

Abstract
Familial hyperkalemic hypertension (FHHt) is caused by pathogenic variants in genes of the CUL3-KLHL3-WNK pathway, manifesting clinically as hyperkalemia, metabolic acidosis, and high systolic blood pressure. The ubiquitin E3 ligase CUL3-KLHL3 targets WNK kinases for degradation to limit activation of the thiazide-sensitive Na-Cl cotransporter (NCC). All known variants in CUL3 lead to exon 9 skipping (CUL3Δ9) and typically result in severe FHHt and growth disturbances in patients. Whether other variants in CUL3 cause FHHt is unknown. Here we identify a novel de novo heterozygous CUL3 variant (CUL3Δ474–477) in a pediatric FHHt patient with multiple congenital anomalies and reveal molecular mechanisms by which the CUL3Δ474–477 leads to dysregulation of CUL3-KLHL3-WNK signaling axis. Using patient-derived urinary extracellular vesicles (uEVs) and dermal fibroblasts, in vitro assays, and cultured kidney cells, we demonstrate that CUL3Δ474–477 causes reduced total CUL3 levels due to increased auto-ubiquitination. The CUL3Δ474–477 that escapes auto-degradation shows enhanced modification with NEDD8 and increased formation of CUL3-KLHL3 complexes that are impaired in ubiquitinating WNK4. Proteomic analysis of CUL3 complexes revealed that, in addition to increased KLHL3 binding, the CUL3Δ474–477 variant also exhibits increased interactions with other BTB substrate adaptors, providing a rationale for the patient’s diverse phenotypes. We conclude that the pathophysiological effects of CUL3Δ474–477 are caused by reduced CUL3 levels and formation of catalytically impaired CUL3 ligase complexes.
Keywords: Familial hyperkalemic hypertension, CUL3, CRL3, developmental disease, ubiquitination, neddylation
INTRODUCTION
Hypertension, hyperkalemia, and metabolic acidosis are the fundamental clinical manifestations of Familial hyperkalemic hypertension (FHHt), also known as Pseudohypoaldosteronism type II (PHAII, OMIM: 145260) or Gordon’s Syndrome.1 The electrolyte imbalances and hypertension of FHHt patients are caused by hyperphosphorylation and overactivation of the thiazide-sensitive sodium-chloride cotransporter (NCC) in the nephron’s distal convoluted tubule (DCT).2 Thiazide diuretics improve FHHt conditions of all severities.3–5 Two kinases, i.e., sterile 20/SPS-1 related proline/alanine-rich kinase (SPAK) and oxidative stress response kinase-1 (OSR1), phosphorylate NCC.6–8 The activity of SPAK/OSR1 is up-regulated through phosphorylation, mediated by the lysine-deficient protein kinases, i.e., WNKs (with no-lysine (K) kinase).9, 10 WNK activity, in turn, is controlled by ubiquitination and subsequent proteasomal degradation. This process requires the interaction of CULLIN-3 (CUL3) with a BTB domain (Bric-a-brac, Tramtrack, and Broad complex) containing substrate adaptor kelch-like-family-member-3 (KLHL3).11–13
CUL3 is a critical subunit of the Cullin 3-RING ubiquitin ligase (CRL3) complex. It acts as a protein scaffold for one of ~90 BTB proteins that serves as substrate-specific adaptors to facilitate the ubiquitination of CUL3 substrates.14–16 In combination with BTB proteins, CUL3 controls many independent, non-renal physiological processes.17, 18 Full activity of the CRL3 complex depends upon site-specific modification of CUL3 with NEDD8 (“neddylation”), which mediates multivalent interactions within CRL assemblies.19 Correspondingly, the removal of NEDD8 (“deneddylation”) reduces or “turns off” the active, neddylated CRLs.20 The deneddylation of CUL3 is carried out by the multi-subunit protein complex, COP9 Signalosome (CSN). While the CSN complex as a whole contains 8 subunits, its catalytic deneddylase activity operates solely through the CSN5/JAB1 subunit.21 Specific deletion of CSN/Jab1 along the mouse nephron regulates the substrate abundance of CUL3.22
Individuals with pathogenic variants in WNK1,9 WNK4,9 and KLHL323, 24 typically produce FHHt symptoms in adulthood with rare cases demonstrating pediatric onset.25 In contrast, CUL3 variants are commonly presented in infants and children and induce FHHt symptoms and growth failure.24, 26–28All known FHHt-causing CUL3 variants are autosomal dominant and lead to exon 9 skipping (CUL3Δ9) resulting in a deletion of 57 amino acids (403–459) within the 4-helix bundle (4HB) domain of CUL3.24 This deletion results in aberrant CUL3 ubiquitin ligase activity, increased activation of the WNK-SPAK/OSR1-NCC cascade, and electrolyte imbalances of FHHt.29, 30 The 403–459 deletion is thought to induce structural flexibility in the CUL3 scaffold, which, compared to WT CUL3, increases NEDD8-conjugated CUL3 relative to the unmodified CUL3 pool (“hyperneddylation”). CUL3Δ403–459 hyperneddylation is known to induce either enhanced auto-ubiquitination30 or increased KLHL3 binding and ubiquitination of this substrate adaptor29. Whether other variants in CUL3 can lead to FHHt is unknown.
In this report, we identify a novel de novo heterozygous CUL3 variant (CUL3Δ474–477) in a pediatric FHHt patient with global developmental delay, short stature, and dysmorphic facial features. Using patient-derived urinary extracellular vesicles and dermal fibroblasts, in vitro assays, and cultured kidney cells, we demonstrate that the reduced CUL3 levels and formation of catalytically impaired ubiquitin ligation complexes are molecular causes of FHHt and other developmental malformations in the patient.
METHODS
All supporting data are available within the article and its online supplementary files. A detailed description of methods including whole exome and Sanger sequencing, cell culture, site-directed mutagenesis, and transfection, in vitro assays, Quantitative PCR, and identification of CUL3 interacting proteins by mass spectrometry is available in the Data Supplement.
Clinical Protocols and Consents
The patient and family members were enrolled in protocol 76-HG-0238, “Diagnosis and Treatment of Patients with Inborn Errors of Metabolism and Other Genetic Disorders” approved by the Institutional Review Board (IRB) of the National Human Genome Research Institute (NHGRI). ClinicalTrials.gov Identifier: NCT00369421. The patient’s family gave written informed consent, and the patient was evaluated through the National Institutes of Health (NIH) Undiagnosed Diseases Program (UDP)31–33 at the NIH Clinical Center.
Urinary extracellular vesicles (uEVs)
First morning urine voids were collected from patient and family members to maximize urine volume for uEVs processing. The patient was taken off thiazide diuretics and sodium citrate medications for 36 hours prior to the collection of urine. Urine was then treated with sodium azide (1.67 ml of 100 mM per 50 mL) in addition to protease and phosphatase inhibitor cocktails. Treated urine was then centrifuged at 17000 × g at 25°C for 10 min. The supernatant was then pooled and underwent ultracentrifugation at 200,000 × g for 1 h at 25°C. The uEVs were present in the pellet of this ultracentrifugation. However, to disrupt and remove the Tamm-Horsfall uromodulin’s entrapment of proteins present in the distal convoluted tubule, each pellet was resuspended in isolation solution [250 mM sucrose, 10 mM triethanolamine (pH 7.6)] containing dithiothreitol (200 mg/ml), vortexed and boiled at 95°C for 2 min34. The total volume of resuspended pellet was made up to 7 ml with isolation solution and again underwent ultracentrifugation at 200,000 × g for 1 hour at 4°C. uEVs were suspended in SDS buffer (50 mM Tris Hcl,1.5% SDS) containing protease and phosphatase inhibitors and stored at −80°C until further analysis.
Immunoblotting and coimmunoprecipitation
Cells were harvested from 70–90% confluent flasks of dermal fibroblasts or HEK293T cells. Cells were washed twice with PBS and lysed using RIPA lysis buffer (Sigma-Aldrich) with Protease (Roche) and Phosphatase (Roche) Inhibitors. Lysates were kept on ice for 45 min followed by the sonication. After centrifugation, to pellet insoluble material, supernatants were transferred to a new tube and protein concentrations were measured using the DC protein assay (Bio-Rad, Hercules, CA). 20–30 μg of total protein was resolved through SDS-PAGE on 4–15% Tris-Glycine Gels (BIO-RAD, Hercules, CA) and transferred to PVDF or nitrocellulose membranes via a Trans-Blot Turbo Transfer System (BIO-RAD). Immunoblotting was carried out as described.35 Images were captured on a LI-COR Odyssey CLx imaging system and analyzed using Image Studio software (LI-COR Biosciences).
For coimmunoprecipitation experiments, cells were lysed in ice-cold RIPA or Triton X-100 lysis buffer [50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, protease, and phosphatase inhibitors] and kept on ice for 45 min with occasional mixing, followed by centrifugation to pellet the insoluble material. Equal concentrations of lysates were then incubated with specific primary antibodies for 2 hours. Antigen-antibody complexes were immunoprecipitated using Protein A Sepharose Fast Flow or Protein G Sepharose Fast Flow beads. After incubation at 4°C for 2 hours or overnight, immunoprecipitates were washed three times with 1 ml of lysis buffer and subsequently resuspended in 2XSDS sample buffer with beta-mercaptoethanol and evaluated by immunoblotting.
Purification of CUL3/RBX1 complexes and in vitro assays
CUL3/RBX1 WT and CUL3/RBX1 Δ474–477 complex proteins were purified as GST fusions from E.coli according to a previously established pGEX-based split-N-co-expression system.36, 37 A detailed description of purification method and in vitro ubiquitination, neddylation, and de-neddylation of CUL3/RBX1 complexes is provided in the Data Supplement.
His-ubiquitin pull-down
HEK293T cells were transfected with pCS2-His-ubiquitin and expression vectors as indicated. 48 h after transfection, cells were washed twice with PBS containing 20 μM deubiquitinase inhibitor PR-619 and resuspended in the urea lysis buffer [8 M Urea, 50 mM Tris HCl (pH 7.5) 20 mM imidazole, pH 8.0] and sonicated. After centrifugation at 12000 rpm for 10 min to pellet insoluble material, equal amounts of cell lysates were added to equilibrated Ni-NTA agarose beads followed by incubation for 2 h at room temperature. Beads were then washed three times using urea lysis buffer. The protein conjugates were eluted with 2X Urea sample buffer [6 M Urea, 6% SDS, 150 mM TRIS HCl (pH 6.5), 25% glycerol and bromophenol blue] and analyzed by western blotting.
Identification of CUL3 interacting proteins by mass spectrometry
Identification of CUL3-interacting proteins by mass spectrometry and compPASS analysis was performed as previously described.38, 39 For a detailed description, please see the Data Supplement.
