

Abstract
The ubiquity and potency of antibiotics may give the false impression that infection is a solved problem. Unfortunately, even bacterial infections, the target of antibiotics, remain a major cause of illness and death. Several major unmet needs persist: biofilms, such as those on implanted hardware, largely resist antibiotics; the inflammatory host response to infection often produces more damage than the infection itself; and systemic antibiotics often decimate the gut microbiome, which can predispose to additional infections and even predispose to non-infectious diseases. Additionally, there is an increasing threat from multi-drug resistant microorganisms, though market forces may continue to inhibit innovation in this realm. These numerous unmet infection-related needs provide attractive goals for innovation of targeted drug delivery technologies, especially those of nanomedicine. Here we review several of those innovations in pre-clinical development, the two such therapies which have made it to clinical use, and the opportunities for further technology development for treating infections.
Keywords: Nanoparticle, antibiotics, bacterial resistance, biofilm, host inflammatory response, chronic infection
Graphical Abstract
1. Introduction
Infections remain a public health concern with significant associated costs. Our example for this review is bacterial pneumonia, a class of infections with serious morbidity and mortality worldwide. In the United States, it is in the top 10 causes of mortality each year[1]. This translates to an estimated yearly cost in excess of $17 billion in the US[2]. Pneumonia is the leading infectious cause of death in children worldwide, accounting for 15% of all deaths of children under five years old in 2017[3]. These statistics persist despite widespread use of antibiotics.
These disappointing statistics are in part due to three pharmacokinetic and biodistribution problems of antibiotics. First, as small molecule drugs, many antibiotics have unfavorable pharmacokinetics and thus require very frequent dosing. Second, many antibiotics have trouble reaching the anatomic source of bacteria in some infections, such as the central nervous system and abscesses (infectious pockets walled off by the body’s response to large concentrations of bacteria). Finally, unwanted side-effects on off-target organ systems can lead to dose limiting toxicity, and may be so severe an antibiotic cannot be used clinically.
Compounding the issues associated with antibiotic pharmacokinetic and biodistribution, antibiotic resistance is a growing problem. There are greater than 2.8 million antibiotic-resistant infections each year in the US, resulting in more than 35,000 deaths[4]. The 2014 World Health Organization Antimicrobial Resistance Global Report on Surveillance estimated that those antibiotic-resistant infection numbers come at a yearly cost to the US health system upwards of $34 billion dollars, with an associated projected fall in real gross domestic product (GDP) of 0.4% to 1.6%, which translates into many billions of dollars globally[5]. To date, resistance to every known class of antibiotics has been shown[6]. Worsening the antibiotic resistance crisis is the fact that no new antibiotic classes have been discovered in more than 30 years[7]. There is currently an urgent need for antimicrobials with a novel mechanism of action[8].
1.1. The Beginning of Antibiotics
For much of history, infections represented a disproportionate amount of population deaths. The search for infectious treatment modalities dates back to the ancient civilizations of Egypt, Greece, and China[9]. The first natural antibiotic was mycophenolic acid, isolated from Penicillium galucum, and reported by Italian physician and microbiologist Bartolomeo Gosio in 1893[10]. Gosio observed that mycophenolic acid was sufficient to inhibit the growth of Bacillus anthracis. The first modern, man-made antimicrobial agent was created in 1911 by Paul Ehrlich’s lab[11]. Salvarsan, or the “magic bullet” as it was referred to in that time, is an organoarsenic compound used to treat syphilis and is regarded as the introduction of targeted chemotherapy[12]. This was followed in 1928 by the discovery of penicillin by Alexander Fleming[13]. Since this work, many antibiotics have been discovered and synthesized to inhibit several key bacterial functions.
1.2. Limitations of Antibiotics
Despite their long history of revolutionizing treatment and outcome of infections, antibiotics also have glaring limitations. To begin with, antibiotics have the typical pharmacokinetics problems common to most small molecule drugs. First, antimicrobial efficacy is dependent on a bacteria’s exposure and susceptibility to the antibiotic, which directly relates to the dose needed and the individual patient’s physiology, which affects a drug’s pharmacokinetics. Despite the knowledge that patient factors impact the pharmacokinetics of antibiotics, these are rarely used to guide clinical practice, with most dosing recommendations based on population data[14]. Second, antibiotic metabolism can lead to chemically active compounds that cause toxicities, such as hypersensitivity reaction and teratogenicity[15]. Third, individual pharmacokinetics can change over time, especially across trajectories of critical illness. This leads to expensive drug level monitoring as antibiotic dosage must be altered to achieve levels of perceived efficacy while avoiding levels that could cause further morbidity[16]. These simple pharmacokinetics problems leave many opportunities for nanomedicine to improve antibiotic delivery.
In addition to these pharmacokinetic problems common to most small molecule drugs, antibiotics also suffer from the problem that penetration of the drug into the infected space is often highly limited (Figure 1). In the example of pneumonia, the lung’s alveoli are filled with purulent material, which makes it difficult for inhaled and intravenous antibiotics to reach the bacteria within the alveoli. Furthermore, complications of pneumonia, including cavity formation in necrotizing pneumonia and empyema (infection of the pleural space), create settings in which antibiotic penetration into the cavities is severely compromised. The former requires prolonged systemic antibiotic therapy, while the latter often requires surgical treatment with associated morbidity. Other infectious compartments associated with difficult antibiotic delivery include: the bone in osteomyelitis and the cerebrospinal fluid in meningitis. Creative solutions to antibiotic delivery may improve their ability to target these body compartments.
Figure 1. Limitations to current antibiotic delivery in pneumonia infections.
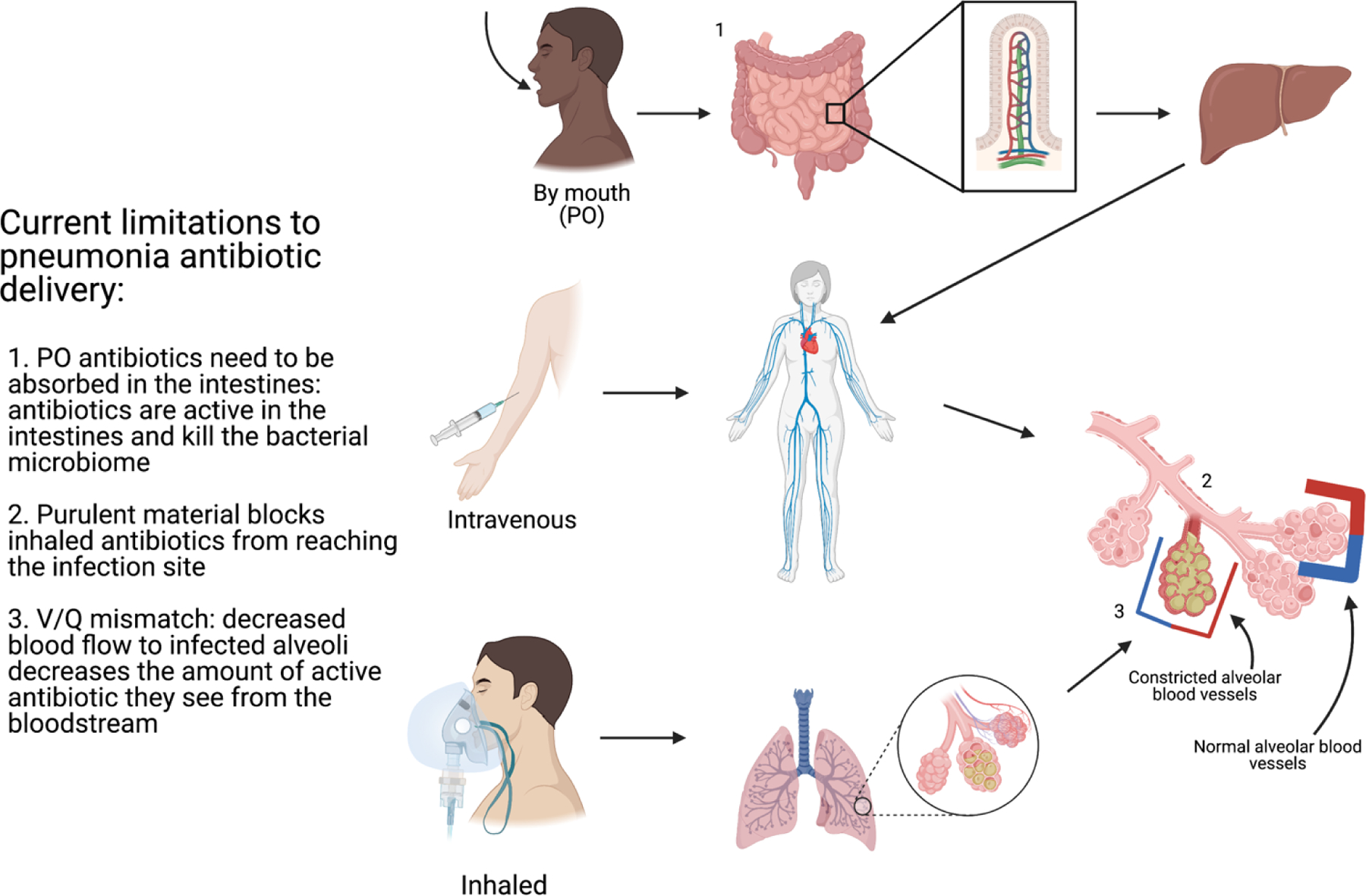
There are three current macrodelivery routes of antibiotic administration: oral, intravenous, and inhaled. Each has a limitation to effective delivery of antibiotics to the site of infection.
