

Abstract
Severe diseases such as the ongoing COVID-19 pandemic, as well as the previous SARS and MERS outbreaks, are the result of coronavirus infections and have demonstrated the urgent need for antiviral drugs to combat these deadly viruses. Due to its essential role in viral replication and function, 3CLpro (main coronaviruses cysteine-protease) has been identified as a promising target for the development of antiviral drugs. Previously reported SARS-CoV 3CLpro non-covalent inhibitors were used as a starting point for the development of covalent inhibitors of SARS-CoV-2 3CLpro. We report herein our efforts in the design and synthesis of submicromolar covalent inhibitors when the enzymatic activity of the viral protease was used as a screening platform.
Keywords: SARS-CoV2, 3CLpro, Mpro, covalent inhibitors
Graphical abstract

1. Introduction
Coronaviruses. Coronaviruses (CoV) are a large family of viruses associated with some forms of common colds (together with rhinoviruses, respiratory syncytial virus, adenoviruses and others), as well as far more serious diseases including Severe Acute Respiratory Syndrome (SARS, caused by SARS-CoV infection), which made headlines worldwide in 2002–2003 with over 700 deaths including 43 in Canada [1], and the Middle East Respiratory Syndrome (MERS, caused by MERS-CoV infection), which was reported in Saudi Arabia in 2012 and killed over 900 [2]. The current outbreak of novel coronavirus (COVID-19, caused by SARS-CoV-2 infection), its numerous variants, and the discovery of animal reservoirs provide significant motivation for the development of potent therapeutics against these viruses to prevent future outbreaks [3,4].
SARS, MERS, and COVID-19 are respiratory illnesses characterized by fever, cough, and shortness of breath, posing significant danger to patients. The case fatality rates for those infected with SARS-CoV and MERS-CoV were estimated at about 10% and 35%, respectively [1,2]. Estimates for SARS-CoV-2 are ranging anywhere from of 0.1–25% depending on the age group, the country and the stage of the pandemic, although this number could change substantially as more accurate information on the numbers of infections and deaths becomes available [5,6]. In contrast to SARS and MERS, COVID-19 has rapidly spread worldwide despite the severe restrictions imposed in many countries, and the official number of deaths now exceeds 5.0 million [7] (which is a well underestimated number as shown by excess mortality studies [8]).
Vaccines and therapeutics. While vaccines are a central pillar of our efforts to end our current deadly phase of the COVID-19 pandemic, therapeutics offer a complementary approach with many distinct advantages. For example, oral therapeutics tend to be easy to store and administer and need only be given to the small minority of patients suffering more serious symptoms. In contrast, a large proportion of the population must be inoculated for vaccines to be effective and mRNA-based vaccines require complex logistics to maintain the cold chain, leading to enormous challenges in production, supply and administration. In addition, large vaccine campaigns require public compliance and amplifies the number of people suffering from adverse reactions to medication. To add to these difficulties, Pfizer recently announced that the immunity of their vaccine drops after about 6 months suggesting that regular injections would be needed, further amplifying the public compliance issue and burden to public health systems [9,10]. Importantly, vaccines primarily induce an immune response against the spike protein [11], while future variants of concern may have mutations in this protein that could allow them to evade immunity. In contrast, antiviral therapeutics can target a wide range of proteins including viral proteases (3CLpro, PLpro), the RNA-dependent RNA polymerase (RdRp) and RNA helicase. Therefore, they can be equally effective against strains of the virus with mutations that escape spike-based vaccination or herd immunity. Overall, it is clear that effective therapeutics would be complementary to mass vaccination. Finally, some groups (pregnant and breastfeeding women, people with allergies, young children, immunocompromised patients or people with other conditions) may be at risk or not responsive to vaccines and alternative treatments (e.g., oral therapeutics) must be available [12]. Consequently, major efforts from a large number of research groups focused on the development of small molecules as antivirals against SARS-CoV-2 which culminated in the recent approval of Molnupiravir in the United Kingdom [13].
Coronavirus (CoV) and 3-Chymotrypsin-like Protease Inhibition. Coronaviruses express 3-chymotrypsin-like cysteine proteases (3CLpro), also referred to as the main proteases (Mpro) or nsp5 (non-structural protein 5), which feature a Cys-His catalytic dyad (Cys145, His41) and are required for viral replication and infection. 3CLpro enzymes were identified early on as attractive targets for antiviral development, resulting in several inhibitors and structures of SARS-3CLpro-inhibitor complexes (eg, PDB codes: 4TWY, 2ZU5, 2ALV [14]). The 3CLpro enzymes from SARS-CoV and SARS-CoV-2 share nearly 80% sequence identity [15,16], suggesting that many of the lessons learned for developing SARS therapeutics can be applied to COVID-19. As a note, 3CLpro is not limited to coronaviruses but is also a drug target for the development of antivirals against noroviruses (such as the one involved in gastroenteritis [17]) or antivirals against enteroviruses (e.g., antiviral drug 3CLpro-1 [18] targeting the hand, foot, and mouth disease enterovirus 71 and Rupintrivir – Fig. 1 - originally developed to fight rhinoviruses [19]).
Fig. 1.

Reported covalent SARS-CoV-2 3CLpro inhibitors 1, GC376 [26], PF-07321332, PF-0730814 [32,35,36] and an analogue [38], and reported non-covalent inhibitors Masitinib [46] and Walcyrin B [47]. Reported inhibitors of SARS-CoV 3CLpro2 [43] and 3 [45]. Orange spheres indicate the warheads for covalent binding.
Covalent Inhibitors. The quest for novel antivirals against SARS-CoV and, more recently, SARS-CoV-2 has been intense, and several viral enzyme inhibitors and crystal structures of enzyme-inhibitor complexes have quickly been reported (e.g., PDB codes: 6LU7 [20], 6M2N [21], 6XQU [22], 6WQF [23]) [[24], [25], [26]]. The presence of a catalytic cysteine residue in the active site makes 3CLpro amenable to covalent inhibition, a strategy that was successfully employed following the SARS-CoV pandemic (SARS). In fact, many of the reported SARS-CoV inhibitors feature an electrophilic group, such as an α-ketoamide, epoxide, aziridine, α,β-unsaturated ester (Michael acceptor), or α-fluoroketone, which forms a covalent bond with the catalytic cysteine residue (Cys145), as confirmed by X-ray crystallography (e.g., PDB code: 5N19) [24]. A crystal structure of the SARS-CoV-2 3CLpro with a covalent peptidic inhibitor bound to Cys145 was quickly elucidated (PDB code: 6LU7). This pseudo-peptidic inhibitor, an analogue of Rupintrivir (tested on SARS [28] and COVID-19 [29]), has been the starting point for a number of drug discovery campaigns [[30], [31], [32], [33], [34]]. 3CLpro inhibitor (PF-0730814 and PF-07321332 – Paxlovid -, Fig. 1) entered clinical trials [32,35,36] and some encouraging phase 2/3 results were reported [37]. Investigations from a group of Canadian researchers identified other warheads for this lead molecule with potential for further development [38,39]. The identification of a potent warhead was also the focus of Hilgenfeld and co-workers [40].
The structurally similar GC376 was originally identified as active against a feline coronavirus [41] and more recently confirmed as a SARS-CoV-2 3CLpro inhibitor [26], and structure-activity relationship studies led to improved analogues [42]. Smaller, more drug-like inhibitors such as the isatin derivative 2 have been devised [43,44]. More recently, Jorgensen and co-workers converted Perampanel, a known antiepileptic drug that is also a weak 3CLpro inhibitor, into potent inhibitors (3, Fig. 1) using a combination of computational and experimental investigation [45].
As described in our recent review [48], covalent drugs can be extremely effective and useful pharmaceuticals, yet they have been largely ignored in most drug design endeavours and particularly in those concerning structure-based drug design. Concerns about their potential off-target reactivity and toxicity have often been raised [49]. Despite these concerns, there are many examples of covalent drugs on the market, including two of the ten most widely prescribed medications in the U.S., as well as several other common drugs like aspirin and penicillin [48]. The advantages of covalent drugs are becoming increasingly recognized: they have extremely high potencies, long residence times, and high levels of specificity [50]. Although skepticism persists, many pharmaceutical companies are embracing covalent drugs as exemplified by Neratinib (Nerlynx®, Pfizer) and Afatinib (Gilotrif®, Boehringer-Ingelheim).
3CLproinhibitor design. Many of the structure-based studies related to COVID-19 to date have employed virtual screening and machine learning techniques. Several potential 3CLpro inhibitors have been identified, however experimental verification has lagged [[51], [52], [53]]. As of today, much of the research has focused on peptidic substrate-like inhibitors (Fig. 1). There is currently a need for the development of drug-like inhibitors with synthetically accessible scaffolds that will allow for more thorough investigations of structure-activity relationships. We thought to benefit from our team's expertise in covalent inhibition and from our software that enables automated docking and virtual screening of covalent inhibitors, which is not possible with most commercial packages. We present herein our efforts towards the development of novel potent covalent inhibitors of 3CLpro.
2. Chemistry
Inhibitor design through covalent docking. In the past years, we have successfully applied covalent docking to the design and discovery of prolyl oligopeptidase inhibitors [54,55] and thought to apply a similar strategy to develop SARS-CoV-2 3CLpro inhibitors. An investigation of the crystal structure of a non-covalent inhibitor (X77, Fig. 2 ) bound to 3CLpro of SARS-CoV-2 (PDB code: 6W63) suggested that it might be possible to modify this inhibitor by incorporating a covalent warhead in proximity to the catalytic cysteine residue. As shown in Fig. 2, the sulphur atom of Cys145 is positioned at 3.2 Å from the imidazole moiety and at the same location as the covalent warhead of PF-00835231. Thus, replacement of the imidazole with a covalent warhead appeared to be a promising strategy to improve the inhibitory potency of this non-covalent inhibitor. Additionally, this scaffold could be prepared via a 4-component Ugi reaction [56], enabling a combinatorial approach that would provide an efficient synthetic method for preparing diverse analogues. This would provide a significant advantage in exploring structure-activity relationships when compared to previously reported inhibitors, as a wide range of covalent warheads could be readily incorporated into the same inhibitor scaffold. As a note, a consortium of research groups including a group at the Weizmann Institute of Science in Rehovot (Israel) took a very similar strategy although focusing primarily on non-covalent inhibitors [57].
Fig. 2.

Binding site interactions of inhibitors X77, ML188 and PF-00835213 (PDB codes: 6W63 [58], 3V3M [56], 6XHM [33]).
To validate the design strategy, a virtual library of modified inhibitors was prepared based on incorporation of covalent warheads that could be accessed via a traditional or modified Ugi 4 component coupling (4CC) reaction (Fig. 3 ). These compounds were docked to 3CLpro (PDB code: 6W63) using our docking program, Fitted [59]. The docked poses (Fig. 4 ) suggested that many of these modified inhibitors would be able to maintain the same non-covalent interactions as the original non-covalent inhibitor while also positioning the warhead close enough to Cys145 to facilitate the formation of a covalent bond. Based on the promising docking results with multiple warheads, a small library of analogues was synthesized for experimental testing.
Fig. 3.

Selected covalent 3CLpro inhibitors for synthesis. Compound X77 is the original non-covalent lead compound.
Fig. 4.

Selected docked binding modes of design covalent inhibitors (pink) overlaid with the non-covalent inhibitor (co-crystallized) X77 (grey). Top left: 6a, top right: 13a, bottom left: 16a and bottom right: 14a.
Synthesis. Following a protocol reported by Jacobs et al. [56], a 4-component Ugi reaction was used to prepare analogues bearing four different classes of covalent warheads (alkene Michael acceptor, α-halo ketone, alkyne Michael acceptor, and α-ketoamide, Scheme 1 a). This synthetic strategy was later employed to probe some of the features of this class of inhibitors by modifying the R2, R3 and R4 groups.
Scheme 1.

a) carboxylic acid (1.0 mmol, 1.0 eq.), 4-tert-butylaniline (1.0 mmol, 1.0 eq.), 3-pyridinecarboxaldehyde (1.0 mmol, 1.0 eq.), cyclohexyl isocyanide (0.9 mmol, 0.9 eq.), MeOH (5 mL, 0.2 M), r.t., overnight, X77 (30%), ML188 (84%), 4a (76%), 6a (32%), 7a (94%), 8a (32%), 9a (73%), 10a (68%), 11a (80%), 12a (37%), 13a (92%), 15a (70%), 16a (93%), 17a (52%), 18a (36%), 20a (84%), 21a (68%), 22a (29%), 23a (43%), 8b (54%), 13b (82%), 6b (42%), 4b (84%), 13c (94%), 13d (86%), 16b (88%), 16c (91%), 11b (67%), 11c (79%), 11d (59%), 11e (26%), 11f (71%), 11g (29%), 11h (78%), 11i (37%), 8c (74%), 16d (87%), 16f (34%), 13e (90%), 13f (69%), 13g (91%), 13h (92%), 13i (22%), 13j (47%), 18b (37%), 13k (83%), 13l (57%), 13m (82%), 13n (89%), 13o (88%), 13p (89%), 13q (35%). b) 3-pyridinecarboxaldehyde (2.0 mmol, 2.0 eq.), glycinamide hydrochloride (2.0 mmol, 2.0 eq.), triethylamine (2.0 mmol, 2.0 eq.), acetic acid (2.0 mmol, 2.0 eq.), cyclohexyl isocyanide (2.0 mmol, 2.0 eq.), MeOH (5 mL, 0.2 M), r.t., overnight, 24a (79%). c) 3-pyridinecarboxaldehyde (1.0 mmol, 1.0 eq.), β-alanine (1.0 mmol, 1.0 eq.), cyclohexyl isocyanide (1.0 mmol, 1.0 eq.), MeOH (5 mL, 0.2 M), r.t., overnight, 25a (54%). D) phosphoric acid, MeOH, r.t., 68%.
A 3-component Ugi reaction was used to prepare additional analogues bearing nitrile [60] and β-lactam [61] covalent warheads following reported procedures (Scheme 1b, c and d).
In order to complete the synthesis of the analogues featuring warheads not accessible through the 4-component Ugi, an intermediate was prepared and used to incorporate vinylsulfonamide and ethyl carbamate warheads (Scheme 2 ). This intermediate was also used to probe the importance of having a group at this position.
Scheme 2.

a) H3PO4, MeOH, rt, 5a (96%); b) Cl–CH2–CH2–SO2Cl, pyridine, DCM, 0 °C to rt, 14a (35%); c) Cl–CO2Et, pyridine, DCM, 0 °C to rt, 19a (76%).
In order to further probe the R4 group of the potential inhibitors, a recently reported strategy to make isocyanides [62,63] was employed (Scheme 3 ). The strategy illustrated in Scheme 2 was then used to prepare these analogues.
Scheme 3.

a) HCO2Et, 60 °C, 26b (95%), 26c (98%), 26d (40%), 26e (99%), 26f (99%), 26g (99%); b) POCl3, Et3N, DCM, 27b (73%), 27c (83%), 27d (79%), 27e (71%), 27f (50%), 27g (74%); c) H3PO4, MeOH, rt, 28b (42%), 28c (45%), 28d (48%), 28e (26%), 28f (25%), 28g (40%); c) Cl–CH2–CH2–SO2Cl, pyridine, DCM, 0 °C, 14b (66%), 14c (45%), 14d (73%), 14e (80%), 14f (42%), 14 g/h (75%).
3. Results and discussions
3CLproinhibition – covalent warheads. Twenty potential warheads (compounds 6a-25a, Fig. 3) and four non-covalent analogues (X77, ML188, 4a and 5a) were synthesized and evaluated for their inhibitory potency using a fluorescence inhibition assay. The compounds were initially screened at 50 μM and IC50 values were subsequently determined for compounds displaying greater than 80% inhibition (Table 1 ).
Table 1.
Inhibitory potency against SARS-CoV-2 3CLpro. Evaluation of warheads (R1)[].
| Entry | Scaffold | Cmpd | R1 | Inhibition (%)a | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | – | GC376 | – | >95 | 0.11 ± 0.06 |
| 2 | I | X77 | 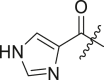 |
>95 | 4.1 ± 1.2 |
| 3 | Ic | ML188 | 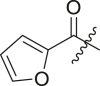 |
>95 | 1.4 ± 0.4 |
| 4 | I | 4a | 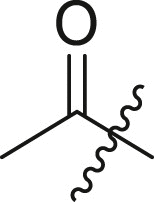 |
25 ± 8 | ndb |
| 5 | I | 5a |  |
28 ± 3 | ndb |
| 6 | I | 6a | 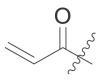 |
84 ± 1 | 11.1 ± 1.5 |
| 7 | I | 7a | 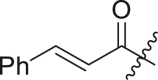 |
44 ± 6 | ndb |
| 8 | I | 8a | 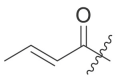 |
63 ± 5 | ndb |
| 9 | I | 9a | 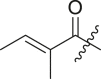 |
47 ± 2 | ndb |
| 10 | I | 10a | 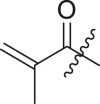 |
30 ± 1 | ndb |
| 11 | I | 11a | 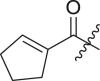 |
55 ± 10 | ndb |
| 12 | I | 12a | 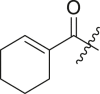 |
52 ± 1 | ndb |
| 13 | I | 13a | 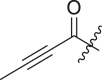 |
>95 | 5.3 ± 0.8 |
| 14 | I | 14a | 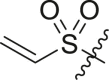 |
>95 | 0.42 ± 0.11 |
| 15 | I | 15a | 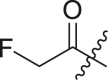 |
30 ± 7 | ndb |
| 16 | I | 16a | 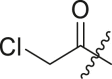 |
>95 | 0.41 ± 0.13 |
| 17 | I | 17a | 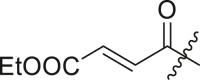 |
59 ± 6 | ndb |
| 18 | I | 18a | 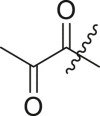 |
92 ± 1 | 5.2 ± 1.2 |
| 19 | I | 19a | 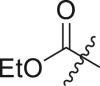 |
35 ± 3 | ndb |
| 20 | I | 20a | 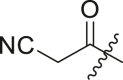 |
74 ± 1 | 7.0 ± 0.2 |
| 21 | I | 21a | 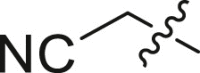 |
<5 | ndb |
| 22 | I | 22a | 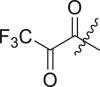 |
77 ± 4 | 12.4 ± 5.2 |
| 23 | I | 23a | 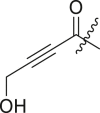 |
>95 | 0.85 ± 0.42 |
| 24 | II | 24a | 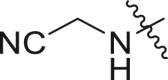 |
25 ± 1 | ndb |
| 25 | II | 25a | 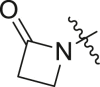 |
<5 | ndb |

a The enzyme activity was measured with 150 nM 3CLpro (114 nM after inhibitor addition) and 50 μM of each potential inhibitor with incubation time of 30 min b not determined. c cyclohexyl replaced by tert-butyl in scaffold I.
Confirmation of covalent binding. To evaluate the covalent inhibition hypothesis, the time dependence of our most potent inhibitors, 16a and 14a, were measured (Fig. S4). The level of inhibition increased with incubation time, an observation that is consistent with the formation of a covalent adduct. Additionally, the presence of 3CLpro -16a and -14a adducts were confirmed by LC-MS (Fig. S4a/b). As a definitive proof of covalent inhibition and binding mode, crystal structures of 3CLpro co-crystallized with 16a and 14a were obtained (Fig. 5 ).
Fig. 5.

a) Crystal structure of 16a (PDB ID: 7MLF) bound to 3CLpro and b) crystal structure of 14a (PDB ID: 7MLG) bound to 3CLpro.
Analysis of covalent warheads. We observed that GC376, X77 and ML188, previously reported SARS-CoV 3CLpro inhibitors (X77: IC50 = 3.4 μM [64] and ML188: IC50 = 4.8 μM [56] respectively) and SARS-CoV2 inhibitor (GC376: 139 nM), also inhibit SARS-CoV-2 3CLpro with similar potencies with our assay. (Table 1, entries 1 to 3). Gratifyingly, low micromolar to sub-micromolar potencies were also observed for the covalent analogues containing acrylamide (6a), alkynylamide (13a), vinyl sulfonamide (14a), α-chloroamide (16a) and α-ketoamide (18a) warheads. Interestingly, our two most potent inhibitors 16a and 14a (IC50 = 0.4 and 0.5 μM) were an order of magnitude more potent than the original non-covalent hit molecule (X77: IC50 = 4.1 μM).
An increase in potency was observed when increasing the electrophilicity of the warhead – the α-chloroamide (16a) was more active than the corresponding α-fluoroamide (15a), and the vinyl sulfonamide (14a) was more active than the corresponding acrylamide (6a). However, this trend was not observed when increasing the electrophilicity of the ketoamide (18a) with a CF3 group (22a), potentially due to an increase in the steric bulk and/or electrostatic properties of the warhead that are not tolerated in the active site. Similarly, while acrylamides are typically more reactive with cysteine than the corresponding alkynylamides (when tested in glutathione or cysteine binding assays [65]), the aklynylamide warhead was more active against 3CLpro. A possible explanation could be that the sp geometry of the aklynylamide warhead positions the electrophile more favorably to the cysteine residue to facilitate covalent bond formation.
The binding pocket also seems to favor smaller warheads – any steric bulk around the acrylamide warhead resulted in a decrease in potency, regardless of electronics. As mentioned previously, a similar effect was observed when comparing the activity of inhibitor 22a and 18a.
The position of the covalent bond formation also appears to influence inhibitor activity. Minimal inhibition was observed with compounds 19a and 25a which both require the formation of a covalent bond directly with the carbonyl carbon. This carbon is positioned slightly further from the cysteine residue (3.4 Å in PDB 6W63) and therefore covalent bond formation may result in the loss of other non-covalent interactions. Covalent bond formation appeared to be equally tolerated at either the alpha position (16a and 18a) or beta position (6a, 13a, and 14a). However, the co-crystallized structure of 16a (Fig. 5a) shows that covalent bond formation at the alpha position resulted in a slight shift of the inhibitor towards the cysteine residue that resulted in the loss of a hydrogen bond interaction with Glu166. This interaction was maintained in the co-crystallized structure of 14a (Fig. 5b), suggesting that covalent bond formation at the beta position is preferable for non-covalent binding affinity.
