

Abstract
Pentamidine, an FDA-approved antiparasitic drug, was recently identified as an outer membrane disrupting synergist that potentiates erythromycin, rifampicin, and novobiocin against Gram-negative bacteria. The same study also described a preliminary structure–activity relationship using commercially available pentamidine analogues. We here report the design, synthesis, and evaluation of a broader panel of bis-amidines inspired by pentamidine. The present study both validates the previously observed synergistic activity reported for pentamidine, while further assessing the capacity for structurally similar bis-amidines to also potentiate Gram-positive specific antibiotics against Gram-negative pathogens. Among the bis-amidines prepared, a number of them were found to exhibit synergistic activity greater than pentamidine. These synergists were shown to effectively potentiate the activity of Gram-positive specific antibiotics against multiple Gram-negative pathogens such as Acinetobacter baumannii, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Escherichia coli, including polymyxin- and carbapenem-resistant strains.
Keywords: antibiotic synergy, outer membrane disruption, bis-amidines, checkerboard assays
The growing threat of antimicrobial resistance (AMR) has led to projections that by 2050 the world may be confronted with as many as 10 million annual AMR-associated deaths.1 Society is already dealing with the rising tide posed by this global health challenge: each year, 700,000 people die due to infections with drug-resistant pathogens.2 At present, the most critical threats are presented by Gram-negative pathogens, including Acinetobacter baumannii (carbapenem-resistant), Pseudomonas aeruginosa (carbapenem-resistant), and the Enterobacteriaceae (carbapenem-resistant and ESBL-producing strains), such as Escherichia coli and Klebsiella pneumoniae, according to the World Health Organization (WHO).3
In treating infections due to Gram-negative bacteria, there is an increased interest in strategies aimed at disrupting the outer membrane (OM) so as to potentiate a number of clinically used antibiotics that on their own are only effective against Gram-positive bacteria.4−6 In an elegant approach recently reported by Brown and coworkers, a panel of 1440 previously approved drugs were screened to identify compounds capable of disrupting the OM of Gram-negative bacteria.7 The assay used in the screen was based on findings that at low temperatures, OM synthesis is altered in E. coli making it more susceptible to vancomycin.8,9 This led to the hypothesis that compounds that antagonize vancomycin in E. coli grown at 15 °C would likely also impact the OM integrity.7,10 Among the hits identified using this innovative screen, the small-molecule bis-amidine pentamidine (1) (Figure 1) exhibited the most effective capacity to antagonize the activity of vancomycin.7
Figure 1.

Structures of pentamidine (1) and analogues 2 and 3 previously found to exhibit synergy with Gram-positive antibiotics against Gram-negative species.7
Pentamidine is used clinically to treat Pneumocystis jiroveci pneumonia, trypanosomiasis, and leishmaniasis.11−13 Apart from its antiprotozoal activity, pentamidine is also known to have moderate antibacterial activity against Gram-positive species.14,15 Furthermore, pentamidine has also been shown to have anti-cancer activity by restoring the tumor-suppressing activity of p53, is capable to bind A/T-rich regions of double-stranded DNA, and can non-specifically bind and disrupt tRNA secondary structures.16−19 Unsurprisingly, this broadly active compound has a high incidence of side effects such as nephrotoxicity, hypotension, hypoglycaemia, or local reactions to the injection.11−13 The Brown group’s discovery that pentamidine potentiates the anti-Gram-negative activity of rifampicin, erythromycin, and novobiocin further highlights the multifaceted nature of the compound.7
It is well established that the disruption of the Gram-negative OM, for example, with the well-studied polymyxin B nonapeptide (PMBN), can potentiate the activity of hydrophobic, Gram-positive specific antibiotics.7,20 In keeping with these findings, it is also known that polymyxin-resistance also reduces the synergistic potential of PMBN.7,20 In this regard, it is notable that the synergistic activity of pentamidine in combination with novobiocin, when evaluated against wild-type and polymyxin-resistant strains of A. baumannii, was observed both in vitro and in vivo.7
In addition to pentamidine, Brown and co-workers also examined the synergistic activity of other commercially available bis-amidines by performing checkerboard assays, from which the fractional inhibitory concentration index (FICI) was derived, serving as a measure of synergistic activity.7,21 These studies highlighted the necessity of two amidine groups for effective potentiation of Gram-positive antibiotics against an E. coli indicator strain.7 In addition, the linker used to connect the benzamidine moieties was also found to play a key role in the determining the activity of the compounds evaluated.7 Based on these studies, two analogues were identified as having enhanced synergistic activities relative to pentamidine (compounds 2 and 3, Figure 1). The conclusions drawn from these studies suggest that increased linker length and hydrophobicity, along with decreased linker flexibility, contributes to an increase in synergistic activity for these bis-amidines.7
Inspired by these findings, we here describe structure–activity relationship (SAR) studies designed to provide a broad understanding of the structural features required for potent and selective synergy by bis-amidines. While the previous study of Brown and coworkers evaluated the synergistic potential of commercially available bis-amidines, we here report the design, synthesis, and evaluation of a number of novel bis-amidines. In addition to screening for synergistic activity, the new compounds here studied were also assessed for their capacity to selectively target the Gram-negative OM membrane rather than act as non-specific membrane disruptors. Our findings serve to both validate published accounts, while also revealing new, more potent, and selective bis-amidine-based synergists.
Results and Discussion
Synthesis and Initial Screening
Linear Linkers
To further explore the correlation between linker length and synergistic activity, a set of linear pentamidine analogues was selected. In addition to the previously reported nonamidine (2) and propamidine (9), we also synthesized heptamidine (10), octamidine (11), and undecamidine (12) analogues (Scheme 1A). Pentamidine (1) was also synthesized by the same route to allow for comparison with the commercial material (Supporting Information, Scheme S1), which subsequently revealed no difference in the synergistic activity of the in-house prepared and commercial materials (data not shown).
Scheme 1. Synthesis of Pentamidine Analogues Containing Different Linear Spacers between the Benzamidine Groups.
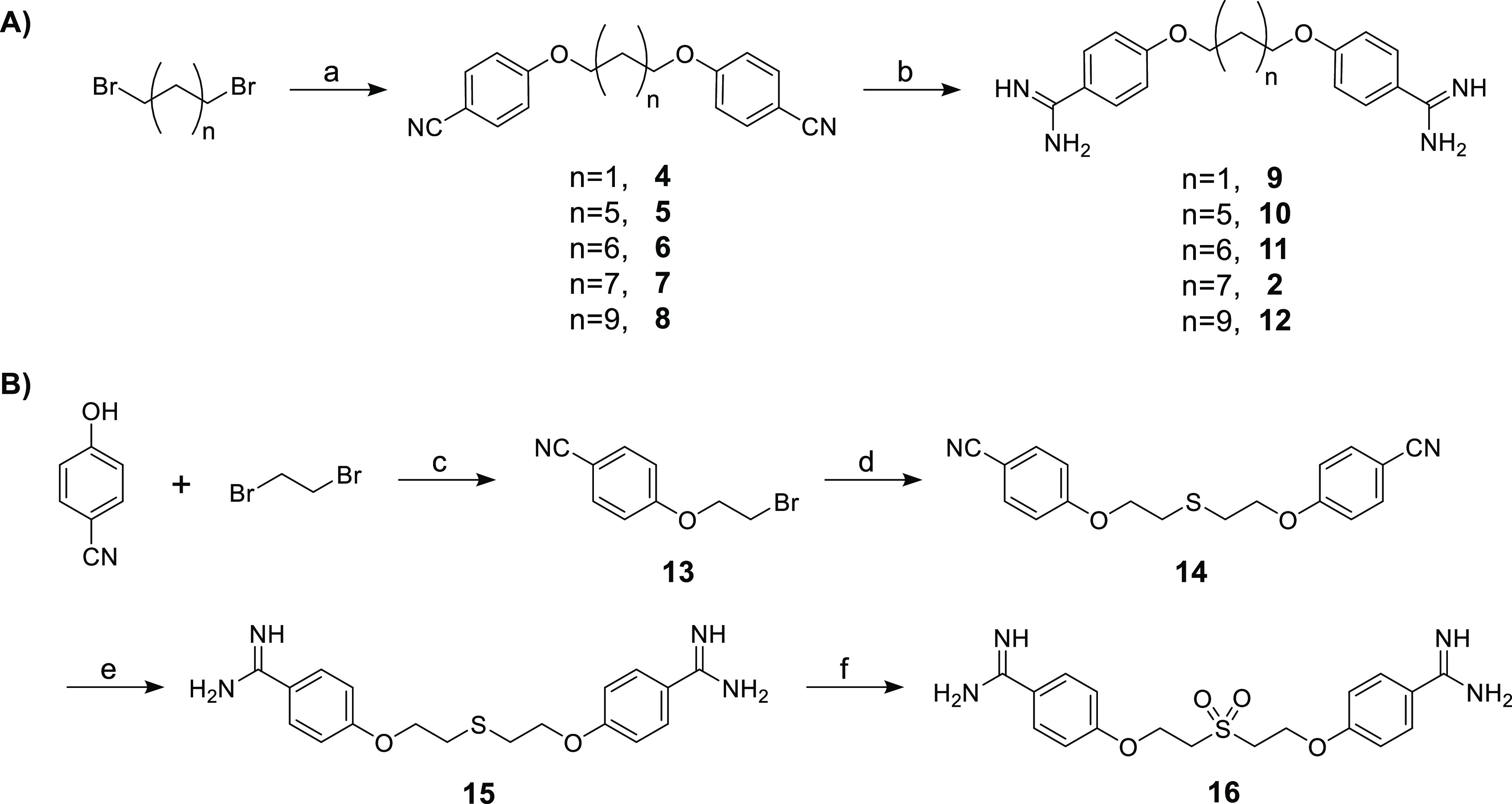
Reagents and conditions: (a) 4-cyanophenol, NaH, DMF, 80 °C, 1 h (59%-quant.); (b) (i) LHMDS, THF, 48 h, rt, (ii) HCl, 0 °C to rt, overnight (49%-quant.); (c) K2CO3, DMF, 100 °C, 5 h (43%); (d) Na2S·9H2O, DMSO, 115 °C, 1 h (93%); (e) (i) LHMDS, THF, rt, 48 h; (ii) HCl, rt, overnight (64%); (f) m-CPBA, DCM, 0 °C, 2 h (32%).
As shown in Scheme 1A, the dibenzonitrile intermediates were prepared from the commercially available α,ω-dibromo-alkanes via a Williamson ether synthesis according to literature protocols.22 Crystallization from ethanol resulted in the pure intermediates 4–8 in good to excellent yields. The transformation of the nitrile groups into the corresponding amidine is classically performed via the Pinner reaction followed by treatment with ammonia.23−27 However, recent publications have described the same transformation by the more convenient use of a lithium bis(trimethylsilyl)amide (LHMDS) solution followed by an acidic quench.28−31 In the synthesis of pentamidine we therefore evaluated the treatment of the corresponding bis-nitrile precursor with LHMDS [1 M in tetrahydrofuran (THF)] followed by a quench with saturated ethanolic HCl, 4 M HCl in dioxane, or 1 M HCl (aq) (see Supporting Information, Scheme S1 and S2). These trial experiments revealed that quenching with 4 M HCl in dioxane resulted in the highest yield, and these conditions were therefore also applied in the preparation of the bis-amidines 2, 9–12, which were subsequently isolated in good yields after high-performance liquid chromatography (HPLC) purification. In addition to probing linker length, we also explored the impact of heteroatom substitution in the linker. Notably, thioether analogue 15 has been previously prepared and tested for antimicrobial activity.15,32 Thioether 15 was therefore synthesized, as indicated in Scheme 1B, also providing ready access to the more hydrophilic sulfone analogue 16 obtained by m-CPBA treatment of 15.
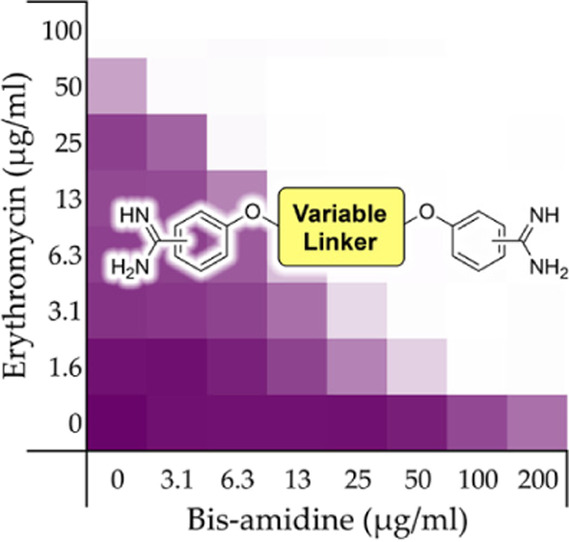
The inherent antibacterial activities of pentamidine (1) and the bis-amidines 2, 3, 9–12, 15, and 16 were first assessed against an indicator strain E. coli BW25113. This revealed a trend wherein compounds containing linkers of eight or more carbons exhibited moderate antibacterial activity with minimum inhibitory concentration (MIC) values of 50 μg/mL (see Table 1). Neither the thioether linked species 15 or sulfone linked 16 showed any inherent activity up to the maximum concentration tested (200 μg/mL). Next, the synergistic activity of the compounds was assessed in combination with both erythromycin and rifampicin using the same indicator E. coli strain. Checkerboard assays were performed in which a dilution series of the synergist was evaluated in combination with the antibiotic of interest, also serially diluted. The resulting “checkerboard” or 2-dimensional MIC readout makes it possible to identify the lowest concentration of both components that results in the most potent synergistic effect. The highest concentrations tested among the synergists correspond to their inherent MIC values (or up to 200 μg/mL in the case where no antibacterial activity was observed). For erythromycin, the highest concentration tested was 200 μg/mL and for rifampicin it was 12 μg/mL.
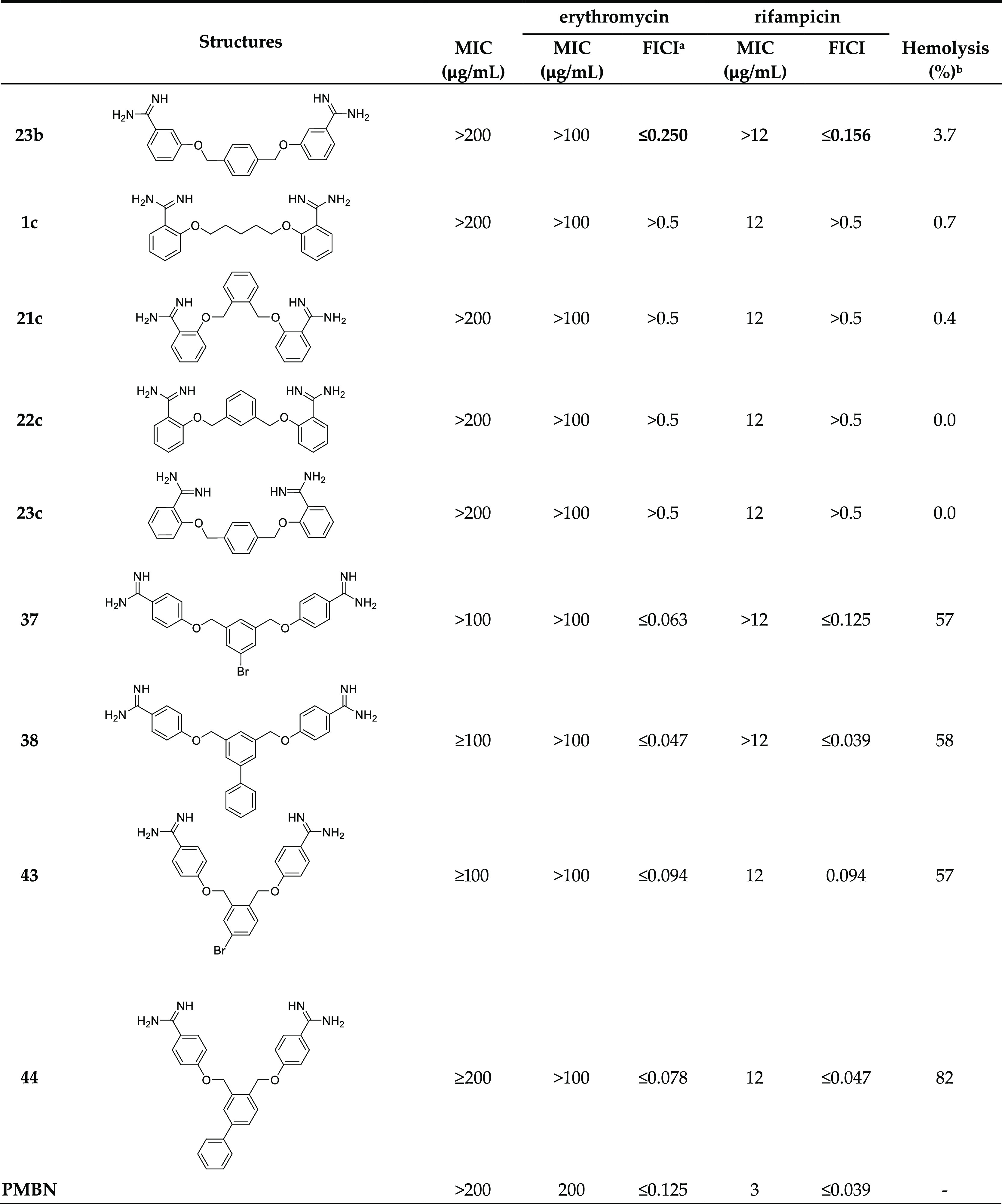
Table 1. Overview of Synergy with Erythromycin against E. coli BW25113 and Hemolysis Data.