Statistical Analysis
Microsoft Excel was used for the calculation of mean values and standard deviation (SD). Results were expressed as mean ± SD or ± SEM and statistical significance was determined by a two-tailed unpaired t-test and by multiple t-test, respectively. A probability (P) value of < 0.05 was considered significant. Results were plotted using GraphPad Prism software. FIJI imaging software (ver. 1.52p) was used for data collection.
RESULTS
A novel de novo heterozygous CUL3 deletion in a boy with severe FHHt
Through the NIH’s Undiagnosed Diseases Program (UDP), we evaluated a 34-month-old male of non-consanguineous European descent with a history of repeated hospitalizations during the first three years of life. Clinical findings during these early admissions showed failure to thrive and atelectasis of both lower lung lobes and multiple pulmonary infections (See supplemental text for a full clinical description). Notably, serum chemistries during these hospitalizations showed hyperkalemia and metabolic acidosis. Although lacking an official genetic diagnosis, the patient was treated with chlorothiazide and bicarbonate which improved the patient’s electrolyte imbalances. During his inpatient evaluation at the NIH, the boy presented with persistent hyperkalemia, hypertension, and metabolic acidosis when off diuretics; he also showed global developmental delay, short stature, dysmorphic facial features, speech dyspraxia, inability to follow verbal commands, and increased drooling with performance of fine motor skills. Systolic blood pressures were consistently > 110 mmHg with a mean systolic pressure of 125 mmHg over the course of his evaluation; 99th percentile is 111 mmHg for a 36-month-old male with height at the 5th percentile (Table 1A). Serum potassium levels were between 5 and 6.7 mmol/L and serum aldosterone levels were within normal limits (Table 1B). The boy was acidotic with a normal non-anion gap and mild hyperchloremia.
Table 1A. Blood pressure and serum electrolytes values in the proband.
Blood pressure of a 34-month-old FHHt patient prior to his diagnosis and thiazide diuretics (Diuril) administration were measured three separate times during the inpatient admission at the NIH’s Clinical Center through the Undiagnosed Diseases Program (UDP). These values were averaged and compared to reference pediatric blood pressure ranges, from 90th (pre-hypertensive) to 99th (stage II hypertension) percentiles normalized by height (5th percentile) and age (3 years old); results reveal severe pediatric hypertension in the patient. Reference ranges were drawn from the National Heart, Lung and Blood Institute (NHLBI/NIH) Boys and Girls Blood Pressure Levels Guidelines.
| Blood Pressure | Proband | 90–99% Reference Range |
|---|---|---|
| Systolic (mmHg) | 125±6 | 100–111 |
| Diastolic (mmHg) | 73±10 | 59–71 |
Table 1B.
Serum values were compiled over the first 6 years of the patient’s life, prior to prescription of thiazide diuretics. Mean values confirmed elevated serum potassium levels, mildly elevated chloride levels, metabolic acidosis, and normal aldosterone levels.
| Serum Values | Proband | Reference Range |
|---|---|---|
| Na [mmol/L] | 138±3.0 | 135–144 |
| K [mmol/L] | 5.9±0.5 | 3.5–5.1 |
| Cl [mmol/L] | 108±3.5 | 97–106 |
| Ca [[mmol/L] | 2.3±0.6 | 2.05–2.5 |
| Total CO2 (HCO3) [mmol/L] | 19±2.7 | 22–30 |
| Creatinine [mg/dL] | 0.28±0.06 | 0.3–0.7 |
| Aldosterone [ng/dL] | 12±1.41 | <40 |
| Renin [ng/mL/h] | 2.8 | 0.9–2.9 |
| pH | 7.4±0.07 | 7.35–7.45 |
Whole exome and follow-up Sanger sequencing revealed a de novo, heterozygous variant in the patient’s CUL3 gene (NM_003590.5: c.1420_1431del12; p. Phe474_Met477del) (Figure 1A). No sequence variants were found in WNK1, WNK4 or KLHL3. The combined annotation dependent depletion (CADD) Phred score, which is a tool for scoring the deleteriousness of variants in the human genome, was 23.2 for the CUL3 variant, indicating likely pathogenicity. According to the American College of Medical Genetics and Genomics guidelines40, the CUL3 variant was ranked as pathogenic with evidence codes PS2, PS3, PM2, PM4, PP3 and PP4. The deletion is not present in the population frequency gnomAD database.
Figure 1: Identification and effects of a novel CUL3 variant in a pediatric FHHt patient.
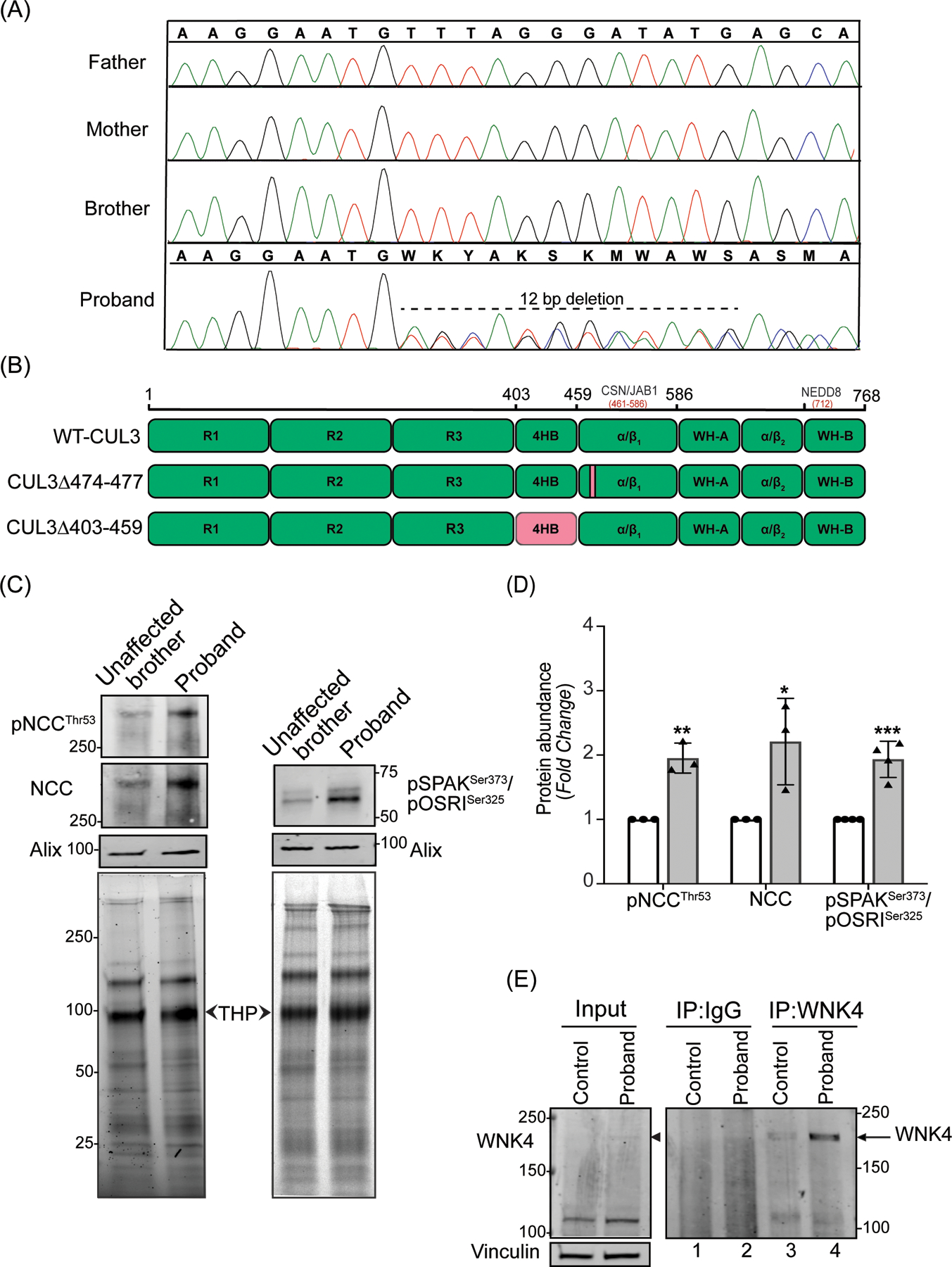
(A) Sanger sequencing chromatograms using genomic DNA from patient (proband) blood samples revealed a de novo heterozygous variant (NM_003590.5: c.1420_1431del12; p. Phe474_Met477del) in the CUL3 gene. CUL3 wild-type genotypes were conserved in the patient’s father, mother, and brother. (B) Domain structure of CUL3 protein shows that the four amino acid deletion (CUL3Δ474–477) in the patient was located within the α/β1 domain of CUL3. This patient’s variant differs from other FHHt-causing CUL3 variants (CUL3Δ403–459), which are located within the 4 helix-bundle (4HB) domain of CUL3. CSN binding site (aa 461–586) is in the α/β1 domain and Nedd8 binding site (K712) is in the WH-B domain of CUL3.
(C) Effects of the CUL3 variant in patient. Immunoblot analysis of uEVs. The patient’s (proband) and his unaffected brother’s urine (first morning void) were processed via ultracentrifugation. Resultant pellets containing uEVs were then treated with dithiothreitol (DTT) to remove Tamm-Horsfall Protein matrices entrapping distal convoluted tubule proteins and lysed in SDS buffer. 10–30 μg of uEVs lysates were separated on 4–15% Tris-Glycine stain-free gels and immunoblotted with antibodies that recognize phosphorylated NCC (pThr53) and total NCC (left panels), and with an antibody that recognizes both phospho SPAK at Ser373 and phospho OSR1 at Ser325 (right panels). Alix was used as a standard marker for uEVs. Imaging of stain-free gels before transfer was used as a loading control. The position of each molecular weight marker in kilodaltons is shown. (D) Quantitative analysis revealed significantly increased phosphorylated NCC, total NCC, and phosphorylated SPAK/OSR1 in the patient’s uEVs when compared to the patient’s unaffected brother’s uEVs. (Samples are technical replicates, error bars denote ± SD, n =3–4, *= P<0.05, **= P<0.01, ***= P<0.001, student t -test). (E) Increased WNK4 abundance in patient’s fibroblasts. Protein lysates from fibroblasts from an unaffected individual (control) and patient were immunoprecipitated and immunoblotted with polyclonal anti-WNK4 antibody. Normal rabbit IgG was used as isotype control antibody to confirm non-specific binding. Immunoblotting of lysates with anti-Vinculin confirmed equal loading of samples.
