

SUMMARY
Legionella pneumophila grows intracellularly within a replication vacuole via action of Icm/Dot-secreted proteins. One such protein, SdhA, maintains the integrity of the vacuolar membrane, thereby preventing cytoplasmic degradation of bacteria. We show here that SdhA binds and blocks the action of OCRL (OculoCerebroRenal syndrome of Lowe), an inositol 5-phosphatase pivotal for controlling endosomal dynamics. OCRL depletion results in enhanced vacuole integrity and intracellular growth of a sdhA mutant, consistent with OCRL participating in vacuole disruption. Overexpressed SdhA alters OCRL function, enlarging endosomes, driving endosomal accumulation of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2), and interfering with endosomal trafficking. SdhA interrupts Rab guanosine triphosphatase (GTPase)-OCRL interactions by binding to the OCRL ASPM-SPD2-Hydin (ASH) domain, without directly altering OCRL 5-phosphatase activity. The Legionella vacuole encompassing the sdhA mutant accumulates OCRL and endosomal antigen EEA1 (Early Endosome Antigen 1), consistent with SdhA blocking accumulation of OCRL-containing endosomal vesicles. Therefore, SdhA hijacking of OCRL is associated with blocking trafficking events that disrupt the pathogen vacuole.
Graphical Abstract
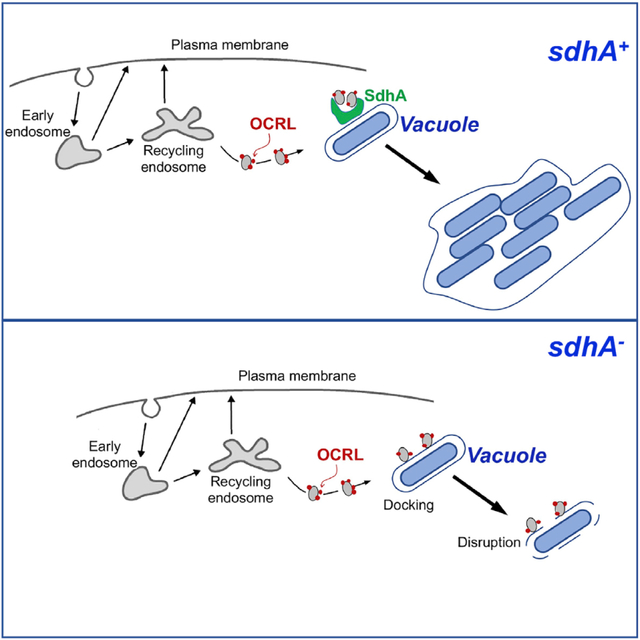
In brief
The Legionella SdhA protein supports intracellular growth in both mammalian macrophages and amoebae by preventing disintegration of the membrane-encompassed replication compartment. Choi et al. show that SdhA binds OculoCerebroRenal syndrome of Lowe (OCRL) protein, preventing early endosomal compartments of the host cell from docking and destabilizing the replication compartment.
INTRODUCTION
Legionella pneumophila is the causative agent of the potentially fatal Legionnaire’s disease, growing within alveolar macrophages as a central step in its pathogenesis (Copenhaver et al., 2014; Nash et al., 1984). As an environmental bacterium, the primary selective force for intracellular growth is its ability to infect amoebae, which can contaminate a variety of plumbing and cooling systems that act as disease reservoirs (Muder et al., 1986; Rowbotham, 1980). Human infection occurs by accidental inhalation or aspiration of contaminated aerosolized water followed by intracellular growth of Legionella in alveolar macrophages (Horwitz and Silverstein, 1980).
The intracellular growth of L. pneumophila depends on the construction of the Legionella-containing vacuole (LCV). Once internalized, about 300 different effectors are injected by the bacterium into the host via the Icm/Dot type IV secretion system (T4SS) (Huang et al., 2011; Luo and Isberg, 2004; Zhu et al., 2011). The secretion of bacterial effector proteins into the host cell allows hijacking of host-membrane trafficking pathways to remodel the LCV into a membranous compartment that supports intracellular replication (Berger and Isberg, 1993; Segal, 2013; Segal and Shuman, 1999). In contrast to phagocytic uptake of nonpathogens, which is characterized by interactions with the endocytic pathway and subsequent targeting to lysosomal compartments, the LCV recruits components of the early secretory pathway, allowing direct interaction with the endoplasmic reticulum (ER) (Clemens et al., 2000; Kagan and Roy, 2002; Swanson and Isberg, 1995; Tilney et al., 2001). This ER-encompassed compartment, protected from lysosomal degradation, also sequesters the bacterium from the cytoplasmic innate immune sensing system in mammalian hosts. The extreme restriction of bacteria that enter the mammalian cell cytosol was first demonstrated by the behavior of L. pneumophila sdhA mutants, which have disrupted vacuoles that result in bacterial exposure to the host cytosol (Aachoui et al., 2013; Creasey and Isberg, 2012; Ge et al., 2012).
The SdhA protein is a T4SS substrate essential for intracellular growth of L. pneumophila in primary macrophages (Laguna et al., 2006). Release of bacteria into the mammalian cytosol in the absence of SdhA occurs via an unknown pathway and results in recognition by cytosol-localized interferon (IFN)-stimulated anti-microbial GBPs (guanylate binding proteins), leading to bacterial degradation (Liu et al., 2018; Pilla et al., 2014). The degraded bacteria release bacterial components, such as LPS (lipopolysaccharide) and DNA, which in turn activate AIM2 (Absent in melanoma 2), caspase-11, and caspase-1 inflammasomes, causing pyroptotic death of the infected host cells (Creasey and Isberg, 2012; Ge et al., 2012; Pilla et al., 2014). Therefore, even if the vacuole avoids entry into the lysosomal pathway, disruption of the vacuole can lead to cytosolic bacterial degradation. RNAi depletion of Rab5, Rab11, and Rab8, all guanosine triphosphatases (GTPases) involved in endocytic and recycling pathways, partially reverses loss of vacuole integrity observed in sdhA mutants. Consistent with these results, the absence of SdhA results in LCV accumulation of EEA1 and Rab11FIP1, downstream effectors of these GTPases (Anand et al., 2020b; Christoforidis et al., 1999; Hales et al., 2001). Therefore, it is likely that SdhA interferes with components of the early endocytic network that are likely to disrupt vacuole integrity.
One protein involved in controlling the identities of compartments associated with the endocytic network is OCRL (OculoCerebroRenal syndrome of Lowe), a polyphosphoinositide-5-phosphatase that regulates the dynamics of early and recycling endosomes as well as autophagosome-lysosomal fusion (De Matteis et al., 2017; Sharma et al., 2015). The protein has an N-terminal pleckstrin-homology (PH) domain (Mao et al., 2009), a central 5-phosphatase catalytic core (Tsujishita et al., 2001), a C-terminal ASH (ASPM-SPD2-Hydin) domain (Erdmann et al., 2007; McCrea et al., 2008), and a catalytically inactive RhoGAP (RhoGTPase activating protein)-like domain (Pirruccello and De Camilli, 2012). The C-terminal RhoGAP-like domain interacts with Rho family GTPases, allowing recruitment to actin-rich membrane regions (Faucherre et al., 2003, 2005). The ASH/RhoGAP domain of OCRL interacts with the endocytic proteins APPL1 and Ses1 (also called IPIP27), associated with endocytosis and receptor recycling, respectively (Diggins and Webb, 2017; Noakes et al., 2011; Swan et al., 2010). Among the proteins that interact with OCRL, the Rab GTPases, which bind to the ASH domain, are most numerous. Interactions with Rab5 and Rab6 target OCRL to endosomes and the TGN (trans-Golgi network), respectively (Hyvola et al., 2006). Loss of OCRL function increases the amount of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) on endosomes, impairing membrane trafficking events, such as endocytosis and recycling of multiple classes of receptors and M6PR retrograde trafficking (Vicinanza et al., 2011).
Here, we demonstrate that SdhA prevents endocytic and recycling vesicles from merging with the LCV by targeting OCRL. We found that sdhA mutants accumulate high levels of endocytic and recycling vesicles on the vacuole in an OCRL-dependent manner. In the process, SdhA interrupts OCRL interactions with Rab GTPases.
RESULTS
SdhA contains multiple eukaryotic protein binding motifs
In search of host targets of SdhA, we found that its amino acid sequence predicted that the protein was connected to control of host cell endocytic dynamics (Figure 1A). Sequence analysis found two putative “clathrin box” consensus sequences, an endocytic sorting motif for binding the AP2β subunit associated with adaptor complexes (Edeling et al., 2006), as well as an OCRL-binding F&H motif (FxxxHxxØ) (Ø-bulky hydrophobic; Table S1). This OCRL-binding motif is found in other endocytosis-associated proteins, such as APPL1, Ses1, and Ses2 (latter two also called IPIP27A and IPIP27B; Figure 1A; Swan et al., 2010; Erdmann et al., 2007). Interestingly, SdhA and OCRL have similar arrangements of motifs predicting endocytic pathway association (Ungewickell et al., 2004; Mao et al., 2009). Most notable is the fact that a sequence motif search of the entire L. pneumophila Philadelphia 1 genome revealed that only SdhA has this combination of three motifs. In fact, the two-motif combination consisting of F&H and AP2β subunit binding is solely found in SdhA (Table S2; Data S1). Furthermore, among the Icm/Dot effector substrates, only five have as many as two of the three motifs together (Data S1). Given the presence of a potential OCRL binding site and the presence of an array of sites uniquely found in SdhA that would direct it toward endocytic transport intermediates, we reasoned that SdhA might associate with OCRL (Figure 1A). Such association could modulate endocytic processes that threaten the integrity of the LCV (Anand et al., 2020b).
Figure 1. SdhA directly interacts with OCRL but independently of F&H motif.

(A) Conserved motifs of SdhA and OCRL and maps of GST or His fusions. Motifs identified by Eukaryotic Linear Motif resource (http://www.ELM.eu.org) are shown. 5 PPase, 5 inositol polyphosphate phosphatase; ASH, ASPM-SPD2-Hydin; PH, pleckstrin homology; RhoGAP, Rho GTPase activating protein.
(B) Top: HA-mCherry or HA-mCherry-SdhA overexpressed in HEK cells were immunoprecipitated (IP) with anti-HA. AP(B), AP complex β subunit. The amount of input (0.2% lysates) and IP (20%) is shown. Bottom: densitometry of coIP is shown. Average of two sets of independent experiments is shown with error bars showing maximum of two determinations.
(C) Purified GST fusions were used in pull-downs (GST PDs) as described (STAR Methods). The amount of input (1% of total lysate) and resulting precipitate (20% of total) are shown. Right: ratio of the pull-down from this gel by densitometry is shown.
(D) GST PDs as in (C) using SdhA-CBM2 mutations in F&H motif (F1195A H1199A; FH*) or clathrin box motif (L1177A L1178A; LL*).
(E) 96-well plates coated with ASH/RhoGAP domain challenged with GST-tagged SdhA peptides (STAR Methods). SdhA binding to OCRL detected using anti-GST antibodies and chromogenic substrate is shown (mean ± SD; n = 3 wells).
(F) Competition test of SdhA-CBM2 versus F&H motif peptide of Ses1 and OCRL binding-defective point mutation (Swan et al., 2010). Data and error bars are means ± SD (n = 3 determinations).