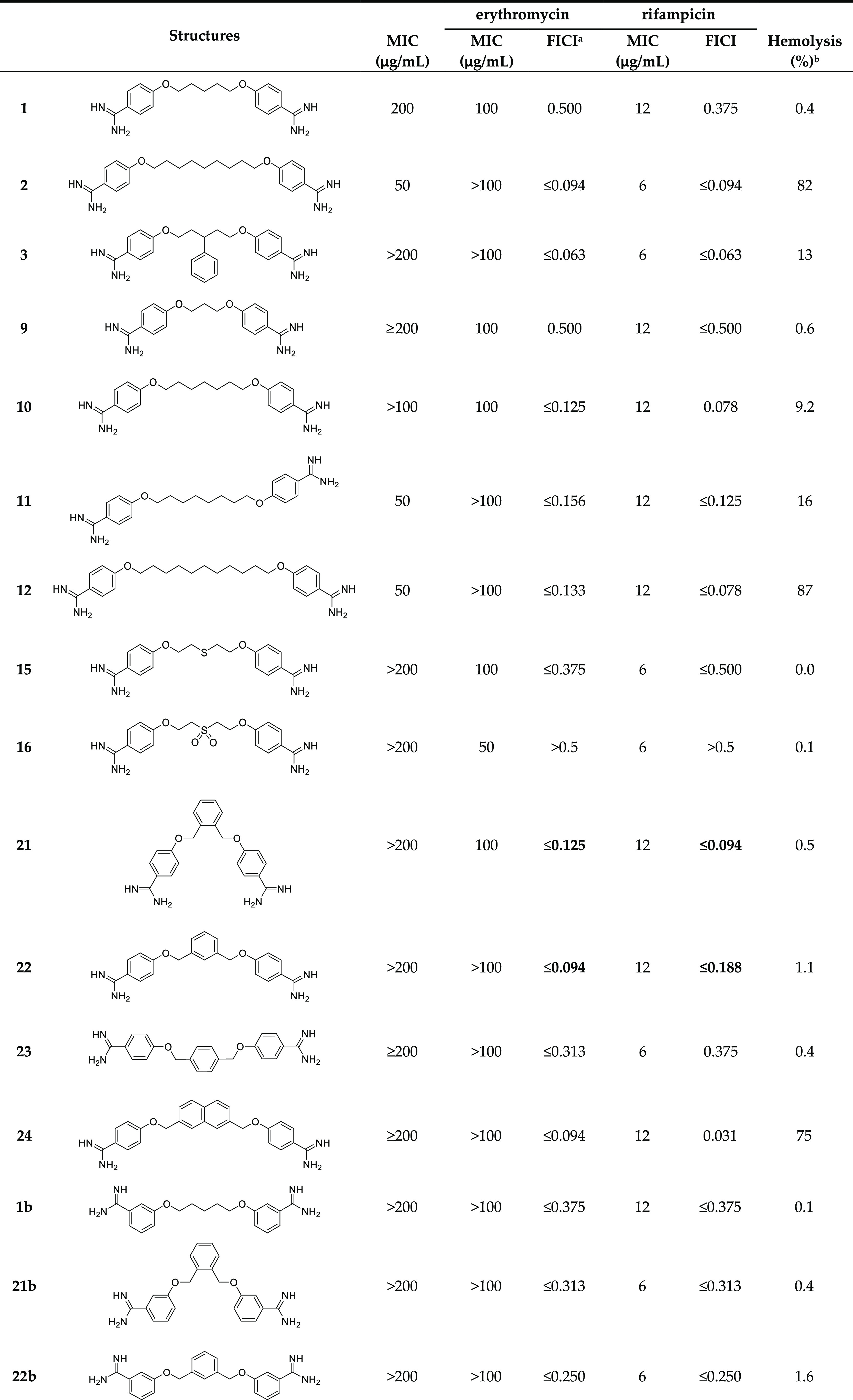
In general, a trend was observed wherein bis-amidines with longer linker lengths showed a great capacity to potentiate the activity of erythromycin (Table 1). Compared with pentamidine (FICI 0.500), nonamidine (2), and heptamidine (10) were found to be the most effective synergists with FICI values of 0.094 and 0.125, respectively, while the shorter propamidine (9) exhibited activity on par with pentamidine (Figure 2). The synergistic activities observed when the same panel of bis-amidines was evaluated with rifampicin corroborates the findings with erythromycin (Table 1 and Supporting Information, Figure S2). These findings highlight the importance of linker length and hydrophobicity for synergistic activity. All analogues containing linkers greater than five carbon atoms demonstrated more potent synergy than observed for pentamidine. By comparison, propamidine (9), containing a three carbon spacer and thioether 15 (isosteric to pentamidine) exhibited synergistic activities comparable to pentamidine. It is also interesting to note that the introduction of the more polar sulfone-linker as in 16 led a complete loss of synergistic activity (Table 1 and Supporting Information, Figures S1 and S2, and Tables S1 and S2).
Figure 2.
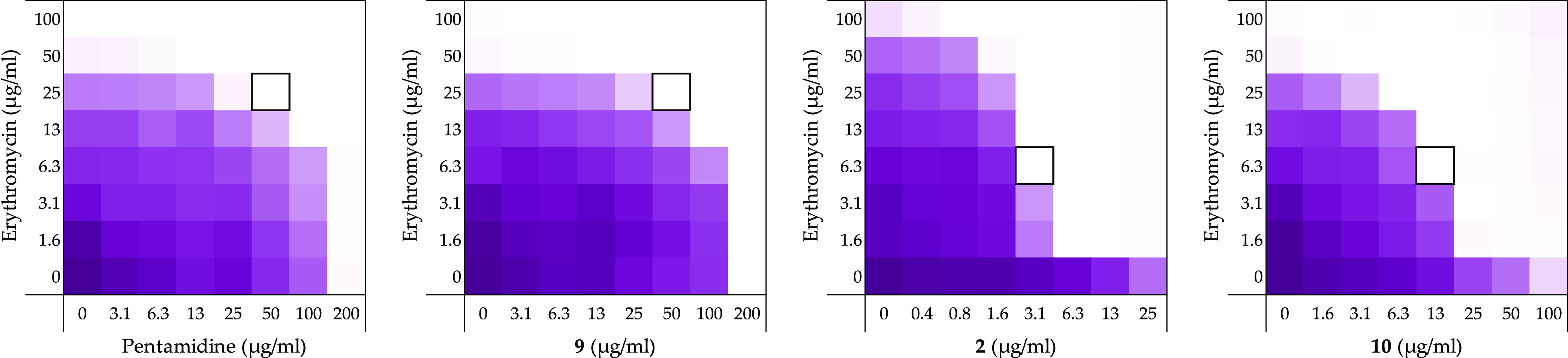
Representative checkerboard assays for pentamidine (1), propamidine (9), nonamidine (2), and heptamidine (10) in combination with erythromycin vsE. coli BW25113. In each case, the bounded box in the checkerboard assays indicates the combination of compound and antibiotic resulting in the lowest FICI (see Table 1). OD600 values were measured using a plate reader and transformed to a gradient: purple represents growth, white represents no growth. An overview of all checkerboard assays with erythromycin can be found in Supporting Information, Figure S1.
Examination of the effect of these bis-amidines on red blood cells revealed another feature that correlates with linker length. Specifically, the enhanced antimicrobial activity and synergistic potential in combination with erythromycin observed for analogues containing longer linkers is accompanied by an increase in hemolytic activity (Table 1 and Supporting Information, Figures S17 and S18 and Table S17). While propamidine (9) and pentamidine (1) have little inherent antibacterial activity (MIC of 200 μg/mL or higher) and are moderate synergists with erythromycin (FICI of 0.500), they are also non-hemolytic (erythrocytes treated with compounds at 200 μg/mL for 20 h at 37 °C, non-hemolytic defined as <10%33). By comparison, the slightly longer heptamidine (10) has an inherent antimicrobial activity (MIC 200 μg/mL) along with enhanced synergistic activity with erythromycin (FICI ≤ 0.125) but also a slight increase in hemolytic activity to 9.2%. However, the longer octamidine (11), nonamidine (2), and undecamidine (12) exhibit very significant levels of hemolysis (16–87%), suggesting that both the inherent antimicrobial activity (MIC 50 μg/mL) and potent synergistic activity in combination with erythromycin (FICI ≤ 0.094–0.156) of these analogues are driven by a general membrane disruption mechanism and not a selective disruption of the Gram-negative OM. Based on these findings, it appears that the “tipping point” associated with the desirable synergistic effects versus the unwanted hemolytic activity appears to be for C7-spaced bis-amidine analogue heptamidine (10). These findings served to inform the design of the next series of analogues.
Linkers with Reduced Flexibility
Building from our initial findings with the linear bis-amidines, we next examined the effect of reducing the rotational flexibility of the linker. In the Brown group’s earlier study, it was noted that phenyl-substituted bis-amidine 3 (Figure 1) was an extremely effective synergist, an effect that was attributed in part to its decreased molecular flexibility.7 To this end, we prepared a series of bis-amidines (Scheme 2, compounds 21–24) that incorporate linkers comprising different planar, aromatic motifs as a means of even further restricting flexibility. For purposes of comparison, we also prepared compound 3 (Supporting Information, Scheme S3) and confirmed its synergistic activity (Table 1, Supporting Information, Figures S1 and S2). Notable, however, was the finding that compound 3 also exhibits significant hemolytic activity (above 10%33) (See Table 1 and Supporting Information, Figure S18 and Table S17) suggesting that impressive synergistic activity associated with the compound is not selective for the Gram-negative OM and is due instead to general membrane disruption. The synthetic route used to access bis-amidines 21–24 is shown in Scheme 2 and was based largely on the published preparation of these and similar compounds previously evaluated as anti-parasitic agents.22,34−39 The meta-oriented linker in compound 22 most closely mimics the 5-carbon spacer found in pentamidine, while analogues 21 and 23 differ slightly due to the ortho- and para-orientations of the benzene core. In the case of compound 24, a 2,7-disubstituted naphthalene motif was envisioned to mimic the 7-carbon spacer found in heptamidine (10). The synthesis of compounds 21–24 started from the corresponding commercially available dibromo-xylenes or 2,7-bis(bromomethyl)naphthalene, which were transformed into the corresponding bis-nitriles 17–20 by treatment with 4-cyanophenol and NaH in dimethylformamide (DMF) at 80 °C. In this case, recrystallization of the intermediates 17, 19, and 20 from ethanol was not successful. However, based on an acceptable purity (as assessed by NMR), the crude bis-nitriles 19 and 20 could be used directly without a need for further purification, while bis-nitrile 17 was purified using column chromatography. Transformation into the corresponding bis-amidines was in turn performed by treatment with LHMDS34 followed by acidic quench with 4 M HCl in dioxane to provide compounds 21–24 in acceptable yields after HPLC purification.
Scheme 2. Synthesis of Bis-Amidines Containing Rigid Aromatic Spacers.
Reagents and conditions: (a) 4-cyanophenol, NaH, DMF, 80 °C, 1 h (79%-quant.); (b) (i) LHMDS, THF, 48 h; (ii) HCl, 0 °C to rt, overnight (19–83%).
Evaluation of the inherent antimicrobial activity of compounds 21–24 as well as their ability to synergize with erythromycin revealed 22 and 24 to be the most effective of these four of compounds (FICI of ≤0.094 with erythromycin) (Figure 3 and Table 1). o-Xylene analogue 21 also exhibited enhanced synergistic activity relative to pentamidine (≤0.125 vs 0.500) while p-xylene analogue 23 showed less activity (FICI ≤ 0.313). Interestingly, while none of compounds 21–24 showed any inherent antibacterial activity up to 200 μg/mL, the 2,7-naphthalene linked analogue 24 was found to exhibit significant hemolytic activity (75%) (see Table 1). These findings are in line with previous studies in which compound 24 was evaluated as an anti-protozoal, where it was also found to exhibit significant toxicity against a rat L6 muscle cell line.38 By comparison, compounds 21 and 22 were found to be non-hemolytic and demonstrate potent synergy when combined with erythromycin with FICI values of ≤0.125 and ≤0.094, respectively (Table 1). Similarly, 21 and 22 were also found to significantly potentiate the activity of rifampicin against the same E. coli indicator strain with FICI values of ≤0.094 and ≤0.188, respectively (Table 1). These findings support the hypothesis that reduced linker flexibility is beneficial for synergistic activity and also reveal the importance of the orientation of the benzamidines on the aromatic nucleus. This is most clearly demonstrated by the potent synergy exhibited by the ortho- and meta-xylene analogues 21 and 22 (FICI ≤ 0.094–0.188) in contrast to the much less active para-xylene linked 23 (FICI ≤ 0.313–0.375).
Figure 3.
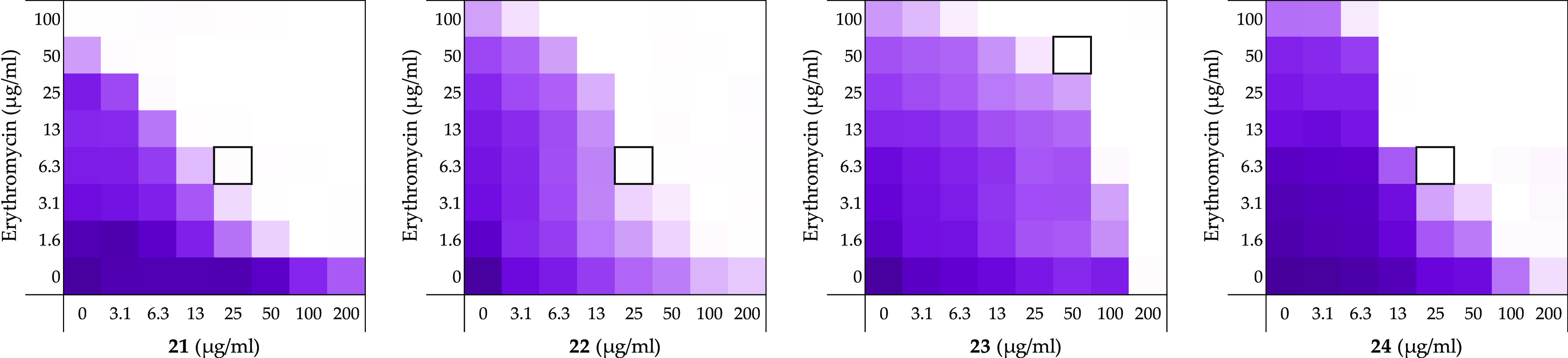
Checkerboard assays for compounds 21–24 in combination with erythromycin vsE. coli BW25113. In each case, the bounded box in the checkerboard assays indicates the combination of compound and antibiotic resulting in the lowest FICI (see Table 1). OD600 values were measured using a plate reader and transformed into a gradient: purple represents growth, white represents no growth. An overview of all checkerboard assays with erythromycin can be found in Supporting Information, Figure S1.
Altering the Position of the Amidine Moiety
The rigidity of the xylene-based linkers described above not only affects the spacing but also the positioning of the amidine groups. In the case of pentamidine (1) and compounds 21–23, the amidine moieties are positioned para relative to the linker. We, therefore, next prepared a series of analogues wherein the positioning of the amidine groups was shifted to either the meta- or ortho-positions (Scheme 3). While the meta-amidine analogues 1b, 21b–23b are known in the literature,24,35,38−41ortho-amidine analogues 1c, 21c–23c have not been previously described. The synthesis of the meta-amidine analogues was performed following the same protocol employed for the preparation of the corresponding para-amidines but using 3-cyanophenol in place of 4-cyanophenol (Scheme 3). For the preparation of the ortho-amidine analogues, the intermediate bis-nitriles were prepared in an analogous fashion, however, conversion to the product bis-amidines required a different set of conditions. Unlike the route used in the preparation of the para- and meta-bis-amidines, treatment of the ortho-bis-nitrile intermediates 29–32 with LHMDS failed to yield the expected amidine product. For this reason, an alternative, previously reported three-step procedure for the conversion of nitriles to amidines, was instead employed.42 In doing so, the nitrile is first converted to the corresponding N-hydroxyamidine by treatment with hydroxylamine hydrochloride. The N-hydroxy group is then acetylated with Ac2O followed by reduction to the amidine product using zinc powder (Scheme 3). After HPLC purification, the ortho-bis-amidines (1c, 21c–23c) were obtained in yields suitable for subsequent evaluation.
Scheme 3. Synthesis of bis-amidine analogues 1b, 21b–23b and 1c, 21c–23c.

Reagents and conditions: (a) 3-cyanophenol, NaH, DMF, 80 °C, 1 h (63%-quant.); (b) (i) LHMDS, THF, 48 h, (ii) HCl, 0 °C to rt, overnight (72%-quant.); (c) 2-cyanophenol, NaH, DMF, 80 °C, 1 h (83–99%); (d) (i) DIPEA, NH2OH·HCl, EtOH, 85 °C, 6 h; (ii) Ac2O, AcOH, rt, 4 h; (iii) Zn powder, AcOH, 35 °C, 6 h (12–48%).
As for pentamidine (1) and the other para-bis-amidines 21–23, no inherent antimicrobial activity or hemolysis was observed for the meta-substituted analogues 1b, 21b–23b or the ortho-substitute analogues 1c, 21c–23c (Table 1). Assessment of synergy with erythromycin showed that the meta-bis-amidines maintain a reasonable degree of synergistic activity (Figure 4) while the ortho-bis-amidines show no such ability (Table 1).
Figure 4.
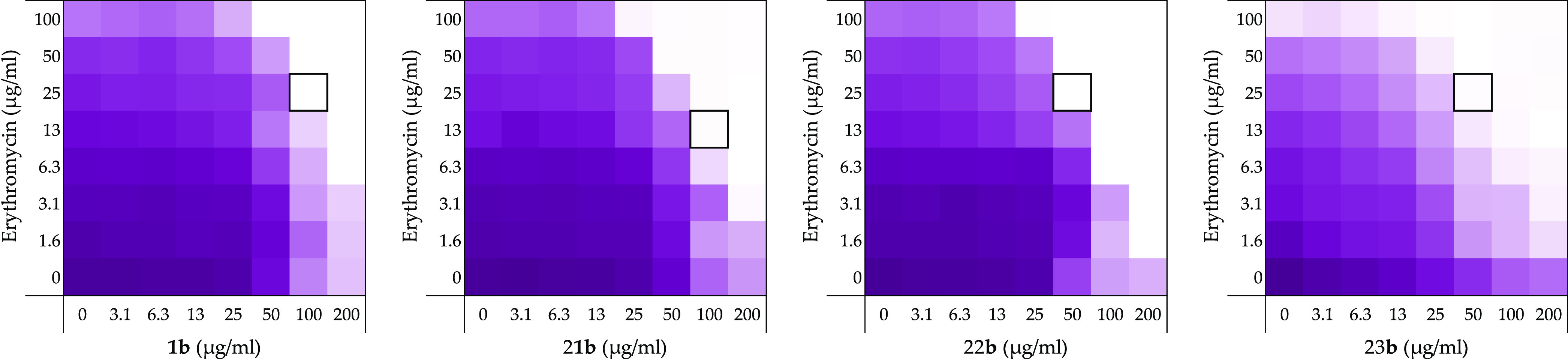
Checkerboard assays for compounds 1b, 21b–23b in combination with erythromycin vsE. coli BW25113. In each case, the bounded box in the checkerboard assays indicates the combination of the compound and antibiotic resulting in the lowest FICI (see Table 1). OD600 values were measured using a plate reader and transformed into a gradient: purple represents growth, white represents no growth. An overview of all checkerboard assays with erythromycin can be found in Supporting Information, Figure S1.
In general, the meta-orientated bis-amidines are less effective synergists than the corresponding para-oriented compounds, a trend also observed in synergy studies with rifampicin (Table 1). An exception to this was observed for compounds 23 and 23b both containing the p-xylene linker. In this case, the placement of the amidine groups at the meta-position relative to the linker results in a slight decrease in FICI from 0.313 for compound 23 to 0.250 for 23b when tested in combination with erythromycin. An even more pronounced potentiation effect was seen when these compounds where evaluated with rifampicin. In this case, compound 23 was found to have an FICI value of 0.375 while for 23b, the FICI value calculated was 0.156, making it one of the most potent, non-hemolytic, rifampicin synergists identified (Table 1). Collectively, these findings indicate that both the geometry of the linker and the positioning of the amidines in the benzamidine moieties are interrelated structural features that play a key role in dictating optimal synergistic activity.