Based upon clinical symptoms and the discovery of a pathogenic variant in CUL3, the patient was diagnosed with FHHt (PHAIIE). Treatment with thiazide diuretics markedly improved the patient’s blood pressure and serum electrolytes (Table 1C and 1D), thus raising the possibility that CUL3Δ474–477 affects the NCC cotransporter in the DCT.
Table 1C.
Blood pressure values of the patient at 9 years of age were measured after administration of thiazide diuretics, which improved electrolyte and blood pressure abnormalities and helped confirm the patient’s FHHt diagnosis. Blood pressure values were averaged and compared to reference pediatric blood pressure ranges from 90th (pre-hypertensive) to 99th (stage II hypertension) percentiles based on reference ranges of the patient’s height (10th percentile) and age (9 years old). The patient became normotensive after administration of thiazide diuretics.
| Blood Pressure | Proband | 90–99% Reference Range |
|---|---|---|
| Systolic (mmHg) | 104±1 | 110–121 |
| Diastolic (mmHg) | 61±6 | 73–85 |
Table 1D.
Serum values were compiled over 4 years after prescription of thiazide diuretics. Mean values show that the patient’s electrolyte imbalances improved after the administration of thiazide diuretics.
| Serum Values | Proband | Reference Range |
|---|---|---|
| Na [mmol/L] | 139±2.0 | 135–144 |
| K [mmol/L] | 5.2±0.5 | 3.5–5.1 |
| Cl [mmol/L] | 102±3.0 | 97–106 |
| Ca [[mmol/L] | 2.4±0.1 | 2.05–2.5 |
| Total CO2 (HCO3) [mmol/L] | 24±2.0 | 22–30 |
| Creatinine [mg/dL] | 0.35±0.08 | 0.3–0.7 |
CUL3Δ474–477 increases WNK4-SPAK/OSR1-NCC signaling
The activity of the NCC cotransporter in the DCT of the kidney is regulated by the WNK-SPAK/OSR1 kinase pathway. We evaluated levels of phosphorylated and total NCC in uEVs from the patient and his unaffected brother. Immunoblot analyses of the uEVs, using antibodies that recognize NCC phosphorylated specifically at Threonine 53 and total NCC, showed a significant increase in the abundance of both phosphorylated and total NCC in the patient compared to his unaffected brother (Figure 1C and 1D).
Increased phosphorylation of NCC likely results from diminished WNK4 proteasomal degradation and subsequent hyperphosphorylation/activation of the WNK4-SPAK/OSR1-NCC axis. We first tested SPAK and OSR1 along this axis by immunoblotting uEVs extracts with antibodies for SPAK phosphorylation at Ser373 and OSR1 phosphorylation at Ser325. This analysis revealed a significantly increased abundance of phosphorylated SPAK and OSR1 in the patient compared to his unaffected brother (Figure 1C and 1D). However, WNK4 is not normally present in uEVs (https://esbl.nhlbi.nih.gov/UrinaryExosomes/) and could not be measured through this approach. To determine the relative abundance of WNK4, we immunoprecipitated WNK4 from the protein extracts prepared from the patient-derived dermal fibroblasts and age-matched control. Immunoblotting revealed increased immunoprecipitation of WNK4 in the patient (Figure 1E, compare lanes 3 and 4). The specificity of the WNK4 antibody was confirmed by comparing western blots of protein extracts from HEK293T cells over-expressing WNK4 (supplementary figure S1). Taken together, these results indicate that CUL3Δ474–477 increases WNK4-SPAK/OSR1-NCC signaling in the patient.
The 474–477 deletion results in reduced CUL3 levels in patient cells and increased CUL3 autoubiquitination activity
To determine whether the increased abundance of WNK4 is due to Δ474–477’s impact on overall CUL3 function, we first explored this variant’s effects on the canonical CUL3 substrate, Cyclin E. Previous studies have shown that deletion of Cul3 led to increased accumulation of Cyclin E and increased numbers of cells confined to S phase.41, 42 Indeed, both Cyclin E protein levels and cellular rate of proliferation were increased in the patient’s fibroblasts compared to controls (Figure 2A and 2B). These observations suggest that the Δ474–477 variant increases substrate abundance by reducing total functional CUL3 ligase. To provide direct evidence of this hypothesis, we investigated the transcript and protein levels of CUL3 in patient-derived fibroblasts. All known FHHt-causing CUL3 variants contained a specific deletion in exon 924, which encodes the 4-helix bundle (4HB) domain (403–459). However, the CUL3 variant (CUL3Δ474–477) in our patient is located within the α/β1 domain (461–586) (Figure 1B). We ruled out the possibility of any independent exon 9 deletion by amplifying cDNA from the patient’s fibroblasts (Supplementary figure S2A). Quantitative PCR analysis of fibroblasts cDNA also revealed no significant differences at the transcriptional level between the patient and controls (Supplementary figure S2B). In contrast, direct immunoblotting of fibroblast lysates with anti-CUL3 antibody revealed significantly reduced total CUL3 levels in the patient’s fibroblasts compared to controls (Figure 2C and 2D), suggesting that, similar to the larger deletion in CUL3Δ403–459, the 4 amino acid deletion in the α/β1 domain destabilizes CUL3 protein through increased auto-ubiquitination and degradation.30 Furthermore, we carried out in vitro ubiquitination assays by adding purified CUL3/Rbx1 or CUL3Δ474–477/Rbx1 to recombinant ubiquitin, UBA1, and UBCH5C and followed the addition of ubiquitin moieties at different time intervals. Immunoblotting with anti-CUL3 antibody revealed the addition of one (mono-ubiquitination) and multiple (poly-ubiquitination) ubiquitin moieties to both WT-CUL3 and CUL3Δ474–477 (Figure 2E). While WT-CUL3 was predominantly mono-ubiquitinated, CUL3Δ474–477 showed enhanced ability to add multiple ubiquitin moieties. These results demonstrate that the 474–477 deletion enhances the auto-ubiquitination of CUL3, suggesting that the diminished CUL3 levels in patient cells are likely due to increased auto-ubiquitination-induced proteasomal degradation. Consistent with this, we found that treatment of fibroblasts with the proteasome inhibitor MG132 led to a significant accumulation of total CUL3 in patient’s fibroblasts and equalization of total CUL3 between controls and patient fibroblasts (Figure 2F).
Figure 2: The 474–477 deletion affects CUL3 protein levels and auto-ubiquitination.
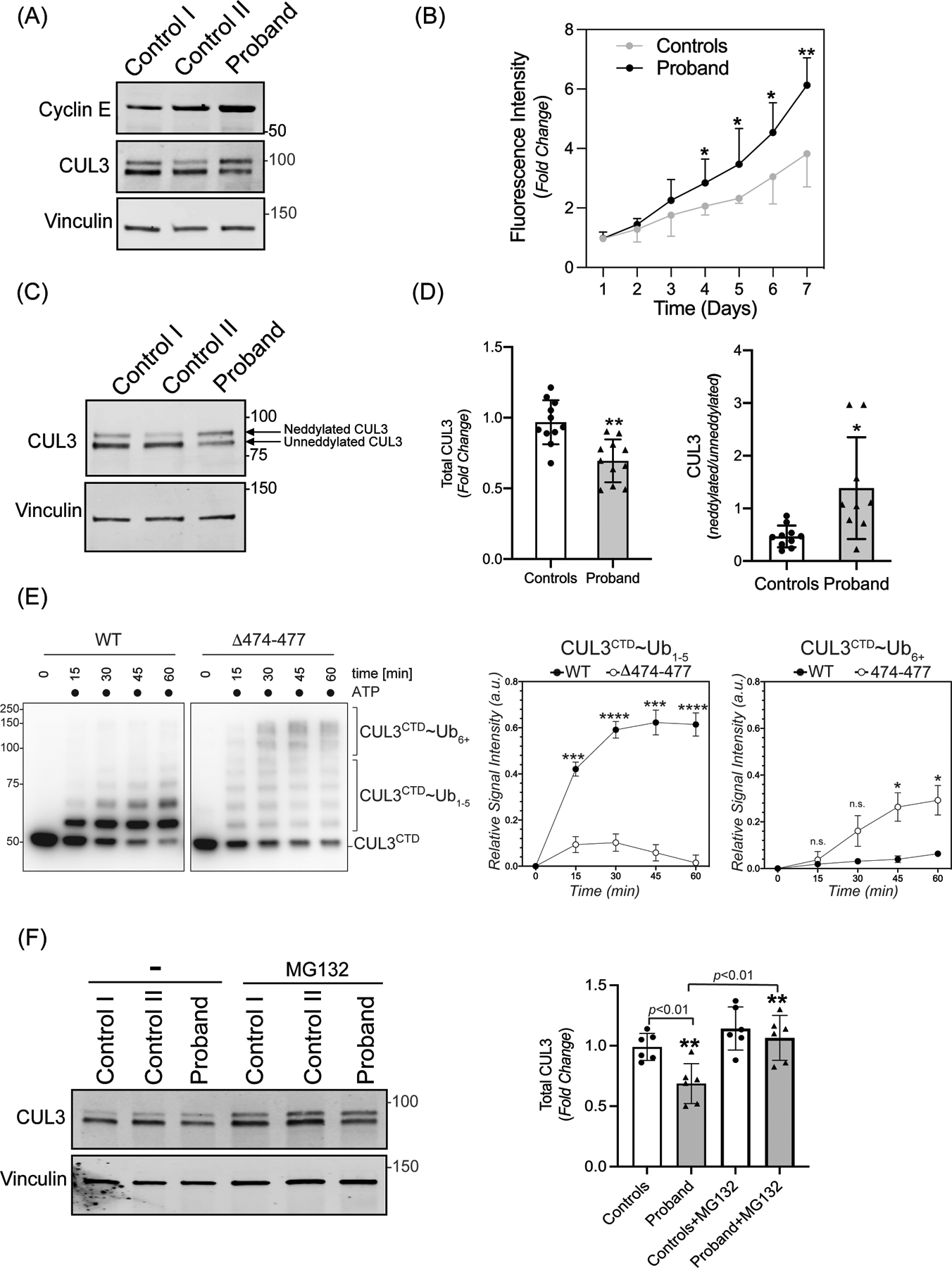
(A) Increased abundance of canonical CRL3 substrate. Fibroblasts from controls and the patient were lysed in RIPA buffer. Levels of Cyclin E were analyzed through immunoblotting of fibroblast lysates from controls and patient fibroblasts using anti-Cyclin E antibodies. Immunoblotting of lysates with anti-Vinculin confirmed equal loading of the samples. (B) Increased rate of proliferation of patient’s fibroblasts. Quadruplicate samples of fibroblasts from patient and two controls were plated into a 96 well plate. Fluorescence measurements of each well were quantified using AlamarBlue dye every 24 hours for 7 days. Patient fibroblasts exhibited significantly increased rates of cellular proliferation compared to controls on days 4–7. Data are represented as fold change of proliferation from day 1. Control represents mean value of two independent controls (error bars indicate ± SD, *= P<0.05, **= P <0.01, student t-test). (C) Total CUL3 abundance and neddylation in fibroblasts. Controls and the patient fibroblasts were lysed in RIPA buffer. Western blot analysis using a polyclonal antibody against CUL3, and subsequent quantitation of bands revealed a reduced abundance of total CUL3 along with a significantly greater ratio of neddylated to unneddylated CUL3 relative to controls. Anti-Vinculin antibody was used to confirm equal loading of the samples. (D) Quantification shows fold change in protein abundance (left) and neddylated/unneddylated CUL3 (right). The control value represents the mean of data from four independent controls (samples are technical replicates, error bars indicate ± SD, n = 9–11 independently performed experiments. **= P<0.01, *= P<0.05, student t -test (E) Left Panel (immunoblots) CUL3/Rbx1 and CUL3Δ474–477/Rbx1 complexes were recombinantly purified using a strategy in which the CUL3 is split into a N-terminal domain (NTD, aa1–309) and C-terminal domain (CTD, aa310–768) and co-expressed with RBX1. These complexes were incubated with recombinant ubiquitin, UBA1, and UBCH5C in the absence or presence of ATP for the indicated time periods. Reactions were stopped by addition of urea sample buffer, separated by SDS-PAGE, and the CTD of CUL3 was detected by immunoblotting with anti-CUL3 antibodies. Right Panel (graphs) Quantification of ubiquitination efficiency for the experiment shown in the left panel. CUL3 modification with 1–5 ubiquitins (CUL3~Ub1–5) or more than 5 ubiquitins (CUL3~Ub6+) was quantified relative to unmodified protein at t=0 min followed by a baseline correction to 0. (n=4 independently performed in vitro experiments, * = P<0.05, *** = P<0.001, **** = P<0.0001, multiple t-test). (F) Left Panel (immunoblots) Control and patient fibroblasts were treated with 10 μM MG132 for 18 hrs. prior to lysis in RIPA buffer and subsequent immunoblotting with anti-CUL3 antibody. Vinculin was used as a loading control. (Right Panel) Quantification shows fold change in total CUL3 abundance. The control value represents the mean of data from two independent controls (error bars indicate ± SD, n = 6 independently performed experiments; samples are technical replicates. **= P<0.01, student t-test).