(E and F) Three independent experiments were performed. Shown is a representative of experiment using replicates from 3 wells from a single experiment. Linked to Tables S1–S3.
SdhA interacts with OCRL
To determine whether SdhA binds OCRL, we first performed co-immunoprecipitation (coIP) with extracts of HEK cells transfected with hemagglutinin (HA)-mCherry-tagged SdhA. The tagged SdhA construct quantitatively coprecipitated OCRL as well as the AP complex beta subunit when compared to mCherry alone, although the input level of HA-mCherry-SdhA was much lower relative to the mCherry-HA control (Figure 1B). Normalizing for relative abundance of the HA-tagged proteins present after immunoprecipitation compared to the input samples, association of OCRL with HA-mCherry-SdhA was approximately 27.5-fold above the control (Figure 1B, bottom left). SdhA also pulled down the AP complex beta subunit with similar efficiency (29.5-fold above control), albeit with higher nonspecific binding (Figure 1B, bottom right). Binding to clathrin was inconclusive due to nonspecific binding to mCherry (data not shown). Based on the strong interaction with OCRL, and potential for SdhA targeting an important regulatory protein involved in multiple endocytic paths, we dissected the interface between these two proteins further and investigated its biological significance.
The interaction between SdhA and OCRL was tested using glutathione S-transferase (GST) pull-down experiments. GST-tagged SdhA fragments were coupled to glutathione beads and incubated with extracts of HEK cells expressing either GFP-tagged, full-length OCRL or GFP. To this end, SdhA fragments containing either N-terminal motifs (clathrin box and AP complex binding motif; CBM1) or C-terminal motifs (clathrin box and F&H motif; CBM2) were tested for binding (Figure 1A; CBM1, 350–402 amino acids [aas]; CBM2, 1,029–1,260 aas). The C-terminal GST-SdhA-CBM2 fragment bound to GFP-OCRL, but not GFP (Figure 1C). The GFP-OCRL association with SdhA-CBM2 was about 47-fold above the control GST protein. In contrast, SdhA-CBM1 showed no binding to OCRL. The conserved amino acid peptide containing F&H motif (FxxxHxxØ) in Ses or APPL1 was shown to be sufficient for OCRL binding (Swan et al., 2010). However, a GST fusion containing the 13-amino-acid F&H motif of SdhA did not bind to OCRL (Figure 1C) and mutations in the putative F&H motif in SdhA-CBM2 did not disrupt OCRL binding (Figure 1D), indicating that OCRL binding by SdhA is independent of the F&H motif.
To discount indirect binding of OCRL to SdhA by a complex series of interactions, we carried out solid-phase binding assays with purified GST-SdhA fragments and the ASH/RhoGAP domain of OCRL (536–901 aas), the binding region for most of the OCRL partners (Figure 1A). Direct binding was monitored by incubating increasing amounts of SdhA fragments with plate-immobilized ASH/RhoGAP domain of OCRL, probing with anti-GST. SdhA-CBM2 showed concentration-dependent binding to immobilized ASH/RhoGAP (Figure 1E). In contrast, neither GST nor GST-CBM1 exhibited binding to ASH/RhoGAP. As predicted from the pull-down assay, binding of a CBM2 ΔF&H motif mutant was equivalent to CBM2, indicating that other sequences are responsible for binding to OCRL, with EC50 = 77.10 nM and 87.43 nM for mutant and wild type (WT), respectively (Figure 1E). SdhA binding to OCRL was further tested by competition with the known OCRL-binding F&H motif (13-mer) of Ses1 (Swan et al., 2010). Addition of Ses1 F&H motif failed to decrease the binding efficiency of SdhA-CBM2, further arguing that the F&H motif of SdhA is not responsible for binding OCRL (Figure 1F).
Mapping the sites responsible for binding of SdhA and OCRL
Because SdhA does not require the F&H motif for OCRL binding, we searched for the OCRL binding site in SdhA-CBM2. The secondary structure of SdhA-CBM2 is predicted to contain 4 coiled-coils by ncoils (Lupas et al., 1991; Figure 2A). Based on this prediction, each of the four coils was individually fused to GST and tested for ASH/RhoGAP binding. SdhA-CBM2A, encompassing residues 1,029–1,080, showed strong binding, with the others showing low but detectable binding to ASH/RhoGAP (Figure 2B). Moreover, SdhA-CBM2A (EC50 ~ 27.2 nM) showed approximately 3-fold higher binding compared to the full SdhA-CBM2 fragment (EC50 ~ 80.7 nM) consistent with other regions in SdhA-CBM2 interfering with SdhA-CBM2A binding to OCRL (Figure 2C). The insolubility of SdhA precluded our ability to purify sufficient quantities to directly test whether deletions in the full-length protein blocked binding to OCRL.
Figure 2. Mapping of the interacting regions of SdhA and OCRL using solid-phase binding assays.

(A) Constructs designed based on the coiled-coils prediction by ncoils (Lupas et al., 1991).
(B) SdhA-CBM2A binds to ASH-RhoGAP domain. Plate coated with ASH/RhoGAP was challenged with indicated SdhA constructs. EC50 was calculated as described (STAR Methods).
(C) High-affinity binding of CBM2A to ASH-RhoGAP. Protocol is as in (B).
(D) SdhA-CBM2 associates with ASH domain specifically. Plate coated with SdhA-CBM2 was challenged with His-tagged OCRL domains. Binding is detected by anti-His antibodies. His-Gαi was used as a negative control.
Linked to Table S3 and Figure S1. Data shown and error bars are means ± SD (n = 3 wells). Shown are representative assays of experiments performed in triplicate wells. Experiments were repeated three times.
The RhoGAP domain of OCRL is the target of binding the F&H motifs in Ses and APPL1 (Pirruccello et al., 2011). To test for binding to the complete CBM2 fragment, we separated the ASH domain from the RhoGAP domain and subjected purified proteins to the solid-phase assay. SdhA showed stronger binding to the ASH domain than to the RhoGAP/ASH derivative, and strikingly, there was no detectable binding to the RhoGAP domain (Figure 2D). Thus, the SdhA-CBM2 fragment binds nearby to the Ses/APPL1 binding region, but not on overlapping sites.
To investigate this interaction further and determine the spectrum of proteins bound by the high-affinity CBM2A region, the experimental approach in Figure 1C was repeated using CBM2A as an affinity probe, followed by analyzing the entire pool of associated proteins. To this end, extracts were prepared and incubated with either GST-CBM2A or GST bound to glutathione beads, washed, and eluted with SDS, followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Limiting the search to cytoplasmic proteins, we identified 21 proteins that were at least 5-fold enriched on the GST-CBM2A beads relative to the GST beads. Two clear patterns emerged. First, at least 12 of these proteins were associated with the ribosome, including initiation and elongation factors tied to GTPase cycles during initiation and elongation of translation (Table S3). Second, GAP domain and GAP-associated domain proteins were identified, including those with nonfunctional GAPs, such as OCRL and IQGAP1. One of the two proteins that did not fall into this pattern was the AP3 complex subunit δ. This is striking, as we previously noted that the AP2 vesicle coat complexes bound to SdhA (Figure 1B). We conclude that CBM2A recognizes a target, or targets, on vesicles, including OCRL, CLASP-2, and AP3 (Figure 1B). Furthermore, the CBM2A region appears to have a predilection for proteins that have GAP domains, such as OCRL, or complexes that associate with GAP-domain-containing proteins, including the ribosome.
The OCRL binding region of SdhA is essential for maintaining LCV integrity and promoting intracellular growth
To address the functional importance of SdhA binding to OCRL, we tested for its role in maintaining LCV integrity during bacterial infection by introducing SdhA deletion derivatives on plasmids into a L. pneumophila ΔsdhA strain. The truncation mutants lacking either CMB2 or CBM2A (Figure 2A) had no distinguishable effect on growth in culture (Figure S1A) or on LCV localization based on immunoprobing with SdhA antibody of macrophages after 3 h incubations (Figure 3A). Based on western blot analysis, the expression levels of SdhA mutants were reduced relative to the levels of overproduced plasmid-borne SdhA-FL during in vitro growth (Figure S1B), so their relative localization properties during infection were measured by scoring SdhA-positive-LCVs at 4 h after infection (Figure S1C). Approximately 90% of LCVs scored positively for SdhA-FL but none with empty vector, indicating the probing is specific. The mutant protein ΔCBM2A showed indistinguishable levels of LCV localization compared to the SdhA-FL WT, although in the ΔCBM2 mutant, about 40% of LCVs were positive. In contrast, a WT strain showing endogenous level of SdhA (not overproduced) did not show sufficient expression to detect SdhA using antibody probing. Therefore, even the most poorly expressed plasmid-borne SdhA mutant resulted in levels of LCV localization that were higher than the endogenously expressed protein.
Figure 3. The OCRL binding region of SdhA is required for maintaining LCV integrity and intracellular growth in macrophages.

BMDMs from the A/J mouse were challenged at MOI = 1 with L. pneumophila WT or ΔsdhA harboring SdhA variants.
(A) SdhA localization on LCV was determined by immunofluorescence using antibodies against SdhA at 3 hpi (scale bar, 2 μm). See also Figure S1.
(B) SdhA mutants do not rescue ΔsdhA vacuole disruption. Macrophages were challenged for 8 h, fixed, and stained for bacteria before and after permeabilization, and internalized bacteria in absence of permeabilization were quantified relative to total infected population (mean ± SD; n = 3).
(C) Examples of cytosolic and vacuolar bacteria. Macrophages were challenged with the ΔsdhA strain, fixed, probed with α-L. pneumophila (Alexa Fluor 594 secondary, red), permeabilized, and reprobed with α-L. pneumophila (Alexa 488 secondary, green; STAR Methods). Cytosolic bacteria are accessible to both antibodies (yellow).
(D) Growth of L. pneumophila strains in macrophages, determined by the number of bacteria per vacuole 16 h post-uptake.
(B and D) Data represent mean ± SD of biological triplicates. More than 100 LCVs were counted per replicate. Linked to Figure S1.
The mutants were next evaluated for vacuole disruption using our previously established immunofluorescence staining, based on antibody accessibility to bacteria in the absence of chemical permeabilization (Creasey and Isberg, 2012). By 8 h after infection, about 40% of ΔsdhA-harboring vacuoles were permeable, compared to approximately 11% for the WT. Complementation of the ΔsdhA strain with FL-sdhA on the plasmid decreased the vacuole disruption to ~10%, indistinguishable from WT strain Lp02 (Figure 3B; see Figure 3C for example of assay). In contrast, both ΔCBM2 and ΔCBM2A showed levels of LCV disruption that were similar to a ΔsdhA strain.
The loss of vacuole integrity triggered by a ΔsdhA strain causes a severe intracellular growth defect (Creasey and Isberg, 2012). As expected, we found that the internal deletions of sdhA resulted in growth defects that were indistinguishable from the total sdhA deletion strain (Figure 3D). Taken together, these data are consistent with binding of OCRL by SdhA being tightly linked to maintaining LCV integrity.