Increasing Linker Hydrophobicity
As described above, bis-amidines with more hydrophobic linkers typically show enhanced synergistic activity but often at the cost of increased hemolysis. In this light, compounds 21 and 22 were deemed to be particularly interesting given that they exhibit potent synergistic activity with both erythromycin and rifampicin while displaying no appreciable hemolytic activity. To examine the possibility of further enhancing these compounds, we next prepared analogues wherein an additional phenyl group, as for compound 3, was added as a substituent to the aromatic linkers in both 21 and 22 to give analogues 38 and 44 (Scheme 4). The synthetic route used also provided ready access to brominated intermediates 35 and 41. Given the hydrophobic character of halogen atoms,43 we opted to also convert these intermediates to the corresponding bis-amidines 37 and 43. The synthesis of meta-linked analogues 37 and 38 started with the reduction of dimethyl 5-bromoisophthalate to give diol 33.44 An Appel reaction was then applied to transform the diol into tribromide 34,45 followed by reaction with 4-cyano phenol to yield bis-nitrile 35.22 A portion of 35 was subsequently used in a Suzuki coupling employing phenylboronic acid, resulting in intermediate 36.46−48 Both 35 and 36 were then converted to the corresponding bis-amidines by treatment with LHMDS followed by HCl quench and HPLC purification to give 37 and 38. The preparation of 43 and 44 followed a similar synthetic strategy but started with the reduction of 4-bromophthalic anhydride using lithium aluminum hydride and ZnCl2.49 The resulting diol 39 was cleanly converted to tribromide 40, which was subsequently transformed into the brominated bis-nitrile intermediate 41. A portion of 41 was then transformed into intermediate 42 using the same Suzuki conditions applied in the previous preparation of 36.46−48 Notably, while bis-nitrile 42 was readily transformed into the desired bis-amidine 44 using the LHMDS protocol, when the same conditions were applied to 41 an unexpected dehalogenation occurred. As an alternative, the same three-step process, described above for the preparation of 21b–23b, was successfully applied to convert the bis-nitrile to the desired bis-amidine 43.42
Scheme 4. Synthesis of (A) Meta-Linked or (B) Ortho-Linked Bis-Amidines Containing Bromo (37, 43) or Phenyl Substitution (38, 44) on the Central Aromatic Core.

Reagents and conditions: (a) (i) DIBALH, DCM, 0 °C, 1 h; (ii) Rochelle salt (quench), rt, overnight (96%); (b) PPh3, CBr4, DCM, rt, 2 h (55–74%); (c) 4-cyanophenol, NaH, DMF, 80 °C, 1 h (87–99%); (d) phenylboronic acid, Pd(dppf)Cl2·DCM, THF/Na2CO3, 65 °C, 8–18 h (8–80%); (e) (i) LHMDS, THF, rt, 48 h; (ii) HCl (quench), 0 °C–rt, overnight (17–75%); (f) (i) LAH, ZnCl2, THF, rt, 6 h; (ii) Rochelle salt (quench), rt, overnight (95%); (g) (i) DIPEA, NH2OH·HCl, EtOH, 85 °C, 6 h; (ii) Ac2O, AcOH, rt, 4 h; (iii) Zn powder, AcOH, 35 °C, 6 h (7%).
Compounds 37, 38, 43, and 44 were found to show no significant inherent antimicrobial activity when tested against E. coli BW25113 (Table 1). As expected, the introduction of the hydrophobic side chains improved the synergistic activity with FICI values ranging from 0.047 to 0.094 (Figure 5 and Table 1). Unfortunately, however, and not entirely unexpectedly, the increased hydrophobicity of these analogues was also found to result in a severe increase in hemolytic activity (Table 1) indicating that the enhanced synergistic activity observed is likely due to non-specific membrane disruption.
Figure 5.
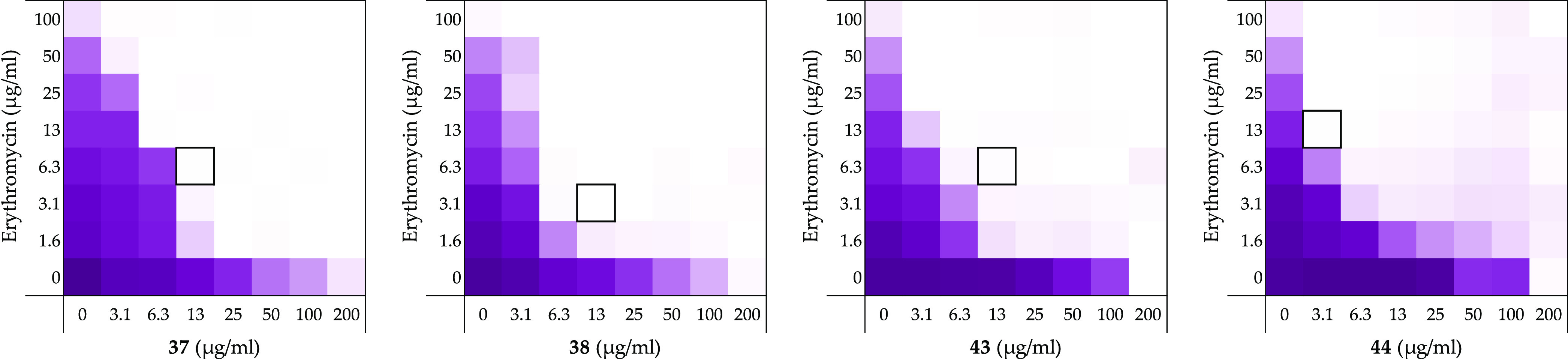
Checkerboard assays for compounds 37, 38, 43, and 44 in combination with erythromycin vsE. coli BW25113. In each case, the bounded box in the checkerboard assays indicates the combination of the compound and antibiotic resulting in the lowest FICI (see Table 1). OD600 values were measured using a plate reader and transformed into a gradient: purple represents growth, white represents no growth. An overview of all checkerboard assays with erythromycin can be found in the Supporting Information, Figure S1.
Exploring the Synergistic Range
Erythromycin, rifampicin, novobiocin, and vancomycin are typically used to treat Gram-positive infections.50−55 However, when combined with OM disrupting agents, these antibiotics can also display efficacy against Gram-negative bacteria.6,20 The Brown group’s recent study with pentamidine showed that erythromycin, rifampicin, and novobiocin were most effectively potentiated by this bis-amidine.7 With this in mind, we next investigated the broader synergy of the most promising compounds identified in our present study, namely, compounds 21, 22, and 23b. As noted above, these three compounds were all found to be more active than pentamidine in potentiating the activity of erythromycin and rifampicin against an indictor E. coli stain while showing no hemolytic activity. To this end, compounds 21, 22, and 23b were evaluated against an expanded panel of organisms, including several E. coli strains (including carbapenem- and polymyxin-resistant strains) and ATCC strains of A. baumannii, K. pneumoniae, and P. aeruginosa. In addition, the well-studied OM disruptor PMBN and pentamidine itself were taken along as benchmarks in the expanded assessment of compounds 21, 22, and 23b.
Synergy with Novobiocin and Vancomycin
Building from the synergy studies with erythromycin and rifampicin described above, compounds 21, 22, and 23b were next tested for the ability to potentiate novobiocin and vancomycin, along with pentamidine (1) and PMBN (Figure 6 and Supporting Information Figures S3 and S4). In agreement with previous studies, novobiocin and vancomycin showed no antimicrobial activity against the indicator E. coli BW25113 strain at the highest concentration tested of 200 μg/mL.7,56 Checkerboard assays with compounds 21, 22, and 23b in combination with novobiocin revealed the compounds to be superior synergists compared to pentamidine (Table 2, Figure 6), a finding in line with the results obtained when the same bis-amidines were evaluated with erythromycin and rifampicin. In general, PMBN was found to be a more potent synergist than the bis-amidines with the exception of compound 22 in combination with erythromycin which resulted in very effective growth prevention of the E. coli indicator strain. In line with expectation, when tested in combination with vancomycin, none of the bis-amidines showed any synergistic activity, while PMBN maintained a potent effect (Table 2). These findings are in line with previously reported observations in which pentamidine was found not to synergize with vancomycin.7
Figure 6.
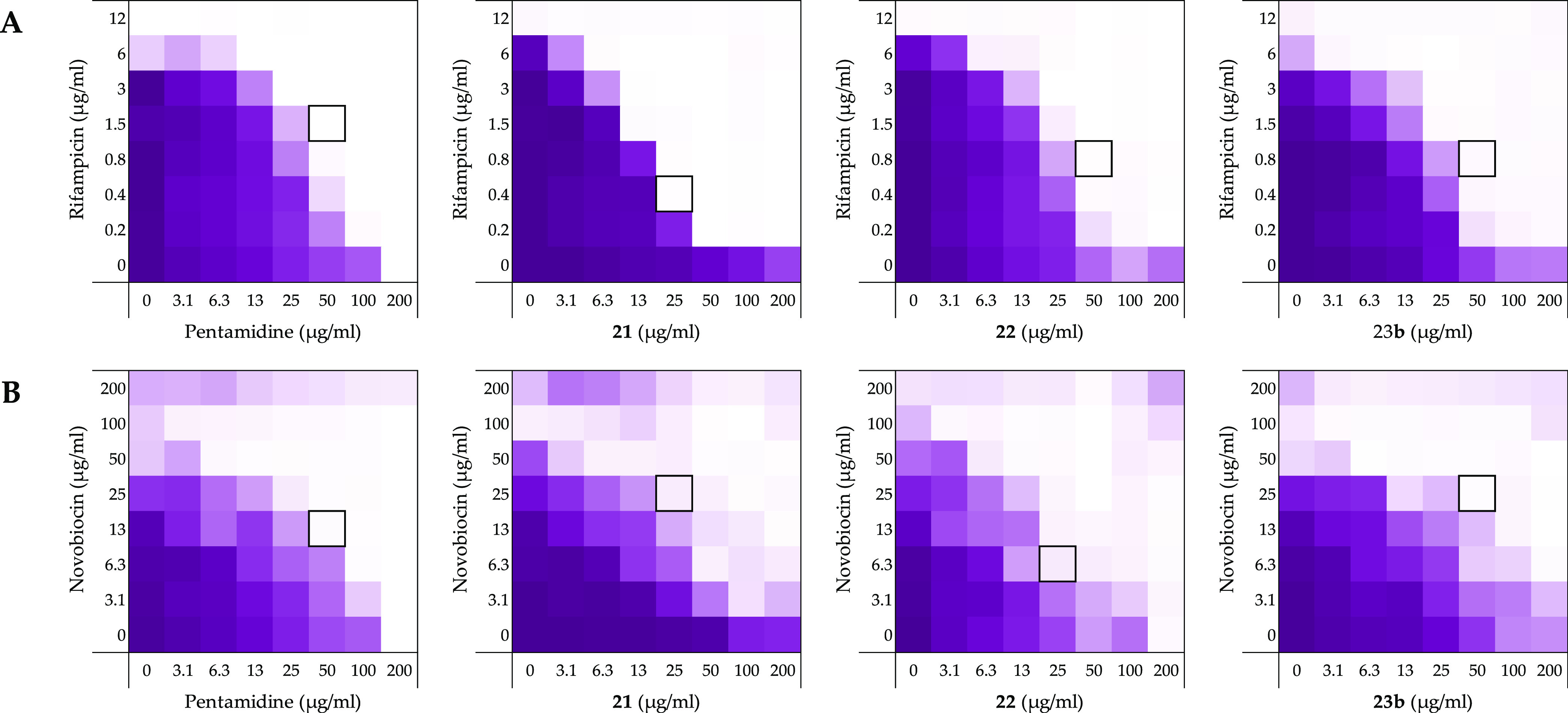
Checkerboard assays of compounds pentamidine (1), 21, 22, and 23b in combination with (A) rifampicin and (B) novobiocin against E. coli BW25113. In each case, the bounded box in the checkerboard assays indicates the combination of the compound and antibiotic resulting in the lowest FICI (see Table 2). OD600 values were measured using a plate reader and transformed into a gradient: purple represents growth, white represents no growth. The poor aqueous solubility of novobiocin results in the background signal observed in the OD600 read-out at when tested at concentrations ≥100 μg/mL. An overview of all checkerboard assays with rifampicin, novobiocin, and vancomycin can be found in Supporting Information, Figures S2–S4.
Table 2. FICI Values of Pentamidine (1), 21, 22, 23b, and PMBN against E. coli BW25113 in Combination with Gram-Positive-Specific Antibiotics Rifampicin, Novobiocin, and Vancomycina.
| erythromycin | rifampicin | novobiocin | vancomycin | |
|---|---|---|---|---|
| pentamidine (1) | 0.500 | 0.375 | ≤0.281 | >0.5b |
| 21 | ≤0.125 | ≤0.094 | ≤0.125 | >0.5b |
| 22 | ≤0.094 | ≤0.188 | ≤0.078 | >0.5b |
| 23b | ≤0.250 | ≤0.156 | ≤0.188 | >0.5b |
| PMBN | ≤0.125 | ≤0.039 | ≤0.047 | ≤0.156 |
MIC and minimal synergistic concentrations (MSCs) data can be found in Supporting Information, Tables S1–S4.
Synergy defined as an FICI ≤ 0.5.21
Synergy against Other E. coliStrains
The next phase of our investigation involved assessing the synergistic activity of the most promising compounds identified against an expanded panel of E. coli strains. For these screens, we opted to focus on rifampicin as the companion antibiotic given that it is bactericidal while erythromycin is considered to be bacteriostatic.11,57 In our initial screens, a more clear-cut distinction of growth versus no growth was indeed observed for rifampicin, possibly due to its bactericidal nature (see Figures 3 and 6A). Furthermore, given that the MIC of rifampicin is significantly lower against the Gram-negative strains used versus the MICs of erythromycin or novobiocin, potential solubility issues at the highest antibiotic concentrations tested were not a problem.
In selecting an expanded panel of E. coli strains, we sought to examine a variety of features ranging from the OM composition to resistance profile. In the case of E. coli, the structure of the lipopolysaccharide (LPS) layer is known to affect their susceptibility to antibiotics58 and we therefore reasoned that it could also play a role in the synergistic activity of compounds targeting the OM. This was seen as particularly relevant for the pentamidine analogues investigated here, given that previous studies have suggested that pentamidine interacts with lipid A.7 With this in mind, E. coli ATCC25922 (smooth LPS) and E. coli W3110 (rough LPS) were selected, along with the indicator lab strain E. coli BW25113 also known to possess a rough LPS layer.59−61 Additionally, a clinical isolate E. coli 552060.1 was included, which, like most clinical isolates, has a smooth LPS layer.58,62 The inherent antimicrobial activity of rifampicin, pentamidine (1), compounds 21, 22, 23b, and PMBN was first established against these E. coli strains (Supporting Information, Figures S5–S7 and Tables S5–S7). In keeping with our initial checkerboard assays with rifampicin and the E. coli BW25113 strain (Table 1), compound 21 in nearly all cases showed the lowest FICI values among the bis-amidines evaluated against the expanded E. coli panel (Figure 7A and Table 3). In general, the bis-amidines tested all showed effective synergy with little difference observed for the rough or smooth LPS strains.
Figure 7.
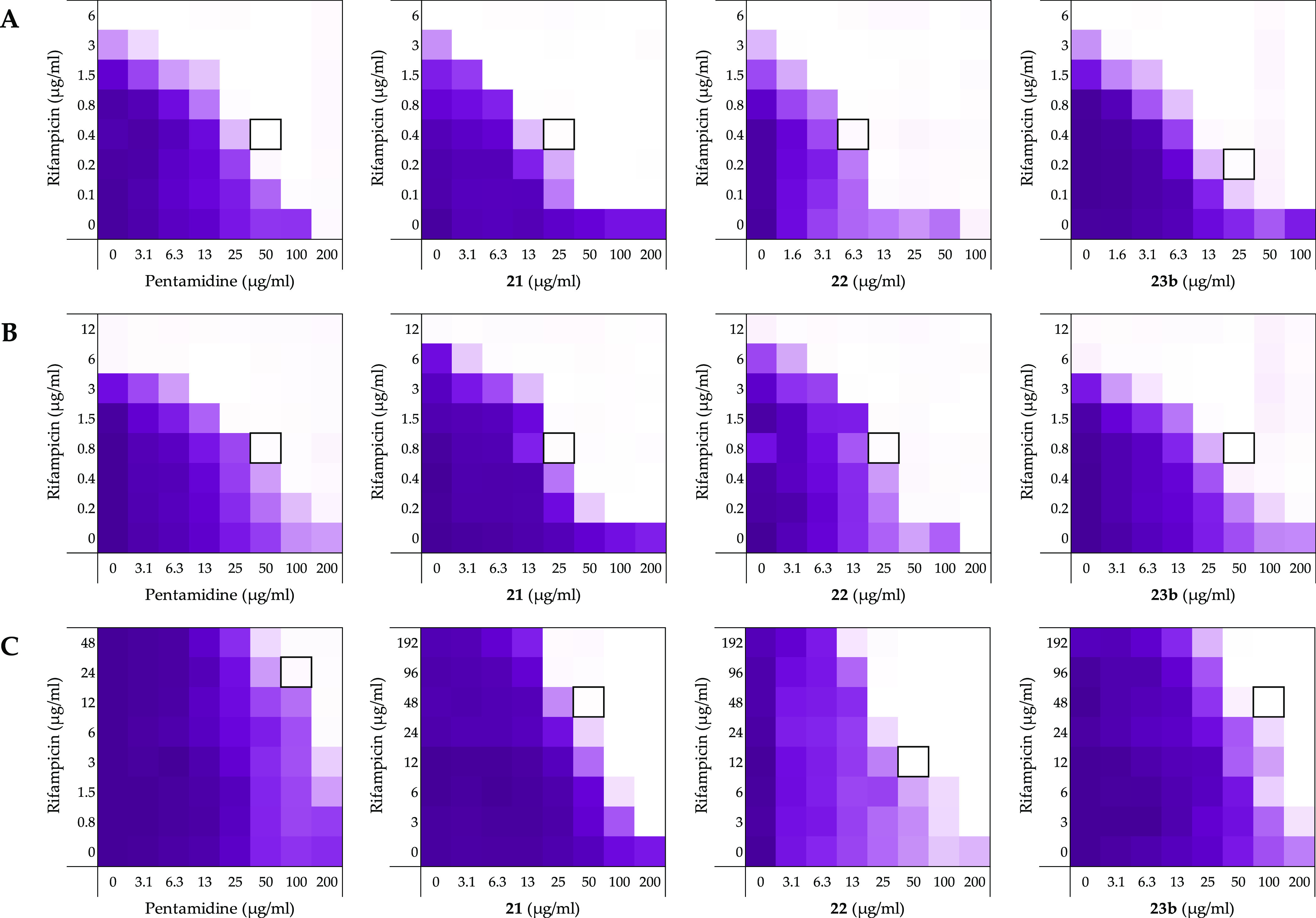
Checkerboard assays of compounds pentamidine (1), 21, 22, and 23b in combination with rifampicin vs (A) E. coli ATCC25922, (B) E. coli EQASmcr-1, and (C) E. coli RC0089. In each case, the bounded box in the checkerboard assays indicates the combination of the compound and antibiotic resulting in the lowest FICI (see Table 3). OD600 values were measured using a plate reader and transformed into a gradient: purple represents growth, white represents no growth. An overview of all checkerboard assays with rifampicin with the E. coli strains can be found in Supporting Information, Figures S5–S13.
