

Abstract
Purpose
Age-related cataracts affect the majority of older adults and are a leading cause of blindness worldwide. Treatments that delay cataract onset or severity have the potential to delay cataract surgery, but require relevant animal models that recapitulate the major types of cataracts for their development. Unfortunately, few such models are available. Here, we report the lens phenotypes of aged mice lacking the critical antioxidant transcription factor Nfe2l2 (designated as Nrf2 −/−).
Methods
Three independent cohorts of Nrf2 −/− and wild-type C57BL/6J mice were evaluated for cataracts using combinations of slit lamp imaging, photography of freshly dissected lenses, and histology. Mice were fed high glycemic diets, low glycemic diets, regular chow ad libitum, or regular chow with 30% caloric restriction.
Results
Nrf2 −/− mice developed significant opacities between 11 and 15 months and developed advanced cortical, posterior subcapsular, anterior subcapsular, and nuclear cataracts. Cataracts occurred similarly in male mice fed high or low glycemic diets, and were also observed in 21-month male and female Nrf2 −/− mice fed ad libitum or 30% caloric restriction. Histological observation of 18-month cataractous lenses revealed significant disruption to fiber cell architecture and the retention of nuclei throughout the cortical region of the lens. However, fiber cell denucleation and initiation of lens differentiation was normal at birth, with the first abnormalities observed at 3 months.
Conclusions
Nrf2 −/− mice offer a tool to understand how defective antioxidant signaling causes multiple forms of cataract and may be useful for screening drugs to prevent or delay cataractogenesis in susceptible adults.
Keywords: age-related cataract, antioxidant, nutrition, glycemic index, caloric restriction
Cataracts account for 51% of global blindness and are especially prevalent in the aging population with 68.3% of Americans over the age of 80 years developing cataracts.1–3 Cataracts are broadly defined as the opacification of the ocular lens and involve a myriad of pathogenic mechanisms. Leading risk factors for cataractogenesis include age, genetic predisposition, diabetes mellitus, and oxidative damage.4 Age-related cataracts are the most prevalent form of cataract and are commonly present in three forms, as visualized by slit-lamp imaging: nuclear, posterior subcapsular, and cortical cataracts.5–7 These various forms of cataracts imply variable susceptibility to cataracts in different areas of the lens and can present separately or often as a mixed cataract.4,5 Given the growing global aging population, the personal burden of lost sight, burgeoning public health costs associated with loss of sight and remediation, along with the dearth of sufficient ophthalmologic capacity to remediate cataracts, there is a dire need for appropriate models of age-related cataracts and their variable presentations to develop interventions to prevent cataract formation.
It is widely accepted that oxidative stress is a leading cause of protein aggregation and subsequent lens opacification causing vision loss.8–13 There is strong evidence demonstrating an age-related decline in antioxidant mechanisms, which when coupled with diminished proteolytic capacities to remove them, increases the risk for cataract development.5,6,14–17 Oxidative stress involves the production of excess reactive oxygen species (ROS), which can lead to numerous biochemical changes, including dysregulation of proteostasis and apoptosis.12,15,18–23 ROS production can occur from a variety of environmental triggers, including diminished antioxidant reserves, light exposure, mitochondrial activity, and hyperglycemia, as observed in diabetes mellitus.22,24 This association between oxidative stress and disease has led to a better understanding of the cellular mechanisms of protection against oxidative stress and means of enhancing these activities.
Nrf2 (nuclear factor erythroid 2-related factor 2 [Nfe2l2]) is a well-characterized transcription factor that acts as a primary cellular defense mechanism against oxidative stress. Under normal conditions, Nrf2 is located in the cytoplasm, bound to Kelch-like ECH-associated protein 1 (Keap1), ubiquitinated, and targeted for degradation by the proteasome.25,26 Upon oxidative stress, Nrf2 is released from Keap1, translocates to the nucleus, binds to the promoter for a set of antioxidant response element (ARE) genes, and initiates their transcription to facilitate a rapid response to oxidative stress.26,27 Genes activated by Nrf2 are largely cytoprotective, involving pathways like glutathione synthesis, detoxification, and elimination of ROS, and drug excretion.26 Thus, it is not surprising that the inhibition of Nrf2 subsequently decreases the capacity for the cell to respond to oxidative stress, which is exacerbated with increasing age.26,28 The association between reduced downstream antioxidant effects due to decreased Nrf2 activity, evident with age and in age-related cataracts, has garnered attention for Nrf2 as a therapeutic target for cataract treatment and prevention.27
Several studies have used human lens epithelial cells (HLECs), treated with suppressors of Nrf2 activity, to recapitulate molecular markers of protein aggregation as expected for cataractogenesis.18,19,29–33 In animal cataract models, treatment with antioxidant compounds, including those that induce Nrf2 transcription and activation protect against selenite-induced cataracts.31,34 Similarly, activators of Nrf2 can protect HLEC from features of cataract that are induced under conditions of oxidative stress with high glucose, hydrogen peroxide, or homocysteine treatments.20,32,33,35–37 Analysis of age-related cataractous lenses from human diabetic or non-diabetic samples showed decreased methylation of the Keap1 promoter, which in turn upregulates Keap1 expression, and inhibits Nrf2 nuclear translocation and antioxidant activity.28,38 Similarly, Nrf2 gene variants in humans have been associated with premature surgical treatment of cortical and posterior subcapsular cataracts.39
At present, there are several mouse cataract models, but the majority of these exhibit early onset lens defects or congenital cataracts (i.e. those present at birth).40,41 These include genetic models for cataracts that contain well-characterized human cataract mutations, altered functionality of critical protein quality control pathways, like ubiquitin proteolysis pathway, autophagy, mitochondrial dynamics, and unfolded protein response, and manipulation of developmental regulatory pathways.42–52 Other models for cataracts use chemical induction, the most common being sodium selenite that induces acute oxidative stress, triggering cataract formation.31 These models are suitable for studying severe cataracts but do not capture the gradual and varied onset that occurs with aging.
Wild-type mouse lines of various strains develop age-related cataracts when aged out to 30 months; these animals demonstrate a variety of opacifications, including anterior opacities, ring cataracts, and cortical haziness.53 However, it is difficult and expensive to keep a majority of mice out to such old ages. Therefore, there is a need for an accelerated timeline to visualize this same range. Several genetic models for age-related cataracts have been developed, although these models often develop one subtype of age-related cataracts. For example, βB2-crystallin (Crybb2) knockout mice are born with transparent lenses but develop cortical cataracts with age and show decreased oxidative stress response capacity.54 In the LEGSKO model, in which de novo glutathione (GSH) production is abolished specifically in the lens, it was reported that mice develop severe nuclear cataracts by 9 months.55 GSH is a critical lens antioxidant whose synthesis is transcriptionally controlled by Nrf2 in response to oxidative stress.26 GSH is also responsible for the regulation of the major protein quality control machinery.56 Similarly, a mouse knockout for glutaredoxin 2 (Grx2), a redox sensor that is activated under oxidative stress, also develops nuclear cataracts at an accelerated rate and severity by 13 to 16 months.57 Although the majority of human cataracts present during the process of aging, there is no “gold standard” mouse model for age-related cataracts that presents the variety of subtypes seen in humans.
Given that many of these cataract models result in a decreased oxidative stress response capacity and that Nrf2 is an important transcription factor in activating cellular protection mechanisms against oxidative stress, we hypothesized that alterations to Nrf2 expression would lead to cataract development. A whole-body Nrf2 knockout (Nrf2 −/−) streptozotocin-induced diabetic animal model showed accelerated cataract development compared to diabetic Nrf2 +/+ control animals.58 However, the possibility of cataracts in non-diabetic Nrf2 −/− mice was not reported. Animals lacking Nrf2 are viable but experience a shortened lifespan compared to wild-type mice, which can be extended by caloric restriction.59 We recently described the protection of mouse retina from accelerated age-related macular degeneration-like features with the feeding of a low glycemic diet in an Nrf2 −/− context.60 Here, we characterize the lens phenotypes of aged Nrf2 −/− mice and propose them to represent an accelerated model of age-related cataracts that captures a wide range of pathologies observed in humans.