Ectopically expressed SdhA associates with OCRL-containing vesicles
To determine whether SdhA-OCRL binding results in shared distribution through the cell, localization of the two proteins was examined by immunofluorescence microscopy in COS-7 cells transfected with mCherry-tagged SdhA and GFP-tagged OCRL. mCherry-SdhA was associated with membranous vesicles, mostly as giant ring-like structures (SdhA in Figure 4A; SdhA-NOCO in Figure S2), although mCherry alone localized to the nucleus and cytoplasm (Figure 4A; CTR in Figure S2). Surprisingly, overexpression of SdhA dramatically altered the distribution of GFP-OCRL. SdhA overexpression caused OCRL aggregation around the perinuclear region, with SdhA trapped within the OCRL-positive vacuoles, disturbing OCRL association with the Golgi and its normal association with punctate cytoplasmic vesicles (Figure 4A, compare mCherry and SdhA). When we treated cells with the microtubule depolymerization drug nocodazole (Figure S2, +NOCO), the aggregated OCRL-encompassed structures were distributed into small puncta that overlapped with SdhA signal, linking the aberrant morphology to microtubule function.
Figure 4. SdhA overexpression phenocopies loss of OCRL function.

(A) Representative micrographs of fixed COS-7 cells co-expressing mCherry-SdhA (red) and GFP-OCRL (green). DNA labeled by Hoechst stain (blue; scale bar, 10 μm). See also Figure S2.
(B) mCherry-SdhA localizes to endocytic vesicles, resulting in enlarged compartments. COS-7 cells expressing mCherry-SdhA (right panel) or mCherry (CTR, left panel) were either immunostained for EEA1 (green) or transfected with GFP-Rab7 or YFP-Rab11 (green). DNA labeled with Hoechst is shown (blue). Scale bar, 10 μm.
(C) COS-7 cells co-expressing mCherry-SdhA and GFP-PLCδ-PH. Scale bar, 10 μm. Additional images are in Figure S3.
(D and E) SdhA impairs endocytosis (D) and recycling of Tf (E). COS-7 cells were transfected with indicated expression vectors, mCherry, or mCherry-SdhA. At 24 h after transfection, cells were incubated with Alexa 488-Tf.
(D) Uptake of Tf was measured as mean fluorescence intensities at 15 min.
(E) For Tf recycling, the cells were loaded with Tf for 1 h at 37°C (load) and chased in complete medium for 40 and 60 min (chase). Arrows indicate SdhA-transfected cells. The fluorescence intensity remaining in cell was quantified and expressed as percentages of the loaded Tf.
(D and E) Data are mean values ± SD (25 cells per replicate), experiments performed in biological triplicate (*p < 0.05; **p < 0.01; ***p < 0.001; unpaired two-tailed Student’s t test). Scale bar represents 10 μm.
Linked to Figures S2 and S3.
SdhA overproduction phenocopies loss of OCRL function
To investigate whether the aberrant vacuoles generated by SdhA were the result of aggregated endosomes, we probed for endosomal markers in cells overexpressing mCherry-SdhA. Strikingly, SdhA-positive structures adopted unique morphologies, associating with endosomal markers of diverse origins, such as the early endosomal EEA1, late endosomal Rab7, and recycling endosome-derived Rab11 (Figure 4B). In each case, SdhA redistributed into giant aggregated structures that were enveloped by a mixture of endosomal compartments. The abnormal vacuoles were strongly reminiscent of enlarged endosomal structures observed in cells defective for OCRL function (Vicinanza et al., 2011; Ben El Kadhi et al., 2011). Based on this result, we then determined whether SdhA disrupts OCRL control of its preferential substrate, PI(4,5)P2, and whether it interferes with endosomal trafficking.
To this end, PI(4,5)P2 localization was probed using binding by GFP-PLCδ-PH. In control cells, PI(4,5)P2 associated with the plasma membrane (PM), particularly in regions of ruffling, as previously reported ((Watt et al., 2002); Figure 4C). Remarkably, with SdhA-transfected cells, we found accumulation of PI(4,5)P2 on large vacuoles as well as depletion of PI(4,5)P2 in the PM, indicating dysfunctional PI(4,5)P2 homeostasis (Figures 4C and S3). The altered subcellular distribution of PI(4,5)P2 that we observed appeared to closely phenocopy previous observations in OCRL-depleted cultured mammalian cells, OCRL-depleted Drosophila, and OCRL-deficient zebrafish (Vicinanza et al., 2011; Ben El Kadhi et al., 2011; Ramirez et al., 2012), consistent with SdhA overproduction interfering with OCRL function.
It has been reported that OCRL knockdown impairs uptake of transferrin (Tf) and slower recycling of internalized Tf from the PM (Vicinanza et al., 2011). Therefore, to probe for effects of SdhA on endosomal trafficking, recycling of transferrin receptor (TfR) was analyzed in cells overproducing SdhA by measuring internalization or recycling of Alexa 488 (A488)-Tf. Compared with mCherry-transfected cells, transfection with SdhA showed a significant defect in uptake of Tf (Figure 4D). The internalized pool of Tf also showed defective recycling to the PM, as Tf-preloaded cells chased for 1 h in the absence of probe lost approximately 50% of the accumulated Tf in SdhA-transfected cells, although 90% of the probe was lost from the control mCherry-transfected cells during the same time period (Figure 4E). In addition, Tf accumulated in abnormal aggregated endosomes and was retained in clustered SdhA-containing structures, consistent with SdhA having direct disruptive effects on endosomal dynamics (Figure 4E).
SdhA inhibits Rab5 binding without interfering with the OCRL 5-phosphatase activity
We tested two models for how SdhA could antagonize OCRL function: altering OCRL association with target proteins or its catalytic function. The ASH domain of OCRL binds various Rab GTPases, most notably Rab5, Rab1, and Rab6 (Hyvola et al., 2006). As SdhA also binds to the ASH domain of OCRL, we tested whether binding to SdhA fragments could block Rab5 association with OCRL. The binding affinities of constitutively active Rab5a (Q79L) and SdhA constructs were first tested using the solid-phase binding assay in which the ASH/RhoGAP domain of OCRL was immobilized and incubated with increasing amounts of each protein (Figure 5A). SdhA-CBM2 or SdhA-CBM2A exhibited a higher affinity for ASH/RhoGAP than Rab5a (Q79L). SdhA-CBM2A (EC50 ~ 8.1 nM) showed approximately 12-fold higher binding compared to the Rab5a (Q79L) (EC50 ~ 97.3 nM), arguing that SdhA may act by competing with known binding partners of OCRL (Figure 5A). We further examined potential competition between SdhA fragments and Rab5a (Q79L) for OCRL binding. The ASH or ASH/RhoGAP domains were immobilized and challenged with 22 nM Rab5 in the presence of increasing amounts of SdhA-CBM2 or SdhA-CBM2A. Interestingly, the lower affinity SdhA-CBM2 fragment disrupted Rab5a (Q79L) binding to either the ASH or ASH/RhoGAP domains in a dose-dependent manner (Figures 5B and 5C). Of note, the smaller fragment, SdhA-CBM2A, did not affect Rab5a (Q79L) binding to OCRL, even though it has a higher apparent binding affinity for OCRL than SdhA-CBM2 (Figure 2C). This is consistent with SdhA and Rab5 binding nonoverlapping sites on OCRL, with the larger CBM2 fragment blocking binding of Rab5 due to steric effects. As would be expected with the higher affinity interactions with SdhA, when either 22 nM of the CBM2 or 7.4 nM of CBM2A fragments were challenged for ASH or ASH/RhoGAP binding in the presence of increasing amounts of Rab5a (Q79L), the Rab protein had no effect on SdhA fragment binding to the OCRL domains (Figures 5D and 5E). Thus, under the conditions tested here, the SdhA CBM2 fragment outcompetes Rab5 for binding to OCRL. This is consistent with our assays showing more efficient SdhA:OCRL binding than we observed for Rab5:OCRL interaction (Figure 5A).
Figure 5. SdhA interrupts Rab5 binding.

(A) High-affinity binding of SdhA variants to OCRL. ASH-RhoGAP was immobilized and challenged with indicated GST-tagged proteins. EC50 were calculated and expressed as nM (STAR Methods).
(B) Competitive binding of Rab5 and SdhA to ASH domain. Immobilized ASH domain was challenged with 22 nM Rab5a (Q79L) in combination with increasing amounts of GST-fused SdhA constructs.
(C) Same as in (B), except ASH/RhoGAP domain was immobilized.
(D) SdhA binding to ASH domain of OCRL was not affected by challenge with Rab5a. Same as in (B), but constant amounts of SdhA (CBM2 = 22 nM; CBM2A = 7.4 nM) and increasing amounts of Rab5a (Q79L) are shown.
(E) Same as in (D), but ASH/RhoGAP domain was immobilized.
(A–E) Data represent mean values ± SD of three wells (technical replicates) for each dilution. Shown is representative of two experiments.
(F) SdhA-CBM2 does not affect the Ses1- and Pacsin2-stimulated 5-phosphatase activity of OCRL. Phosphatase activity was measured as described (STAR Methods). His-OCRL (50 nM) was incubated with indicated proteins and with 200 μM of PI(4,5)P2-containing liposomes. Ses1:SdhA refers to concentration of SdhA-CBM2 relative to Ses1. Data expressed as percentage of 5-phosphatase activity compared to incubation of OCRL with liposomes are shown. Data represent mean values ± SE (standard error) of three independent experiments (statistical significance was tested using two-tailed unpaired Student’s t test; *p < 0.05; **p < 0.01; ***p < 0.001).
(G) SdhA-CBM2 binding to OCRL during 5-phosphatase assay in (F) demonstrated by pull-down assay. His-OCRL and SdhA-CBM2 were collected with Ni2+ resin and analyzed by Coomassie staining. The amount input (10%), unbound FT (flowthrough) (20%), and bound PD (50%) are displayed.
We next analyzed whether SdhA directly affects the catalytic activity of OCRL as a consequence of binding the ASH domain, which is proximal to the 5-phosphatase domain of OCRL (Figure 1A). To this end, the 5-phosphatase activity was assayed using purified OCRL incubated with PI(4,5)P2-containing liposomes. The 5-phosphatase activity of OCRL is inherently weak in published assays in the absence of a source of stimulation, making inhibitory effects difficult to detect (Billcliff et al., 2016). It has been shown that the activity is stimulated by formation of tripartite complex with Ses1 and Pacsin2 (Billcliff et al., 2016), so we used these components to test whether SdhA interferes with the 5-phosphatase activity. Consistent with previous results, the 5-phosphatase activity was dramatically stimulated in the presence of both Ses1 and Pacsin2. Addition of SdhA-CBM2 in the presence of this complete reaction mix, however, showed no significant depression of the stimulated activity (Figure 5F). SdhA-CBM2 was clearly competent to bind OCRL using these assay conditions, as His-OCRL efficiently pulled down GST-SdhA-CBM2, but not GST alone (Figure 5G). These results indicate that SdhA likely hijacks OCRL, interrupting binding of cellular partners without disrupting its phosphatase activity.
Cellular depletion of OCRL enhances the integrity of LCVs harboring the sdhA mutant
To assess the role of OCRL in modulating LCV integrity, OCRL was efficiently RNAi-depleted in COS-7 cells (Figure 6A). The OCRL-depleted cells were next challenged for either 4 or 8 h with ΔsdhA or WT strains, and the relative levels of disrupted LCVs was determined by immunostaining (STAR Methods). Cells depleted of OCRL showed a significant enhancement in the integrity of vacuoles harboring the ΔsdhA strain at both time points, indicating that the absence of OCRL stabilized the sdhA-containing vacuole (Figure 6B). OCRL knockdown also resulted in enhanced intracellular growth of ΔsdhA as well as WT in COS-7 cells (Figure 6C).