Table 3. FICI Values of Pentamidine (1), 21, 22, 23b, and PMBN in Combination with Rifampicin against Different E. coli Strains Including Polymyxin- and Carbapenem-Resistant Strainsa.
| strain | pentamidine (1) | 21 | 22 | 23b | PMBN |
|---|---|---|---|---|---|
| wild-type | |||||
| BW25113 | 0.375 | ≤0.094 | ≤0.188 | ≤0.156 | ≤0.039 |
| ATCC25922 | 0.313 | ≤0.125 | 0.094 | 0.156 | ≤0.047 |
| W3110 | ≤0.188 | ≤0.188 | 0.313 | ≤0.188 | ≤0.031 |
| 552060.1 | 0.375 | ≤0.094 | 0.250 | ≤0.188 | ≤0.047 |
| polymyxin-resistant | |||||
| BW25113 mcr-1 | ≤0.250 | ≤0.094 | ≤0.156 | ≤0.188 | ≤0.156 |
| mcr-1 | ≤0.188 | ≤0.188 | ≤0.188 | ≤0.188 | ≤0.094 |
| EQASmcr-1 | ≤0.250 | ≤0.125 | 0.188 | ≤0.188 | ≤0.125 |
| EQASmcr-2 | 0.375 | ≤0.125 | 0.313 | ≤0.125 | ≤0.156 |
| EQASmcr-3 | ≤0.188 | ≤0.125 | ≤0.188 | ≤0.188 | ≤0.094 |
| carbapenem-resistant | |||||
| RC0089 | ≤0.375 | ≤0.250 | ≤0.156 | ≤0.375 | ≤0.188 |
MIC and MSCs data can be found in Supporting Information, Table S2, S5–S13.
The expanded screening was continued with E. coli bearing mcr-1, mcr-2, and mcr-3 genotypes known to confer polymyxin resistance. For this purpose, a lab strain E. coli BW25113 mcr-1, transformed with the pGDP2 plasmid, was also included to directly assess the effect of the phosphoethanolamine transferase responsible for lipid A modification.63−65 The bis-amidines displayed synergy with rifampicin against all mcr-positive strains evaluated (Figure 7B, Table 3, Supporting Information, Figures S8–S12, and Tables S8–S12). Again, in nearly all cases, compound 21 gave the lowest FICI values among the bis-amidines evaluated, with synergy comparable to that of PMBN, which was found to be generally less effective against mcr-positive strains than non-mcr strains (Table 3).
In addition, carbapenem-resistant E. coli RC0089, a clinical isolate producing New Delhi β-lactamase 1 (NDM-1), was also evaluated to assess whether this resistance mechanism affected the synergistic activity of the bis-amidines here studied. Notably, the MIC of rifampicin was significantly elevated against this strain (MIC of >192 μg/mL, see Supporting Information, Figure S13 and Table S13). While the bis-amidines were again found to synergize with rifampicin, the FICI values calculated were elevated, with the exception of compound 22 (Figure 7C and Table 3). Interestingly, this strain also resulted in an increased FICI for PMBN.
Synergy against A. baumannii, K. pneumoniae, and P. aeruginosa
In addition to studying the synergistic activity of the selected bis-amidines against the E. coli strains described above, we also investigated their capacity to potentiate the activity of rifampicin against the selected strains of A. baumannii, K. pneumoniae, and P. aeruginosa (Figure 8, Table 4). As for the E. coli strains, the inherent antimicrobial activities of rifampicin, pentamidine (1), compounds 21, 22, 23b, and PMBN were first established against each strain (Supporting Information, Tables S14–S16). Full checkerboard assays with the A. baumannii and K. pneumoniae strains tested showed the bis-amidines and PMBN to be effective synergists. In general, compounds 21, 22, and 23b were found to be more potent than pentamidine (1), while PMBN was found to be an even more effective synergist. Among the bis-amidines tested, compound 22 displayed the most effective potentiation of rifampicin. Interestingly, when tested against P. aeruginosa, the FICIs determined for pentamidine and compounds 21, 22, and 23b were significantly elevated while PMBN maintained potent synergistic activity.
Figure 8.
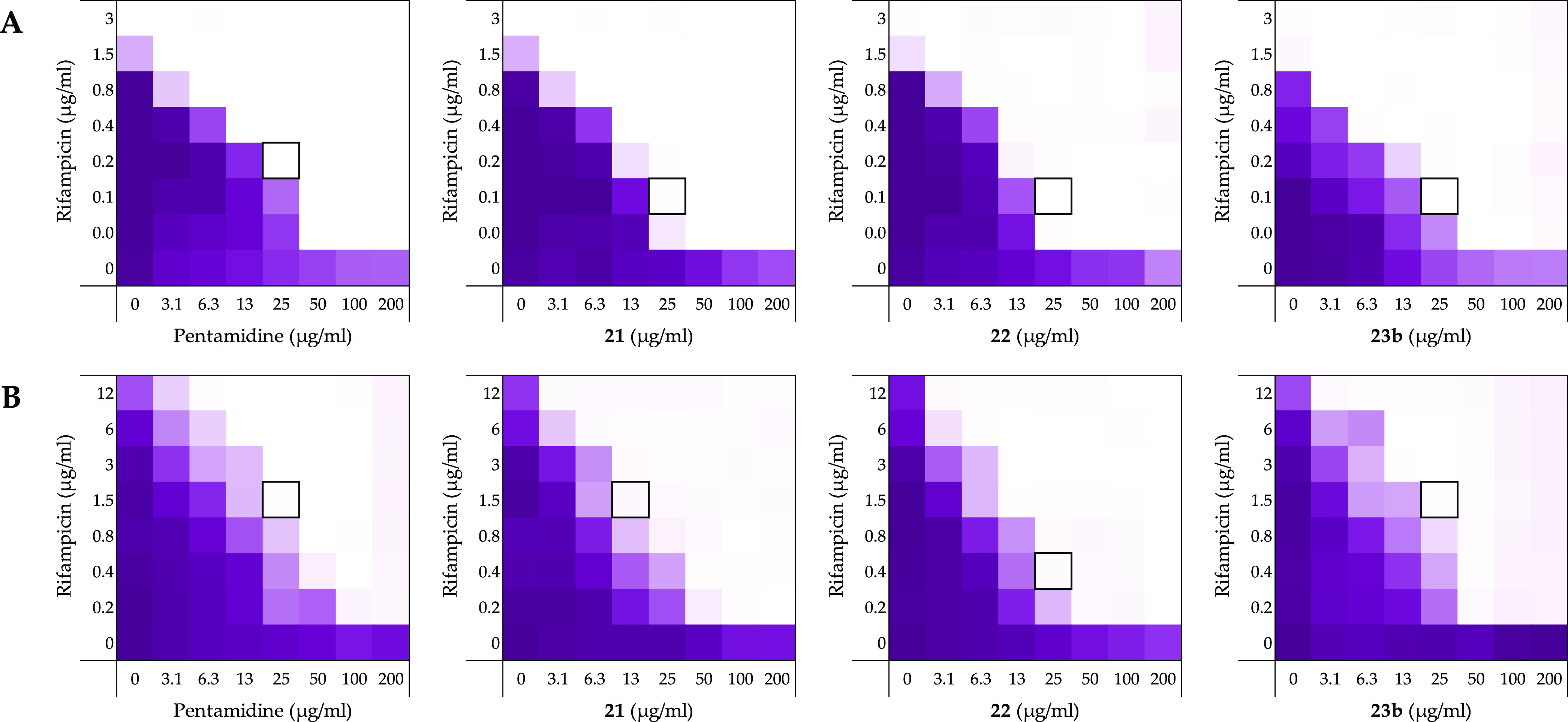
Checkerboard assays of pentamidine (1), 21, 22, and 23b in combination with rifampicin and vs (A) A. baumannii ATCC17978 and (B) K. pneumoniae ATCC13883. In each case, the bounded box in the checkerboard assays indicates the combination of compound and antibiotic resulting in the lowest FICI (see Table 4). OD600 values were measured using a plate reader and transformed into a gradient: purple represents growth, white represents no growth. An overview of all checkerboard assays with rifampicin with the E. coli strains can be found in Supporting Information, Figures S14–S16.
Table 4. FICI Values of Pentamidine (1), 21, 22, 23b, and PMBN in Combination with Rifampicin against Different Gram-Negative Pathogensa.
| strain | pentamidine (1) | 21 | 22 | 23b | PMBN |
|---|---|---|---|---|---|
| A. baumannii ATCC17978 | ≤0.125 | ≤0.094 | ≤0.094 | ≤0.094 | ≤0.023 |
| K. pneumoniae ATCC13883 | ≤0.125 | ≤0.094 | ≤0.078 | ≤0.125 | ≤0.070 |
| P. aeruginosa ATCC27853 | ≤0.500 | ≤0.313 | ≤0.250 | ≤0.375 | 0.031 |
MIC and MSCs data can be found in Supporting Information, Tables S14–S16.
Mechanistic Studies
To characterize the mechanism of action of the bis-amidines here studied, we next investigated the capacity of the most active compounds to disrupt the Gram-negative OM. This line of investigation was based in part on the previously noted interaction of pentamidine with lipid A and also on the knowledge that the potentiation of antibiotics like erythromycin, rifampicin, and novobiocin generally relies on OM disruption.7,20,66 To this end, we employed an established assay relying on the fluorescent properties of N-phenyl-napthalen-1-amine (NPN) allowing for the real-time monitoring and quantification of OM disruption.67 In the presence of intact bacterial cells, NPN exhibits relatively low levels of fluorescence. However, in the event that the OM is disrupted, NPN can gain entry to the phospholipid layer resulting in a detectable increase in fluorescence that can, in turn, be measured.67 For this assay, we selected compounds 21 and 22 based on their consistently potent activity in the various synergy assays described above. The bacterial strain used was E. coli BW25113 and pentamidine (1) and PMBN were taken along as benchmarks. As illustrated in Figure 9, a clear, dose-dependent increase in the fluorescent signal is observed for both 21 and 22, indicating effective OM disruption. In general, both compounds appear to outperform pentamidine in their ability to disrupt the OM with compound 22 also exhibiting a stronger effect than PMBN (see Supporting Information, Figure S19 for NPN fluorescence at higher concentrations of bis-amidines and PMBN).
Figure 9.

OM permeabilization assay of pentamidine (1), compounds 21, 22, and PMBN with E. coli BW25113 using N-phenyl-1-naphthylamine (NPN) as a fluorescent probe. The read-out was performed after 60 min of incubation using a plate reader with λex 355 nm and λem 420 nm. The NPN uptake values shown are relative to the uptake signal obtained upon treating the cells with 100 μg/mL colistin as previously reported.68 All values corrected for the background signal of the negative control. Error bars represent the standard deviation based on n = 3 technical replicates.
Conclusions
We here describe SAR studies aimed at delivering new insights into the capacity for small-molecule bis-amidines to potentiate the activity of Gram-positive specific antibiotics against Gram-negative bacteria. Inspired by the finding that anti-parasitic drug pentamidine disrupts the Gram-negative OM to synergize with antibiotics like erythromycin, rifampicin, and novobiocin, we prepared a number of structurally similar bis-amidines and characterized their synergistic potential with the same antibiotics. Our studies confirm that the length, rigidity, and hydrophobicity of the linker unit present in these bis-amidines play an important role in determining their ability to potentiate Gram-positive specific antibiotics.7 Also of note, however, is the finding that the potent synergy exhibited by bis-amidines containing long, hydrophobic linkers is likely driven by nonspecific membrane disruption as indicated by the strong hemolytic activity associated with these analogues. Further assessment of the linker motif also revealed that, in general, a single aromatic ring provides a desirable balance of enhanced synergistic activity relative to pentamidine, without introducing hemolytic activity. Further examination of the relative positioning of the benzamidine groups on the aromatic linker and as well as the ortho-, meta-, and para-geometry of the amidine moieties themselves identified compounds 21, 22, and 23b as most promising. These compounds were found to consistently outperform pentamidine in their ability to potentiate the activity of erythromycin, rifampicin, and novobiocin against a number of E. coli strains including polymyxin-resistant and carbapenem-resistant variants. Additional screening showed that among the bis-amidines here studied, compounds 21, 22, and 23b maintain their superior synergistic activity against other Gram-negative pathogens including A. baumannii, K. pneumoniae, and P. aeruginosa. Mechanistic studies also confirm that these bis-amidines effectively induce Gram-negative OM disruption. Taken together, the findings here reported provide a broader understanding of the potential for bis-amidines to be used as synergists in expanding the activity of Gram-positive specific antibiotics against Gram-negative bacteria.
Methods
General Procedures
All reagents employed were of American Chemical Society (ACS) grade or finer and were used without further purification unless otherwise stated. For compound characterization, 1H NMR spectra were recorded at 400 MHz with chemical shifts reported in parts per million (ppm) downfield relative to CHCl3 (7.26) or dimethyl sulfoxide (DMSO) (δ 2.50). 1H NMR data are reported in the following order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet and m, multiplet), coupling constant (J) in hertz (Hz), and the number of protons. Where appropriate, the multiplicity is preceded by br, indicating that the signal was broad. 13C NMR spectra were recorded at 101 MHz with chemical shifts reported relative to CDCl3 (δ 77.16) or DMSO (δ 39.52). HRMS analysis was performed on a Shimadzu Nexera X2 UHPLC system with a Waters Acquity HSS C18 column (2.1 × 100 mm, 1.8 μm) at 30 °C and equipped with a diode array detector. The following solvent system, at a flow rate of 0.5 mL/min, was used: solvent A, 0.1% formic acid in water and solvent B, 0.1% formic acid in acetonitrile. Gradient elution was as follows: 95:5 (A/B) for 1 min, 95:5 to 15:85 (A/B) over 6 min, 15:85 to 0:100 (A/B) over 1 min, 0:100 (A/B) for 3 min, and then reversion back to 95:5 (A/B) for 3 min. This system was connected to a Shimadzu 9030 QTOF mass spectrometer (ESI ionization) calibrated internally with an Agilent’s API-TOF reference mass solution kit (5.0 mM purine, 100.0 mM ammonium trifluoroacetate and 2.5 mM hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazine) diluted to achieve a mass count of 10,000. Compounds 13, 14, 33, and 34 were synthesized as previously described and had NMR spectra and mass spectra consistent with the assigned structures.32,69 Compounds 1, 2, 4–6, 8–11, 15, 18, 19, 21–23, 1b, 21b–23b, 39, and 40 were synthesized using optimized protocols as described below and gave NMR spectra and mass spectra consistent for the same compounds previously described in the literature.22,29,32,34,38,39,70−72 Purity of the final compounds 1–3, 9–12, 15, 16, 21–24, 1b, 21b–23b, 1c, 21c–23c, 37, 38, 43, and 44 was confirmed to be ≥95% by analytical RP-HPLC using a Shimadzu Prominence-i LC-2030 system with a Dr. Maisch ReproSil Gold 120 C18 column (4.6 × 250 mm, 5 μm) at 30 °C and equipped with a UV detector monitoring at 214 nm. The following solvent system, at a flow rate of 1 mL/min, was used: solvent A, 0.1% trifluoroacetic acid (TFA) in water/acetonitrile, 95/5 and solvent B, 0.1% TFA in water/acetonitrile, 5/95. Gradient elution was as follows: 95:5 (A/B) for 2 min, 95:5 to 0:100 (A/B) over 30 min, 0:100 (A/B) for 1 min, then reversion back to 95:5 (A/B) over 1 min, 95:5 (A/B) for 3 min. The compounds were purified via preparative HPLC using a BESTA-Technik system with a Dr. Maisch Reprosil Gold 120 C18 column (25 × 250 mm, 10 μm) and equipped with a ECOM Flash UV detector monitoring at 214 nm. The following solvent system, at a flow rate of 12 mL/min, was used: solvent A, 0.1% TFA in water/acetonitrile 95/5 and solvent B, 0.1% TFA in water/acetonitrile 5/95. Unless stated otherwise in the protocol, the gradient elution was as follows: 100:0 (A/B) to 0:100 (A/B) over 25 min, 0:100 (A/B) for 3 min, then reversion back to 100:0 (A/B) over 1 min, 100:0 (A/B) for 1 min.
Synthesis
4,4′-(Pentane-1,5-diylbis(oxy))dibenzimidamide/Pentamidine (1)
This protocol was based on the synthesis of structurally similar amidine containing compounds previously described in the literature.28−31 4,4′-(pentane-1,5-diylbis(oxy))dibenzonitrile (94 mg, 0.3 mmol) was dissolved in dry THF (2 mL) under an argon atmosphere and LHMDS (1.2 mL, 1 M THF solution, 4.0 equiv) was added. The reaction was stirred at room temperature for 48 h or longer until complete conversion to the bis-amidine [monitored by liquid chromatography–mass spectrometry (LCMS)]. The solution was cooled to 0 °C and quenched with HCl (4.5 mL, 4 M dioxane solution, 60 equiv). The mixture was stirred at room temperature overnight, then diluted with diethyl ether, and filtered. The precipitate was purified by preparative HPLC with the gradient 0–100% in 30 min to give pentamidine (1) (120 mg, quant). 1H NMR (400 MHz, DMSO-d6): δ 9.14 (s, 4H), 9.06 (s, 4H), 7.81 (d, J = 8.9 Hz, 4H), 7.15 (d, J = 8.9 Hz, 4H), 4.12 (t, J = 6.4 Hz, 4H), 1.88–1.75 (m, 4H), 1.65–1.52 (m, 2H). 13C NMR (101 MHz, DMSO): δ 164.70, 163.06, 130.19, 119.50, 114.79, 68.05, 28.21, 22.09. HRMS (ESI): calcd for C19H24N4O2 [M + H]+, 341.1977; found, 341.1977.