The 474–477 deletion results in increased CUL3 neddylation in cells
In addition to reduced overall CUL3 levels in patient cells, immunoblot analysis also revealed a marked increase in the higher molecular weight form of CUL3 (neddylated CUL3) (Figure 2C). Quantitative analysis of the bands revealed a significantly higher ratio of neddylated/unneddylated CUL3 in the patient’s fibroblasts compared to controls (Figure 2D), suggesting an altered post-translational cycle of conjugation/deconjugation of CUL3. To corroborate this finding of CUL3 hyperneddylation in patient cells, we transiently transfected HEK293T cells with vectors for wild type (WT)-myc-CUL3 or variant CUL3 (myc-CUL3Δ474–477), followed by immunoprecipitation with anti-Myc antibody. Immunoblotting with anti-NEDD8 antibody showed a substantially higher level of immunoprecipitated NEDD8 with CUL3Δ474–477 (Figure 3A), demonstrating CUL3Δ474–477 hyperneddylation.
Figure 3: The 474–477 deletion alters CUL3 neddylation.
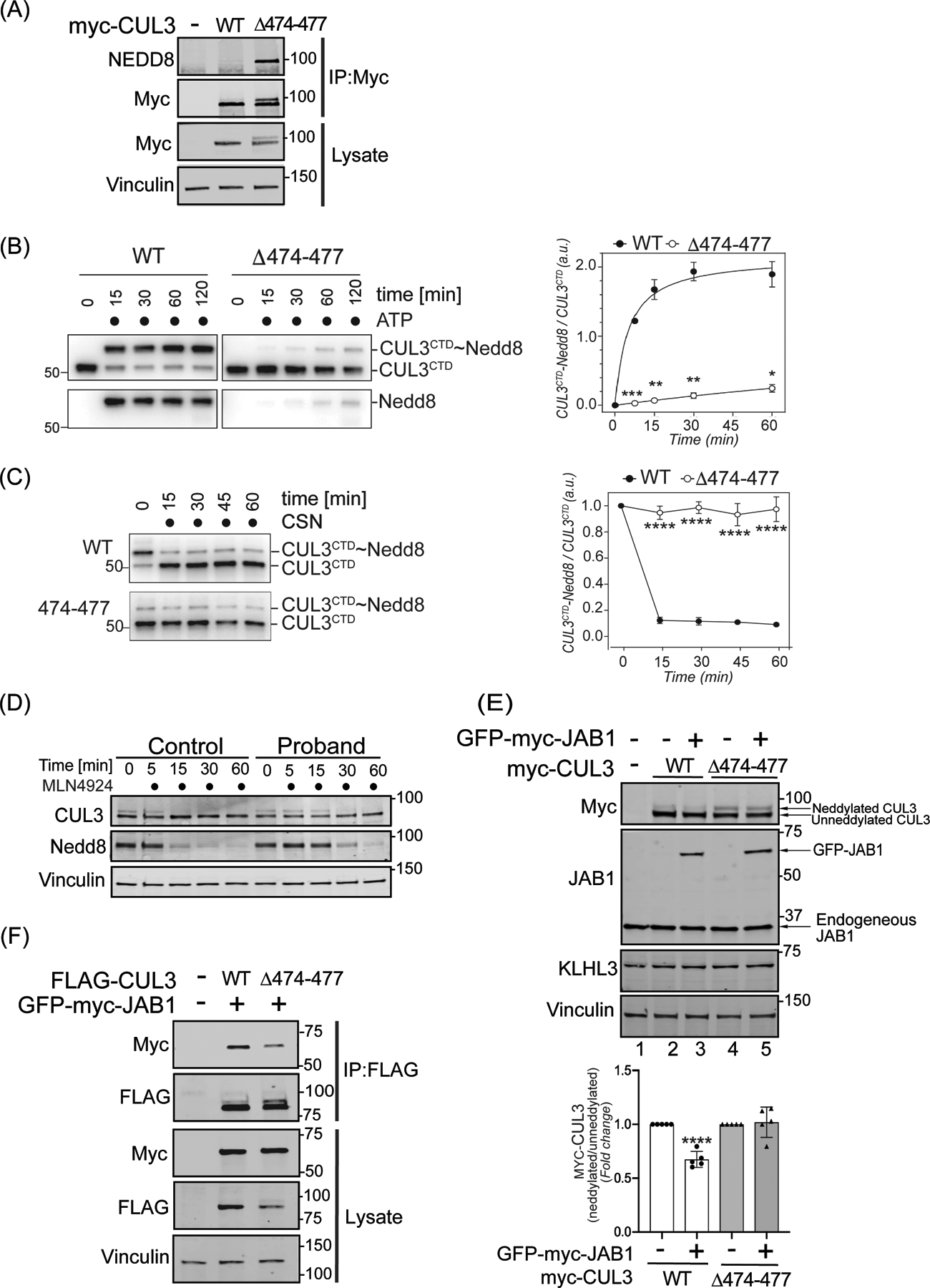
(A) CUL3Δ474–477 is hyperneddylated in HEK293T cells. Whole cell extracts of HEK293T cells overexpressing WT-myc-CUL3 or myc-CUL3Δ474–477 were immunoprecipitated with anti-Myc antibody and immunoblotted with both anti-NEDD8 and anti-Myc antibodies. Immunoblotting with anti-Vinculin was used to confirm equivalent loading of input lysates. (B-C) CUL3Δ474–477 is deficient in reversible modification with NEDD8 in vitro. (B) Left Panel (immunoblots) CUL3/Rbx1 and CUL3Δ474–477/Rbx1 complexes were recombinantly purified using a strategy in which the CUL3 is split into a N-terminal domain (NTD, aa1–309) and C-terminal domain (CTD, aa310–768) and co-expressed with RBX1. Purified Cul3/Rbx1 and Cul3Δ474–477 /Rbx1 complexes were incubated with recombinant NEDD8, NEDD8 E1, and NEDD8 E2 in the absence or presence of ATP for the indicated time periods. Reactions were stopped by addition of urea sample buffer, separated by SDS-PAGE, and subjected to immunoblotting with anti-CUL3 antibodies (detecting the CTD of CUL3) and anti-NEDD8 antibodies. Right Panel (graph) Quantification of neddylation efficiency for the experiment shown in the left panel. The ratio of neddylated CUL3 over unmodified CUL3 is plotted over time (n=5 independently performed in vitro experiments, * = P<0.05, ** = P<0.01, *** = P<0.001, multiple t-test). (C) Left Panel (immunoblots) CUL3/Rbx1 and CUL3Δ474–477 /Rbx1 complexes were maximally neddylated and ATP was quenched by addition of phosphatase. Neddylated CUL3/Rbx1 and CUL3Δ474–477 /Rbx1 were then incubated with recombinant COP9 signalosome (CSN) for the indicated time periods. Reactions were stopped by addition of urea sample buffer, separated by SDS-PAGE, and subjected to immunoblotting with anti-Cul3 antibodies. Right Panel (graph) Quantification of neddylation efficiency for the experiment shown in the left panel. The ratio of neddylated CUL3 over unmodified CUL3 is plotted over time (n=6 independently performed in vitro experiments, **** = P<0.0001, multiple t-test). (D) Neddylation of CUL3 was blocked by the treatment of 500 nM of nedd8-E1 enzyme inhibitor MLN4924 in control and patient fibroblasts. Cells were lysed in RIPA buffer at the indicated time points and lysates were immunoblotted with anti-CUL3 antibody. Vinculin was used to confirm equivalent loading of samples. (E) Upper Panel (immunoblots), JAB1 deneddylation efficiency was evaluated by co-transfecting WT-myc-CUL3 or myc-CUL3Δ474–477 without or with 100 ng GFP-myc-JAB1 in HEK293T cells. Western blot analysis with ant-Myc antibodies showed no obvious change in the deneddylation of CUL3Δ474–477-myc-CUL3, while WT-CUL3 was effectively deneddylated. Anti-JAB1 immunoblot shows both endogenous and over-expressed GFP-JAB1. Equal loading of lysate samples was verified by anti-Vinculin antibody. (Lower Panel) Quantification of deneddylation efficiency of JAB1. The ratio of neddylated/unneddylated CUL3 was calculated in the absence or presence of exogenous JAB1 and shown as fold change. Transfection of 100 ng of JAB1 leads to a significant reduction in the neddylated/unneddylated ratio of WT-CUL3, while neddylated/unneddylated CUL3Δ474–477 remains unaffected (error bars indicate ± SD, n = 5 independent experiments, ****= P<<0.0001, student’s t-test. (F) Reduced interaction of CUL3Δ474–477 with CSN subunit JAB1. HEK293T cells underwent transient co-transfection of WT-FLAG-CUL3 or FLAG-CUL3Δ474–477 along with GFP-myc-JAB1. Immunoprecipitation of lysates with anti-FLAG antibody and subsequent immunoblotting with anti-Myc antibody revealed reduced interaction between JAB1 and CUL3Δ474–477.