Given the above pharmacokinetic and drug penetration issues, antibiotics often are delivered in high and prolonged doses, leading to off-target side effects. Systemic antibiotics lead to perturbations in the microbiota, which can have both short-term and long-term consequences. The systemic exposure to antibiotics alters the microbiota and can make critically-ill patients more susceptible to further infections, and can select for drug-resistant bacteria[17]. Antibiotic exposure has been linked to morbidities such as C. difficile infection in adults[18] and necrotizing enterocolitis in neonates[19]. There are emerging studies suggesting longer term morbidities, such as development of asthma, obesity, and depression, may be associated with systemic antibiotic exposure in neonates and pediatric populations; and therefore, directed antibiotic delivery may lead to improved outcomes.
1.3. Bacterial resistance patterns
Beyond the host-factor and delivery limitations of antibiotics, bacteria have developed mechanisms to evade antibiotic action. As early as 1948, research suggested that bacterial genetic changes were one method of escaping antibiotic action, which continues to be one of bacteria’s effective resistance strategies[20][21]. The three main methods bacteria employ to resist antibiotic action are: membrane changes to alter antibiotic permeability (including efflux pumps), producing enzymes to degrade antibiotics, and altering the bacterial protein target[22]. Bacterial resistance genes can be spread by plasmid transfer, amplifying resistance as it develops[23]. These strategies have been so effective that there has now been antibiotic resistance documented to nearly every available antibiotic[24].
The lowest concentration of an antimicrobial that will inhibit the visible growth of a microorganism after overnight incubation is known as the minimum inhibitory concentration (MIC), and is an important measure of bacterial sensitivity to specific antibiotics[25]. As bacteria develop the aforementioned resistance mechanisms, initially they can be overcome by increasing concentrations of antibiotics. This is seen as rising measurements of the MIC[26]. Eventually the antibiotic dosing required to reach MIC in a patient will result in intolerable off-target effects limiting its clinical utility.
Bacteria’s propensity for developing resistance patterns in conjunction with inappropriate and overuse of antibiotics has driven the emergence of multi-drug resistant (MDR) bacteria[27]. The rapidly increasing population of MDR bacteria is a serious threat to public health with significant morbidity and mortality, making new antibiotic development a global priority[28]. Current strategies have included improved antibiotic stewardship, combination antibiotic therapies[29][30], which require use of 2 or more antibiotics to treat infections, and adjuvant therapies[31][32][33], where pharmaceuticals that have their own antibacterial properties or target bacterial resistance mechanisms are given in addition to traditional antibiotics[34]. Despite these efforts, there continues to be an exponential increase in multidrug resistant bacteria that continues to challenge clinical care[35].
Nanomedicine is moving into the area of MDR treatment, which requires more than just development of new classes of antibiotics. MDR treatment will additionally need to harness the antimicrobial properties of compounds outside of traditional antibiotics. The antimicrobial potential of heavy metals, such as a silver and gold, has been recognized for many years[36]. Silver has a long history of topical application, being used to decrease infection risk of burns and wounds[37]. It has not been proven as an effective therapy when administered orally or parenterally, and has poor excretion once absorbed, leading to the FDA removing its approval for use[38–40].
Heavy metal nanoparticles hold the potential of clinically harnessing the antimicrobial properties of these metal for clinical effect. On particular study by Figueiredo et al. obtained silver nanoparticles (AgNP) by fungus-mediated synthesis, average size 77.68±33.95 nm. These were coupled with Simvastatin, a member of the statin family, which are known for their antihyperlipidemic effects by competitively inhibiting the enzyme HMG-CoA reductase, decreasing cholesterol biosynthesis. Statins have also been found to have antimicrobial effects, which is believed to be related to their immunomodulary and anti-inflammatory effects[41]. Coupling of AgNPs to Simvastatin resulted in a synergistic effect of bactericidal antibacterial activity against several resistant organisms, including Methicillin Resistant S. aureus and Extended Spectrum Beta-Lactamase producing E. coli[42]. To date, studies on the antimicrobial properties of heavy metal nanoparticles have largely been in vitro studies. Clinical use of heavy metal nanoparticles has been limited by the: 1. expensive and hazardous synthesis[43] and 2. human toxicity[44]. As advances are made on synthesis techniques and biocompatibility, the use of metal nanoparticles should be a future focus for development of MDR infection treatment.
2. Nanomedicine: modernizing antibiotics
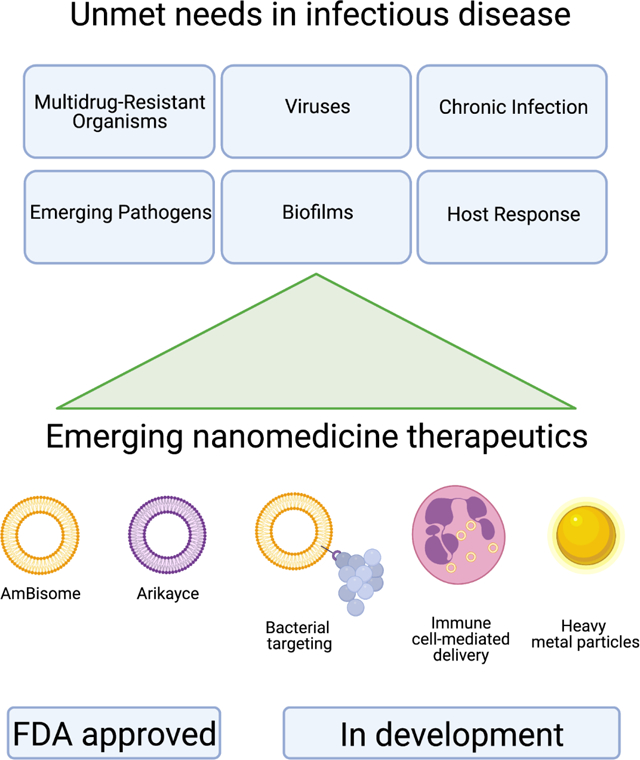
A modern approach to antimicrobial therapy will address the many limitations and disadvantages of traditional antibiotics by utilizing nano-scale carriers (Table 1). Incorporating antibiotics into nanoparticles has many potential advantages. Particles can be engineered to specifically target an organ, tissue type, cell type, or bacteria directly. Targeting has the benefits of directing high antimicrobial concentrations at the site where bacterial killing is needed, which should lower the overall effective antibiotic dose, and therefore, decrease the side effect profile. The nanoparticle preparation additionally has the ability to change the solubilization of antimicrobial pharmaceuticals. There is also potential to alter the pharmacokinetics which may allow for a more favorable side effect profile. These benefits of nanocarrier preparations have the potential to address current resistance concerns, as well as, introduce more antibiotics to clinical use.
Table 1. Unmet needs in infectious disease treatment.
These are broken down into several sub-categories, with future areas of innovation to address each need.
| Need | What it is | Current drug delivery limitations | Opportunities for drug development | Notable Examples |
|---|---|---|---|---|
| Multidrug-Resistant Organisms | Bacteria that have developed resistance to many antimicrobial drugs. |
|
Ghosh et al. show the mixed ligand analog of the natural Acinetobacter baumannii selective siderophore coupled to daptomycin, a Gram-positive only antibiotic, resulted in antibacterial activity against multidrug resistant strains of A. baumannii both in vitro and in vivo[47]. | |
| Viruses | Infectious microorganism that needs a host living cell to replicate. |
|
Cao et al. reported that gold nanoparticles could be functionalized with RNAse A and then modified with anti-HCV oligonucleotides to effectively mimic the function of the cellular RNA-induced silencing complex machinery for guiding target RNA cleavage[50]. | |
| Chronic Infection | A bacterial or viral infection that is resistant to treatment or has slow growth rates, resulting in long-term infection. |
|
Dou et al. demonstrated a depot-style release of indinavir, an antiretroviral drug, from macrophages over 14 continuous days which suppressed viral load significantly in the brain after intravenous administration in an HIV-1 encephalitis mouse model[53]. | |
| Emerging Pathogens | Infectious diseases that have newly appeared in a population or have existed, but are rapidly increasing in incidence or geographic range. |
|
|
Lok et al. showed that ~9nm spherical Ag nanoparticles were able to exert antibacterial effects through destabilizing the outer membrane, collapsing the plasma membrane potential and depleting the levels of intracellular ATP[55]. |
| Biofilms | Surface-attached microbes enclosed in an extracellular polymeric substance matrix. Biofilms are commonly seen on teeth and foreign bodies (i.e. catheters, replacement heart valves, joint replacement) | Nanoparticles that:
|
Qiu et al. recently showed that ~250nm drug-free cationic nanoparticles (CNP) were capable of time-and concentration-dependent activity against MRSA growth. These CNP were able to cause significant killing of planktonic bacteria, and completely inhibit biofilm formation[58]. | |
| Host Response | The inflammatory immune response to infection can ultimately cause damage to the host itself. |
|
|
Yang et al. co-loaded intercellular adhesion molecule-1 (I-CAM) targeted poly(lactide-co-glycolide acid) (PLGA) nanoparticles with Sparfloxacin, an antimicrobial, and Tacrolimus, an immunosuppressant drug. This combination nanoparticle preparation was able to decrease bacterial load and improve survival in a mouse pneumonia model[60]. |
2.1. Nanoparticle antibiotic preparations currently in clinical use
While nanocarrier antibiotic preparations could be the future of antibiotics, there are currently just a few that have already been brought to clinical use (Table 2). AmBisome, a <100nm unilamellar liposome preparation of amphotericin B, is the first clinically approved nano-scale carrier packaged antimicrobial. The AmBisome liposome is made of 61% hydrogenated soy phosphatidylcholine, 24% distearoyl phosphatidylglycerol, and 15% cholesterol, and has a lipid-to-drug mass ratio of 0.14[61]. The liposomal preparation decreases the volume of distribution of amphotericin B, which in turn leads to an 8- to 10-fold increase in the peak concentration (Cmax) and areas under concentration-time curves (AUC) for the antifungal[62]. As compared to amphotericin B, AmBisome has increased concentrations of drug in the blood, liver, and spleen but decreased concentrations in the kidney and lung[63]. The decreased kidney concentration in conjunction with changes in pharmacokinetic clearance with markedly decreased excretion of unchanged drug in urine accounts for AmBisome’s decreased kidney toxicity associated with conventional amphotericin B deoxycholate formulations[64]. It additionally offers some fungal cell wall targeting[65]. For these reasons, AmBisome is the preferred amphotericin preparation in clinical use, commonly employed for life-threatening fungal infections[66].