4,4′-(Nonane-1,9-diylbis(oxy))dibenzimidamide/Nonamidine (2)
Following the procedure as described for compound 1, using compound 7 (100 mg, 0.28 mmol), LHMDS (1.5 mL, 1 M THF solution, 5.4 equiv), and HCl (5 mL, 4 M dioxane solution, 71 equiv) afforded the crude product. Purification by preparative HPLC with the gradient 20–100% in 30 min afforded compound 2 (86 mg, 84%). 1H NMR (400 MHz, DMSO-d6): δ 9.14 (d, J = 6.2 Hz, 8H), 7.81 (d, J = 8.9 Hz, 4H), 7.13 (d, J = 9.0 Hz, 4H), 4.07 (t, J = 6.5 Hz, 4H), 1.78–1.67 (m, 4H), 1.48–1.27 (m, 10H). 13C NMR (101 MHz, DMSO): δ 164.82, 163.12, 130.21, 119.50, 114.82, 68.16, 29.01, 28.77, 28.52, 25.47. HRMS (ESI): calcd for C23H32N4O2 [M + H]+, 397.2604; found, 397.2597.
4,4′-((3-Phenylpentane-1,5-diyl)bis(oxy))dibenzimidamide (3)
4,4′-((3-Phenylpentane-1,5-diyl)bis(oxy))dibenzonitrile (109 mg, 0.28 mmol) was dissolved in the LHMDS solution (1.1 mL, 1 M THF solution, 4.0 equiv) under an argon atmosphere. The reaction mixture was stirred at room temperature for 48 h or longer until complete conversion to the bis-amidine (monitored by LCMS). The solution was cooled to 0 °C and quenched with HCl (4.5 mL, 4 M dioxane solution, 60 equiv). The mixture was stirred at room temperature overnight, then diluted with diethyl ether, and filtered. The precipitate was purified by preparative HPLC with the gradient 20–100% in 30 min to give compound 3 (27.4 mg, 23%). 1H NMR (400 MHz, DMSO-d6): δ 9.11 (d, J = 12.6 Hz, 8H), 7.77 (d, J = 8.9 Hz, 4H), 7.34–7.16 (m, 5H), 7.05 (d, J = 9.0 Hz, 4H), 4.00–3.90 (m, 2H), 3.83 (dd, J = 15.0, 8.9 Hz, 2H), 3.14–3.04 (m, 1H), 2.29–2.16 (m, 2H), 2.13–2.00 (m, 2H). 13C NMR (101 MHz, DMSO): δ 164.81, 162.92, 143.38, 130.21, 128.62, 127.69, 126.58, 119.64, 66.21, 38.31, 35.10. HRMS (ESI): calcd for C25H28N4O2 [M + H]+, 417.2291; found, 417.2287.
4,4′-(Propane-1,3-diylbis(oxy))dibenzonitrile (4)
These conditions were based on literature protocols.22 4-cyanophenol (0.29 g, 2.4 mmol, 2.4 equiv) was suspended in dry DMF (3 mL) under an argon atmosphere. The suspension was cooled to 0 °C using an ice bath and NaH (96 mg, 60% dispersion in mineral oil, 2.4 equiv) was slowly added. The reaction mixture was stirred until a clear solution appeared, the ice bath was removed and 1,3-dibromopropane (202 mg, 1 mmol) was added. The reaction mixture was heated to 80 °C for 1 h and then cooled to room temperature. Water (10 mL) was added to the mixture to obtain precipitation. The precipitate was filtered, washed with water, and recrystallized from EtOH to give compound 4 as white crystals (164 mg, 59%). 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 8.9 Hz, 4H), 6.95 (d, J = 8.9 Hz, 4H), 4.21 (t, J = 6.0 Hz, 4H), 2.37–2.27 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 162.09, 134.19, 119.26, 115.29, 104.39, 64.56, 28.96.
4,4′-(Heptane-1,7-diylbis(oxy))dibenzonitrile (5)
Following the procedure as described above for compound 4, using 1,7-dibromoheptane (0.60 mL, 3.5 mmol) afforded compound 5 (1.17 g, quant). 1H NMR (400 MHz, CDCl3): δ 7.56 (d, J = 8.8 Hz, 4H), 6.92 (d, J = 8.8 Hz, 4H), 3.99 (t, J = 6.4 Hz, 4H), 1.89–1.76 (m, 4H), 1.55–1.40 (m, 6H). 13C NMR (101 MHz, CDCl3): δ 162.49, 134.09, 119.42, 115.26, 103.82, 68.38, 29.14, 29.03, 26.00.
4,4′-(Octane-1,8-diylbis(oxy))dibenzonitrile (6)
Following the procedure as described above for compound 4, using 1,8-dibromooctane (0.64 mL, 3.5 mmol) afforded compound 6 (1.10 g, 90%). 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 8.9 Hz, 4H), 6.93 (d, J = 8.9 Hz, 4H), 3.99 (t, J = 6.5 Hz, 4H), 1.84–1.77 (m, 4H), 1.51–1.43 (m, 4H), 1.43–1.35 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 162.54, 134.12, 119.45, 115.29, 103.86, 68.46, 29.37, 29.11, 26.04.
4,4′-(Nonane-1,9-diylbis(oxy))dibenzonitrile (7)
Following the procedure as described above for compound 4, using 1,9-dibromononane (0.71 mL, 3.5 mmol) afforded compound 7 (1.26 g, 99%). 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 8.9 Hz, 4H), 6.93 (d, J = 8.8 Hz, 4H), 3.99 (t, J = 6.5 Hz, 4H), 1.86–1.75 (m, 4H), 1.51–1.41 (m, 4H), 1.40–1.30 (m, 6H). 13C NMR (101 MHz, CDCl3): δ 162.55, 134.11, 119.46, 115.29, 103.82, 68.49, 29.56, 29.39, 29.11, 26.07.
4,4′-(Undecane-1,11-diylbis(oxy))dibenzonitrile (8)
Following the procedure as described above for compound 4, using 1,11-dibromoundecane (0.82 mL, 3.5 mmol) afforded compound 8 (1.24 g, 92%). 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 8.9 Hz, 4H), 6.93 (d, J = 8.9 Hz, 4H), 3.99 (t, J = 6.5 Hz, 4H), 1.84–1.75 (m, 4H), 1.49–1.40 (m, 4H), 1.39–1.28 (m, 10H). 13C NMR (101 MHz, CDCl3): δ 162.57, 134.11, 119.47, 115.30, 103.80, 68.53, 29.65, 29.63, 29.46, 29.12, 26.08.
4,4′-(Propane-1,3-diylbis(oxy))dibenzimidamide/Propamidine (9)
Following the procedure as described above for pentamidine (1), using compound 4 (60 mg, 0.2 mmol). After LCMS analysis of the reaction mixture at 48 h, LHMDS (0.2 mL, 1 M THF solution, 1 eq.) was added. The HCl quench was therefore also increased (4 mL, 4 M dioxane solution, 75 equiv). Compound 9 was obtained after HPLC purification (33 mg, 49%). 1H NMR (400 MHz, DMSO-d6): δ 9.15 (d, J = 9.4 Hz, 8H), 7.82 (d, J = 8.9 Hz, 4H), 7.18 (d, J = 8.9 Hz, 4H), 4.27 (t, J = 6.2 Hz, 4H), 2.24 (p, J = 6.2 Hz, 2H). 13C NMR (101 MHz, DMSO): δ 164.73, 162.82, 130.22, 119.76, 114.84, 64.84, 28.26. HRMS (ESI): calcd for C17H20N4O2 [M + H]+, 313.1664; found, 313.1662.
4,4′-(Heptane-1,7-diylbis(oxy))dibenzimidamide/Heptamidine (10)
Following the procedure as described above for compound 3, using compound 5 (100 mg, 0.3 mmol), LHMDS (1.5 mL, 1 M THF solution, 5 equiv), and HCl (5 mL, 4 M dioxane solution, 67 equiv) afforded compound 10 (95.3 mg, 86%). 1H NMR (400 MHz, DMSO-d6): δ 9.13 (d, J = 17.8 Hz, 8H), 7.81 (d, J = 8.9 Hz, 4H), 7.14 (d, J = 9.0 Hz, 4H), 4.08 (t, J = 6.5 Hz, 4H), 1.81–1.69 (m, 4H), 1.49–1.36 (m, 6H). 13C NMR (101 MHz, DMSO): δ 164.80, 163.12, 130.21, 119.50, 114.82, 68.14, 28.51, 28.47, 25.43. HRMS (ESI): calcd for C21H28N4O2 [M + H]+, 369.2290; found, 369.2290.
4,4′-(Octane-1,8-diylbis(oxy))dibenzimidamide/Octamidine (11)
Following the procedure as described above for compound 3, using compound 6 (100 mg, 0.29 mmol). After LCMS analysis of the reaction mixture at 48 h, LHMDS (0.3 mL, 1 M THF solution, 1 equiv) was added, bringing the total of equivalents to 5. After an acidic quench with HCl (5 mL, 4 M dioxane solution, 69 equiv), the reaction mixture was stirred overnight. HPLC purification afforded product 11 (41 mg, 41%). 1H NMR (400 MHz, DMSO-d6): δ 8.95 (br, 8H), 7.78 (d, J = 8.9 Hz, 4H), 7.13 (d, J = 8.9 Hz, 4H), 4.06 (t, J = 6.5 Hz, 4H), 1.79–1.67 (m, 4H), 1.46–1.31 (m, 8H). 13C NMR (101 MHz, DMSO): δ 164.75, 163.19, 130.29, 119.49, 114.88, 68.22, 28.81, 28.55, 25.50. HRMS (ESI): calcd for C22H30N4O2 [M + H]+, 383.2447; found, 383.2446.
4,4′-(Undecane-1,11-diylbis(oxy))dibenzimidamide/Undecamidine (12)
Following the procedure as described above for compound 3, using compound 8 (98 mg, 0.25 mmol), LHMDS (1 mL, 1 M THF solution, 4 equiv), and HCl (2 mL, 4 M dioxane solution, 32 equiv) afforded product 12 (68 mg, 64%). 1H NMR (400 MHz, DMSO-d6): δ 8.96 (br, 8H), 7.79 (d, J = 8.8 Hz, 4H), 7.14 (d, J = 8.9 Hz, 4H), 4.07 (t, J = 6.5 Hz, 4H), 1.78–1.67 (m, 4H), 1.44–1.26 (m, 14H). 13C NMR (101 MHz, DMSO): δ 164.81, 163.12, 130.20, 119.48, 114.81, 68.15, 29.06, 29.02, 28.82, 28.52, 25.48. HRMS (ESI): calcd for C25H36N4O2 [M + H]+, 425.2916; found, 425.2919.
4-(2-Bromoethoxy)benzonitrile (13)
The protocol is as described in literature.69 1,2-dibromoethane (4.3 mL, 50 mmol, 5 equiv), 4-cyanophenol (1.2 g, 10 mmol), and K2CO3 (4.2 g, 30 mmol, 3 equiv) were suspended in dry DMF (20 mL) under an argon atmosphere. The mixture was stirred at 100 °C for 5 h, cooled to room temperature, and EtOAc and water were added. The organic layer was separated, washed with brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography (petroleum ether/EtOAc = 9:1) to afford compound 13 (0.97 g, 43%). 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 9.0 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 4.33 (t, J = 6.1 Hz, 2H), 3.66 (t, J = 6.1 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ 161.44, 134.24, 119.11, 115.44, 104.88, 68.08, 28.47.
4,4′-((Thiobis(ethane-2,1-diyl))bis(oxy))dibenzonitrile (14)
The protocol is as described in literature.32 Compound 13 (0.96 g, 4.3 mmol, 2 equiv) and Na2S·9H2O (0.51 g, 2.1 mmol) were dissolved in DMSO (5 mL), and the mixture was heated to 115 °C under an argon atmosphere. After 1 h, the mixture was poured into ice water (25 mL) and left for 24 h in a fridge. The precipitate was filtered, washed with cold water, and recrystallized from EtOH to obtain compound 14 (0.65 g, 93%). 1H NMR (400 MHz, CDCl3): δ 7.58 (d, J = 8.9 Hz, 4H), 6.94 (d, J = 8.9 Hz, 4H), 4.23 (t, J = 6.4 Hz, 4H), 3.05 (t, J = 6.4 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 161.73, 134.20, 119.15, 115.31, 104.61, 68.35, 31.71.
4,4′-((Thiobis(ethane-2,1-diyl))bis(oxy))dibenzimidamide (15)
Following the procedure as described above for compound 3, using compound 14 (100 mg, 0.31 mmol), LHMDS (1.55 mL, 1 M THF solution, 5 equiv) and quenched with HCl (5.2 mL, 4 M dioxane solution, 67 equiv) afforded the product 15 (71 mg, 64%). 1H NMR (400 MHz, DMSO-d6): δ 9.00 (s, 6H), 7.81 (d, J = 8.9 Hz, 4H), 7.18 (d, J = 8.9 Hz, 4H), 4.29 (t, J = 6.4 Hz, 4H), 3.04 (t, J = 6.4 Hz, 4H). 13C NMR (101 MHz, DMSO): δ 164.61, 162.61, 130.24, 119.80, 114.86, 68.01, 30.54. HRMS (ESI): calcd for C18H22N4O2S [M + H]+, 359.1541; found, 359.1541.
4,4′-((Sulfonylbis(ethane-2,1-diyl))bis(oxy))dibenzimidamide (16)
Compound 15 (100 mg, 0.22 mmol) was dissolved in dry dichloromethane (DCM) (10 mL) under an argon atmosphere. The solution was cooled to 0 °C using an ice bath and m-CPBA (54 mg, 77% aqueous solution, 1.1 equiv) was added. The mixture was stirred at 0 °C for 2 h and then concentrated in vacuo. After HPLC purification with a 0–100% gradient in 30 min to obtain compound 16 (27 mg, 32%). 1H NMR (400 MHz, DMSO-d6): δ 9.18 (s, 4H), 8.99 (s, 4H), 7.83 (d, J = 8.9 Hz, 4H), 7.21 (d, J = 9.0 Hz, 4H), 4.52 (t, J = 5.5 Hz, 4H), 3.79 (t, J = 5.5 Hz, 4H). 13C NMR (101 MHz, DMSO): δ 164.65, 162.00, 130.29, 120.39, 114.94, 62.19, 53.38. HRMS (ESI): calcd for C18H22N4O4S [M + H]+, 391.1441; found, 391.1434.
4,4′-((1,2-Phenylenebis(methylene))bis(oxy))dibenzonitrile (17)
Following the procedure as described above for compound 4, using 1,2-bis(bromomethyl)benzene (1.0 g, 3.8 mmol) afforded the title compound as a crude product. No precipitation occurred upon the addition of water. Therefore, the mixture was concentrated under reduced pressure and the crude product was purified by column chromatography (petroleum ether/EtOAc = 19:1) to obtain compound 17 (1.2 g, 96%). 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 8.0 Hz, 4H), 7.46 (dd, 4H), 6.99 (d, J = 8.0 Hz, 4H), 5.21 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 161.76, 134.26, 134.12, 129.56, 129.27, 119.12, 115.57, 104.79, 68.46.
4,4′-((1,3-Phenylenebis(methylene))bis(oxy))dibenzonitrile (18)
Following the procedure as described above for compound 4, using 1,3-bis(bromomethyl)benzene (0.92 g, 3.5 mmol) afforded compound 18 (0.94 g, 79%). 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 8.8 Hz, 4H), 7.50–7.37 (m, 4H), 7.02 (d, J = 8.8 Hz, 4H), 5.13 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 161.92, 136.51, 134.19, 129.37, 127.60, 126.52, 119.21, 115.66, 104.54, 70.09.
4,4′-((1,4-Phenylenebis(methylene))bis(oxy))dibenzonitrile (19)
Following the procedure as described above for compound 4, using 1,4-bis(bromomethyl)benzene (0.92 g, 3.5 mmol) afforded compound 19 as a crude product. The crude product was not recrystallized due to insolubility issues and was used in the next step without further purification based on a purity assessment (NMR) (1.2 g, 97%). 1H NMR (400 MHz, DMSO-d6): δ 7.78 (d, J = 8.8 Hz, 4H), 7.48 (s, 4H), 7.18 (d, J = 8.9 Hz, 4H), 5.22 (s, 4H). 13C NMR (101 MHz, DMSO): δ 161.74, 136.11, 134.24, 128.08, 119.13, 115.92, 103.05, 69.36.
4,4′-((2-Benzylpropane-1,3-diyl)bis(oxy))dibenzonitrile (20)
Following the procedure as described above for compound 4, using 2,7-bis(bromomethyl)naphthalene (0.20 g, 0.64 mmol) afforded compound 20 as a crude product. The crude product was not recrystallized due to insolubility issues and was used in the next step without further purification based on a purity assessment (NMR) (0.25 g, quant). 1H NMR (400 MHz, CDCl3): δ 7.90 (d, J = 8.5 Hz, 2H), 7.87 (s, 2H), 7.60 (d, J = 8.9 Hz, 4H), 7.54 (dd, J = 8.5, 1.7 Hz, 2H), 7.06 (d, J = 8.9 Hz, 4H), 5.29 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 134.23, 134.04, 133.08, 128.74, 126.61, 125.65, 119.26, 115.77, 104.56, 70.42.
4,4′-((1,2-Phenylenebis(methylene))bis(oxy))dibenzimidamide (21)
Following the procedure as described above for compound 3, using compound 17 (102 mg, 0.3 mmol), LHMDS (1.5 mL, 1 M THF solution, 5 equiv), and HCl (5.0 mL, 4 M dioxane solution, 67 equiv) afforded product 21 (63 mg, 56%). 1H NMR (400 MHz, DMSO-d6): δ 9.14 (s, 4H), 9.04 (s, 4H), 7.81 (d, J = 8.9 Hz, 4H), 7.55 (dd, J = 5.6, 3.4 Hz, 2H), 7.40 (dd, J = 5.7, 3.3 Hz, 2H), 7.24 (d, J = 9.0 Hz, 4H), 5.38 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.70, 162.45, 134.57, 130.19, 128.82, 128.44, 120.04, 115.18, 67.51. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1821.
4,4′-((1,3-Phenylenebis(methylene))bis(oxy))dibenzimidamide (22)
Following the procedure as described above for compound 3, using compound 18 (100 mg, 0.29 mmol), LHMDS (2.35 mL, 1 M THF solution, 8 equiv), and HCl (4.35 mL, 4 M dioxane solution, 60 equiv) afforded product 22 (91 mg, 83%). 1H NMR (400 MHz, DMSO-d6): δ 9.13 (s, 4H), 8.85 (s, 4H), 7.80 (d, J = 8.9 Hz, 4H), 7.56 (s, 1H), 7.44 (d, J = 1.3 Hz, 3H), 7.23 (d, J = 9.0 Hz, 4H), 5.24 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.60, 162.61, 136.67, 130.24, 127.66, 127.19, 119.90, 115.15, 69.54. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1821.