Materials and Methods
Animals
Study 1: Eleven to 18-month male Nrf2 −/−: Nrf2 +/−, and Nrf2 −/− mice (allele: Nfe2l2tm1Ywk),61 maintained for at least 10 generations on a C57BL/6J genetic background, were obtained from Michael Freeman (Vanderbilt University) and The Jackson Laboratory (Bar Harbor, ME), respectively, and were interbred to generate Nrf2 −/− mice. The Nrf2 −/− gene targeting resulted in replacement of part of exon 4 and exon 5, containing the DNA binding domain and leucine zipper motif, with an in-frame LacZ-neomycin cassette.61
Animals were fed standard chow ad libitum (Teklad 7012; Harlan Laboratories, Madison, WI, USA) until 3 months of age, at which time they were placed on a study diet. Thirty-nine Nrf2 −/− male mice were randomized into groups of 20 high glycemic (HG)-fed mice and 19 low glycemic (LG)-fed mice. At the time that slit lamp imaging was performed, 17 HG-fed mice and 18 LG-fed mice were still alive. A full description of dietary treatment is published elsewhere.60 Diets contained identical macronutrient compositions with the exception that the HG starch was composed of 100% amylopectin (Amioca starch; Ingredion Inc., Bridgewater, NJ, USA), whereas the LG starch was composed of 70% amylose/30% amylopectin (Hylon VII starch; Ingredion Inc.). For comparisons with wild-type C57BL/6J mice, a separate cohort of 3 male mice fed standard chow ad libitum was studied at 18 months. All animal work was performed at the Tufts University Human Nutrition Research Center of Aging (HNRCA) and approved by the Tufts University Institutional Animal Care & Use Committee (IACUC) in adherence with the ARVO statement for the use of animals in ophthalmic and vision research.
Study 2: Neonatal and 3-month old wild-type and Nrf2 −/− mice: wild-type (Nrf2+/+) and Nrf2 −/− mice (allele: Nfe2l2tm1Mym)62 on a C57BL/6J genetic background were housed at a temperature range of 20 to 23°C under 12:12-hour light-dark cycles with free access to water and food, as described previously.63 The Nrf2 −/− mice were generated by replacing a portion of exon 5 of Nrf2 with lacZ cDNA, resulting in the deletion of the conserved carboxyl 280 amino acid residues DNA binding domain and producing a nonfunctional Nrf2-LacZ fusion protein.62 Mice were used in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University School of Medicine, and mouse studies adhered to the ARVO statement for the use of animals in ophthalmic and vision research.
Study 3: Twenty-one month male and female Nrf2 −/− mice: Nrf2 −/− mice (allele: Nfe2l2tm1Ywk), maintained for at least 10 generations on a C57BL/6J genetic background, were obtained from The Jackson Laboratory (Bar Harbor, ME) and were backcrossed to C57BL/6J mice. Mice were bred at the National Institute on Aging (NIA) and fed chow diets (NIH-31 diet; #T.9717.15, Envigo) ad libitum or were given a 30% caloric restriction, beginning at 9 months of age until 21 months of age, as described previously.59 Mice were used in accordance with protocols approved by the Institutional Animal Care and Use Committee of the NIA, and mouse studies adhered to the ARVO statement for the use of animals in ophthalmic and vision research.
Statistical Analyses
Analysis for age-diet and age-genotype interactions was performed in SPSS as a univariate general linear model using a type III sum-of-squares method. Pairwise comparisons of cortical nuclear depth were performed in SPSS using Student's t-test. Cataract frequency was compared using Fisher's exact test.
Cataract Imaging and Scoring
Study 1: Morphological changes in the lenses of the mice were monitored by a slit-lamp (Zeiss Slit Lamp Biomicroscopy, Santa Rosa, CA, USA). After dilation with eye drops containing 1% tropicamide and 0.5% phenylephrine hydrochloride, the animals were handheld unanesthetized and were presented to the slit lamp observer (author S.J.) randomly. The slit lamp was first frontal view, followed by 30 degree angle viewing. The degree of lens opacity in each eye was graded and classified according to the LOCS II system.64 Total cataract scores of 0 to 4 were based on The Framingham Eye Study, where scores of 4 and 5 were combined into a single score due to limitations of resolution due to the smaller eye size.65 Scores are reported per animal, with individual eyes combined. The examiner was blinded to the age and diet groups of the mice.
Front view slit lamp photography was carried out using a Zeiss slit lamp microscope. Narrow focal slit lamp photography was carried out using the Micron III system with an attached slit lamp light source (Phoenix Research Laboratories). The Micron III system was used for fundus photography. Freshly dissected lenses were photographed on a Leica dissection microscope.
Study 3: Dissected lenses were imaged on a dissecting microscope, either mounted against a 300-mesh electron microscopy grid (Electron Microscopy Sciences, Hatsfield, PA, USA, Catalog No. 6300H-Cu), a wire grid, or placed on a dark background. Lenses were evaluated from two large cohorts of mice. The first cohort of mouse lenses, representing mostly male mice, was evaluated and scored at the University of Delaware, whereas the second cohort of mice, consisting of mostly female mice, was evaluated and scored at the National Eye Institute (NEI). Opacities were determined ex vivo and scored in a binary fashion. Lenses with visible opacification and distorted appearance in grid imaging were considered as defective.
Histology and Lens Immunofluorescence
Study 1: Eyes (n = 5, Nrf2 −/− mice [3 HG and 2 LG]; n = 3 wild-type mice [regular chow]) were fixed by immersion in fixative (2.5% glutaraldehyde, 2% paraformaldehyde, with 0.025% [w/v] CaCl2 in 0.1 M sodium cacodylate buffer, pH 7.4) and lenses were dissected. Fixed lenses were processed by freeze-substitution followed by embedment in paraffin, as described in ref. 66. In brief, eyes were plunged into dry iced-cooled liquid propane for 1 minute, very rapidly transferred to 20 mL 97% methanol, 3% acetic acid, and cooled on dry ice. Freeze-substitution was allowed to proceed for 48 hours at −80°C. Samples were gradually warmed up, and then processed through graded ethanols to three changes of 100% ethanol. Samples were then processed conventionally into paraffin. The 5 µm sections were harvested, deparaffinized, and hydrated. To visualize membranes, sections were blocked in 10% normal goat serum in PBS, then exposed to Wheat Germ Agglutinin, Alexa Fluor 488, diluted 1:20, for 1 hour (Thermo Fisher).
Study 2: Heads of P1 pups (n = 3, Nrf2 −/− mice and n = 3 wild-type mice) were fixed by immersion in 4% paraformaldehyde for 2 hours at 4°C and transferred to PBS. Tissue was transferred to 30% sucrose for cryopreservation and then embedded in OCT. Cryosections were obtained at a thickness of 12 µm, dried overnight, and stored at −80°C. For immunofluorescence, tissue sections were rehydrated in PBS containing 0.1% (v/v) Triton X-100 (PBT). Next, slides were blocked using normal donkey serum (Jackson Immunoresearch), incubated with primary antibodies (goat anti-γ-crystallins; Santa Cruz Biotechnology) for 2 hours, washed with PBT, and incubated with secondary antibody conjugated to either Cy3 or Alexa Fluor 488 (Jackson Immunoresearch). Slides were mounted in Prolong Gold Antifade with DAPI (Molecular Probes) and photographed on a Zeiss Axiovert fluorescence microscope and digital camera.
Study 3: Intact ocular globes (n = 37, Nrf2 −/− mice and n = 13, wild-type mice) were fixed by immersion in formalin and embedded in methacrylate for conventional hematoxylin and eosin stain (H&E) histology.