Another observation is the significant loss of potency when removing the heterocyclic ring of X77 or ML188 (X77/ML188 vs. 4a, Table 1). As illustrated in Fig. 2, the basic imidazole nitrogen of X77 forms a hydrogen bond interaction with the backbone of Gly143, an interaction that is also observed with the furan ring of ML188 (PDB code: 3V3M) or other heterocycles of the same chemical series [56,66]. Gly143, together with the backbone amides of Ser144 and Cys145, forms an oxyanion hole that contributes to the catalytic activity of this enzyme. Substitution of this heterocycle with a carbocycle of similar size but no hydrogen bonding groups (compounds 11a vs. X77) does not preserve the inhibitory potency even when this ring was converted to a warhead for covalent binding. Additionally, no difference in activity was observed when comparing the activity of compounds 4a and 5a, suggesting that the carbonyl group does not contribute significantly to the inhibitor's binding affinity.
As shown in Fig. 2, both X77 and PF-00835231 interact with the catalytic His41, either through a direct hydrogen bond in the case of PF-00835231, or though a water-mediated hydrogen bond (X77) via a conserved water molecule. In an attempt to reproduce this interaction, longer covalent groups were designed by incorporating an ethyl ester to an acrylamide warhead (17a) and by incorporating a hydroxyl group to an alkynylamide warhead (23a). While 17a resulted in a loss of potency, 23a led to a nearly 10-fold improvement in potency over the alkynylamide analogue 13a. Although our initial hypothesis was that the increase in potency was due to the formation of a hydrogen-bond interaction with His41, docking suggests that the hydroxyl group of 23a instead occupies the oxyanion hole and interacts with the backbone amides of Gly143, Ser144 and Cys145 (Fig. S7).
Isothermal Titration Calorimetry (ITC). ITC was employed to validate the initial fluorescence inhibition assay and to gain information of the kinetics of covalent bond formation. ITC provides unique insights into the kinetics and thermodynamics of inhibitor binding that are not available in traditional enzyme assays [67]. ITC experiments measure the heat produced by catalysis in real time (Fig. 6 , y- and x-axis, respectively), as one component is titrated into another. The power is proportional to the enzyme velocity, with larger deflections from the baseline corresponding to higher velocities, and exothermic and endothermic reactions giving downward and upward deflections of the ITC signal, respectively. Since the instrument detects the heat released by the native reaction, unlabeled substrates can be used in the experiments, unlike the fluorescence assay which requires substrates modified with hydrophobic dyes and quenchers.
Fig. 6.

ITC enzyme activity assay in the presence of a) no inhibitor, b) 16a, c) 14a, d) 14c; each successive injection is shown in separate color. ITC simulations corresponding to the minimized kinetic parameters from fitting a covalent inhibition model are shown as black.
We performed activity assays where 3CLpro was titrated into the sample cell, which contained either substrate alone or a mixture of substrate and inhibitor. We then fit the resulting kinetic traces to determine both the kinetic parameters of the enzyme and the rate and mechanism of inhibitor binding. Fig. 6a shows the ITC trace obtained when 3CLpro was injected into substrate alone, with the first (0.2 μL) and second (1.5 μL) injections shown in red and yellow. The signal was deflected downwards, indicating an exothermic reaction, and gradually returned to the baseline over about 1000 s as all the substrate was cleaved. The data were fit to the Michaelis Menten equation to extract the turnover number, k cat, and the Michaelis constant, K m. Panels b–d show the ITC traces obtained when 3CLpro was injected into a mixture of substrate and inhibitor. In all cases, the total amounts of heat (areas of the peaks) were less than that obtained in the absence of inhibitor (panel a), indicating that the enzyme was inactivated before all the substrate was cleaved. To obtain quantitative information on inhibition, the traces were fit to three different models: (i) a reversible (rapid equilibrium) mechanism, E↔EI characterized by an equilibrium affinity constant, K i; (ii) an irreversible mechanism, E→EI characterized by a second order rate constant k inact; and (iii) a two-step mechanism, E↔EI∗→EI, consisting of a rapid pre-equilibrium described by K i, followed by irreversible inhibition described by a first-order rate constant k inact. In models (ii) and (iii), k inact can be tentatively assigned to the rate of covalent bond formation with the enzyme. The Michaelis Menten enzymatic parameters were held fixed at the values obtained in (a) and the parameters of the inhibition models were varied to minimize the residual-sum-of-squared-deviations (RSS). In all cases, the pre-equilibrium irreversible model (iii) gave the best agreement with data. The improvements in RSS given by model (iii) compared to models (ii) and (i) were calculated using F-test statistics [68] and found to be significant at levels of p ≤ 10−2. The extracted values of K i and k inact are listed in Table S1. 14c had both the fastest rate of covalent bond formation (largest k inact) and tightest binding in the initial non-covalent step (smallest K i) of the three inhibitors, tested. Both 14a and 14c have similar values of K i to the parent non-covalent scaffold, which is consistent with the two-step binding model (iii), since the first step corresponds to non-covalent binding.
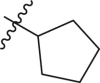
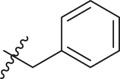
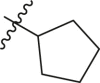
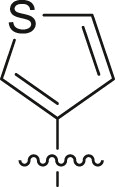
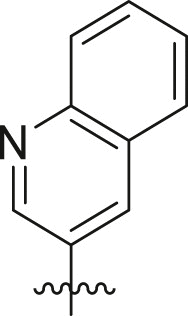
Structure-Activity Relationship. Following our search for an optimal warhead, several modifications were made to the core of the molecule (Table 2, Table 3, Table 4 ). Previously, it was shown that replacement of the tert-butylphenyl group (R2) by smaller groups or differently substituted phenyl groups modulates the potency against SARS-CoV 3CLpro with slight improvements in some cases, while various hydrophobic groups were tolerated as R3 [64]. We thought this information could help further improve these inhibitors for the highly homologous SARS-CoV-2 3CLpro. However, all of our attempts to replace the tert-butylphenyl and pyridyl groups have proven unsuccessful to date. More specifically, the use of a variety of aromatic heterocycles such as thiophene, benzothiazole, pyrimidine, pyrazine and benzothiophene led to loss of potency (Table 3). Additionally, replacement of the heterocyclic ring with a hydantoin moiety also led to a loss in potency. Similarly, diversely functionalized phenyl groups in place of the tert-butylphenyl group were detrimental for the activity and replacement of the phenyl group with a benzyl moiety led to a complete loss of activity (Table 4).
Table 2.
Inhibitory potency against SARS-CoV-2 3CLpro. Optimization of R4.
| Entry | Cmpd | R1 | R4 | Inhibition (%)a | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 8b | 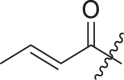 |
 |
33 ± 1 | ndb |
| 2 | 13b | 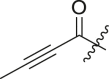 |
 |
93 | 15.0 ± 9.3 |
| 3 | 6b | 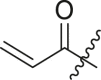 |
 |
68 ± 1 | ndb |
| 4 | 4b | 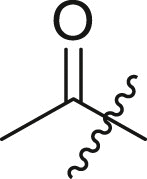 |
 |
<5 | ndb |
| 5 | 13c | 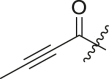 |
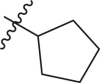 |
88 | 9.7 ± 3.8 |
| 6 | 13d | 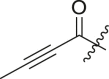 |
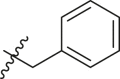 |
69 | >30 |
| 7 | 16b | 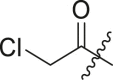 |
 |
>95 | 0.38 ± 0.09 |
| 8 | 16c | 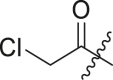 |
 |
>95 | 0.92 ± 0.24 |
| 9 | 11b | 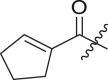 |
 |
16 | ndb |
| 9 | 14b | 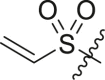 |
 |
>95 | 0.28 ± 0.10 |
| 10 | 14c | 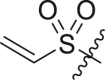 |
 |
>95 | 0.17 ± 0.07 |
| 11 | 14d | 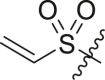 |
 |
>95 | 0.24 ± 0.15 |
| 12 | 14e | 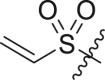 |
 |
>95 | 0.52 ± 0.16 |
| 13 | 14f | 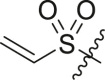 |
 |
>95 | 0.22 ± 0.08 |
| 14 | 14g (R,S) | 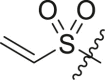 |
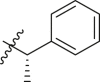 |
>95 | 0.32 ± 0.10 |
| 15 | 14h (S,S) | 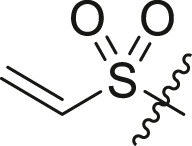 |
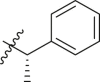 |
>95 | 6.0 ± 2.7 |
The enzyme activity was measured with 150 nM 3CLpro (114 nM after inhibitor addition) and 50 μM of each potential inhibitor with incubation time of 30 min.
Not determined.
Table 3.
Inhibitory potency against SARS-CoV-2 3CLpro. Optimization of R3.
| Entry | Cmpd | R1 | R3 | R4 | Inhibition (%)a | IC50 (μM) |
|---|---|---|---|---|---|---|
| 1 | 11c | 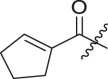 |
 |
cHex | 22 ± 8 | ndb |
| 2 | 11d | 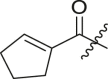 |
 |
cHex | 18 ± 8 | ndb |
| 3 | 11e | 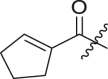 |
 |
cHex | <5 | ndb |
| 4 | 11f | 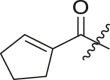 |
 |
cHex | 19 ± 8 | ndb |
| 5 | 11g | 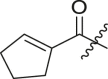 |
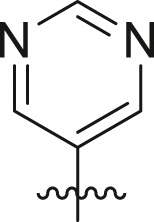 |
cHex | 38 ± 8 | ndb |
| 6 | 11h | 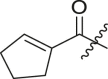 |
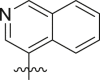 |
cHex | <5 | ndb |
| 7 | 11i | 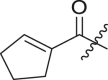 |
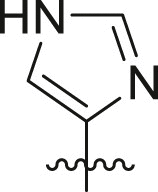 |
cHex | 24 ± 8 | ndb |
| 8 | 8c | 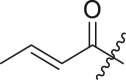 |
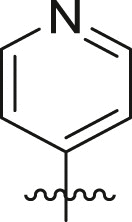 |
tBu | 24 ± 8 | ndb |
| 9 | 16d | 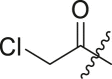 |
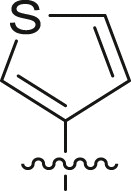 |
cHex | 22 ± 11 | ndb |
| 10 | 16e | 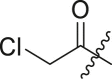 |
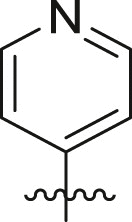 |
cHex | >95 | 0.84 ± 0.30 |
| 11 | 16f | 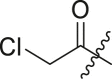 |
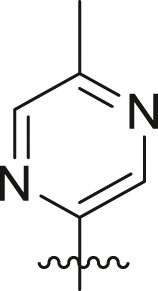 |
cHex | >95 | 0.98 ± 0.35 |
| 12 | 13e | 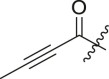 |
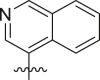 |
cHex | >95 | 5.0 ± 2.3 |
| 13 | 13f | 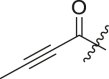 |
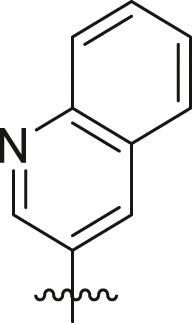 |
cHex | 80 ± 10 | ndb,c |
| 14 | 13g | 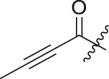 |
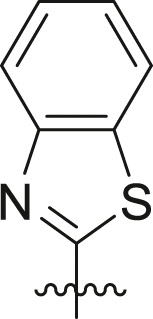 |
cHex | 54 ± 15 | ndb |
| 15 | 13h | 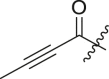 |
 |
cHex | 57 ± 6 | ndb |
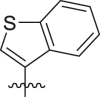
| 16 | 13i | 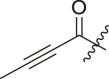 |
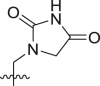 |
cHex | 10 ± 10 | ndb |
| 17 | 13j | 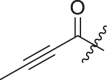 |
 |
cHex | 19 ± 5 | ndb |
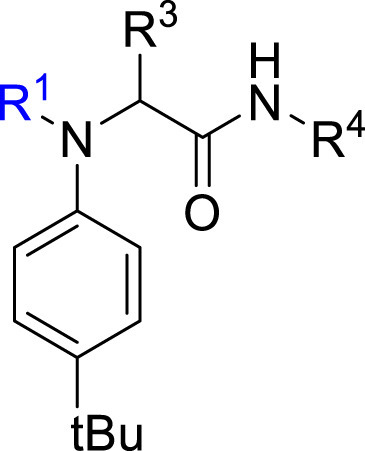
Table 4.
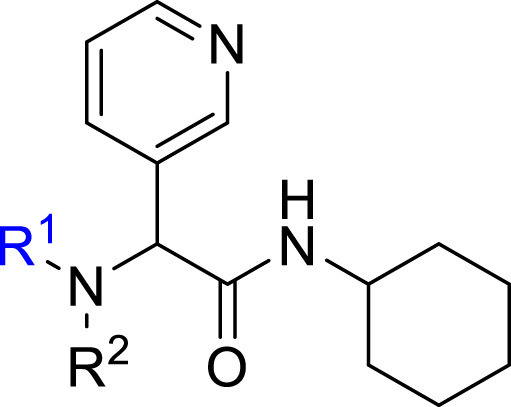
Inhibitory potency against SARS-CoV-2 3CLpro. Optimization of R2.
| Entry | Cmpd | R1 | R2 | Inhibition (%)a | IC50 (μM) |
|---|---|---|---|---|---|
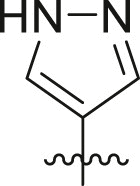
| 1 | 18b | 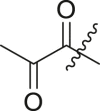 |
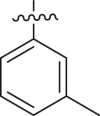 |
< 5% | ndb |
| 2 | 13k | 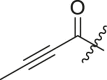 |
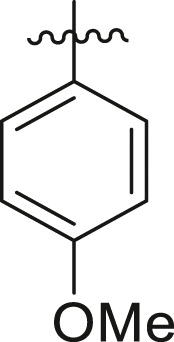 |
39 ± 1 | ndb |
| 3 | 13l | 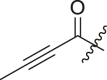 |
-H | 10 ± 15 | ndb |
| 4 | 13m | 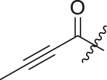 |
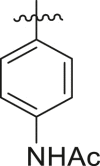
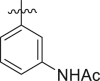 |
43 ± 3 | ndb |
| 5 | 13n | 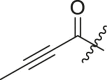 |
 |
23 ± 13 | ndb |
| 6 | 13o | 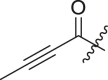 |
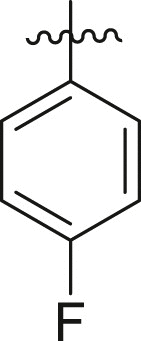 |
65 ± 3 | ndb |
| 7 | 13p | 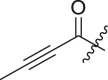 |
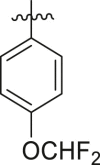 |
77 ± 2 | 45.1 ± 18.3 |
| 8 | 13q | 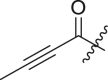 |
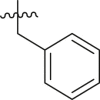 |
< 5% | ndb |
The enzyme activity was measured with 150 nM 3CLpro (114 nM after inhibitor addition) and 50 μM of each potential inhibitor with incubation time of 30 min.
not determined.
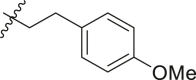
Replacement of the cyclohexyl group with groups of similar sizes did not improve the potency (Table 2, entries 1–7 and 9). However, we have found that substitution of the cyclohexyl (R4 group in Table 2) for longer groups (14b-l) improved the inhibitory potency over that of 16a and 14a. The longer chains likely enable the inhibitor to form a hydrophobic interaction with Pro168, while the amine nitrogen of analogue 14f may facilitate a hydrogen-bond interaction with Gln189 or the carbonyl of Glu166. Based on these optimizations, our current most potent compound (14c) has a potency similar to the previously reported inhibitor GC376. It is expected that one enantiomer is more potent that the other one. To confirm this hypothesis, we used a chiral isocyanide leading to easily separable diastereomers 14h and 14g. Diastereomer 14g was found to be approximately 20 times more potent that 14h in line with what has been observed previously [69].
aThe enzyme activity was measured with 150 nM 3CLpro (114 nM after inhibitor addition) and 50 μM of each potential inhibitor with incubation time of 30 min b not determined. c IC50 was not determined as poor solubility did not allow us to accurately measure activity beyond 50 μM.
aThe enzyme activity was measured with 150 nM 3CLpro (114 nM after inhibitor addition) and 50 μM of each potential inhibitor with incubation time of 30 min b not determined.
Off-target Cathepsin L inhibition. Lysosomal ubication of cysteine proteases could have cross-reactivity with the potential viral inhibitors, decreasing the therapeutical effect against SARS-CoV-2. [70]. For example, the increase of the cathepsin L levels, a ubiquitous human protease, in plasma of patients with SARS-CoV-2 severe infections generates a target competence for the protease antiviral inhibitors [71]. To assess this potential undesired effect of our most potent 3CLpro inhibitors, we measured the inhibitor effect against cathepsin L. The low inhibition of cathepsin L activity for our inhibitors at 50 μM (25% ± 2, 19% ± 1 and 15% ± 2 for 16a, 14a and 14c, respectively, Figs. S3a and S3b) indicates an excellent selectivity of these inhibitors against 3CLpro over cathepsin L.
4. Conclusion
Covalent inhibition of SARS-CoV-2 3CLpro is a promising strategy for the treatment of COVID-19. Our strategy relied on a previously reported imidazole-containing inhibitor of the similar coronavirus SARS-CoV responsible for the epidemic of SARS in the early 2000's. We first used our docking program Fitted, specifically modified to accommodate covalent inhibitors, and screened a set of covalent warheads. The docked poses confirmed that replacing the imidazole ring by a reactive group should lead to potent covalent inhibition. Gratifyingly, while the imidazole of X77 was known to be essential for the inhibitory potency, replacing it with many warheads maintained and even improved the potency, with our lead compounds 16a and 14a being an order of magnitude more potent. Both the inhibition pattern of enzymatic activity and the biophysical data first suggested that these inhibitors bind covalently to the viral protease, a binding mode later confirmed by crystallography; thus, the robustness of in silico rational-drug design was validated using in vitro detection of protein processing.
5. Experimental section
5.1. Synthesis and characterization
General Considerations. All other reagents were purchased from commercial suppliers and used without further purification. All 1H, 13C and 19F NMR spectra were acquired Bruker Avance 500 MHz spectrometer. Chemical shifts are reported in ppm using the residual of deuterated solvents as an internal standard. Chromatography was performed on silica gel 60 (230-40 mesh) or using the Biotage One Isolera with ZIP cartridges. High resolution mass spectrometry was performed by ESI on a Bruker Maxis Impact API QqTOF mass spectrometer at McGill University. Reversed-phase HPLC (water and MeCN or MeOH gradient) was used to verify the purity of compounds on an Agilent 1100 series instrument equipped with VWD-detector, C18 reverse column (Agilent, Zorbax Eclipse XDBC18 150 mm 4.6 mm, 5 μm), and UV detection at 254 nm. Measured purities for all tested compounds are listed in Table S3 in the supporting information.
General Procedures A, B and C for 4-Component Ugi Reaction. In a 6-dram vial equipped with a stir bar aldehyde (1.0 mmol, 1.0 eq.), aniline (1.0 mmol, 1.0 eq.) and carboxylic acid (1.0 mmol, 1.0 eq.) were combined in MeOH (4 mL). The obtained reaction mixture was stirred for 30 min at room temperature. Afterwards cyclohexyl isocyanide (0.9 mmol, 0.9 eq.) was added to the reaction mixture and the walls of the vial were washed with 1 mL of MeOH. The reaction mixture was continued to stir at room temperature overnight. The crude reaction mixture was evaporated in vacuo. Purification procedure A) The crude product was triturated with hexanes (5 mL) and filtered. The obtained product was further washed with hexanes (3 x 3 mL). Purification procedure B) The crude product recrystallized from CHCl3/hexanes mixture, filtered and the obtained product was further washed with hexanes (3 x 3 mL). Purification procedure C) The crude product was redissolved in DCM. The obtained crude solution was deposited on silica. It was then purified using flash column chromatography using DCM/MeOH (gradient 0 → 5%) as eluent.
General Procedure D to Prepare Formamides. The synthesis of formamides was derived from known literature [62]. In a 1-dram vial equipped with a stir bar, 5 mmol (1 eq.) of amine was mixed with 15 mmol (3 eq.) of ethyl formate and stirred at 60 °C until completion (monitored using TLC - 1:1 EtOAc:hexanes or 2:1 EtOAc:hexanes). Once the amine was fully converted, ethyl formate was removed in vacuo and the product was used in the next step without further purification.
General procedure E to Prepare Isocyanides. The synthesis of isocyanides was derived from known literature [63]. In a 6-dram vial equipped with a stir bar, 2 mmol (1 eq.) of formamide was dissolved in 1 mL DCM with 10 mmol (5 eq.) of Et3N. The mixture was cooled to 0 °C and 2 mmol (1 eq.) of POCl3 was added dropwise. The reaction mixture was stirred at 0 °C for 10 min then quickly purified through a silica pad (2 g of silica 230–400 mesh, washing with a gradient of mixture of Et2O and DCM (100% Et2O →, 100% DCM). The fractions containing the desired product were collected and concentrated in vacuo. This procedure (silica pad, washing and concentration) is repeated once.
General Procedures F for 3-Component Ugi Reaction. In a 6-dram vial equipped with a stir bar, 1 mmol (1.0 eq.) of 4-tert-butylaniline and 1 mmol (1.0 eq.) of 3-pyridinecarboxaldehyde were dissolved in 5 mL MeOH with 10 μL of 85% H3PO4. 1 mmol (1.0 eq.) of isocyanide was then added. The mixture was stirred at rt overnight. The reaction mixture was then concentrated in vacuo. The crude product was purified using flash column chromatography using EtOAc/hexanes (33% → 80%) as eluent.