Figure 6. OCRL is linked to vacuole disruption of L. pneumophila sdhA mutants.

(A) COS-7 cells treated with OCRL-targeting (siOCRL) or non-targeting control siRNA (siCon) for 72 h were gel fractionated and immunoprobed with antibodies directed against noted proteins.
(B) COS-7 cells were depleted by siRNA (3 days) and then challenged at MOI = 5 with noted L. pneumophila strains. Cells were fixed and stained for bacteria before and after permeabilization, as in Figure 4B. Data are mean values ± SD of three biological replicates. 100 LCVs per replicate were quantified (**p < 0.01 using two-tailed unpaired Student’s t test).
(C) L. pneumophila lux+ strains were incubated with COS-7 cells at MOI = 20, and bacterial yield was measured 48 h post-infection by relative luminescence (RLU). The replication-deficient dotA null mutant Lp03 was used as a negative control. Data are mean values ± SD of triplicate biological infections (**p < 0.01 and ***p < 0.001 using two-tailed unpaired Student’s t test).
(D) Confocal image showing a section of vacuoles isolated from infected U937 cells (MOI = 10; 3 h) with ΔsdhA mutant harboring pSdhA or pJB vector. The presence of OCRL and SdhA on LCVs was assessed by immunofluorescence using antibodies directed against OCRL, SdhA, and L. pneumophila (scale bar, 4 μm). See also Figure S4.
(E) Quantification of OCRL-positive LCVs containing ΔsdhA mutants with pSdhA or pJB vector at 3 hpi. 85 LCVs were quantified for each infection, and OCRL-positive vacuoles were displayed as percent of total vacuoles analyzed. Data are mean percentage ± SD of biological triplicate infections.
(F) Plot of OCRL intensity associated with LCV, with medians displayed. More than 70 LCVs were quantified per experiment, and data were pooled from 3 experiments (****p < 0.0001; Mann-Whitney U test).
(G) Plot comparing the OCRL intensity on LCVs harboring pSdhA compared to strain harboring empty pJB vector, divided into impermeable and permeable LCVs with medians displayed. More than 70 LCVs were quantified per experiment, and data were pooled from 3 experiments (****p < 0.0001; Mann-Whitney U test).
(H and I) Challenge of D. discoideum strain AX3 (WT) with L. pneumophila-GFP. Fixed cells were probed with α-L. pneumophila followed by secondary (Alexa Fluor 594, red) and analyzed in absence of permeabilization.
(H) Example of vacuolar (intact LCV) bacteria not detected by antibody.
(I) Example of cytosolically exposed bacteria.
(J) D. discoideum AX3 (WT) and Dd5p4 (OCRL−) were challenged for 6 h with either L. pneumophila LP02-GFP (WT) or isogenic ΔsdhA-GFP strains, fixed, and probed with α-L. pneumophila to identify disrupted vacuoles. More than 50 LCVs were imaged per experiment, and disrupted vacuoles were shown as percentage of total number of imaged vacuoles. Biological triplicates were performed with two to three technical replicates each time. Data shown are mean ± SD from all biological and technical replicates pooled to give the individual datapoints displayed (n = 6–9 replicates). Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons. ****p < 0.0001.
OCRL accumulates on the disrupted vacuole of sdhA mutants
Previous work has shown that OCRL localizes to LCVs in D. discoideum and RAW264.7 macrophages (Weber et al., 2009). To determine whether SdhA is involved in the localization of OCRL on LCVs, we challenged U937 macrophages for 3 h with a ΔsdhA strain harboring either a plasmid overproducing SdhA or an empty vector control. After homogenization of infected macrophages, the localization of SdhA and endogenous OCRL on LCVs in postnuclear supernatants was determined by immunofluorescence microscopy. Approximately 90% of the LCVs stained positively for OCRL, regardless of the presence of SdhA (Figures 6D and 6E). The pattern of OCRL localization on the LCVs, however, could clearly be differentiated between the two strains. In the presence of SdhA, OCRL formed punctate structures associated with LCVs that appeared to be enveloped by SdhA. In contrast, there was dense circumferential accumulation of OCRL about the LCV in absence of sdhA (Figure 6D; more images in Figure S4). Based on image analysis, the median fluorescence intensity of OCRL accumulation about vacuoles was approximately 1.4 times greater in the absence of SdhA than in its presence, with a broad distribution of intensities observed for the mutant, pointing toward two populations of LCVs (p < 0.0001; Mann-Whitney U test; Figure 6F). To investigate this distribution further and test whether vacuole disruption was related to accumulated OCRL, we quantified OCRL on intact or disrupted LCVs (Figure 6G). There was a significant correlation between OCRL intensity and vacuole disruption. For the strain lacking SdhA, the median OCRL fluorescence intensity of disrupted LCVs was 1.6-fold greater compared to those with intact LCVs (p < 0.0001; Mann-Whitney U test). The OCRL intensity of intact LCVs containing the empty vector strain was significantly higher than the OCRL intensity of LCVs containing the pSdhA strains, indicating OCRL accumulation occurs before vacuole disruption and then continues to accumulate (Figure 6G). Therefore, SdhA interferes with the accumulation of OCRL on LCVs and is associated with vacuole disruption.
To determine whether OCRL antagonism of L. pneumophila intracellular growth was specific to mammalian cells or evolutionarily conserved, the role of the protein after uptake into the genetically tractable amoebal species Dictyostelium discoideum was analyzed. To this end, vacuole integrity in the D. discoideum WT AX3 strain was compared to the isogenic Dd5p4 strain, which has a null mutation in the amoebal ortholog of OCRL (Li et al., 2018). After challenge of D. discoideum AX3, the ΔsdhA showed significantly higher levels of disrupted vacuoles than was observed with L. pneumophila WT (Figures 6H–6J). When either bacterial strain was used to challenge the Dd5p4 (OCRL−) strain, there was a large decrease in permeable vacuoles (greater than 2× for WT and about 3.5× for ΔsdhA). Furthermore, vacuole permeability for the L. pneumophila WT and ΔsdhA strains was indistinguishable after challenge of Dd5p4 (Figure 6J). This indicates that OCRL antagonizes vacuole integrity in D. discoideum, and SdhA is dispensable for maintaining vacuole integrity in the absence of OCRL function in this amoebal species.
SdhA inhibits the accumulation of OCRL-dependent endosomal traffic to LCVs
The localization of several well-characterized host proteins associated with OCRL-controlled endosomal traffic was assessed by immunofluorescence microscopy of LCVs from postnuclear supernatants of U937 cells (Vicinanza et al., 2011). We quantified the number of LCVs positive for endosomal EEA1, retrograde cargo trafficking cation-independent mannose 6-phosphate receptor (CIMPR), and TfR, linked to recycling cargo (Figure 7A). For EEA1 and CIMPR, there was a significant increase in LCV association for each marker after infection with a ΔsdhA strain for 3 h when compared to WT. The increase was particularly striking with EEA1, supporting our previous analysis of the behavior of ΔsdhA strains with murine bone-marrow-derived macrophages ((Anand et al., 2020b); Figure 7A).
Figure 7. OCRL-dependent accumulation of endosomal compartments on vacuole surrounding ΔsdhA mutant.

(A) U937 macrophages were challenged for 3 h with L. pneumophila WT and ΔsdhA (MOI = 10). The presence of endosomal EEA1, retrograde trafficking cargo CIMPR, and recycling endosomal TfR on LCVs was evaluated by immunofluorescence microscopy in postnuclear supernatants of infected cells (STAR Methods). Scale bar represents 4 μm.
(B) Left: effect of siOCRL on protein expression in U937 cells. GAPDH was used as a loading control. Gel fractionated samples were immunoprobed with indicated antibodies. Right: the presence of EEA1 on LCVs was quantified from the infected U937 cell lysates as in (A). 50 vacuoles were quantified per condition from 3 separate infections. Data shown are mean ± SD (n = 3 biological replicate infections). Percent LCVs and percent EEA1-positive LCVs were compared within the groups using unpaired two-tailed Student’s t test. Statistical significance is represented as *p < 0.05 and **p < 0.01.
To assess whether enhanced accumulation of EEA1 on LCVs harboring sdhA mutants is dependent on OCRL, OCRL was depleted by small interfering RNA (siRNA) prior to infection. In comparison to the control with scrambled siRNA, EEA1-positive LCVs harboring the ΔsdhA strain decreased to a level that was indistinguishable from WT (Figure 7B). This indicates that aberrant trafficking of EEA1 to LCVs harboring the ΔsdhA mutant appeared largely dependent on OCRL function, consistent with SdhA controlling OCRL function for the purpose of blockading early endosomal vesicles.
DISCUSSION
Mammalian OCRL protein and its Dictyostelium discoideum homolog Dd5P4 localize to the LCV, limiting intracellular replication of L. pneumophila (Weber et al., 2009; Finsel et al., 2013). In our study, we argue that OCRL promotes events that disrupt LCV integrity, with SdhA protein being a key player in preventing OCRL restriction of pathogen growth. OCRL antagonism of vacuole integrity is evolutionarily conserved, with its presence being a major contributor to vacuole disruption in the amoebal species D. discoideum. As SdhA is one of the few Icm/Dot translocated substrates required for survival in primary macrophages, the interface of SdhA with this host inositol polyphosphate 5-phosphatase is likely to be a critical step in controlling the balance between host innate restriction and proliferation of the pathogen. The OCRL phosphatase activity is known to control a number of membrane-trafficking steps. The results documented here argue that SdhA blocks an OCRL-regulated step in the movement of vesicles from an endosomal compartment, resulting in disruption of the LCV membrane. Presumably, the blockade occurs by SdhA hijacking of OCRL.
We have found that RNAi depletion of OCRL in cells challenged with the sdhA mutant significantly increases the number of intact LCVs, consistent with a role for OCRL in driving vacuole membrane disruption. OCRL depletion does not completely rescue the intracellular replication defect no the loss in vacuole integrity resulting from the absence of SdhA. We think it likely that SdhA has functions other than binding OCRL. In fact, evidence for interaction with multiple vesicle coat and adaptor proteins was obtained during affinity chromatography with a SdhA fragment, and binding to one of these (AP2; Figure 1) was verified. Therefore, SdhA appears to interfere with multiple host proteins that act to destabilize the replication compartment.
We think it likely that there are likely two modes of OCRL interface with the vacuole based on immunofluorescence analysis. On encounter with vacuoles harboring WT L. pneumophila, OCRL-staining compartments target to SdhA-rich regions. In contrast, loss of SdhA function enhances OCRL circumferential recruitment about the LCV, and this recruitment appears particularly robust in vacuoles undergoing disruption. Taken together, these results argue that the LCV surrounding the sdhA mutant vacuole merges with OCRL-containing compartments, resulting in eventual vacuole disruption. In cells harboring the WT strain, vacuole integrity is likely maintained because these compartments are diverted by SdhA.
In previous work, we presented evidence that SdhA likely acts by preventing access of endosome-derived compartments to the LCV (Anand et al., 2020b). This model was based on RNAi screens demonstrating that disruption of ΔsdhA-mutant-containing vacuoles can be reversed by depletion of endocytic Rab GTPases. A primary consequence of SdhA loss was shown to be the accumulation of early endosomal protein EEA1 on the defective vacuole in primary macrophages, dependent on the function of Rab5 and Rab11 (Anand et al., 2020b). Interestingly, a number of GTPases involved in endosome dynamics, such as Rab5 and Rab8, are known interacting partners of OCRL (Grant and Donaldson, 2009). Depletion of Rab5, Rab11, and OCRL all have similar phenotypes in that reduced function of each results in dramatic decreases in EEA1 association with the ΔsdhA vacuole, arguing that these factors act in similar steps that drive vacuole disruption.
