

Abstract
The cellular responses to hypoxia or hypoxia-ischemia (HI) are governed largely by the hypoxia-inducible factor (HIF) family of transcription factors. Our previous studies show that HIF-1α induction is an important factor that mediates protective effects in the brain after neonatal HI. In the present study, we investigated the contribution of another closely related HIF α isoform, HIF-2α, specifically the neuronal HIF-2α, to brain HI injury. Homozygous transgenic mice with a floxed exon 2 of HIF-2α were bred with CaMKIIα-Cre mice to generate a mouse line with selective deletion of HIF-2α in forebrain neurons. These mice, along with their wildtype littermates, were subjected to HI at postnatal day 9. Brain injury at different ages was evaluated by the levels of cleaved caspase-3 and spectrin breakdown products at 24 h; and histologically at 6 days or 3 months after HI. Multiple behavioral tests were performed at 3 months, prior to sacrifice. Loss of neuronal HIF-2α exacerbated brain injury during the acute (24 h) and subacute phases (6 days), with a trend towards more severe volume loss in the adult brain. The long-term brain function for coordinated movement and recognition memory, however, were not impacted in the neuronal HIF-2α deficient mice. Our data suggest that, similar to HIF-1α, neuronal HIF-2α promotes cell survival in the immature mouse brain. The two HIF alpha isoforms may act through partially overlapping or distinct transcriptional targets to mediate their intrinsic protective responses against neonatal HI brain injury.
Keywords: Hypoxic-ischemic encephalopathy, Brain development, Transcription factor, Hypoxia-inducible factor 2α, RRID: AB_10002593, RRID: AB_10000633, RRID: AB_11214057, RRID: AB_2070042, RRID: AB_626632, RRID: AB_2149209, RRID: AB_2883116, RRID: AB_143157
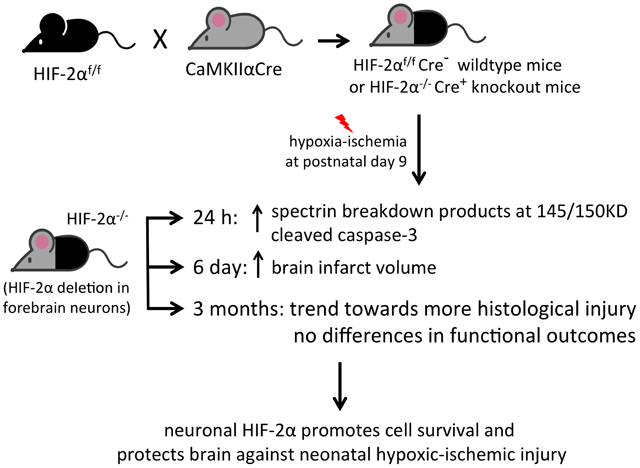
Graphical Abstract:
Using CaMKIIαCre and floxed-HIF-2α mice, we generated mice with specific deletion of HIF-2α gene in the forebrain neurons. We showed that neuronal deficiency of HIF-2α increased cell death and brain injury following neonatal hypoxia-ischemia suggesting its involvement in neuroprotection.
Introduction
Hypoxic-ischemic encephalopathy (HIE), due to fetal or neonatal asphyxia, is an important cause of death or permanent neurological impairments among full-term or late premature infants (Douglas-Escobar & Weiss, 2015; Millar, Shi, Hoerder-Suabedissen, & Molnar, 2017). As the current interventions, including therapeutic hypothermia, only provide partial protection (Natarajan, Pappas, & Shankaran, 2016; Shankaran et al., 2016), research endeavors focus on identifying the endogenous protective and repair mechanisms in order to develop additional therapies for translation to the clinic. Over the last decades, we have found that hypoxia-inducible factor 1-α (HIF-1α), a transcriptional factor that mediates acute adaptation to hypoxia, plays a critical role in protecting neonatal mice and immature primary neurons against hypoxic-ischemic (HI) brain injury in vivo and in vitro (Hamrick et al., 2005; Liang et al., 2019; Sheldon, Lee, Jiang, Knox, & Ferriero, 2014; Sheldon et al., 2009).