Table 2.
Current FDA approved nanocarrier antibiotics.
| Existing Nanomedicines to treat Infectious Diseases | |||
|---|---|---|---|
| Drug name | Drug type | Target | Benefit |
| AmBisome | A unilamellar liposome preparation of amphotericin B[61] | Fungi | The liposomal preparation decreases the drug volume of distribution leading to a dual benefit:
|
| Arikayce | An amikacin liposome inhalation suspension[67] | Nontuberculous mycobacterium | Inhalation of liposomal preparation offers the advantage of directed delivery to the lung macrophages. The amikacin-laden macrophages then act as a drug depot, allowing increased intracellular and extracellular bacterial killing. Arikayce has a 3-fold increased conversion to NTM negative sputum rate, as compared to standard multidrug antibiotic regimen[71]. |
The second antimicrobial nanomedicine introduced was Arikayce, an amikacin liposome inhalation suspension, which was approved for clinical use within the last few years. Arikayce is an approximately 300nm liposome made of cholesterol and DPPC with a lipid-to-amikacin ratio of 0.6 to 0.79[67]. Arikayce was developed as a response to poor treatment options for pulmonary infections with nontuberculous mycobacteria. Amikacin has been used to treat nontuberculous mycobacteria infections with variable efficacy, and its use has been significantly limited by off-target systemic toxicities. Intravenous liposomal amikacin has been able to decrease side effects and achieve effective nontuberculous mycobacteria treatment; however, the inhibition of bacterial proliferation from this preparation is brief and followed by bacterial growth after completion of treatment. Arikayce has been shown to produce 274-fold higher amikacin levels in lung tissues, airways, and pulmonary macrophages compared with intravenous amikacin[68]. This concentration of antibiotic at the site of infection, in conjunction with fewer toxic side effects observed, makes Arikayce a promising treatment for nontuberculous mycobacterial infections[69].
Pneumonia special considerations
Two clinical outcomes of pneumonia discussed in the introduction, formation of a cavitary lesion and empyema, pose significant barriers to drug delivery for effective treatment. Cavitary pneumonia can develop in cases of antibiotic-resistant bacteria[72], immunocompromised hosts, or non-adherence to outpatient treatment regimen[73]. Lung tissue with cavitary lesions has decreased blood supply, which makes it difficult for systemic intravenous antibiotics to reach these tissues[74]. Cavitary lesions are often not in continuity with the airway, which additionally makes it difficult for inhaled antibiotics to reach the site of infection, resulting in frequent need for surgical debridement to address this complication[75].
While diffusion of antibiotics into the pleural space is usually good, infection with empyema presents other difficulties. The antibiotics can be inactivated by the purulent material in the pleural space, the low pH of infected material in the pleural space, and beta-lactamase enzymes produced by some bacteria[76]. For these reasons, despite antibiotic therapy, definite treatment of empyema often includes surgical debridement and decortication[77].
3. Infection treatment targets beyond bacterial killing
Infection treatment has not been limited to antimicrobials with the goal of bacterial killing. Other areas of interest include: antivirals, biofilm perturbation, sequestration of bacterial toxins, and alteration of host response to infection.
3.1. Antivirals
Antivirals have been employed with varying degrees of success. In the case of Hepatitis C, direct-acting antiviral drugs have led to a more than 90% cure rate[78][79]. Despite the great success seen with Hepatitis C, direct-acting antiviral technology has not continued to develop due to a significant roadblock. For many other viral infections, direct targeting of the virus has been largely unsuccessful due to the rapid emergence of antiviral drug resistance[80][81][82][83][84].
Recent work has change focused from pharmaceuticals with a direct-acting viral target. Rather, emerging technologies focus on modifiable host factors that can help clear viral infection. One possible target is host cell factors that are required for viral replication, but dispensable for the host[85][86][87]. These targets offer the benefit of decreasing viral load by blocking replication, while also having minimal effects to host functioning. These therapies are limited by the identification of dispensable host-cell targets and viral escape through secondary pathways.
The host damage inflicted by viral infections is two-pronged. The intrinsic viral pathogenicity is the first cause of viral morbidity and mortality. The second host is due directly to the innate immune response, which can lead to tissue injury in the process of viral clearance[88]. This leads to a second possible target for infection treatment: the host immune response, with a goal of enhancing host-directed viral control while limiting tissue damaging immunopathology[89]. Future antiviral treatments will need include a combination of a direct-acting antiviral with a focus on host immunomodulation to address both etiologies of morbidity and mortality.
3.2. Biofilms
Biofilms are formed when bacteria attach to a surface, create a colony, and form a matrix of sticky extracellular polymeric substances[90]. Biofilms are estimated to be responsible for approximately 80% of human infections[91]. Biofilm formation can lead to persistent infection as the biofilm eludes innate and adaptive host defenses, and the extracellular polymeric matrix can shelter bacteria from antimicrobial drugs[92]. Biofilm resistance to treatment is through a combination of several modalities including: decreased antibiotic penetration, extracellular DNA release, bacteriophage production, quorum sensing, and horizontal transfer of resistance genes[93]. Due to the frequency with which biofilms produce persistent infections, they represent a significant challenge to clinical care.
Biofilms form more readily on damaged tissues and implantable devices, causing their resistance to treatment to be especially worrisome in the medical device space. Bacterial biofilms are the principal source of central line associated infections, and persist despite advances in catheter engineering and sterile placement techniques[94]. Biofilms have been implicated in the difficulty with treating infective endocarditis for both native and prosthetic heart valves, and have led to poor treatment outcomes and frequent need for surgical intervention[95][96]. Prosthetic joint biofilm infection is especially devastating as it almost always requires surgical revision, sometimes with joint replacement[97][98], at an estimated cost to the US of $1.62 billion by 2020[99]. Between the clinical impact and economic implications, it is imperative to discover more effective biofilm treatment modalities.
Given the many limitations of traditional antibiotic treatment to eradicating biofilms, nanomaterials have been engineered to specifically address biofilms with some success in the lab. Particles that are smaller than 350nm can diffuse through the biofilm pores[100], allowing them to deliver antibiotic cargo to bacteria within the biofilm. This method of antibiotic delivery has the potential to overcome the protective extracellular polymeric matrix, which has traditionally contributed to biofilm resistance to antibiotic treatment.
As mentioned in section 1.3, bacterial efflux pumps are a common resistance mechanism, which cause antibiotic levels in bacteria to remain below the minimum inhibitory concentration. Metal nanoparticles, like zinc oxide nanoparticles, can inhibit bacterial efflux pumps, which allows adequate antibiotic levels to accumulate in the bacteria and enhance antibacterial effects in biofilms[101]. Cationic nanoparticles have enhanced antibacterial activity due to increased interaction with biofilms as bacterial cell membranes display higher levels of anionic lipids than mammalian cells[102]. Individually these preparations show promise for future clinical use for biofilm infections, and in combination they have the potential prospect of saving implantable devices.
3.3. Bacterial toxins
Bacterial toxins can cause broad morbidity, including diarrhea, rash, peeling skin, fever, hypotension, and more[103], even in the absence of replicating bacteria. Many of these toxins need to interact with the cell membrane to exhibit their virulence. Cell membrane mimicking particles have been created to neutralize toxins by sequestering these virulence factors. While these particles do not aid in bacterial clearance, they can improve morbidity associated with infection[104]. This has already been shown in a Gram-positive infection mouse model, where engineered toxin binding liposomes were able to rescue mice from fatal septicemia[105]. Used in conjunction with a bacteriostatic or bactericidal pharmaceutical, they can improve infection clinical outcomes.
Another particle with dual functionality is the biomimetic nanoparticle, which has been developed with dual action of neutralizing endotoxin while decreasing proinflammatory cytokines[106]. The polymeric core is wrapped with macrophage cell membrane to act as a macrophage-like decoy. These particles first bind and neutralize bacterial endotoxin, and then sequester proinflammatory cytokines. They have been shown to inhibit bacterial proliferation and reduce proinflammatory cytokine levels in mice, leading to improved survival in E. coli sepsis. Through several mechanisms, the addition of these anti-toxin nanopreparations to antibacterial treatment has the potential to decrease morbidity associated with infections.
3.4. Host inflammatory response
The human immune response to infection is inflammation, which can cause damage to the host itself. Unchecked inflammation leads to clinical syndromes such as sepsis and ARDS that can be even more damaging than the initial infection. Modulation of the immune response has been an area of interest to improve patient outcomes.