Based on these results, we hypothesize that, in the absence of SdhA, OCRL and Rab GTPase interactions recruit endosomal compartments to the ΔsdhA vacuole and disrupt the pathogen niche. In the vacuole harboring WT bacteria, SdhA can bind OCRL and interfere with Rab binding to the phosphatase, preventing OCRL-containing compartments from directly targeting the vacuolar membrane. The fact that SdhA binds to the ASH domain of OCRL and sterically blocks Rab protein interaction with OCRL may be significant in this regard. OCRL mutants defective in Rab binding have been shown to result in aberrant OCRL targeting, and depletions of Rab1 or Rab6 show similar defects (Hyvola et al., 2006). Therefore, association of OCRL-containing vesicles with SdhA and consequent disruption of Rab protein binding could prevent Rab effectors from promoting efficient docking of either OCRL or its associated endosomes with the LCV surface.
The relatively small CBM2A region in the C-terminal of SdhA (aas 1,029–1080), predicted to form one of several coiled-coil structures throughout the protein, is functionally important, as overproduction of a variant of the protein that precisely deletes this region fails to complement the ΔsdhA mutation. Consistent with the defect being tightly associated with loss of OCRL control, OCRL is profoundly altered in transfectants overexpressing SdhA, resulting in giant OCRL-encompassed vacuoles that surround SdhA-rich regions (Figure 4). Loss of OCRL function has been documented to generate large vacuoles harboring endosomal components, thought to result from blockading of endosomal traffic to the Golgi (Vicinanza et al., 2011; Choudhury et al., 2005; Ben El Kadhi et al., 2011). As a consequence, disrupted OCRL causes accumulation of PI(4,5)P2 in endosomal compartments and disruption of TfR recycling (Vicinanza et al., 2011; Choudhury et al., 2005; Ben El Kadhi et al., 2011). Overexpression of SdhA exactly phenocopies the functional loss of OCRL, as we have demonstrated that SdhA transfectants cause both mislocalization of PI(4,5)P2 and dysfunctional TfR recycling.
It seems counterintuitive that OCRL function should be associated with LCV disruption. The LCV is rich in PI4P, so it might be thought that OCRL would play a collaborative role in LCV biogenesis, as its inositol polyphosphate 5-phosphatase activity can generate PI4P, which in turn anchors a wide swath of Icm/Dot effector proteins to the LCV cytoplasmic surface (Weber et al., 2009; Hsu et al., 2012). L. pneumophila has a number of well-characterized translocated inositol phosphate kinases and phosphatases capable of modulating PI4P dynamics, consistent with maintaining PI4P homeostasis in the LCV (Dong et al., 2016; Hsu et al., 2012; Toulabi et al., 2013). It seems likely that OCRL plays a surprising negative role by stimulating the biogenesis, recruitment, or docking of forbidden membrane compartments that act to destabilize the LCV rather than directly modifying the LCV. Alternatively, the presence of OCRL on the vacuolar membrane could disrupt the PI4P homeostatic balance provided by Legionella Icm/Dot effectors, resulting in overload of this lipid and hypersensitivity to phospholipases.
As we have previously argued, SdhA belongs to a larger class of pathogen proteins called vacuole guards that act to prevent intravacuolar microbial pathogens from being attacked by disruptive membrane components largely derived from endosomes and recycling compartments (Anand et al., 2020b). Although it may be predicted that such interactions would lead to trafficking of the LCV to the lysosome, interaction of endocytic compartments with pathogen-containing vacuoles has consistently resulted in disruption of the vacuole (Anand et al., 2020a; Mellouk et al., 2014). This is likely because pathogen proteins inserted into the vacuole result in specific phospholipid requirements for maintaining vacuole homeostasis, with docking of endocytic vesicles disrupting this homeostasis. SdhA fits in well with this class of proteins involved in maintaining vacuolar phospholipid homeostasis, as blocking vesicular transit of these compartments to the LCV increases vacuole stability (Anand et al., 2020b). By binding OCRL, SdhA may play a role in modulating self-nonself recognition by the LCV. Forbidden compartments harboring OCRL may have membrane compositions rich in PI4P that are similar to that of the LCV. This, in turn, could direct targeting and fusion of these disruptive compartments with the LCV. SdhA acts to “guard” the pathogen-containing vacuole by binding and diverting OCRL, preventing either direct interaction with disruptive compartments or blocking association of OCRL with the LCV.
Although the described SdhA biochemical activity has not been observed previously, the connection of OCRL to the endocytic and recycling compartments increases documented parallels between SdhA and the Salmonella SifA protein (Beuzón et al., 2000; McGourty et al., 2012). Both SifA and SdhA are required to maintain the integrity of their respective vacuoles, with failure to prevent host-cell-mediated disruption resulting in release of bacteria into the cytosol and activation of a caspase-4/11-gasdermin-D-dependent pyroptotic response in phagocytes (Aachoui et al., 2013; Casson et al., 2015; Pilla et al., 2014; Shi et al., 2015). An intimate connection between maintaining vacuole integrity and preserving appropriate lipid content of the vacuolar membrane has long been suspected in both cases, primarily based on suppressor mutation analysis (Creasey and Isberg, 2012; Kolodziejek et al., 2019). Finally, our work argues that SdhA regulates the host cell endocytic and recycling pathways, reminiscent of the demonstrated role of SifA in hijacking retrograde cellular transit and controlling egress of CIMPR (Dumont et al., 2010; McEwan et al., 2015; McGourty et al., 2012). This argues that components of the endosomal pathway act as important disruptive forces that can block pathogen growth without directly targeting the organism into a lysosomal compartment. The details of the nature of these disruptive forces, and the phospholipid composition that results in destabilization of these compartments, remain to be determined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Ralph R. Isberg (ralph.isberg@tufts.edu).
Materials availability
The materials generated in this study are available upon request.
Data and code availability
All data reported in this paper will be shared by the lead contract upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The mice used in this study were female A/J mice. Bone marrow-derived macrophages (BMDM) were isolated from the femurs of 7 weeks old mice. Animals were maintained at 26°C under a 12 h light-dark cycle with free access to food and water prior to euthanasia and isolation of femurs. Maintenance, care of animals and euthanasia procedure were approved by the Tufts University School of Medicine Institutional Animal Care and Use Committee after review of specific protocols detailing the use of mice for the purpose of BMDM isolation.
Cell culture
Bone marrow-derived macrophages (BMDMs) from female A/J mice femurs (7 weeks old) were isolated and cultured in RPMI 1640 (GIBCO) containing 10% heat-inactivated fetal bovine serum (FBS; GIBCO), 30% L-cell-conditioned medium and 100U of penicillin-streptomycin (GIBCO) per mL (Swanson and Isberg, 1995). COS-7 cells and HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO) supplemented with 10% heat-inactivated FBS (GIBCO). U937 cells were maintained in RPMI 1640 containing 10% heat-inactivated FBS and 1 mM glutamine (Invitrogen) and differentiated using 10 ng/mL 12-tetradecanoyl phorbol 13-acetate (TPA) for 48 hr (Losick et al., 2010). All cells were cultured at 37C in a humidified atmosphere containing 5% CO2.
METHOD DETAILS
Cloning and mutagenesis
The primers used for this work are listed in Table S4. His-Tagged-full length SdhA was constructed in pQE80L while appropriate truncations (CBM1, CBM2, CBM2A, CBM2B, CBM2C, and CBM2D) were generated in pGEX-6P-1. Mutations were generated by PCR using Quikchange (Stratagene). SdhA deletions were generated by inverse PCR (Ochman et al., 1988). For insect cell expression of human OCRL, the full-length cDNA with an amino-terminal 6xHis tag was inserted into pFastBac vector. His-tagged ASH/RhoGAP and ASH domain of human OCRL cDNA were inserted into pQE80L. The amino-terminal-6xHis fused RhoGAP domain of OCRL was inserted into pGEX-6P-1 to generate GST-6His (internal tag) for purposes of improving solubility of the recombinant protein. All constructs were verified by DNA sequencing (Genewiz).
Co-immunoprecipitation
HEK293T cells were co-transfected with GFP-OCRL and either HA-mcherry or HA-mcherry-SdhA using Lipofectamine 2000 for 2 days. Lysates were made on ice for 1hr (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2% Octylglucoside, protease inhibitor cocktail) and clarified by centrifugation at 16,000 g for 20min at 4°C. Immune complexes from the supernatants were adsorbed on anti-HA affinity beads for 2 hr at 4°C. After washing (50 mM Tris-HCl, pH 7.4, 40 mM NaCl, 0.2% Octylglucoside, protease inhibitor cocktail), proteins were fractionated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore), and probed with the indicated antibodies.
Pulldown experiments
Pulldown experiments with purified GST or GST-SdhA truncations were performed with the lysates of HEK cells transfected with GFP or GFP-OCRL after 24 hr transfection. The lysates were prepared from a 10 cm dish in 1ml of pull down/lysis buffer (25 mM HEPES–KOH (pH 7.2), 125 mM potassium acetate, 2.5 mM magnesium acetate, 0.4% Triton X-100, and protease inhibitor cocktail), followed by incubation for 3 hr at 4°C with 250 μg of GST-fusion protein coupled to glutathione-agarose (Thermo Scientific). Beads were then washed four times (pull down buffer containing 0.1% Triton X-100) and resuspended with SDS-PAGE sample buffer followed by SDS-PAGE and western blotting. For MS analysis, proteins were eluted with SDS sample buffer followed by fractionation on SDS-PAGE, then excised from the gel, and analyzed by LC/MS/MS by the Taplin Mass Spectrometry Facility (Harvard Medical School).
Protein preparations
6xHis-OCRL was prepared from Sf9 insect cells using a baculovirus expression system (Invitrogen) according to the manufacturer’s specification. Cells were lysed (20mM Tris, pH 8, 150mM NaCl, 5mM MgCl2, 5mM β-mercaptoethanol, 1% NP40, 10% glycerol, and protease inhibitor) and purified by nickel affinity chromatography (GE healthcare Lifesciences). E. coli BL21 (DE3) was used for bacterially-expressed protein, inducing overnight with 0.1 mM IPTG at 18°C. GST fusion proteins were purified on glutathione superflow agarose according to the manufacturer’s protocol (Thermo Scientific). The GST tag of N-terminal GST-6xHis-RhoGAP domain was removed by addition of PreScission protease (GE healthcare Lifesciences) directly to the glutathione beads followed by incubation overnight at 4°C to obtain His-tagged RhoGAP. His-tagged proteins were purified by nickel affinity chromatography. The proteins were then concentrated by ultrafiltration using Amicon filters (EMD Millipore).
