Chromosome Biology 24(1):105–26. doi: 10.1007/s10577-015-9505-5. [DOI] [PubMed] [Google Scholar]
- Duensing Anette, Liu Ying, Tseng Michelle, Malumbres Marcos, Barbacid Mariano, and Duensing Stefan. 2006. “Cyclin-Dependent Kinase 2 Is Dispensable for Normal Centrosome Duplication but Required for Oncogene-Induced Centrosome Overduplication.” Oncogene 25(20):2943–49. doi: 10.1038/sj.onc.1209310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Crum CP, and Münger K. 2001. “Human Papillomavirus Type 16 E7 Oncoprotein-Induced Abnormal Centrosome Synthesis Is an Early Event in the Evolving Malignant Phenotype.” Cancer Research 61(6):2356–60. [PubMed] [Google Scholar]
- Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, and Munger K. 2000. “The Human Papillomavirus Type 16 E6 and E7 Oncoproteins Cooperate to Induce Mitotic Defects and Genomic Instability by Uncoupling Centrosome Duplication from the Cell Division Cycle.” Proceedings of the National Academy of Sciences of the United States of America 97(18):10002–7. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing Stefan, and Münger Karl. 2002. “Human Papillomaviruses and Centrosome Duplication Errors: Modeling the Origins of Genomic Instability.” Oncogene 21(40):6241–48. doi: 10.1038/sj.onc.1205709. [DOI] [PubMed] [Google Scholar]
- Fonseca-Moutinho José Alberto. 2011. “Smoking and Cervical Cancer.” ISRN Obstetrics and Gynecology 2011:847684. doi: 10.5402/2011/847684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontham Elizabeth T. H., Wolf Andrew M. D., Church Timothy R., Etzioni Ruth, Flowers Christopher R., Herzig Abbe, Guerra Carmen E., Oeffinger Kevin C., Ya-Chen Tina Shih Louise C. Walter, Kim Jane J., Andrews Kimberly S., DeSantis Carol E., Fedewa Stacey A., Manassaram-Baptiste Deana, Saslow Debbie, Wender Richard C., and Smith Robert A.. 2020. “Cervical Cancer Screening for Individuals at Average Risk: 2020 Guideline Update from the American Cancer Society.” CA: A Cancer Journal for Clinicians 70(5):321–46. doi: 10.3322/caac.21628. [DOI] [PubMed] [Google Scholar]
- Ganem Neil J., Godinho Susana A., and Pellman David. 2009. “A Mechanism Linking Extra Centrosomes to Chromosomal Instability.” Nature 460(7252):278–82. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacinti C, and Giordano A. 2006. “RB and Cell Cycle Progression.” Oncogene 25(38):5220–27. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- Godinho SA, and Pellman D. 2014. “Causes and Consequences of Centrosome Abnormalities in Cancer.” Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 369(1650). doi: 10.1098/rstb.2013.0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho Susana A., Picone Remigio, Burute Mithila, Dagher Regina, Su Ying, Leung Cheuk T., Polyak Kornelia, Brugge Joan S., Théry Manuel, and Pellman David. 2014. “Oncogene-like Induction of Cellular Invasion from Centrosome Amplification.” Nature 510(7503):167–71. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González Erik, Rother Marion, Kerr Markus C., Al-Zeer Munir A., Abu-Lubad Mohammad, Kessler Mirjana, Brinkmann Volker, Loewer Alexander, and Meyer Thomas F.. 2014. “Chlamydia Infection Depends on a Functional MDM2-P53 Axis.” Nature Communications 5(1):5201. doi: 10.1038/ncomms6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber Scott S., Grieshaber Nicole A., Miller Natalie, and Hackstadt Ted. 2006. “Chlamydia Trachomatis Causes Centrosomal Defects Resulting in Chromosomal Segregation Abnormalities.” Traffic (Copenhagen, Denmark) 7(8):940–49. doi: 10.1111/j.1600-0854.2006.00439.x. [DOI] [PubMed] [Google Scholar]
- Hart Madeleine, Adams Sophie D., and Draviam Viji M.. 2021. “Multinucleation Associated DNA Damage Blocks Proliferation in P53-Compromised Cells.” Communications Biology 4(1):1–11. doi: 10.1038/s42003-021-01979-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske Kevin, and Stephens Richard S.. 2007. “Mechanisms of Host Cell Exit by the Intracellular Bacterium Chlamydia.” Proceedings of the National Academy of Sciences 104(27):11430–35. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Collaboration of Epidemiological Studies of Cervical Cancer, Appleby Paul, Beral Valerie, de González Amy Berrington, Colin Didier, Franceschi Silvia, Goodhill Adrian, Green Jane, Peto Julian, Plummer Martyn, and Sweetland Siân. 2007. “Cervical Cancer and Hormonal Contraceptives: Collaborative Reanalysis of Individual Data for 16,573 Women with Cervical Cancer and 35,509 Women without Cervical Cancer from 24 Epidemiological Studies.” Lancet (London, England) 370(9599):1609–21. doi: 10.1016/S0140-6736(07)61684-5. [DOI] [PubMed] [Google Scholar]