One immune modulation goal is to decrease inflammatory cell activity in infection. Platelet-derived extracellular vesicles have been engineered for pneumonia-targeted drug delivery. When loaded with anti-inflammatory factors, they have been shown to decrease inflammatory cell migration into the lung and decrease the cytokine levels within lung tissue in pneumonia[107]. Intercellular adhesion molecule-1 (I-CAM) targeted poly(lactide-co-glycolide acid) (PLGA) nanoparticles co-loaded with Sparfloxacin, an antimicrobial, and Tacrolimus, an immunosuppressant drug were able to decrease bacterial load and host inflammatory factors in a pneumonia mouse model[60]. This led to a 6-fold increase in survival with treatment, demonstrating effective improvement in treatment by immune modulation.
Another area for immune modulation is the amplification signaling associated with activation of immune cells and cytokine release. Doxorubicin-conjugated albumin nanoparticles were able to selectively target activated neutrophils using pH-labile bonding leading to targeted Doxorubicin-induced apoptosis of activated neutrophils. This treatment inhibited neutrophil transmigration into tissues, decreased inflammatory cytokine levels, and led to improved survival in sepsis[108]. Siglec receptors are immunomodulatory receptors found in both mice and humans. PLGA nanoparticles have also been engineered with Siglec-binding ligands, and were shown to block macrophage production of inflammatory cytokines. These particles demonstrate therapeutic benefit in both mouse and human models of lung infection and injury[109]. Another approach has been macrophage or neutrophil membrane coating of nanoparticles to bind and neutralize inflammatory cytokines[110]. As treatment modalities improve, the modulation of the host inflammatory response will be an important area of growth for treatment of infections.
4. Shifting Focus to Targeted Drug Delivery Systems
Future innovation for antibiotics will certainly center on the delivery of small molecule which can alter their pharmacokinetics, volume of distribution, efficacy, and off-target effects. There is currently research on many different delivery targets: to organ tissues, to specific cells, and directly to bacteria (Figure 2).
Figure 2. Potential classes of target for nanocarrier antibiotic delivery.
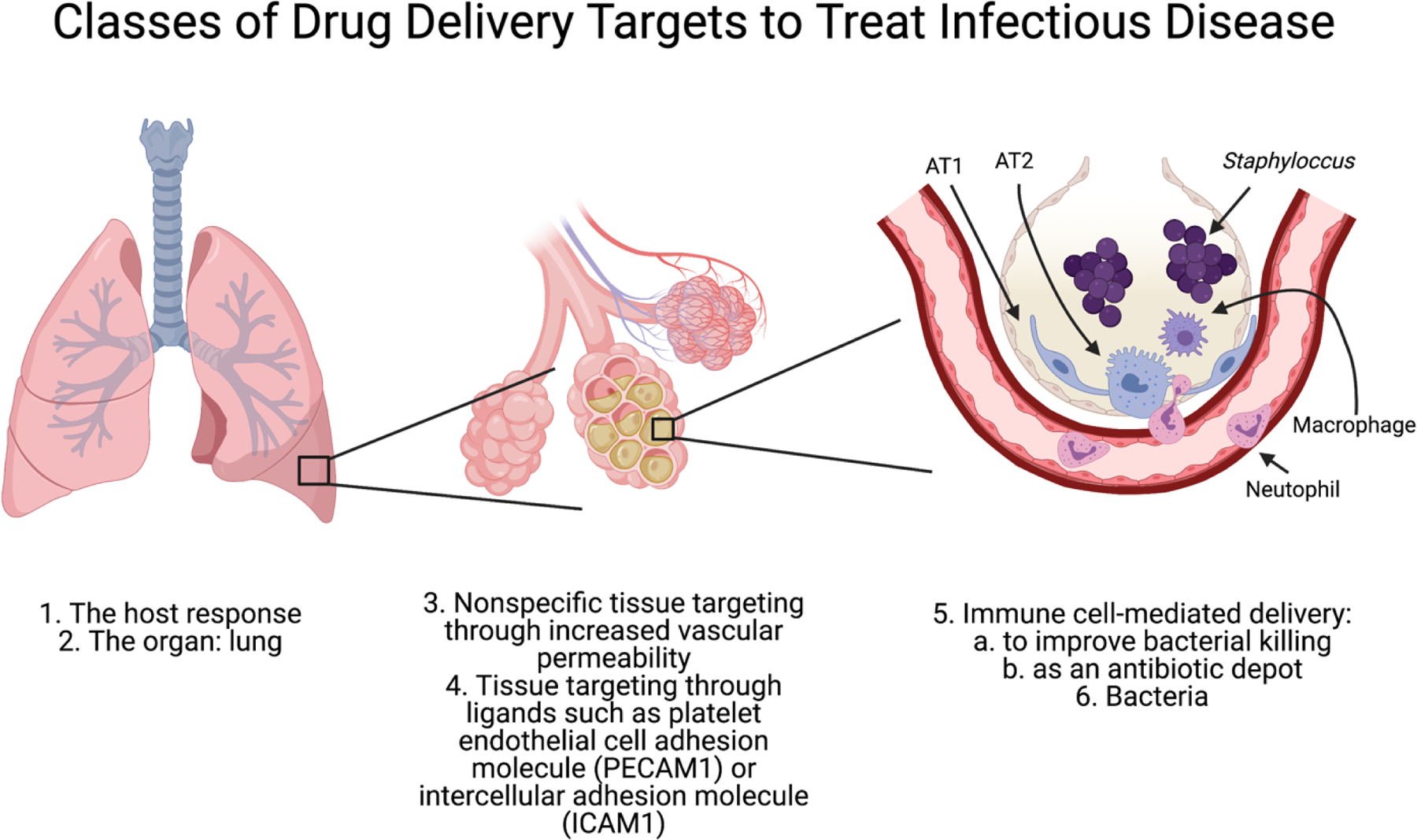
There are many options of target for nanocarrier delivery to treat infectious disease. Ligands on nanoparticles can be used to target different organs, tissues, and cells, and are discussed in detail in section 4. AT1 = alveolar type 1 cell, AT2 = alveolar type 2 cell.
4.1. Tissue and cell passive targeting
Local biochemical changes in response to bacterial infection can be used for tissue targeting of antibiotics. The accumulation of bacterial components and products, from both Gram-positive and Gram-negative organisms, triggers rapid vasodilatation with subsequent recruitment of additional capillaries[111]. Bacterial components additionally stimulate immune cells to release inflammatory mediators resulting in endothelial gap widening, barrier dysfunction, and increased vascular permeability[112]. Macromolecules will preferentially accumulate at sites of enhance vascular permeability, known as the enhanced permeability and retention (EPR) effect[113]. Laverman et al. were able to show that Polyethylene glycol (PEG) liposomes accumulate in the soft tissue of localized S. aureus infection without any added targeting moieties on the liposomes[114]. They found a maximum of 3.78±0.62% injected dose at the site of abscess as compared to uptake in the non-infected contralateral muscle, which remained low. The abscess-to-muscle ratios reached values as high as 39.7±9.5 at 24 hours post-injection based on the relative uptake calculated in %ID/g. These particles were mainly localized in the vicinity of blood vessels, and TEM showed these liposomes were localized mainly to macrophages. This passive targeting allows for antibiotic concentration at sites of leaky vasculature, with the benefit of no bioengineering needed to see the effect. The EPR effect is not an ideal modality as widespread vascular dysfunction, which is frequently seen in sepsis, results in diffuse delivery of antibiotic, rather than a concentration at the site of infection.
4.2. Tissue and cell active targeting
The addition of tissue- and cell-specific targeting moieties on nanoparticles has the potential to improve bacterial clearance with decreased off-target side effects. Two particularly beneficial targets are neutrophils and macrophages, as uptake of antibiotics may improve their bacterial killing capacity. Macrophage targeting is already being used in one of the two currently FDA approved anti-infective nanoparticle preparations, Arikayce (table 2)[71].
Wang et al. utilized the addition of a reactive oxygen species-responsive material to nanoparticle design to direct antibiotic delivery to infected lung tissue and pulmonary macrophages. By encapsulating moxifloxacin with 4-(hydroxymethyl) phenylboronic acid pinacol ester-modified α-cyclodextrin (Oxi-αCD), there was effective internalization by bacteria-infected macrophages in in vitro models, which decreased bacterial load in macrophages as compared to non-targeted moxifloxacin nanoparticles. Additionally, it resulted in better antibacterial activity and prolonged survival in an in vivo mouse model[115].
Treatment of intracellular infections may require drug targeting to macrophages and monocytes. Prior et al. utilized end-group capped and uncapped poly(lactide) (PLA) and PLGA microspheres loaded with gentamicin for their recent study. They showed these were readily phagocytosed by macrophages and monocytes, with improved affinity associated with increasing polymer hydrophobicity[116]. Microsphere phagocytosis caused activation of human monocytes, resulting in an oxidative burst. Not only do these particles serve to deliver antibiotics directly to the bacterial-fighting cells. They also have a secondary activating effect, which may serve to improve bacterial killing activity of these monocytes. Because of the multiple benefits discussed above, directed antibiotic delivery into immune cells should be an important focus for future nanotechnology development.
4.3. Bacterial targeting
Another option is targeting the pathogenic bacteria itself. Bacterial cell targeting has been used with some success in in vitro studies, and is one of the main areas of study for anti-microbial heavy metal particles. One antibody-antibiotic conjugate utilized a highly abundant S. aureus cell wall protein, β-N-acetylglucosamine cell-wall teichoic acid (β-GlcNAc-WTA), which is not present on mammalian cells[117]. The particles were designed with a potent antibiotic conjugated to an intracellular protease-sensitive linker, leading to antibiotic release only with uptake into the phagolysosome. The design allowed for targeting directly to S. aureus, with directed intracellular bacterial killing.
