

Abstract
Inactivation of DNA mismatch repair by mutation or by transcriptional silencing of the MLH1 gene results in genome instability and cancer predisposition. We recently found (P. V. Shcherbakova and T. A. Kunkel, Mol. Cell. Biol. 19:3177–3183, 1999) that an elevated spontaneous mutation rate can also result from increased expression of yeast MLH1. Here we investigate the mechanism of this mutator effect. Hybridization of poly(A)+ mRNA to DNA microarrays containing 96.4% of yeast open reading frames revealed that MLH1 overexpression did not induce changes in expression of other genes involved in DNA replication or repair. MLH1 overexpression strongly enhanced spontaneous mutagenesis in yeast strains with defects in the 3′→5′ exonuclease activity of replicative DNA polymerases δ and ɛ but did not enhance the mutation rate in strains with deletions of MSH2, MLH1, or PMS1. This suggests that overexpression of MLH1 inactivates mismatch repair of replication errors. Overexpression of the PMS1 gene alone caused a moderate increase in the mutation rate and strongly suppressed the mutator effect caused by MLH1 overexpression. The mutator effect was also reduced by a missense mutation in the MLH1 gene that disrupted Mlh1p-Pms1p interaction. Analytical ultracentrifugation experiments showed that purified Mlh1p forms a homodimer in solution, albeit with a Kd of 3.14 μM, 36-fold higher than that for Mlh1p-Pms1p heterodimerization. These observations suggest that the mismatch repair defect in cells overexpressing MLH1 results from an imbalance in the levels of Mlh1p and Pms1p and that this imbalance might lead to formation of nonfunctional mismatch repair complexes containing Mlh1p homodimers.
Mismatch repair (MMR) is an important and powerful mutation avoidance mechanism in prokaryotes and eukaryotes. A major function of MMR proteins is to correct mismatches arising during DNA replication and recombination (reviewed in references 2, 20, 29, and 44). In Escherichia coli, the early steps in MMR require the products of the mutS and mutL genes. These genes are conserved in eukaryotes, which contain multiple MutS and MutL homologs that are proposed to function early in MMR. Repair of mismatches in DNA involves either of two complexes of MutS homologs. The MSH2-MSH6 heterodimer participates in the repair of base-base mismatches and insertion-deletion mismatches involving a small number of nucleotides, while MSH2-MSH3 is responsible mainly for repair of insertion-deletion mismatches involving a larger number of nucleotides. MMR also requires complexes of MutL homologs, three of which have been described for both yeast and humans. One is a heterodimer of Mlh1p and Pms1p in yeast (36) or of MLH1 and PMS2 in humans (26) that has been designated hMutLα. The Saccharomyces cerevisiae Mlh1p-Pms1p heterodimer forms a complex with Msh2p-Msh6p or Msh2p-Msh3p bound to mismatched DNA in vitro (12, 13). This suggests that the Mlh1p-Pms1p heterodimer participates in correcting mismatches recognized by both Msh2p-Msh6p and Msh2p-Msh3p. Yeast Mlh1p has been also shown to interact with two other MutL homologs, Mlh3p and Mlh2p, presumably forming heterodimers (46), and human MLH1 protein has been shown to interact with the human MLH3, a homolog of yeast Mlh3p (27). The yeast Mlh1p-Mlh3p complex was suggested by genetic data to participate in Msh2p-Msh3p-dependent repair of insertion-deletion mispairs (10). A minor role in correcting frameshift intermediates has been also proposed for the yeast Mlh1p-Mlh2p heterodimer (14). Human MLH1 protein also interacts with human PMS1 to form a heterodimer designated hMutLβ (24, 37), although the function of this complex in MMR or other processes remains to be established.
Mutations in MMR genes can inactivate MMR and thus strongly elevate spontaneous mutation rates, especially addition and deletion mutations in short, repetitive (microsatellite) sequences. These MMR gene mutations also predispose humans and mice to tissue-specific cancers. In addition to inactivation via gene mutation, MMR activity can also be modulated by changes in expression of MMR genes. For example, hypermethylation of the human MLH1 promoter results in lack of the MLH1 gene expression, loss of MMR activity, and microsatellite instability (16, 18). An increasing number of reports describe a correlation between human MLH1 promoter hypermethylation and sporadic colon, gastric, and endometrial cancers with microsatellite instability (see, for example, references 9, 16, 23, and 45). Conversely, MMR activity can be reduced by increased expression of MMR genes. Thus, methotrexate-resistant human cell lines that overproduce MSH3 have a base substitution mutator phenotype (8). Extracts of these cells are defective in repairing base-base mismatches, and repair can be restored by addition of purified hMSH2-hMSH6 complex. The MMR defect was suggested to result from diminished MSH2-MSH6 levels due to sequestration of MSH2 by excess MSH3 (8, 28).
Elevated mutation rates have also been reported in yeast strains overproducing either of two eukaryotic MutL homologs. Expression of the PMS1 gene from a multicopy plasmid in a wild-type yeast strain produced a 10-fold increase in the reversion rate of the hom3-10 frameshift mutation (21). More recently, we found that increased expression of the yeast MLH1 gene produced a strong mutator phenotype for several genetic markers, with the mutation rate approaching that of a mlh1 deletion strain (39). These mutator effects are in contrast to results with the yeast MSH2 gene, which does not confer hypermutability when overexpressed in wild-type yeast strains (7).
In this study, we explore the mechanism of the Mlh1p-induced mutator effect. Using hybridization to DNA microarrays, we show that overexpression of MLH1 does not induce changes in expression of other genes involved in DNA repair or replication. While Mlh1p is required to repair replication errors, it also participates in other DNA transactions including recombination and transcription-coupled nucleotide excision repair (17, 22, 46) which can modulate the mutation rate. To determine if the Mlh1p-induced mutator phenotype was due to failure to correct DNA replication errors, here we have examined effects of MLH1 overexpression in strains defective in genes controlling either DNA replication or MMR. The data suggest that the mutator phenotype of MLH1-overexpressing strains results from inactivation of MMR dependent on MSH2, MLH1, and PMS1 genes. We also show that the Mlh1p-induced mutator phenotype can be suppressed by overexpression of PMS1 or by a missense mutation in the MLH1 gene that disrupts Mlh1p interaction with Pms1p. Finally, we show that purified Mlh1p is capable of forming a homodimer in solution, a finding which suggests that MMR in cells overexpressing MLH1 could be disrupted by nonfunctional MMR complexes containing Mlh1p homodimers.
MATERIALS AND METHODS
Strains.
S. cerevisiae strain E134 (MATα ade5 lys2::InsEA14 trp1–289 his7-2 leu2-3,112 ura3-52) and its mlh1Δ derivative have been described previously (39). A deletion of the PMS1 gene was constructed in the E134 strain as described elsewhere (31). DAG60 is isogenic to E134 but contains a msh2::kanMX disruption (4). Wild-type strains CG379-3-29RL (MATα ade5 lys2-Tn5-13 trp1-289 his7-2 leu2-3,112 ura3Δ bik1::ura3-29RL) and 8C-YUNI101 (MATa ade2-1 trp1-1 his7-2 leu2-3,112 ura3Δ bik1::ura3-29RL) will be described elsewhere (Y. I. Pavlov, P. V. Shcherbakova, and T. A. Kunkel, submitted for publication). The pol2-4 mutant of CG379-3-29RL was constructed as previously described (30). The pol3-01 mutant of 8C-YUNI101 constructed as described elsewhere (31) was kindly provided by Y. I. Pavlov (National Institute of Environmental Health Sciences). A diploid heterozygous for the pol3-01 mutation and an isogenic wild-type diploid were constructed by crossing the E134 strain to the pol3-01 mutant of 8C-YUNI101 or the original 8C-YUNI101 strain. Haploid strains with missense mutations in the MLH1 gene were constructed by replacing the chromosomal wild-type MLH1 gene of the E134 strain with the mutant alleles as previously described (39). Strains CG1945 and Y187 for the two-hybrid analysis were purchased from Clontech Laboratories, Inc.
Plasmids.