Results
Cataract phenotypes have not previously been reported in aged Nrf2 −/− mice. Using slit lamp or fundus imaging, we observed a full range of LOCSII cataract scores in these 11 to 15 month old Nrf2 −/− mice. The grades ranged from 1 (mild) to 4 (severe; Fig. 1).64 Cataracts scoring a grade of 3 or 4 sufficiently obscured the light to the back of the eye such that fundus images did not reveal the retina or its vasculature. None of the Nrf2 −/− mice had completely transparent lenses at 11 to 15 months, which were observed in wild-type C57BL/6J mice of the same age (see Fig. 1).
Figure 1.

Mature 11 to 15 month Nrf2 −/− mice develop cataracts of various severities. Top panels are slit lamp images of cataracts representative of different LOCSII grades. Arrows indicate regions of opacification. Bottom panels show effects of different grade cataracts on fundus images, taken from different mice.
Because epidemiologic data suggested a correlation between HG diets and risk for cataracts,67,68 we evaluated cataracts in our Nrf2 −/− cohort that had consumed HG or LG diets.60 We have previously shown that dietary treatment with an LG diet ameliorated retinal phenotypes.60 Cataract grades were lowest in the youngest mice we scored (11 months of age) and increased with aging such that almost all of the mice had cataract grades of 4 at 15 months (Fig. 2). The trend to increased cataract score with age was highly significant (P = 2.3 × 10−4). In contrast, the effect of diet on cataract score was not significant (P = 0.97). Cataracts were present bilaterally, with no animals scoring more than one grade apart between eyes. Cataract prevalence was also determined based on a cutoff grade of two (Table 1). Again, cataract frequency increased with age but was not associated with diet (see Table 1).
Figure 2.
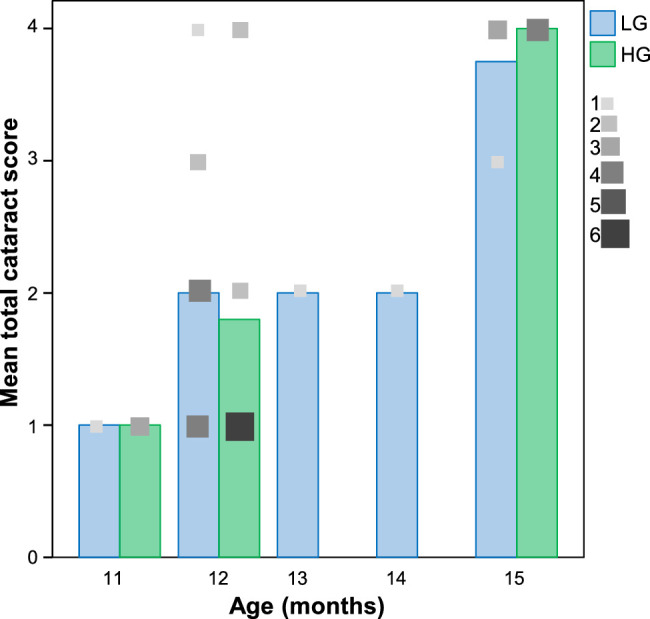
Nrf2 −/− cataract scores get progressively worse with age irrespective of diet. Bar graphs for the diet groups indicate mean total cataract score. Lenses with a respective cataract score are indicated by grayscale squares that are scaled to the number of lenses with that LOCSII grade (total sample size: N = 18, low glycemic diet [LG]; N = 17, high glycemic diet [HG]).
Table 1.
Summary Data for Cataract Frequency in Male Nrf2 −/− Mice Scored Between 11 and 15 Months
| 11 mo. | 12 mo. | 13 mo. | 14 mo. | 15 mo. | PAge | |
|---|---|---|---|---|---|---|
| HG | 0/3 | 4/10 | 4/4 | 0.029 | ||
| LG | 0/1 | 7/11 | 1/1 | 1/1 | 4/4 | 0.11 |
| Total | 0/4 | 11/21 | 1/1 | 1/1 | 8/8 | 0.0022 |
| PDiet | 1 | 0.39 | N/A | N/A | 1 |
Cataract was defined as a LOCSII grade of ≥2. Ages were compared between 11 and 12 months versus 13 to 15 months.
HG, high glycemic diet; LG, low glycemic diet.
Further analysis of 15 month Nrf2 −/− mouse lenses using slit lamp photography revealed that cataracts resembled the 3 major human age-related cataracts: cortical cataracts (Figs. 3A, 3B), posterior subcapsular cataract (see Figs. 3C, 3D), and nuclear cataract (see Figs. 3E, 3F). Cataracts were further scored for cataract subtype and total cataract score using the modified LOCSII scoring system. The majority of cataracts presented with significant (defined as grade 2 or higher) cortical cataracts and/or posterior subcapsular cataracts (19/35). Nuclear cataracts were observed in 5 of the 35 mice. Most of the cataracts and all of the nuclear cataracts consisted of at least two kinds of cataracts simultaneously. Ex vivo analysis of cataractous lenses at 18 months revealed significant and heterogeneous opacification of the anterior capsule (see Figs. 3G, 3H) and the posterior capsule (see Figs. 3I, 3J).
Figure 3.

Nrf2 −/− mice develop multiple types of cataracts. Slit lamp images from 15 month old mice reveal cortical cataracts (A, B), posterior subcapsular cataracts (C, D), and nuclear cataracts (E, F). Extracted fresh lenses reveal heterogeneous anterior and posterior subcapsular cataracts (G–J). Arrows indicate the diagnostic opacities associated with each cataract type.
Lenses from 18 month old mice were histologically evaluated using wheat germ agglutinin to visualize the shape of lens cells (Fig. 4, green) and DAPI to visualize the nuclei (see Fig. 4, red). In these older animals, cells in the anterior lens (see Figs. 4A–C), cortex (see Figs. 4D–F), and posterior (see Figs. 4G–I) revealed widespread disruptions to the lens fiber architecture, in association with opacification. The most severe abnormalities were observed in the cortical region, consistent with the degree of cortical cataract (see Figs. 4D, 4E). The lens nucleus architecture was largely intact, also consistent with nuclear cataract being a less penetrant phenotype. Alongside changes in lens cell architecture, we also observed aggregates throughout the lens. Strikingly, we observed nuclei abnormally present in Nrf2 −/− mouse lenses well beyond the outer cortical region. Ectopic nuclei were observed in the posterior part of Nrf2 −/− mouse lenses (see Figs. 4G, 4H) but not wild-type mouse lenses (see Fig. 4I). Many of these nuclei appeared pyknotic or rounded-up (see Figs. 4D–F, insets). We did not observe ectopic nuclei in the deeper parts of the lens nucleus in either Nrf2−/− or wild-type mice lenses, suggesting that nuclear degradation occurred normally in primary fiber cells.
Figure 4.

Histological analysis of cataracts in 18-month Nrf2 −/− or wild-type lenses. Samples from the anterior region (A–C), cortical region (D–F), posterior region (G–I), or the nuclear region (J–L) are stained with wheat germ agglutinin (green) and DAPI (red) from representatives of severe cataracts (A, D, G, J), milder cataracts (B, E, H, K), or age-matched wild-type clear lenses (C, F, I, L). Abnormalities are observed in all parts of the Nrf2 −/− lens, especially the cortex, which shows severe fiber cell disorganization. Aggregates are also observed (yellow arrows) as are ectopic nuclei, which mostly appear pyknotic (white arrowheads – see insets in D–F), in contrast to the normal nuclei of the lens epithelium and bow region (white arrows). Asterisks indicate tears in the lens that are likely histological artifacts. Abbreviations: b, bow region; c, lens capsule. Scale bar in (A) is 100 µm.
The process of lens fiber cell denucleation (LFCD), whereby the nucleus of the fiber cell is systematically disassembled and degraded, begins during mid-gestation in the center of the lens, and continues throughout the life of the organism in a stereotypical pattern beginning at the lens outer cortex and proceeding to the inner cortex.69,70 Usually, retention of lens nuclei is noted in congenital cataracts and is most obvious in the center of the lens.46,47,71 In order to determine whether embryonic LFCD occurred normally in Nrf2 −/− mice, we evaluated lenses at P1, when LFCD has already occurred in wild-type mice (Supplementary Fig. S1A). Nrf2 −/− mouse lenses at P1 showed appropriately timed LFCD (see Supplementary Fig. S1B). Lens differentiation also occurred appropriately, with normal timing and levels of gamma crystallins expression in Nrf2 −/− mouse lenses (see Supplementary Figs. S1C, S1D). We did observe some fracturing of the Nrf2 −/− mouse lens upon sectioning, which may indicate some defective packing of primary fiber cells (see Supplementary Fig. S1D).