Gold-silver nanoparticles have been successfully targeted to bacteria by incorporation of anti-methicillin resistant S. aureus (MRSA) antibodies[118]. Antibody modification resulted in more than a 10-fold enhancement in targeting with significantly lower concentrations needed to achieve bacteriostasis.
Microbe-targeting is not limited to intravenous preparations, and has been developed in some oral forms[119]. Addition of a chitosan-binding peptide to PLGA nanoparticles resulted in Cryptococcus neoformans targeting, and subsequent incubation in free chitosan enhanced oral absorption of these particles. Itraconazole loading of these particles resulted in clearance of C. neoformans pneumonia and improved survival in mouse models.
5. Market Innovation
While it is clear that there are multiple unmet needs in the infection treatment space, it is less clear that market forces will support such development. Several factors collaborate to make the antibiotic market unattractive to large pharmaceutical companies and to investors[120]. First, most bacterial infections are short-lived, so each prescribed patient does not generate the large revenues of chronic disease patients. Second, there are cheap, generic drugs that work in most patients (relatively few infections are multi-drug resistant). Third, and most importantly, healthcare systems intentionally ration the use of new extended-spectrum antibiotics, to prevent the development of resistance. All these factors combined to cause a 90% decline in new antibiotic development in the 40 years[121].
However, all is not lost. Many national governments are now introducing policies that could encourage new antibiotic development, such as more government research grants and extended patent rights[120]. While waiting for those policies to solidify, makers of novel antibiotics might benefit from comparing Table 1 with the 3 leading factors listed above for why there is poor investment in novel antibiotics. From such a comparison, it would seem that the economically riskiest area to innovate in is acute multi-drug resistant infections, as their use will (and should) be rationed to prevent development of new bacterial resistance. By contrast, drugs that focus on the other unmet needs listed in Table 1 do not face such economic headwinds. Intriguingly, it is possible that some drug delivery technologies could be developed address the unmet needs of Table 1, and incidentally also be used to treat multi-drug resistant infection. For example, a technology that localizes a drug to the site of infection (e.g., pneumonia) to prevent decimation of the gut microbiome could have a viable market from that more widespread indication, but also secondarily be used for the rarer cases of refractory, multi-drug resistant pneumonia. This approach has already been proven out with Arikayce, which is an inhaled formulation of liposomes loaded with the effective but toxic antibiotic amikacin[67]. Arikayce is approved for chronic, non-tuberculous mycobacterial infection of the lungs. It’s localization to the lungs prevents a number of side effects from the cargo drug (e.g., gut microbiome elimination and kidney failure), while also allowing it to treat some of the most multi-drug resistant bacteria outside of hospitals. Considering examples like Arikayce, and the above economic realities and practicalities, may help improve the innovation process for antimicrobial drug delivery.
6. Summary
Antibiotics are a marvel of modern medicine that have changed the outcome of infection for over 100 years. Nonetheless, major limitations remain, in key areas such as treating biofilms, the host response, viral infections, and preventing decimation of the gut microbiome. Additionally, multi-drug resistant organisms are becoming a greater concern, though market forces may delay innovation in that space until governmental incentive programs are rolled out. Targeted drug delivery holds great potential for solving these unmet needs, and may eventually achieve the original promise of antibiotics, that infections will no longer be a major cause of significant illness and death.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Xu J, Murphy SL, Kockanek KD, Arias E, Mortality in the United States, 2018, NCHS Data Brief. (2020) 1–8. [PubMed] [Google Scholar]
- [2].File TM, Marrie TJ, Burden of community-acquired pneumonia in North American adults, Postgrad. Med 122 (2010) 130–141. 10.3810/pgm.2010.03.2130. [DOI] [PubMed] [Google Scholar]
- [3].Pneumonia, (n.d.). https://www.who.int/news-room/fact-sheets/detail/pneumonia (accessed March 4, 2021).
- [4].U. Centers for Disease Control, Antibiotic Resistance Threats in the United States, 2019, (n.d.). 10.15620/cdc:82532. [DOI] [Google Scholar]
- [5].ANTIMICROBIAL RESISTANCE Global Report on Surveillance, n.d.
- [6].Reardon S, Spread of antibiotic-resistance gene does not spell bacterial apocalypse — yet, Nature. (2015). 10.1038/nature.2015.19037. [DOI] [Google Scholar]
- [7].Silver LL, Challenges of antibacterial discovery, Clin. Microbiol. Rev 24 (2011) 71–109. 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J, Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America, Clin. Infect. Dis 48 (2009) 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- [9].Sengupta S, Chattopadhyay MK, Grossart H-P, Walsh F, Lin J, Cytryn E, The multifaceted roles of antibiotics and antibiotic resistance in nature, (1940). 10.3389/fmicb.2013.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nicolaou KC, Rigol S, A brief history of antibiotics and select advances in their synthesis, J. Antibiot. (Tokyo) 71 (2018) 153–184. 10.1038/ja.2017.62. [DOI] [PubMed] [Google Scholar]
- [11].Zaffiri L, Gardner J, Toledo-Pereyra LH, History of antibiotics. from salvarsan to cephalosporins, J. Investig. Surg 25 (2012) 67–77. 10.3109/08941939.2012.664099. [DOI] [PubMed] [Google Scholar]
- [12].Lloyd NC, Morgan HW, Nicholson BK, Ronimus RS, The Composition of Ehrlich’s Salvarsan: Resolution of a Century-Old Debate, Angew. Chemie Int. Ed 44 (2005) 941–944. 10.1002/anie.200461471. [DOI] [PubMed] [Google Scholar]
- [13].F.R.C.S. FLEMING ALEXANDER, ON THE ANTIBACTERIAL ACTION OF CULTURES OF A PENICILLIUM, WITH SPECIAL REFERENCE TO THEIR USE IN THE ISOLATION OF B. INFLUENZ?1E., Br. J. Exp. Pathol (1929). [PMC free article] [PubMed] [Google Scholar]
- [14].de Velde F, Mouton JW, de Winter BCM, van Gelder T, Koch BCP, Clinical applications of population pharmacokinetic models of antibiotics: Challenges and perspectives, Pharmacol. Res 134 (2018) 280–288. 10.1016/j.phrs.2018.07.005. [DOI] [PubMed] [Google Scholar]
- [15].Rouveix B, Antibiotic safety assessment, Int. J. Antimicrob. Agents 21 (2003) 215–221. 10.1016/S0924-8579(02)00354-0. [DOI] [PubMed] [Google Scholar]
- [16].Roberts JA, Kirkpatrick CMJ, Lipman J, Monte Carlo simulations: maximizing antibiotic pharmacokinetic data to optimize clinical practice for critically ill patients, (n.d.). 10.1093/jac/dkq449. [DOI] [PubMed] [Google Scholar]
- [17].De Biasi S, Coloretti I, Cossarizza A, Multidrug resistant bacteria in critically ill patients: a step further antibiotic therapy, (2018). 10.21037/jeccm.2018.11.08. [DOI] [Google Scholar]
- [18].Deshpande A, Pasupuleti V, Thota P, Pant C, Rolston DDK, Sferra TJ, Hernandez AV, Donskey CJ, Community-associated Clostridium difficile infection and antibiotics: a meta-analysis, J. Antimicrob. Chemother 68 (2013) 1951–1961. 10.1093/jac/dkt129. [DOI] [PubMed] [Google Scholar]
- [19].Raba AA, O’Sullivan A, Semberova J, Martin A, Miletin J, Are antibiotics a risk factor for the development of necrotizing enterocolitis—case-control retrospective study, Eur. J. Pediatr 178 (2019) 923–928. 10.1007/s00431-019-03373-0. [DOI] [PubMed] [Google Scholar]
- [20].Demerec M, ORIGIN OF BACTERIAL RESISTANCE TO ANTIBIOTICS’, n.d. http://jb.asm.org/. [DOI] [PMC free article] [PubMed]
- [21].Sköld O, Sulfonamide resistance Sulfonamide resistance: mechanisms and trends, (2000). 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- [22].Dever LA, Dermody TS, Mechanisms of Bacterial Resistance to Antibiotics, n.d. https://jamanetwork.com/. [PubMed]
- [23].Clewell DB, Plasmids, Drug Resistance, and Gene Transfer in the Genus Streptococcus, 1981. http://mmbr.asm.org/. [DOI] [PMC free article] [PubMed]
- [24].Lee Ventola C, The Antibiotic Resistance Crisis Part 1: Causes and Threats, 2015. [PMC free article] [PubMed]
- [25].Andrews JM, Determination of minimum inhibitory concentrations, J. Antimicrob. Chemother 48 (2001) 5–16. 10.1093/JAC/48.SUPPL_1.5. [DOI] [PubMed] [Google Scholar]
- [26].Cojutti P, Sartor A, Bassetti M, Scarparo C, Pea F, Is meropenem MIC increase against KPC-producing Klebsiella pneumoniae correlated with increased resistance rates against other antimicrobials with Gram-negative activity?, J. Glob. Antimicrob. Resist 14 (2018) 238–241. 10.1016/J.JGAR.2018.05.005. [DOI] [PubMed] [Google Scholar]
- [27].Medina E, Pieper DH, Tackling threats and future problems of multidrug-resistant bacteria, Curr. Top. Microbiol. Immunol 398 (2016) 3–33. 10.1007/82_2016_492. [DOI] [PubMed] [Google Scholar]