Plasmid pMMR75, containing the MLH1 gene under the control of the ADH1 promoter, and the URA3-based integrative plasmid YIpMLH1 have been described previously (39). A control vector for MLH1 overexpression experiments was constructed by digestion of pMMR75 with SacI and BamHI that cut out the MLH1 open reading frame (ORF) followed by self-ligation of the blunt-ended vector. Missense mutations in the MLH1 gene were made in pMMR75 and YIpMLH1 by site-directed PCR mutagenesis as previously described (39). The presence of mutations was confirmed by partial sequencing of the MLH1 gene as described elsewhere (39). We used the pAM58 plasmid (31) for disruption of the PMS1 gene and the pJB1 plasmid (30) for making the pol2-4 mutation. Plasmid pAC12 (4) was used for MSH2 overexpression. Plasmid pMMR84, containing the PMS1 ORF, was a gift from L. Prakash (University of Texas). To create a plasmid overexpressing PMS1 from the galactose-inducible GAL10 promoter, the PMS1 ORF from pMMR84 was amplified by PCR using primers 5′-TTA GGT ACC ATG ACA CAA ATT CAT CAG-3′ (forward) and 5′-TTT GGT ACC TCA TAT TTC GTA ATC C-3′ (reverse). The primers introduced a KpnI restriction enzyme site at each end. After digestion with KpnI, the PCR product was ligated into the KpnI site of YEp181SPGAL (4) to generate pMH8.
Plasmids pGAD424 and pGBT9 were purchased from Clontech Laboratories, Inc. To construct pGAD424-yPMS1, the PMS1 ORF was amplified by PCR as described above using primers 5′-TTT GGA TCC GCA TGA CAC AAA TTC ATC AG -3′ (forward) and 5′-TTT TTT GTC GAC TCA TAT TTC GTA ATC C-3′ (reverse), which introduced a BamHI restriction site at the 5′ end and a SalI restriction site at the 3′ end. Following digestion with BamHI and SalI, the PCR product was ligated into the corresponding sites of pGAD424 to create a translational fusion between PMS1 and the GAL4 transcriptional activation domain. In a similar fashion, the MLH1 ORF was amplified by PCR from pMMR75 using primers 5′-TTT GGA TCC TTA TGT CTC TCA GAA TAA AAG C-3′ (forward) and 5′-CAT TCA GTC GAC TTA ACA CCT CTC AAA AAC-3′ (reverse) and ligated into the BamHI and SalI sites of pGBT9 to create a translational fusion between MLH1 and the GAL4 DNA binding domain. To confirm that no mutations were introduced during PCR, the entire coding regions of MLH1 and PMS1 in all constructs were sequenced using an ABI Prism 377 DNA sequencer.
Microarray hybridizations.
cDNA microarray chips containing 6,226 yeast ORFs were prepared as previously described (5, 15). Briefly, primers specific for each ORF (Research Genetics, Birmingham, Ala.) were used to amplify yeast cDNAs from genomic DNA in a 100-μl PCR mixture using AmpliTaq (PE Biosystems, Foster City, Calif.) and Pfu polymerases (Stratagene, La Jolla, Calif.). The PCR products were analyzed on 2% agarose gels to ensure amplification of the desired product, which was then ethanol precipitated. The overall success rate of the reactions based on the size and purity of PCR products was approximately 99.5%. The purified cDNAs were resuspended in ArrayIt buffer (Telechem, San Jose, Calif.) and spotted onto poly-l-lysine-coated glass slides using a modified, robotic DNA arrayer (Beecher Instruments, Bethesda, Md.). Total RNA was extracted from the E134 strain containing the pMMR75 plasmid or control vector by hot acid phenol extraction. A cell pellet from ∼100 to 150 ml of culture was resuspended in 4 ml of buffer containing 10 mM Tris-HCl (pH 7.5), 10 mM EDTA, and 0.5% sodium dodecyl sulfate (SDS); mixed with 4 ml of acid (pH 4.5) phenol; and incubated at 65°C for 1 h with occasional vigorous vortexing. The mixture was then cooled on ice for 10 min and centrifuged at 4°C for 10 min. The aqueous phase was reextracted with phenol at room temperature and extracted once with chloroform, and RNA was ethanol precipitated. A poly(A)+ RNA-enriched fraction was isolated from total RNA by using the Oligotex mRNA Midi kit from Qiagen. RNA (2 to 4 μg) was labeled with Cyanine 3 (Cy3)- and Cy5-conjugated dUTP (Amersham, Piscataway, N.J.) using a reverse transcription reaction and hybridized to a yeast cDNA microarray chip (4). cDNA chips were scanned using an Axon scanner (Axon Instruments, Foster City, Calif.), and images were analyzed using the Array Suite software (Scanalytics, Fairfax, Va.). The relative fluorescence intensity was measured for each labeled RNA, and a ratio of the intensity of each fluor bound to each probe was calculated. The amount of autofluorescence generated in the Cy3 channel was measured, and a minimum intensity cutoff was set just above this value. Hybridizations were conducted on two independently obtained pools of RNA, two times on one pool and once on another pool.
Mutation rate measurements.
Reversion of his7-2 and lys2::InsEA14 alleles was used to monitor changes in the spontaneous mutation rate. The his7-2 frameshift mutation reverts mainly via +1 insertions and −2 deletions in the wild-type strain and almost exclusively via +1 insertions in a run of seven A · T base pairs in the mlh1 strain (39). The lys2::InsEA14 mutation reverts via loss of a single base pair in a run of 14 A · T base pairs (41). Rates of His+ and Lys+ reversion were measured by fluctuation analysis as previously described (39). To estimate the effect of PMS1 or MSH2 overexpression on the mutation rate, strains containing corresponding plasmids were grown in medium containing 2% galactose instead of glucose.
Antibodies to yeast MMR proteins.
A part of the yeast MLH1 gene corresponding to 200 C-terminal amino acids and a part of the yeast PMS1 gene corresponding to 125 N-terminal amino acids were amplified by PCR using primers that introduced an XhoI restriction site at the 5′ end and a KpnI restriction site at the 3′ end. Following digestion with XhoI and KpnI, the PCR products were ligated into the corresponding sites of pRSET (Invitrogen). Expression of the resulting constructs in Escherichia coli BL21(DE3) produced histidine-tagged insoluble polypeptides that were purified using Ni-nitrilotriacetic acid (Ni-NTA) chromatography (Qiagen) under denaturing conditions with minor modifications. Briefly, cells from a 1-liter culture were disrupted by sonication, and the pellet was separated from soluble proteins by centrifugation and solubilized in buffer containing 8 M urea, 100 mM NaH2PO4, and 10 mM Tris-HCl (pH 8.0). The solution was then clarified by centrifugation and applied to an NTA-Ni2+ agarose column (1.79 cm2 by 5 cm). Further purification steps were the same as those in the Qiagen protocol. Fractions containing purified peptides were diluted stepwise to a urea concentration of 2 M and used to raise polyclonal antiserum in rabbits. Polyclonal antibodies to yeast Msh2p have been described previously (7).
Immunoblot analysis of yeast extracts.
Transformants of the E134 strain with pMMR75, pMH8, pAC12, or combinations of these plasmids were grown to an optical density at 600 nm of 0.8 to 0.9 in 50 ml of standard dropout media (38) selective for the plasmids. To induce PMS1 and MSH2 expression, strains containing corresponding plasmids were grown in medium containing 2% galactose instead of glucose. Cells were harvested by centrifugation, resuspended in an equal volume of lysis buffer (25 mM Tris-HCl [pH 7.6], 1 mM EDTA, 100 mM NaCl, 10 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride), and disrupted by vortexing with an equal volume of 0.5-mm glass beads (BioSpec Products, Inc.). Cell debris was pelleted by centrifugation, and supernatants were stored at −80°C. The extracts were subjected to electrophoresis in a 4 to 20% gradient polyacrylamide gel (Novex) and transferred to a polyvinylidene difluoride membrane (Novex). The blots were incubated in 1% nonfat dry milk for 30 min; probed with a 1:1,000 dilution of anti-Mlh1p, anti-Pms1p, or anti-Msh2p polyclonal serum for 1 h; washed three times with 1% nonfat dry milk plus 0.5% bovine serum albumin; and incubated with a 1:1,000 dilution of alkaline phosphatase-conjugated goat anti-rabbit immunoglobulin G (Oncogene Research Products) for 30 min. The bands were visualized by using a Western Blue-stabilized substrate (Promega).