4,4′-((1,4-Phenylenebis(methylene))bis(oxy))dibenzimidamide (23)
Following the procedure as described above for compound 3, using compound 19 (102 mg, 0.3 mmol), LHMDS (1.5 mL, 1 M THF solution, 5 equiv), and HCl (5 mL, 4 M dioxane solution, 67 equiv) afforded product 23 (21 mg, 19%). 1H NMR (400 MHz, DMSO-d6): δ 9.15 (s, 4H), 9.04 (s, 4H), 7.81 (d, J = 8.9 Hz, 4H), 7.50 (s, 4H), 7.23 (d, J = 9.0 Hz, 4H), 5.25 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.71, 162.55, 136.21, 130.20, 128.05, 119.93, 115.19, 69.34. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1820.
4,4′-((Naphthalene-2,7-diylbis(methylene))bis(oxy))dibenzimidamide (24)
Following the procedure as described above for compound 3, using compound 20 (117 mg, 0.3 mmol) and LHMDS (1.5 mL, 1 M THF solution, 5 equiv). After LCMS analysis of the reaction mixture at 48 h, LHMDS (0.5 mL, 1 M THF solution, 1.7 equiv) was added. The reaction was quenched using HCl (6 mL, 4 M dioxane solution, 80 equiv). Compound 24 was obtained in a 26% yield (33 mg). 1H NMR (400 MHz, DMSO-d6): δ 9.14 (s, 4H), 9.07 (s, 4H), 8.04–7.95 (m, 4H), 7.82 (d, J = 9.0 Hz, 4H), 7.61 (dd, J = 8.4, 1.7 Hz, 2H), 7.29 (d, J = 9.0 Hz, 4H), 5.42 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.74, 162.58, 134.48, 132.52, 132.25, 130.21, 128.15, 126.59, 126.04, 119.98, 115.27, 69.68. HRMS (ESI): calcd for C26H24N4O2 [M + H]+, 425.1977; found, 425.1977.
3,3′-(Pentane-1,5-diylbis(oxy))dibenzonitrile (25)
Following the procedure as described above for compound 4, using 1,5-dibromopentane (0.48 mL, 3.5 mmol) and 3-cyanophenol (1 g, 8.4 mmol) afforded compound 25 (0.68 g, 63%). 1H NMR (400 MHz, CDCl3): δ 7.39–7.34 (m, 2H), 7.25–7.21 (m, 2H), 7.15–7.10 (m, 4H), 4.00 (t, J = 6.3 Hz, 4H), 1.93–1.83 (m, 4H), 1.72–1.61 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 159.19, 130.48, 124.59, 119.92, 118.91, 117.46, 113.32, 68.20, 28.90, 22.79.
3,3′-((1,2-Phenylenebis(methylene))bis(oxy))dibenzonitrile (26)
Following the procedure as described above for compound 4, using 1,2-bis(bromomethyl)benzene (1.0 g, 3.8 mmol) and 3-cyanophenol (1.1 g, 9.1 mmol, 2.4 equiv) afforded the title compound as a crude product. The crude product did not precipitate but had very high viscosity. During filtration, a minimal amount of acetone was used to prevent clogging. The precipitate was collected and the filtrate was concentrated under reduced pressure to evaporate the acetone. The precipitate in aqueous solution was filtered again with a minimal amount of acetone. This process was repeated three times to obtain compound 26 (1.1 g, 85%). 1H NMR (400 MHz, CDCl3): δ 7.53–7.47 (m, 2H), 7.45–7.35 (m, 4H), 7.28–7.27 (m, 1H), 7.26–7.25 (m, 1H), 7.20–7.16 (m, 4H), 5.18 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 158.65, 134.27, 130.67, 129.54, 129.22, 125.21, 120.18, 118.71, 117.74, 113.49, 68.55.
3,3′-((1,3-Phenylenebis(methylene))bis(oxy))dibenzonitrile (27)
Following the procedure as described above for compound 4, using 1,3-bis(bromomethyl)benzene (0.92 g, 3.5 mmol) and 3-cyanophenol (1.0 g, 8.4 mmol, 2.4 equiv) afforded compound 27 as a crude product. The crude product was not recrystallized due to insolubility issues and was used in the next step without further purification based on a purity assessment (NMR) (1.2 g, quant). 1H NMR (400 MHz, CDCl3): δ 7.51–7.34 (m, 6H), 7.28–7.24 (m, 2H), 7.23–7.17 (m, 4H), 5.11 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 158.73, 136.65, 130.56, 129.34, 127.48, 126.41, 125.02, 120.22, 118.77, 117.91, 113.38, 70.14.
3,3′-((1,4-Phenylenebis(methylene))bis(oxy))dibenzonitrile (28)
Following the procedure as described above for compound 4, using 1,4-bis(bromomethyl)benzene (0.9 g, 4 mmol) and 3-cyanophenol (1.1 g, 9.6 mmol, 2.4 equiv) produced compound 28 (1.3 g, 97%). 1H NMR (400 MHz, CDCl3): δ 7.46 (s, 4H), 7.41–7.35 (m, 2H), 7.29–7.24 (m, 3H), 7.20 (m, 4H), 5.10 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 158.78, 136.21, 130.58, 127.96, 125.03, 120.27, 118.80, 117.93, 113.42, 70.08.
3,3′-(Pentane-1,5-diylbis(oxy))dibenzimidamide (1b)
Following the procedure as described above for compound 3, using compound 25 (92 mg, 0.3 mmol). LHMDS (1.5 mL, 1 M THF solution, 5 equiv) was added, and after LCMS analysis of the reaction mixture at 48 h, LHMDS (3.0 mL, 1 M THF solution, 10 equiv) was additionally added. A quench with HCl (5.0 mL, 4 M dioxane solution, 67 equiv), afforded the crude product. The crude product was purified using HPLC affording compound 1b (93 mg, 91%). 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 4H), 9.32 (s, 4H), 7.52 (t, J = 8.1 Hz, 2H), 7.38 (d, J = 6.6 Hz, 4H), 7.29 (d, J = 8.8 Hz, 2H), 4.09 (t, J = 6.3 Hz, 4H), 1.90–1.74 (m, 4H), 1.68–1.52 (m, 2H). 13C NMR (101 MHz, DMSO): δ 165.55, 158.72, 130.35, 129.48, 119.92, 113.80, 67.87, 28.29, 22.22. HRMS (ESI): calcd for C19H24N4O2 [M + H]+, 341.1977; found, 341.1977.
3,3′-((1,2-Phenylenebis(methylene))bis(oxy))dibenzimidamide (21b)
Following the procedure as described above for compound 3, using compound 26 (102 mg, 0.3 mmol). LHMDS (1.5 mL, 1 M THF solution, 5 equiv) was added, and after LCMS analysis of the reaction mixture at 48 h, LHMDS (3.0 mL, 1 M THF solution, 10 equiv) was additionally added. A quench with HCl (5.0 mL, 4 M dioxane solution, 67 equiv), afforded the crude product. The crude product was purified using HPLC affording compound 21b (80.4 mg, 72%). 1H NMR (400 MHz, DMSO-d6): δ 9.45 (s, 4H), 9.33 (s, 4H), 7.59–7.54 (m, 2H), 7.53 (s, 1H), 7.52–7.46 (m, 3H), 7.45–7.35 (m, 6H), 5.34 (s, 4H). 13C NMR (101 MHz, DMSO): δ 165.43, 158.25, 134.76, 130.37, 129.52, 128.84, 128.44, 120.46, 119.94, 114.63, 67.52. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1821.
3,3′-((1,3-Phenylenebis(methylene))bis(oxy))dibenzimidamide (22b)
Following the procedure as described above for compound 3, using compound 27 (102 mg, 0.3 mmol). LHMDS (1.5 mL, 1 M THF solution, 5 equiv) was added, and after LCMS analysis of the reaction mixture at 48 h, LHMDS (2.0 mL, 1 M THF solution, 6.7 equiv) was additionally added. A quench with HCl (5.0 mL, 4 M dioxane solution, 67 equiv), afforded the crude product. The crude product was purified using HPLC affording compound 22b (88 mg, 78%). 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 4H), 9.34 (s, 4H), 7.62–7.52 (m, 3H), 7.50 (s, 2H), 7.47 (s, 3H), 7.44–7.36 (m, 4H), 5.23 (s, 4H). 13C NMR (101 MHz, DMSO): δ 165.44, 158.38, 136.84, 130.41, 129.52, 128.84, 127.59, 127.13, 120.41, 120.03, 114.43, 69.59. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1821.
3,3′-[1,4-Phenylenebis(methyleneoxy)]dibenzenecarboximidamide (23b)
Following the procedure as described above for compound 3, using compound 28 (102 mg, 0.3 mmol), LHMDS (2.4 mL, 1 M THF solution, 8 equiv), and HCl (4.5 mL, 4 M dioxane solution, 60 equiv) produced compound 23b (114 mg, quant). 1H NMR (400 MHz, DMSO-d6): δ 9.30 (d, J = 19.8 Hz, 8H), 7.54–7.38 (m, 8H), 7.38–7.26 (m, 4H), 5.16 (s, 4H). 13C NMR (101 MHz, DMSO): δ 165.40, 158.35, 136.33, 130.41, 129.51, 128.04, 120.40, 120.03, 114.47, 69.42. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1818.
2,2′-(Pentane-1,5-diylbis(oxy))dibenzonitrile (29)
Following the procedure as described above for compound 4, using 1,5-dibromopentane (0.54 mL, 4 mmol) and 2-cyanophenol (1.14 g, 9.6 mmol, 2.4 equiv) afforded compound 29 (1.20 g, 97%). 1H NMR (400 MHz, CDCl3): δ 7.61–7.45 (m, 4H), 7.04–6.92 (m, 4H), 4.12 (t, J = 6.2 Hz, 4H), 1.95 (m, 4H), 1.85–1.69 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 160.85, 134.49, 133.85, 120.77, 116.70, 112.37, 102.08, 68.88, 28.58, 22.70.
2,2′-((1,2-Phenylenebis(methylene))bis(oxy))dibenzonitrile (30)
Following the procedure as described above for compound 4, using 1,2-bis(bromomethyl)benzene (0.53 g, 2 mmol) and 2-cyanophenol (0.57 g, 4.8 mmol, 2.4 equiv) afforded compound 30 (0.56 g, 83%). 1H NMR (400 MHz, CDCl3): δ 7.62–7.46 (m, 6H), 7.45–7.36 (m, 2H), 7.15 (d, J = 8.4 Hz, 2H), 6.99 (td, J = 7.6, 0.9 Hz, 2H), 5.39 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 160.13, 134.72, 133.97, 133.87, 129.30, 129.05, 121.32, 116.66, 112.91, 102.09, 69.45.
2,2′-((1,3-Phenylenebis(methylene))bis(oxy))dibenzonitrile (31)
Following the procedure as described above for compound 4, using 1,3-bis(bromomethyl)benzene (0.53 g, 2 mmol) and 2-cyanophenol (0.57 g, 4.8 mmol, 2.4 equiv) afforded compound 31 (0.60 g, 88%). 1H NMR (400 MHz, CDCl3): δ 7.58 (dd, J = 8.1, 1.7 Hz, 2H), 7.55–7.48 (m, 3H), 7.44 (m, 3H), 7.05–6.96 (m, 4H), 5.23 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 160.29, 136.37, 134.51, 134.00, 129.40, 127.02, 125.20, 121.31, 116.57, 111.95, 102.53, 77.48, 77.16, 76.84, 70.49.
2,2′-((1,4-Phenylenebis(methylene))bis(oxy))dibenzonitrile (32)
Following the procedure as described above for compound 4, using 1,4-bis(bromomethyl)benzene (0.92 g, 3.5 mmol), 2-cyanophenol (1.1 g, 9.6 mmol, 2.6 equiv), and NaH (0.38 g, 60% dispersion in mineral oil, 2.6 equiv) afforded compound 32 (1.2 g, 99%). 1H NMR (400 MHz, CDCl3): δ 7.59 (dd, J = 7.6, 1.7 Hz, 2H), 7.55–7.44 (m, 6H), 7.08–6.95 (m, 4H), 5.22 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 160.31, 135.86, 134.46, 134.04, 127.49, 121.30, 116.55, 112.99, 102.58, 70.37.
2,2′-(Pentane-1,5-diylbis(oxy))dibenzimidamide (1c)
These conditions were based on literature protocols.42 To a suspension of compound 29 (190 mg, 0.62 mmol) and DIPEA (0.56 mL, 3.2 mmol, 5 equiv) in EtOH (10 mL) was added NH2OH·HCl (208 mg, 3 mmol, 4.8 equiv). The reaction mixture was stirred at 85 °C overnight. The mixture was concentrated in vacuo and the residue was dissolved in AcOH (4.2 mL) and Ac2O (0.29 mL, 3 mmol, 4.8 equiv) was added. The reaction mixture was stirred for 4 h and then concentrated in vacuo. The residue was co-evaporated with toluene three times and then suspended in AcOH (7.5 mL) under an argon atmosphere. Zinc powder (60 mg, 0.92 mmol, 1.5 equiv) was added and the mixture was stirred at 35 °C overnight. Upon completion, the reaction mixture was filtered through Celite, the Celite was rinsed with acetone and all collected fractions were concentrated in vacuo. The crude product purified by preparative HPLC (gradient 20–100%, 30 min) to afford final compound 1c (102 mg, 48%). 1H NMR (400 MHz, DMSO-d6): δ 9.32 (s, 4H), 9.12 (s, 4H), 7.60 (t, J = 7.9 Hz, 2H), 7.51 (d, J = 7.5 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.11 (t, J = 7.5 Hz, 2H), 4.09 (t, J = 6.4 Hz, 4H), 1.81 (p, J = 6.9 Hz, 4H), 1.56 (p, J = 7.6 Hz, 2H). 13C NMR (101 MHz, DMSO): δ 164.64, 156.10, 133.82, 129.53, 120.35, 118.55, 113.07, 68.28, 28.01, 21.76. HRMS (ESI): calcd for C19H24N4O2 [M + H]+, 341.1977; found, 341.1972.
2,2′-((1,2-Phenylenebis(methylene))bis(oxy))dibenzimidamide (21c)
Following the procedure as described above for compound 1c, using compound 30 (211 mg, 0.62 mmol) afforded compound 21c (43 mg, 18%). 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 4H), 9.24 (s, 4H), 7.67–7.58 (m, 4H), 7.55 (dd, J = 7.6, 1.7 Hz, 2H), 7.44–7.31 (m, 4H), 7.15 (td, J = 7.5, 0.8 Hz, 2H), 5.35 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.75, 155.41, 134.33, 133.66, 129.61, 128.31, 128.22, 120.79, 119.07, 113.41, 67.37. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1815.
2,2′-((1,3-Phenylenebis(methylene))bis(oxy))dibenzimidamide (22c)
Following the procedure as described above for compound 1c, using compound 31 (210 mg, 0.62 mmol) afforded compound 22c (49 mg, 21%). 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 4H), 9.22 (s, 4H), 7.62 (ddd, J = 8.8, 7.4, 1.7 Hz, 2H), 7.58–7.42 (m, 6H), 7.33 (d, J = 8.4 Hz, 2H), 7.15 (td, J = 7.6, 0.8 Hz, 2H), 5.23 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.69, 155.69, 136.70, 133.76, 129.65, 128.80, 127.31, 126.80, 120.77, 118.98, 113.46, 69.95. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1816.
2,2′-((1,4-Phenylenebis(methylene))bis(oxy))dibenzimidamide (23c)
Following the procedure as described above for compound 1c, using compound 32 (211 mg, 0.62 mmol) afforded compound 23c (27 mg, 12%). 1H NMR (400 MHz, DMSO-d6): δ 9.33 (s, 4H), 9.21 (s, 4H), 7.62 (ddd, J = 8.8, 7.4, 1.8 Hz, 2H), 7.57–7.49 (m, 6H), 7.33 (d, J = 8.8 Hz, 2H), 7.14 (td, J = 7.6, 0.9 Hz, 2H), 5.23 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.62, 155.66, 136.23, 133.75, 129.65, 127.79, 120.72, 118.92, 113.39, 69.75. HRMS (ESI): calcd for C22H22N4O2 [M + H]+, 375.1821; found, 375.1816.
(5-Bromo-1,3-phenylene)dimethanol (33)
The protocol is as described in literature.44 Dimethyl 5-bromoisophthalate (2.3 g, 8.3 mmol) was dissolved in dry DCM (25 mL) under an argon atmosphere. The solution was then cooled to 0 °C using an ice bath and DIBALH (40 mL, 1 M hexane solution, 4.8 equiv) was added dropwise. The mixture was stirred from 0 °C to room temperature for 1 h. The reaction was quenched with Rochelle salt (60 mL, sat. aq) and the biphasic mixture was stirred at room temperature overnight. The layers were separated and the aqueous layer was two times extracted with diethyl ether. The organic layers were combined, washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified using column chromatography (DCM/EtOAc = 1:1) and afforded compound 33 (1.8 g, 96%). 1H NMR (400 MHz, MeOD): δ 7.42 (s, 2H), 7.28 (s, 1H), 4.58 (s, 4H), 3.35 (s, 2H). 13C NMR (101 MHz, MeOD): δ 145.51, 129.42, 124.82, 123.31, 64.29.
1-Bromo-3,5-bis(bromomethyl)benzene (34)
The protocol is as described in literature.45 To a solution of compound 33 (1.0 g, 4.6 mmol) in dry DCM (50 mL) was added PPh3 (2.5 g, 9.7 mmol, 2.1 equiv) and CBr4 (3.2 g, 9.7 mmol, 2.1 equiv), and the mixture was stirred at room temperature for 2 h under an argon atmosphere. The reaction was quenched with water (30 mL) and the product was extracted from the aqueous layer with DCM three times. The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography (petroleum ether 100%) to give compound 34 (0.87 g, 55%). 1H NMR (400 MHz, CDCl3): δ 7.51–7.45 (m, 2H), 7.34 (s, 1H), 4.53 (d, J = 4.0 Hz, 1H), 4.41 (d, J = 4.2 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 140.42, 140.11, 132.11, 132.09, 131.64, 128.40, 127.90, 122.83, 44.89, 31.64, 31.59.
4,4′-(((5-Bromo-1,3-phenylene)bis(methylene))bis(oxy))dibenzonitrile (35)
Following the procedure as described above for compound 4, using compound 34 (0.82 g, 2.4 mmol) afforded compound 35 as a crude product. The crude product was not recrystallized due to insolubility issues and was used in the next step without further purification based on a purity assessment (NMR) (1.0 g, quant). 1H NMR (400 MHz, CDCl3): δ 7.66–7.57 (m, 4H), 7.57–7.53 (m, 2H), 7.43–7.33 (m, 1H), 7.04–6.96 (m, 4H), 5.15–5.05 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 161.57, 138.62, 134.25, 130.33, 124.72, 123.30, 119.09, 115.62, 104.87, 69.18.