In order to gain further insight into the early steps of cataractogenesis in Nrf2 −/− mice, we also evaluated young adult lenses in 3 month wild-type and Nrf2 −/− mice. Nrf2 −/− mouse lenses were transparent with no obvious gross phenotypes. Histological evaluation of the lenses showed that lenses were properly formed and lens fiber cell differentiation occurred normally at the transition zone (Supplementary Figs. S2D–F). However, the precise geometry of the bow region appeared subtly different in Nrf2 −/− mouse lenses (see Supplementary Fig. S2F). We also observed some abnormalities in the anterior part of the lens, where delaminated nucleated cells were observed in conjunction with disorganization of the lens fiber cells in the region, a possible initiating event in the formation of an anterior subcapsular cataract (see Supplementary Figs. S2H, S2I).
The abnormalities in 18 month Nrf2 −/− mouse lenses in the bow region prompted us to evaluate potential connections between retained cortical nuclei, age, and genotype. Figure 5 shows measurements of the distance from the lens equator to the furthest intact nucleus. We observed both age-related increases in nuclear depth and genotype-related increase, but only in older Nrf2 −/− mice. These findings indicate a statistically significant age-genotype interaction in equatorial nuclear depth that may underlie some of the opacities observed in older Nrf2 −/− mouse lenses.
Figure 5.
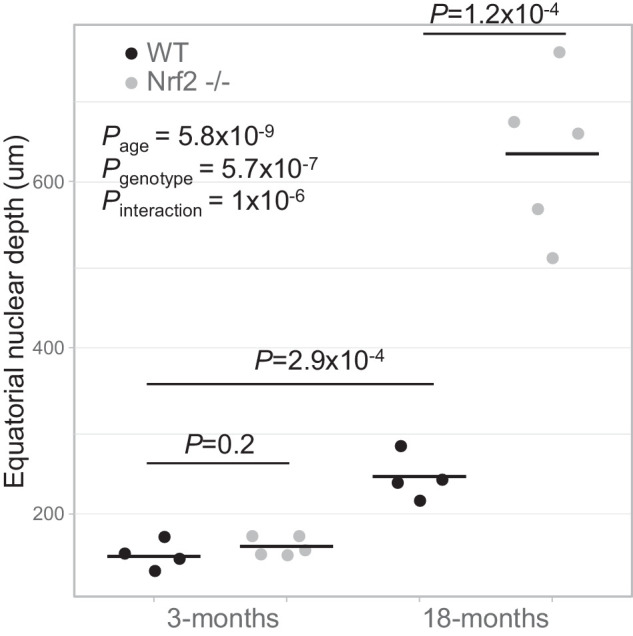
Age and genotype affect equatorial nuclear depth. Pair-wise comparisons show statistically significant differences between 3 month old and 18 month old lenses and between wild-type and Nrf2 −/− 18 month old mouse lenses. A generalized linear model was used to show statistically significant age and genotype effects on equatorial nuclear depth, as well as an age-genotype interaction.
Given the increasing severity in cataracts between 11 and 15 months (see Fig. 2) and the severity of cataract phenotype at 18 months in male Nrf2 −/− mice (see Figs. 3G–J, Fig. 4), we tested whether further aging to 21 months would lead to increased severity of cataracts in an independent cohort of male and female Nrf2 −/− mice. This cohort also allowed us to test whether delaying aging via a 30% caloric restriction compared to ad libitum feeding would impact cataractogenesis in Nrf2 −/− mice,59 a finding we observed using Emory mice.72,73 Cataracts were observed at high frequency in both male and female mice (Supplementary Fig. S3, Tables 2–3, Figs. 6A–D). Furthermore, observed cataracts were heterogeneous, sometimes leading to complete opacification of the lens (see Supplementary Fig. S3B, Fig. 6B) or varying degrees of regional opacities (see Supplementary Figs. S3C, S3D; Figs. 6C, 6D). Both male and female mouse Nrf2 −/− mice showed a significantly higher frequency of cataracts than wild-type mice, and CR did not significantly reduce the frequency of cataracts relative to ad libitum feeding (see Tables 2, 3).
Table 2.
Summary Data for Cataract Incidence in Female 21-Month-Old Mice
| AL | CR | Total | Pdiet | |
|---|---|---|---|---|
| Wild-type | 3/11 | 1/13 | 4/24 | 0.30 |
| Nrf2 −/− | 17/19 | 14/20 | 31/39 | 0.24 |
| Pgenotype | 9.7 × 10−4 | 8.8 × 10−4 | 1.2 × 10−6 |
Wild-type and Nrf2 −/− mouse lenses, as scored via photography of dissected lenses mounted on grids.
Table 3.
Summary Data for Cataract Incidence in Male 21-Month-Old Mice
| AL | CR | Total | Pdiet | |
|---|---|---|---|---|
| Wild-type | 1/5 | 3/12 | 4/17 | 1 |
| Nrf2 −/− | 11/11 | 12/15 | 23/26 | 0.24 |
| Pgenotype | 2.8 × 10−3 | 7.1 × 10−3 | 4.4 × 10−5 |
Wild-type and Nrf2 −/− mouse lenses, as scored via photography of dissected lenses mounted on grids.
Figure 6.

Appearance and histology of cataracts from female 21-month old mice. (A–D) Freshly dissected lenses from wild-type mice (A) or Nrf2 −/− mice (B–D) reveal a diverse array of cataracts with different degrees of severity in Nrf2 −/− mouse lenses. Cataracts shown are from mice fed their diets ad libitum. (E–N) Histological images are shown from wild-type mouse lenses (E, G, I, L) or Nrf2 −/− mouse lenses (F, H, J, K, M, N). High-magnification images from (E) are shown in (G, I, L), as indicated by green boxed regions, covering the lens equatorial region (G, H), posterior capsular region (I–K), or anterior capsular regions (L–N). Asterisks indicate histological abnormalities (e.g. cellular disorganization, vacuolation) corresponding to lens opacities. Arrows point to lens epithelial cells and arrowheads point to the lens capsule. Scale bar in (E, G, I, L) is 100 µm.
Histological analysis was performed on contralateral eyes from 21 month female wild-type and Nrf2 −/− mice. Whereas wild-type mouse lenses were clear and showed no histologic abnormalities (see Figs. 6E, 6G, 6I, 6L), nuclear cataracts were observed both in lens sections from Nrf2 −/− mice (see Fig. 6F), consistent with the images from whole lenses (see Figs. 6B, 6C). Cortical lens defects were also observed histologically (see Fig. 6H). These cortical defects showed aberrant fiber cell differentiation, similar to that observed in 18 month mouse lenses (see Fig. 4D). Most Nrf2 −/− mouse lenses exhibited posterior and anterior subcapsular defects (see Figs. 6J, 6K, 6M, 6N). Posterior subcapsular defects were associated with cellular (see Fig. 6J) or acellular plaques (see Fig. 6K). Anterior subcapsular defects were associated with degeneration of the lens epithelium (see Fig. 6M), normally a monolayer of flat cells (see Fig. 6L), or vacuolation of the subcapsular region (see Fig. 6N).
Discussion
There are few highly reproducible models of age-related human cataracts. Because cataracts in humans often occur in combinations of different types and they appear later in life, the property of Nrf2 −/− mice to similarly exhibit different types of cataracts in an age-dependent manner (see Tables 12–3), present them as a good model of human cataracts, while also providing a useful and suitable model for age-related cataracts of various specific types.