General Procedure G for Synthesis of Vinyl Sulfonamides. The synthesis of vinyl sulfonamides was derived from known literature [72]. In a 6-dram vial equipped with a stir bar, 0.2–0.3 mmol (1.0 eq.) of the previously made acetamide (see general procedure F) was dissolved in 5 mL of DCM with 0.7–3. 5 mmol (from 0.5 eq. to 3 eq.) of Et3N. The mixture was cooled to 0 °C and 0.3–0.45 mmol (1.5 eq.) of 2-chloroethanesulfonyl chloride was added dropwise. The solution was stirred at 0 °C for 2 h. The solution was then diluted with 5 mL DCM and washed with 10 mL sat. NaHCO3. The aqueous layer was extracted with 10 mL DCM and the combined organic layer was washed with 10 mL sat. NaCl solution and further dried with anhydrous Na2SO4. The crude product was purified using flash column chromatography using DCM/EtOAc (gradient 0% → 50%) as eluent.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-1H-imidazole-5-carboxamide (X77). Compound was made and purified using general procedure B, white solid 30% yield, 125 mg. 1H NMR (500 MHz, MeOD) δ 8.37 (s, 1H), 8.33 (dd, J = 4.9, 1.5 Hz, 1H), 7.66–7.57 (m, 2H), 7.31 (d, J = 7.8 Hz, 2H), 7.22 (dd, J = 7.9, 4.9 Hz, 1H), 6.27 (s, 1H), 5.46 (s, 1H), 3.71 (td, J = 10.5, 9.3, 3.9 Hz, 1H), 1.93 (d, J = 12.3 Hz, 1H), 1.80–1.72 (m, 2H), 1.65 (ddt, J = 30.9, 12.9, 3.8 Hz, 2H), 1.27 (s, 12H). 13C NMR (126 MHz, MeOD) δ 169.00, 152.54, 150.62, 148.24, 138.83, 136.36, 131.58, 131.07, 125.72, 123.31, 62.74, 34.16, 32.16, 32.12, 30.25, 25.22, 24.72, 24.64. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C27H33N5NaO2 482.2526; found 482.2535.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)furan-2-carboxamide (ML188). Compound was made and purified using general procedure A, white solid 84% yield, 290 mg. 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 2.3 Hz, 1H), 8.46 (dd, J = 4.9, 1.7 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.39 (dd, J = 1.7, 0.7 Hz, 1H), 7.24 (d, J = 6.6 Hz, 2H), 7.06 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 6.98 (s, 2H), 6.19–6.13 (m, 2H), 6.10 (s, 1H), 5.38 (dd, J = 3.6, 0.8 Hz, 1H), 1.37 (s, 9H), 1.28 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 167.96, 159.80, 152.62, 151.59, 149.72, 146.36, 145.12, 138.32, 136.63, 130.56, 130.30, 126.22, 122.94, 117.26, 111.35, 63.83, 51.92, 34.84, 31.41, 28.80. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C26H31N3NaO3 456.2258; found 456.2245.
2-(N-(4-(tert-butyl)phenyl)acetamido)-N-cyclohexyl-2-(pyridin-3-yl)acetamide (4a). Compound was made and purified using general procedure A, white solid 76% yield, 280 mg. 1H NMR (500 MHz, CDCl3) δ 8.46–8.43 (m, 1H), 8.42 (s, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.23 (d, J = 8.5 Hz, 2H), 7.03 (dd, J = 8.0, 4.8 Hz, 1H), 6.93 (s, 1H), 6.03 (s, 2H), 3.94–3.65 (m, 1H), 1.98 (d, J = 16.8 Hz, 1H), 1.89–1.81 (m, 4H), 1.75–1.63 (m, 2H), 1.59 (dt, J = 13.0, 4.3 Hz, 1H), 1.43–1.28 (m, 2H), 1.25 (s, 9H), 1.23–1.04 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 171.83, 168.21, 151.89, 151.44, 149.66, 138.09, 137.42, 130.87, 129.60, 126.28, 122.92, 62.42, 48.88, 34.73, 32.95 (d, J = 10.4 Hz), 31.37, 25.61, 24.87 (d, J = 6.9 Hz), 23.34. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C25H33N3NaO2 430.2465; found 430.2464.
N-cyclohexyl-2-(pyridin-3-yl)-2-(p-tolylamino)acetamide (5a). To a solution of 4-tBu-aniline (1.01 mmol, 0.16 mL) and 3-pyridinecarboxaldehyde (1.01 mmol, 0.09 mL) in MeOH (5 mL) was added cyclohexyl isocyanide (1.01 mmol, 0.12 mL) and phosphoric acid (0.2 mmol, 0.01 mL, 85%), and the solution stirred at room temperature overnight. The solvent was evaporated under a stream of air, and the crude reaction mixture was suspended in a small amount of EtOAc. Hexanes was added and the precipitate was collected by filtration and rinsed with hexanes and acetone. The precipitate was dried over vacuum to afford the desired product (353 mg, 96% yield) as a white powder. 1H NMR (500 MHz, DMSO) δ 8.69 (dd, J = 2.4, 0.9 Hz, 1H), 8.46 (dd, J = 4.8, 1.7 Hz, 1H), 8.19 (d, J = 7.9 Hz, 1H), 7.85 (dt, J = 7.9, 2.0 Hz, 1H), 7.35 (ddd, J = 7.8, 4.8, 0.9 Hz, 1H), 7.10–7.04 (m, 2H), 6.61–6.55 (m, 2H), 6.04 (d, J = 8.1 Hz, 1H), 5.02 (d, J = 8.0 Hz, 1H), 3.56–3.47 (m, 1H), 1.74 (dd, J = 10.5, 4.8 Hz, 1H), 1.70–1.63 (m, 1H), 1.62–1.48 (m, 3H), 1.31–1.19 (m, 2H), 1.18 (s, 10H), 1.17–1.03 (m, 1H). 13C NMR (126 MHz, DMSO) δ 169.29, 148.65, 148.56, 144.47, 138.99, 135.44, 134.52, 125.39, 123.45, 112.89, 58.39, 47.58, 33.46, 32.24, 32.06, 31.38, 25.11, 24.37, 24.26. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C23H31N3NaO 388.2359; found 388.2352.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl) acrylamide (6a). Compound was made and purified using general procedure C, white solid 32% yield, 120 mg. 1H NMR (500 MHz, CDCl3) δ 8.41–8.39 (m, 1H), 8.38–8.37 (m, 1H), 7.42–7.35 (m, 1H), 7.19 (d, J = 8.1 Hz, 2H), 7.01 (dd, J = 8.0, 4.8 Hz, 1H), 6.91 (s, 1H), 6.49 (d, J = 8.0 Hz, 1H), 6.33 (dd, J = 16.8, 2.0 Hz, 1H), 6.11 (s, 1H), 5.93 (dd, J = 16.8, 10.3 Hz, 1H), 5.49 (dd, J = 10.4, 2.0 Hz, 1H), 3.84–3.73 (m, 1H), 1.93 (s, 1H), 1.81 (dd, J = 13.1, 4.1 Hz, 1H), 1.67–1.53 (m, 3H), 1.37–1.26 (m, 2H), 1.22 (s, 9H), 1.17–1.06 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 168.04, 166.50, 151.79, 151.28, 149.45, 137.93, 136.14, 130.83, 129.93, 128.64, 128.52, 126.08, 122.85, 62.69, 48.76, 34.64, 32.86, 32.80, 31.27, 25.52, 24.83, 24.77. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C26H33N3NaO2 442.2465; found 442.2456.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl) cinnamamide (7a). Compound was made and purified using general procedure A, pale white solid 94% yield, 420 mg. 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 2.3 Hz, 1H), 8.47 (dd, J = 4.8, 1.7 Hz, 1H), 7.71 (d, J = 15.6 Hz, 1H), 7.50 (dt, J = 7.9, 2.0 Hz, 1H), 7.27 (s, 6H), 7.07 (dd, J = 8.0, 4.8 Hz, 1H), 6.98 (s, 1H), 6.33 (d, J = 8.1 Hz, 1H), 6.24 (d, J = 15.5 Hz, 1H), 6.14 (s, 1H), 3.91–3.80 (m, 1H), 1.94 (dd, J = 52.0, 13.0 Hz, 2H), 1.70 (ddd, J = 18.3, 11.4, 6.7 Hz, 2H), 1.59 (dd, J = 8.9, 4.1 Hz, 1H), 1.44–1.32 (m, 2H), 1.29 (s, 9H), 1.26–1.09 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 168.16, 167.14, 151.97, 151.36, 149.60, 143.22, 138.03, 136.60, 135.07, 130.93, 129.91, 129.86, 128.82, 128.13, 126.35, 122.95, 118.61, 63.32, 48.85, 34.81, 33.02, 32.96, 31.38, 25.62, 24.90, 24.86. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C32H37N3NaO2 518.2778; found 518.2790.
(E)-N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-enamide (8a). Compound was made and purified using general procedure B, pale white solid 32% yield, 126 mg. 1H NMR (500 MHz, CDCl3) δ 8.46–8.41 (m, 2H), 7.44 (dt, J = 7.9, 2.0 Hz, 1H), 7.24 (d, J = 8.2 Hz, 2H), 7.04 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 6.99–6.91 (m, 3H), 6.36 (d, J = 8.1 Hz, 1H), 6.09 (s, 1H), 5.65 (dd, J = 15.0, 1.7 Hz, 1H), 3.87–3.76 (m, 1H), 1.97 (dd, J = 11.7, 4.6 Hz, 1H), 1.92–1.82 (m, 1H), 1.72 (dd, J = 7.0, 1.7 Hz, 3H), 1.69–1.63 (m, 1H), 1.60–1.56 (m, 1H), 1.42–1.30 (m, 2H), 1.27 (s, 9H), 1.22–1.09 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 168.25, 166.99, 151.78, 151.24, 149.41, 143.08, 138.09, 136.62, 131.00, 129.80, 126.22, 122.88, 122.71, 62.96, 48.78, 34.75, 32.97, 32.91, 31.37, 25.62, 24.88, 24.84, 18.21. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C27H35N3NaO2 456.2621; found 456.2630.
(E)-N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-2-methylbut-2-enamide (9a). Compound was made and purified using general procedure A, white solid 73% yield, 294 mg. 1H NMR (500 MHz, CDCl3) δ 8.50 (d, J = 2.3 Hz, 1H), 8.49–8.45 (m, 1H), 7.55 (dt, J = 7.9, 2.0 Hz, 1H), 7.17 (d, J = 8.8 Hz, 2H), 7.10 (ddd, J = 7.9, 4.8, 0.8 Hz, 1H), 6.87 (d, J = 8.1 Hz, 2H), 6.29 (d, J = 8.2 Hz, 1H), 5.98 (s, 1H), 5.77 (dddd, J = 8.5, 6.9, 5.5, 1.6 Hz, 1H), 3.90–3.79 (m, 1H), 1.96 (d, J = 9.1 Hz, 1H), 1.92–1.87 (m, 1H), 1.74–1.55 (m, 3H), 1.51 (s, 3H), 1.45 (dd, J = 6.9, 1.2 Hz, 3H), 1.44–1.30 (m, 2H), 1.24 (s, 9H), 1.23–1.08 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 174.17, 168.17, 151.06, 150.92, 149.49, 138.40, 137.72, 132.44, 131.30, 131.17, 128.98, 125.80, 123.03, 64.28, 34.67, 33.00, 32.98, 31.36, 25.63, 24.86, 24.81, 14.17, 13.46. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C28H37N3NaO2 470.2778; found 470.2766.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl) methacrylamide (10a). Compound was made and purified using general procedure B, pale white solid 68% yield, 265 mg. 1H NMR (500 MHz, MeOD) δ 8.33 (d, J = 2.3 Hz, 1H), 8.31 (dd, J = 4.9, 1.6 Hz, 1H), 7.56 (dt, J = 7.9, 2.0 Hz, 1H), 7.23–7.15 (m, 3H), 7.05 (s, 2H), 6.10 (s, 1H), 5.01 (dt, J = 6.9, 1.3 Hz, 2H), 3.70 (tt, J = 10.9, 3.9 Hz, 1H), 1.90 (dd, J = 10.7, 3.8 Hz, 1H), 1.73 (s, 5H), 1.70–1.59 (m, 2H), 1.41–1.27 (m, 3H), 1.21 (s, 9H), 1.19–1.06 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 172.82, 167.95, 151.29, 151.19, 149.55, 140.42, 137.86, 137.64, 130.88, 129.32, 125.77, 122.97, 119.77, 63.47, 48.78, 34.63, 32.90 (d, J = 10.4 Hz), 31.32, 25.57, 24.83, 24.77, 20.42. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C27H35N3NaO2 456.2621; found 456.2620.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)cyclopent-1-ene-1-carboxamide (11a). Compound was made and purified using general procedure A, pale yellow solid 80% yield, 333 mg. 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 2.3 Hz, 1H), 8.46 (dd, J = 4.8, 1.7 Hz, 1H), 7.51 (dt, J = 8.0, 2.0 Hz, 1H), 7.21–7.16 (m, 2H), 7.08 (ddd, J = 7.9, 4.8, 0.8 Hz, 1H), 6.90 (d, J = 8.0 Hz, 2H), 6.28 (d, J = 8.0 Hz, 1H), 6.04 (s, 1H), 5.82 (d, J = 2.3 Hz, 1H), 3.89–3.78 (m, 1H), 2.19 (ddt, J = 7.7, 5.1, 2.5 Hz, 2H), 2.12 (tt, J = 6.7, 2.8 Hz, 2H), 2.02–1.93 (m, 1H), 1.92–1.84 (m, 1H), 1.73–1.54 (m, 4H), 1.44–1.29 (m, 2H), 1.25 (s, 9H), 1.23–1.07 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 168.92, 168.16, 151.57, 151.22, 149.46, 140.13, 139.09, 137.96, 137.69, 130.98, 129.50, 125.81, 122.94, 63.92, 48.74, 34.69, 33.80, 33.22, 32.95, 32.92, 31.35, 25.60, 24.84, 24.79, 23.29. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C29H37N3NaO2 482.2778; found 482.2781.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)cyclohex-1-ene-1-carboxamide (12a). Compound was made and purified using general procedure A, white solid 37% yield, 157 mg. 1H NMR (500 MHz, CDCl3) δ 8.50 (d, J = 2.3 Hz, 1H), 8.47 (dd, J = 4.8, 1.6 Hz, 1H), 7.55 (dt, J = 8.0, 2.0 Hz, 1H), 7.18 (d, J = 8.8 Hz, 2H), 7.11 (ddd, J = 7.9, 4.8, 0.8 Hz, 1H), 6.88 (d, J = 8.1 Hz, 2H), 6.30 (d, J = 8.2 Hz, 1H), 6.00 (s, 1H), 5.84 (dt, J = 3.8, 2.0 Hz, 1H), 3.90–3.79 (m, 1H), 1.99–1.80 (m, 5H), 1.75–1.54 (m, 6H), 1.44–1.29 (m, 4H), 1.25 (s, 9H), 1.24–1.13 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 173.52, 168.18, 151.11, 151.05, 149.50, 138.33, 137.79, 134.48, 133.17, 131.12, 129.08, 125.69, 123.02, 63.95, 48.72, 34.68, 33.01, 32.97, 31.37, 26.14, 25.64, 25.00, 24.85, 22.04, 21.45. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C30H39N3NaO2 496.2934; found 496.2931.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-ynamide (13a). Compound was made and purified using general procedure A, white solid 92% yield, 357 mg. 1H NMR (500 MHz, CDCl3) δ 8.44 (d, J = 4.9 Hz, 1H), 8.41 (d, J = 2.3 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 7.05 (dd, J = 8.0, 4.8 Hz, 1H), 6.97 (d, J = 8.0 Hz, 2H), 6.20 (s, 1H), 6.03 (s, 1H), 3.80 (dtd, J = 10.8, 7.2, 4.0 Hz, 1H), 1.96 (dq, J = 13.2, 4.8 Hz, 1H), 1.84 (d, J = 16.6 Hz, 1H), 1.75–1.62 (m, 5H), 1.58 (dd, J = 13.1, 4.1 Hz, 1H), 1.42–1.26 (m, 2H), 1.25 (s, 9H), 1.25–1.05 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.43, 155.41, 151.87, 151.20, 149.62, 138.15, 136.38, 130.34, 129.90, 125.70, 122.98, 92.12, 73.86, 62.23, 34.71, 32.87, 32.80, 31.31, 25.56, 24.85, 24.80. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C27H33N3NaO2 454.2465; found 454.2458.
2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-N-cyclohexyl-2-(pyridin-3-yl)acetamide (14a). To a solution of 5a (0.41 mmol, 150 mg) in DCM (4 mL) was added pyridine (0.56 mmol, 0.04 mL) and the solution cooled to 0 °C. 2-chloroethanesulfonyl chloride (0.49 mmol, 0.05 mL) was added dropwise and stirred for 1 h, after which pyridine (0.56 mmol, 0.04 mL) was added, and the solution was warmed to r.t. and stirred overnight. The reaction was monitored by TLC (1:1 DCM:EtOAc). The reaction was quenched with water and extracted with DCM (x2). The combined organic layers were washed with sat. NH4Cl, sat. NaHCO3 and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was further purified by column chromatography (0–4% (MeOH + 1%NH4OH)/DCM) to afford the pure product (66 mg, 35% yield) as a white powder. 1H NMR (500 MHz, DMSO) δ 8.35 (dd, J = 4.9, 1.6 Hz, 1H), 8.31 (s, 0H), 8.10 (d, J = 7.6 Hz, 1H), 7.36 (dt, J = 8.0, 2.0 Hz, 1H), 7.17–7.09 (m, 4H), 6.98 (dd, J = 16.5, 9.9 Hz, 1H), 6.06 (d, J = 9.9 Hz, 1H), 5.98 (d, J = 16.5 Hz, 1H), 5.81 (s, 1H), 3.57 (tdt, J = 11.0, 7.6, 3.7 Hz, 1H), 1.79–1.72 (m, 1H), 1.67 (dt, J = 12.9, 4.0 Hz, 1H), 1.64–1.47 (m, 2H), 1.33–1.19 (m, 2H), 1.17 (s, 9H), 1.16–0.92 (m, 2H). 13C NMR (126 MHz, DMSO) δ 167.43, 150.53, 150.51, 148.97, 136.88, 136.07, 133.56, 131.92, 131.00, 127.18, 124.92, 122.94, 63.05, 47.95, 34.19, 32.07, 31.88, 30.94, 25.09, 24.41, 24.31. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C25H33N3NaO3S 478.2135; found 478.2127.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-2-fluoroacetamide (15a). Compound was made and purified using general procedure A, pale yellow solid 70% yield, 270 mg. 1H NMR (500 MHz, CDCl3) δ 8.47 (dd, J = 4.9, 1.7 Hz, 1H), 8.43 (d, J = 2.3 Hz, 1H), 7.43 (dt, J = 7.9, 2.0 Hz, 1H), 7.24 (s, 2H), 7.06 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 6.04 (s, 1H), 5.87 (d, J = 7.8 Hz, 1H), 4.64 (d, J = 3.1 Hz, 1H), 4.55 (d, J = 3.3 Hz, 1H), 3.86–3.75 (m, 1H), 2.02–1.95 (m, 1H), 1.89–1.81 (m, 1H), 1.74–1.55 (m, 3H), 1.44–1.28 (m, 2H), 1.25 (s, 9H), 1.22–1.02 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 168.05, 167.89, 167.48, 153.00, 151.45, 149.99, 138.13, 133.76, 129.97, 129.82, 126.64, 123.12, 78.70 (d, J = 178.1 Hz), 62.38, 49.16, 34.84, 32.94 (d, J = 9.3 Hz), 31.31, 25.57, 24.90 (d, J = 6.9 Hz). HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C25H32FN3NaO2 448.2371; found 448.2366.
N-(4-(tert-butyl)phenyl)-2-chloro-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl) acetamide (16a). Compound was made and purified using general procedure A, yellow solid 93% yield, 370 mg. 1H NMR (500 MHz, CDCl3) δ 8.47 (dd, J = 4.8, 1.6 Hz, 1H), 8.43 (d, J = 2.3 Hz, 1H), 7.44 (dt, J = 7.9, 2.0 Hz, 1H), 7.26 (s, 3H), 7.07 (ddd, J = 7.8, 4.9, 0.8 Hz, 1H), 5.99 (s, 1H), 5.88 (s, 1H), 3.85 (s, 2H), 3.84–3.78 (m, 1H), 1.98 (dd, J = 12.6, 4.2 Hz, 1H), 1.85 (dd, J = 12.6, 4.2 Hz, 1H), 1.74–1.63 (m, 2H), 1.59 (dt, J = 12.8, 3.8 Hz, 1H), 1.43–1.28 (m, 2H), 1.26 (s, 9H), 1.22–1.03 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.43, 167.42, 152.83, 151.27, 149.75, 138.27, 135.35, 130.28, 129.73, 126.61, 123.16, 63.16, 49.11, 42.58, 34.84, 32.97, 32.90, 31.33, 25.57, 24.91, 24.85. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C25H32ClN3NaO2 464.2075; found 464.2087.
Ethyl (E)-4-((4-(tert-butyl)phenyl)(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)amino)-4-oxobut-2-enoate (17a). Compound was made and purified using general procedure A, white powder 52% yield, 170 mg. 1H NMR (500 MHz, CDCl3) δ 8.51–8.38 (m, 2H), 7.49 (dt, J = 8.1, 2.0 Hz, 1H), 7.23 (d, J = 8.1 Hz, 2H), 7.09 (dd, J = 8.0, 4.8 Hz, 1H), 6.85 (d, J = 15.3 Hz, 1H), 6.72 (d, J = 15.3 Hz, 1H), 6.24 (d, J = 8.1 Hz, 1H), 6.10 (s, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.80 (dtd, J = 10.8, 7.2, 4.0 Hz, 1H), 1.98–1.90 (m, 1H), 1.89–1.79 (m, 1H), 1.72–1.53 (m, 3H), 1.33 (ddd, J = 13.0, 10.0, 3.3 Hz, 1H), 1.25 (s, 9H), 1.20 (t, J = 7.1 Hz, 3H), 1.10 (ddt, J = 23.0, 15.4, 10.8 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 167.37, 165.46, 165.04, 152.42, 150.54, 148.89, 138.83, 135.41, 133.89, 132.15, 130.95, 129.77, 126.48, 123.26, 63.08, 61.13, 49.00, 34.77, 32.90, 32.84, 31.28, 25.53, 24.86, 24.80, 14.11. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C29H37N3NaO4 514.2676; found 514.2691.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-2-oxopropanamide (18a). To a solution of 4-tert-butylaniline (0.10 mL, 0.67 mmol, 1.0 eq.) in MeOH (2 mL) was added 3-pyridine carboxaldehyde (0.06 mL, 0.67 mmol, 1.0 eq.) and the solution stirred at room temperature for 30 min. The solution was cooled to 0 °C, and pyruvic acid (0.06 mL, 0.80 mmol, 1.2 eq.) and cyclohexyl isocyanide (0.10 mL, 0.80 mmol, 1.2 eq.) were added in quick succession. The solution was slowly warmed to room temperature and stirred overnight. The crude reaction mixture was evaporated in vacuo and purified by column chromatography (1:1 Hex:EtOAc) to afford the product (105 mg, 36%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 8.69 (s, 1H), 8.56 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 7.7 Hz, 1H), 7.23 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 8.1 Hz, 2H), 6.49 (d, J = 8.0 Hz, 1H), 6.15 (s, 1H), 3.80 (tdd, J = 10.7, 6.7, 4.0 Hz, 1H), 2.19 (s, 3H), 1.97–1.79 (m, 2H), 1.69 (ddt, J = 17.1, 13.1, 4.0 Hz, 2H), 1.59 (dt, J = 12.8, 3.9 Hz, 1H), 1.41–1.25 (m, 2H), 1.24 (s, 9H), 1.16 (dtd, J = 16.2, 13.6, 12.7, 9.9 Hz, 2H).13C NMR (126 MHz, CDCl3) δ 197.49, 168.14, 166.68, 152.69, 150.19, 148.63, 139.25, 134.15, 130.64, 129.82, 126.29, 123.61, 62.36, 49.22, 34.77, 32.85, 32.81, 31.26, 27.84, 25.51, 24.87, 24.81. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C26H33N3NaO3 458.2412; found 458.2421.