To explore this further, we carried out in vitro neddylation reactions using purified CUL3/Rbx1 or CUL3Δ474–477/Rbx1, NEDD8, NEDD8 E1, and NEDD8 E2 enzymes. The addition of ATP to the reaction led to robust NEDD8 modification of WT-CUL3. Surprisingly, CUL3Δ474–477 was modified with NEDD8, with significantly lower efficiency compared to WT-CUL3 (Figure 3B). To explain this apparent discrepancy with our findings in cells, we performed in vitro deneddylation reactions by first maximally neddylating CUL3/Rbx1 and CUL3Δ474–477/Rbx1 complexes followed by the addition of recombinant COP9 signalosome (CSN). While CSN carried out nearly all of its deneddylation of WT-CUL3 within the first 15 min of incubation, it did not significantly deneddylate CUL3Δ474–477 during 60 minutes of incubation (Figure 3C). This suggests that once CUL3Δ474–477 is neddylated, it becomes resistant to deneddylation by CSN in vitro. In support of this notion, treatment of fibroblasts with the neddylation inhibitor MLN4924 resulted in a markedly slower loss of neddylated CUL3 in patient cells compared to control cells (Figure 3D). In addition, experiments in HEK293T cells revealed that ectopic expression of JAB1 significantly reduced the neddylation of WT-myc-CUL3 (Figure 3E, top panel, lanes 2 and 3), but not neddylation of myc-CUL3Δ474–477 (Figure 3E, top panel, lanes 4 and 5). Because aa474–477 of CUL3 are located within the binding site of CSN43, we considered whether CSN’s impaired ability to deneddylate CUL3Δ474–477 was due to diminished JAB1-CUL3Δ474–477 interaction. To assess this, we transiently transfected vectors for WT-FLAG-CUL3 or FLAG-CUL3Δ474–477 with GFP-myc-JAB1 into HEK293T cells and immunoprecipitated CUL3 using anti-FLAG antibody. Immunoblotting of samples with anti-Myc antibody revealed that JAB1 maintains binding to myc-CUL3Δ474–477, albeit to a lower extent compared to WT-myc-CUL3 (Figure 3F). Taken together, these results provide evidence that Δ474–477 deletion results in hyperneddylation of CUL3 in cells, likely due to the CSN’s restricted ability to hydrolyze the NEDD8 isopeptide bond.
CUL3Δ474–477 is defective in ubiquitinating WNK4 and forms abnormal ubiquitin ligation complexes
We considered whether the fraction of CUL3 (Figure 2C) that is hyperneddylated and escapes auto-degradation in patient fibroblasts also contributes to increased WNK4 accumulation (Figure 1E). To first investigate CUL3Δ474–477’s ability to ubiquitinate WNK4, we transiently transfected vectors for HIS-ubiquitin, FLAG-WNK4, HA-KLHL3, and WT-myc-CUL3 or myc-CUL3Δ474–477 in HEK293T. Co-expression of KLHL3 with WT-CUL3 robustly degrades WNK4 (Figure 4A, lysates, anti-FLAG, lane 2), however, CUL3Δ474–477 is less efficient at this process (Figure 4A, lysates, anti-FLAG, lane 3). Next, we blocked the degradation of ubiquitinated proteins with the proteasome inhibitor MG132 and purified ubiquitinated proteins using Ni-NTA resin. Immunoblotting of Ni-NTA eluted proteins with anti-FLAG antibody revealed an accumulation of high molecular weight ubiquitinated FLAG-WNK4 when co-expressed with WT-myc-CUL3 as compared to myc-CUL3Δ474–477 (Figure 4A, top panel, lanes 4 and 5), suggesting that despite its increased neddylation, CUL3Δ474–477 is impaired in ubiquitinating WNK4. To investigate the molecular basis for this impairment, we transiently transfected HEK293T cells with vectors for WT-myc-CUL3 or myc-CUL3Δ474–477 with FLAG-WNK4. We immunoprecipitated WNK4 using anti-FLAG antibody and immunoblotted the immunoprecipitants with anti-Myc antibody. Surprisingly, this showed that CUL3Δ474–477 not only retained the ability to interact with WNK4 but appeared to bind to WNK4 with greater efficiency than wild-type CUL3 (Figure 4B, top panel, lanes 2 and 3), despite the reduced abundance of total CUL3Δ474–477 in the lysate (Figure 4B, lysate samples, anti-Myc, lane 3). The interaction between CUL3 and WNK4 in these experiments is likely mediated by the endogenous substrate adaptor KLHL3, as ectopic expression of KLHL3 markedly increased CUL3-WNK4 interaction (supplementary figure S3). To directly explore the effect of the Δ474–477 deletion on CUL3-KLHL3 interaction in the patient’s fibroblasts, we immunoprecipitated CUL3 from patient fibroblast lysates and immunoblotted the immunoprecipitants with anti-KLHL3 antibody. Quantitative analysis of co-immunoprecipitated KLHL3 and total CUL3 bands revealed a higher KLHL3/CUL3 ratio in patient fibroblasts (Figure 4C), suggesting that the CUL3Δ474–477 variant increases the interaction of CUL3 with KLHL3. In these experiments, direct immunoblotting of the fibroblast lysates with anti-KLHL3 antibody did not reveal any significant difference in KLHL3 abundance between patient and control (Figure 4C, lysates). Next, we transiently transfected HEK293T cells with vectors for WT-myc-CUL3 or myc-CUL3Δ474–477 with FLAG-KLHL3. We immunoprecipitated myc-CUL3 with anti-Myc antibody and immunoblotted the immunoprecipitants with anti-FLAG antibody. This analysis also revealed an increased interaction of FLAG-KLHL3 with CUL3Δ474–477 (Figure 4D, IP), with no significant difference in the abundance of FLAG-KLHL3, when co-transfected with WT or CUL3Δ474–477 (Figure 4D, lysates). Thus, deletion of aa474–477 of CUL3 results in increased interactions with KLHL3 and WNK4, without increased degradation of substrate adaptor KLHL3 or effective ubiquitination of WNK4. These results further suggest that the fraction of CUL3Δ474–477 that is not auto-degraded in patient cells engages in abnormal CRL complexes through increased KLHL3 and WNK4 binding. Given that CUL3Δ474–477 was able to heterodimerize with wildtype CUL3 (supplementary figure S4), it is also possible that these abnormal CRL complexes may sequester wildtype CUL3 into catalytically impaired ubiquitin ligation assemblies.
Figure 4: CUL3Δ474–477 affects WNK4 ubiquitination without altering KLHL3 abundance.
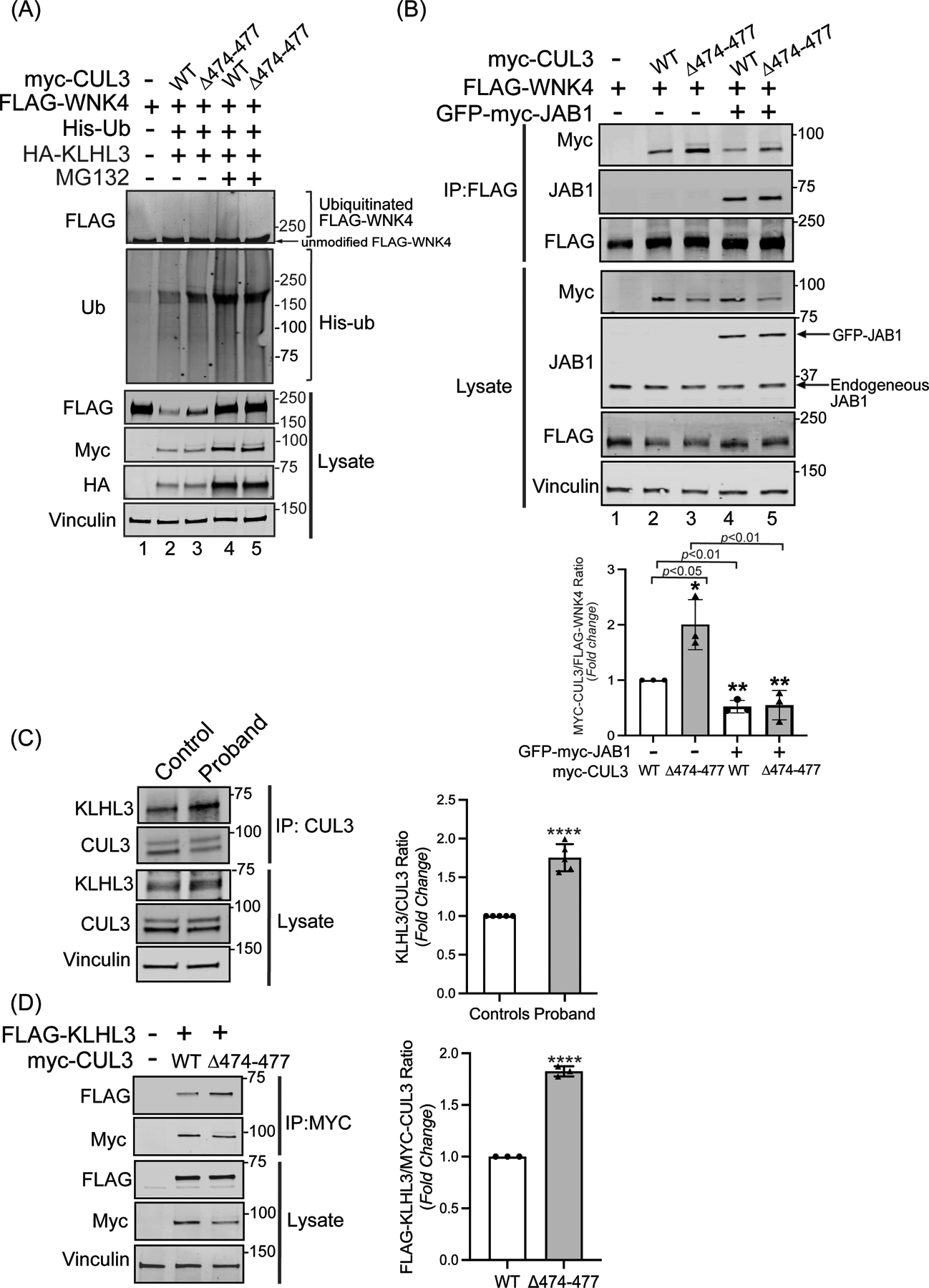