- [28].Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, Ouellette M, Outterson K, Patel J, Cavaleri M, Cox EM, Houchens CR, Grayson ML, Hansen P, Singh N, Theuretzbacher U, Magrini N, Aboderin AO, Al-Abri SS, Awang Jalil N, Benzonana N, Bhattacharya S, Brink AJ, Burkert FR, Cars O, Cornaglia G, Dyar OJ, Friedrich AW, Gales AC, Gandra S, Giske CG, Goff DA, Goossens H, Gottlieb T, Guzman Blanco M, Hryniewicz W, Kattula D, Jinks T, Kanj SS, Kerr L, Kieny MP, Kim YS, Kozlov RS, Labarca J, Laxminarayan R, Leder K, Leibovici L, Levy-Hara G, Littman J, Malhotra-Kumar S, Manchanda V, Moja L, Ndoye B, Pan A, Paterson DL, Paul M, Qiu H, Ramon-Pardo P, Rodríguez-Baño J, Sanguinetti M, Sengupta S, Sharland M, Si-Mehand M, Silver LL, Song W, Steinbakk M, Thomsen J, Thwaites GE, van der Meer JW, Van Kinh N, Vega S, Villegas MV, Wechsler-Fördös A, Wertheim HFL, Wesangula E, Woodford N, Yilmaz FO, Zorzet A, Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis, Lancet Infect. Dis 18 (2018) 318–327. 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- [29].Ahmed A, Azim A, Gurjar M, Baronia AK, Current concepts in combination antibiotic therapy for critically ill patients, Indian J. Crit. Care Med 18 (2014) 310–314. 10.4103/0972-5229.132495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Karaiskos I, Lagou S, Pontikis K, Rapti V, Poulakou G, The “Old” and the “New” antibiotics for MDR Gram-negative pathogens: For whom, when, and how, Front. Public Heal 7 (2019) 151. 10.3389/fpubh.2019.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Song M, Liu Y, Huang X, Ding S, Wang Y, Shen J, Zhu K, A broad-spectrum antibiotic adjuvant reverses multidrug-resistant Gram-negative pathogens, Nat. Microbiol 5 (2020) 1040–1050. 10.1038/s41564-020-0723-z. [DOI] [PubMed] [Google Scholar]
- [32].Geitani R, Ayoub Moubareck C, Touqui L, Karam Sarkis D, Cationic antimicrobial peptides: Alternatives and/or adjuvants to antibiotics active against methicillin-resistant Staphylococcus aureus and multidrug-resistant Pseudomonas aeruginosa, BMC Microbiol 19 (2019) 1–12. 10.1186/s12866-019-1416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ammeter D, Idowu T, Zhanel GG, Schweizer F, Development of a nebramine-cyclam conjugate as an antibacterial adjuvant to potentiate β-lactam antibiotics against multidrug-resistant P. aeruginosa, J. Antibiot. (Tokyo) 72 (2019) 816–826. 10.1038/s41429-019-0221-9. [DOI] [PubMed] [Google Scholar]
- [34].Worthington RJ, Melander C, Combination approaches to combat multidrug-resistant bacteria, Trends Biotechnol. 31 (2013) 177–184. 10.1016/j.tibtech.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bassetti M, Righi E, Vena A, Graziano E, Russo A, Peghin M, Risk stratification and treatment of ICU-acquired pneumonia caused by multidrug-resistant/extensively drug-resistant/pandrug-resistant bacteria, Curr. Opin. Crit. Care 24 (2018) 385–393. 10.1097/MCC.0000000000000534. [DOI] [PubMed] [Google Scholar]
- [36].Clement JL, Jarrett PS, ANTIBACTERIAL SILVER, n.d. [DOI] [PMC free article] [PubMed]
- [37].Brouillard C, Bursztejn A-C, Latarche C, Cuny J-F, Truchetet F, Goullé J-P, Schmutz J-L, Silver absorption and toxicity evaluation of silver wound dressings in 40 patients with chronic wounds, J. Eur. Acad. Dermatology Venereol 32 (2018) 2295–2299. 10.1111/JDV.15055. [DOI] [PubMed] [Google Scholar]
- [38].Fung MC, Bowen DL, Silver products for medical indications: Risk-benefit assessment, J. Toxicol. - Clin. Toxicol 34 (1996) 119–126. 10.3109/15563659609020246. [DOI] [PubMed] [Google Scholar]
- [39].Lansdown ABG, A Pharmacological and Toxicological Profile of Silver as an Antimicrobial Agent in Medical Devices, Adv. Pharmacol. Sci 2010 (2010) 16. 10.1155/2010/910686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lara HH, Ayala-Núñez NV, del C L. Turrent Ixtepan, Rodríguez Padilla C, Bactericidal effect of silver nanoparticles against multidrug-resistant bacteria, World J. Microbiol. Biotechnol 26 (2010) 615–621. 10.1007/s11274-009-0211-3. [DOI] [Google Scholar]
- [41].J. S, C. J, Unexpected antimicrobial effect of statins, J. Antimicrob. Chemother 61 (2008) 362–364. 10.1093/JAC/DKM496. [DOI] [PubMed] [Google Scholar]
- [42].Figueiredo EP, Ribeiro JM, Nishio EK, Scandorieiro S, Costa AF, Cardozo VF, Oliveira AG, Durán N, Panagio LA, Kobayashi R, Nakazato G, New Approach For Simvastatin As An Antibacterial: Synergistic Effect With Bio-Synthesized Silver Nanoparticles Against Multidrug-Resistant Bacteria, (2019). 10.2147/IJN.S211756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].G. S, P. JH, H. JW, K. JH, Comparative assessment of the apoptotic potential of silver nanoparticles synthesized by Bacillus tequilensis and Calocybe indica in MDA-MB-231 human breast cancer cells: targeting p53 for anticancer therapy, Int. J. Nanomedicine 10 (2015) 4203–4223. 10.2147/IJN.S83953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liao C, Li Y, Tjong SC, Bactericidal and Cytotoxic Properties of Silver Nanoparticles, (n.d.). 10.3390/ijms20020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kim S, Covington A, Pamer EG, The intestinal microbiota: Antibiotics, colonization resistance, and enteric pathogens, Immunol. Rev 279 (2017) 90. 10.1111/IMR.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ghai I, Ghai S, Understanding antibiotic resistance via outer membrane permeability, Infect. Drug Resist 11 (2018) 523. 10.2147/IDR.S156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ghosh M, Miller PA, Mö U, Claypool WD, Schroeder VA, Wolter WR, Suckow M, Yu H, Li S, Huang W, Zajicek J, Miller MJ, Targeted Antibiotic Delivery: Selective Siderophore Conjugation with Daptomycin Confers Potent Activity against Multidrug Resistant Acinetobacter baumannii Both in Vitro and in Vivo, (2017). 10.1021/acs.jmedchem.7b00102. [DOI] [PubMed] [Google Scholar]
- [48].B. D, S. A, E. RI, Cellular immunity and lung injury in respiratory virus infection, Viral Immunol. 19 (2006) 147–155. 10.1089/VIM.2006.19.147. [DOI] [PubMed] [Google Scholar]
- [49].CM H, MA M, Viral pathogens and acute lung injury: investigations inspired by the SARS epidemic and the 2009 H1N1 influenza pandemic, Semin. Respir. Crit. Care Med 34 (2013) 475–486. 10.1055/S-0033-1351122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang Z, Liu H, Yang SH, Wang T, Liu C, Charles Cao Y, Nanoparticle-based artificial RNA silencing machinery for antiviral therapy, (n.d.). 10.1073/pnas.1207766109/-/DCSupplemental. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Landersdorfer CB, Bulitta JB, Sörgel F, Pharmacokinetics and Pharmacodynamics of Antibiotics in Bone, (2015). 10.1002/9781118581742.ch3. [DOI] [Google Scholar]
- [52].Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace J. Richard J., Winthrop K, An Official ATS/IDSA Statement: Diagnosis, Treatment, and Prevention of Nontuberculous Mycobacterial Diseases, Https://Doi.Org/10.1164/Rccm.200604-571ST. 175 (2012) 367–416. 10.1164/RCCM.200604-571ST. [DOI] [PubMed] [Google Scholar]
- [53].Dou H, Grotepas CB, McMillan JM, Destache CJ, Chaubal M, Werling J, Kipp J, Rabinow B, Gendelman HE, Macrophage Delivery of Nanoformulated Antiretroviral Drug to the Brain in a Murine Model of NeuroAIDS, J. Immunol 183 (2009) 661–669. 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang P, Liu Y, Xu Y, Xu Z, Staphylococcus saccharolyticus infection: case series with a PRISMA-compliant systemic review, Medicine (Baltimore). 99 (2020) e20686. 10.1097/MD.0000000000020686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].†,§,‖ Lok Chun-Nam, †,‡ Ho Chi-Ming, †,‡ Chen Rong, †,‡ He Qing-Yu, †,‡ Yu Wing-Yiu, †,‡ Sun Hongzhe, ⊥,# Tam Paul Kwong-Hang, *,†,§,‖ and Chiu Jen-Fu, †,‡ Che Chi-Ming *, Proteomic Analysis of the Mode of Antibacterial Action of Silver Nanoparticles, J. Proteome Res 5 (2006) 916–924. 10.1021/PR0504079. [DOI] [PubMed] [Google Scholar]
- [56].Hall CW, Mah T-F, Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria, FEMS Microbiol. Rev 010 (2017) 276–301. 10.1093/femsre/fux010. [DOI] [PubMed] [Google Scholar]
- [57].M. JS, B. M, H. LH, S. SJ, The interconnection between biofilm formation and horizontal gene transfer, FEMS Immunol. Med. Microbiol 65 (2012) 183–195. 10.1111/J.1574-695X.2012.00960.X. [DOI] [PubMed] [Google Scholar]
- [58].Qiu Y, Wu Y, Lu B, Zhu G, Gong T, Wang R, Peng Q, Li Y, Inhibition of methicillin-resistant Staphylococcus aureus (MRSA) biofilm by cationic poly (D, L-lactide-co-glycolide) nanoparticles, Https://Doi-Org.Proxy.Library.Upenn.Edu/10.1080/08927014.2020.1740687. 36 (2020) 159–168. 10.1080/08927014.2020.1740687. [DOI] [PubMed] [Google Scholar]
- [59].Branchett WJ, Lloyd CM, Regulatory cytokine function in the respiratory tract, Mucosal Immunol. 2019 123. 12 (2019) 589–600. 10.1038/s41385-019-0158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yang Y, Ding Y, Fan B, Wang Y, Mao Z, Wang W, Wu J, Inflammation-targeting polymeric nanoparticles deliver sparfloxacin and tacrolimus for combating acute lung sepsis, (2020). 10.1016/j.jconrel.2020.02.030. [DOI] [PubMed] [Google Scholar]
- [61].Mechanism of action of AmBisome® (amphotericin B) liposome for injection, (n.d.).