4,4′-(([1,1′-Biphenyl]-3,5-diylbis(methylene))bis(oxy))dibenzonitrile (36)
Conditions were based on protocols described in literature.47,48 Dibenzonitrile intermediate 35 (0.30 g, 0.72 mmol) was dissolved in a 3:1 mixture of THF and 2 M Na2CO3 (aq) of 8 mL, respectively. Phenylboronic acid (0.13 g, 1.1 mmol, 1.5 equiv) and Pd(dppf)Cl2·DCM (58 mg, 0.07 mmol, 0.1 equiv) were added. The reaction mixture was heated to 65 °C for 18 h and then partitioned between DCM and NaHCO3 (sat. aq). The aqueous layer was three times extracted with DCM, the organic layers were combined and dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified using column chromatography (petroleum ether/EtOAc = 4:1) to obtain compound 36 (0.28 g, 94%). 1H NMR (400 MHz, CDCl3): δ 7.66–7.57 (m, 8H), 7.50–7.36 (m, 4H), 7.05 (d, J = 8.8 Hz, 4H), 5.19 (s, 4H). 13C NMR (101 MHz, CDCl3): δ 161.90, 142.59, 140.22, 137.05, 134.21, 129.06, 128.03, 127.31, 126.40, 125.32, 119.19, 115.67, 104.57, 70.12.
4,4′-(((5-Bromo-1,3-phenylene)bis(methylene))bis(oxy))dibenzimidamide (37)
Following the procedure as described above for compound 3, using compound 35 (126 mg, 0.3 mmol), LHMDS (3.0 mL, 1 M THF solution, 10 equiv), and HCl (10 mL, 4 M dioxane solution, 133 equiv) afforded product 37 (23 mg, 17%). 1H NMR (400 MHz, DMSO-d6): δ 9.17 (s, 3H), 9.09 (s, 3H), 7.83 (d, J = 8.9 Hz, 4H), 7.67 (d, J = 1.1 Hz, 2H), 7.58 (s, 1H), 7.24 (d, J = 9.0 Hz, 4H), 5.27 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.78, 162.35, 139.43, 130.28, 130.05, 125.93, 121.85, 120.20, 115.20, 68.57. HRMS (ESI): calcd for C22H21BrN4O2 [M + H]+, 453.0926; found, 453.0924.
4,4′-(([1,1′-Biphenyl]-3,5-diylbis(methylene))bis(oxy))dibenzimidamide (38)
Following the procedure as described above for compound 3, using compound 36 (0.28 g, 0.67 mmol), LHMDS (5.4 mL, 1 M THF solution, 8 equiv), and HCl (10 mL, 4 M dioxane solution, 60 equiv). HPLC purification using a 30–100% gradient for 30 min afforded compound 38 (0.23 g, 74%). 1H NMR (400 MHz, DMSO-d6): δ 9.15 (d, J = 15.3 Hz, 8H), 7.83 (d, J = 9.0 Hz, 4H), 7.76 (s, 2H), 7.69 (d, J = 7.2 Hz, 2H), 7.58 (s, 1H), 7.50 (t, J = 7.6 Hz, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.27 (d, J = 9.0 Hz, 4H), 5.33 (s, 4H). 13C NMR (101 MHz, DMSO): δ 164.83, 162.61, 140.77, 139.54, 137.54, 130.26, 129.14, 127.94, 126.84, 126.19, 126.03, 120.04, 115.25, 69.51. HRMS (ESI): calcd for C28H26N4O2 [M + H]+, 451.2135; found, 451.2130.
(4-Bromo-1,2-phenylene)dimethanol (39)
Conditions were based on a protocol reported in literature.49 LAH (15 mL, 1 M THF solution, 2 equiv) and ZnCl2 (0.61 g, 4.5 mmol, 0.6 equiv) were suspended in dry THF (30 mL) and cooled to 0 °C, then 4-bromophthalic anhydride (1.7 g, 7.5 mmol) was slowly added. The mixture was stirred at room temperature for 6 h under an argon atmosphere. The mixture was cooled to 0 °C and quenched with Rochelle salt (30 mL, sat. aq) and the biphasic mixture was stirred at room temperature overnight. The layers were separated and the aqueous layer was extracted with diethyl ether two times and the combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography (DCM/EtOAc = 1:1) to give compound 39 (1.5 g, 95%). 1H NMR (400 MHz, CDCl3): δ 7.48 (d, J = 2.1 Hz, 1H), 7.42 (dd, J = 8.0, 2.1 Hz, 1H), 7.18 (d, J = 8.0 Hz, 1H), 4.62 (d, J = 2.6 Hz, 4H), 3.20 (s, 2H). 13C NMR (101 MHz, CDCl3): δ 141.49, 138.18, 129.88, 128.77, 127.92, 122.30, 64.53, 64.40, 64.31, 63.49, 63.47, 31.08, 23.80.
4-Bromo-1,2-bis(bromomethyl)benzene (40)
Following the procedure described for compound 34, using compound 39 (1.5 g, 7.0 mmol) as a starting material afforded compound 40 (1.8 g, 74%). 1H NMR (400 MHz, CDCl3): δ 7.52 (d, J = 2.1 Hz, 1H), 7.43 (dd, J = 8.2, 2.1 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H), 4.59 (s, 2H), 4.58 (s, 2H). 13C NMR (101 MHz, CDCl3): δ 138.65, 135.67, 134.02, 132.69, 132.58, 131.24, 129.60, 123.17, 66.00, 42.46, 42.32, 30.14, 29.32, 29.12, 29.00, 28.83, 15.43.
4,4′-(((4-Bromo-1,2-phenylene)bis(methylene))bis(oxy))dibenzonitrile (41)
Following the procedure as described above for compound 4, using compound 40 (0.80 g, 2.3 mmol) afforded compound 41 as a crude product. The crude product was not recrystallized due to insolubility issues and was used in the next step without further purification (1.1 g, quant). 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 2.1 Hz, 1H), 7.62–7.57 (m, 4H), 7.54 (dd, J = 8.1, 2.1 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 6.99 (dd, J = 8.9, 4.6 Hz, 4H), 5.15 (d, J = 6.0 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 161.46, 136.30, 134.29, 134.27, 132.83, 132.15, 132.10, 131.05, 129.52, 129.23, 123.21, 118.99, 115.52, 105.06, 104.99, 67.83, 67.51.
4,4′-(([1,1′-Biphenyl]-3,4-diylbis(methylene))bis(oxy))dibenzonitrile (42)
Following the procedure as described above for compound 36, using compound 41 (0.33 g, 0.79 mmol), a 3:1 mixture of THF and 2 M Na2CO3 (aq) (8.0 mL), phenylboronic acid (0.13 g, 1.1 mmol, 1.5 equiv), and Pd(dppf)Cl2·DCM (58 mg, 0.07 mmol, 0.1 equiv) afforded compound 42 (0.28 g, 84%). 1H NMR (400 MHz, CDCl3): δ 7.72 (s, 1H), 7.66–7.57 (m, 8H), 7.49–7.44 (m, 2H), 7.41–7.36 (m, 1H), 7.04–7.00 (m, 4H), 5.25 (d, J = 7.1 Hz, 4H). 13C NMR (101 MHz, CDCl3): δ 161.76, 142.35, 140.07, 134.61, 134.27, 132.94, 130.17, 129.54, 129.26, 129.09, 128.37, 128.04, 127.82, 127.27, 119.11, 115.58, 104.84, 104.80, 68.55, 68.27.
4,4′-(((4-Bromo-1,2-phenylene)bis(methylene))bis(oxy))dibenzimidamide (43)
Following the procedure as described above for compound 1c, using compound 41 (172 mg, 0.41 mmol) afforded compound 43 (72 mg, 39%). 1H NMR (400 MHz, DMSO-d6): δ 9.14 (d, J = 18.5 Hz, 8H), 7.82 (dd, J = 9.0, 3.2 Hz, 4H), 7.77 (d, J = 2.1 Hz, 1H), 7.62 (dd, J = 8.2, 2.1 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.33–7.20 (m, 4H), 5.37 (d, J = 10.1 Hz, 4H). 13C NMR (101 MHz, DMSO-d6): δ 164.76, 164.74, 162.24, 162.17, 137.22, 133.96, 131.16, 131.01, 130.76, 130.25, 130.21, 121.48, 120.32, 120.23, 115.22, 66.83, 66.59, 40.15, 39.94, 39.73, 39.52, 39.31, 39.10, 38.89. HRMS (ESI): calcd for C22H21BrN4O2 [M + H]+, 453.0926; found, 453.0923.
4,4′-(([1,1′-Biphenyl]-3,4-diylbis(methylene))bis(oxy))dibenzimidamide (44)
Following the procedure as described above for compound 3, using compound 42 (0.28 g, 0.66 mmol), LHMDS (5.4 mL, 1 M THF solution, 8 equiv), and HCl (10 mL, 4 M dioxane solution, 60 equiv) afforded the crude product. The crude product was purified using HPLC with a 30–100% gradient for 30 min to obtain pure compound 44 (35 mg, 12%). 1H NMR (400 MHz, DMSO-d6): δ 9.14 (d, J = 5.2 Hz, 4H), 9.00 (d, J = 4.8 Hz, 4H), 7.88 (d, J = 2.0 Hz, 1H), 7.81 (dd, J = 8.9, 3.2 Hz, 4H), 7.72–7.61 (m, 4H), 7.48 (t, J = 7.6 Hz, 2H), 7.39 (t, J = 7.3 Hz, 1H), 7.28 (t, J = 8.6 Hz, 4H), 5.44 (d, J = 10.4 Hz, 4H). 13C NMR (101 MHz, DMSO): δ 164.72, 162.49, 162.45, 140.20, 139.34, 135.20, 133.89, 130.25, 129.56, 129.11, 127.37, 126.75, 120.10, 120.08, 115.26, 115.23, 67.64, 67.31. HRMS (ESI): calcd for C28H26N4O2 [M + H]+, 451.2135; found, 451.2129.
Antimicrobial Assays
All compounds were screened for antimicrobial activity against E. coli BW25113. A select group of the pentamidine analogues was further tested against E. coli ATCC25922, E. coli W3110, E. coli 552060.1, and E. coli BW25113 transformed with pGDP2-mcr-1 (the plasmid was a gift from Gerard Wright (Addgene plasmid # 118404; http://www.n2t.net/addgene:118404; RRID: Addgene_118404)63), E. coli mcr-1, E. coli EQASmcr-1 (EQAS 2016 412016126), E. coli EQASmcr-2 (EQAS 2016 KP37), E. coli EQASmcr-3 (EQAS 2017 2013-SQ352), E. coli RC0089, K. pneumoniae ATCC13883, P. aeruginosa ATCC27853, and A. baumannii ATCC17978. The antimicrobial assay was performed according to CLSI guidelines. Bacteria were plated out directly from their glycerol stocks on blood agar plates, incubated overnight at 37 °C, and then kept in the fridge. The blood agar plates were only used for 2 weeks and then replaced.
MIC Assays
A single colony from a blood agar plate was inoculated in Lysogeny Broth (LB) at 37 °C until a 0.5 optical density at 600 nm (OD600) was reached (compared to the sterility control of LB). The bacterial suspension was diluted in fresh LB to 2.0 × 106 CFU/mL. The serial dilutions were prepared in polypropylene microtiter plates: a stock of the test compounds was prepared with a 2× final concentration in LB. 100 μL of the stock was added to the wells of the top row of which 50 μL was used for the serial dilution. The bottom row of each plate was used as the positive (50 μL of LB) and negative controls (100 μL of LB) (6 wells each). 50 μL of the 2.0 × 106 CFU/mL bacterial stock was added to each well except for the negative controls, adding up to a total volume of 100 μL per well. The plates were sealed with a breathable seal and incubated for 20 h at 37 °C and 600 rpm. The MIC was visually determined after centrifuging the plates for 2 min at 3000 rpm.
Checkerboard Assays
Dilution series of both the test compound and antibiotic to be evaluated was prepared in LB media. To evaluate synergy, 25 μL of the test compound solutions were added to wells containing 25 μL of the antibiotic solution. This was replicated in three columns for each combination so as to obtain triplicates. To the resulting 50 μL volume of the antibiotic + test compound was next added 50 μL of the bacterial stock (see MIC Assays), and the plates were sealed. After incubation for 20 h at 37 °C while shaking at 600 rpm, the breathable seals were removed, and the plates shaken using a bench top shaker to ensure an even suspension of the bacterial cells as established by visual inspection. The plates were then transferred to a Tecan Spark plate reader and following another brief shaking (20 s), the density of the bacterial suspensions was measured at 600 nm (OD600). The resulting OD600 values were transformed into a 2D gradient to visualize the growth/no-growth results. The FICI was calculated using eq 1, with an FICI ≤ 0.5 indicating synergy.21
| 1 |
Equation 1 calculation of FICI. MSCant = MIC of antibiotic in combination with synergist; MICant = MIC of antibiotic alone; MSCsyn = MIC of synergist in combination with antibiotic; and MICsyn = MIC of synergist alone. In the cases, where the MIC of the antibiotic or synergist was found to exceed the highest concentration tested, the next highest concentration in the dilution series was used in determining the FICI, and the result reported as ≤ the calculated value.
Hemolysis Assays
The hemolytic activity of each analogue was assessed in triplicate. Red blood cells from defibrinated sheep blood obtained from Thermo Fisher were centrifuged (400 g for 15 min at 4 °C) and washed five times with phosphate-buffered saline containing 0.002% Tween 20 (buffer). Then, the red blood cells were normalized to obtain a positive control read-out between 2.5 and 3.0 at 415 nm to stay within the linear range with the maximum sensitivity. A serial dilution of the compounds (200–6.25 μg/mL, 75 μL) was prepared in a 96-well plate. The outer border of the plate was filled with 75 μL buffer. Each plate contained a positive control (0.1% Triton-X final concentration, 75 μL) and a negative control (buffer, 75 μL) in triplicate. The normalized blood cells (75 μL) were added, and the plates were incubated at 37 °C for 1 or 20 h while shaking at 500 rpm. A flat-bottom plate of polystyrene with 100 μL of buffer in each well was prepared. After incubation, the plates were centrifuged (800g for 5 min at room temperature) and 25 μL of the supernatant was transferred to their respective wells in the flat-bottom plate. The values obtained from a read out at 415 nm were corrected for the background (negative control) and transformed into a percentage relative to the positive control.
Membrane Permeability Assay Using N-Phenylnaphthalen-1-amine
The assay was performed based on protocols adapted from those described in literature.67,68 Bacteria were inoculated overnight at 37 °C in LB, diluted the next day 50x in LB, and grown to OD600 of 0.5. The bacterial suspension was then centrifuged for 10 min at 1000g at 25 °C. The pellet of bacteria was suspended in 5 mM HEPES buffer containing 20 mM glucose to a final concentration of OD600 of 1.0. The compounds were serial diluted (25 μL) in triplicate in a black 1/2 area clear-bottom 96-well plate. 100 μg/mL final concentration of colistin in triplicate served as the positive control. Three wells were filled with 25 μL buffer to serve as the negative control. Additional controls of the compounds were made in triplicate using 25 μL of the highest concentration to detect interactions of the compounds with NPN in the absence of bacteria. A stock of 0.5 mM of NPN in acetone was prepared and diluted 12.5× in the buffer. 25 μL of the NPN solution was added to each well. 50 μL of the 1.0 OD600 bacterial stock was then added to each well except for the controls of the compounds with NPN. To these wells, 50 μL of buffer was added. After 60 min, the plate was measured using a Tecan plate reader with λex 355 ± 20 nm and λem 420 ± 20 nm. The fluorescence values obtained were then transformed into a NPN uptake percentage using following eq 2
| 2 |
Equation 2: NPN uptake. The observed fluorescence (Fobs) is corrected for background using the negative control (F0). This value is divided by the positive control corrected for background (F100 – F0) and multiplied by 100% to obtain the percentage NPN uptake.73
Sample Availability
Samples of all compounds reported are available from the authors upon request.
Acknowledgments
We kindly thank Dr. Paolo Innocenti for performing HRMS measurements. The contribution of Dr. Bart W. Bardoel of transforming the E. coli BW25113 strain with the pGDP2 MCR-1 plasmid is gratefully acknowledged. Financial support is provided by the European Research Council (ERC consolidator grant to N.I.M., grant agreement no. 725523).
Glossary
Abbreviations
- Ac2O
acetic anhydride
- AcOH
acetic acid
- ACS
American Chemical Society
- AMR
antimicrobial resistance
- ATCC
American Type Culture Collection
- CBr4
carbon tetrabromide
- CLSI
Clinical & Laboratory Standards Institute
- DiPEA
N,N-diisopropylethylamine
- EtOAc
ethyl acetate
- FICI
fractional inhibitory concentration index
- LB
Lysogeny Broth
- LPS
lipopolysaccharide
- MIC
minimum inhibitory concentration
- MSC
minimal synergistic concentration
- NDM-1
New Delhi β-lactamase 1
- NPN
N-phenyl-napthalen-1-amine
- OM
outer membrane
- PMBN
polymyxin B nonapeptide
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00466.
Synthesis protocols and schemes for compounds 1 and 3; MIC, checkerboard assay, and FICI results of all compounds in combination with erythromycin and rifampicin for E. coli BW25113; MIC, checkerboard assay, and FICI results of compounds 1, 3, 21, 22, 23b, 37, 38, 43, and 44, and PMBN in combination with novobiocin and vancomycin for E. coli BW25113; MIC, checkerboard assay, and FICI results of compounds 1, 3, 21, 22, 23b, 37, 38, 43, and 44, and PMBN in combination with rifampicin for all screened strains and pathogens; hemolysis data of all compounds; NPN data read out at 60 min; HRMS data; 1H and 13C NMR spectra of all final compounds; HPLC purity traces of all compounds; and sources of the bacterial strains (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Amann S.; Neef K.; Kohl S. Antimicrobial resistance (AMR). Eur. J. Hosp. Pharm. 2019, 26, 175–177. 10.1136/ejhpharm-2018-001820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UN Interagency Coordination Group on Antimicrobial Resistance . No time to wait: securing the future from drug-resistant infections. Report to the Secretary-General of the United Nations, Geneva; WHO, 2019. https://www.who.int/publications/i/item/no-time-to-wait-securing-the-future-from-drug-resistant-infections (date accessed Oct 24, 2021).