Ethyl (4-(tert-butyl)phenyl)(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)carbamate (19a). To a solution of 5a (0.41 mmol, 150 mg) in DCM (8 mL) was added pyridine (0.82 mmol, 0.06 mL) and the solution was cooled to 0 °C. Ethyl chloroformate (0.49 mmol, 0.05 mL) was added dropwise and stirred at 0 °C for 1 h, then room temperature overnight. The reaction was quenched with water and extracted twice with DCM. The combined organic layers were washed with sat. NH4Cl, sat. NaHCO3 and brine, then dried over Na2SO4 and concentrated in vacuo. The crude residue was further purified by column chromatography (0–80% EtOAc/Hex) to afford the desired product (136 mg, 76% yield) as a white powder. 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 8.51–8.46 (m, 1H), 7.66 (dt, J = 8.1, 1.9 Hz, 1H), 7.23 (td, J = 6.0, 5.5, 2.3 Hz, 3H), 6.97 (d, J = 8.5 Hz, 2H), 6.24 (d, J = 8.1 Hz, 1H), 5.72 (s, 1H), 4.15 (qd, J = 7.1, 2.7 Hz, 2H), 3.84 (dddd, J = 14.5, 10.5, 7.9, 3.9 Hz, 1H), 1.99–1.83 (m, 2H), 1.69 (tt, J = 12.5, 3.9 Hz, 2H), 1.60 (dt, J = 12.9, 3.9 Hz, 1H), 1.43–1.30 (m, 2H), 1.26 (s, 9H), 1.22–1.08 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 167.76, 156.29, 150.83, 148.98, 147.37, 139.54, 137.15, 132.52, 128.33, 126.00, 123.58, 64.95, 62.65, 48.91, 34.67, 32.97, 32.93, 31.38, 25.57, 24.84, 24.82, 14.64. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C26H35N3NaO3 460.2571; found 460.2579.
N-(4-(tert-butyl)phenyl)-2-cyano-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)acetamide (20a). Compound was made and purified using general procedure B, white solid 84% yield, 328 mg. 1H NMR (500 MHz, CDCl3) δ 8.52–8.45 (m, 1H), 8.41 (s, 1H), 7.44–7.38 (m, 2H), 7.10–7.04 (m, 2H), 6.47 (s, 1H), 5.98 (d, J = 2.1 Hz, 1H), 5.68 (s, 1H), 3.86–3.75 (m, 1H), 3.27–3.20 (m, 2H), 1.97 (d, J = 12.6 Hz, 1H), 1.84 (d, J = 13.0 Hz, 2H), 1.68–1.56 (m, 2H), 1.40–1.28 (m, 2H), 1.25 (s, 9H), 1.23–0.99 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 166.87, 162.92, 152.91, 151.14, 149.87, 137.79, 135.02, 129.75, 129.52, 123.02, 113.69, 62.87, 49.04, 34.61, 32.70, 32.64, 31.04, 26.19, 25.28, 24.67, 24.60.
2-((4-(tert-butyl)phenyl)(cyanomethyl)amino)-N-cyclohexyl-2-(pyridin-3-yl)acetamide (21a). To a solution of 2-((4-(tert-butyl)phenyl)amino)acetonitrile (0.27 mmol, 50 mg) and 3-pyridinecarboxaldehyde (0.27 mmol, 0.03 mL) in MeOH (3 mL) was added cyclohexyl isocyanide (0.27 mmol, 0.04 mL) and phosphoric acid (0.05 mmol, 0.004 mL, 85%), and the solution stirred at room temperature overnight. The solvent was evaporated under a stream of air, and the crude reaction mixture was suspended in a small amount of EtOAc. Hexanes was added and the precipitate was collected by filtration and rinsed with hexanes and acetone. The precipitate was dried over vacuum to afford the desired product (74 mg, 68% yield) as a white powder. 1H NMR (500 MHz, CDCl3) δ 9.07 (s, 1H), 8.61 (s, 1H), 8.32 (d, J = 7.9 Hz, 1H), 7.61 (t, J = 6.6 Hz, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 7.03 (d, J = 7.8 Hz, 1H), 5.57 (s, 1H), 4.16 (d, J = 17.9 Hz, 1H), 4.01 (d, J = 17.8 Hz, 1H), 1.98–1.67 (m, 2H), 1.68–1.49 (m, 4H), 1.45–1.28 (m, 1H), 1.27 (s, 9H), 1.24–0.64 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 166.94, 147.84, 143.94, 126.95, 125.47, 120.77, 115.57, 65.98, 48.79, 42.14, 34.47, 32.71, 32.32, 31.42, 31.21, 25.44, 24.68, 24.61.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-3,3,3-trifluoro-2-oxopropanamide (22a). To a solution of 4-tBu-aniline (0.62 mmol, 0.1 mL) in MeOH was added 3-Py-carboxaldehyde (0.62 mmol, 0.06 mL) and stirred for 30 min. The solution was cooled to 0 °C and trifluoropyruvic acid (0.62 mmol, 100 mg) and cyclohexyl isocyanide (0.62 mmol, 0.08 mL) were added. The solution was slowly warmed to r.t. and stirred overnight. The solvent was evaporated under a stream of air, and the crude reaction mixture was suspended in a small amount of EtOAc. Hexanes was added and the precipitate was collected by filtration and rinsed with hexanes and acetone. The precipitate was dried over vacuum to afford the desired product (87 mg, 29% yield) as a white powder. 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1H), 8.55–8.51 (m, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.23 (d, J = 8.6 Hz, 3H), 7.03 (s, 2H), 6.10 (s, 2H), 3.82 (tdt, J = 10.9, 7.8, 3.9 Hz, 1H), 1.90 (dd, J = 49.5, 12.9 Hz, 2H), 1.75–1.54 (m, 4H), 1.43–1.27 (m, 1H), 1.23 (s, 9H), 1.21–1.02 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 165.76, 162.63, 153.82, 150.12, 148.74, 139.63, 132.24, 130.28, 130.01, 126.64, 123.76, 62.36, 49.38, 34.89, 32.81, 32.79, 31.33, 31.21, 31.17, 25.49, 24.88, 24.83. 19F NMR (471 MHz, CDCl3) δ −74.82.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-4-hydroxybut-2-ynamide (23a). To a solution of 4-tBu-aniline (0.31 mmol, 0.05 mL) in MeOH was added 3-Py-carboxaldehyde (0.31 mmol, 0.03 mL) and stirred for 30 min. 4-hydroxy-butynoic acid (0.31 mmol, 31 mg) and cyclohexyl isocyanide (0.31 mmol, 0.04 mL) were added and the solution stirred at room temperature overnight. The solvent was evaporated under a stream of air, and the crude reaction mixture was suspended in a small amount of EtOAc. Hexanes was added and the precipitate was collected by filtration and further rinsed with hexanes. The precipitate was dried over vacuum to afford the desired product (60 mg, 43% yield) as a white powder. 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 2.3 Hz, 1H), 8.44 (dd, J = 4.8, 1.7 Hz, 1H), 7.44 (dt, J = 8.0, 2.0 Hz, 1H), 7.22 (d, J = 8.8 Hz, 2H), 7.06 (dd, J = 8.0, 4.8 Hz, 1H), 7.03 (d, J = 7.9 Hz, 2H), 6.17 (d, J = 8.0 Hz, 1H), 6.07 (s, 1H), 4.05 (s, 2H), 3.84–3.73 (m, 1H), 2.45 (s, 1H), 1.96 (dd, J = 12.5, 4.1 Hz, 1H), 1.84 (dd, J = 12.1, 4.2 Hz, 1H), 1.74–1.53 (m, 2H), 1.43–1.27 (m, 3H), 1.25 (s, 9H), 1.23–1.00 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.25, 154.75, 152.38, 151.32, 149.75, 138.19, 135.94, 130.27, 130.25, 125.80, 123.11, 92.47, 78.94, 62.16, 50.59, 49.09, 34.78, 32.88, 32.83, 31.36, 25.57, 24.92, 24.85. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C27H33N3O3 448.2595; found 448.2592.
N-cyclohexyl-2-(2-oxoazetidin-1-yl)-2-(pyridin-3-yl)acetamide (24a). In a 6-dram vial equipped with a stir bar 3-Pyridinecarboxaldehyde (214 mg, 2.0 mmol, 1.0 eq.), glycinamide hydrochloride (221 mg, 2.0 mmol, 1.0 eq.), and triethylamine (202 mg, 2.0 mmol, 1.0 eq.), were mixed in methanol (5 mL). The obtained solution was stirred for 15 min at room temperature. Cyclohexyl isocyanide (218 mg, 2.0 mmol, 1.0 eq.), and acetic acid (120 mg, 2.0 mmol, 1.0 eq.) were then added to the reaction mixture and the walls of the vial were washed with additional MeOH (5 mL). The reaction mixture was stirred at room temperature overnight. The crude reaction mixture was evaporated in vacuo and redissolved in EtOAc (50 mL). The organic layer was extracted with water (3 x 100 mL). The obtained organic layer was dried over Na2SO4 and evaporated in vacuo. The obtained crude solid was triturated with hexanes (5 mL) and filtered. The obtained powder was further washed with hexanes (2 x 5 mL). The product was obtained as pale-yellow solid, 428 mg 79%. 1H NMR (500 MHz, CDCl3) δ 8.59 (s, 2H), 7.75 (dd, J = 8.0, 2.1 Hz, 1H), 7.32 (dd, J = 8.0, 4.9 Hz, 1H), 6.40 (d, J = 7.9 Hz, 1H), 5.34 (s, 0H), 3.76 (dtt, J = 11.4, 8.5, 4.0 Hz, 1H), 3.61 (td, J = 5.6, 2.8 Hz, 1H), 3.20 (td, J = 5.7, 2.8 Hz, 1H), 3.05–2.97 (m, 1H), 2.95–2.87 (m, 1H), 1.94–1.83 (m, 2H), 1.73–1.55 (m, 2H), 1.43–1.26 (m, 3H), 1.22–0.98 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 168.10, 166.82, 150.04, 149.62, 135.82, 130.98, 123.90, 58.10, 48.97, 39.28, 36.53, 32.89, 32.85, 29.84, 25.52, 24.82, 24.78.
2-((cyanomethyl)amino)-N-cyclohexyl-2-(pyridin-3-yl)acetamide (25a). In a 6-dram vial equipped with a stir bar 3-pyridinecarboxaldehyde (107 mg,1.0 mmol, 1.0 eq.) and β-alanine (89 mg, 1.0 mmol, 1.0 eq.) were mixed together in MeOH (4 mL). The obtained solution was stirred for 30 min at room temperature. Cyclohexyl isocyanide (109 mg, 1.0 mmol, 1.0 eq.) was added to the reaction mixture and the walls of the vial were washed with 1 mL of MeOH. The obtained reaction mixture was stirred at room temperature overnight. The crude reaction mixture was evaporated in vacuo and redissolved in DCM. The obtained crude solution was deposited on silica. It was then purified using flash column chromatography using DCM/MeOH (gradient 0 → 5%) as eluent. The product was obtained as colorless oil, 156 mg 54%. 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 2.4 Hz, 1H), 8.62 (dd, J = 4.8, 1.7 Hz, 1H), 7.75 (dt, J = 7.9, 2.0 Hz, 1H), 7.34 (dd, J = 7.9, 4.8 Hz, 1H), 6.02 (d, J = 8.4 Hz, 1H), 4.40 (s, 1H), 3.82–3.71 (m, 1H), 3.64 (d, J = 17.5 Hz, 1H), 3.45 (d, J = 17.3 Hz, 1H), 2.53 (s, 1H), 1.85 (ddd, J = 17.0, 12.4, 4.4 Hz, 2H), 1.64 (dtd, J = 25.9, 9.0, 4.7 Hz, 4H), 1.40–1.28 (m, 2H), 1.20–1.02 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 168.45, 150.57, 149.43, 135.66, 133.25, 124.24, 116.87, 63.55, 48.67, 35.50, 33.06, 33.02, 25.49, 24.83. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C15H20N4NaO 295.1529; found 295.1523.
(E)-N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-enamide (8b). Compound was made and purified using general procedure A, white solid 54% yield, 198 mg. 1H NMR (500 MHz, CDCl3) δ 8.48–8.41 (m, 2H), 7.43 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 8.2 Hz, 2H), 7.05 (dd, J = 8.0, 4.8 Hz, 1H), 7.01–6.95 (m, 1H), 6.91 (s, 1H), 6.35 (s, 1H), 6.07 (s, 1H), 5.68 (dd, J = 15.1, 1.7 Hz, 1H), 1.75 (dd, J = 7.1, 1.7 Hz, 3H), 1.39 (s, 9H), 1.30 (s, 9H).13C NMR (126 MHz, CDCl3) δ 168.38, 166.89, 151.78, 151.38, 149.47, 142.97, 138.10, 136.53, 130.94, 129.85, 126.20, 122.77, 63.16, 51.73, 34.76, 31.38, 28.81, 18.21. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C25H33N3NaO2 430.2438; found 430.2450.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-ynamide (13b). Compound was made and purified using general procedure A, white solid 82% yield, 300 mg. 1H NMR (500 MHz, CDCl3) δ 8.45 (dd, J = 4.8, 1.5 Hz, 1H), 8.43 (d, J = 2.3 Hz, 1H), 7.43 (dt, J = 8.1, 2.0 Hz, 1H), 7.25–7.20 (m, 2H), 7.05 (dd, J = 8.0, 4.8 Hz, 1H), 6.97 (d, J = 8.0 Hz, 2H), 6.09 (s, 1H), 5.96 (s, 1H), 1.68 (s, 3H), 1.36 (s, 9H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 167.54, 155.43, 151.94, 151.27, 149.67, 138.25, 136.37, 130.29, 129.91, 125.77, 122.95, 92.21, 73.90, 62.66, 51.98, 34.76, 31.35, 28.75. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C25H31N3NaO2 428.2308; found 428.2307.
N-(4-(tert-butyl)phenyl)-N-(2-(tert-butylamino)-2-oxo-1-(pyridin-3-yl)ethyl)acrylamide (6b). Compound was made and purified using general procedure C, white solid 42% yield, 150 mg. 1H NMR (500 MHz, CDCl3) δ 8.46–8.42 (m, 2H), 7.43 (dt, J = 8.0, 2.0 Hz, 1H), 7.24 (d, J = 8.3 Hz, 2H), 7.04 (ddd, J = 8.0, 4.9, 0.9 Hz, 1H), 6.91 (s, 1H), 6.40 (dd, J = 16.8, 2.0 Hz, 1H), 6.20 (s, 1H), 6.06 (s, 1H), 5.98 (dd, J = 16.8, 10.3 Hz, 1H), 5.55 (dd, J = 10.3, 2.0 Hz, 1H), 1.37 (s, 9H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.13, 166.58, 152.00, 151.38, 149.58, 138.15, 136.19, 130.77, 129.89, 128.77, 128.60, 126.25, 122.87, 63.18, 51.84, 34.77, 31.37, 28.80. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C24H31N3NaO2 416.2308; found 416.2291.
N-(tert-butyl)-2-(N-(4-(tert-butyl)phenyl)acetamido)-2-(pyridin-3-yl)acetamide (4b). Compound was made and purified using general procedure A, white solid 84% yield, 290 mg. 1H NMR (500 MHz, CDCl3) δ 8.43 (dd, J = 4.8, 1.6 Hz, 1H), 8.40 (s, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.22 (d, J = 8.2 Hz, 2H), 7.01 (dd, J = 7.6, 5.3 Hz, 1H), 6.91 (s, 1H), 6.04 (s, 1H), 5.98 (s, 1H), 1.87 (s, 3H), 1.36 (s, 9H), 1.25 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 171.75, 168.32, 151.86, 151.45, 149.59, 138.13, 137.31, 130.88, 129.65, 126.24, 122.84, 62.64, 51.80, 34.72, 31.36, 28.79, 23.36. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C23H31N3NaO2 404.2308; found 404.2307.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclopentylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-ynamide (13c). Compound was made and purified using general procedure A, white solid 94% yield, 354 mg. 1H NMR (500 MHz, CDCl3) δ 8.46 (dd, J = 4.9, 1.6 Hz, 1H), 8.44 (d, J = 2.3 Hz, 1H), 7.50–7.44 (m, 1H), 7.23 (d, J = 8.8 Hz, 2H), 7.07 (dd, J = 8.0, 4.8 Hz, 1H), 6.97 (d, J = 8.0 Hz, 2H), 6.20 (s, 1H), 6.02 (s, 1H), 4.23 (h, J = 6.8 Hz, 1H), 2.02 (dd, J = 13.0, 6.2 Hz, 1H), 1.95 (q, J = 2.7 Hz, 1H), 1.68 (s, 3H), 1.64–1.56 (m, 4H), 1.49–1.42 (m, 1H), 1.39–1.34 (m, 1H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 167.92, 155.47, 151.97, 151.06, 149.54, 138.34, 136.39, 130.32, 129.87, 125.79, 123.06, 92.26, 73.85, 62.23, 51.89, 34.76, 33.08, 33.01, 31.34, 23.87, 23.84, 4.06. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C26H32N3O2 418.24890; found 418.24854.
N-(2-(benzylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-(4-(tert-butyl)phenyl)but-2-ynamide (13d). Compound was made and purified using general procedure A, pale yellow solid 86% yield, 340 mg. 1H NMR (500 MHz, CDCl3) δ 8.43 (d, J = 4.7 Hz, 1H), 8.40 (t, J = 1.6 Hz, 2H), 7.45 (d, J = 7.8 Hz, 1H), 7.33–7.16 (m, 6H), 7.05 (dd, J = 8.0, 4.8 Hz, 1H), 6.94 (d, J = 8.1 Hz, 2H), 6.66 (d, J = 8.5 Hz, 1H), 6.05 (s, 1H), 4.55–4.43 (m, 2H), 1.66 (s, 3H), 1.25 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.38, 155.47, 151.96, 151.08, 149.59, 138.41, 137.91, 136.33, 130.18, 129.93, 128.85, 127.84, 127.64, 125.78, 123.11, 92.27, 73.83, 62.35, 44.02, 34.74, 31.65, 31.33, 4.04. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C28H30N3O2 440.23325; found 440.23303.
N-(4-(tert-butyl)phenyl)-2-chloro-N-(2-(cyclopentylamino)-2-oxo-1-(pyridin-3-yl)ethyl)acetamide (16b). Compound was made and purified using general procedure A, pale yellow solid 88% yield, 340 mg. 1H NMR (500 MHz, CDCl3) δ 8.46 (dd, J = 4.9, 1.7 Hz, 1H), 8.42 (d, J = 2.3 Hz, 1H), 7.44 (dd, J = 8.0, 2.1 Hz, 1H), 7.26 (s, 3H), 7.06 (dd, J = 8.0, 4.8 Hz, 1H), 6.06 (d, J = 7.3 Hz, 1H), 5.99 (s, 1H), 4.23 (h, J = 6.8 Hz, 1H), 3.85 (s, 2H), 2.10–1.99 (m, 1H), 1.98–1.90 (m, 1H), 1.70–1.52 (m, 4H), 1.51–1.40 (m, 1H), 1.33 (dq, J = 13.6, 6.8, 5.8 Hz, 1H), 1.25 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 167.93, 167.42, 152.82, 151.20, 149.68, 138.30, 135.29, 130.25, 129.75, 126.59, 123.16, 63.03, 51.94, 42.61, 34.83, 33.08, 33.03, 31.32, 23.88, 23.86. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C24H31N3O2Cl 428.20993; found 428.21001.
N-benzyl-2-(N-(4-(tert-butyl)phenyl)-2-chloroacetamido)-2-(pyridin-3-yl)acetamide (16c). Compound was made and purified using general procedure A, yellow solid 91% yield, 370 mg. 1H NMR (500 MHz, CDCl3) δ 8.45 (dd, J = 4.8, 1.6 Hz, 1H), 8.40 (d, J = 2.3 Hz, 1H), 7.43 (dt, J = 8.0, 2.1 Hz, 1H), 7.35–7.13 (m, 9H), 7.04 (ddd, J = 8.1, 4.9, 0.9 Hz, 1H), 6.55 (s, 1H), 6.06 (s, 1H), 4.49 (qd, J = 14.8, 5.8 Hz, 2H), 3.85 (d, J = 1.5 Hz, 2H), 1.25 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.45, 167.46, 152.84, 151.36, 149.93, 138.24, 137.84, 135.18, 129.95, 129.79, 128.86, 127.83, 127.70, 126.59, 123.14, 63.10, 44.07, 42.67, 34.82, 31.31. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C26H29N3O2Cl 450.19428; found 450.19392.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclopentylamino)-2-oxo-1-(pyridin-3-yl)ethyl)cyclopent-1-ene-1-carboxamide (11b). Compound was made and purified using general procedure B, white solid 67% yield, 270 mg. 1H NMR (500 MHz, CDCl3) δ 8.51 (d, J = 2.3 Hz, 1H), 8.49 (dd, J = 4.8, 1.7 Hz, 1H), 7.54 (dt, J = 8.0, 2.0 Hz, 1H), 7.25–7.19 (m, 2H), 7.11 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 6.92 (d, J = 8.0 Hz, 2H), 6.35 (d, J = 7.4 Hz, 1H), 6.05 (s, 1H), 5.85 (p, J = 2.2 Hz, 1H), 4.28 (q, J = 6.7 Hz, 1H), 2.22 (ddd, J = 7.6, 6.1, 2.5 Hz, 2H), 2.15 (tt, J = 6.4, 2.2 Hz, 2H), 2.10–1.93 (m, 2H), 1.65–1.64 (m, 6H), 1.53–1.35 (m, 2H), 1.28 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.81, 168.55, 151.49, 151.12, 149.40, 140.09, 138.96, 137.84, 137.57, 130.79, 129.37, 125.73, 122.83, 63.78, 51.62, 34.60, 33.71, 33.13, 33.01, 32.97, 31.25, 23.73, 23.71, 23.18. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C28H36N3O2 446.28020; found 446.28024.