- [62].H. V, B. D, J. U, K. B, W. K, D. A, S. P, K. HJ, W. W, Pharmacokinetics of liposomal amphotericin B (Ambisome) in critically ill patients, Antimicrob. Agents Chemother 41 (1997) 1275–1280. 10.1128/AAC.41.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].van E. EW, O.-L. M, van V. W, ten K. MT, B.-W. AJ, Biodistribution of liposomal amphotericin B (AmBisome) and amphotericin B-desoxycholate (Fungizone) in uninfected immunocompetent mice and leucopenic mice infected with Candida albicans, J. Antimicrob. Chemother 35 (1995) 509–519. 10.1093/JAC/35.4.509. [DOI] [PubMed] [Google Scholar]
- [64].Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ, Pharmacokinetics, Excretion, and Mass Balance of Liposomal Amphotericin B (AmBisome) and Amphotericin B Deoxycholate in Humans, Antimicrob. Agents Chemother 46 (2002) 828. 10.1128/AAC.46.3.828-833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Walker L, Sood P, Lenardon MD, Milne G, Olson J, Jensen G, Wolf J, Casadevall A, Adler-Moore J, Gow NAR, The viscoelastic properties of the fungal cell wall allow traffic of ambisome as intact liposome vesicles, MBio. 9 (2018). 10.1128/mBio.02383-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Aversa F, Busca A, Candoni A, Cesaro S, Girmenia C, Luppi M, Nosari AM, Pagano L, Romani L, Rossi G, Venditti A, Novelli A, Liposomal amphotericin B (AmBisome®) at beginning of its third decade of clinical use, J. Chemother 29 (2017) 131–143. 10.1080/1120009X.2017.1306183. [DOI] [PubMed] [Google Scholar]
- [67].FDA, ARIKAYCE [package insert], (n.d.). https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207356s000lbl.pdf (accessed September 3, 2021).
- [68].Zhang J, Leifer F, Rose S, Chun DY, Thaisz J, Herr T, Nashed M, Joseph J, Perkins WR, DiPetrillo K, Amikacin liposome inhalation suspension (ALIS) penetrates non-tuberculous mycobacterial biofilms and enhances amikacin uptake into macrophages, Front. Microbiol 9 (2018). 10.3389/fmicb.2018.00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Khan O, Chaudary N, The use of amikacin liposome inhalation suspension (Arikayce) in the treatment of refractory nontuberculous mycobacterial lung disease in adults, Drug Des. Devel. Ther 14 (2020) 2287–2294. 10.2147/DDDT.S146111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Drug Approval Package: Ambisome (Amphotericin B) NDA# 050740, (n.d.). https://www.accessdata.fda.gov/drugsatfda_docs/nda/97/050740_ambisome_toc.cfm (accessed September 3, 2021).
- [71].Griffith DE, Eagle G, Thomson R, Aksamit TR, Hasegawa N, Morimoto K, Addrizzo-Harris DJ, O’Donnell AE, Marras TK, Flume PA, Loebinger MR, Morgan L, Codecasa LR, Hill AT, Ruoss SJ, Yim J-J, Ringshausen FC, Field SK, Philley JV, Wallace J. Richard J., van Ingen J, Coulter C, Nezamis J, Winthrop KL, Amikacin Liposome Inhalation Suspension for Treatment-Refractory Lung Disease Caused by Mycobacterium avium Complex (CONVERT). A Prospective, Open-Label, Randomized Study, Https://Doi.Org/10.1164/Rccm.201807-1318OC. 198 (2018) 1559–1569. 10.1164/RCCM.201807-1318OC. [DOI] [PubMed] [Google Scholar]
- [72].Holt MR, Chan ED, Chronic Cavitary Infections Other than Tuberculosis, in: J. Thorac. Imaging, Lippincott Williams and Wilkins, 2018: pp. 322–333. 10.1097/RTI.0000000000000345. [DOI] [PubMed] [Google Scholar]
- [73].Cotton JL, Ali-Dinar T, Navas-Nazario A, Cavitary Pneumonia: A Complication of Antibiotic Noncompliance, Case Rep. Pulmonol 2020 (2020) 1–5. 10.1155/2020/5971348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Krutikov M, Rahman A, Tiberi S, Necrotizing pneumonia (aetiology, clinical features and management), Curr. Opin. Pulm. Med 25 (2019) 225–232. 10.1097/MCP.0000000000000571. [DOI] [PubMed] [Google Scholar]
- [75].C. N, F. D, B. KJ, Management of necrotizing pneumonia and pulmonary gangrene: A case series and review of the literature, Can. Respir. J (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hughes CE, Van Scoy RE, Antibiotic therapy of pleural empyema, Semin. Respir. Infect 6 (1991) 94–102. https://europepmc.org/article/med/1771308 (accessed March 14, 2021). [PubMed] [Google Scholar]
- [77].Reichert M, Hecker M, Witte B, Bodner J, Padberg W, Weigand MA, Hecker A, Stage-directed therapy of pleural empyema, Langenbeck’s Arch. Surg 402 (2017) 15–26. 10.1007/s00423-016-1498-9. [DOI] [PubMed] [Google Scholar]
- [78].Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH, From non-A, non-B hepatitis to hepatitis C virus cure, J. Hepatol 62 (2015) S87–S99. 10.1016/j.jhep.2015.02.006. [DOI] [PubMed] [Google Scholar]
- [79].Baumert TF, Berg T, Lim JK, Nelson DR, Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges, Gastroenterology. 156 (2019) 431–445. 10.1053/j.gastro.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li DK, Chung RT, Overview of direct-acting antiviral drugs and drug resistance of hepatitis C virus, in: Methods Mol. Biol, Humana Press Inc., 2019: pp. 3–32. 10.1007/978-1-4939-8976-8_1. [DOI] [PubMed] [Google Scholar]
- [81].Duwe S, Influenza viruses - antiviral therapy and resistance., GMS Infect. Dis 5 (2017) Doc04. 10.3205/id000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Locarnini S, Bowden S, Drug resistance in antiviral therapy, Clin. Liver Dis 14 (2010) 439–459. 10.1016/j.cld.2010.05.004. [DOI] [PubMed] [Google Scholar]
- [83].Pillay D, Zambon M, Antiviral drug resistance, Br. Med. J 317 (1998) 660–662. 10.1136/bmj.317.7159.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].J.S. OXFORD, Drug resistance and antiviral agents, J. Antimicrob. Chemother 2 (1976) 223–224. 10.1093/jac/2.3.223. [DOI] [PubMed] [Google Scholar]
- [85].Rasmussen AL, Host factors involved in ebola virus replication, in: Curr. Top. Microbiol. Immunol, Springer Verlag, 2018: pp. 113–150. 10.1007/82_2017_27. [DOI] [PubMed] [Google Scholar]
- [86].Delpeut S, Noyce RS, Siu RW, Richardson CD, Host factors and measles virus replication, Curr. Opin. Virol 2 (2012) 773–783. 10.1016/j.coviro.2012.10.008. [DOI] [PubMed] [Google Scholar]
- [87].Mehle A, Doudna JA, A Host of Factors Regulating Influenza Virus Replication, Viruses. 2 (2010) 566–573. 10.3390/v2020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Herold S, Becker C, Ridge KM, Budinger GRS, Influenza virus-induced lung injury: pathogenesis and implications for treatment, Eur. Respir. J 45 (2015) 1463–1478. 10.1183/09031936.00186214. [DOI] [PubMed] [Google Scholar]
- [89].Kumar N, Sharma S, Kumar R, Tripathi BN, Barua S, Ly H, Rouse BT, Host-Directed Antiviral Therapy, Clin. Microbiol. Rev 33 (2020). 10.1128/CMR.00168-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Donlan RM, Biofilms: Microbial Life on Surfaces, 2002. http://www.microbelibrary.org/. [DOI] [PMC free article] [PubMed]
- [91].Davies D, Understanding biofilm resistance to antibacterial agents, Nat. Rev. Drug Discov 2 (2003) 114–122. 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- [92].Arciola CR, Campoccia D, Montanaro L, Implant infections: Adhesion, biofilm formation and immune evasion, Nat. Rev. Microbiol 16 (2018) 397–409. 10.1038/s41579-018-0019-y. [DOI] [PubMed] [Google Scholar]
- [93].Hall CW, Mah T-F, Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria, FEMS Microbiol. Rev 010 (2017) 276–301. 10.1093/femsre/fux010. [DOI] [PubMed] [Google Scholar]
- [94].Percival SL, Kite P, Intravascular catheters and biofilm control., J. Vasc. Access 8 (n.d.) 69–80. http://www.ncbi.nlm.nih.gov/pubmed/17534791. [PubMed] [Google Scholar]