- Johnson Kirsten A., Tan Ming, and Sütterlin Christine. 2009. “Centrosome Abnormalities during a Chlamydia Trachomatis Infection Are Caused by Dysregulation of the Normal Duplication Pathway.” Cellular Microbiology 11(7):1064–73. doi: 10.1111/j.1462-5822.2009.01307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton Andrea E., Fowler Larry J., Patel Rahul K., Wallet Shannon M., and Grieshaber Scott S.. 2013. “Chlamydia Induces Anchorage Independence in 3T3 Cells and Detrimental Cytological Defects in an Infection Model.” PloS One 8(1):e54022. doi: 10.1371/journal.pone.0054022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski Nina, Treat Benjamin, and Duensing Stefan. 2011. “The HPV-16 E7 Oncoprotein Induces Centriole Multiplication through Deregulation of Polo-like Kinase 4 Expression.” Molecular Cancer 10(1):61. doi: 10.1186/1476-4598-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, and Salisbury JL. 1998. “Centrosome Hypertrophy in Human Breast Tumors: Implications for Genomic Stability and Cell Polarity.” Proceedings of the National Academy of Sciences of the United States of America 95(6):2950–55. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Hoi Tang, and Poon Randy Y. C.. 2017. “Synchronization of HeLa Cells.” Methods in Molecular Biology (Clifton, N.J.) 1524:189–201. doi: 10.1007/978-1-4939-6603-5_12. [DOI] [PubMed] [Google Scholar]
- Meyer Thomas. 2016. “Diagnostic Procedures to Detect Chlamydia Trachomatis Infections.” Microorganisms 4(3). doi: 10.3390/microorganisms4030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikule Keith, Delaval Benedicte, Kaldis Philipp, Jurcyzk Agata, Hergert Polla, and Doxsey Stephen. 2007. “Loss of Centrosome Integrity Induces P38—P53—P21-Dependent G1—S Arrest.” Nature Cell Biology 9(2):160–70. doi: 10.1038/ncb1529. [DOI] [PubMed] [Google Scholar]
- Münger Karl, Baldwin Amy, Edwards Kirsten M., Hayakawa Hiroyuki, Nguyen Christine L., Owens Michael, Grace Miranda, and Huh Kyungwon. 2004. “Mechanisms of Human Papillomavirus-Induced Oncogenesis.” Journal of Virology 78(21):11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz Nubia, Bosch F. Xavier, de Sanjosé Silvia, Herrero Rolando, Castellsagué Xavier, Shah Keerti V., Snijders Peter J. F., Meijer Chris J. L. M., and International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. 2003. “Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer.” The New England Journal of Medicine 348(6):518–27. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Murall Carmen Lía, Jackson Robert, Zehbe Ingeborg, Boulle Nathalie, Segondy Michel, and Alizon Samuel. 2019. “Epithelial Stratification Shapes Infection Dynamics.” PLOS Computational Biology 15(1):e1006646. doi: 10.1371/journal.pcbi.1006646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, and Doxsey SJ. 1998. “Centrosome Defects and Genetic Instability in Malignant Tumors.” Cancer Research 58(17):3974–85. [PubMed] [Google Scholar]
- Pinidis Petros, Tsikouras Panagiotis, Iatrakis Georgios, Zervoudis Stefanos, Koukouli Zacharoula, Bothou Anastasia, Galazios Georgios, and Vladareanu Simona. 2016. “Human Papilloma Virus’ Life Cycle and Carcinogenesis.” Mædica 11(1):48–54. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Salisbury Jeffrey L., Whitehead Clark M., Lingle Wilma L., and Barrett Susan L.. 1999. “Centrosomes and Cancer.” Biology of the Cell 91(6):451–60. doi: 10.1111/j.1768-322X.1999.tb01100.x. [DOI] [PubMed] [Google Scholar]
- Schaeffer Anthony J., Nguyen Marie, Liem Amy, Lee Denis, Montagna Cristina, Lambert Paul F., Ried Thomas, and Difilippantonio Michael J.. 2004. “E6 and E7 Oncoproteins Induce Distinct Patterns of Chromosomal Aneuploidy in Skin Tumors from Transgenic Mice.” Cancer Research 64(2):538–46. doi: 10.1158/0008-5472.can-03-0124. [DOI] [PubMed] [Google Scholar]