Cataracts in these Nrf2−/− mice can be compared with the few prior models of age-related cataracts. The Emory mouse develops progressive cortical and nuclear cataracts but penetrance is low and severity is limited, making it a costly and difficult model.72,74 The Grx2 knockout first develops a nuclear opacity, which progresses to affect cortical and subcapsular regions, but this does not model the typical human cataract pattern.57 Few rodent models recapitulate highly prevalent posterior subcapsular cataracts, which are best modeled in chemically treated rabbits or ionizing radiation-treated mice.75,76 The LEGSKO mouse, lacking the glutathione synthesis enzyme, GCLC, develop age-related nuclear cataracts.40 Similarly, mice lacking Gpx1, glutathione peroxidase 1, develop age-related nuclear cataracts, as did mice lacking Grx2, glutaredoxin 2.57,77 Nrf2 directly activates the rate-limiting enzymes responsible for glutathione synthesis, GCLC, and GCLM, and regulates glutathione regeneration.78 Thus, it is somewhat surprising that Nrf2 −/− mouse cataracts do not develop nuclear cataracts at a higher prevalence. Genetic redundancy with Nrf1, also present at high levels in the lens, may be an additional factor.79,80 Whether phenotypic disparities relate to different genetic backgrounds, genetic redundancies, or baseline levels of glutathione should be sorted out as we determine which are the best models of human age-related cataracts.
Nrf2 −/− mice offer an opportunity to understand the sequence of events behind cataract development, especially considering that Nrf2 functions as a nuclear transcription factor, and thus likely performs its critical functions in lens epithelial cells and/or newly developing cortical fiber cells prior to LFCD, when the lens cell nucleus is disassembled and degraded.69 Our observation that young Nrf2 −/− mouse lenses, although transparent, progress to cataracts suggests that they may have underlying defects in fiber cell formation, findings hinted at in histological analysis of 3 month old Nrf2 −/− mouse lenses. Careful analysis of Nrf2 −/− mouse lens epithelial cells may reveal aspects of accelerated aging, altered stress response pathways, or abnormal metabolism and homeostasis. Future studies also warrant the study of Nrf2 binding partners like MafG and MafK, whose deficiency is also associated with cataracts.80 The notion of subtle alterations in fiber cells escalating into progressive cataracts is supported by the Crybb2 knockout mouse, where the loss of a β-crystallin manifests as age-related cortical cataracts,54 and in Bfsp2 knockout mice, where loss of the intermediate protein filensin results in more rapid age-related cataracts.81
Our observation of large numbers of abnormal nuclei through the inner and outer cortex of Nrf2 −/− mouse lenses indicates abrogation or truncation of secondary LFCD. Some cataract models have shown delayed secondary LFCD, with nuclei observed anteriorly, such as mutations in βB2-crystallin or loss of Connexin 50, but the fiber cells eventually do undergo denucleation.82,83 More commonly, impaired LFCD occurs in primary fiber cells, thus preferentially affecting the central part of the lens.47,84 Impaired primary LFCD is observed in many congenital cataract models, as well as certain age-related cataracts like the Grx2 knockout mouse.85 The finding of normal primary LFCD may explain why we do not observe more significant nuclear cataracts. However, retention of fiber cell nuclei in itself does not explain the degree of cortical opacification, as milder cataracts also retained fiber cell nuclei (see Figs. 4B, 4E, 4H, 4K). Our finding that LFCD occurred normally at birth (see Supplementary Figs. S1A, S1B) and was not different between wild-type and Nrf2 −/− mouse lenses at 3 months (see Fig. 2) raises the possibility that there may be alternative regulatory mechanisms for LFCD during adulthood. Absent or incomplete secondary LFCD may be a more general feature of age-related cataracts than previously described. Our observation of increased nuclear depth with age, even in wild-type mice (see Fig. 5), confirms results found during normal aging in many organisms.86 Our data suggest that the rate-limiting steps in nuclear removal are impacted by normal aging and further exacerbated by a deficiency in Nrf2. The recent transcriptomic characterization of the aging mouse lens may offer potential mechanisms linked to cortical LFCD.87
The presence of anterior and posterior subcapsular cataracts in Nrf2 −/− mice offers the potential to determine the etiology of these cataracts as well. Some subcapsular cataracts were associated with the presence of nucleated cells, as is typical during congenital TGFβ-induced cataracts, and associated with pathological epithelial-to-mesenchymal transition (EMT).88 Evidence of potential EMT was observed in 18 month cataracts (see Fig. 4H), as well as in young 3 month lenses (see Supplementary Figs. S2H, S2I). Nevertheless, other posterior and anterior opacities did not show such cellular plaques and had severely disrupted fiber cell architecture (see Figs. 4A, 4B, 4G; Figs. 6K, 6M, 6N). Our data support a model where subcapsular plaques consist of populations of fiber cells and myofibroblastic cells that may eventually die or degenerate.89,90 Plaque formation may be further linked to specific tissue damage, including breakage of the posterior lens capsule (see Fig. 6J), which may activate a subsequent fibrotic immune response.91
Our finding that Nrf2 −/− mice developed severe age-related cataracts emphasizes the importance of the Nrf2 pathway in lens morphogenesis and cataractogenesis. In human cataractogenesis, disruption of the Nrf2 pathway appears to occur via altered epigenetic regulation of KEAP1, leading to a diminished Nrf2 response.38 During the normal process of lens aging, downstream functions of Nrf2, including glutathione synthesis, thioredoxin reductase, glutathione reductase, thioltransferases, and levels of antioxidant enzymatic function, all decrease in association with cataractogenesis.5,6,8,14,15,92 Whether those decreases are caused directly or indirectly by loss of Nrf2 function remains to be determined.
This research also informs about potential ways to alleviate or remediate cataracts using Nrf2-related biology. The Nrf2 −/− mouse cataract model presented here should be useful to screen anti-cataractogenic treatments, preferably in combination with other Nrf2 +/+ models. Existing approaches, including small molecule Nrf2 activators or plant-derived Nrf2 activators, like sulforaphane, should be studied further in Nrf2 +/+ and Nrf2 −/− cataract models. In vitro data in lens epithelial cells or aging human lenses suggests that sulforaphane treatment can confer improved antioxidant levels and response and therefore may protect against cataracts.93,94 It will be beneficial to identify compounds that can protect the lens in an Nrf2-independent fashion, because human cataracts are associated with epigenetically diminished Nrf2 responses over the course of aging. Because multiple types of cataracts are observed, this Nrf2 −/− mouse model may reveal treatments that preferentially improve different parts of the lens.
Prior laboratory and epidemiology studies indicated that LG diets protect against age-related ocular conditions, including age-related macular degeneration and some cataracts, and that caloric restriction delayed cataracts in Emory mice and some pigmented lines of mice and rats.60,68,72,73,95–97 We therefore explored the possibility of a dietary cataract intervention by feeding Nrf2 −/− mice LG or caloric restriction diets. However, we did not find a significant difference in cataract onset, prevalence, or type of cataract in Nrf2 −/− mice fed LG or caloric restriction diets. There was some hint from cataract frequency data that caloric restriction may reduce cataract frequency in 21 month male and female Nrf2 −/− mice (approximate 20–25% reduction; see Tables 2, 3), but we were likely underpowered for this difference to reach statistical significance. This reduction may indicate separate roles for Nrf2 in regulating aging events related to cataracts, which are reduced by caloric restriction, separately from cell autonomous requirements for Nrf2 in the lens.
Our study design has strengths and limitations. Our comparison of multiple Nrf2 −/− alleles, dietary treatments, ages, and cataract assessments may appear as a limitation. However, one strength of utilizing multiple cohorts of mice, reared and analyzed at different centers, is that we can be more confident of the reproducibility of cataracts in Nrf2 −/− mice in both sexes of mice fed multiple dietary treatments, and whose cataracts are observable in slit-lamp imaging, gross appearance, and histology. We evaluated mice with two different knockout alleles of Nrf2, although we did not evaluate mice with the Nfe2l2tm1Mym allele at older ages. It is conceivable that different alleles of Nrf2 bred to different genetic backgrounds may develop cataracts at different frequencies than what we report here. Future studies will evaluate proteomic and transcriptomic changes in Nrf2 −/− lenses and direct measurements of antioxidant status in mild and severe cataracts.