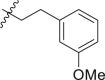
N-[2-(4-methoxy) ethyl] formamide (26b). Compound was made and purified using general procedure D. Light yellow liquid with 95% yield, 852.0 mg. Rf = 0.07 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 8.10 (d, J = 1.7 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.92 (bs, 1H), 3.79 (s, 3H), 3.52 (q, J = 6.2 Hz, 2H), 2.78 (t, J = 7.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 164.55, 161.26, 158.35, 129.70, 114.09, 55.28, 39.40, 34.60. HRMS (ESI) m/z: [M + Na]+ calculated for C10H13NNaO2 202.0838; found 202.0841.
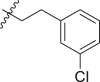
N-[2-(3-chlorophenyl) ethyl] formamide (26c). Compound was made and purified using general procedure D. Light yellow liquid with 98% yield, 899.3 mg. Rf = 0.12 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 8.09 (d, J = 1.8 Hz, 1H), 7.31–7.15 (m, 3H), 7.09 (dt, J = 7.2, 1.5 Hz, 1H), 6.10 (bs, 1H), 3.53 (q, J = 6.8 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 161.35, 140.67, 134.36, 129.95, 128.87, 126.98, 126.82, 38.64, 35.20. HRMS (ESI) m/z: [M + Na]+ calculated for C9H10ClNNaO 206.0343; found 206.0346.
N-[2-(3-methoxy) ethyl] formamide (26d). Compound was made and purified using general procedure D. Light yellow liquid with 40% yield, 331.0 mg. Rf = 0.18 (2:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 8.15 (d, J = 1.7 Hz, 1H), 7.27–7.24 (m, 1H), 6.82–6.79 (m, 2H), 6.78–6.75 (m, 1H), 5.64 (bs, 1H), 3.82 (s, 3H), 3.59 (q, J = 6.8 Hz, 2H), 2.84 (t, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 161.12, 159.89, 140.09, 129.75, 121.05, 114.54, 111.93, 55.21, 39.05, 35.54. HRMS (ESI) m/z: [M + Na]+ calculated for C10H13NNaO2 202.0838; found 202.0831.
N-(3-phenylpropyl)formamide (26e). Compound was made and purified using general procedure D. Colourless liquid with 99% yield, 803 mg. Rf = 0.31 (4:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 8.15 (s, 1H), 7.33–7.28 (m, 2H), 7.23–7.17 (m, 3H), 6.07 (bs, 1H), 3.32 (q, J = 7.2 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 1.89 (p, J = 7.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 161.43, 141.23, 128.51, 128.38, 126.09, 37.80, 33.17, 31.10. HRMS (APCI) m/z: [M + H]+ calculated for C10H14NO 164.1070; found 164.1070.
N-(1-benzylpiperidin-4-yl)formamide (26f). Compound was made and purified using general procedure D. Orange viscous liquid with more than 99% yield, 1270 mg. Rf = 0.05 (4:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 8.12 (s, 1H), 7.35–7.30 (m, 4H), 7.28–7.25 (m,1H), 5.71 (bs, 1H), 3.94–3.87 (m, 1H), 3.51 (s, 2H), 2.83 (d, J = 12.2 Hz, 2H), 2.14 (t, J = 11.4 Hz, 2H), 1.93 (d, J = 12.7 Hz, 2H), 1.51 (dtd, J = 12.7, 11.0, 3.8 Hz, 2H).). 13C NMR (126 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 160.55, 138.18, 129.13, 128.26, 127.12, 63.00, 52.11, 45.42, 32.13. HRMS (ESI) m/z: [M + H]+ calculated for C13H19N2O 219.1492; found 219.1491.
(S)–N-(1-phenylethyl)formamide (26g). Compound was made and purified using general procedure D. Light yellow liquid with more than 99% yield, 763.7 mg. Rf = 0.27 (2:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 8.15 (s, 1H), 7.40–7.25 (m, 5H), 6.17 (s, 1H), 5.21 (p, J = 7.1 Hz, 1H), 1.52 (dd, J = 7.0, 1.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) mixture of rotamers is observed, major rotamer is given: δ 160.34, 142.63, 128.74, 127.53, 126.16, 47.61, 21.76. HRMS (ESI) m/z: [M + Na]+ calculated for C9H11NNaO 172.0733; found 172.0735.
1-(2-isocyanoethyl)-4-methoxybenzene (27b). Compound was made and purified using general procedure E. Dark red liquid with 73% yield, 228.6 mg. Rf = 0.87 (3:1 Et2O:DCM). 1H NMR (500 MHz, CDCl3): δ 7.17 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.6 Hz, 2H), 3.82 (s, 3H), 3.59 (tt, J = 7.1, 1.8 Hz, 2H), 2.95 (tt, J = 7.1, 2.1 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 158.83, 156.43 (t, J = 5.4 Hz), 129.74, 128.72, 114.20, 55.30, 43.28 (t, J = 6.3 Hz), 34.88. HRMS (ESI) m/z: [M + Na]+ calculated for C10H11NNaO 184.0733; found 184.0730.
1-chloro-3-(2-isocyanoethyl)benzene (27c). Compound was made and purified using general procedure E. Dark red liquid with 83% yield, 273 mg. Rf = 0.84 (3:1 Et2O:DCM). 1H NMR (500 MHz, CDCl3): δ 7.32–7.27 (m, 2H), 7.25 (s, 1H), 7.15–7.11 (m, 1H),f 3.62 (tt, J = 7.0, 1.9 Hz, 2H), 2.96 (tt, J = 7.0, 2.1 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 157.14 (t, J = 5.4 Hz), 138.54, 134.61, 130.10, 128.81, 127.58, 126.96, 42.67 (t, J = 6.6 Hz), 35.23. HRMS (ESI) m/z: [M + Na]+ calculated for C9H8ClNNa 188.0237; found 188.0232.
1-(2-isocyanoethyl)-3-methoxybenzene (27d). Compound was made and purified using general procedure E. Orange liquid with 79% yield, 256 mg. Rf = 0.95 (2:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 7.28 (t, J = 8.0 Hz, 1H), 6.86–6.83 (m, 2H), 6.79 (t, J = 2.1 Hz, 1H), 3.83 (s, 3H), 3.63 (tt, J = 7.2, 1.9 Hz, 2H), 2.99 (tt, J = 7.1, 2.0 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 159.89,156.62 (t, J = 5.3 Hz), 138.19, 129.84, 120.94, 114.52, 112.56, 55.24, 42.89 (t, J = 6.6 Hz), 35.74. HRMS (ESI) m/z: [M + Na]+ calculated for C10H11NNaO 184.0733; found 184.0730.
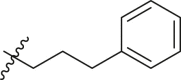
(3-isocyanopropyl)benzene (27e). Compound was made and purified using general procedure E. Orange liquid with 71% yield, 196 mg. Rf = 0.89 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 7.35–7.32 (m, 2H), 7.27–7.21 (m, 3H), 3.39 (tt, J = 6.5, 1.8 Hz 2H), 2.82 (t, J = 7.4 Hz, 2H), 2.07–1.98 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 156.28 (t, J = 5.6 Hz), 139.82, 128.67, 128.49, 126.46, 40.70 (t, J = 6.3 Hz), 32.20, 30.56. HRMS (APCI) m/z: [M + H]+ calculated for C10H12N 146.0964; found 146.0967.
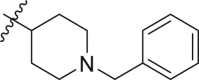
1-benzyl-4-isocyanopiperidine (27f). Compound was made and purified using general procedure E. Orange viscous liquid with 50% yield, 300 mg (3 mmol 2 g used). Rf = 0.74 (3:1 Et2O:DCM). 1H NMR (500 MHz, CDCl3): δ 7.47–7.21 (m, 5H), 3.68 (s, 1H), 3.53 (s, 2H), 2.67 (s, 2H), 2.35 (s, 2H), 2.01–1.95 (m, 2H), 1.93–1.83 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 155.19 (t, J = 5.5 Hz), 138.12, 129.01, 128.31, 127.18, 62.98, 49.74, 31.91. HRMS (ESI) m/z: [M + H]+ calculated for C13H17N2 201.1386; found 201.1389.
(S)-(1-isocyanoethyl)benzene (27g). Compound was made and purified using general procedure E. Dark red liquid with 74% yield, 198.5 mg. Rf = 0.92 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 7.48–7.33 (m, 5H), 4.85 (qt, J = 6.9, 1.9 Hz, 1H), 1.71 (dt, J = 6.9, 2.2 Hz 3H). 13C NMR (126 MHz, CDCl3): δ 156.37 (t, J = 4.9 Hz), 138.57, 128.96, 128.31, 125.41, 53.88, 53.83 (t, J = 6.1 Hz), 53.78, 25.15. HRMS (APCI) m/z: [M + H]+ calculated for C9H10N 132.0808; found 132.0810.
2-((4-(tert-butyl)phenyl)amino)-N-(4-methoxyphenethyl)-2-(pyridin-3-yl)acetamide (28b). Compound was made and purified using general procedure F. White solid with 42% yield, 176 mg. Rf = 0.2 (2:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 8.66 (d, J = 2.3 Hz, 1H), 8.60 (dd, J = 4.9, 1.6 Hz, 1H), 7.60 (dt, J = 7.9, 1.8 Hz, 1H), 7.27 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 6.84 (t, J = 6.2 Hz, 1H), 6.77 (d, J = 8.5 Hz, 2H), 6.60 (d, J = 8.6 Hz, 2H), 4.74 (d, J = 2.3 Hz, 1H), 4.28 (s, 1H), 3.80 (s, 3H), 3.56 (ddt, J = 46.9, 13.6, 6.9 Hz, 2H), 2.73 (ddt, J = 59.3, 14.1, 6.7 Hz, 2H), 1.31 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 170.25, 158.27, 149.90, 149.08, 143.78, 142.65, 134.86, 134.70, 130.38, 129.71, 126.25, 123.94, 114.05, 113.67, 62.23, 55.26, 40.66, 34.72, 34.03, 31.49. HRMS (ESI) m/z: [M + Na]+ calculated for C26H31N3NaO2 440.2308; found 440.2324.
2-((4-(tert-butyl)phenyl)amino)-N-(3-chlorophenethyl)-2-(pyridin-3-yl)acetamide (28c). Compound was made and purified using general procedure F. Off-white solid with 45% yield, 165 mg. Rf = 0.25 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 8.64 (d, J = 20.0 Hz, 1H), 7.63 (dt, J = 7.9, 2.0 Hz, 1H), 7.32–7.30 (m,1H), 7.27 (d, J = 8.7 Hz, 2H), 7.21–7.19 (m, 1H), 7.15 (t, J = 7.7 Hz, 1H), 7.09 (t, J = 1.9 Hz, 1H), 6.92–6.87 (m, 2H), 6.60 (d, J = 8.6 Hz, 2H), 4.74 (d, J = 2.3 Hz, 1H), 4.46 (s, 1H), 3.59 (ddt, J = 45.0, 14.0, 6.8 Hz, 2H), 2.79 (ddt, J = 31.0, 13.2, 6.6 Hz, 2H), 1.31 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 170.47, 150.00, 149.03, 143.74, 142.78, 140.53, 134.78, 134.56, 134.39, 129.89, 128.82, 126.94, 126.82, 126.31, 124.01, 113.67, 62.35, 40.28, 35.27, 34.04, 31.48. HRMS (ESI) m/z: [M + Na]+ calculated for C25H28ClN3NaO 444.1813; found 444.1800.
2-((4-(tert-butyl)phenyl)amino)-N-(3-methoxyphenethyl)-2-(pyridin-3-yl)acetamide (28d). Compound was made and purified using general procedure F. White solid with 48% yield, 202 mg. Rf = 0.23 (2:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3): δ 8.64 (d, J = 3.3 Hz, 1H), 8.60 (dd, J = 4.8, 1.6 Hz, 1H), 7.61 (dt, J = 7.9, 2.0 Hz, 1H), 7.30–7.24 (m, 2H), 7.15 (t, J = 7.8 Hz, 1H), 6.87 (t, J = 5.9 Hz, 1H), 6.77 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 6.67 (t, J = 5.9 Hz, 1H), 6.61–6.58 (m, 3H), 4.74 (d, J = 2.6 Hz, 1H), 4.29 (d, J = 2.5 Hz, 1H), 3.77 (s, 3H), 3.60 (q, J = 6.7 Hz, 2H), 2.79 (ddt, J = 49.2, 14.0, 6.9 Hz, 1H), 1.31 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 170.34, 159.85, 149.90, 149.08, 143.83, 142.63, 140.01, 134.83, 134.69, 129.64, 126.26, 123.97, 121.08, 114.24, 113.67, 112.05, 62.31, 55.15, 40.40, 35.61, 34.02, 31.48. HRMS (ESI) m/z: [M + Na]+ calculated for C26H31N3NaO2 440.2308; found 440.2294.
2-((4-(tert-butyl)phenyl)amino)-N-(3-phenylpropyl)-2-(pyridin-3-yl)acetamide (28e). Compound was made and purified using general procedure F. Pale yellow solid with 26% yield, 105 mg. Rf = 0.16 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) δ 8.71 (d, J = 2.3 Hz, 1H), 8.62 (dd, J = 4.8, 1.6 Hz, 1H), 7.75 (dt, J = 7.9, 2.0 Hz, 1H), 7.33 (dd, J = 7.8, 4.7 Hz, 1H), 7.28–7.25 (m, 3H), 7.22–7.19 (m, 1H), 7.11 (d, J = 6.7 Hz, 2H), 6.89 (t, J = 6.1 Hz, 1H), 6.64 (d, J = 8.6 Hz, 1H), 4.76 (s, 1H), 4.32 (s, 1H), 3.36 (q, J = 7.2 Hz, 2H), 2.59 (td, J = 8.3, 7.6, 2.4 Hz, 2H), 1.88–1.81 (m, 2H), 1.30 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.35, 149.93, 148.96, 143.76, 142.70, 141.20, 134.96, 134.82, 128.50, 128.34, 126.30, 126.06, 124.00, 113.67, 62.29, 39.19, 34.02, 33.20, 31.47, 31.13. HRMS (ESI) m/z: [M + H]+ calculated for C26H32N3O 402.2540; found 402.2535.
N-(1-benzylpiperidin-4-yl)-2-((4-(tert-butyl)phenyl)amino)-2-(pyridin-3-yl)acetamide (28f). Compound was made and purified using general procedure F. Pale yellow solid with 25% yield, 346 mg (3 mmol 3 g used). Rf = 0.31 (5% MeOH in DCM). 1H NMR (500 MHz, CDCl3) δ 8.71 (d, J = 2.4 Hz, 1H), 8.61 (dd, J = 4.8, 1.6 Hz, 1H), 7.75 (dt, J = 7.9, 2.0 Hz, 1H), 7.36–7.29 (m, 4H), 7.28–7.22 (m, 3H), 6.78 (d, J = 8.3 Hz, 1H), 6.62 (d, J = 8.7 Hz, 2H), 4.75 (d, J = 2.5 Hz, 1H), 4.27 (d, J = 2.6 Hz, 1H), 3.92–3.77 (m, 1H), 3.48 (s, 2H), 2.77 (dd, J = 24.3, 11.3 Hz, 2H), 2.13 (dq, J = 11.3, 2.1 Hz, 2H), 1.90 (dddd, J = 16.2, 11.7, 4.0, 2.0 Hz, 2H), 1.70 (bs, 1H), 1.51 (qd, J = 11.3, 4.0 Hz, 1H), 1.41 (qd, J = 11.1, 4.0 Hz, 1H), 1.30 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 169.70, 149.94, 149.00, 143.76, 142.74, 134.93, 134.77, 129.06, 128.23, 127.07, 126.25, 123.97, 113.71, 62.99, 62.47, 52.11, 52.00, 46.66, 34.02, 32.02, 31.77, 31.47. HRMS (ESI) m/z: [M + H]+ calculated for C29H37N4O 457.2962; found 457.1959.
2-((4-(tert-butyl)phenyl)amino)-N-((S)-1-phenylethyl)-2-(pyridin-3-yl)acetamide (28g). Compound was made and purified using general procedure F. Pale yellow solid with 40% yield, 157 mg. Rf = 0.16 (1:1 EtOAc:hexanes). 1H NMR (500 MHz, CDCl3) two diastereomers were observed, both diastereomers are described: δ 8.73 (s, 1H), 8.66 (s, 1H), 8.61 (d, J = 5.2 Hz, 1H), 8.58 (d, J = 4.9 Hz, 1H), 7.77 (dt, J = 7.9, 2.0 Hz, 1H), 7.69 (dt, J = 7.9, 2.0 Hz, 1H), 7.35–7.11 (m, 20H), 6.64 (d, J = 8.7 Hz, 2H), 6.58 (d, J = 8.7 Hz, 2H), 5.26–5.11 (m, 2H), 4.80 (dd, J = 3.9, 2.3 Hz, 2H), 4.41 (s, 1H), 4.35 (s, 1H), 1.51 (d, J = 7.0 Hz, 3H), 1.46 (d, J = 6.9 Hz, 3H), 1.31 (s, 9H), 1.30 (s, 9H). 13C NMR (126 MHz, CDCl3) two diastereomers were observed, both diastereomers are described: δ 169.64, 169.55, 149.90, 149.84, 148.98, 148.92, 143.86, 143.54, 142.60, 135.04, 134.95, 134.83, 134.74, 128.78, 128.50, 127.58, 127.31, 126.27, 126.17, 126.00, 124.00, 123.98, 113.84, 113.74, 62.44, 62.15, 48.80, 34.03, 34.01, 31.50, 31.48, 21.86, 21.23, 14.22. HRMS (ESI) m/z: [M + Na]+ calculated for C25H29N3NaO 410.2203; found 410.2199.
2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-N-(4-methoxyphenethyl)-2-(pyridin-3-yl)acetamide (14b). Compound was made and purified using general procedure G. White solid with 66% yield, 61 mg. Rf = 0.38 (1:1 EtOAc:DCM). 1H NMR (500 MHz, CDCl3): δ 8.49 (dd, J = 4.7, 1.6 Hz, 1H), 8.35 (d, J = 2.3 Hz, 1H), 7.22–7.18 (m, 3H), 7.08–7.04 (m, 3H), 6.94 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 6.11 (d, J = 16.6 Hz, 1H), 6.02 (t, J = 5.8 Hz, 1H), 5.94 (d, J = 9.9 Hz, 1H), 5.80 (s, 1H), 3.80 (s, 3H), 3.59 (ddt, J = 29.2, 13.2, 6.5 Hz, 2H), 2.87–2.74 (m, 2H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.34, 159.85, 149.90, 149.08, 143.83, 142.63, 140.01, 134.83, 134.69, 129.64, 126.26, 123.97, 121.08, 114.24, 113.67, 112.05, 62.31, 55.15, 40.40, 35.61, 34.02, 31.48. HRMS (ESI) m/z: [M + Na]+ calculated for C28H33N3NaO4S 530.2084; found 530.2087.
2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-N-(3-chlorophenethyl)-2-(pyridin-3-yl)acetamide (14c). Compound was made and purified using general procedure G. White solid with 45% yield, 46 mg. Rf = 0.38 (1:1 EtOAc:DCM). 1H NMR (500 MHz, CDCl3): δ 8.50 (dd, J = 4.7, 1.7 Hz, 1H), 8.35 (d, J = 2.3 Hz, 1H), 7.27–7.18 (m, 4H), 7.16 (s, 1H), 7.11–7.03 (m, 2H), 6.95 (d, J = 8.6 Hz, 2H), 6.82 (dd, J = 16.6, 9.9 Hz, 1H), 6.12 (d, J = 16.6 Hz, 1H), 6.03 (t, J = 5.8 Hz, 1H), 5.95 (d, J = 9.9 Hz, 1H), 5.80 (s, 1H), 3.62 (ddt, J = 62.7, 13.5, 6.6 Hz, 2H), 2.86 (t, J = 6.9 Hz, 2H), 1.27 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 168.80, 152.28, 151.22, 150.04, 140.44, 137.86, 135.54, 134.44, 133.10, 131.41, 130.20, 130.00, 128.89, 127.59, 127.02, 126.91, 126.05, 123.02z, 65.52, 40.93, 35.12, 34.63, 31.19. HRMS (ESI) m/z: [M + Na]+ calculated for C27H30ClN3NaO3S 534.1589; found 534.1584.
2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-N-(3-methoxyphenethyl)-2-(pyridin-3-yl)acetamide (14d). Compound was made and purified using general procedure G. White solid with 73% yield, 96 mg. Rf = 0.36 (1:1 EtOAc:DCM). 1H NMR (500 MHz, CDCl3): δ 8.47 (d, J = 4.8 Hz, 1H), 8.30 (s, 1H), 7.25–7.13 (m, 3H), 7.05 (dd, J = 8.0, 4.8 Hz, 1H), 6.95 (d, J = 8.6 Hz, 2H), 6.86–6.68 (m, 4H), 6.12 (s, 1H), 6.10 (d, J = 16.5 Hz, 1H), 5.94 (d, J = 9.9 Hz, 1H), 5.80 (s, 1H), 3.79 (s, 3H), 3.62 (ddt, J = 40.3, 13.1, 6.4 Hz, 2H), 2.84 (td, J = 6.8, 4.7 Hz, 2H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.66, 159.89, 152.19, 151.25, 149.93, 139.91, 137.90, 135.59, 133.12, 131.45, 130.27, 129.75, 127.50, 125.99, 122.98, 121.04, 114.37, 112.14, 65.49, 55.18, 41.07, 35.44, 34.61, 31.19. HRMS (ESI) m/z: [M + Na]+ calculated for C28H33N3NaO4S 530.2084; found 530.2101.