Hypoxia-inducible factors are heterodimeric transcription factors comprised of an α subunit that can be HIF-1α, HIF-2α or HIF-3α, and a β subunit (Semenza, 1998, 2001b). The activity and expression of the α subunit are closely modulated by cellular oxygen availability, whereas the β subunit is insensitive to oxygen and steadily expressed (Semenza, 2001a, 2007, 2012). HIF α subunit is rapidly degraded under normoxia; but is stabilized during hypoxia and translocates into the nucleus, where it dimerises with the β subunit. Together with other transcriptional coactivators, the HIF complex binds to the hypoxia response element (HRE) motif of their target DNA to initiate the transcription and expression of genes that are involved in a wide variety of cellular functions, including glucose and energy metabolic processes (Ke & Costa, 2006; Koyasu, Kobayashi, Goto, Hiraoka, & Harada, 2018; Semenza, 2012). Therefore, the HIF pathways are best understood as the master regulators of oxygen homeostasis since their discovery in 1995.
In models of neonatal brain ischemia, our previous work suggests that HIF-1α mediated-neuroprotection is associated with the induction of HIF-1α target genes, such as erythropoietin (Epo) and vascular endothelial growth factor (VEGF), which are implicated in angiogenesis and neurogenesis after HI (Liang et al., 2019; Mu, Chang, Vexler, & Ferriero, 2005; Mu et al., 2003; Sheldon et al., 2014). Recent studies have revealed that some putative HIF-1α targets, including brain Epo, are under the control of another closely related HIF α isoform, HIF-2α (Chavez, Baranova, Lin, & Pichiule, 2006; Ruscher et al., 2002; Yeo, Cho, Kim, & Park, 2008). Although HIF-2α is also recognized as an essential regulator of oxygen homeostasis, its function is underestimated in gene ontology databases, and less well characterized in pathological conditions. HIF-1α and HIF-2α share high sequence homology (≈ 48%) (Tian, McKnight, & Russell, 1997) and many target genes. It is increasingly clear that they are not functionally redundant and may regulate unique transcriptional targets in the context of different tissue and cell types, injury paradigms and timing (Downes, Laham-Karam, Kaikkonen, & Yla-Herttuala, 2018; Hu, Wang, Chodosh, Keith, & Simon, 2003; Keith, Johnson, & Simon, 2011).
The majority of studies concerning the distinct roles and substrates of HIF-1α versus HIF-2α have been performed in cancer (Mole et al., 2009; Smythies et al., 2019) or endothelial cells (Bartoszewski et al., 2019), and not brain cells, especially the developing brain following HI. A clear dissection of the distinct roles of HIF-1α and HIF-2α will provide important insights into the functional differences between these two HIF homologs and how the HIF system orchestrates diverse responses following neonatal HI. We have published important findings of HIF-1α in neonatal HI in vivo and in primary neurons; however, the role of HIF-2α in brain HI is still unexplored. In this study, we investigate the contribution of neuron-specific HIF-2α to neonatal brain HI injury by using a Cre-mediated conditional knockout strategy to selectively delete HIF-2α predominately in mouse forebrain neurons. Our data show that loss of neuronal HIF-2α exacerbates brain injury at 24 h and 6 days after HI, indicating its role in promoting neuronal survival. HIF-2α-associated neuroprotection did not translate to long-term functional benefits as evaluated by behavioral testing at 3 months, but at this age, HIF-2α knockout mice showed a tendency towards more severe brain volume loss than their wildtype littermates.
Materials and methods
Animal experiments
All animal procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animals of National Institutes of Health and were approved by the University of California San Francisco (UCSF) Institutional Animal Care and Use Committee. The animals were housed five mice per cage in humidity (55±10%)- and temperature (22±1°C)- controlled room with a light cycle of 12 hr on (6a.m.–6p.m.) and 12 hr off (6p.m.–next day 6a.m.) at UCSF barrier facility. The animals were fed ad libitum with a standard rodent diet and had free access to water. The standard physical enrichment including nestlets, shredded paper twist (ENVIRO-DRI), and a paper hut was provided in each cage. Animals are handled during cages change and if health issues arise that require handling. All animal handling practices and procedures, including animal health monitoring, diet, primary enclosures, environmental control, and means of identification met current recommendations of the American Veterinary Medical Association. All experiments were reported according to the Animal Research: Reporting of In Vivo Experiments guidelines. Both sexes were included in the animal procedures. The mice were randomized and the investigator/operator was blinded to the respective mouse genotypes.
Generation of neuron-specific HIF-2α knockout mice
Transgenic mice with two loxP sites flanking exon 2 of HIF-2α (Gruber et al., 2007) were purchased from the Jackson Laboratory (stock No: 008407, Sacramento, CA). The homozygous floxed-HIF-2α f/f mice were bred with another mouse line expressing Cre recombinase under the control of the Ca2+/calmodulin-dependent protein kinase IIα promoter (CaMKIIα-Cre) (Dragatsis & Zeitlin, 2000) to generate mice with selective deletion of HIF-2α in forebrain neurons. The mice were on C57Bl/6 background. The pups positive for the Cre gene were considered as HIF-2α knockout (KO, HIF-2α−/−) and their littermates with negative Cre expression were wildtype (WT, HIF-2α f/f) control. Mice were genotyped using primers (Integrated DNA Technologies, Inc., Coralville, IA) listed in Table 1.