- Schmidt Kerstin N., Kuhns Stefanie, Neuner Annett, Hub Birgit, Zentgraf Hanswalter, and Pereira Gislene. 2012. “Cep164 Mediates Vesicular Docking to the Mother Centriole during Early Steps of Ciliogenesis.” Journal of Cell Biology 199(7):1083–1101. doi: 10.1083/jcb.201202126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz Elisabeth, Ulrich Karl Freese Lutz Gissmann, Mayer Wolfgang, Roggenbuck Birgit, Stremlau Armin, and zur Hausen Harald. 1985. “Structure and Transcription of Human Papillomavirus Sequences in Cervical Carcinoma Cells.” Nature 314(6006):111–14. doi: 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- Siegl Christine, Prusty Bhupesh K., Karunakaran Karthika, Wischhusen Jörg, and Rudel Thomas. 2014. “Tumor Suppressor P53 Alters Host Cell Metabolism to Limit Chlamydia Trachomatis Infection.” Cell Reports 9(3):918–29. doi: 10.1016/j.celrep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Silva Jani, Cerqueira Fátima, and Medeiros Rui. 2014. “Chlamydia Trachomatis Infection: Implications for HPV Status and Cervical Cancer.” Archives of Gynecology and Obstetrics 289(4):715–23. doi: 10.1007/s00404-013-3122-3. [DOI] [PubMed] [Google Scholar]
- Smith Jennifer S., Muñoz Nubia, Herrero Rolando, Eluf-Neto José, Ngelangel Corazon, Franceschi Silvia, Bosch F. Xavier, Walboomers Jan M. M., and Peeling Rosanna W.. 2002. “Evidence for Chlamydia Trachomatis as a Human Papillomavirus Cofactor in the Etiology of Invasive Cervical Cancer in Brazil and the Philippines.” The Journal of Infectious Diseases 185(3):324–31. doi: 10.1086/338569. [DOI] [PubMed] [Google Scholar]
- Spardy Nicole, Covella Kathryn, Cha Elliot, Hoskins Elizabeth E., Wells Susanne I., Duensing Anette, and Duensing Stefan. 2009. “Human Papillomavirus 16 E7 Oncoprotein Attenuates DNA Damage Checkpoint Control by Increasing the Proteolytic Turnover of Claspin.” Cancer Research 69(17):7022–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurgeon Megan E., and Lambert Paul F.. 2017. “Human Papillomavirus and the Stroma: Bidirectional Crosstalk during the Virus Life Cycle and Carcinogenesis.” Viruses 9(8). doi: 10.3390/v9080219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun He Song, Sin Alex T. W., Poirier Mathieu B., and Harrison Rene E.. 2016. “Chlamydia Trachomatis Inclusion Disrupts Host Cell Cytokinesis to Enhance Its Growth in Multinuclear Cells.” Journal of Cellular Biochemistry 117(1):132–43. doi: 10.1002/jcb.25258. [DOI] [PubMed] [Google Scholar]
- Thomas JT, and Laimins LA. 1998. “Human Papillomavirus Oncoproteins E6 and E7 Independently Abrogate the Mitotic Spindle Checkpoint.” Journal of Virology 72(2):1131–37. doi: 10.1128/JVI.72.2.1131-1137.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin Keng-Ling, Wiklund Fredrik, Luostarinen Tapio, Angström Tord, Anttila Tarja, Bergman Frank, Hallmans Göran, Ikäheimo Irma, Koskela Pentti, Lehtinen Matti, Stendahl Ulf, Paavonen Jorma, and Dillner Joakim. 2002. “A Population-Based Prospective Study of Chlamydia Trachomatis Infection and Cervical Carcinoma.” International Journal of Cancer 101(4):371–74. doi: 10.1002/ijc.10639. [DOI] [PubMed] [Google Scholar]
- Wong Yao Liang, Anzola John V., Davis Robert L., Yoon Michelle, Motamedi Amir, Kroll Ashley, Seo Chanmee P., Hsia Judy E., Kim Sun K., Mitchell Jennifer W., Mitchell Brian J., Desai Arshad, Gahman Timothy C., Shiau Andrew K., and Oegema Karen. 2015. “Reversible Centriole Depletion with an Inhibitor of Polo-like Kinase 4.” Science (New York, N.Y.) 348(6239):1155–60. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, and Sen S. 1998. “Tumour Amplified Kinase STK15/BTAK Induces Centrosome Amplification, Aneuploidy and Transformation.” Nature Genetics 20(2):189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- Zhu Haiyan, Shen Zhaojun, Luo Hui, Zhang Wenwen, and Zhu Xueqiong. 2016. “Chlamydia Trachomatis Infection-Associated Risk of Cervical Cancer: A Meta-Analysis.” Medicine 95(13):e3077. doi: 10.1097/MD.0000000000003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.