Solid phase binding assay
96-well ELISA plates (Thermo Fisher) were coated with 1ug of recombinant proteins (20 mM NaHCO3/Na2CO3, pH 9.4), washed four times (Tris (pH = 7.4)-buffered saline (TBS), 0.1% Tween 20 (TBST)), and blocked in TBS + 5% BSA. Wells were then probed with GST- or His-tagged proteins diluted in TBST containing 0.5% BSA for 1 hr. Wells were washed again with TBST and incubated with appropriate primary antibodies and HRP-conjugated secondary antibodies. Protein interaction was detected by incubating with TMB (Thermo Fisher) as a chromogenic substrate. The reactions were stopped (1M HCl and 5M acetic acid in water) and the resulting absorbance was measured at 450 nm in a microtiter plate spectrophotometer. EC50 was calculated using the GraphPad Prism 8 software for windows applying the nonlinear curve fit module.
Bacterial challenge of mammalian cells
The analysis of intracellular replication at a single cell level and analysis of cytosolic bacteria in BMDMs were performed as previously described (Creasey and Isberg, 2012). To quantify SdhA localization on LCV, SdhA and L. pneumophila were probed with anti-SdhA and anti-Legionella, respectively, incubated with fluorescent secondary antibodies, and 100 vacuoles were counted. Isolation of vacuoles from L. pneumophila-infected U937 was performed as described previously (Vogel et al., 1998). Briefly, at the noted infection times, cells were washed 3 times with 10 mL of pre-warmed PBS and lifted from the dish using 10 mL of cold PBS. The cells were pelleted for 5 min at 1000 rpm and 4°C, and were washed once with 5 mL of homogenization buffer (20 mM HEPES/KOH pH = 7.2, 250 mM sucrose, 5 mM EGTA). Washed cells were resuspended in 2 mL of homogenization buffer and lysed in a Dounce homogenizer until 90% of the cells were broken (five or less plunges). The cell lysate was transferred to microfuge tubes and unbroken cells and nuclei were pelleted for 3 min. at 1500 rpm and 4°C. The post-nuclear supernatant (PNS) containing L. pneumophila vacuoles was collected and analyzed by immunofluorescence microscopy.
For intracellular growth in COS-7 cells, 2×10^4 cells were seeded in 96-well plates, challenged with L. pneumophila lux (MOI 20) for 1 hr, washed three times with Dulbecco’s modified Eagle’s medium (no phenol red) supplemented with 10% FBS, and luciferase production was measured at 48 hr post infection in a microtiter luminometer.
Imaging
Fluorescence microscopy was performed following standard procedures (Losick and Isberg, 2006) and all antibodies were used according to manufacturer’s procedures. Nuclei were stained using Hoechst stain (Invitrogen). Cells were transfected using Lipofectamine 2000 (Invitrogen) for 24 hr according to the manufacturer’s instructions and fixed with 4% formaldehyde in PBS. For nocodazole treatment, 0.1 μg/ml of nocodazole (Sigma) was added to cells for 20 hr. Cells were imaged by either Zeiss observer Z1 or Leica Falcon SP8 microscopy. Images were processed using ImageJ or Volocity software (Improvision). The fluorescence intensity of OCRL covering a single LCV and transferrin in the cytoplasm was quantified using ImageJ software after background correction.
Determining vacuole integrity after L. pneumophila challenge of D. discoideum
Dictyostelium discoideum strains were grown in 100 mm dishes containing HL5 medium (Solomon et al., 2000), washed, harvested, pelleted for 5 min at 500 × g by centrifugation and resuspended in low fluorescence axenic medium (LoFlo; http://dictybase.org/techniques/media/lowflo_medium.html), containing 1 mM IPTG, followed by seeding onto poly-L-lysine coated-coverslips at 2 × 105/well in 24 well plates. Cells were allowed to adhere for 2 h at 25.5°C, prior to challenge with L. pneumophila strains expressing GFP under the Ptac promoter at MOI = 1. At 1h post-infection, the cells were rinsed 3 times with LoFlo and replenished with 400 μL of fresh LoFlo medium. At 6h post-infection, 400 μL of 2x fixing medium (LoFlo containing 8% PFA, 10.5% sucrose and 0.5% glutaraldehyde) was added to the cell culture medium and incubated in the dark for 10 mins at room temperature. Fixed cells were gently washed with 1xPBS, and autofluorescence was quenched with PBS containing 0.1M glycine by performing three successive 1 min. incubations. Cells were then washed 3 times with 1XPBS and incubated in blocking buffer (PBS, 4% BSA) at 4°C overnight. Bacterial staining was performed as with mammalian cells.
Transferrin uptake and recycling
COS-7 cells-transfected with SdhA or with control vector were incubated in serum-free medium for 1 hr and then exposed to 50 μg/ml Alexa Fluor-488-Tf (Invitrogen) on ice for 30 min. The cells were transferred to 37°C and incubated for the times indicated. External Tf was removed by washing with PBS and bound Tf was removed by an acid wash (150 mM NaCl, and 10 mM acetic acid, pH 3.5) followed by washes with PBS. The fluorescence intensity of internalized Tf was quantified by image capturing using a Zeiss Observer Z1 microscope (63x oil objective) and analysis using ImageJ. To measure recycling, cells were incubated first in serum-free medium and subsequently in medium containing fluorescent Tf for 1 h at 37°C to saturate the receptor population. After extensive washing with HEPES-buffered DMEM, the recycling of Tf was followed by incubating the cells in the presence of complete medium for 40 and 60 min, at 37°C. The cells were then acid washed before fixing.
Lipid phosphatase assay
To measure phosphatase activity, lipid vesicles containing PI(4,5)P2 were generated by extrusion with polycarbonate membranes with pore size of 200-nm diameter as described in Billcliff et al. (2016). Lipids DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), DOPS (1,2-dioleoyl-sn-glycero-3-phospho-l-serine), and natural PI(4,5)P2 were purchased from Avanti Polar Lipids. His-tagged OCRL (50 nM) was incubated with indicated proteins on ice for 20 min in reaction buffer (50mM Tris-HCl, pH7.4, 5mM MgCl2). Ses1 C-terminal fragment (192–249) and GST-Pacsin2 were added at 20-fold molar excess of OCRL. The phosphatase reaction was started by addition of 200 μM of PI(4,5)P2-containing liposomes. After incubation at 37°C for 20 min, the reaction was stopped by the addition of malachite green solution (Sigma-Aldrich) and the resulting absorbance was read at 620nm.
RNA interference
COS-7 cells were transfected using Lipofectamine RNAiMAX (ThermoFisher Scientific) according to the manufacturer’s instructions. The siRNA SMARTpool for OCRL and non-targeting siRNAs were purchased from Dharmacon. For OCRL depletion in U937 cells, siRNA was transfected with Amaxa cell line nucleofector kit and Nucleofector II (Lonza) according to the manufacturer’s protocol.
Endocytic pathway sequence motif search
All L. pneumophila coding sequences (CDS) were divided into an effector set (Data S1, S10; 290 effectors) and a noneffector set (Data S1, 2652 sequences) based on presence of effector translocation signals (Lifshitz et al., 2013). The protein sequences from each category were analyzed for the presence of each motif of interest (Data S1: Search patterns) using ScanProsite (de Castro et al., 2006). The total number of the detected motifs for each individual protein sequence was then summarized in a Table (Data S1). The percentage of effectors and noneffectors containing motifs was calculated and displayed in Table S2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis and software used
Data are presented as mean ± standard deviation unless otherwise stated. Statistical analysis was performed using Prism GraphPad 8 with experiment-dependent tests as described below.
Densitometry analysis
The densitometry of co-immunoprecipitation (Figures 1B and 1C) was quantified from western blot data using ImageJ (https://imagej.nih.gov/ij/) analysis. For Figure 1B, two independent experiments of co-immunoprecipitations were performed, and one representative western blot result was shown. Densitometry data shown are average of two sets of independent experiments with error bars showing the maximum of two determinations, n = 2; n = number of independent experiments.
Protein binding assay (Figures 1E, 1F, and 2B–2D)
Protein binding affinity was measured using absorbance at 450nm wavelength. Three biological replicates were performed with three technical replicates each time (3 wells per sample on a 96-well plate). Results from one representative biological replicated experiment were plotted as mean absorbance with standard deviation calculated using Prism GraphPad. n = 3; n represents number of technical replicates (wells) per experiment.
Percent cytosolic bacteria and bacterial yield (Figures 3B and 3D)
Percent cytosolic bacteria was calculated for all groups. More than 100 LCVs were quantified per replicate. Biological triplicate experiments were performed in both B and D. n = 3; n represents number of biological replicated independent experiments.
Transferrin (Tf) recycling (Figures 4D and 4E)
The percentages of the loaded Tf were compared between SdhA-transfected cells and control cells using two-tailed unpaired Student’s t test. In total, 25 cells were quantified per sample per replicate and biologically triplicate experiments were performed. Statistical significance is expressed as: * p < 0.05; ** p < 0.01; *** p < 0.001. n = 3; n represents number of independent experiments (biological replicates).
Competitive binding assay (Figures 5A–5E)
Protein binding affinity was measured using absorbance at 450nm wavelength. Two independent experiments of proteins added to triplicate wells were performed, and data plotted are mean ± SD of three wells from one representative experiment, using Prism GraphPad for analysis. n = 3; n represents number of wells per experiment.
5 Phosphatase activity of OCRL (Figure 5F)
5-phosphatase activities of OCRL from various conditions were compared to control group containing both Ses1 and Pacsin2 using two-tailed unpaired Student’s t test. Three independent experiments were performed and data shown are mean ± standard error (SE). n = 3; n represents number of independent experiments. Statistical significance is expressed as: * p < 0.05; ** p < 0.01; *** p < 0.001.
Percent cytosolic bacteria and bacterial yield (Figures 6B and 6C)
Figure 6B: Percent cytosolic bacterial amount was compared between siOCRL- and siCon- infected COS-7 cells at either 4hrs or 8hrs post-infection and statistical analysis was performed using two-tailed unpaired Student’s t test. Three biological replicate experiments were performed and 100 cells were quantified per experiment. ** represents p < 0.01. n = 3; n represents number of independent experiments.
Figure 6C: Bacterial yield at 48hrs post infection was compared between siOCRL- and siCon- infected COS-7 cells for various L. pneumophila strains using two-tailed unpaired Student’s t test. Biological triplicate experiments were performed. ** represents p < 0.01, *** represents p < 0.001. n = 3; n represents number of independent experiments.
OCRL fluorescent intensity (Figures 6E–6G)
Figure 6E: Percent OCRL-positive LCVs harboring pSdhA was compared to strains harboring the empty vector. 85 LCVs were quantified per experiment and biological triplicate experiments were performed. n = 3; n represents number of independent experiments.
Figures 6F and 6G: OCRL intensity on LCVs conataining ΔsdhA strain harboring pSdhA was compared to ΔsdhA strain harboring pJB vector (total, permeable subpopulation or impermeable subpopulation) using Mann-Whitney U-test. More than 70 LCVs were captured per experiment for strains carrying pSdhA or pJB vector. Infections were performed three separate times and data displayed are pooled from these three experiments along with median. Statistical significance of p < 0.0001 was expressed as ****. n > 210; n represents total number of LCVs quantified or number of samples taken.
Percent disrupted vacuoles in D. discoideum (Figure 6J)
Percent disrupted vacuoles from various conditions were compared within or among the groups using one-way ANOVA with Tukey’s multiple comparisons. Three infections with two to three technical replicates from each infection were performed for all conditions. More than 50 LCVs were imaged per technical replicate. Data shown are mean ± SD. **** represents p < 0.0001; ns represents not significant. n = 6–9; n represents pooled data from the technical replicates of the three infections.