(A) The CUL3Δ474–477 variant leads to reduced ubiquitination of WNK4. HEK293T cells expressing WT-myc-CUL3 or myc-CUL3Δ474–477 were cotransfected with FLAG-WNK4, HA-KLHL3, and His-ubiquitin. Cells were treated with 50 μM MG132 for 5 hrs. before harvesting. Ubiquitin conjugates were purified under denaturing conditions and immunoblotted with an anti-FLAG antibody. Panels labeled ‘Lysates’ indicate samples not subjected to HIS purification and directly immunoblotted with designated antibodies. (B) Upper Panel: Interaction of CUL3Δ474–477 with substrate WNK4. HEK293T cells were co-transfected with WT-myc-CUL3 or myc-CUL3Δ474–477 along with FLAG-WNK4 in the absence or presence of 250 ng of GFP-myc-JAB1. Cells lysates were subjected to immunoprecipitation with anti-FLAG antibody. Immunoblotting of precipitates with anti-Myc antibody revealed interactions of WNK4 with both WT-CUL3 and CUL3Δ474–477, however, the interaction with CUL3Δ474–477 is significantly higher compared to WT-CUL3. Immunoblotting of precipitates with anti-JAB1 antibody also revealed interactions of WNK4 with JAB1 in the presence of WT-CUL3 or CUL3Δ474–477. Immunoblotting of lysates, not subjected to immunoprecipitation, showed the reduced abundance of myc-CUL3Δ474–477 as compared to WT-myc-CUL3. Immunoblotting of lysates with anti-Vinculin antibody confirmed the equal loading of samples. Lower Panel: Quantitative analysis of myc-CUL3 and FLAG-WNK4 bands in IP samples showed significantly higher interaction (shown by increased CUL3/WNK4 ratio) of CUL3Δ474–477-WNK4 compared to WT-CUL3-WNK4 (error bars indicate ± SD, n = 3 independent experiments, *= P<0.05, student’s t-test). The addition of JAB1 significantly reduced the interaction of WNK4 with both WT-myc-CUL3 and MYC-CUL3Δ474–477(error bars indicate ± SD, n = 3 independent experiments, **= P<0.01, student’s t-test) (C) Interaction of KLHL3 with CUL3 is increased in patient’s fibroblasts. Total cellular extracts of patient and control fibroblasts were prepared using RIPA lysis buffer and subsequently immunoprecipitated using an anti-CUL3 antibody. Immunoblotting with anti-KLHL3 antibody (left) and quantitation (right) of KLHL3 and CUL3 bands in IP samples showed significantly higher KLHL3/CUL3 ratio in the proband (samples are technical replicates, error bars indicate ± SD, n = 5 independent experiments, ****= P<0.0001, student’s t-test). Direct immunoblotting of lysates with anti-KLHL3 antibody demonstrated no significant difference in abundance between proband and control fibroblasts. Immunoblotting of lysates with anti-Vinculin antibody showed sample loading. (D) Interaction of KLHL3 with CUL3 was analyzed in HEK293T cells overexpressing FLAG-KLHL3 cotransfected with either WT-myc-CUL3 or myc-CUL3Δ474–477. Immunoprecipitation with anti-Myc antibody and subsequent immunoblotting of lysates with anti-FLAG antibody revealed increased interaction between FLAG-KLHL3 and MYC-CUL3Δ474–477 compared to WT-MYC-CUL3 (upper panels, IP). Quantitation of bands (right) in IP samples showed significant difference (error bars indicate ± SD, n = 3 independent experiments, ****= P<0.0001, student’s t-test). Direct immunoblotting of lysates with anti-FLAG antibody demonstrated no change in FLAG-KLHL3 abundance between WT-myc-CUL3 and myc-CUL3Δ474–477 over-expressed lysates. Vinculin was used to confirm the equivalent loading of the samples.
The fraction of CUL3Δ474–477 that immunoprecipitated with WNK4 remained hyperneddylated (Figure 4B, top panel, lanes 2 and 3), likely due to CSN’s restricted ability to deneddylate CUL3Δ474–477. To investigate this further, we measured CUL3-WNK4 interactions in the presence of JAB1 ectopically expressed at the level approximately equivalent to intracellular JAB1 (Figure 4B, input, anti-JAB1). This addition of JAB1 significantly reduced WNK4 binding with both WT-CUL3 and CUL3Δ474–477 (Figure 4B, top panel, lanes 4 and 5), marginally increased the abundance of FLAG-WNK4 (Figure 5B, input, anti-FLAG), and showed a significant reduction of CUL3Δ474–477 levels (43±24% of untreated CUL3Δ474–477, mean± SD, P<0.05) (Figure 4B, input, anti-Myc, compare lanes 3 and 5). Interestingly, JAB1 also co-immunoprecipitated with FLAG-WNK4 (Figure 4B, second panel, lanes 4 and 5). Taken together, these findings suggest that increased deneddylation of CUL3Δ474–477 by ectopically expressed JAB1 reduces CUL3-WNK4 complex formation, ultimately leading to further auto-ubiquitination and degradation of CUL3.
Figure 5. The 474–477 deletion results in global changes in the CUL3 interaction network and promotes association with BTB substrate adaptors.
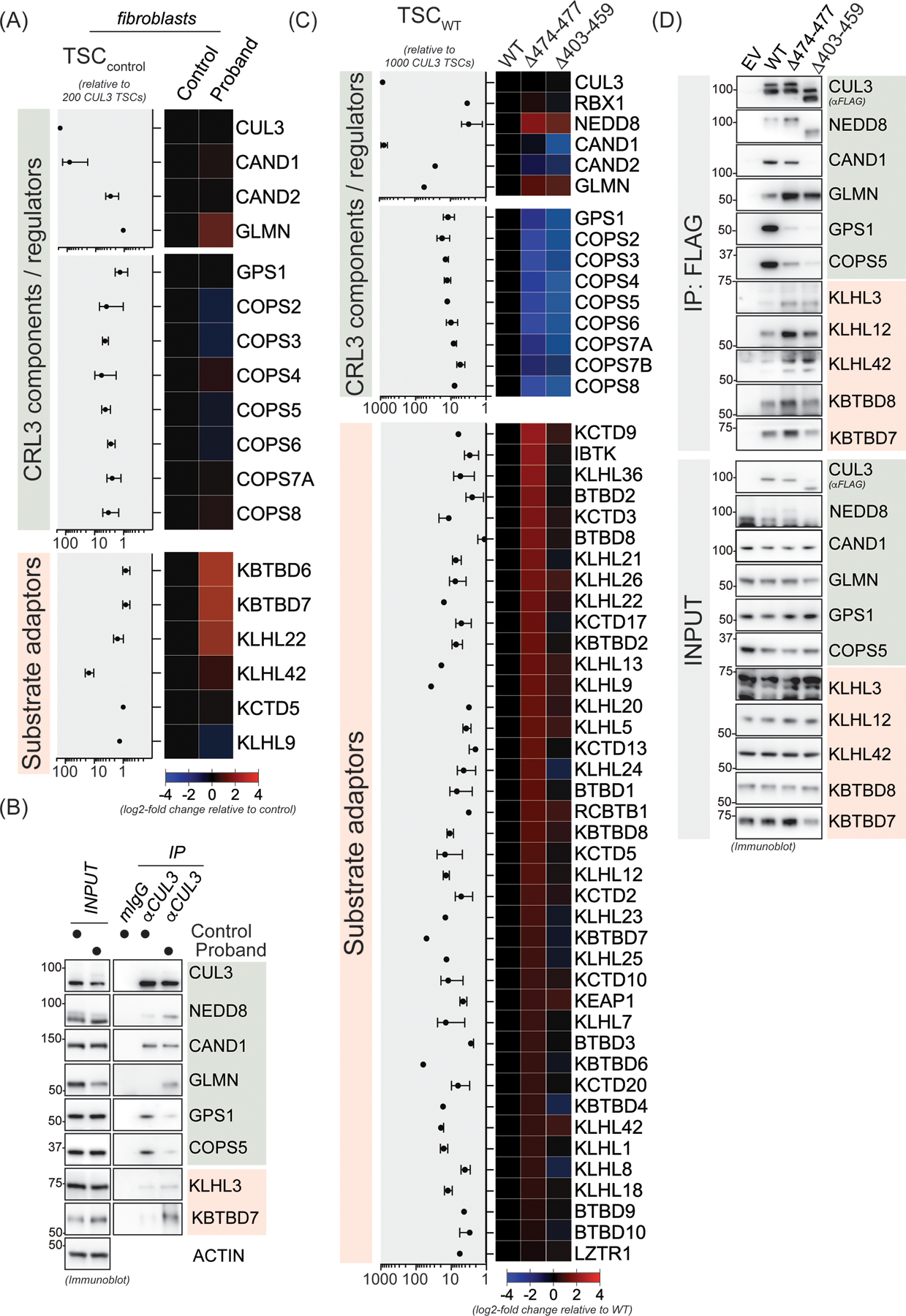
(A) CUL3Δ474–477 leads to aberrant interactions in patient fibroblasts. Control or patient (proband) fibroblasts were lysed and subjected to anti-CUL3 immunoprecipitation as described in the material and methods section followed by mass spectrometry analysis of endogenous CUL3 complexes. Relative total spectral counts (rTSCs) for each CUL3 interactor in control fibroblasts are shown on the left and log2-fold changes in interactions between control and patient fibroblasts are depicted in the heatmap on the right. rTSCcontrol indicate ± SD, n=4 (2 independent IP experiments with 2 technical replicates each). (B) Validation of the changes in the interaction landscape in patient fibroblasts as determined in panel A. Control or patient fibroblasts were lysed and subjected to anti-CUL3 immunoprecipitation as described in the material and methods section followed by immunoblot analysis with indicated antibodies against representative COP9 signalosome components, CRL3 regulators, and BTB substrate adaptors. (C) HEK293T cells transiently expressing FLAG-CUL3-wild-type (WT), or FLAG-CUL3Δ474–477 or FLAG-CUL3Δ403–459 variant, were lysed and lysates were subjected to anti-FLAG immunoprecipitation followed by mass spectrometry analysis for interacting proteins. Relative total spectral counts (rTSCs) for each FLAG-CUL3-WT interactor are shown on the left and log2-fold changes in interactions between WT and the Δ474–477 or Δ403–459 variants are depicted in the heatmap on the right. rTSCWT indicate ± SD, n=4 (2 independent IP experiments with 2 technical replicates each). (D) Validation of the changes in the CUL3Δ474–477 and CUL3Δ403–459 interaction landscape as determined in panel C. HEK293T cells transiently expressing an empty control vector (EV), FLAG-CUL3-WT, FLAG-CUL3Δ474–477, or FLAG-CUL3Δ403–459 were lysed and lysates were subjected to anti-FLAG immunoprecipitation followed by immunoblot analysis with indicated antibodies against representative COP9 signalosome components, CRL3 regulators, and BTB substrate adaptors.