In summary, aged Nrf2 −/− mice develop all major types of human age-related cataracts at a high prevalence. Nrf2 −/− lenses retain cortical fiber cell nuclei, suggesting a role for Nrf2 in terminal differentiation of late-born lens fiber cells. Understanding the roles of Nrf2 in normal lens homeostasis will generate new insights into the processes of cataractogenesis and will generate new leads into the development of anti-cataract treatments. This new model of highly prevalent age-related cataracts should facilitate drug discovery and testing.
Supplementary Material
Acknowledgments
The authors are grateful to Jennifer Cho and Jonathan Morrison for assistance with animal feeding, Michael Freeman and Swati Biswas for Nrf2 +/− mice, and Elizabeth Whitcomb and Eloy Bejarano for critical reading of the manuscript.
Supported by NIH RO1EY021212, RO1EY028559, and RO1EY026979 (to A.T.); NIH EY022383 and EY022683 (to E.J.D.); NIH RO1EY021505 and RO1029770 (to S.A.L.); USDA NIFA 2016-08885 (to A.T. and S.R.); USDA 8050-51000-089-01S and 8050-51000-101-01S (to A.T.); Thome Memorial Foundation (to A.T.); BrightFocus Foundation (to S.R.); the National Natural Science Foundation of China (NSFC) 81760177 (to S.J.); NIH Grant #Fi2GM123963 from NIGMS (to L.C.D.P). This study was supported by the NIH Intramural Research Program of the NIA and NEI.
This material is based upon work supported by the US Department of Agriculture – Agricultural Research Service (ARS), under Agreement Nos. 58-1950-4-003 and 58-8050-9-004.
Disclosure: S. Rowan, None; S. Jiang, None; S.G. Francisco, None; L.C.D. Pomatto, None; Z. Ma, None; X. Jiao, None; M.M. Campos, None; S. Aryal, None; S.D. Patel, None; B. Mahaling, None; S.A. Riazuddin, None; E.J. Duh, None; S.A. Lachke, None; J.F. Hejtman, None; R. de Cabo, None; P.G. FitzGerald, None; A. Taylor, None
References
- 1. Congdon N, O'Colmain B, Klaver CC, et al.. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004; 122(4): 477–485. [DOI] [PubMed] [Google Scholar]
- 2. Pascolini D, Mariotti SP.. Global estimates of visual impairment: 2010. Br J Ophthalmol. 2012; 96(5): 614–618. [DOI] [PubMed] [Google Scholar]
- 3. World Health Organization. Blindness and vision impairment; 2019. [updated October 8, 2019]. Available from: https://www.who.int/news-room/fact-sheets/detail/blindness-and-visual-impairment.
- 4. Liu YC, Wilkins M, Kim T, Malyugin B, Mehta JS.. Cataracts. Lancet. 2017; 390(10094): 600–612. [DOI] [PubMed] [Google Scholar]
- 5. Michael R, Bron AJ.. The ageing lens and cataract: a model of normal and pathological ageing. Philos Trans R Soc Lond B Biol Sci. 2011; 366(1568): 1278–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vrensen GF. Aging of the human eye lens–a morphological point of view. Comp Biochem Physiol A Physiol. 1995; 111(4): 519–532. [DOI] [PubMed] [Google Scholar]
- 7. Merin S. Congenital cataracts. In: Goldberg MF, ed. Genetic and metabolic eye disease. New York, NY: Little, Brown, and Co.; 1974. p. 337–355. [Google Scholar]
- 8. Giblin FJ. Glutathione: a vital lens antioxidant. J Ocul Pharmacol Ther. 2000; 16(2): 121–135. [DOI] [PubMed] [Google Scholar]
- 9. Lou MF. Redox regulation in the lens. Prog Retin Eye Res. 2003; 22(5): 657–682. [DOI] [PubMed] [Google Scholar]
- 10. Clark JI. Self-assembly of protein aggregates in ageing disorders: the lens and cataract model. Philos Trans R Soc Lond B Biol Sci. 2013; 368(1617): 20120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pande A, Pande J, Asherie N, Lomakin A, Ogun O, King J, et al.. Crystal cataracts: human genetic cataract caused by protein crystallization. Proc Natl Acad Sci USA. 2001; 98(11): 6116–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Palsamy P, Shinohara T.. Age-related cataracts: Role of unfolded protein response, Ca(2+) mobilization, epigenetic DNA modifications, and loss of Nrf2/Keap1 dependent cytoprotection. Prog Retin Eye Res. 2017; 60: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shang F, Taylor A.. Oxidation and protein degradation in the lens. Mol Aspects Med. 1997; 18: 339–352. [Google Scholar]
- 14. Lou MF, Dickerson JE Jr.. Protein-thiol mixed disulfides in human lens. Exp Eye Res. 1992; 55(6): 889–896. [DOI] [PubMed] [Google Scholar]
- 15. Vinson JA. Oxidative stress in cataracts. Pathophysiology. 2006; 13(3): 151–162. [DOI] [PubMed] [Google Scholar]
- 16. Shang F, Gong X, Palmer HJ, Nowell TR Jr., Taylor A. Age-related decline in ubiquitin conjugation in response to oxidative stress in the lens. Exp Eye Res. 1997; 64(1): 21–30. [DOI] [PubMed] [Google Scholar]
- 17. Jahngen-Hodge J, Cyr D, Laxman E, Taylor A.. Ubiquitin and ubiquitin conjugates in human lens. Exp Eye Res. 1992; 55(6): 897–902. [DOI] [PubMed] [Google Scholar]
- 18. Elanchezhian R, Palsamy P, Madson CJ, Mulhern ML, Lynch DW, Troia AM, et al.. Low glucose under hypoxic conditions induces unfolded protein response and produces reactive oxygen species in lens epithelial cells. Cell Death & Disease. 2012; 3(4): e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zheng XY, Xu J, Chen XI, Li W, Wang TY. Attenuation of oxygen fluctuation-induced endoplasmic reticulum stress in human lens epithelial cells. Exp Ther Med. 2015; 10(5): 1883–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X, Zhao X, Cheng R, Huang Y.. Autophagy attenuates high glucose-induced oxidative injury to lens epithelial cells. Biosci Rep. 2020; 40(4): BSR20193006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shang F, Gong X, Taylor A.. Changes in ubiquitin conjugation activities in young and old lenses in response to oxidative stress. Inv Ophthalmol Vis Sci. 1995; 36: S528. [Google Scholar]
- 22. Shang F, Taylor A.. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radical Biology & Medicine. 2011; 51(1): 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jahngen JH, Lipman RD, Eisenhauer DA, Jahngen EG Jr., Taylor A. Aging and cellular maturation cause changes in ubiquitin-eye lens protein conjugates. Arch Biochem Biophys. 1990; 276(1): 32–37. [DOI] [PubMed] [Google Scholar]
- 24. Klein BE, Klein R, Wang Q, Moss SE. Older-onset diabetes and lens opacities. The Beaver Dam Eye Study. Ophthalmic Epidemiol. 1995; 2: 49–55. [DOI] [PubMed] [Google Scholar]
- 25. Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K.. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules. 2020; 10(2): 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto M, Kensler TW, Motohashi H.. The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev. 2018; 98(3): 1169–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu XF, Hao JL, Xie T, Malik TH, Lu CB, Liu C, et al.. Nrf2 as a target for prevention of age-related and diabetic cataracts by against oxidative stress. Aging Cell. 2017; 16(5): 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palsamy P, Ayaki M, Elanchezhian R, Shinohara T.. Promoter demethylation of Keap1 gene in human diabetic cataractous lenses. Biochemical and Biophysical Research Communications. 2012; 423(3): 542–548. [DOI] [PubMed] [Google Scholar]
- 29. Gong W, Zhu G, Li J, Yang X.. LncRNA MALAT1 promotes the apoptosis and oxidative stress of human lens epithelial cells via p38MAPK pathway in diabetic cataract. Diabetes Res Clin Pract. 2018; 144: 314–321. [DOI] [PubMed] [Google Scholar]