Yeast two-hybrid assays.
Yeast strains CG1945 and Y187 were cotransformed with pGAD424 containing the wild-type PMS1 gene and pGBT9 containing wild-type or mutant alleles of the MLH1 gene. Transformants were selected on media lacking tryptophan and leucine. Interactions were determined by the ability of the corresponding combination of proteins to induce expression of the HIS3 reporter gene in CG1945 or the lacZ reporter gene in Y187. For the HIS3 expression assay, CG1945 transformants were plated onto medium lacking tryptophan, leucine, and histidine and supplemented with 5 mM 3-amino-1,2,4-triazole. For the β-galactosidase activity assay, Y187 transformants were grown to saturation in medium lacking tryptophan and leucine, and the cultures were then dispensed into the wells of a 96-well microtiter plate. Aliquots of the cultures from each well were transferred using a 48-pin replicator onto a nitrocellulose filter (Schleicher & Schuell BA85) placed on the surface of a petri dish containing Trp− Leu− dropout medium. The plate was incubated at 30°C for 2 days. The filter was then frozen in liquid nitrogen and incubated at 30°C on filter paper soaked in buffer containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) and 2-mercaptoethanol, as recommended by the supplier (Clontech Laboratories, Inc.).
Analytical ultracentrifugation.
The associative behavior of Mlh1p and the Mlh1p-Pms1p complex was studied by equilibrium analytical ultracentrifugation in a BeckmanCoulter XL-A analytical ultracentrifuge. The studies were carried out at 4°C using 12-mm optical path length carbon-filled Epon double-sector centerpieces and quartz windows in the cells in a four-place rotor. Equilibrium was considered to have been attained when scans taken 24 h apart did not vary. The time to equilibrium was significantly extended because of the presence of 5% glycerol in the buffer. The buffer contained 150 mM NaCl, 5% glycerol, 0.1 mM EDTA, 15 mM 2-mercaptoethanol, and 25 mM NaPO4 (pH 7.6). The buffer density, 1.02207 g cm−3, was measured in an Anton Paar DMA 5000 density meter. The compositional partial specific volumes and the extinction coefficients at 280 nm were calculated from the amino acid compositions using the consensus values of Perkins (35). Values of 0.734 and 0.726 cm3 g−1 for the partial specific volumes and 48,820 and 49,570 M−1 cm−1 for the extinction coefficients were obtained for the Mlh1p and Pms1p monomers, respectively. The rotor speeds used were 8,000 and 10,000 rpm for the study of the Mlh1p monomer-dimer equilibrium and 15,000 rpm for the study of the more complex Mlh1p-Pms1p homodimer-heterodimer equilibrium reaction. Data were analyzed by fitting the equilibrium concentration distributions as functions of the radius with several mathematical models by nonlinear least-squares curve fitting using the mathematical modeling system MLAB (Civilized Software, Silver Spring, Md.).
A model for the Mlh1p equilibrium distribution is as follows:
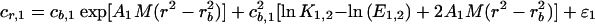 |
1 |
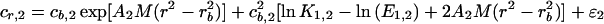 |
2 |
where the subscripts 1 and 2 for cr, cb, A, and ɛ refer to the two different rotor speeds; cr is the total concentration expressed as absorbency at 280 nm; cb is the concentration of monomer at the radial position of the cell bottom, rb, similarly expressed and is a local fitting parameter; M is the molar mass of monomer, 87,060 Da; ɛ is a small baseline error term and is a local fitting parameter; and ln K1,2 is the natural logarithm of the molar equilibrium constant for dimer formation and is a global fitting parameter, common to both data sets. E1,2 = 1.2E/2, where E is the molar extinction coefficient of Mlh1p, 48,820 M−1 cm−1, and ln K1,2 − ln E1,2 = ln k1,2, the equilibrium constant on an absorbency scale. E is multiplied by 1.2 because the optical path length is 1.2 cm. A = (1 − φ*ρ)ω2/2RT, where φ* is the compositional partial specific volume, ρ is the solvent density, ω is the rotor angular velocity in radians per second, R is the gas constant, and T is the absolute temperature.
A model for the Mlh1p-Pms1p equilibrium distribution is as follows:
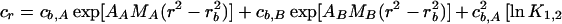 |
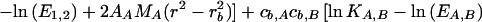 |
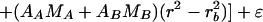 |
3 |
In this model, the subscript A refers to Mlh1p and the subscript B refers to Pms1p. AA, MA, E1,2, and ln K1,2 have the same values as those used for Mlh1p alone; EA,B = (1.2EMlh1)(1.2EPms1)/(1.2EMlh1 + 1.2EPms1); and EMlh1 and MA have the same values as above, while EPms1 = 49,570 M−1 cm−1 and MB = 99,343 Da. Thus, cb,A, cb,B, ln KAB, and ɛ are the fitting parameters. A model similar to this but eliminating cb,A as a fitting parameter has been developed by invoking conservation of mass (25). Given that the Mlh1p-Pms1p complex exists in a 1:1 molar stoichiometry, this is a particularly useful model, since the elimination of a parameter reduces the returned errors of the other parameter values and is a more highly constrained model; for these reasons it was applied here.
RESULTS
Genome-wide analysis of gene expression in strains overproducing Mlh1p.
Overexpression of the yeast MLH1 gene in a wild-type strain causes a strong increase in the mutation rate that is dependent on the MLH1 expression level (39). This mutator effect could be caused by excess of Mlh1p per se or indirectly through changes in regulation of other genes controlling DNA repair or replication. We therefore used hybridization to DNA microarrays containing 96.4% of the yeast ORFs to determine if Mlh1p-overproducing strains displayed any changes in expression of genes other than MLH1. The mRNA from each strain—E134 containing pMMR75 or E134 containing the control vector—was labeled with either Cy3- or Cy5-deoxyuridine and hybridized in a competitive reaction on a single cDNA array. Gene expression differences between the two strains were expressed as ratios of the intensities of the hybridized cDNAs. The complete data set is available on the Internet at http://dir.niehs.nih.gov/microarray/datasets. Analysis of plots of Cy3 intensity versus Cy5 intensity showed that there was a very tight distribution of the ratios of all of the targets (Fig. 1), indicating that minimal change in gene expression occurred upon overexpression of MLH1. In fact, of ∼6,200 ORFs, only 9 showed a more than twofold increase or decrease in mRNA level in three replicate experiments (Table 1). The greatest elevation of mRNA level (∼20-fold) was observed for the MLH1 gene and was consistent with the increase in protein level (see below). None of the other known MMR genes was induced or repressed in the strain overproducing Mlh1p (Table 1).
FIG. 1.

Genome-wide analysis of gene expression in a yeast strain overproducing Mlh1p and an isogenic wild-type strain. The mRNA of strain E134 containing plasmid pMMR75 was labeled with Cy3-deoxyuridine, and mRNA of strain E134 containing the control vector was labeled with Cy5-deoxyuridine. Cy5 intensity values are plotted against Cy3 intensity values for each hybridized cDNA. Open circles correspond to individual cDNAs. The dashed lines indicate twofold differences in hybridization intensity. The R value represents a Pearson correlation coefficient calculated for the log2(Cy3) values versus the log2(Cy5) values.
TABLE 1.