Percent LCVs (Figure 7)
Percent LCVs and percent EEA1 positive LCVs were compared within the groups using unpaired two tailed Student’s t test. 50 vacuoles were quantified per condition from three separate infections. Statistical significance is represented as: * p < 0.05; ** p < 0.01. n = 3; n represents number of independent infections (biological replicates).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-HA | Santa Cruz | Cat# SC7392; RRID:AB_627809 |
| Rabbit polyclonal anti-OCRL | Sigma | Cat# O7640, RRID:AB_1851805 |
| Goat polyclonal anti-AP2B | Santa Cruz | Cat# SC6425, RRID:AB_2227237 |
| Mouse monoclonal anti-OCRL | Pietro De Camilli | N/A |
| Rabbit polyclonal anti-GFP | Life Technologies | Cat# A11122, RRID:AB_221569 |
| Mouse monoclonal anti-GST | Santa Cruz | Cat# SC138, RRID:AB_627677 |
| Mouse monoclonal anti-polyHis | Sigma | Cat# H1029, RRID:AB_260015 |
| Rabbit polyclonal anti-Rab5A | Santa Cruz | Cat# SC-309, RRID:AB_632295 |
| Mouse monoclonal anti-EEA1 | BD Transduction | Cat# 610456, RRID:AB_397829 |
| Mouse monoclonal anti-GAPDH | Santa Cruz | Cat# SC32233, RRID:AB_627679 |
| Mouse monoclonal anti-CIMPR | Novusbio | Cat# NB300-514, RRID:AB_2139105 |
| Mouse monoclonal anti-TfR | Abcam | Cat# AB9179, RRID:AB_2110055 |
| Mouse monoclonal anti-β-Tubulin | Sigma | Cat# T4026, RRID:AB_477577 |
| HRP-conjugated rabbit anti-goat | Life Technologies | Cat# 611620, RRID:AB_87867 |
| HRP-conjugated goat anti-mouse | Life Technologies | Cat# 626520, RRID:AB_88369 |
| HRP-conjugated goat anti-rabbit | Life Technologies | Cat# 65-6120, RRID:AB_2533967 |
| Alexa594-conjugated goat anti-mouse | Life Technologies | Cat# A-11005, RRID:AB_2534073 |
| Alexa488-conjugated donkey anti-rabbit | Jackson ImmunoResearch | Cat# 711-545-152, RRID:AB_2313584 |
| Alexa488-conjugated donkey anti-mouse | Jackson ImmunoResearch | Cat# 715-545-150, RRID:AB_2340846 |
| Alexa 594-conjugated donkey anti-rat | Jackson ImmunoResearch | Cat# 712-585-153, RRID:AB_2340689 |
| Alexa594-conjugated donkey anti-rabbit | Jackson ImmunoResearch | Cat# 711-585-152, RRID:AB_2340621 |
| Dylight 405-conjugated donkey anti-Rat | Jackson ImmunoResearch | Cat# 712-475-153, RRID:AB_2340681 |
| Rabbit polyclonal anti-SdhA | (Laguna et al., 2006) | N/A |
| Rabbit polyclonal anti-ICDH | (Duménil et al., 2004) | N/A |
| Rat monoclonal anti-Legionella | (Isaac et al., 2015) | N/A |
| Rabbit polyclonal anti-Legionella | (Isaac et al., 2015) | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| L. pneumophila Lp02 | (Berger and Isberg, 1993) | N/A |
| L. pneumophila Lp02 ΔsdhA | (Zhu et al., 2011) | N/A |
| L. pneumophila Lp03 (Lp02 dotA03) | (Berger and Isberg, 1993) | N/A |
| L. pneumophila Lp02 lux+ (PahpC::lux) | (Ensminger et al., 2012) | N/A |
| L. pneumophila Lp02 ΔsdhA lux+ (kanR PahpC::lux) | (Anand et al., 2020b) | N/A |
| L. pneumophila Lp03 lux+ (PahpC::lux) | (Ensminger et al., 2012) | N/A |
|
| ||
| Biological samples | ||
|
| ||
| Dictyostelium discoideum Ax3 | (Li et al., 2018) | N/A |
| Dictyostelium discoideum Ax3 Dd5p4− | (Li et al., 2018) | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Western Lightning Plus-ECL | PerkinElmer | NEL105001EA |
| Nocodazole | Sigma | M1404 |
| DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) | Avanti Polar Lipids | Cat# 850375 |
| DOPS (1,2-dioleoyl-sn-glycero-3-phospho-l-serine) | Avanti Polar Lipids | Cat# 840035 |
| Brain PI(4,5)P2 | Avanti Polar Lipids | Cat# 840046 |
| Malachite green phosphate assay kit | Sigma | Cat# MAK307 |
| Peptide Ses1-F&H 13-mer (PFARLHECYGQEI) | Tufts University Core Facility | N/A |
| Peptide Ses1–FA (PAARLHECYGQEI) | Tufts University Core Facility | N/A |
| Protease inhibitor cocktail | Roche | Cat# 11873580001 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668030 |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778075 |
| Alexa Fluor-488-Tf | Invitrogen | Cat# T13342 |
| Amaxa cell line nucleofector kit | Lonza | Cat# VCA-1004 |
| TMB substrate kit | Pierce | Cat# 34021 |
| Anti-HA affinity beads | Sigma | Cat# E6779 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| HEK293T | ATCC | CRL-11268, RRID:CVCL_1926 |
| Mouse bone marrow-derived macrophages | This study | N/A |
| COS-7 | ATCC | CRL-1651, RRID:CVCL_0224 |
| U937 | ATCC | CRL-1593.2, RRID:CVCL_0007 |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse: A/J | The Jackson laboratory | Cat# 000646, RRID:CVCL_ZL15 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S4 for the list of oligonucleotides | IDT | N/A |
| Non-targeting siRNA pools | Dharmacon | Cat# D-001810-10-05 |
| siRNA SMARTpool for human OCRL | Dharmacon | Cat# L-010026-00-0005 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pMMB207Δ267-GFP | (Anand et al., 2020b) | N/A |
| pHA-mcherry-SdhA | This study | N/A |
| pGEX-6P-1-SdhA-CBM1 (350-402aa) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2 (1029-1260aa) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2 FH* (F1195A H1199A) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2 LL* (L1177A L1178A) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2 LL* (L1177A L1178A) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2 ΔFH (Δ1193-1209aa) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2A (1029-1080aa) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2B (1075-1110aa) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2C (1140-1185aa) | This study | N/A |
| pGEX-6P-1-SdhA-CBM2D (1225-1260aa) | This study | N/A |
| pGFP-OCRL | (Mao et al., 2009) | N/A |
| pQE80L-ASH/RhoGAP of OCRL (536aa-901aa) | This study | N/A |
| pQE80L-ASH of OCRL (536aa-678aa) | This study | N/A |
| pGEX6P-1-RGS-6xHis-RhoGAP of OCRL (678-901aa) | This study | N/A |
| pGEX-Rab5a (Q79L) | (Gaspar and Machner, 2014) | N/A |
| pJB908 | (Sexton et al., 2004) | N/A |
| pJB908-sdhA | (Zhu et al., 2011) | N/A |
| pJB908-3xflag-sdhA | This study | N/A |
| pJB908-3xflag-sdhA Δ1029-1260aa (ΔCBM2) | This study | N/A |
| pJB908-3xflag-sdhA Δ1029-1260aa (ΔCBM2) | This study | N/A |
| pGFP-PLCδ-PH | (Sarantis et al., 2012) | N/A |
| pGFP-Rab7 | Sina Mohammadi | N/A |
| pHA-mYFP-Rab11 | (Mohammadi and Isberg, 2013), | N/A |
| pGEX-4T-2-192-249aa of IPIP27A (Ses1) | (Noakes et al., 2011) | N/A |
| pGEX-4T-2-Pacsin 2 | (Billcliff et al., 2016) | N/A |
| pFastBac-6xHis-OCRL | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism | GraphPad | https://graphpad.com:443/scientific-software/prism/ |
| ImageJ | NIH ImageJ | https://imagej.nih.gov/ij/ |
| Eukaryotic linear motif resource | ELM | http://Elm.eu.org |
| ncoils | Bio.tools | http://bio.tools/ncoils |
| Volocity | PerkinElmer | https://www.perkinelmer.com/volocity |
| ScanProsite | (de Castro et al., 2006) | https://prosite.expasy.org/scanprosite/ |
Highlights.
The Legionella SdhA protein binds mammalian OCRL protein
OCRL drives replication compartment disintegration, blocking intracellular growth
SdhA stabilizes the compartment by blocking the action of OCRL
OCRL destabilization of the replication compartment is evolutionarily conserved
ACKNOWLEDGMENTS
This work was supported by HHMI and NIAID grants R01 AI113211 and R01 AI146245 to R.R.I. P.A. was funded by DFG grant AU550/1-1. M.E. is supported by NIH grant 1R15GM143733 from NIGMS. We thank Ila Anand for scientific discussions throughout the course of this work and Kristen Davis and Erion Lipo for review of the manuscript. We thank Dr. Pietro De Camilli for providing OCRL antibody and GFP-OCRL construct and Dr. Matthias Machner for Rab5a (Q79L) construct. We thank Ross Tomaino of Taplin Mass Spectrometry Facility at Harvard Medical School for performing MS/MS analysis as well as for numerous consultations regarding data analysis. We also thank Drs. Elizabeth Draganova and Ellen White for patient help setting up the lipid extrusion assays and with baculovirus expression.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109894.