- 30. Palsamy P, Bidasee KR, Shinohara T.. Valproic acid suppresses Nrf2/Keap1 dependent antioxidant protection through induction of endoplasmic reticulum stress and Keap1 promoter DNA demethylation in human lens epithelial cells. Exp Eye Res. 2014; 121: 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palsamy P, Bidasee KR, Shinohara T.. Selenite cataracts: activation of endoplasmic reticulum stress and loss of Nrf2/Keap1-dependent stress protection. Biochim Biophys Acta. 2014; 1842(9): 1794–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu Z-m, Yin X-x, Ji L, Gao Y-y, Pan Y-m, Lu Q, et al.. Ginkgo biloba extract prevents against apoptosis induced by high glucose in human lens epithelial cells. Acta Pharmacologica Sinica. 2008; 29(9): 1042–1050. [DOI] [PubMed] [Google Scholar]
- 33. Yang SP, Yang XZ, Cao GP.. Acetyl-l-carnitine prevents homocysteine-induced suppression of Nrf2/Keap1 mediated antioxidation in human lens epithelial cells. Molec Med Rep. 2015; 12(1): 1145–1150. [DOI] [PubMed] [Google Scholar]
- 34. Fang W, Ye Q, Yao Y, Xiu Y, Gu F, Zhu Y.. Protective Effects of Trimetazidine in Retarding Selenite-Induced Lens Opacification. Current Eye Research. 2019; 44(12): 1325–1336. [DOI] [PubMed] [Google Scholar]
- 35. Elanchezhian R, Palsamy P, Madson CJ, Lynch DW, Shinohara T.. Age-related cataracts: homocysteine coupled endoplasmic reticulum stress and suppression of Nrf2-dependent antioxidant protection. Chem Biol Interact. 2012; 200(1): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu Y, Luo W, Luo X, Yong Z, Zhong X. Effects of Rosa laevigata Michx. extract on reactive oxygen species production and mitochondrial membrane potential in lens epithelial cells cultured under high glucose. Int J Clin Exp Med. 2015; 8(9): 15759–15765. [PMC free article] [PubMed] [Google Scholar]
- 37. Park JY, Kang KA, Kim KC, Cha JW, Kim EH, Hyun JW.. Morin Induces Heme Oxygenase-1 via ERK-Nrf2 Signaling Pathway. J Cancer Prev. 2013; 18(3): 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao Y, Yan Y, Huang T.. Human age related cataracts: epigenetic suppression of the nuclear factor erythroid 2related factor 2mediated antioxidant system. Molecular Medicine Reports. 2015; 11(2): 1442–1447. [DOI] [PubMed] [Google Scholar]
- 39. von Otter M, Landgren S, Nilsson S, et al.. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer's disease and age-related cataract. Mechanisms of Ageing and Development. 2010; 131(2): 105–110. [DOI] [PubMed] [Google Scholar]
- 40. Graw J. Mouse models of cataract. J Genet. 2009; 88(4): 469–486. [DOI] [PubMed] [Google Scholar]
- 41. Shiels A, Bennett TM, Hejtmancik JF.. Cat-Map: putting cataract on the map. Molecular Vision. 2010; 16: 2007–2015. [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao J, Wu X, Wu D, et al.. Embryonic Surface Ectoderm-specific Mitofusin 2 Conditional Knockout Induces Congenital Cataracts in Mice. Sci Rep. 2018; 8(1): 1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andley UP, Hamilton PD, Ravi N, Weihl CC.. A knock-in mouse model for the R120G mutation of alphaB-crystallin recapitulates human hereditary myopathy and cataracts. PLoS One. 2011; 6(3): e17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Molnar KS, Dunyak BM, Su B, et al.. Mechanism of Action of VP1-001 in cryAB(R120G)-Associated and Age-Related Cataracts. Invest Ophthalmol Vis Sci. 2019; 60(10): 3320–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vicart P, Caron A, Guicheney P, et al.. A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nature Genetics. 1998; 20(1): 92–95. [DOI] [PubMed] [Google Scholar]
- 46. Caceres A, Shang F, Wawrousek E, et al.. Perturbing the ubiquitin pathway reveals how mitosis is hijacked to denucleate and regulate cell proliferation and differentiation in vivo. PLoS One. 2010; 5: e13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lyu L, Whitcomb EA, Jiang S, et al.. Unfolded-protein response-associated stabilization of p27(Cdkn1b) interferes with lens fiber cell denucleation, leading to cataract. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2016; 30(3): 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Firtina Z, Danysh BP, Bai X, Gould DB, Kobayashi T, Duncan MK.. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. The Journal of Biological Chemistry. 2009; 284(51): 35872–35884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morishita H, Eguchi S, Kimura H, et al.. Deletion of Autophagy-related 5 (Atg5) and Pik3c3 Genes in the Lens Causes Cataract Independent of Programmed Organelle Degradation. The Journal of Biological Chemistry. 2013; 288(16): 11436–11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shiels A, Hejtmancik JF.. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2017; 156: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu K, Lyu L, Chin D, et al.. Altered ubiquitin causes perturbed calcium homeostasis, hyperactivation of calpain, dysregulated differentiation, and cataract. Proc Natl Acad Sci. 2015; 112(4): 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shiels A, Hejtmancik JF.. Biology of Inherited Cataracts and Opportunities for Treatment. Annu Rev Vis Sci. 2019; 5: 123–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cheng C, Parreno J, Nowak RB, et al.. Age-related changes in eye lens biomechanics, morphology, refractive index and transparency. Aging. 2019; 11(24): 12497–12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang J, Li J, Huang C, et al.. Targeted knockout of the mouse betaB2-crystallin gene (Crybb2) induces age-related cataract. Invest Ophthalmol Vis Sci. 2008; 49(12): 5476–5483. [DOI] [PubMed] [Google Scholar]
- 55. Fan X, Liu X, Hao S, Wang B, Robinson ML, Monnier VM.. The LEGSKO mouse: a mouse model of age-related nuclear cataract based on genetic suppression of lens glutathione synthesis. PLoS One. 2012; 7(11): e50832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jahngen-Hodge J, Obin MS, Gong X, et al.. Regulation of ubiquitin-conjugating enzymes by glutathione following oxidative stress. The Journal of Biological Chemistry. 1997; 272: 28218–28226. [DOI] [PubMed] [Google Scholar]
- 57. Wu H, Yu Y, David L, Ho YS, Lou MF.. Glutaredoxin 2 (Grx2) gene deletion induces early onset of age-dependent cataracts in mice. The Journal of Biological Chemistry. 2014; 289(52): 36125–36139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Palsamy P, Bidasee KR, Ayaki M, Augusteyn RC, Chan JY, Shinohara T.. Methylglyoxal induces endoplasmic reticulum stress and DNA demethylation in the Keap1 promoter of human lens epithelial cells and age-related cataracts. Free Radical Biology & Medicine. 2014; 72: 134–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pomatto LCD, Dill T, Carboneau B, et al.. Deletion of Nrf2 shortens lifespan in C57BL6/J male mice but does not alter the health and survival benefits of caloric restriction. Free Radical Biology & Medicine. 2020; 152: 650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rowan S, Jiang S, Chang ML, et al.. A low glycemic diet protects disease-prone Nrf2-deficient mice against age-related macular degeneration. Free Radic Biol Med. 2020; 150: 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chan K, Lu R, Chang JC, Kan YW.. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1996; 93(24): 13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Itoh K, Chiba T, Takahashi S, et al.. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications. 1997; 236(2): 313–322. [DOI] [PubMed] [Google Scholar]
- 63. Xu Z, Wei Y, Gong J, et al.. NRF2 plays a protective role in diabetic retinopathy in mice. Diabetologia. 2014; 57(1): 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chylack LT, Leske MC, McCarthy D, Khu P, Kashiwagi T, Sperduto R.. Lens opacities classification system II (LOCS II). Arch Ophthalmol. 1989; 107(4125): 991–997. [DOI] [PubMed] [Google Scholar]