Relative mRNA levels for MMR genes and ORFs that showed increased or reduced expression in an Mlh1p-overproducing strain
| ORF | Gene | Gene producta | E134 pMMR75/E134 vector ratiob |
|---|---|---|---|
| Genes induced in MLH1-overexpressing strainc | |||
| YMR167W | MLH1 | MMR protein | 19.6 |
| YLR162W | ? | Hypothetical protein | 4.3 |
| YMR046Cd | TY1A | Ty1A protein | 3.3 |
| YOL149W | DCP1 | mRNA decapping enzyme | 2.2 |
| Genes repressed in MLH1-overexpressing strainc | |||
| YDR007W | TRP1 | Phosphoribosylanthranilate isomerase | 0.42 |
| YDR008C | ? | 0.39 | |
| Genes known or suggested to control MMR | |||
| YNL082W | PMS1 | MutL homolog | 1.2 |
| YPL164C | MLH3 | MutL homolog | 1.1 |
| YLR035C | MLH2 | MutL homolog | 1.2 |
| YOL090W | MSH2 | MutS homolog | 1.6 |
| YCR092C | MSH3 | MutS homolog | 1.0 |
| YDR097C | MSH6 | MutS homolog | 1.0 |
| YBR088C | POL30 | PCNA | 0.71 |
| YOR033C | EXO1 | 5′→3′ exonuclease | 1.8 |
| YAR007C | RFA1 | Replication factor A, 69-kDa subunit | 1.3 |
| YDL102W | POL3 | DNA polymerase δ, catalytic subunit | 1.1 |
| YNL262W | POL2 | DNA polymerase ɛ, catalytic subunit | 0.90 |
Descriptions were derived from the MIPS and SGD databases.
Average for three experiments.
Genes for which mRNA levels were increased or reduced more than twofold in all three replicate experiments are listed as induced or repressed, respectively.
In addition to YMR046C, three other ORFs encoding Ty1Ap showed more than twofold increases in mRNA level in all three experiments with average E134 pMMR75-to-E134 vector ratios of 2.8 to 3.2. Two additional ORFs encoding Ty1Ap showed more than twofold increases in mRNA levels in two of the three experiments.
Of the eight other genes that showed altered mRNA level, none was expected to control DNA replication or repair. An increase in mRNA level was observed for the YLR162W ORF, which encodes a protein of 118 amino acids with no known function or significant similarity to any known protein (Table 1). We have found that the 3′ half of this ORF is 96% identical to a part of the gene encoding 25S rRNA. The yeast rRNA genes are organized in a cluster containing about 100 to 200 repeating units. The number of repeats depends on the strain and can also vary within a single strain (47). Since rRNA was abundant in the RNA preparations used for the hybridizations, the 4.3-fold increase in hybridization intensity for the YLR162W ORF likely reflects changes in the number and/or expression of the rRNA genes rather than induction of expression of this ORF.
Four Ty1 ORFs corresponding to TYA proteins showed more than twofold increases in mRNA level in all three experiments (Table 1). Two additional TYA ORFs showed more than twofold-increased hybridization intensities in two of the three experiments. Since different Ty1 elements are nearly identical, the primers designed to amplify specific TYA ORFs for the microarray chips likely amplified a mixture of ORFs, which could hybridize to virtually any Ty1 transcript. Thus, increased levels of Ty1 transcripts in the MLH1-overexpressing strain could not be interpreted as induction of expression of any particular ORFs but rather indicate general changes in Ty1 distribution and/or expression.
Two ORFs, TRP1 and YDR008C, showed more than twofold reductions in mRNA level in the strain overexpressing MLH1. The TRP1 gene was present both on the MLH1-overexpressing plasmid pMMR75 and on the control vector. The 2.4-fold decrease in the TRP1 mRNA level could reflect a lower plasmid copy number in cells overexpressing MLH1. The YDR008C ORF, which is described as a questionable ORF in the MIPS yeast database, overlaps with the TRP1 ORF. Thus, the 2.5-fold reduction in hybridization intensity for this ORF could result from the reduced level of the TRP1 transcript in cells overproducing Mlh1p.
MMR is inactivated in strains overexpressing MLH1.
To determine if the Mlh1p-induced mutator phenotype results from suppression of MMR, we overexpressed MLH1 in strains with disruptions of genes known to be required for MMR and in strains with mutations that reduce the intrinsic 3′→5′ exonuclease activities of replicative DNA polymerases δ and ɛ. These exonucleases proofread replication errors, and this proofreading and MMR act in series to correct mismatches (31, 32). If the Mlh1p-induced mutator effect results from inhibition of MMR, then the MLH1-overexpressing plasmid should not elevate the mutation rate in strains which are MMR deficient due to mutations in MLH1, PMS1, or MSH2. However, Mlh1p-induced suppression of MMR would lead to a synergistic increase in the mutation rate in strains with the exonuclease defects because in these strains more polymerase errors would remain uncorrected by proofreading.
We introduced plasmid pMMR75 expressing the MLH1 gene from the ADH1 promoter into strains bearing deletions of MLH1, PMS1, or MSH2 or mutations that reduce the 3′→5′ exonuclease activity of DNA polymerases ɛ (pol2-4) or δ (pol3-01). Reversion of the his7-2 frameshift mutation was used to estimate the effects of MLH1 overexpression on the mutation rate in these strains. The his7-2 mutation is a deletion of one nucleotide in a run of eight adenines in the HIS7 gene (39). It reverts mainly via +1 insertions and −2 deletions in wild-type strains and predominantly via +1 insertions in the A7 run in MMR-deficient strains (39). Deletion of MLH1, PMS1, or MSH2 led to an ∼200-fold elevation in the His+ reversion rate (Table 2), consistent with earlier observations (19, 32, 39). The presence of the plasmid expressing MLH1 had negligible effects on the reversion rates of the mlh1, pms1, and msh2 strains (Table 2). It is interesting that the plasmid overexpressing wild-type MLH1 does not complement the MMR defect in the mlh1 deletion strain, although this defect could be readily complemented by a single copy of the wild-type MLH1 gene in diploid strains (39).
TABLE 2.
Effect of MLH1 overexpression on mutation rate in strains with defects in MMR or exonucleolytic proofreading
| Straina | His+ reversion rate (10−8)b | Fold increasec |
|---|---|---|
| Expt with MMR mutants | ||
| Wild type | 0.71 (0.49–0.97) | 1 |
| Wild type + [ADH1::MLH1] | 94 (78–110) | 130 |
| mlh1Δ | 120 (82–200) | 170 |
| mlh1Δ + [ADH1::MLH1] | 90 (62–110) | 130 |
| pms1Δ | 180 (140–540) | 250 |
| pms1Δ + [ADH1::MLH1] | 190 (86–460) | 270 |
| msh2Δ | 130 (110–280) | 180 |
| msh2Δ + [ADH1::MLH1] | 180 (120–260) | 250 |
| Expt with pol2-4 mutant | ||
| Wild type | <1 | 1 |
| Wild type + [ADH1::MLH1] | 83 (57–150) | >80 |
| pol2-4 | 8.7 (6.7–14) | >8 |
| pol2-4 + [ADH1::MLH1] | 1,300 (350–2,400) | >1,300 |
| Expt with POL3/pol3-01 diploid | ||
| POL3/POL3 | 2.5 (1.3–4.3) | 1 |
| POL3/POL3 + [ADH1::MLH1] | 160 (100–220) | 64 |
| POL3/pol3-01 | 41 (29–65) | 16 |
| POL3/pol3-01 + [ADH1::MLH1] | 2,300 (1,300–3,600) | 920 |
Strains contained pMMR75 or the control vector.
Median for at least nine cultures, with 95% confidence limits in parentheses.
Relative to the corresponding wild type strain.
A mutation in the exonuclease domain of DNA polymerase ɛ, pol2-4, increased the His+ reversion rate more than eightfold, in agreement with the reported mutator phenotype of the pol2-4 strain (30). Overexpression of MLH1 in this strain resulted in a 150-fold increase in the reversion rate (Table 2; 1,300 × 10−8 versus 8.7 × 10−8). This effect is similar to the Mlh1p-induced increase in the reversion rate observed with wild-type strains. Likewise, the 15-fold mutator effect of the pol2-4 mutation in the presence of the MLH1-overexpressing plasmid (Table 2; 1,300 × 10−8 versus 83 × 10−8) is similar to the previously reported 10- to 20-fold effect of this mutation in isogenic wild-type strains (30, 32) and consistent with the more than eightfold effect of pol2-4 in the wild-type strain observed in this study (Table 2). The His+ reversion rate in the pol2-4 mutant in the presence of the MLH1-overexpressing plasmid (1,300 × 10−8) was indistinguishable from the previously described His+ reversion rate in an isogenic pol2-4 pms1 double mutant (1,000 × 10−8 [32]). The multiplicity of the mutator effects of the pol2-4 mutation and MLH1 overexpression suggests that both error-correcting mechanisms have been inactivated, the latter by overexpression of MLH1.