REFERENCES
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, and Miao EA (2013). Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand I, Choi W, and Isberg RR (2020a). The vacuole guard hypothesis: how intravacuolar pathogens fight to maintain the integrity of their beloved home. Curr. Opin. Microbiol. 54, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand IS, Choi W, and Isberg RR (2020b). Components of the endocytic and recycling trafficking pathways interfere with the integrity of the Legionella-containing vacuole. Cell. Microbiol. 22, e13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben El Kadhi K, Roubinet C, Solinet S, Emery G, and Carréno S (2011). The inositol 5-phosphatase dOCRL controls PI(4,5)P2 homeostasis and is necessary for cytokinesis. Curr Biol 21, 1074–1079. [DOI] [PubMed] [Google Scholar]
- Berger KH, and Isberg RR (1993). Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19. [DOI] [PubMed] [Google Scholar]
- Beuzón CR, Méresse S, Unsworth KE, Ruíz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, and Holden DW (2000). Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19, 3235–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billcliff PG, Noakes CJ, Mehta ZB, Yan G, Mak L, Woscholski R, and Lowe M (2016). OCRL1 engages with the F-BAR protein pacsin 2 to promote biogenesis of membrane-trafficking intermediates. Mol. Biol. Cell 27, 90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, Nguyen HT, Collman RG, and Shin S (2015). Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. USA 112, 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury RR, Hyvola N, and Lowe M (2005). Phosphoinositides and membrane traffic at the trans-Golgi network. Biochem Soc Symp 72, 31–38. [DOI] [PubMed] [Google Scholar]
- Christoforidis S, McBride HM, Burgoyne RD, and Zerial M (1999). The Rab5 effector EEA1 is a core component of endosome docking. Nature 397, 621–625. [DOI] [PubMed] [Google Scholar]
- Clemens DL, Lee BY, and Horwitz MA (2000). Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 68, 2671–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver AM, Casson CN, Nguyen HT, Fung TC, Duda MM, Roy CR, and Shin S (2014). Alveolar macrophages and neutrophils are the primary reservoirs for Legionella pneumophila and mediate cytosolic surveillance of type IV secretion. Infect. Immun. 82, 4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasey EA, and Isberg RR (2012). The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl. Acad. Sci. USA 109, 3481–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, and Hulo N (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis MA, Staiano L, Emma F, and Devuyst O (2017). The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat. Rev. Nephrol. 13, 455–470. [DOI] [PubMed] [Google Scholar]
- Diggins NL, and Webb DJ (2017). APPL1 is a multifunctional endosomal signaling adaptor protein. Biochem. Soc. Trans. 45, 771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N, Niu M, Hu L, Yao Q, Zhou R, and Shao F (2016). Modulation of membrane phosphoinositide dynamics by the phosphatidylinositide 4-kinase activity of the Legionella LepB effector. Nat. Microbiol. 2, 16236. [DOI] [PubMed] [Google Scholar]
- Duménil G, Montminy TP, Tang M, and Isberg RR (2004). IcmR-regulated membrane insertion and efflux by the Legionella pneumophila IcmQ protein. J. Biol. Chem. 279, 4686–4695. [DOI] [PubMed] [Google Scholar]
- Dumont A, Boucrot E, Drevensek S, Daire V, Gorvel JP, Poüs C, Holden DW, and Méresse S (2010). SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 11, 899–911. [DOI] [PubMed] [Google Scholar]
- Edeling MA, Mishra SK, Keyel PA, Steinhauser AL, Collins BM, Roth R, Heuser JE, Owen DJ, and Traub LM (2006). Molecular switches involving the AP-2 beta2 appendage regulate endocytic cargo selection and clathrin coat assembly. Dev Cell 10, 329–342. [DOI] [PubMed] [Google Scholar]
- Ensminger AW, Yassin Y, Miron A, and Isberg RR (2012). Experimental evolution of Legionella pneumophila in mouse macrophages leads to strains with altered determinants of environmental survival. PLoS Pathog. 8, e1002731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann KS, Mao Y, McCrea HJ, Zoncu R, Lee S, Paradise S, Modregger J, Biemesderfer D, Toomre D, and De Camilli P (2007). A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev. Cell 13, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucherre A, Desbois P, Satre V, Lunardi J, Dorseuil O, and Gacon G (2003). Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. Hum. Mol. Genet. 12, 2449–2456. [DOI] [PubMed] [Google Scholar]
- Faucherre A, Desbois P, Nagano F, Satre V, Lunardi J, Gacon G, and Dorseuil O (2005). Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum. Mol. Genet. 14, 1441–1448. [DOI] [PubMed] [Google Scholar]
- Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, van Rahden VA, Johannes L, and Hilbi H (2013). The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe 14, 38–50. [DOI] [PubMed] [Google Scholar]
- Gaspar AH, and Machner MP (2014). VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc. Natl. Acad. Sci. USA 111, 4560–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Gong YN, Xu Y, and Shao F (2012). Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc. Natl. Acad. Sci. USA 109, 6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, and Donaldson JG (2009). Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, Griner R, Hobdy-Henderson KC, Dorn MC, Hardy D, Kumar R, Navarre J, Chan EK, Lapierre LA, and Goldenring JR (2001). Identification and characterization of a family of Rab11-interacting proteins. J. Biol. Chem. 276, 39067–39075. [DOI] [PubMed] [Google Scholar]
- Horwitz MA, and Silverstein SC (1980). Legionnaires’ disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Invest. 66, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, and Mao Y (2012). Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc. Natl. Acad. Sci. USA 109, 13567–13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Boyd D, Amyot WM, Hempstead AD, Luo ZQ, O’Connor TJ, Chen C, Machner M, Montminy T, and Isberg RR (2011). The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 13, 227–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, and Lowe M (2006). Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 25, 3750–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac DT, Laguna RK, Valtz N, and Isberg RR (2015). MavN is a Legionella pneumophila vacuole-associated protein required for efficient iron acquisition during intracellular growth. Proc. Natl. Acad. Sci. USA 112, E5208–E5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, and Roy CR (2002). Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4, 945–954. [DOI] [PubMed] [Google Scholar]
- Kolodziejek AM, Altura MA, Fan J, Petersen EM, Cook M, Brzovic PS, and Miller SI (2019). Salmonella translocated effectors recruit OSBP1 to the phagosome to promote vacuolar membrane integrity. Cell Rep. 27, 2147–2156.e5. [DOI] [PubMed] [Google Scholar]
- Laguna RK, Creasey EA, Li Z, Valtz N, and Isberg RR (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. USA 103, 18745–18750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Edwards M, Swaney KF, Singh N, Bhattacharya S, Borleis J, Long Y, Iglesias PA, Chen J, and Devreotes PN (2018). Mutually inhibitory Ras-PI(3,4)P2 feedback loops mediate cell migration. Proc. Natl. Acad. Sci. USA 115, E9125–E9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, and Segal G (2013). Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl. Acad. Sci. USA 110, E707–E715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BC, Sarhan J, Panda A, Muendlein HI, Ilyukha V, Coers J, Yamamoto M, Isberg RR, and Poltorak A (2018). Constitutive interferon maintains GBP expression required for release of bacterial components upstream of pyroptosis and anti-DNA responses. Cell Rep. 24, 155–168.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick VP, and Isberg RR (2006). NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick VP, Haenssler E, Moy MY, and Isberg RR (2010). LnaB: a Legionella pneumophila activator of NF-kappaB. Cell. Microbiol. 12, 1083–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ, and Isberg RR (2004). Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. USA 101, 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, and Stock J (1991). Predicting coiled coils from protein sequences. Science 252, 1162–1164. [DOI] [PubMed] [Google Scholar]
- Mao Y, Balkin DM, Zoncu R, Erdmann KS, Tomasini L, Hu F, Jin MM, Hodsdon ME, and De Camilli P (2009). A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J. 28, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea HJ, Paradise S, Tomasini L, Addis M, Melis MA, De Matteis MA, and De Camilli P (2008). All known patient mutations in the ASH-Rho-GAP domains of OCRL affect targeting and APPL1 binding. Biochem. Biophys. Res. Commun. 369, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Richter B, Claudi B, Wigge C, Wild P, Farhan H, McGourty K, Coxon FP, Franz-Wachtel M, Perdu B, et al. (2015). PLEKHM1 regulates Salmonella-containing vacuole biogenesis and infection. Cell Host Microbe 17, 58–71. [DOI] [PubMed] [Google Scholar]
- McGourty K, Thurston TL, Matthews SA, Pinaud L, Mota LJ, and Holden DW (2012). Salmonella inhibits retrograde trafficking of mannose-6-phosphate receptors and lysosome function. Science 338, 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellouk N, Weiner A, Aulner N, Schmitt C, Elbaum M, Shorte SL, Danckaert A, and Enninga J (2014). Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16, 517–530. [DOI] [PubMed] [Google Scholar]
- Mohammadi S, and Isberg RR (2013). Cdc42 interacts with the exocyst complex to promote phagocytosis. J. Cell Biol. 200, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muder RR, Yu VL, and Woo AH (1986). Mode of transmission of Legionella pneumophila. A critical review. Arch. Intern. Med. 146, 1607–1612. [PubMed] [Google Scholar]
- Nash TW, Libby DM, and Horwitz MA (1984). Interaction between the legionnaires’ disease bacterium (Legionella pneumophila) and human alveolar macrophages. Influence of antibody, lymphokines, and hydrocortisone. J. Clin. Invest. 74, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes CJ, Lee G, and Lowe M (2011). The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol. Biol. Cell 22, 606–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Gerber AS, and Hartl DL (1988). Genetic applications of an inverse polymerase chain reaction. Genetics 120, 621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, and Coers J (2014). Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc. Natl. Acad. Sci. USA 111, 6046–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello M, and De Camilli P (2012). Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochem. Sci. 37, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirruccello M, Swan LE, Folta-Stogniew E, and De Camilli P (2011). Recognition of the F&H motif by the Lowe syndrome protein OCRL. Nat Struct Mol Biol 18, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez IB, Pietka G, Jones DR, Divecha N, Alia A, Baraban SC, Hurlstone AF, and Lowe M (2012). Impaired neural development in a zebrafish model for Lowe syndrome. Hum Mol Genet 21, 1744–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbotham TJ (1980). Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33, 1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarantis H, Balkin DM, De Camilli P, Isberg RR, Brumell JH, and Grinstein S (2012). Yersinia entry into host cells requires Rab5-dependent dephosphorylation of PI(4,5)P2 and membrane scission. Cell Host Microbe 11, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal G (2013). Identification of legionella effectors using bioinformatic approaches. Methods Mol. Biol. 954, 595–602. [DOI] [PubMed] [Google Scholar]
- Segal G, and Shuman HA (1999). Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67, 2117–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton JA, Pinkner JS, Roth R, Heuser JE, Hultgren SJ, and Vogel JP (2004). The Legionella pneumophila PilT homologue DotB exhibits ATPase activity that is critical for intracellular growth. J. Bacteriol. 186, 1658–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Skowronek A, and Erdmann KS (2015). The role of the Lowe syndrome protein OCRL in the endocytic pathway. Biol. Chem. 396, 1293–1300. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Solomon JM, Rupper A, Cardelli JA, and Isberg RR (2000). Intracellular growth of Legionella pneumophila in Dictyostelium discoideum, a system for genetic analysis of host-pathogen interactions. Infect. Immun. 68, 2939–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan LE, Tomasini L, Pirruccello M, Lunardi J, and De Camilli P (2010). Two closely related endocytic proteins that share a common OCRL-binding motif with APPL1. Proc. Natl. Acad. Sci. USA 107, 3511–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, and Isberg RR (1995). Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63, 3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Harb OS, Connelly PS, Robinson CG, and Roy CR (2001). How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114, 4637–4650. [DOI] [PubMed] [Google Scholar]
- Toulabi L, Wu X, Cheng Y, and Mao Y (2013). Identification and structural characterization of a Legionella phosphoinositide phosphatase. J. Biol. Chem. 288, 24518–24527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujishita Y, Guo S, Stolz LE, York JD, and Hurley JH (2001). Specificity determinants in phosphoinositide dephosphorylation: crystal structure of an archetypal inositol polyphosphate 5-phosphatase. Cell 105, 379–389. [DOI] [PubMed] [Google Scholar]
- Ungewickell A, Ward ME, Ungewickell E, and Majerus PW (2004). The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci U S A 101, 13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, Di Campli A, Polishchuk E, Santoro M, Di Tullio G, Godi A, Levtchenko E, De Leo MG, Polishchuk R, Sandoval L, et al. (2011). OCRL controls trafficking through early endosomes via PtdIns4,5P2-dependent regulation of endosomal actin. EMBO J. 30, 4970–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel JP, Andrews HL, Wong SK, and Isberg RR (1998). Conjugative transfer by the virulence system of Legionella pneumophila. Science 279, 873–876. [DOI] [PubMed] [Google Scholar]
- Watt SA, Kular G, Fleming IN, Downes CP, and Lucocq JM (2002). Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J 363, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SS, Ragaz C, and Hilbi H (2009). The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell Microbiol 11, 442–460. [DOI] [PubMed] [Google Scholar]
- Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, and Luo ZQ (2011). Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS ONE 6, e17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contract upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.