2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-N-(3-phenylpropyl)-2-(pyridin-3-yl)acetamide (14e). Compound was made and purified using general procedure G. White solid with 80% yield, 103.0 mg. Rf = 0.50 (1:1 EtOAc:DCM). 1H NMR (500 MHz, CDCl3): δ 8.52 (d, J = 4.2 Hz, 1H), 8.39 (s, 1H), 7.34 (dt, J = 8.0, 2.0 Hz, 1H), 7.31–7.26 (m, 3H), 7.22 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 6.9 Hz, 2H),7.12 (dd, J = 8.0, 4.8 Hz, 1H), 7.04 (d, J = 8.6 Hz, 2H), 6.84 (dd, J = 16.6, 9.9 Hz, 1H), 6.12 (d, J = 16.5 Hz, 1H), 6.02 (s, 1H), 5.95 (d, J = 9.9 Hz, 1H), 5.82 (s, 1H), 3.38 (dt, J = 7.3, 5.4 Hz, 2H), 2.67 (td, J = 7.4, 1.3 Hz, 2H), 1.89 (p, J = 7.4 Hz, 2H), 1.26 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 168.64, 152.27, 151.26, 150.03, 141.06, 138.01, 135.58, 133.12, 131.45, 130.30, 128.54, 128.34, 127.54, 126.13, 126.04, 123.06, 65.45, 39.83, 34.62, 33.23, 31.18, 30.99. HRMS (ESI) m/z: [M + H]+ calculated for C28H34N3O3S 493.2315; found 492.2313.
N-(1-benzylpiperidin-4-yl)-2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-2-(pyridin-3-yl)acetamide (14f). Compound was made and purified using general procedure G. Pale yellow solid with 42% yield, 116.5 mg. Rf = 0.24 (5% MeOH in DCM). 1H NMR (500 MHz, CDCl3): δ 8.51 (dd, J = 4.9, 1.6 Hz, 1H), 8.39 (d, J = 2.3 Hz, 1H), 7.35–7.30 (m, 4H), 7.31–7.24 (m, 1H), 7.21 (d, J = 8.6 Hz, 2H), 7.11 (dd, J = 8.0, 4.8 Hz, 1H), 7.03 (d, J = 8.6 Hz, 2H), 6.82 (dd, J = 16.5, 9.9 Hz, 1H), 6.12 (d, J = 16.6 Hz, 1H), 6.00 (d, J = 7.9 Hz, 1H), 5.94 (d, J = 9.9 Hz, 1H), 5.85 (s, 1H), 3.93–3.85 (m, 1H), 3.52 (s, 2H), 2.82 (q, J = 11.0 Hz, 2H), 2.17 (q, J = 11.8 Hz, 2H), 2.03–1.95 (m, 1H), 1.95–1.88 (m, 1H), 1.52 (dqd, J = 41.1, 11.0, 3.8 Hz, 2H), 1.25 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 167.96, 152.26, 151.28, 150.01, 137.98, 135.57, 133.06, 131.46, 130.29, 129.05, 128.27, 127.51, 127.12, 126.01, 123.03, 65.29, 62.92, 52.00, 47.46, 34.62, 31.90, 31.81, 31.18. HRMS (ESI) m/z: [M + H]+ calculated for C31H39N4O3S 547.2737; found 547.2736.
(S)- and (R)-2-(N-(4-(tert-butyl)phenyl)vinylsulfonamido)-N-((S)-1-phenylethyl)-2-(pyridin-3-yl)acetamide (14g and 14h). Compound was made and purified using general procedure G. White solid with 75% yield, 276.8 mg. Rf = 0.63 (1:1 EtOAc:DCM). The diastereomer was separated by reverse-phase HPLC using the ZORBAX Rx-C18 Semi-Preparative column with 50% ACN in water with 3 mL/min flow rate. The absolute stereochemistry was inferred based on previously published crystal structures and the structure-activity relationship between different diastereomers. 14g: 1H NMR (500 MHz, CDCl3) δ 8.51 (dd, J = 4.8, 1.6 Hz, 1H), 8.42 (d, J = 2.2 Hz, 1H), 7.43–7.31 (m, 5H), 7.16 (d, J = 8.6 Hz, 2H), 7.11 (dd, J = 8.0, 4.8 Hz, 1H), 6.98–6.88 (m, 2H), 6.69 (dd, J = 16.5, 9.9 Hz, 1H), 6.38 (d, J = 8.1 Hz, 1H), 6.08 (d, J = 16.6 Hz, 1H), 5.88 (d, J = 10.0 Hz, 1H), 5.86 (s, 1H), 5.17 (p, J = 7.1 Hz, 1H), 1.51 (d, J = 7.0 Hz, 3H), 1.24 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 167.64, 152.21, 151.21, 150.03, 142.49, 138.09, 135.43, 132.87, 131.44, 130.12, 128.87, 127.69, 127.56, 126.21, 125.96, 123.01, 65.03, 49.79, 34.60, 31.17, 21.68. HRMS (ESI) m/z: [M + Na]+ calculated for C27H31N3NaO3S 500.1978; found 500.1968. 14h: 1H NMR (500 MHz, CDCl3) δ 8.49 (dd, J = 4.7, 1.7 Hz, 1H), 8.39 (d, J = 2.3 Hz, 1H), 7.35–7.30 (m, 2H), 7.25 (ddd, J = 11.8, 7.8, 1.7 Hz, 3H), 7.19 (d, J = 8.6 Hz, 2H), 7.09–7.04 (m, 1H), 7.04–6.98 (m, 2H), 6.82 (dd, J = 16.5, 9.9 Hz, 1H), 6.34 (d, J = 7.7 Hz, 1H), 6.12 (d, J = 16.5 Hz, 1H), 5.94 (d, J = 9.9 Hz, 1H), 5.90 (s, 1H), 5.18 (p, J = 7.1 Hz, 1H), 1.57 (d, J = 6.9 Hz, 3H), 1.25 (s, 8H). 13C NMR (126 MHz, CDCl3) δ 167.78, 152.24, 151.29, 150.02, 142.55, 138.05, 135.56, 132.90, 131.54, 130.00, 128.74, 127.54, 125.99, 125.97, 122.94, 65.11, 49.83, 34.61, 31.18, 22.00. HRMS (ESI) m/z: [M + Na]+ calculated for C27H31N3NaO3S 500.1978; found 500.1982.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(thiophen-3-yl)ethyl)cyclopent-1-ene-1-carboxamide (11c). Compound was made and purified using general procedure B, off white solid 79% yield, 330 mg. 1H NMR (500 MHz, CDCl3) δ 7.27–7.24 (m, 1H), 7.20–7.12 (m, 3H), 6.94–6.87 (m, 3H), 6.09 (d, J = 8.1 Hz, 1H), 6.06 (s, 1H), 5.81 (q, J = 2.2 Hz, 1H), 3.87–3.76 (m, 1H), 2.22–2.06 (m, 4H), 2.01–1.84 (m, 2H), 1.71–1.53 (m, 5H), 1.42–1.28 (m, 2H), 1.26 (s, 9H), 1.15 (ddt, J = 16.1, 12.0, 8.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.73, 168.53, 151.12, 139.46, 139.40, 138.34, 135.52, 129.16, 129.08, 126.40, 125.51, 125.33, 61.62, 48.59, 34.68, 33.83, 33.19, 32.97, 32.93, 31.42, 25.67, 24.88, 24.83, 23.36. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C28H36N2NaO2S 487.2390; found 487.2404.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(quinolin-3-yl)ethyl)cyclopent-1-ene-1-carboxamide (11d). Compound was made and purified using general procedure B, pale yellow solid 59% yield, 270 mg. 1H NMR (500 MHz, CDCl3) δ 8.73 (d, J = 2.1 Hz, 1H), 8.03 (d, J = 8.4 Hz, 1H), 8.00 (s, 1H), 7.74–7.66 (m, 1H), 7.65–7.61 (m, 1H), 7.52–7.45 (m, 1H), 7.13 (d, J = 8.2 Hz, 2H), 6.39 (s, 2H), 6.27 (s, 1H), 5.86 (p, J = 2.3 Hz, 1H), 3.93–3.82 (m, 1H), 2.23–2.09 (m, 4H), 2.00 (dd, J = 12.2, 4.1 Hz, 2H), 1.89 (dd, J = 12.8, 4.2 Hz, 1H), 1.66 (p, J = 7.7 Hz, 5H), 1.36 (ddd, J = 16.2, 13.1, 11.4 Hz, 2H), 1.19 (s, 9H), 1.18–1.08 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 169.02, 168.29, 151.91, 151.58, 147.65, 140.28, 139.13, 138.06, 137.59, 130.00, 129.59, 129.15, 128.23, 127.98, 127.47, 126.87, 125.83, 63.71, 48.83, 34.69, 33.87, 33.26, 33.02, 32.98, 31.31, 25.62, 24.90, 24.85, 23.32. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C33H39N3NaO2 532.2934; found 532.2946.
N-(1-(benzo[b]thiophen-3-yl)-2-(cyclohexylamino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)cyclopent-1-ene-1-carboxamide (11e). Compound was made and purified using general procedure B, pale yellow solid 26% yield, 120 mg. 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 7.6 Hz, 1H), 7.78–7.72 (m, 1H), 7.53 (s, 1H), 7.37 (p, J = 7.0 Hz, 2H), 7.03 (d, J = 8.2 Hz, 2H), 6.69 (d, J = 19.7 Hz, 3H), 6.20 (d, J = 8.2 Hz, 1H), 5.84–5.79 (m, 1H), 2.17–2.00 (m, 4H), 1.97–1.90 (m, 2H), 1.65 (pt, J = 14.3, 6.5 Hz, 5H), 1.36 (ddt, J = 15.3, 11.9, 6.0 Hz, 3H), 1.19 (s, 12H). 13C NMR (126 MHz, CDCl3) δ 169.16, 168.29, 151.15, 139.68, 139.60, 139.30, 138.74, 137.04, 129.40, 129.26, 129.10, 125.16, 124.61, 124.59, 122.92, 121.61, 57.25, 48.65, 34.61, 33.87, 33.18, 33.04, 32.98, 31.36, 25.65, 24.94, 24.89, 23.36. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C32H38N2NaO2S 537.2546; found 537.2549.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-4-yl)ethyl)cyclopent-1-ene-1-carboxamide (11f). Compound was made and purified using general procedure B, white solid 71% yield, 90 mg. 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 6.2 Hz, 2H), 7.30–7.19 (m, 4H), 6.97 (d, J = 8.1 Hz, 2H), 6.45 (d, J = 8.1 Hz, 1H), 5.88 (p, J = 2.2 Hz, 1H), 5.82 (s, 1H), 3.89–3.78 (m, 1H), 2.21 (ddt, J = 7.6, 5.0, 2.6 Hz, 2H), 2.12 (dtt, J = 9.9, 5.0, 2.2 Hz, 2H), 1.98–1.84 (m, 3H), 1.67 (p, J = 7.4 Hz, 4H), 1.43–1.29 (m, 2H), 1.26 (s, 9H), 1.19 (dddd, J = 26.8, 22.7, 10.8, 4.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.03, 167.76, 151.65, 149.80, 144.29, 140.77, 139.08, 138.60, 128.71, 126.05, 124.23, 67.01, 48.74, 34.76, 33.69, 33.27, 32.92, 32.88, 31.39, 25.62, 24.80, 23.34. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C29H38N3O2 460.29585; found 460.29546.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyrimidin-5-yl)ethyl)cyclopent-1-ene-1-carboxamide (11g). Compound was made and purified using general procedure B, off white solid 29% yield, 120 mg. 1H NMR (500 MHz, CDCl3) δ 9.06 (s, 1H), 8.55 (s, 2H), 7.25 (d, J = 8.9 Hz, 2H), 6.85 (dd, J = 15.8, 5.0 Hz, 2H), 6.62 (d, J = 8.1 Hz, 1H), 6.18 (s, 1H), 5.84 (td, J = 2.7, 1.3 Hz, 1H), 3.89–3.78 (m, 1H), 2.55 (dd, J = 25.2, 2.4 Hz, 2H), 2.20 (ddd, J = 7.8, 6.0, 2.5 Hz, 2H), 2.12 (ddd, J = 10.3, 5.6, 2.3 Hz, 2H), 2.01–1.86 (m, 1H), 1.76–1.57 (m, 5H), 1.46–1.31 (m, 2H), 1.27 (s, 9H), 1.26–1.14 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 169.14, 167.31, 158.59, 158.11, 152.37, 145.94, 141.02, 138.68, 136.78, 129.38, 128.84, 126.31, 61.37, 48.85, 34.82, 33.84, 33.70, 33.32, 32.99, 32.91, 31.36, 31.32, 25.60, 24.79, 23.31, 23.29. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C28H36N4NaO2 483.2730; found 483.2746.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-1-(isoquinolin-4-yl)-2-oxoethyl)cyclopent-1-ene-1-carboxamide (11h). Compound was made and purified using general procedure A, off white solid 78% yield, 358 mg. 1H NMR (500 MHz, CDCl3) δ 9.08 (d, J = 1.9 Hz, 1H), 8.28 (s, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.77 (dd, J = 8.5, 6.9 Hz, 1H), 7.66–7.59 (m, 1H), 7.06 (s, 1H), 6.93 (d, J = 8.1 Hz, 2H), 6.78 (s, 1H), 5.81 (q, J = 2.1 Hz, 1H), 5.67 (s, 1H), 3.91 (dt, J = 7.5, 3.5 Hz, 1H), 2.21–2.09 (m, 4H), 1.97 (d, J = 16.2 Hz, 2H), 1.72–1.62 (m, 5H), 1.34 (tdd, J = 12.3, 8.3, 3.8 Hz, 2H), 1.13 (d, J = 0.9 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 168.96, 168.39, 153.31, 151.05, 145.51, 139.71, 139.19, 136.80, 135.07, 131.47, 129.61, 128.57, 128.14, 127.44, 125.06, 124.84, 122.71, 58.84, 49.11, 34.52, 33.87, 33.23, 33.04, 33.00, 31.28, 25.61, 25.00, 24.89, 23.34. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C33H39N3NaO2 532.2934; found 532.2932.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-1-(1H-imidazole-5-yl)-2-oxoethyl)cyclopent-1-ene-1-carboxamide (11i). Compound was made and purified using general procedure B, off white solid 37% yield, 150 mg. 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 1.0 Hz, 1H), 7.28 (d, J = 8.6 Hz, 2H), 7.11 (s, 1H), 7.00 (d, J = 8.2 Hz, 2H), 6.81 (d, J = 2.5 Hz, 1H), 5.86 (p, J = 2.3 Hz, 1H), 5.75 (s, 1H), 3.79 (td, J = 6.3, 3.4 Hz, 1H), 2.60 (ddd, J = 10.0, 4.9, 2.3 Hz, 2H), 2.52 (ddt, J = 7.7, 5.2, 2.6 Hz, 2H), 2.21 (ddt, J = 7.6, 5.0, 2.5 Hz, 2H), 1.85 (d, J = 14.6 Hz, 2H), 1.73–1.65 (m, 4H), 1.37–1.22 (m, 15H). 13C NMR (101 MHz, CDCl3) δ 169.46, 167.62, 158.91, 158.43, 152.68, 146.26, 141.34, 139.00, 137.10, 129.70, 129.15, 126.63, 61.69, 49.16, 35.14, 34.15, 34.02, 33.64, 33.31, 33.22, 31.67, 31.64, 25.92, 25.11, 23.63, 23.60. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C27H37N4O2 449.29110; found 449.29124.
(E)-N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-4-yl)ethyl)but-2-enamide (8c). Compound was made and purified using general procedure A, white solid 74% yield, 287 mg. 1H NMR (500 MHz, CDCl3) δ 8.49–8.45 (m, 2H), 7.28 (d, J = 8.9 Hz, 2H), 7.19–7.12 (m, 2H), 7.03–6.92 (m, 3H), 6.47 (d, J = 7.9 Hz, 1H), 5.84 (s, 1H), 5.70 (dd, J = 15.0, 1.8 Hz, 1H), 4.03–3.75 (m, 1H), 1.95 (dd, J = 12.3, 4.3 Hz, 1H), 1.91–1.85 (m, 1H), 1.78–1.74 (m, 3H), 1.70–1.64 (m, 2H), 1.58 (dt, J = 12.9, 3.9 Hz, 1H), 1.37–1.31 (m, 2H), 1.29 (s, 9H), 1.25–1.08 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.84, 167.10, 151.87, 149.73, 144.20, 143.48, 137.54, 129.06, 126.42, 124.35, 122.64, 66.12, 48.75, 34.80, 32.89, 31.39, 25.62, 24.83, 24.80, 18.25. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C27H35N3NaO2 456.2621; found 456.2614.
N-(4-(tert-butyl)phenyl)-2-chloro-N-(2-(cyclohexylamino)-2-oxo-1-(thiophen-3-yl)ethyl)acetamide (16d). Compound was made and purified using general procedure A, white solid 87% yield, 352 mg. 1H NMR (500 MHz, CDCl3) δ 7.28 (s, 2H), 7.25–7.21 (m, 1H), 7.17 (dd, J = 5.0, 3.0 Hz, 1H), 6.84 (dd, J = 5.0, 1.3 Hz, 1H), 6.01 (s, 1H), 5.79 (d, J = 8.2 Hz, 1H), 3.87 (s, 2H), 3.82 (ddt, J = 14.7, 6.7, 3.9 Hz, 1H), 1.98–1.92 (m, 1H), 1.91–1.85 (m, 1H), 1.74–1.60 (m, 3H), 1.38 (dqd, J = 8.3, 3.3, 1.6 Hz, 2H), 1.30 (s, 9H), 1.23–1.05 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.83, 166.99, 152.39, 136.30, 134.59, 129.25, 128.85, 126.93, 126.34, 125.82, 61.28, 48.89, 42.69, 34.81, 32.95 (d, J = 10.4 Hz),31.57, 31.38, 25.62, 24.90, 24.84. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C24H32N2O2ClS 447.18675; found 447.18675.
N-(4-(tert-butyl)phenyl)-2-chloro-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-4-yl)ethyl)acetamide (16e). Compound was made and purified using general procedure A, white solid 94% yield, 375 mg. 1H NMR (500 MHz, CDCl3) δ 8.47 (d, J = 6.1 Hz, 2H), 7.26 (s, 2H), 7.16–7.11 (m, 2H), 5.88 (d, J = 8.0 Hz, 1H), 5.83 (s, 1H), 3.87 (d, J = 1.9 Hz, 2H), 3.85–3.77 (m, 1H), 2.00–1.94 (m, 1H), 1.86 (dd, J = 12.9, 4.0 Hz, 1H), 1.75–1.53 (m, 3H), 1.41–1.28 (m, 2H), 1.26 (s, 9H), 1.23–1.03 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.57, 166.93, 152.92, 149.88, 143.19, 135.93, 129.26, 126.68, 124.90, 65.47, 49.11, 42.49, 34.87, 32.90, 32.85, 31.33, 25.56, 24.88, 24.82. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C25H33N3O2Cl 442.22558; found 442.22525.
N-(4-(tert-butyl)phenyl)-2-chloro-N-(2-(cyclohexylamino)-1-(5-methylpyrazin-2-yl)-2-oxoethyl)acetamide (16f). Compound was made and purified using general procedure B, pale yellow solid 34% yield, 140 mg. 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1H), 8.39 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.29–7.06 (m, 2H), 5.88 (s, 1H), 3.89 (q, J = 13.6 Hz, 2H), 3.82–3.72 (m, 1H), 1.92–1.85 (m, 3H), 1.77–1.53 (m, 5H), 1.32 (s, 13H), 1.22–1.12 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 167.11, 165.73, 153.11, 152.59, 147.62, 144.81, 142.87, 136.87, 128.87, 126.66, 65.99, 48.50, 42.31, 34.75, 32.59, 32.49, 31.60, 31.24, 25.48, 24.57, 24.50, 22.66, 21.31, 14.13. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C25H34N4O2Cl 457.23648; found 457.23621.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-1-(isoquinolin-4-yl)-2-oxoethyl)but-2-ynamide (13e). Compound was made and purified using general procedure A, pale white solid 90% yield, 392 mg. 1H NMR (500 MHz, CDCl3) δ 9.11 (d, J = 0.7 Hz, 1H), 8.24 (s, 1H), 8.00–7.94 (m, 2H), 7.82–7.73 (m, 1H), 7.65 (ddd, J = 7.9, 6.9, 1.0 Hz, 1H), 7.00 (d, J = 8.1 Hz, 2H), 6.95 (s, 1H), 6.86 (s, 2H), 5.62 (d, J = 8.1 Hz, 1H), 3.89 (dtd, J = 10.8, 7.8, 7.2, 3.9 Hz, 1H), 2.01–1.94 (m, 1H), 1.93 (s, 1H), 1.65 (s, 6H), 1.31 (d, J = 18.7 Hz, 2H), 1.15 (s, 9H), 1.10–0.97 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.30, 155.31, 153.07, 151.22, 144.79, 135.59, 134.71, 131.56, 129.62, 128.48, 127.90, 127.41, 124.89, 124.21, 122.31, 91.66, 73.65, 57.76, 48.95, 34.33, 32.74, 32.68, 30.99, 25.32, 24.73, 24.63, 3.79, 3.63. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C31H36N3O2 482.28020; found 482.28025.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(quinolin-3-yl)ethyl)but-2-ynamide (13f). Compound was made and purified using general procedure A, pale white solid 69% yield, 298 mg. 1H NMR (500 MHz, CDCl3) δ 8.70 (d, J = 2.2 Hz, 1H), 8.07 (dd, J = 8.4, 1.0 Hz, 1H), 8.00 (d, J = 2.4 Hz, 1H), 7.73–7.70 (m, 1H), 7.65 (dd, J = 8.2, 1.4 Hz, 1H), 7.52 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.21 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.0 Hz, 2H), 6.27 (s, 1H), 6.23–6.18 (m, 1H), 3.95–3.83 (m, 1H), 2.05–1.99 (m, 1H), 1.92–1.84 (m, 1H), 1.78–1.58 (m, 6H), 1.47–1.35 (m, 3H), 1.23 (s, 9H), 1.21–1.10 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.79, 155.80, 152.21, 151.92, 147.77, 138.75, 136.61, 130.53, 130.17, 129.23, 128.49, 127.69, 127.51, 127.29, 126.06, 92.59, 74.16, 49.31, 35.00, 33.22, 33.15, 31.54, 25.85, 25.15, 25.11, 23.05, 14.51, 4.35. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C31H36N3O2 482.28020; found 482.28007.