The pol3-01 mutation in the exonuclease domain of DNA polymerase δ is incompatible with pms1 or msh2 deletions in haploid strains (31, 42), which has been suggested (31) to reflect a catastrophically high mutation rate. In line with the hypothesis that MLH1 overexpression confers an MMR defect, the pMMR75 plasmid transformed a pol3-01 haploid strain with low efficiency in comparison to transformation of this strain with a control vector or transformation of an isogenic wild-type strain with pMMR75 (data not shown). To overcome this incompatibility, we overexpressed the MLH1 gene in a diploid strain that is heterozygous for the pol3-01 mutation. Heterozygosity for pol3-01 leads to a moderate mutator phenotype (31) (Table 2; 41 × 10−8 versus 2.5 × 10−8). Similar to the effect of MLH1 overexpression in the pol2-4 mutant, the presence of the pMMR75 plasmid in the POL3/pol3-01 diploid caused a synergistic increase in the His+ reversion rate (Table 2). The 56-fold increase observed upon MLH1 overexpression in the heterozygous strain (Table 2; 2,300 × 10−8 versus 41 × 10−8) is similar to the 64-fold increase in the wild-type diploid strain, and the 14-fold mutator effect of heterozygosity for pol3-01 in the presence of the MLH1-overexpressing plasmid (Table 2; 2,300 × 10−8 versus 160 × 10−8) is similar to its effect in the control diploid (16-fold; Table 2; 41 × 10−8 versus 2.5 × 10−8), again consistent with suppression of two error-correcting mechanisms acting in series.
Suppression of the mutator phenotype by overexpression of PMS1
One hypothesis to explain the Mlh1p-induced suppression of MMR is that excess Mlh1p interferes with protein-protein interactions that are needed for efficient MMR. A prime candidate is Pms1p, since Mlh1p interacts with Pms1p to form the MutLα heterodimer that functions in MMR (36). To test if the Mlh1p-induced mutator effect can be suppressed by increased production of Pms1p, we introduced plasmid pMH8 expressing the PMS1 gene from the GAL10 promoter into the strain containing pMMR75 or a control vector. Overproduction of both Mlh1p and Pms1p in the transformants was confirmed by immunoblot analysis (Fig. 2, left and middle panels).
FIG. 2.

Overexpression of yeast MLH1, PMS1, and MSH2. (Left) Extracts (40 μg) from strain E134 (wild type), its mlh1 deletion mutant, and E134 containing plasmid pMMR75 were probed with anti-Mlh1p antibodies. (Middle) Extracts (20 μg) from strain E134, its pms1 deletion mutant, and E134 containing plasmid pMH8 were probed with anti-Pms1p antibodies. (Right) Extracts (40 μg) from strain E134, its msh2 deletion mutant, and E134 containing plasmid pAC12 were probed with anti-Msh2p antibodies.
It has been reported previously (21) that expression of PMS1 from a high-copy-number plasmid in a wild-type yeast strain produced a 10-fold increase in the rate of reversion of the hom3-10 allele, which scores single-base deletions in a run of seven A · T base pairs. Similarly, we observed that the pMH8 plasmid alone conferred an 8-fold increase in the his7-2 reversion rate, which measures single-base additions in a run of seven A · T base pairs, and a 180-fold increase in the rate of lys2::InsEA14 reversion, which measures single-base deletions in a run of 14 A · T base pairs (Table 3). Thus, the mutator effect of PMS1 overexpression was much weaker than that of MLH1 overexpression (39) (Tables 2 and 3). Both proteins were produced at much higher levels in cells harboring the expression plasmids than in control cells (Fig. 2). Since the specific activity of the antibodies is not known, direct comparison of Mlh1p and Pms1p levels could not be done in these experiments. Thus, the differences in Mlh1p- and Pms1p-induced mutator effects may or may not reflect different levels of overexpression.
TABLE 3.
Effects of PMS1 and MSH2 overexpression on the mutator phenotype of an MLH1-overexpressing straina
| Gene(s) overexpressed | Mutation rate (10−8)b
|
|
|---|---|---|
| Lys+ | His+ | |
| None | 13 (9.2–30) | 0.55 (0.30–0.96) |
| MLH1 | 55,000 (43,000–87,000) | 99 (67–260) |
| PMS1 | 950 (440–2,200) | 4.4 (3.0–8.1) |
| MLH1 + PMS1 | 590 (350–650) | 6.5 (5.5–9.1) |
| MSH2 | 15 (12–25)c | NDd |
| MLH1 + MSH2 | 110,000 (53,000–140,000) | 200 (170–520) |
Mutation rates were measured in strain E134 containing control vectors or plasmids overexpressing MMR genes. Plasmids pMMR75, pMH8, and pAC12 were used to overexpress MLH1, PMS1, and MSH2, respectively.
Median for at least nine independent cultures, with 95% confidence limits in parentheses.
From reference 6.
ND, not determined.
Overexpressing PMS1 in the Mlh1p-overproducing strain strongly reduced the mutator effect due to Mlh1p overproduction. The decrease was 93-fold for Lys+ reversion (Table 3; 590 × 10−8 versus 55,000 × 10−8) and 15-fold for His+ reversion (Table 3; 6.5 × 10−8 versus 99 × 10−8). However, concomitant overproduction of both MutL homologs did not return the reversion rate to that of the wild-type yeast strain but rather yielded a rate similar to that observed in the strain overexpressing Pms1p alone.
In contrast to suppression of the Mlh1p-induced mutator phenotype by overexpression of PMS1, overexpressing MSH2 from the GAL1 promoter does not suppress the mutator phenotype conferred by MLH1 overexpression (Table 3). Overproduction of Msh2p in the cells bearing both MLH1- and MSH2-overexpressing plasmids was confirmed by immunoblot analysis (Fig. 2, right panel). The Mlh1p-induced mutator phenotype was also unaffected by the presence of a yeast MSH6 expression plasmid (data not shown). Thus, the ability to suppress Mlh1p-induced spontaneous mutagenesis is specific for PMS1.
Effect of mlh1 missense mutations on the mutator phenotype.
To investigate the functional requirements for the Mlh1p-induced mutator phenotype, we next examined the effects of three mlh1 missense mutations on the ability of Mlh1p to confer a mutator phenotype (Fig. 3). By homology to the E. coli MutL protein, four conserved motifs in the N-terminal region of Mlh1p are suggested to be important for ATP hydrolysis involved in MMR function (1, 33). We previously showed (39) that a missense mutation that changes amino acid residue 65 from isoleucine to asparagine in motif II (Fig. 3A and B) results in a strong mutator phenotype characteristic of complete loss of MMR function. Also, a homologous change in human MLH1 is associated with hereditary nonpolyposis colorectal cancer (HNPCC) (40). When the I65N mutation was introduced into the MLH1 gene on the pMMR75 plasmid, it did not have a significant effect on the Mlh1p-induced mutator phenotype (Fig. 3C). This strongly implies that the Mlh1p-induced mutator phenotype does not require intact Mlh1p function. It also implies that the proposed ATPase activity of Mlh1p is not important for the mutator effect.
FIG. 3.


Effect of mlh1 missense mutations on the Mlh1p-induced mutator phenotype. (A) Schematic representation of the yeast Mlh1 protein. The location of the putative ATP-binding domain is indicated by homology to the E. coli MutL, with regions I, II, III, and IV corresponding to the conserved ATP binding motifs (1, 33). The Pms1p interaction region and the C-terminal homology (CTH) motif were described by Pang et al. (34). The location of the missense mutations that were made in the MLH1 overexpression vector is indicated. (B) Alignment of the amino acid sequences of the E. coli MutL, the S. cerevisiae Mlh1p, and human MLH1. Invariant amino acids are boxed. The amino acid substitutions studied here are indicated below the alignments. (C) Mutator phenotypes of strains overexpressing mlh1 missense alleles. The data are the rates of Lys+ reversion in strain E134 containing pMMR75 or its derivatives with mutations in MLH1 and are the medians for at least nine independent cultures with error bars indicating 95% confidence limits.