- 65. Leibowitz HM, Krueger DE, Maunder LR, et al.. The Framingham Eye Study monograph: An ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973-1975. Survey of Ophthalmology. 1980; 24(Suppl): 335–610. [PubMed] [Google Scholar]
- 66. Sun N, Shibata B, Hess JF, FitzGerald PG.. An alternative means of retaining ocular structure and improving immunoreactivity for light microscopy studies. Molecular Vision. 2015; 21: 428–442. [PMC free article] [PubMed] [Google Scholar]
- 67. Tan JS, Wang JJ, Flood V, et al.. Carbohydrate nutrition, glycemic index and the 10-year incidence of cataract. Am J Clin Nutr. 2007; 86(5): 1502–1508. [DOI] [PubMed] [Google Scholar]
- 68. Chiu CJ, Milton RC, Gensler G, Taylor A. Dietary carbohydrate intake and glycemic index in relation to cortical and nuclear lens opacities in the Age-Related Eye Disease Study. Am J Clin Nutr. 2006; 83(5): 1177–1184. [DOI] [PubMed] [Google Scholar]
- 69. Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res. 2009; 88: 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rowan S, Chang ML, Reznikov N, Taylor A.. Disassembly of the lens fiber cell nucleus to create a clear lens: the p27 descent. Exp Eye Res. 2017; 156: 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Siddam AD, Gautier-Courteille C, Perez-Campos L, et al.. The RNA-binding protein Celf1 post-transcriptionally regulates p27Kip1 and Dnase2b to control fiber cell nuclear degradation in lens development. PLoS Genetics. 2018; 14(3): e1007278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Taylor A, Zuliani AM, Hopkins RE, et al.. Moderate caloric restriction delays cataract formation in the Emory mouse. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 1989; 3(6): 1741–1746. [DOI] [PubMed] [Google Scholar]
- 73. Taylor A, Lipman RD, Jahngen-Hodge J, et al.. Dietary calorie restriction in the Emory mouse: effects on lifespan, eye lens cataract prevalence and progression, levels of ascorbate, glutathione, glucose, and glycohemoglobin, tail collagen breaktime, DNA and RNA oxidation, skin integrity, fecundity, and cancer. Mechanisms of Ageing and Development. 1995; 79(1): 33–57. [DOI] [PubMed] [Google Scholar]
- 74. Kuck JF, Kuwabara T, Kuck KD.. The Emory mouse cataract: an animal model for human senile cataract. Current Eye Research. 1981; 1(11): 643–649. [DOI] [PubMed] [Google Scholar]
- 75. Gwon A, Mantras C, Gruber L, Cunanan C.. Concanavalin A-induced posterior subcapsular cataract: a new model of cataractogenesis. Invest Ophthalmol Vis Sci. 1993; 34(13): 3483–3488. [PubMed] [Google Scholar]
- 76. Pawliczek D, Fuchs H, Gailus-Durner V, et al.. On the Nature of Murine Radiation-Induced Subcapsular Cataracts: Optical Coherence Tomography-Based Fine Classification [published online ahead of print February 25, 2021], In: Vivo Dynamics and Impact on Visual Acuity. Radiat Res, 10.1667/RADE-20-00163.1. [DOI] [PubMed] [Google Scholar]
- 77. Reddy VN, Giblin FJ, Lin LR, et al.. Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice. Invest Ophthalmol Vis Sci. 2001; 42(13): 3247–3255. [PubMed] [Google Scholar]
- 78. Tonelli C, Chio IIC, Tuveson DA.. Transcriptional Regulation by Nrf2. Antioxid Redox Signal. 2018; 29(17): 1727–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kakrana A, Yang A, Anand D, et al.. iSyTE 2.0: a database for expression-based gene discovery in the eye. Nucleic Acids Res. 2018; 46(D1): D875–D885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Agrawal SA, Anand D, Siddam AD, et al.. Compound mouse mutants of bZIP transcription factors Mafg and Mafk reveal a regulatory network of non-crystallin genes associated with cataract. Hum Genet. 2015; 134(7): 717–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Alizadeh A, Clark J, Seeberger T, Hess J, Blankenship T, FitzGerald PG.. Targeted deletion of the lens fiber cell-specific intermediate filament protein filensin. Invest Ophthalmol Vis Sci. 2003; 44(12): 5252–5258. [DOI] [PubMed] [Google Scholar]
- 82. Graw J, Loster J, Soewarto D, et al.. Aey2, a new mutation in the betaB2-crystallin-encoding gene of the mouse. Invest Ophthalmol Vis Sci. 2001; 42(7): 1574–1580. [PubMed] [Google Scholar]
- 83. Rong P, Wang X, Niesman I, et al.. Disruption of Gja8 (alpha8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development. 2002; 129(1): 167–174. [DOI] [PubMed] [Google Scholar]
- 84. Chaffee BR, Shang F, Chang ML, et al.. Nuclear removal during terminal lens fiber cell differentiation requires CDK1 activity: appropriating mitosis-related nuclear disassembly. Development. 2014; 141(17): 3388–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lou MF, Basu S, Yu Y, Wu H, Menko AS.. Glutaredoxin (Grx2) Gene Knockout Suppresses Fiber Cell Differentiation and Delays De-nucleation of the Mouse Lens. Invest Ophth Vis Sci. 2012; 53(14): Abstract 5592-. [Google Scholar]
- 86. Pendergrass W, Zitnik G, Urfer SR, Wolf N.. Age-related retention of fiber cell nuclei and nuclear fragments in the lens cortices of multiple species. Molecular Vision. 2011; 17: 2672–2684. [PMC free article] [PubMed] [Google Scholar]
- 87. Faranda AP, Shihan MH, Wang Y, Duncan MK.. The aging mouse lens transcriptome. Exp Eye Res. 2021; 209: 108663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lovicu FJ, Schulz MW, Hales AM, et al.. TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. Br J Ophthalmol. 2002; 86(2): 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lovicu FJ, Steven P, Saika S, McAvoy JW.. Aberrant lens fiber differentiation in anterior subcapsular cataract formation: a process dependent on reduced levels of Pax6. Invest Ophthalmol Vis Sci. 2004; 45(6): 1946–1953. [DOI] [PubMed] [Google Scholar]
- 90. Gerhart J, Greenbaum M, Scheinfeld V, et al.. Myo/Nog cells: targets for preventing the accumulation of skeletal muscle-like cells in the human lens. PLoS One. 2014; 9(4): e95262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Menko AS, DeDreu J, Logan CM, Paulson H, Levin AV, Walker JL.. Resident immune cells of the avascular lens: Mediators of the injury and fibrotic response of the lens. FASEB J. 2021; 35(4): e21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wei M, Xing KY, Fan YC, Libondi T, Lou MF.. Loss of thiol repair systems in human cataractous lenses. Invest Ophthalmol Vis Sci. 2014; 56(1): 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu H, Smith AJ, Lott MC, et al.. Sulforaphane can protect lens cells against oxidative stress: implications for cataract prevention. Invest Ophthalmol Vis Sci. 2013; 54(8): 5236–5248. [DOI] [PubMed] [Google Scholar]
- 94. Kubo E, Chhunchha B, Singh P, Sasaki H, Singh DP.. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci Rep. 2017; 7(1): 14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rowan S, Jiang S, Korem T, et al.. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc Natl Acad Sci USA. 2017; 114(22): E4472–E4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tan J, Wang JJ, Flood V, et al.. Carbohydrate nutrition, glycemic index, and the 10-y incidence of cataract. Am J Clin Nutr. 2007; 86(5): 1502–1508. [DOI] [PubMed] [Google Scholar]
- 97. Wolf NS, Li Y, Pendergrass W, Schmeider C, Turturro A.. Normal mouse and rat strains as models for age-related cataract and the effect of caloric restriction on its development. Exp Eye Res. 2000; 70(5): 683–692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.