N-(1-(benzo[d]thiazol-2-yl)-2-(cyclohexylamino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)but-2-ynamide (13g). Compound was made and purified using general procedure A, pale white solid 91% yield, 398 mg. 1H NMR (500 MHz, CDCl3) δ 9.61 (d, J = 7.9 Hz, 1H), 8.11–8.00 (m, 1H), 7.95–7.84 (m, 1H), 7.56–7.51 (m, 1H), 7.47 (d, J = 1.2 Hz, 1H), 7.46–7.35 (m, 2H), 7.29–7.22 (m, 2H), 6.15 (d, J = 1.6 Hz, 1H), 4.04–3.94 (m, 1H), 1.99 (d, J = 1.6 Hz, 3H), 1.91–1.64 (m, 4H), 1.55–1.34 (m, 6H), 1.24 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 171.84, 169.60, 163.31, 158.61, 152.14, 148.14, 135.47, 134.80, 126.83, 126.70, 126.56, 126.18, 124.24, 123.56, 122.32, 121.64, 121.47, 50.84, 49.26, 34.67, 32.77, 32.67, 31.72, 31.59, 31.37, 25.94, 25.14, 24.84, 24.68, 24.60, 13.39, 4.08. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C29H34N3O2S 488.23662; found 488.23672.
N-(1-(benzo[b]thiophen-3-yl)-2-(cyclohexylamino)-2-oxoethyl)-N-(4-(tert-butyl)phenyl)but-2-ynamide (13h). Compound was made and purified using general procedure A, grey solid 92% yield, 402 mg. 1H NMR (500 MHz, CDCl3) δ 7.86–7.82 (m, 1H), 7.77–7.71 (m, 1H), 7.50 (s, 1H), 7.45–7.34 (m, 2H), 7.14–7.08 (m, 2H), 6.82 (s, 2H), 6.65 (s, 1H), 6.04 (d, J = 8.2 Hz, 1H), 3.88 (dddd, J = 14.5, 10.5, 7.9, 3.9 Hz, 1H), 2.01–1.91 (m, 1H), 1.92–1.87 (m, 1H), 1.79–1.60 (m, 6H), 1.47–1.28 (m, 3H), 1.23 (s, 9H), 1.21–1.07 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 167.45, 155.48, 151.34, 139.47, 138.29, 136.01, 129.42, 129.35, 128.26, 125.05, 124.60, 124.58, 122.81, 121.41, 91.83, 73.83, 56.30, 48.74, 34.53, 32.83, 32.79, 31.19, 25.49, 24.79, 24.75, 3.93. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C30H35N2O2S 487.24138; found 487.24187.
N-(4-(tert-butyl)phenyl)-N-(1-(cyclohexylamino)-3-(2,5-dioxoimidazolidin-1-yl)-1-oxopropan-2-yl)but-2-ynamide (13i). To a solution of 4-tBu-aniline (0.56 mmol, 0.09 mL) in MeOH was added 2-(2,4-dioxoimidazolidin-1-yl)acetaldehyde (0.56 mmol, 80 mg) and stirred for 30 min. Butynoic acid (0.56 mmol, 47 mg) and cyclohexyl isocyanide (0.56 mmol, 0.07 mL) were added and the solution stirred at room temperature overnight. The resulting white precipitate was collected by filtration and rinsed with hexanes. The precipitate was dried over vacuum to afford the desired product (58 mg, 22% yield) as a white precipitate. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.39 (d, J = 8.5 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.0 Hz, 1H), 5.24 (dd, J = 9.3, 5.3 Hz, 1H), 4.07 (d, J = 18.0 Hz, 1H), 3.77 (d, J = 18.0 Hz, 1H), 3.70 (dd, J = 14.0, 5.3 Hz, 1H), 3.28 (dd, J = 13.9, 9.4 Hz, 1H), 1.89 (t, J = 16.9 Hz, 2H), 1.70 (s, 5H), 1.65–1.54 (m, 0H), 1.44–1.28 (m, 11H), 1.22 (q, J = 10.5 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 170.49, 168.02, 156.59, 155.84, 152.37, 135.31, 128.88, 126.21, 93.11, 73.57, 56.15, 52.78, 48.72, 42.35, 34.89, 32.75, 32.71, 31.38, 25.59, 24.78, 24.74, 4.08. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C26H34N4O4 489.2472; found 489.2475.
N-(4-(tert-butyl)phenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(1H-pyrazol-4-yl)ethyl)propiolamide (13j). 1H-pyrazole-4-carbaldehyde (100 mg, 1.04 mmol) and 4-tert-butylaniline (0.17 mL, 1.04 mmol) were stirred in MeOH (4.16 mL) and for 30 min at rt. A white precipitate was observed. But-2-ynoic acid (87.4 mg, 1.04 mmol) and cyclohexyl isocyanide (0.12 mL, 0.936 mmol) were added and the solution stirred at rt overnight. Upon addition of the isocyanide, the white precipitate dissolved. A stream of air was used to evaporate the MeOH. EtOAc was added to dissolve the impurities and the mixture was sonicated. The product was finally triturated with hexanes and EtOAc. Vacuum filtration was used to collect the precipitate. The white powder precipitate was dried using the vacuum filtration and the product (186 mg, 47% yield) was obtained. Rf (5% (MeOH + 1% NH4OH) in DCM): 0.45. 1H NMR (500 MHz, CDCl3) δ 8.63–8.39 (m, 2H), 7.50 (s, 2H), 7.28 (s, 1H), 7.00–6.93 (m, 2H), 6.38 (d, J = 8.1 Hz, 1H), 6.09 (s, 1H), 3.78 (tdd, J = 10.4, 7.1, 4.0 Hz, 1H), 1.96–1.82 (m, 2H), 1.69 (d, J = 8.5 Hz, 4H), 1.59 (dq, J = 13.0, 4.0 Hz, 1H), 1.41–1.31 (m, 1H), 1.28 (s, 9H), 1.24–1.12 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 167.63, 155.30, 151.92, 136.43, 134.76, 129.41, 126.03, 125.75, 114.45, 92.11, 73.85, 55.72, 48.82, 34.78, 32.86, 32.80, 31.37, 25.61, 24.80, 4.07. HRMS (ESI/Q-TOF) m/z: [M+Na]¬+ calculated for C25H32N4NaO2 443.2417, found 443.2426.
N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-2-oxo-N-(m-tolyl)propanamide (18b). Compound was prepared according to the procedure for 6m. Pale yellow solid, 134 mg, 37% yield. 1H NMR (500 MHz, CDCl3) δ 8.63 (d, J = 5.7 Hz, 2H), 7.69 (dd, J = 5.8, 3.8 Hz, 1H), 7.30 (dd, J = 8.0, 4.8 Hz, 1H), 7.25–7.14 (m, 2H), 7.07 (s, 1H), 7.02 (s, 1H), 6.22 (d, J = 8.0 Hz, 1H), 6.14 (s, 1H), 4.06–3.86 (m, 1H), 2.35 (s, 3H), 2.32 (s, 3H), 2.13–1.94 (m, 3H), 1.88–1.68 (m, 2H), 1.55–1.40 (m, 2H), 1.37–1.16 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 197.36, 168.01, 166.76, 150.59, 149.11, 139.55, 138.91, 136.94, 130.81, 130.27, 130.10, 129.10, 127.29, 123.56, 62.46, 49.25, 32.88, 32.84, 27.84, 25.51, 24.88, 24.81, 21.22. HRMS (ESI/Q-TOF) m/z: [M + Na]+ calculated for C23H27N3O3 416.1945; found 416.1952.
N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-(4-methoxyphenyl)but-2-ynamide (13k). Compound was made and purified using general procedure A, grey solid 83% yield, 302 mg. 1H NMR (500 MHz, CDCl3) δ 8.50 (dd, J = 4.8, 1.7 Hz, 1H), 8.45 (d, J = 2.3 Hz, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.12 (ddd, J = 8.0, 4.9, 0.9 Hz, 1H), 6.98 (s, 2H), 6.79–6.70 (m, 2H), 6.10 (s, 1H), 6.00 (d, J = 8.0 Hz, 1H), 3.88–3.81 (m, 1H), 3.79 (s, 3H), 1.94–1.84 (m, 2H), 1.74 (s, 4H), 1.61 (s, 1H), 1.42–1.11 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 167.75, 159.81, 155.97, 151.57, 149.93, 138.74, 132.10, 130.52, 123.44, 114.20, 92.59, 61.98, 55.74, 49.28, 33.21, 33.14, 25.85, 25.10, 4.39. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C24H28N3O3 406.21252; found 406.21250.
N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-ynamide (13l). Compound was made and purified using general procedure C, white solid 57% yield, 153 mg. 1H NMR (500 MHz, CDCl3) δ 8.58 (dd, J = 2.4, 0.9 Hz, 1H), 8.50 (dd, J = 4.8, 1.6 Hz, 1H), 7.77–7.72 (m, 1H), 7.62 (d, J = 7.5 Hz, 1H), 7.32–7.20 (m, 2H), 5.65 (d, J = 7.5 Hz, 1H), 3.74–3.63 (m, 1H), 1.93 (s, 3H), 1.75–1.64 (m, 3H), 1.63–1.52 (m, 2H), 1.36–1.16 (m, 2H), 1.14–0.98 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.88, 153.20, 149.46, 148.97, 135.07, 134.31, 124.12, 85.46, 74.64, 54.99, 49.14, 32.84, 32.68, 25.70, 25.02, 24.90, 3.98.
N-(3-acetamidophenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-ynamide (13m). Compound was made and purified using general procedure A, white solid 82% yield, 320 mg. 1H NMR (500 MHz, CDCl3) δ 8.42 (s, 2H), 7.96 (s, 1H), 7.83–7.76 (m, 1H), 7.49 (dt, J = 7.9, 2.1 Hz, 2H), 7.17–7.04 (m, 2H), 6.74 (d, J = 7.8 Hz, 1H), 6.34 (d, J = 8.1 Hz, 1H), 6.03 (s, 1H), 3.78–3.68 (m, 1H), 2.13 (s, 3H), 1.92 (d, J = 12.2 Hz, 1H), 1.82 (s, 1H), 1.69 (s, 3H), 1.63–1.51 (m, 2H), 1.33–1.22 (m, 2H), 1.11 (dtd, J = 35.2, 11.2, 3.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.57, 167.51, 155.06, 151.09, 149.64, 139.27, 138.81, 137.84, 130.10, 129.22, 125.98, 123.24, 121.38, 119.91, 92.30, 77.29, 77.04, 76.79, 73.69, 62.29, 49.04, 32.69, 25.40, 24.76, 24.71, 24.54, 3.99. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C25H29N4O3 433.22342; found 433.22345.
N-(4-acetamidophenyl)-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)but-2-ynamide (13n). Compound was made and purified using general procedure A, light brown solid 89% yield, 345 mg. 1H NMR (500 MHz, CDCl3) δ 8.48–8.44 (m, 1H), 8.41 (s, 1H), 7.50 (dt, J = 7.6, 1.9 Hz, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.14 (dd, J = 8.0, 4.8 Hz, 1H), 7.03 (d, J = 7.5 Hz, 2H), 6.24 (s, 1H), 6.09 (s, 1H), 3.81 (dp, J = 11.0, 4.0, 3.6 Hz, 1H), 2.15–2.12 (m, 3H), 2.01–1.91 (m, 1H), 1.89–1.82 (m, 1H), 1.78–1.64 (m, 4H), 1.64–1.52 (m, 1H), 1.45–1.05 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 168.74, 167.40, 155.36, 151.06, 149.50, 138.70, 138.23, 134.05, 131.10, 130.16, 123.29, 119.30, 92.50, 77.30, 77.05, 76.79, 73.67, 61.88, 49.01, 32.77, 32.71, 25.42, 24.75, 24.70, 24.57, 4.00. HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C25H29N4O3 433.22342; found 433.22352.
N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-(4-fluorophenyl)but-2-ynamide (13o). Compound was made and purified using general procedure A, light brown solid 88% yield, 311 mg. 1H NMR (500 MHz, CDCl3) δ 8.49 (dd, J = 4.8, 1.6 Hz, 1H), 8.44 (d, J = 2.3 Hz, 1H), 7.46 (dt, J = 8.0, 2.0 Hz, 1H), 7.11 (dt, J = 10.7, 5.3 Hz, 2H), 6.91 (t, J = 8.6 Hz, 2H), 6.10 (s, 1H), 6.02 (s, 1H), 3.82 (dtd, J = 10.8, 7.3, 4.0 Hz, 1H), 2.04–1.93 (m, 1H), 1.89–1.82 (m, 1H), 1.72 (s, 3H), 1.67–1.56 (m, 2H), 1.45–1.29 (m, 2H), 1.27–1.04 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.24, 163.25, 161.27, 155.15, 151.13, 149.73, 138.05, 134.72, 134.69, 132.76, 132.69, 130.05, 123.21, 115.69, 115.51, 92.45, 73.58, 61.42, 49.01, 32.78, 32.72, 25.43, 24.77, 24.71, 3.89.
N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)-N-(4-(difluoromethoxy)phenyl)but-2-ynamide (13p). Compound was made and purified using general procedure A, light brown solid 89% yield, 355 mg. 1H NMR (500 MHz, CDCl3) δ 8.51 (dd, J = 4.8, 1.6 Hz, 1H), 8.46 (d, J = 2.3 Hz, 1H), 7.50 (dt, J = 8.0, 2.0 Hz, 1H), 7.20–7.10 (m, 2H), 7.03–6.93 (m, 2H), 6.50 (t, J = 73.4 Hz, 1H), 6.10 (s, 1H), 5.93 (d, J = 8.1 Hz, 1H), 3.83 (dtd, J = 10.8, 7.7, 7.3, 4.0 Hz, 1H), 1.98 (s, 1H), 1.87 (dd, J = 12.1, 4.0 Hz, 1H), 1.76–1.71 (m, 4H), 1.70–1.57 (m, 2H), 1.45–1.28 (m, 2H), 1.24–1.03 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.53, 155.46, 151.37, 150.03, 138.52, 132.78, 130.44, 123.65, 119.71, 115.88, 92.99, 73.95, 61.89, 49.41, 33.17, 33.11, 25.81, 25.14, 25.09, 4.30, 4.16. 19F NMR (471 MHz, CDCl3) δ −81.48 (d, J = 73.5 Hz). HRMS (ESI/Q-TOF) m/z: [M + H]+ calculated for C24H26N3O3F2 442.19367; found 442.19361.
N-benzyl-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-3-yl)ethyl)propiolamide (13q). Pyridine-3-carboxaldehyde (0.09 mL, 0.93 mmol) and benzylamine (0.10 mL, 0.93 mmol) were stirred in MeOH (3.7 mL) and for 30 min at rt. 2-Butynoic acid (78.5 mg, 0.93 mmol) and cyclohexyl isocyanide (0.10 mL, 0.84 mmol) were added and the solution stirred at rt overnight. The reaction was concentrated in vacuo and the crude product was dissolved in EtOAc washed with water and brine. The resulting aqueous phases were extracted three times for residual compound. The combined organic phases were washed with brine and dried over Na2SO4. The crude product was purified using column chromatography with a gradient of mixture of (MeOH + 1% NH4OH) and DCM (0% → 5%). The reaction was further purified using column chromatography with a gradient of EtOAc in hexanes (0% → 100%). White powder (114 mg, 35% yield). Rf (5% (MeOH + 1% NH4OH) in DCM) = 0.39. 1H NMR (500 MHz, CDCl3) δ 8.57–8.49 (m, 0.4H), 8.43 (t, J = 2.9 Hz, 1.6H), 7.75 (dt, J = 8.1, 1.9 Hz, 0.8H), 7.64 (d, J = 7.9 Hz, 0.2H), 7.25–7.16 (m, 3H), 7.11 (dd, J = 7.4, 2.0 Hz, 1.4H), 7.03 (dd, J = 6.7, 2.8 Hz, 0.3H), 6.14 (s, 0.3H), 6.02 (d, J = 8.0 Hz, 0.7H), 5.73 (d, J = 8.1 Hz, 0.3H), 5.64 (s, 0.7H), 5.06 (d, J = 16.2 Hz, 0.7H), 4.79 (d, J = 16.2 Hz, 0.7H), 4.58 (d, J = 15.3 Hz, 0.3H), 4.52 (d, J = 15.3 Hz, 0.3H), 3.75–3.64 (m, 1H), 2.04 (d, J = 9.0 Hz, 0.7H), 1.98 (s, 2.1H), 1.85–1.77 (m, 2H), 1.71–1.52 (m, 4H), 1.37–1.23 (m, 2H), 1.21–0.98 (m, 2H). 13C NMR (126 MHz, CDCl3, ∗: major isomer) δ 167.05, 166.83∗, 156.12, 150.98∗, 150.72, 149.83∗, 149.60, 137.46∗, 137.30∗, 137.19, 136.84, 130.54∗, 130.44, 128.92∗, 128.67, 128.25∗, 127.75∗, 127.70, 127.43, 123.43∗, 123.26, 92.78, 91.19, 73.52, 73.15, 64.66∗, 60.61, 52.48, 48.81, 47.03∗, 32.78, 32.72, 32.66, 25.57, 25.46, 24.80, 24.76, 4.39∗, 4.27. HRMS (ESI/Q-TOF) m/z: [M+Na]+ calculated for C24H27N3NaO2 412.1995, found 412.1995.
5.1.1. In vitro assays
Protein production. The DNA sequence encoding 3CL SARS-CoV-2 protease (GI: 1831502838) was synthetized (optimized for expression in Escherichia coli) and cloned into a pGEX-6-1 vector between BamHI and XhoI restriction sites, by GenScript (Piscataway, NJ, USA), based on Zhang et al. methodology. [25]. The protein sequence was preceded by an N-terminal GST sequence and the HRV3C PreScission protease site (SAVLQ↓SGFRK). Autocleavage activity of the enzyme leaves its N-terminus starting with the serine after the QS cleavage site (Q↓S). At the C-terminus, based on Xue et al. [73], and Zhang et al. [25], the protein was designed to end at the last glutamine comprising the natural C-terminus of the enzyme, followed by a glycine, a proline residue and six histidine residues. The protein this has a 6-His purification tag that can be cleaved using a modified-PreScission protease approach (SGVTFQ↓GP).
E. coli BL21 (DE3) cells were transformed using the plasmid described above and the CaCl2 method [74] and colonies were selected on LB-agar-ampicillin plates. A single colony was picked and grew over night in LB media and then used to inoculate a 400 mL LB culture. This was grown at 37 °C, 250 rpm, until an optical density at 600 nm of 0.7 (O.D.600) was reached and then induced for 18 h at 16 °C (250 rpm) by adding 0.25 mM IPTG. The bacterial pellet was collected after 4000 rpm centrifugation and resuspended in 30 mL of 50 mM Tris-HCl pH 8.0, 150 mM NaCl (resuspension buffer). An Ultrasonic Cell Disruption Sonifier 450 (Branson) was used to sonicate the cells (on ice) with 6 pulses of 2 min (Output Control 7 and 50% Duty Cycle).
Buffer A (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 20 mM imidazole) and Buffer B (50 mM Tris-HCl, 150 mM NaCl, 500 mM imidazole) were used for purifying 3CLpro after the clarified supernatant (14,000 rpm centrifugation) was loaded into a 1 mL HisTrap FF (GE Healthcare, IL, USA). 5 mL fractions were collected by washing with single column volumes of increasing imidazole concentration (0, 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50% Buffer B).
After running SDS-PAGE for confirmation (Mini-PROTEAN® TGX™ Precast Gels, Bio-Rad), two sets of fractions were pooled independently. One of them contained 3CLpro with a high molecular weight contaminant (5 and 10% Buffer B) while the other contained only the pure enzyme (15, 20 and 25% buffer B). Imidazole was eliminated from pooled fractions by multiple washes with resuspension buffer using 10K cut-off Amicon® Ultra-15 Centrifugal Filter Units (Millipore Sigma, Burlington, MA, USA). Absorbance at 280 nm was recorded for the pure sample and the protein concentration was estimated to be 8.5 μM (around 0.3 g/L). Based on SDS-PAGE results, the same concentration was assumed for the other sample. These samples were aliquoted and stored at −20 Celsius degrees. In all the experiments (unless specified) the pure preparation was used. See Fig. S1 for more information.
Bulk amount of 3CLpro for crystallization was purified from cell pellets resuspended in a buffer (50 mM Tris, 150 mM NaCl, 20 mM imidazole, pH 8.0). Cell lysate was subsequently centrifuged at 15,000g at 4 °C for 20 min. The supernatant was further loaded on a HisTrap HP column (Cytiva) and then purified using a second buffer (50 mM Tris, 150 mM NaCl, 500 mM imidazole, pH 8.0) on an AKTA Avant purification system (Cytiva). The fractions were pooled, the his-tag removed with Pierce HRV 3C protease (Thermo Scientific) at 4 °C for 24 h and dialyzed in buffer A with TCEP at 0.5 mM. The unlabeled protein was loaded again on a HisTrap HP column (Cytiva), the flow-through was collected and dialyzed in a third buffer (20 mM Tris, 100 mM NaCl, 0.5 mM TCEP, pH 8.0). A second step of purification was carried out on a HiTrap Q FF column using the third buffer (20 mM Tris, 100 mM NaCl, 0.5 mM TCEP, pH 8.0) The fractions were pooled and concentrated to 12 mg/mL.
Detection of inhibitors by fluorescence spectrophotometry. For all inhibition experiments, reactions were performed in 50 μL assay with 11.76 μM fluorescence substrate DABCYL-KTSAVLQSGFRKME-EDANS from BPS Biosciences (San Diego, CA, USA), which has been previously used for assaying 3CL proteases [75].
38 μL of enzyme (diluted in a 3CLpro Protease Assay Buffer from BPS Bioscience supplemented with 1 mM DTT) were incubated for 30 min at room temperature with 2 μL of 1.25 mM compounds (diluted in DMSO; Sigma). In both cases, screenings (duplicates) and IC50 experiments (duplicates and triplicates), 10 μL of diluted substrate (58.8 μM) were added and reactions monitored by following the fluorescence as a function of time (excitation at 360 nm, emission at 460 nm) using a Synergy™ H4 Hybrid Multi-Mode Microplate Reader (Winooski, VT, USA). Controls were (i) no inhibition: 2 μL of DMSO, (ii) positive inhibition: 2 μL GC376 (500 μM, BPS Bioscience) diluted in distilled water and (iii) blank: 38 μL of the 3CLpro Protease Assay Buffer with 1 mM DTT was added instead of the enzyme, together with 2 μL of DMSO. In all cases the compounds were added as DMSO solutions and their final concentration in the reactions was 50 μM. The reactions ran for at least 1 h, and the linear initial slopes of the progress curves were used to calculate the reaction initial velocity in Relative Fluorescent Units in time (RFU per minute). GraphPad software was used to determine IC50 values.