To determine if Mlh1p-Pms1p interaction is necessary for the mutator effect, we created two different amino acid changes in the C-terminal region of Mlh1p that is involved in the interaction with Pms1p (34). The two mutations R672P and A694T (Fig. 3A and B) were chosen by homology to HNPCC-associated mutations in the human MLH1 which have been shown to cause more than 95% reduction in the ability of MLH1 to interact with PMS2 (11). The A694T substitution in Mlh1p did not have any effect on the Mlh1p-induced mutator phenotype (Fig. 3C). However, the R672P change clearly reduced the mutator effect of MLH1 overexpression, eightfold for Lys+ reversion and fourfold for His+ reversion.
To check if the effects of the two C-terminal mutations on the Mlh1p-induced mutator phenotype correlate with their effects on Mlh1p-Pms1p interaction, we tested the mutant variants of proteins for interaction using the yeast two-hybrid system. The R672P and A694T mutations were introduced into the MLH1 gene fused to the sequence of the Gal4 DNA binding domain in the pGBT9 plasmid, and the interaction was tested in the CG1945 strain containing a HIS3 reporter gene. As shown in Fig. 4A, the R672P substitution in Mlh1p reduced the interaction with Pms1p to a level below detection, while Mlh1p-A694T retained the ability to interact with Pms1p. We also examined the effect of the missense mutations on Mlh1p-Pms1p interaction using the lacZ reporter gene in a Y187 yeast strain. Again, we did not detect any interaction of the Mlh1p-R672P with Pms1p (Fig. 4B). Mlh1p-A694T retained the ability to interact with Pms1p, although this ability was reduced in comparison to the wild-type Mlh1p as judged by slower development of blue color in the β-galactosidase filter assay (Fig. 4B). Thus, the effect of these mlh1 missense mutations on the Mlh1p-induced mutator phenotype reflected their ability to disrupt Mlh1p-Pms1p interaction.
FIG. 4.


Effect of amino acid substitutions in the C-terminal region of Mlh1p on interaction with Pms1p in the yeast two-hybrid system and the spontaneous mutation rate. (A) The CG1945 strain was cotransformed with pGAD424-yPMS1 and pGBT9 containing wild-type or mutant alleles of the MLH1 gene. The transformants were grown on medium lacking tryptophan, leucine, and histidine and supplemented with 5 mM 3-amino-1,2,4-triazole. (-), pGBT9 with no MLH1. (B) The Y187 strain was cotransformed with pGAD424-yPMS1 and pGBT9 containing wild-type or mutant alleles of the MLH1 gene. The transformants were tested for β-galactosidase activity using the color filter assay described in Materials and Methods. (C) Spontaneous mutation rates (relative to the wild type) in strain E134 and its derivatives with mutations in chromosomal MLH1.
To determine whether suppression of the Mlh1p-induced mutator phenotype and disruption of the Mlh1p-Pms1p interaction by the R672P mutation also correlated with the effect of this mutation on MMR efficiency in vivo, we replaced the chromosomal MLH1 allele in the E134 strain with the mlh1-R672P allele and measured the spontaneous mutation rate in this mutant. Similarly, a haploid mlh1-A694T mutant was constructed to look for any possible mutator effect of this mutation that did not suppress the Mlh1p-induced mutator phenotype and only slightly reduced interaction with Pms1p. The R672P mutation increased the rate of Lys+ and His+ reversion and Canr mutation 11,000-fold, 60-fold, and 16-fold, respectively; this effect is similar to the mutator effect of a mlh1 deletion (Fig. 4C) and is characteristic of a complete MMR defect. The A694T mutation did not have a significant effect on mutation rate for any of the three markers, suggesting that MMR was not significantly reduced in this mutant. Taken together, our data show that the R672P substitution in Mlh1p disrupts Mlh1p-Pms1p interaction, resulting in MMR defect, and reduces the ability of the overexpressed protein to interfere with MMR, while the A694T mutation does not have a strong effect on Mlh1p-Pms1p interaction, does not impair MMR, and does not affect the Mlh1p-induced mutator phenotype.
Purified Mlh1p can form homodimers in solution.
The results on suppression of the Mlh1p-induced mutator phenotype by overexpression of PMS1 or by the R672P substitution in Mlh1p led us to hypothesize that excess Mlh1p produces a mutator effect through formation of a Mlh1p-Mlh1p homodimer that could replace the Mlh1p-Pms1p heterodimer in protein-protein interactions, thus resulting in nonfunctional MMR complexes. To determine if Mlh1p is capable of forming homodimers, we studied the associative behavior of Mlh1p by equilibrium analytical ultracentrifugation. For comparison, the assembly state of the Mlh1p-Pms1p complex was also studied. The yeast Mlh1p and the Mlh1p-Pms1p heterodimer were purified as described elsewhere (M. C. Hall and T. A. Kunkel, submitted for publication), and the ultracentrifugation experiments were performed as described in Materials and Methods. The data for Mlh1p were analyzed by globally fitting the data sets at 8,000 and 10,000 rpm with several mathematical models including a monomer, a dimer, monomer-dimer equilibrium, and more complex associations. The data obtained for Mlh1p were most consistent with the monomer-dimer model described by equations 1 and 2 (see Materials and Methods). The results of the joint fit of the data for Mlh1p at 8,000 and 10,000 rpm are shown in Fig. 5A. The value of ln K12 obtained by global fitting is 12.671 ± 0.060, corresponding to a Kd of 3.14 ± 0.19 μM and a ΔG° of −6.98 ± 0.03 kcal mol−1. The excellent distribution of the residuals illustrated in Fig. 5A attests to the quality of the fit and the appropriateness of the model. In contrast, the distribution of residuals and plots themselves showed gross systematic deviations from the data sets when a model describing a monomeric protein was used (data not shown). Similarly to the analysis of Mlh1p data, the more complex Mlh1p-Pms1p association was analyzed by fitting several mathematical models to the data. A model that best describes the Mlh1p-Pms1p data takes into account both Mlh1p-Pms1p heterodimerization and Mlh1p-Mlh1p homodimerization and is given by equation 3 (see Materials and Methods). The fit of this mathematical model to the data of the Mlh1p-Pms1p complex is shown in Fig. 5B. The value of ln KAB obtained by fitting this model is 16.258 ± 0.408, corresponding to a Kd of 86.9 ± 36.4 nM for the Mlh1p-Pms1p heterodimer and a ΔG° of −8.95 ± 0.22 kcal mol−1. The distribution of the residuals illustrated in Fig. 5B indicates that the quality of the fit is adequate and that the model is appropriate. Of particular significance is the fact that there is a difference of almost 2 kcal mol−1 between the free-energy change for the formation of the Mlh1p-Mlh1p homodimer and the free-energy change for the formation of the Mlh1p-Pms1p heterodimer, reflecting the fact that the bond strength for the formation of the heterodimer is 36 times greater than that for the homodimer.
FIG. 5.

Analytical ultracentrifugation of purified Mlh1p and Mlh1p-Pms1p complex. (A) Concentration distributions as functions of radius for Mlh1p at equilibrium at 8,000 rpm (squares) and at 10,000 rpm (triangles) at 4°C. The distributions of the residuals for the two fits are shown at the top. (B) Concentration distribution as a function of radius for the Mlh1p-Pms1p complex at equilibrium at 15,000 rpm at 4°C. The distribution of the residuals is shown at the top.
DISCUSSION
It is now well established that DNA mismatch repair can be inactivated by mutations in any of several different MMR genes, by diminished expression of the human MLH1 gene, or by imbalanced expression of human MutS homologs. This study extends the latter mechanism to eukaryotic MutL homologs, by demonstrating that imbalanced expression of yeast MLH1 and PMS1 genes leads to a spontaneous mutator phenotype that reflects reduced postreplication mismatch repair capacity. We have shown that overexpression of the MLH1 gene does not induce changes in expression of any other genes involved in DNA repair or replication, suggesting that an excess of Mlh1p per se was causing inactivation of MMR. The importance of a proper balance in the relative amounts of eukaryotic MMR proteins was first appreciated in studies of a methotrexate-resistant human cell line that overexpresses MSH3 (8, 28). Those studies provided strong evidence that overproduced MSH3 protein sequestered most of the available MSH2 into a MSH2-MSH3 (MutSβ) complex. This reduced the amount of the MSH2-MSH6 (MutSα) heterodimer, resulting in diminished MutSα-dependent repair of base-base mismatches and a strong base substitution mutator phenotype. Our results suggesting that MMR activity can also be strongly reduced by overexpression of MLH1 further emphasize the importance to genome stability of maintaining the appropriate level of key MMR proteins.