The 474–477 deletion results in increased interactions with BTB adaptors and global changes in the CUL3 interactome network
CRL3 complexes distinct from CRL3-KLHL3 have been shown to control various aspects of human development.18 Given that our patient exhibited diverse developmental phenotypes, we speculated that CUL3Δ474–477 might also affect other CRL3-BTB complexes. To test this hypothesis, we performed mass spectrometry and immunoblot analysis of endogenous CUL3 interacting proteins in patient’s fibroblasts. Consistent with our previous results, we found increased NEDD8 modification in CUL3 immunoprecipitates from patient cells compared to controls (Figure 5A and 5B). Interestingly, we observed increased interaction with the CRL inhibitor glomulin (GLMN)44 and several BTB substrate adaptors. However, due to the low expression of certain BTB adaptors in fibroblast and our overall low yield of endogenous CUL3 complexes, we repeated mass spectrometry analysis with immunoprecipitates of ectopically expressed WT-FLAG-CUL3, FLAG-CUL3Δ474–477, or FLAG-CUL3Δ403–459 from HEK293T cells based on our previously used cellular model.38 Indeed, results from these experiments revealed that compared to WT-CUL3, both CUL3Δ474–477 and CUL3Δ403–459 variants exhibited reduced CSN binding. CSN subunits exhibited greater interaction with CUL3Δ474–477 than CUL3Δ403–459, indicating that CSN maintains a restricted role in CUL3Δ474–477 neddylation/deneddylation cycles. Surprisingly, we did not find any difference in CAND1 interaction between CUL3Δ474–477 and WT, while CAND1 binding in CUL3Δ403–459 was significantly reduced. CUL3Δ474–477 also showed increased interactions with many BTB substrate adaptors including important regulators of neuroectodermal differentiation (i.e., KBTBD8, KCTD13, and KCTD17), while this was less the case for the canonical CUL3Δ403–459. Taken together, our proteomic analyses revealed similarities and differences between CUL3Δ474–477 and CUL3Δ403–459 interaction networks. We speculate that significantly increased interaction between BTBs and CUL3Δ474–477 may reduce the function of wildtype CUL3 by sequestering BTBs, thus diminishing their tissue-specific function and providing a rationale for the diverse developmental phenotypes of our patient.
DISCUSSION
All documented cases of FHHt related to CUL3 variants have been due to a specific deletion of 57 amino acids (CUL3Δ403–459). Several studies have explored the consequences of CUL3Δ403–459 on CUL3 post-translational modifications, substrate adaptors binding, substrate abundance, and the pathophysiological NCC hyperactivation that corresponds with FHHt.29, 30, 43, 45, 46 Here, we identified a novel heterozygous variant (Δ474–477) in CUL3 and investigated the mechanisms causing FHHt. First, the levels of CRL substrates WNK4 and Cyclin E were increased in the patient’s fibroblasts. Increased WNK4 abundance likely accounts for the increased activation (phosphorylation) of the NCC transporter through the WNK-SPAK/OSR1 signaling cascade. Second, we confirmed the presence of heightened NCC phosphorylation in the patient’s uEVs; this elucidated the proximate pathophysiological cause for the electrolyte imbalances seen in the patient. Third, we observed an increased proliferation in the patient’s fibroblasts, which could be a consequence of an increased abundance of Cyclin E. However, it remains unclear if this fibrotic proliferation is affecting other physiological and cellular processes in the patient.
Our observations of increased signaling through the WNK-SPAK/OSRI-NCC axis are consistent with the canonical CUL3Δ403–459 variant. McCormick et al. have shown that WNK4 accumulates because of CUL3Δ403–459’s increased ability to ubiquitinate and degrade KLHL3.29 However, heterozygous Cul3Δ403–459 mice, which display the FHHt phenotype, did not demonstrate decreased KLHL3, despite increased abundance of WNK4.30 In the same study, authors showed that CUL3Δ403–459 induces increased structural flexibility, leading to the enhanced autoubiquitination. This would result in haploinsufficiency of functional wild type CUL3, increasing the total abundance of substrate WNK4.30 Yet, Cul3 heterozygous mice do not display the FHHt phenotype,47 suggesting that the Cul3Δ403–459 allele may be necessary to induce the disease phenotype. Here, our mechanistic studies suggest two distinct mechanisms by which the novel CUL3Δ474–477 variant hyperactivates the WNK-SPAK/OSRI-NCC to cause FHHt. First, increased auto-ubiquitination induced proteasomal degradation of CUL3Δ474–477 results in reduced total CUL3 levels in patient cells. Second, the remaining CUL3Δ474–477 protein exhibits hyperneddylation and enhanced interactions with KLHL3 and WNK4, binding reactions that may sequester CUL3Δ474–477 with KLHL3 and WNK4 in inactive ubiquitin ligation complexes that – in addition to the overall reduction in CUL3 levels - would further reduce the ubiquitination activity of wildtype CRL3 towards WNK4.
In concordance with CUL3Δ403–459, our patient’s variant likely introduces increased CUL3 structural flexibility, which may alter its interaction with ubiquitin-charged E2, leading to abnormal auto-ubiquitination at multiple sites that are normally inaccessible.30 This increased structural flexibility may also hamper the ability of CUL3Δ474–477 to incorporate NEDD8 molecules in vitro and interfere with CSN-mediated CUL3Δ474–477 deneddylation. Impaired CUL3 neddylation/deneddylation cycles would ultimately inhibit the productive formation of Cul3/RBX1-E2~Ub ubiquitin ligation assemblies19 and prevent substrate ubiquitination. However, in contrast to the canonical CUL3Δ403–459, CUL3Δ474–477 exhibits increased interaction with many BTB adaptors and retains its ability to interact with CAND1. While CUL3Δ403–459 loses part of its CAND1 binding region, aa474–477 is located near, but outside the CAND1 binding site of cullin proteins.48, 49 Thus, we propose that the deletion of aa 474–477 does not lead to the same structural rearrangement as suggested for the canonical CUL3Δ403–459.30
Studies have shown that disrupted binding of CSN impairs deneddylation of CUL3Δ403–459.30, 43 Our patient’s CUL3 deletion is located within the CSN binding site in the α/β1 domain (aa 459–586).43 Interestingly, co-immunoprecipitation experiments in HEK293T cells showed reduced but not absent interaction of the CSN subunit JAB1 with CUL3Δ474–477. In addition, overexpression of catalytic subunit JAB1 reduced the aberrant interaction of CUL3Δ474–477 and WNK4, suggesting that the deletion of amino acids 474–477 in the α/β1 domain restricts CSN’s ability to properly deneddylate CUL3Δ474–477 in cells. In these experiments, we also found that JAB1 co-immunoprecipitated with WNK4, likely through interactions between JAB1 and CUL3. Further studies could illuminate the molecular basis of CSN interactions with CUL3Δ474–477 and WNK4 and how its dysregulation could contribute to the disease.
Treatment with thiazide diuretics improved the patient’s FHHt symptoms, likely by specifically inhibiting the NCC cotransporter along with the DCT.4 In addition to FHHt, our patient also displayed short stature, global developmental delay, gastroesophageal reflux disease, frequent pulmonary infections, and dysmorphic facial features. Occurrences of other phenotypes have been reported in FHHt patients.1, 50, 51 A possible explanation for these disparate clinical manifestations outside of the WNK-NCC axis could be that CUL3Δ474–477 sequesters BTB proteins involved in regulating human development; the biochemically defective complexes would be impaired in ubiquitination and would limit the functional CUL3 pool. In support of this notion, our mass spectrometry analyses identified several BTB adaptor proteins with markedly higher binding to CUL3Δ474–477. Indeed, CRL3 complexes using KBTBD8, KCTD13, and KCTD17 as substrate adaptors have been shown to control neural crest specification,38 synaptic transmission52, and ciliogenesis53 respectively. We speculate that overall reduced CUL3 levels together with sequestration of BTB adaptors in catalytically impaired CUL3Δ474–477 complexes interfere with the function of particular CRL3-BTBs, ultimately generating abnormal developmental, neurological, facial, and respiratory manifestations seen in this patient. Additional studies exploring the relationship between CUL3 deletions, and these substrate adaptor binding proteins could further elucidate these mechanisms.
PERSPECTIVES
The discovery of a novel, de novo CUL3 variant (CUL3Δ474–477) in a pediatric FHHt patient and its subsequent characterization in patient-derived cells, in vitro reconstitution systems, and cultured kidney cells offer both confirmation and new findings regarding the FHHt disease mechanism. This study demonstrates molecular defects caused by CUL3Δ474–477 and further expands our understanding of the disease mechanism.
Supplementary Material
Novelty and Significance.
What is New?
A pediatric patient with familial hyperkalemic hypertension (FHHt) has a novel four amino acid deletion located within the COP9 Signalosome (CSN) binding site of CUL3.
This study is the first to use patient-derived cells and urinary extracellular vesicles (uEVs) to explore the mechanism behind FHHt.
What is Relevant?
CUL3Δ474–477 activates WNK-SPAK/OSR1-NCC signaling cascade and causes the electrolyte imbalances of FHHt by dysregulating CUL3 neddylation/deneddylation, substrate adaptor binding, and substrate ubiquitination.
In addition to KLHL3, increased binding with several BTB-substrate adaptors provides a rationale for non-renal phenotypes associated with FHHt.
Summary
The pathophysiological effects of CUL3Δ474–477 are caused by the reduced total CUL3 levels and formation of catalytically impaired CUL3 ligase complexes, culminating in WNK4 accumulation and NCC hyperactivation.
ACKNOWLEDGEMENTS
We thank the patient and his family for their support and participation in this study. We thank the UDP Bioinformatics team for analyzing whole exome data, Yan Huang for establishing primary dermal fibroblast cultures, Dawn Maynard for the assistance in exosomes isolation, and Dr. Yan Wang from the NIDCR Mass Spectrometry Facility (ZIA DE00075) for performing mass spectrometry analysis. We also thank Dr. Mark A Knepper (NHLBI) for providing NCC antibodies and his expertise on urinary exosomes.
SOURCES OF FUNDING
This research work was supported in part by the NIH Office of the Director’s Common Fund and the Intramural Research Program of the National Human Genome Research Institute and the National Institute of Dental and Craniofacial Research.