- WHO . Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterialinfections Including Tuberculosis. WHO/EKP/IAU/2017.12; WHO: Geneva, 2017. [Google Scholar]
- Macnair C. R.; Brown E. D. Outer membrane disruption overcomes intrinsic, acquired, and spontaneous antibiotic resistance. MBio 2020, 11, 1–15. 10.1128/mBio.01615-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H.The role of outer membrane and efflux pumps in the resistance of gram-negative bacteria. Can we improve drug access? Drug Resistance Updates; Churchill Livingstone, 1998; pp 93–98. [DOI] [PubMed] [Google Scholar]
- Vaara M.; Vaara T. Sensitization of Gram-negative bacteria to antibiotics and complement by a nontoxic oligopeptide. Nature 1983, 303, 526–528. 10.1038/303526a0. [DOI] [PubMed] [Google Scholar]
- Stokes J. M.; Macnair C. R.; Ilyas B.; French S.; Côté J.-P.; Bouwman C.; Farha M. A.; Sieron A. O.; Whitfield C.; Coombes B. K.; Brown E. D. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol. 2017, 2, 17028. 10.1038/nmicrobiol.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; Davis J. H.; Mangat C. S.; Williamson J. R.; Brown E. D. Discovery of a small molecule that inhibits bacterial ribosome biogenesis. eLife 2014, 3, 3e03574. 10.7554/eLife.03574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; French S.; Ovchinnikova O. G.; Bouwman C.; Whitfield C.; Brown E. D. Cold stress makes Escherichia coli susceptible to glycopeptide antibiotics by altering outer membrane integrity. Cell Chem. Biol. 2016, 23, 267–277. 10.1016/j.chembiol.2015.12.011. [DOI] [PubMed] [Google Scholar]
- Bean D. C.; Wareham D. W. Pentamidine: a drug to consider re-purposing in the targeted treatment of multi-drug resistant bacterial infections?. J. Lab. Precis. Med. 2017, 2, 49. 10.21037/jlpm.2017.06.18. [DOI] [Google Scholar]
- Waller D. G.; Sampson A. P.. Chemotherapy of infections. Medical Pharmacology and Therapeutics, 5th ed.; Elsevier, 2018; pp 581–629. [Google Scholar]
- Sands M.; Kron M. A.; Brown R. B. Pentamidine: a review. Rev. Infect. Dis. 1985, 7, 625–634. 10.1093/clinids/7.5.625. [DOI] [PubMed] [Google Scholar]
- Goa K. L.; Campoli-Richards D. M. Pentamidine isethionate: A review of its antiprotozoal activity, pharmacokinetic properties and therapeutic use in Pneumocystis carinii pneumonia. Drugs 1987, 33, 242–258. 10.2165/00003495-198733030-00002. [DOI] [PubMed] [Google Scholar]
- Libman M. D.; Miller M. A.; Richards G. K. Antistaphylococcal activity of pentamidine. Antimicrob. Agents Chemother. 1990, 34, 1795–1796. 10.1128/AAC.34.9.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewska D.; Żabiński J.; Kaźmierczak P.; Wójciuk K.; Kruszewski M.; Kruszewska H. In vitro screening of pentamidine analogs against bacterial and fungal strains. Bioorg. Med. Chem. Lett. 2014, 24, 2918–2923. 10.1016/j.bmcl.2014.04.075. [DOI] [PubMed] [Google Scholar]
- Edwards K. J.; Jenkins T. C.; Neidle S. Crystal Structure of a Pentamidine-Oligonucleotide Complex: Implications for DNA-Binding Properties. Biochemistry 1992, 31, 7104–7109. 10.1021/bi00146a011. [DOI] [PubMed] [Google Scholar]
- Sun T.; Zhang Y. Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res. 2008, 36, 1654–1664. 10.1093/nar/gkm1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paracini N.; Clifton L. A.; Skoda M. W. A.; Lakey J. H. Liquid crystalline bacterial outer membranes are critical for antibiotic susceptibility. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E7587–E7594. 10.1073/pnas.1803975115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier M. C.; Ansari M. I.; Pierce A. D.; Wilder P. T.; McKnight L. E.; Raman E. P.; Neau D. B.; Bezawada P.; Alasady M. J.; Charpentier T. H.; Varney K. M.; Toth E. A.; MacKerell A. D.; Coop A.; Weber D. J. Small molecule inhibitors of Ca2+-S100B reveal two protein conformations. J. Med. Chem. 2016, 59, 592–608. 10.1021/acs.jmedchem.5b01369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam C.; Hildebrandt J.; Schütze E.; Wenzel A. F. Membrane-disorganizing property of polymyxin B nonapeptide. J. Antimicrob. Chemother. 1986, 18, 9–15. 10.1093/jac/18.1.9. [DOI] [PubMed] [Google Scholar]
- Odds F. C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- Gromyko A. V.; Popov K. V.; Mosoleva A. P.; Streltsov S. A.; Grokhovsky S. L.; Oleinikov V. A.; Zhuze A. L. DNA sequence-specific ligands: XII. Synthesis and cytological studies of dimeric hoechst 33258 molecules. Russ. J. Bioorg. Chem. 2005, 31, 344–351. 10.1007/s11171-005-0047-z. [DOI] [PubMed] [Google Scholar]
- Turner A. D.; Pizzo S. V.; Rozakis G.; Porter N. A. Photoreactivation of irreversibly inhibited serine proteinases. J. Am. Chem. Soc. 1988, 110, 244–250. 10.1021/ja00209a040. [DOI] [Google Scholar]
- Tidwell R. R.; Jones S. K.; Geratz J. D.; Ohemeng K. A.; Cory M.; Hall J. E. Analogs of 1,5-bis(4-amidinophenoxy)pentane (pentamidine) in the treatment of experimental Pneumocystis carinii pneumonia. J. Med. Chem. 1990, 33, 1252–1257. 10.1021/jm00166a026. [DOI] [PubMed] [Google Scholar]
- Stolić I.; Avdičević M.; Bregović N.; Piantanida I.; Glavaš-Obrovac L.; Bajić M. Synthesis, DNA interactions and anticancer evaluation of novel diamidine derivatives of 3,4-ethylenedioxythiophene. Croat. Chem. Acta 2012, 85, 457–467. 10.5562/cca2141. [DOI] [Google Scholar]
- Ewins A. J.; Barber H. J.; Newbery G.; Ashley J. N.; Self A. D. H.. Verfahren zur Herstellung von Diamidinderivaten. DE 844897 C, 1952.
- Roger R.; Neilson D. G. The chemistry of imidates. Chem. Rev. 2002, 61, 179–211. 10.1021/CR60210A003. [DOI] [Google Scholar]
- Bruncko M.; McClellan W. J.; Wendt M. D.; Sauer D. R.; Geyer A.; Dalton C. R.; Kaminski M. A.; Weitzberg M.; Gong J.; Dellaria J. F.; Mantei R.; Zhao X.; Nienaber V. L.; Stewart K.; Klinghofer V.; Bouska J.; Rockway T. W.; Giranda V. L. Naphthamidine urokinase plasminogen activator inhibitors with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2005, 15, 93–98. 10.1016/j.bmcl.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Qian K.; Yan C.; He M.; Jassim B. A.; Ivanov I.; Zheng Y. G. Discovery of decamidine as a new and potent PRMT1 inhibitor. Medchemcomm 2017, 8, 440–444. 10.1039/c6md00573j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt M. D.; Rockway T. W.; Geyer A.; McClellan W.; Weitzberg M.; Zhao X.; Mantei R.; Nienaber V. L.; Stewart K.; Klinghofer V.; Giranda V. L. Identification of novel binding interactions in the development of potent, selective 2-naphthamidine inhibitors of urokinase. synthesis, structural analysis, and SAR of N-phenyl amide 6-substitution. J. Med. Chem. 2004, 47, 303–324. 10.1021/jm0300072. [DOI] [PubMed] [Google Scholar]
- Abou-Elkhair R. A. I.; Hassan A. E. A.; Boykin D. W.; Wilson W. D. Lithium hexamethyldisilazane transformation of transiently protected 4-Aza/benzimidazole nitriles to amidines and their dimethyl sulfoxide mediated imidazole ring formation. Org. Lett. 2016, 18, 4714–4717. 10.1021/acs.orglett.6b02359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejewska D.; Żabinski J.; Kaźmierczak P.; Rezler M.; Krassowska-Świebocka B.; Collins M. S.; Cushion M. T. Analogs of pentamidine as potential anti-Pneumocystis chemotherapeutics. Eur. J. Med. Chem. 2012, 48, 164–173. 10.1016/j.ejmech.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin K.; Dannenfelser R.-M. In vitro hemolysis: Guidance for the pharmaceutical scientist. J. Pharm. Sci. 2006, 95, 1173–1176. 10.1002/JPS.20627. [DOI] [PubMed] [Google Scholar]
- Wang B.; Boykin D.; Choudhary M.; Kumar A.; Yu B.; Zhu M.. Amidines and amidine analogs for the treatment of bacterial infections and potentiation antibiotics. WO 2019241566 A1, 2019.
- Geratz J. D.; Cheng M. C. F.; Tidwell R. R. Novel bis(benzamidino) compounds with an aromatic central link. Inhibitors of thrombin, pancreatic kallikrein, trypsin, and complement. J. Med. Chem. 1976, 19, 634–639. 10.1021/jm00227a011. [DOI] [PubMed] [Google Scholar]
- Vanden Eynde J. J.; Mayence A.; Huang T. L.; Collins M. S.; Rebholz S.; Walzer P. D.; Cushion M. T. Novel bisbenzamidines as potential drug candidates for the treatment of Pneumocystis carinii pneumonia. Bioorg. Med. Chem. Lett. 2004, 14, 4545–4548. 10.1016/j.bmcl.2004.06.034. [DOI] [PubMed] [Google Scholar]
- Chauhan P. M. S.; Niyer R.; Bhakuni D. S.; Shankhdhar V.; Guru P. Y.; Sen A. B. Antiparasitic agents: Part VI - synthesis of 1,2-, 1,3,- and 1,4-bis(4-substituted aryloxy)benzenes and their biological activities. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1988, 27, 38–42. [Google Scholar]
- Patrick D. A.; Bakunov S. A.; Bakunova S. M.; Suresh Kumar E. V. K.; Chen H.; Jones S. K.; Wenzler T.; Barzcz T.; Werbovetz K. A.; Brun R.; Tidwell R. R. Synthesis and antiprotozoal activities of dicationic bis(phenoxymethyl)benzenes, bis(phenoxymethyl)naphthalenes, and bis(benzyloxy)naphthalenes. Eur. J. Med. Chem. 2009, 44, 3543–3551. 10.1016/j.ejmech.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Bakunova S. M.; Bakunov S. A.; Patrick D. A.; Kumar E. V. K. S.; Ohemeng K. A.; Bridges A. S.; Wenzler T.; Barszcz T.; Jones S. K.; Werbovetz K. A.; Brun R.; Tidwell R. R. Structure–Activity Study of Pentamidine Analogues as Antiprotozoal Agents. J. Med. Chem. 2009, 52, 2016–2035. 10.1021/jm801547t. [DOI] [PubMed] [Google Scholar]
- Ashley J. N.; Barber H. J.; Ewins A. J.; Newbery G.; Self A. D. H. A chemotherapeutic comparison of the trypanocidal action of some aromatic diamidines. J. Chem. Soc. 1942, 103–116. 10.1039/jr9420000103. [DOI] [Google Scholar]
- Hamano S.; Kanazawa T.; Kitamura S.-I.. Bis-(meta-amidinophenoxy)-compounds and pharmacologically acceptable acid addition salts thereof. U.S. Patent 4,034,010 A, 1977.
- Goswami R.; Mukherjee S.; Wohlfahrt G.; Ghadiyaram C.; Nagaraj J.; Chandra B. R.; Sistla R. K.; Satyam L. K.; Samiulla D. S.; Moilanen A.; Subramanya H. S.; Ramachandra M. Discovery of pyridyl bis(oxy)dibenzimidamide derivatives as selective matriptase inhibitors. ACS Med. Chem. Lett. 2013, 4, 1152–1157. 10.1021/ml400213v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong N.; Liu E. A.; Vu B. T.. Cis-imidazolines. U.S. Patent 6,617,346 B1, 2003.
- Huang H.; Li H.; Martásek P.; Roman L. J.; Poulos T. L.; Silverman R. B. Structure-guided design of selective inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 2013, 56, 3024–3032. 10.1021/jm4000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A. Organoborane coupling reactions (Suzuki coupling). Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2004, 80, 359. 10.2183/pjab.80.359. [DOI] [Google Scholar]
- Martino G.; Muzio L.; Riva N.; Gornati D.; Seneci P.; Eleuteri S.. Aminoguanidine hydrazones as retromer stabilizers useful for treating neurological diseases. WO 2020201326 A1, 2020.
- Innocenti P.; Woodward H.; O’Fee L.; Hoelder S. Expanding the scope of fused pyrimidines as kinase inhibitor scaffolds: synthesis and modification of pyrido[3,4-d]pyrimidines. Org. Biomol. Chem. 2014, 13, 893–904. 10.1039/C4OB02238F. [DOI] [PubMed] [Google Scholar]
- García D.; Foubelo F.; Yus M. Selective lithiation of 4- and 5-halophthalans. Heterocycles 2009, 77, 991–1005. 10.3987/COM-08-S(F)85. [DOI] [Google Scholar]
- Washington J. A.; Wilson W. R. Erythromycin: A microbial and clinical perspective after 30 years of clinical use (first of two parts). Mayo Clin. Proc. 1985, 60, 189–203. 10.1016/S0025-6196(12)60219-5. [DOI] [PubMed] [Google Scholar]
- Washington J. A.; Wilson W. R. Erythromycin: A microbial and clinical perspective after 30 years of clinical use (second of two parts). Mayo Clin. Proc. 1985, 60, 271–278. 10.1016/S0025-6196(12)60322-X. [DOI] [PubMed] [Google Scholar]
- Farr B.; Mandell G. L. Rifampin. Med. Clin. North Am. 1982, 66, 157–168. 10.1016/S0025-7125(16)31449-3. [DOI] [PubMed] [Google Scholar]
- Levine J. F. Vancomycin: A review. Med. Clin. North Am. 1987, 71, 1135–1145. 10.1016/S0025-7125(16)30801-X. [DOI] [PubMed] [Google Scholar]
- Kirby W. M. M.; Hudson D. G.; Noyes W. D. Clinical and laboratory studies of novobiocin, a new antibiotic. AMA Arch. Intern. Med. 1956, 98, 1–7. 10.1001/ARCHINTE.1956.00250250007001. [DOI] [PubMed] [Google Scholar]
- Drugs@FDA: FDA-Approved Drugs https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=050339 (accessed July 08, 2021).
- Wesseling C. M. J.; Wood T. M.; Slingerland C. J.; Bertheussen K.; Lok S.; Martin N. I. Thrombin-derived peptides potentiate the activity of Gram-positive-specific antibiotics against Gram-negative bacteria. Molecules 2021, 26, 1954. 10.3390/molecules26071954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornsberry C.; Hill B. C.; Swenson J. M.; McDougal L. K. Rifampin: Spectrum of antibacterial activity. Rev. Infect. Dis. 1983, 5, S412–S417. 10.1093/CLINIDS/5.SUPPLEMENT_3.S412. [DOI] [PubMed] [Google Scholar]
- Ebbensgaard A.; Mordhorst H.; Aarestrup F. M.; Hansen E. B. The role of outer membrane proteins and lipopolysaccharides for the sensitivity of Escherichia coli to antimicrobial peptides. Front. Microbiol. 2018, 9, 2153. 10.3389/fmicb.2018.02153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K.; Mizushima S. A simple method for isolation of lipopolysaccharides from Pseudomonas aeruginosa and some other bacterial strains. Agric. Biol. Chem. 1987, 51, 3107–3114. 10.1080/00021369.1987.10868532. [DOI] [Google Scholar]
- Furevi A.; Ståhle J.; Muheim C.; Gkotzis S.; Udekwu K. I.; Daley D. O.; Widmalm G. Structural analysis of the O-antigen polysaccharide from Escherichia coli O188. Carbohydr. Res. 2020, 498, 108051. 10.1016/j.carres.2020.108051. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wang J.; Ren G.; Li Y.; Wang X. Influence of core oligosaccharide of lipopolysaccharide to outer membrane behavior of Escherichia coli. Mar. Drugs 2015, 13, 3325–3339. 10.3390/md13063325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorduijn D. J.; Heesterbeek D. A. C.; Ruyken M.; de Haas C. J. C.; Stapels D. A. C.; Aerts P. C.; Rooijakkers S. H. M.; Bardoel B. W. Polymerization of C9 enhances bacterial cell envelope damage and killing by membrane attack complex pores. bioRxiv 2021, 10.1101/2021.05.12.443779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox G.; Sieron A.; King A. M.; De Pascale G.; Pawlowski A. C.; Koteva K.; Wright G. D. A common platform for antibiotic dereplication and adjuvant discovery. Cell Chem. Biol. 2017, 24, 98–109. 10.1016/J.CHEMBIOL.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Liu Y.-Y.; Wang Y.; Walsh T. R.; Yi L.-X.; Zhang R.; Spencer J.; Doi Y.; Tian G.; Dong B.; Huang X.; Yu L.-F.; Gu D.; Ren H.; Chen X.; Lv L.; He D.; Zhou H.; Liang Z.; Liu J.-H.; Shen J. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- Nang S. C.; Li J.; Velkov T. The rise and spread of mcr plasmid-mediated polymyxin resistance. Crit. Rev. Microbiol. 2019, 45, 131–161. 10.1080/1040841X.2018.1492902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorio R.; Vaara M. The lipid A biosynthesis mutation lpxA2 of Escherichia coli results in drastic antibiotic supersusceptibility. Antimicrob. Agents Chemother. 1992, 36, 826–829. 10.1128/AAC.36.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander I. M.; Mattila-Sandholm T. Fluorometric assessment of Gram-negative bacterial permeabilization. J. Appl. Microbiol. 2000, 88, 213–219. 10.1046/j.1365-2672.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- MacNair C. R.; Stokes J. M.; Carfrae L. A.; Fiebig-Comyn A. A.; Coombes B. K.; Mulvey M. R.; Brown E. D. Overcoming mcr-1 mediated colistin resistance with colistin in combination with other antibiotics. Nat. Commun. 2018, 9, 458. 10.1038/s41467-018-02875-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Sun C. L.. Piperidine and piperazine derivatives. WO 2007089462 A2, 2007.
- Suzuki N.; Ishikawa T.; Kishimoto K.; Yamazaki K.; Kumamoto T.; Margetić D. Immobilized 1,2-bis(guanidinoalkyl)benzenes: Potentially useful for the purification of arsenic-polluted water. Synlett 2013, 24, 2510–2514. 10.1055/S-0033-1338980. [DOI] [Google Scholar]
- Berglund A. J.; Bodner M. J.. Aryl diamidines and prodrugs thereof for treating myotonic dystrophy. U.S. Patent 20,130,281,462 A1, 2013.
- Lewis J. E. M. Self-templated synthesis of amide catenanes and formation of a catenane coordination polymer. Org. Biomol. Chem. 2019, 17, 2442–2447. 10.1039/C9OB00107G. [DOI] [PubMed] [Google Scholar]
- Wang J.; Chou S.; Xu L.; Zhu X.; Dong N.; Shan A.; Chen Z. High specific selectivity and membrane-active mechanism of the synthetic centrosymmetric α-helical peptides with Gly-Gly pairs. Sci. Rep. 2015, 5, 15963. 10.1038/srep15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.