- [95].Di Domenico EG, Rimoldi SG, Cavallo I, D’Agosto G, Trento E, Cagnoni G, Palazzin A, Pagani C, Romeri F, De Vecchi E, Schiavini M, Secchi D, Antona C, Rizzardini G, Dichirico RB, Toma L, Kovacs D, Cardinali G, Gallo MT, Gismondo MR, Ensoli F, Microbial biofilm correlates with an increased antibiotic tolerance and poor therapeutic outcome in infective endocarditis, BMC Microbiol 19 (2019) 228. 10.1186/s12866-019-1596-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yaban B, Kikhneyid J, Musci M, A.P. 1¤, Schmidt J, Hajduczenia M, Schoenrath F, Falkid V, Moter A, Aerococcus urinae – A potent biofilm builder in endocarditis, (2020). 10.1371/journal.pone.0231827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Jamal M, Ahmad W, Andleeb S, Jalil F, Imran M, Nawaz MA, Hussain T, Ali M, Rafiq M, Kamil MA, Bacterial biofilm and associated infections, J. Chinese Med. Assoc 81 (2018) 7–11. 10.1016/j.jcma.2017.07.012. [DOI] [PubMed] [Google Scholar]
- [98].Haddad FS, Ngu A, Negus JJ, Prosthetic joint infections and cost analysis?, in: Adv. Exp. Med. Biol, Springer; New York: LLC, 2017: pp. 93–100. 10.1007/5584_2016_155. [DOI] [PubMed] [Google Scholar]
- [99].Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J, Economic burden of periprosthetic joint infection in the united states, J. Arthroplasty 27 (2012) 61–65.e1. 10.1016/j.arth.2012.02.022. [DOI] [PubMed] [Google Scholar]
- [100].Peulen TO, Wilkinson KJ, Diffusion of nanoparticles in a biofilm, Environ. Sci. Technol 45 (2011) 3367–3373. 10.1021/es103450g. [DOI] [PubMed] [Google Scholar]
- [101].Banoee M, Seif S, Nazari ZE, Jafari-Fesharaki P, Shahverdi HR, Moballegh A, Moghaddam KM, Shahverdi AR, ZnO nanoparticles enhanced antibacterial activity of ciprofloxacin against Staphylococcus aureus and Escherichia coli, J. Biomed. Mater. Res. Part B Appl. Biomater 93B (2010) 557–561. 10.1002/jbm.b.31615. [DOI] [PubMed] [Google Scholar]
- [102].Palermo EF, Kuroda K, Structural determinants of antimicrobial activity in polymers which mimic host defense peptides, Appl. Microbiol. Biotechnol 87 (2010) 1605–1615. 10.1007/s00253-010-2687-z. [DOI] [PubMed] [Google Scholar]
- [103].Schmitt CK, Meysick KC, O’Brien AD, Bacterial toxins: Friends or foes?, Emerg. Infect. Dis 5 (1999) 224–234. 10.3201/eid0502.990206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Fang RH, Luk BT, Hu CMJ, Zhang L, Engineered nanoparticles mimicking cell membranes for toxin neutralization, Adv. Drug Deliv. Rev 90 (2015) 69–80. 10.1016/j.addr.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Henry BD, Neill DR, Becker KA, Gore S, Bricio-Moreno L, Ziobro R, Edwards MJ, Mühlemann K, Steinmann J, Kleuser B, Japtok L, Luginbühl M, Wolfmeier H, Scherag A, Gulbins E, Kadioglu A, Draeger A, Babiychuk EB, Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice, Nat. Biotechnol 33 (2015) 81–88. 10.1038/nbt.3037. [DOI] [PubMed] [Google Scholar]
- [106].Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, Zhang S, Fang RH, Gao W, Nizet V, Zhang L, Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management, Proc. Natl. Acad. Sci. U. S. A 114 (2017) 11488–11493. 10.1073/pnas.1714267114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ma Q, Fan Q, Xu J, Bai J, Han X, Dong Z, Zhou X, Liu Z, Gu Z, Wang C, Calming Cytokine Storm in Pneumonia by Targeted Delivery of TPCA-1 Using Platelet-Derived Extracellular Vesicles, Matter. 3 (2020) 287–301. 10.1016/j.matt.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zhang CY, Dong X, Gao J, Lin W, Liu Z, Wang Z, Nanoparticle-induced neutrophil apoptosis increases survival in sepsis and alleviates neurological damage in stroke, Sci. Adv 5 (2019) eaax7964. 10.1126/sciadv.aax7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Spence S, Greene MK, Fay F, Hams E, Saunders SP, Hamid U, Fitzgerald M, Beck J, Bains BK, Smyth P, Themistou E, Small DM, Schmid D, O’kane CM, Fitzgerald DC, Abdelghany SM, Johnston JA, Fallon PG, Burrows JF, Mcauley DF, Kissenpfennig A, Scott CJ, Targeting Siglecs with a sialic acid-decorated nanoparticle abrogates inflammation, n.d. www.ScienceTranslationalMedicine.org (accessed March 12, 2021). [DOI] [PubMed]
- [110].Wu B, Lin L, Zhou F, Wang X, Precise engineering of neutrophil membrane coated with polymeric nanoparticles concurrently absorbing of proinflammatory cytokines and endotoxins for management of sepsis, Bioprocess Biosyst. Eng 43 (2020) 2065–2074. 10.1007/s00449-020-02395-5. [DOI] [PubMed] [Google Scholar]
- [111].Kumar V, Abbas A, Fausto N, Aster J, Robbins and Coutran’s Pathologic Basis of Disease, 2009. [Google Scholar]
- [112].Distasi MR, Ley K, Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability, (n.d.). 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Azzopardi EA, Ferguson EL, Thomas DW, The enhanced permeability retention effect: A new paradigm for drug targeting in infection, J. Antimicrob. Chemother 68 (2013) 257–274. 10.1093/jac/dks379. [DOI] [PubMed] [Google Scholar]
- [114].Laverman P, Dams ETM, Storm G, Hafmans TG, Croes HJ, Oyen WJG, Corstens FHM, Boerman OC, Microscopic localization of PEG-liposomes in a rat model of focal infection, J. Control. Release 75 (2001) 347–355. 10.1016/S0168-3659(01)00402-3. [DOI] [PubMed] [Google Scholar]
- [115].Wang Y, Yuan Q, Feng W, Pu W, Ding J, Zhang H, Li X, Yang B, Dai Q, Cheng L, Wang J, Sun F, Zhang D, Targeted delivery of antibiotics to the infected pulmonary tissues using ROS-responsive nanoparticles, J. Nanobiotechnology 17 (2019) 103. 10.1186/s12951-019-0537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Prior S, Gander B, Blarer N, Merkle HP, Subirá ML, Irache JM, Gamazo C, In vitro phagocytosis and monocyte-macrophage activation with poly(lactide) and poly(lactide-co-glycolide) microspheres, Eur. J. Pharm. Sci 15 (2002) 197–207. 10.1016/S0928-0987(01)0021-4. [DOI] [PubMed] [Google Scholar]
- [117].Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Hiroshi Morisaki J, Kim J, Park S, Darwish M, Lee BC, Hernandez H, Loyet KM, Lupardus P, Fong R, Yan D, Chalouni C, Luis E, Khalfin Y, Plise E, Cheong J, Lyssikatos JP, Strandh M, Koefoed K, Andersen PS, Flygare JA, Wah Tan M, Brown EJ, Mariathasan S, Novel antibody-antibiotic conjugate eliminates intracellular S. aureus, Nature. 527 (2015) 323–328. 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- [118].Huo D, Ding J, Cui YX, Xia LY, Li H, He J, Zhou ZY, Wang HW, Hu Y, X-ray CT and pneumonia inhibition properties of gold-silver nanoparticles for targeting MRSA induced pneumonia, Biomaterials. 35 (2014) 7032–7041. 10.1016/j.biomaterials.2014.04.092. [DOI] [PubMed] [Google Scholar]
- [119].Tang Y, Wu S, Lin J, Cheng L, Zhou J, Xie J, Huang K, Wang X, Yu Y, Chen Z, Liao G, Li C, Nanoparticles Targeted against Cryptococcal Pneumonia by Interactions between Chitosan and Its Peptide Ligand, Nano Lett. 18 (2018) 6207–6213. 10.1021/acs.nanolett.8b02229. [DOI] [PubMed] [Google Scholar]
- [120].Renwick MJ, Brogan DM, Mossialos E, A systematic review and critical assessment of incentive strategies for discovery and development of novel antibiotics, J. Antibiot. (Tokyo) 69 (2016) 73–88. 10.1038/ja.2015.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Spellberg B, The future of antibiotics, n.d. http://ccforum.com/content/18/3/228. [DOI] [PMC free article] [PubMed] [Google Scholar]