The ∼4,000-fold increase in reversion of the lys2::InsEA14 allele in the strain overexpressing MLH1 (39) (Table 2) is much stronger than the increases in single msh6 or msh3 mutants, which are only 190-fold and 6-fold, respectively (42). The strong Mlh1p-induced mutator effect is in fact characteristic of strains with deletions of mlh1, pms1, msh2 (Table 2), or both msh3 and msh6 (42). This indicates that overexpression of MLH1 inactivates both the MutSα- and the MutSβ-dependent MMR pathways. This situation is obviously different from overexpression of human MSH3, which selectively inactivates MutSα-dependent repair. Within the framework of existing MMR models (10, 20), inactivation of both pathways is not easily rationalized by sequestration of a known functional partner. Current evidence indicates that Mlh1p is essential for both the MutSα- and the MutSβ-dependent MMR pathways, and Mlh1p is the invariant component of all three MutL-related heterodimers identified to date in yeast: Mlh1-Pms1 (36), Mlh1-Mlh3 (10), and Mlh1-Mlh2 (46). Thus, overexpression of Mlh1p would be expected to permit formation of all three MutL heterodimers.
This suggests that the Mlh1p-induced suppression of MMR results from excess Mlh1p that does not participate in heterodimer formation. The excess Mlh1p could interfere with MMR by nonproductive binding to any of several other essential MMR proteins with which MutLα may interact. These include MutS homologs and PCNA (12, 13, 43). Relevant here is that increased expression of PMS1 (Table 3) and the mlh1-R672P missense mutation (Fig. 3) that disrupts interaction with Pms1p (Fig. 4) both suppress the Mlh1p-induced mutator effect. These data are consistent with possible formation of a Mlh1p-Mlh1p homodimer that could replace the Mlh1p-Pms1p heterodimer in protein-protein interactions, thus resulting in nonfunctional MMR complexes. Indeed, the analytical ultracentrifugation experiments showed that Mlh1p is capable of forming a homodimer in solution (Fig. 5A), although the Kd for the homodimer formation is 36-fold higher than the Kd for Mlh1p-Pms1p heterodimerization. Thus, if the Mlh1p-Mlh1p homodimer inhibiting MMR can be formed in vivo, its abundance in wild-type yeast cells must be low in comparison to the Mlh1p-Pms1p heterodimer, yet increased in cells overexpressing MLH1. Overproduction of Pms1p together with Mlh1p would then shift the homodimer-to-heterodimer ratio and thus restore MMR. The homodimerization could occur via C-terminal interactions similar to those involved in Mlh1p-Pms1p heterodimerization (34) or homodimerization in E. coli MutL (6). The mlh1-R672P mutation impairing Mlh1p-Pms1p heterodimerization might then also affect homodimerization, thus reducing the mutator effect of MLH1 overexpression. We could not directly examine the effect of the R672P mutation on the ability of Mlh1p to form a homodimer because the protein with this substitution became insoluble during the course of purification. It is also possible that dimer formation is not required for the excess Mlh1p to interact with MutS homologs and/or other MMR proteins and that MMR is inhibited by protein complexes containing Mlh1p monomers. However, the effect of suppression of the mutator phenotype by the R672P mutation argues against this possibility.
Overexpression of the PMS1 gene from the strong GAL10 promoter conferred a clear mutator phenotype (Table 3) that is less severe than that produced by MLH1 overexpression from the ADH1 promoter. While both proteins are clearly overexpressed at high levels (Fig. 2), the relative amounts of Mlh1p and Pms1p produced are uncertain since the specific activities of the two antibodies are not known. Thus, it remains possible that Pms1p is produced less efficiently than is Mlh1p. Alternatively, excess Pms1p might inactivate MMR in a manner different from Mlh1p. According to the logic described above for human MSH3, the Pms1p-induced mutator phenotype could result from sequestration of Mlh1p into an Mlh1p-Pms1p heterodimer, thus suppressing Mlh1p-Mlh3p- or Mlh1p-Mlh2p- dependent repair.
The Mlh1p- and Pms1p-induced increases in the spontaneous mutation rate suggest that expression of the partners in MutL complexes required for MMR may be strictly regulated to maintain genome stability. Just as MMR gene mutations or diminished human MLH1 gene expression is associated with cancer susceptibility, so too might inactivation of MMR by imbalanced gene expression. This need not require the high expression levels described here. We previously demonstrated increased mutagenesis induced by substantially lower expression of MLH1 (39). Moreover, the absolute levels of the partners may be less important than their relative ratios, with smaller expression changes but in opposite directions being capable of destabilizing the genome. A recent study of MMR gene regulation in human cells showed that MLH1 is expressed at levels three to five times lower than that for MSH2 or MSH6, and PMS2 is expressed slightly less than MLH1 (3). This might suggest that MutL-related complexes function at a step that is rate limiting in human MMR, and quantitative changes in the MMR components could be a potential source of genome instability. Potentially adverse consequences of excess Mlh1p are implied by a recent report showing that apoptosis is induced in a human cell line when the human MLH1 gene is expressed from the cytomegalovirus (CMV) promoter (48). However, MLH1 overexpression is not necessarily incompatible with survival, since human MLH1 expressed from the same CMV promoter in Mlh1-deficient mouse embryonic fibroblasts reduced spontaneous mutagenesis (2). Further studies will be required to determine if imbalanced expression of human MutL homologs decreases the stability of mammalian genomes and/or is associated with increased cancer susceptibility.
ACKNOWLEDGMENTS
We thank Youri Pavlov for providing yeast strains, Louise Prakash for pMMR84, Michelle Feldman for assistance in plasmid construction, Kate Johnson and Pat Hurban for optimization of the yeast ORFs, Lee Bennett for help in the statistical analysis of the microarray hybridizations, Wilfried Kramer for helpful discussions, and Youri Pavlov and Leroy Worth for critically reading the manuscript.