Footnotes
REFERENCES
- 1.Gordon RD. Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension (Dallas, Tex. : 1979). 1986;8:93–102 [DOI] [PubMed] [Google Scholar]
- 2.Yang CL, Angell J, Mitchell R, Ellison DH. Wnk kinases regulate thiazide-sensitive na-cl cotransport. J Clin Invest. 2003;111:1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon RD, Hodsman GP. The syndrome of hypertension and hyperkalaemia without renal failure: Long term correction by thiazide diuretic. Scottish medical journal. 1986;31:43–44 [DOI] [PubMed] [Google Scholar]
- 4.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type ii: Marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. The Journal of clinical endocrinology and metabolism. 2002;87:3248–3254 [DOI] [PubMed] [Google Scholar]
- 5.Appel LJ. The verdict from allhat--thiazide diuretics are the preferred initial therapy for hypertension. JAMA. 2002;288:3039–3042 [DOI] [PubMed] [Google Scholar]
- 6.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive na+-cl- cotransporter by the wnk-regulated kinases spak and osr1. Journal of cell science. 2008;121:675–684 [DOI] [PubMed] [Google Scholar]
- 7.Delpire E, Gagnon K. Spak and osr1: Ste20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. The Biochemical journal. 2008;409:321–331 [DOI] [PubMed] [Google Scholar]
- 8.Ferdaus MZ, Barber KW, Lopez-Cayuqueo KI, Terker AS, Argaiz ER, Gassaway BM, Chambrey R, Gamba G, Rinehart J, McCormick JA. Spak and osr1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. The Journal of physiology. 2016;594:4945–4966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, et al. Human hypertension caused by mutations in wnk kinases. Science (New York, N.Y.). 2001;293:1107–1112 [DOI] [PubMed] [Google Scholar]
- 10.Vitari AC, Deak M, Morrice NA, Alessi DR. The wnk1 and wnk4 protein kinases that are mutated in gordon’s hypertension syndrome phosphorylate and activate spak and osr1 protein kinases. Biochem J. 2005;391:17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, Alessi DR, Kurz T. The cul3-klhl3 e3 ligase complex mutated in gordon’s hypertension syndrome interacts with and ubiquitylates wnk isoforms: Disease-causing mutations in klhl3 and wnk4 disrupt interaction. Biochem J. 2013;451:111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of wnk4. Proc Natl Acad Sci U S A. 2013;110:7838–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, et al. Impaired klhl3-mediated ubiquitination of wnk4 causes human hypertension. Cell Rep. 2013;3:858–868 [DOI] [PubMed] [Google Scholar]
- 14.Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, et al. The btb protein mel-26 is a substrate-specific adaptor of the cul-3 ubiquitin-ligase. Nature. 2003;425:311–316 [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. Btb proteins are substrate-specific adaptors in an scf-like modular ubiquitin ligase containing cul-3. Nature. 2003;425:316–321 [DOI] [PubMed] [Google Scholar]
- 16.Geyer R, Wee S, Anderson S, Yates J, Wolf DA. Btb/poz domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–790 [DOI] [PubMed] [Google Scholar]
- 17.Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-btb complexes join the family. EMBO J. 2004;23:1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asmar AJ, Beck DB, Werner A. Control of craniofacial and brain development by cullin3-ring ubiquitin ligases: Lessons from human disease genetics. Exp Cell Res. 2020;396:112300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baek K, Krist DT, Prabu JR, Hill S, Klugel M, Neumaier LM, von Gronau S, Kleiger G, Schulman BA. Nedd8 nucleates a multivalent cullin-ring-ube2d ubiquitin ligation assembly. Nature. 2020;578:461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petroski MD, Deshaies RJ. Function and regulation of cullin-ring ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20 [DOI] [PubMed] [Google Scholar]
- 21.Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, Fischer ES, Thoma NH. Crystal structure of the human cop9 signalosome. Nature. 2014;512:161–165 [DOI] [PubMed] [Google Scholar]
- 22.Cornelius RJ, Si J, Cuevas CA, Nelson JW, Gratreak BDK, Pardi R, Yang CL, Ellison DH. Renal cop9 signalosome deficiency alters cul3-klhl3-wnk signaling pathway. J Am Soc Nephrol. 2018;29:2627–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis-Dit-Picard H, Hadchouel J, Jeunemaitre X. [klhl3 and cullin-3: New genes involved in familial hypertension]. Med Sci (Paris). 2012;28:703–706 [DOI] [PubMed] [Google Scholar]
- 24.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glover M, Ware JS, Henry A, Wolley M, Walsh R, Wain LV, Xu S, Van’t Hoff WG, Tobin MD, Hall IP, et al. Detection of mutations in klhl3 and cul3 in families with fhht (familial hyperkalaemic hypertension or gordon’s syndrome). Clin Sci (Lond). 2014;126:721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osawa M, Ogura Y, Isobe K, Uchida S, Nonoyama S, Kawaguchi H. Cul3 gene analysis enables early intervention for pediatric pseudohypoaldosteronism type ii in infancy. Pediatr Nephrol. 2013;28:1881–1884 [DOI] [PubMed] [Google Scholar]
- 27.Tsuji S, Yamashita M, Unishi G, Takewa R, Kimata T, Isobe K, Chiga M, Uchida S, Kaneko K. A young child with pseudohypoaldosteronism type ii by a mutation of cullin 3. BMC Nephrol. 2013;14:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao L, Cui L, Lu J, Lang Y, Bottillo I, Zhao X. A novel mutation in exon 9 of cullin 3 gene contributes to aberrant splicing in pseudohypoaldosteronism type ii. FEBS Open Bio. 2018;8:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCormick JA, Yang CL, Zhang C, Davidge B, Blankenstein KI, Terker AS, Yarbrough B, Meermeier NP, Park HJ, McCully B, et al. Hyperkalemic hypertension-associated cullin 3 promotes wnk signaling by degrading klhl3. J Clin Invest. 2014;124:4723–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, Al Maskari RS, Ferryman JT, Hardege I, Yasmin, et al. Characterisation of the cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7:1285–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gahl WA, Tifft CJ. The nih undiagnosed diseases program: Lessons learned. JAMA. 2011;305:1904–1905 [DOI] [PubMed] [Google Scholar]
- 32.Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, Adams DR, Tifft CJ, Udn. The nih undiagnosed diseases program and network: Applications to modern medicine. Mol Genet Metab. 2016;117:393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, et al. The national institutes of health undiagnosed diseases program: Insights into rare diseases. Genet Med. 2012;14:51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzales PA, Zhou H, Pisitkun T, Wang NS, Star RA, Knepper MA, Yuen PS. Isolation and purification of exosomes in urine. Methods Mol Biol. 2010;641:89–99 [DOI] [PubMed] [Google Scholar]
- 35.Sharma P, Reichert M, Lu Y, Markello TC, Adams DR, Steinbach PJ, Fuqua BK, Parisi X, Kaler SG, Vulpe CD, et al. Biallelic hephl1 variants impair ferroxidase activity and cause an abnormal hair phenotype. PLoS Genet. 2019;15:e1008143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li T, Pavletich NP, Schulman BA, Zheng N. High-level expression and purification of recombinant scf ubiquitin ligases. Methods Enzymol. 2005;398:125–142 [DOI] [PubMed] [Google Scholar]
- 37.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, et al. Structure of the cul1-rbx1-skp1-f boxskp2 scf ubiquitin ligase complex. Nature. 2002;416:703–709 [DOI] [PubMed] [Google Scholar]
- 38.Werner A, Iwasaki S, McGourty CA, Medina-Ruiz S, Teerikorpi N, Fedrigo I, Ingolia NT, Rape M. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature. 2015;525:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin e for ubiquitination and controls s phase in mammalian cells. Genes Dev. 1999;13:2375–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saritas T, Cuevas CA, Ferdaus MZ, Kuppe C, Kramann R, Moeller MJ, Floege J, Singer JD, McCormick JA. Disruption of cul3-mediated ubiquitination causes proximal tubule injury and kidney fibrosis. Sci Rep. 2019;9:4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cornelius RJ, Zhang C, Erspamer KJ, Agbor LN, Sigmund CD, Singer JD, Yang CL, Ellison DH. Dual gain and loss of cullin 3 function mediates familial hyperkalemic hypertension. Am J Physiol Renal Physiol. 2018;315:F1006–F1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duda DM, Olszewski JL, Tron AE, Hammel M, Lambert LJ, Waddell MB, Mittag T, DeCaprio JA, Schulman BA. Structure of a glomulin-rbx1-cul1 complex: Inhibition of a ring e3 ligase through masking of its e2-binding surface. Mol Cell. 2012;47:371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki E, Susa K, Mori T, Isobe K, Araki Y, Inoue Y, Yoshizaki Y, Ando F, Mori Y, Mandai S, et al. Klhl3 knockout mice reveal the physiological role of klhl3 and the pathophysiology of pseudohypoaldosteronism type ii caused by mutant klhl3. Mol Cell Biol. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing mutations in cullin3 protein impair rhoa protein ubiquitination and augment the association with substrate adaptors. J Biol Chem. 2015;290:19208–19217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferdaus MZ, Miller LN, Agbor LN, Saritas T, Singer JD, Sigmund CD, McCormick JA. Mutant cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, Zheng N. Structure of the cand1-cul1-roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119:517–528 [DOI] [PubMed] [Google Scholar]
- 49.Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, Yasuda T, Cavadini S, Wakasugi M, Hanaoka F, et al. The molecular basis of crl4ddb2/csa ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039 [DOI] [PubMed] [Google Scholar]
- 50.Cichon-Kawa K, Mizerska-Wasiak M, Cieslik M, Zaniew M, Panczyk-Tomaszewska M. [does gordon’s syndrome always manifest as hypertension? - a case report of one family]. Pol Merkur Lekarski. 2019;47:193–196 [PubMed] [Google Scholar]
- 51.Farfel A, Mayan H, Melnikov S, Holtzman EJ, Pinhas-Hamiel O, Farfel Z. Effect of age and affection status on blood pressure, serum potassium and stature in familial hyperkalaemia and hypertension. Nephrol Dial Transplant. 2011;26:1547–1553 [DOI] [PubMed] [Google Scholar]
- 52.Escamilla CO, Filonova I, Walker AK, Xuan ZX, Holehonnur R, Espinosa F, Liu S, Thyme SB, Lopez-Garcia IA, Mendoza DB, et al. Kctd13 deletion reduces synaptic transmission via increased rhoa. Nature. 2017;551:227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kasahara K, Kawakami Y, Kiyono T, Yonemura S, Kawamura Y, Era S, Matsuzaki F, Goshima N, Inagaki M. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat Commun. 2014;5:5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.