Liquid chromatography-mass spectrometry (LC-MS). Stored-frozen protein samples were buffer exchanged with 5 mM Tris-HCl pH 8.0, 15 mM NaCl buffer using 4K cut-off Amicon® small Centrifugal Filter Units (Millipore Sigma, Burlington, MA, USA) and prepared at 0.1–0.2 mg/mL 96 μL of this solution was mixed with 4 μL of 2.5 mM 16a or 14a. Pure DMSO was used as a negative control. Protein samples were analyzed by LC-MS using a Dionex Ultimate 3000 UHPLC coupled to a Bruker Maxis Impact Q-TOF in positive ESI mode. Samples were separated on an Agilent PLRP-S column (1000 Å, 5 μM, 2.1 × 50 mm) heated to 80 °C at a flow rate of 0.5 ml/min using a gradient of 80% mobile phase A (0.1% formic acid in H2O) and 20% mobile phase B (0.1% formic acid in ACN) for 2 min, ramped to 60% mobile phase A and 40% mobile phase B in 5 min, and 10% mobile phase A and 90% mobile phase B in 5 min. The data was processed and spectra deconvoluted using the Bruker DataAnalysis software version 4.2.
Isothermal Titration Calorimetry (ITC). All ITC experiments were performed on an ITC200 instrument (Malvern Panalytical Ltd, UK) in high-feedback mode with a 1s signal averaging window, a stirring rate of 750 rpm, pre-injection delay of 120 s, and a reference power of 7. A small first injection of 0.2 μL was followed by a 1.5 μL injection with a wait period of 1400 s. The sample cell contained 0.5 mg/mL of the peptide Cbz-TSAVLQSGFRK (CanPeptide, Montreal, QC, Canada) with either 16.7 μM inhibitor no inhibitor, while the injection syringe contained 54.6 μM 3CLpro. All syringe and sample cell components were dissolved in 3CLpro Protease Assay Buffer from BPS Bioscience.
Cathepsin L fluorescence assays: Cathepsin L activity was measured by Cathepsin L Inhibitor Screening Assay Kit (BPS Bioscience, USA). 3CLpro inhibitor concentrations were tested from 0.1 to 100 μM, following the kit's instructions. E−64 protease inhibitor at 10 μM was used such as positive control. The percentage of inhibition for each molecule was calculated based on slope values versus a blank condition experiment without inhibitor. All the samples were processed in duplicate in a Synergy™ H4 Hybrid Multi-Mode Microplate Reader (Winooski, VT, USA) (excitation at 360 nm, emission at 460 nm) in 100 μL assays. 10 min of incubation at room temperature (inhibitors and enzyme).
Protein crystallization and structure solution. The enzyme was buffer exchanged into 20 mM Tris pH8, 100 mM NaCl, 1 mM DTT and concentrated to 5 mg/ml, and was then incubated with 450 μM of compound 16a for 1 h at room temperature. Following incubation, the sample was filtered using a 0.22 μm filter and used for crystallization trials. Crystals were grown using the sitting drop method at 22 °C. 200 nL enzyme was mixed with 200 nL well solution (30% PEG2000 MME, 0.2 M Potassium thiocyanate) and allowed to equilibrate against 50 μL well solution. The crystals were cryo-protected using well solution supplemented with 20% ethylene glycol and flash frozen in liquid nitrogen. Data was collected at the Canadian Light Source CMCF-BM beamline and processed in space group C 1 2 1. The structure was solved in PHASER [76] with a previously published structure of the enzyme (6WTK) [26] as a search model. Restraints for the covalently bonded inhibitor were generated using AceDRG [77] in CCP4i2 [78], and the model was refined with REFMAC5 [79] and Coot [80].
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We thank the Canadian Institutes of Health Research (CIHR, OV3-170644), the McGill Interdisciplinary Initiative in Infection and Immunity (MI4) and the Faculty of Science for funding.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.114046.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Addition information on LC-MS data, ITC measurement, dose-response curves and 1H and 13C NMR spectra.
References
- 1.https://www.who.int/
- 2.https://www.ecdc.europa.eu/en/publications-data/distribution-confirmed-cases-mers-cov-place-infection-and-month-onset-march-2012
- 3.Schlottau K., Rissmann M., Graaf A., Schön J., Sehl J., Wylezich C., Höper D., Mettenleiter T.C., Balkema-Buschmann A., Harder T., Grund C., Hoffmann D., Breithaupt A., Beer M. SARS-CoV-2 in fruit bats, ferrets, pigs, and chickens: an experimental transmission study. Lancet Micr. 2020;1:E218–E225. doi: 10.1016/S2666-5247(20)30089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi Z., Hu Z. A review of studies on animal reservoirs of the SARS coronavirus. Virus Res. 2008;133:74–87. doi: 10.1016/j.virusres.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdollahi E., Champredon D., Langley J.M., Galvani A.P., Moghadas S.M. Temporal estimates of case-fatality rate for COVID-19 outbreaks in Canada and the United States. Can. Med. Assoc. J. 2020;192:E666–E670. doi: 10.1503/cmaj.200711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.https://www.imperial.ac.uk/mrc-global-infectious-disease-analysis/news--wuhan-coronavirus/
- 7.Organization W.H. COVID-19) Dashboard; 2020. WHO Coronavirus Disease. [Google Scholar]
- 8.Modi C., Böhm V., Ferraro S., Stein G., Seljak U. Estimating COVID-19 mortality in Italy early in the COVID-19 pandemic. Nat. Commun. 2021;12:2729. doi: 10.1038/s41467-021-22944-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stankiewicz K. 2021. Pfizer's CEO Says Covid Vaccine Effectiveness Drops to 84% after Six Months. [Google Scholar]
- 10.Ledford H. Six months of COVID vaccines: what 1.7 billion doses have taught scientists. Nature. 2021;594:164–167. doi: 10.1038/d41586-021-01505-x. [DOI] [PubMed] [Google Scholar]
- 11.Dai L., Gao G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021;21:73–82. doi: 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chodick G., Tene L., Rotem R.S., Patalon T., Gazit S., Ben-Tov A., Weil C., Goldshtein I., Twig G., Cohen D., Muhsen K. The effectiveness of the two-dose BNT162b2 vaccine: analysis of real-world data. Clin. Infect. Dis. 2021 doi: 10.1093/cid/ciab438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merck Merck, Molnupiravir Ridgeback’s. Receives First Authorization in the World; 2021. An Oral COVID-19 Antiviral Medicine. [Google Scholar]
- 14.Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., Zheng X.-S., Zhao K., Chen Q.-J., Deng F., Liu L.-L., Yan B., Zhan F.-X., Wang Y.-Y., Xiao G.-F., Shi Z.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P., Meng J., Zhu Z., Zhang Z., Wang J., Sheng J., Quan L., Xia Z., Tan W., Cheng G., Jiang T. Cell Host & Microbe; 2020. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rathnayake A.D., Kim Y., Dampalla C.S., Nguyen H.N., Jesri A.-R.M., Kashipathy M.M., Lushington G.H., Battaile K.P., Lovell S., Chang K.-O., Groutas W.C. Structure-guided optimization of dipeptidyl inhibitors of norovirus 3CL protease. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.0c01252. [DOI] [PubMed] [Google Scholar]
- 18.Kuo C.-J., Shie J.-J., Fang J.-M., Yen G.-R., Hsu J.T.A., Liu H.-G., Tseng S.-N., Chang S.-C., Lee C.-Y., Shih S.-R., Liang P.-H. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem. 2008;16:7388–7398. doi: 10.1016/j.bmc.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dragovich P.S., Prins T.J., Zhou R., Webber S.E., Marakovits J.T., Fuhrman S.A., Patick A.K., Matthews D.A., Lee C.A., Ford C.E., Burke B.J., Rejto P.A., Hendrickson T.F., Tuntland T., Brown E.L., Meador J.W., Ferre R.A., Harr J.E.V., Kosa M.B., Worland S.T. Structure-based design, synthesis, and biological evaluation of irreversible human Rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 1999;42:1213–1224. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- 20.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 21.Su H.-x., Yao S., Zhao W.-f., Li M.-j., Liu J., Shang W.-j., Xie H., Ke C.-q., Hu H.-c., Gao M.-n., Yu K.-q., Liu H., Shen J.-s., Tang W., Zhang L.-k., Xiao G.-f., Ni L., Wang D.-w., Zuo J.-p., Jiang H.-l., Bai F., Wu Y., Ye Y., Xu Y.-c. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kneller D.W., Galanie S., Phillips G., O'Neill H.M., Coates L., Kovalevsky A. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure. 2020;28:1313–1320. doi: 10.1016/j.str.2020.10.007. e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kneller D.W., Phillips G., O'Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 29.Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westberg M., Su Y., Zou X., Ning L., Hurst B., Tarbet B., Lin M.Z. bioRxiv; 2020. Rational Design of a New Class of Protease Inhibitors for the Potential Treatment of Coronavirus Diseases. 2020.2009.2015.275891. [Google Scholar]
- 31.Iketani S., Forouhar F., Liu H., Hong S.J., Lin F.-Y., Nair M.S., Zask A., Huang Y., Xing L., Stockwell B.R., Chavez A., Ho D.D. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021;12 doi: 10.1038/s41467-021-22362-2. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., Hammond J., Hoffman R., Kadar E.P., Kania R., Kimoto E., Kirkpatrick M.G., Lanyon L., Lendy E.K., Lillis J.R., Luthra S.A., Ma C., Noell S., Obach R.S., O’ Brien M.N., O'Connor R., Ogilvie K., Owen D., Pettersson M., Reese M.R., Rogers T.F., Rossulek M.I., Sathish J.G., Steppan C., Ticehurst M., Updyke L.W., Zhu Y., Wang J., Chatterjee A.K., Mesecar A.D., Anderson A.S., Allerton C. bioRxiv; 2021. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease as a Clinical Candidate for the Potential Treatment of COVID-19. 2020.2009.2012.293498. [Google Scholar]
- 33.Hoffman R., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., Lockner J.W., Lou J., Marra M.T., Mitchell L.J.J., Murraya B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J.I., Solowiej J.E., Steppan C.M., Taggart B. The discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12631496.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., Lockner J.W., Lou J., Marra M.T., Mitchell L.J., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., Solowiej J.E., Steppan C.M., Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Study of PF-07321332 in Healthy Participants - NCT04756531. 2021. [Google Scholar]
- 36.D.R. Owen, C.M.N. Allerton, A.S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R.D. Cardin, A. Carlo, K.J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K.S. Gajiwala, S.A. Gibson, S.E. Greasley, B.L. Hurst, E.P. Kadar, A.S. Kalgutkar, J.C. Lee, J. Lee, W. Liu, S.W. Mason, S. Noell, J.J. Novak, R.S. Obach, K. Ogilvie, N.C. Patel, M. Pettersson, D.K. Rai, M.R. Reese, M.F. Sammons, J.G. Sathish, R.S.P. Singh, C.M. Steppan, A.E. Stewart, J.B. Tuttle, L. Updyke, P.R. Verhoest, L. Wei, Q. Yang, Y. Zhu, An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19, Science, 0 eabl4784. [DOI] [PubMed]
- 37.Pfizer . 2021. Pfizer's Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk of Hospitalization O Death by 89% in Iterim Analysis of Phase 2/3 EPIC-HR Study. [Google Scholar]
- 38.Bai B., Belovodskiy A., Hena M., Kandadai A.S., Joyce M.A., Saffran H.A., Shields J.A., Khan M.B., Arutyunova E., Lu J., Bajwa S.K., Hockman D., Fischer C., Lamer T., Vuong W., van Belkum M.J., Gu Z., Lin F., Du Y., Xu J., Rahim M., Young H.S., Vederas J.C., Tyrrell D.L., Lemieux M.J., Nieman J.A. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai B., Arutyunova E., Khan M.B., Lu J., Joyce M.A., Saffran H.A., Shields J.A., Kandadai A.S., Belovodskiy A., Hena M., Vuong W., Lamer T., Young H.S., Vederas J.C., Tyrrell D.L., Lemieux M.J., Nieman J.A. Peptidomimetic nitrile warheads as SARS-CoV-2 3CL protease inhibitors. RSC Med. Chem. 2021;12:1722–1730. doi: 10.1039/d1md00247c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., Snijder E.J., Liu H., Hilgenfeld R. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 41.Kim Y., Liu H., Galasiti Kankanamalage A.C., Weerasekara S., Hua D.H., Groutas W.C., Chang K.-O., Pedersen N.C. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vuong W., Fischer C., Khan M.B., van Belkum M.J., Lamer T., Willoughby K.D., Lu J., Arutyunova E., Joyce M.A., Saffran H.A., Shields J.A., Young H.S., Nieman J.A., Tyrrell D.L., Lemieux M.J., Vederas J.C. Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021;222:113584. doi: 10.1016/j.ejmech.2021.113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen L.-R., Wang Y.-C., Lin Y.W., Chou S.-Y., Chen S.-F., Liu L.T., Wu Y.-T., Kuo C.-J., Chen T.S.-S., Juang S.-H. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett. 2005;15:3058–3062. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J., Huitema C., Niu C., Yin J., James M.N.G., Eltis L.D., Vederas J.C. Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3C-like proteinase. Bioorg. Chem. 2008;36:229–240. doi: 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang C.-H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., Spasov K.A., Zhang S., Takeo Y., Kudalkar S.N., Liang Z., Isaacs F., Lindenbach B., Miller S.J., Anderson K.S., Jorgensen W.L. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug Perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021 doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drayman N., Jones K.A., Azizi S.-A., Froggatt H.M., Tan K., Maltseva N.I., Chen S., Nicolaescu V., Dvorkin S., Furlong K., Kathayat R.S., Firpo M.R., Mastrodomenico V., Bruce E.A., Schmidt M.M., Jedrzejczak R., Muñoz-Alía M.Á., Schuster B., Nair V., Botten J.W., Brooke C.B., Baker S.C., Mounce B.C., Heaton N.S., Dickinson B.C., Jaochimiak A., Randall G., Tay S. bioRxiv; 2020. Drug Repurposing Screen Identifies Masitinib as a 3CLpro Inhibitor that Blocks Replication of SARS-CoV-2 in Vitro. 2020.2008.2031.274639. [Google Scholar]
- 47.Zhu W., Xu M., Chen C.Z., Guo H., Shen M., Hu X., Shinn P., Klumpp-Thomas C., Michael S.G., Zheng W. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 2020 doi: 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Cesco S., Kurian J., Dufresne C., Mittermaier A., Moitessier N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017;138:96–114. doi: 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 49.Zhou S., Chan E., Duan W., Huang M., Chen Y.Z. Drug Metab. Rev.; 2005. Drug Bioactivation, Covalent Binding to Target Proteins and Toxicity Relevance; pp. 41–213. [DOI] [PubMed] [Google Scholar]
- 50.Guterman L. Covalent drugs form long-lived ties. C&EN. 2011;89:19–26. [Google Scholar]
- 51.Tang B., He F., Liu D., Fang M., Wu Z., Xu D. 2020. AI-aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. bioRxiv. 2020.2003.2003.972133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen D.D., Gao K., Chen J., Wang R., Wei G.-W. bioRxiv; 2020. Potentially Highly Potent Drugs for 2019-nCoV. 2020.2002.2005.936013. [Google Scholar]
- 53.Roy R., Sk M.F., Jonniya N.A., Poddar S., Kar P. ChemRxiv; Preprint: 2020. Finding Potent Inhibitors against SARS-CoV-2 Main Protease through Virtual Screening, ADMET, and Molecular Dynamic Simulation Studies. [DOI] [PubMed] [Google Scholar]
- 54.Mariaule G., De Cesco S., Airaghi F., Kurian J., Schiavini P., Rocheleau S., Huskić I., Auclair K., Mittermaier A., Moitessier N. 3-Oxo-hexahydro-1H-isoindole-4-carboxylic acid as a drug chiral bicyclic scaffold: structure-based design and preparation of conformationally constrained covalent and noncovalent prolyl oligopeptidase inhibitors. J. Med. Chem. 2016;59:4221–4234. doi: 10.1021/acs.jmedchem.5b01296. [DOI] [PubMed] [Google Scholar]
- 55.Plescia J., Dufresne C., Janmamode N., Wahba A.S., Mittermaier A.K., Moitessier N. Discovery of covalent prolyl oligopeptidase boronic ester inhibitors. Eur. J. Med. Chem. 2020;185:111783. doi: 10.1016/j.ejmech.2019.111783. [DOI] [PubMed] [Google Scholar]
- 56.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. Discovery, synthesis, and structure-based optimization of a series of N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaidman D., Gehrtz P., Filep M., Fearon D., Prilusky J., Duberstein S., Cohen G., Owen D., Resnick E., Strain-Damerell C., Lukacik P., Barr H., Walsh M.A., von Delft F., London N. bioRxiv; 2020. An Automatic Pipeline for the Design of Irreversible Derivatives Identifies a Potent SARS-CoV-2 Mpro Inhibitor. 2020.2009.2021.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mesecar A.D. 2020. Structure of COVID-19 Main Protease Bound to Potent Broad-Spectrum Non-covalent Inhibitor X77. [DOI] [Google Scholar]
- 59.Moitessier N., Pottel J., Therrien E., Englebienne P., Liu Z., Tomberg A., Corbeil C.R. Medicinal chemistry projects requiring imaginative structure-based drug design methods. Acc. Chem. Res. 2016;49:1646–1657. doi: 10.1021/acs.accounts.6b00185. [DOI] [PubMed] [Google Scholar]
- 60.Behnke D., Taube R., Illgen K., Nerdinger S., Herdtweck E. Substituted 2-(Cyanomethyl-amino)-acetamides by a novel three-component reaction. Synlett. 2004:688–692. 2004. [Google Scholar]
- 61.Gedey S., Van der Eycken J., Fülöp F. Liquid-phase combinatorial synthesis of alicyclic β-lactams via Ugi four-component reaction. Org. Lett. 2002;4:1967–1969. doi: 10.1021/ol025986r. [DOI] [PubMed] [Google Scholar]
- 62.Dhake K.P., Tambade P.J., Singhal R.S., Bhanage B.M. An efficient, catalyst- and solvent-freeN-formylation of aromatic and aliphatic amines. Green Chem. Lett. Rev. 2011;4:151–157. [Google Scholar]
- 63.Patil P., Ahmadian-Moghaddam M., Dömling A. Isocyanide 2.0. Green Chem. 2020;22:6902–6911. [Google Scholar]
- 64.St John S.E., Mesecar A.D. 2018. Broad Spectrum Non-covalent Coronavirus Protease Inhibitors. USA. [Google Scholar]
- 65.Petri L., Egyed A., Bajusz D., Imre T., Hetényi A., Martinek T., Ábrányi-Balogh P., Keserű G.M. An electrophilic warhead library for mapping the reactivity and accessibility of tractable cysteines in protein kinases. Eur. J. Med. Chem. 2020;207:112836. doi: 10.1016/j.ejmech.2020.112836. [DOI] [PubMed] [Google Scholar]
- 66.Turlington M., Chun A., Tomar S., Eggler A., Grum-Tokars V., Jacobs J., Daniels J.S., Dawson E., Saldanha A., Chase P., Baez-Santos Y.M., Lindsley C.W., Hodder P., Mesecar A.D., Stauffer S.R. Discovery of N-(benzo[1,2,3]triazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg. Med. Chem. Lett. 2013;23:6172–6177. doi: 10.1016/j.bmcl.2013.08.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Di Trani J.M., De Cesco S., O'Leary R., Plescia J., do Nascimento C.J., Moitessier N., Mittermaier A.K. Rapid measurement of inhibitor binding kinetics by isothermal titration calorimetry. Nat. Commun. 2018;9:893. doi: 10.1038/s41467-018-03263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bevington P.R., Robinson D.K. WCB/McGraw-Hill; New York: 1992. Data Reduction and ErrorAnalysis for the Physical Sciences. [Google Scholar]
- 69.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X., Zhang F., Zhang X., Ba M., Szeto T., Kukuljac A., Marty M.T., Schultz D., Cherry S., Xiang Y., Chen Y., Wang J. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steuten K., Kim H., Widen J.C., Babin B.M., Onguka O., Lovell S., Bolgi O., Cerikan B., Neufeldt C.J., Cortese M., Muir R.K., Bennett J.M., Geiss-Friedlander R., Peters C., Bartenschlager R., Bogyo M. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis. 2021 doi: 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao M.-M., Yang W.-L., Yang F.-Y., Zhang L., Huang W.-J., Hou W., Fan C.-F., Jin R.-H., Feng Y.-M., Wang Y.-C., Yang J.-K. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021;6:134. doi: 10.1038/s41392-021-00558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rogachev V.O., Metz P. Thermal and high pressure intramolecular Diels–Alder reaction of vinylsulfonamides. Nat. Protoc. 2006;1:3076–3087. doi: 10.1038/nprot.2006.463. [DOI] [PubMed] [Google Scholar]
- 73.Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 2007;366:965–975. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 75.Chen S., Chen L.-l., Luo H.-b., Sun T., Chen J., Ye F., Cai J.-h., Shen J.-k., Shen X., Jiang H.-l. Enzymatic activity characterization of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer technique. Acta Pharmacol. Sin. 2005;26:99–106. doi: 10.1111/j.1745-7254.2005.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Long F., Nicholls R.A., Emsley P., Graǽulis S., Merkys A., Vaitkus A., Murshudov G.N. Validation and extraction of molecular-geometry information from small-molecule databases. Acta Crystallogr. D Struct. Biol. 2017;73:103–111. doi: 10.1107/S2059798317000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Potterton L., Agirre J., Ballard C., Cowtan K., Dodson E., Evans P.R., Jenkins H.T., Keegan R., Krissinel E., Stevenson K., Lebedev A., McNicholas S.J., Nicholls R.A., Noble M., Pannu N.S., Roth C., Sheldrick G., Skubak P., Turkenburg J., Uski V., von Delft F., Waterman D., Wilson K., Winn M., Wojdyr M. CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr. D Struct. Biol. 2018;74:68–84. doi: 10.1107/S2059798317016035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Skubák P., Murshudov G.N., Pannu N.S. Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2196–2201. doi: 10.1107/S0907444904019079. [DOI] [PubMed] [Google Scholar]
- 80.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.