REFERENCES
- 1.Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95:541–552. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 2.Buermeyer A B, Wilson-Van Patten C, Baker S M, Liskay R M. The human MLH1 cDNA complements DNA mismatch repair defects in Mlh1-deficient mouse embryonic fibroblasts. Cancer Res. 1999;59:538–541. [PubMed] [Google Scholar]
- 3.Chang D K, Ricciardiello L, Goel A, Chang C L, Boland C R. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275:18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- 4.Clark A B, Cook M E, Tran H T, Gordenin D A, Resnick M A, Kunkel T A. Functional analysis of human MutSα and MutSβ complexes in yeast. Nucleic Acids Res. 1999;27:736–742. doi: 10.1093/nar/27.3.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeRisi J L, Iyer V R, Brown P O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 6.Drotschmann K, Aronshtam A, Fritz H J, Marinus M G. The Escherichia coli MutL protein stimulates binding of Vsr and MutS to heteroduplex DNA. Nucleic Acids Res. 1998;26:948–953. doi: 10.1093/nar/26.4.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drotschmann K, Clark A B, Tran H T, Resnick M A, Gordenin D A, Kunkel T A. Mutator phenotypes of yeast strains heterozygous for mutations in the MSH2 gene. Proc Natl Acad Sci USA. 1999;96:2970–2975. doi: 10.1073/pnas.96.6.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drummond J T, Genschel J, Wolf E, Modrich P. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSα/hMutSβ ratio and reduces the efficiency of base-base mismatch repair. Proc Natl Acad Sci USA. 1997;94:10144–10149. doi: 10.1073/pnas.94.19.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esteller M, Levine R, Baylin S B, Ellenson L H, Herman J G. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene. 1998;17:2413–2417. doi: 10.1038/sj.onc.1202178. [DOI] [PubMed] [Google Scholar]
- 10.Flores-Rozas H, Kolodner R D. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc Natl Acad Sci USA. 1998;95:12404–12409. doi: 10.1073/pnas.95.21.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guerrette S, Acharya S, Fishel R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem. 1999;274:6336–6341. doi: 10.1074/jbc.274.10.6336. [DOI] [PubMed] [Google Scholar]
- 12.Habraken Y, Sung P, Prakash L, Prakash S. Enhancement of MSH2-MSH3-mediated mismatch recognition by the yeast MLH1-PMS1 complex. Curr Biol. 1997;7:790–793. doi: 10.1016/s0960-9822(06)00337-x. [DOI] [PubMed] [Google Scholar]
- 13.Habraken Y, Sung P, Prakash L, Prakash S. ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2-MSH6 and MLH1-PMS1 protein complexes. J Biol Chem. 1998;273:9837–9841. doi: 10.1074/jbc.273.16.9837. [DOI] [PubMed] [Google Scholar]
- 14.Harfe B D, Minesinger B K, Jinks-Robertson S. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr Biol. 2000;10:145–148. doi: 10.1016/s0960-9822(00)00314-6. [DOI] [PubMed] [Google Scholar]
- 15.Hauser N C, Vingron M, Scheideler M, Krems B, Hellmuth K, Entian K-D, Hoheisel J D. Transcriptional profiling on all open reading frames of Saccharomyces cerevisiae. Yeast. 1998;14:1209–1221. doi: 10.1002/(SICI)1097-0061(19980930)14:13<1209::AID-YEA311>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Herman J G, Umar A, Polyak K, Graff J R, Ahuja N, Issa J P, Markowitz S, Willson J K, Hamilton S R, Kinzler K W, Kane M F, Kolodner R D, Vogelstein B, Kunkel T A, Baylin S B. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter N, Borts R H. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev. 1997;11:1573–1582. doi: 10.1101/gad.11.12.1573. [DOI] [PubMed] [Google Scholar]
- 18.Kane F M, Loda M, Gaida G M, Lipman J, Mishra R, Goldman H, Jessup J M, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 19.Kirchner J M, Tran H, Resnick M A. A DNA polymerase ɛ mutant that specifically causes +1 frameshift mutations within homonucleotide runs in yeast. Genetics. 2000;155:1623–1632. doi: 10.1093/genetics/155.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 1996;10:1433–1442. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 21.Kramer W, Fartmann B, Ringbeck E C. Transcription of mutS and mutL-homologous genes in Saccharomyces cerevisiae during the cell cycle. Mol Gen Genet. 1996;252:275–283. doi: 10.1007/BF02173773. [DOI] [PubMed] [Google Scholar]
- 22.Leadon S A, Avrutskaya A V. Requirement for DNA mismatch repair proteins in the transcription-coupled repair of thymine glycols in Saccharomyces cerevisiae. Mutat Res. 1998;407:177–187. doi: 10.1016/s0921-8777(98)00007-x. [DOI] [PubMed] [Google Scholar]
- 23.Leung S Y, Yuen S T, Chung L P, Chu K M, Chan A S, Ho J C. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer Res. 1999;59:159–164. [PubMed] [Google Scholar]
- 24.Leung W K, Kim J J, Wu L, Sepulveda J L, Sepulveda A R. Identification of a second MutL DNA mismatch repair complex (hPMS1 and hMLH1) in human epithelial cells. J Biol Chem. 2000;275:15728–15732. doi: 10.1074/jbc.M908768199. [DOI] [PubMed] [Google Scholar]
- 25.Lewis M S. Ultracentrifugal analysis of a mixed association. Appendix to S. P. Becerra, A. Kumar, M. S. Lewis, S. G. Widen, J. Abbotts, E. M. Karawya, S. H. Hughes, J. Shiloach, and S. H. Wilson. Protein-protein interactions of HIV-1 reverse transcriptase: implications of central and C-terminal regions in subunit binding. Biochemistry. 1991;30:11707–11719. doi: 10.1021/bi00114a015. [DOI] [PubMed] [Google Scholar]
- 26.Li G M, Modrich P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci USA. 1995;92:1950–1954. doi: 10.1073/pnas.92.6.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipkin S M, Wang V, Jacoby R, Banerjee-Basu S, Baxevanis A D, Lynch H T, Elliott R M, Collins F S. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. 2000;24:27–35. doi: 10.1038/71643. [DOI] [PubMed] [Google Scholar]
- 28.Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci USA. 1998;95:8568–8573. doi: 10.1073/pnas.95.15.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 30.Morrison A, Bell J B, Kunkel T A, Sugino A. Eukaryotic DNA polymerase amino acid sequence required for 3′→5′ exonuclease activity. Proc Natl Acad Sci USA. 1991;88:9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison A, Johnson A L, Johnston L H, Sugino A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrison A, Sugino A. The 3′→5′ exonucleases of both DNA polymerases δ and ɛ participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet. 1994;242:289–296. doi: 10.1007/BF00280418. [DOI] [PubMed] [Google Scholar]
- 33.Mushegian A R, Bassett D E, Jr, Boguski M S, Bork P, Koonin E V. Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc Natl Acad Sci USA. 1997;94:5831–5836. doi: 10.1073/pnas.94.11.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pang Q, Prolla T A, Liskay R M. Functional domains of the Saccharomyces cerevisiae Mlh1p and Pms1p DNA mismatch repair proteins and their relevance to human hereditary nonpolyposis colorectal cancer-associated mutations. Mol Cell Biol. 1997;17:4465–4473. doi: 10.1128/mcb.17.8.4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perkins S J. Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur J Biochem. 1986;157:169–180. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- 36.Prolla T A, Pang Q, Alani E, Kolodner R D, Liskay R M. MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science. 1994;265:1091–1093. doi: 10.1126/science.8066446. [DOI] [PubMed] [Google Scholar]
- 37.Raschle M, Marra G, Nystrom-Lahti M, Schar P, Jiricny J. Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J Biol Chem. 1999;274:32368–32375. doi: 10.1074/jbc.274.45.32368. [DOI] [PubMed] [Google Scholar]
- 38.Rose D M, Winston F, Hieter P. Methods in yeast genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- 39.Shcherbakova P V, Kunkel T A. Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol Cell Biol. 1999;19:3177–3183. doi: 10.1128/mcb.19.4.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tannergard P, Lipford J R, Kolodner R, Frodin J E, Nordenskjold M, Lindblom A. Mutation screening in the hMLH1 gene in Swedish hereditary nonpolyposis colon cancer families. Cancer Res. 1995;55:6092–6096. [PubMed] [Google Scholar]
- 41.Tran H T, Keen J D, Kricker M, Resnick M A, Gordenin D A. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran H T, Gordenin D A, Resnick M A. The 3′→5′ exonucleases of DNA polymerases δ and ɛ and the 5′→3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2000–2007. doi: 10.1128/mcb.19.3.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Umar A, Buermeyer A B, Simon J A, Thomas D C, Clark A B, Liskay R M, Kunkel T A. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87:65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- 44.Umar A, Kunkel T A. DNA-replication fidelity, mismatch repair and genome instability in cancer cells. Eur J Biochem. 1996;238:297–307. doi: 10.1111/j.1432-1033.1996.0297z.x. [DOI] [PubMed] [Google Scholar]
- 45.Veigl L M, Kasturi L, Olechnowicz J, Ma A H, Lutterbaugh J D, Periyasamy S, Li G M, Drummond J, Modrich P L, Sedwick W D, Markowitz S D. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T F, Kleckner N, Hunter N. Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci USA. 1999;96:13914–13919. doi: 10.1073/pnas.96.24.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woolford J L J, Warner J R. The molecular biology of the yeast Saccharomyces. Vol. 1. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1991. The ribosome and its synthesis; pp. 587–626. [Google Scholar]
- 48.Zhang H, Richards B, Wilson T, Lloyd M, Cranston A, Thorburn A, Fishel R, Meuth M. Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 1999;59:3021–3027. [PubMed] [Google Scholar]