Table 1.
List of primers used for PCR genotyping
| Gene | Forward primer sequence (5′-3′) | Reverse primer sequence (5′-3′) |
|---|---|---|
| Cre | GCGGTCTGGCAGTAA AAACTATC | GTGAAACAGCATTGCTGTCACTT |
| HIF-2α | GAGAGCAGCTTCTCCTGGAA | TGTAGGCAAGGAAACCAAGG |
Neonatal brain HI
Neonatal hypoxic-ischemic brain damage was produced using the Vannucci model as previously described (Sheldon, Windsor, & Ferriero, 2019). At postnatal day 9 (P9), the neuronal HIF-2α WT and KO pups were anesthetized with 2%-3% isoflurane, a vertical midline incision was made on the neck and the left common carotid artery (CCA) was separated and occluded by electrocoagulation. After 1-hour of recovery and feeding with their dam, the pups were subjected to hypoxia for 60 minutes in a homeothermic chamber at 36°C with 10% oxygen/balance nitrogen. For sham control group animals, all the surgical procedures were provided except coagulation of the left CCA and hypoxia. The mortality of this HI procedure was low, approximately 7%.
Extraction of nuclear/cytoplasmic fractions
The brain cortices of HIF-2α WT and KO mice were dissected, snap frozen and preserved at −80 °C before use. The NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL) were used to extract the nuclear and cytoplasmic protein according to the manufacturer’s protocol. The tissue was homogenized in 250 μl of ice-cold CER I buffer containing Halt™ protease and phosphatase inhibitors (Pierce Biotechnology). The samples were then incubated with 13.75 μl of CER II buffer and centrifuged for 10 min at 16,000 x g at 4° C. The resultant supernatant was saved as the cytoplasmic extract. The pellet was resuspended in 100 μl of ice-cold NER buffer, shaken for 20 min at 1,500 rpm at 4° C, followed by centrifugation at 16,000 x g for 30 min at 4° C. The supernatant was collected as the nuclear extract. BCA assay kit (Pierce) was used to determine protein concentrations.
Immunoblotting
Equal amounts of protein (20μg of nuclear extract for the measurement of HIF-2α/HIF-1α expression; and 40μg of cytoplasmic fraction for spectrin, cleaved caspase-3 and β-actin expression) were separated by SDS-PAGE and transferred to PVDF membranes. After blocking for 1 h at room temperature (RT) with TBS-T buffer with 5% non-fat dry milk and 0.05% Tween-20, the membranes were incubated with the following primary antibodies diluted in the blocking buffer overnight at 4°C: rabbit polyclonal anti-HIF-2α antibody (Novus Biologicals, LLC, Centennial, CO, Cat# NB100-122, RRID: AB_10002593, 1:1000); rabbit polyclonal anti-HIF-1α antibody (Novus Biologicals, LLC, Cat# NB100-479, RRID: AB_10000633, developed against a fusion protein including amino acids 530-825 of the mouse HIF-1α protein, 1:1000); mouse monoclonal anti-α-spectrin antibody (MilliporeSigma, Burlington, MA, Cat#: MAB1622, RRID: AB_11214057, 1:5000); rabbit monoclonal anti-cleaved caspase-3 (Cell Signaling Technology Inc.; Danvers, MA, Cat# 9664, RRID: AB_2070042, 1:1000) and mouse monoclonal anti-β-actin antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, Cat# sc-47778, RRID: AB_626632, 1:5000). The membranes were washed with TBS-T buffer with 0.05% Tween-20 and incubated with HRP-conjugated secondary antibodies (Santa Cruz Biotechnology Inc.) for 1h at RT. Enhanced chemiluminescence kit was used to visualize the protein signal, which was then developed with radiographic film. After scanning the film, the mean optical densities (OD) and areas of protein bands were measured with Image J software.
Three to five animals were used to evaluate the time course of HIF-2α protein expression. To quantify spectrin and cleaved caspase-3 levels, 6 HI WT (male=3, female=3) and 5 HI KO (male=2, female=3) were used.
Immunofluorescent staining
At 4 h and 24 h after HI, the mice were perfusion-fixed with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (pH 7.4). Brains were dissected and post-fixed in the same buffer overnight at 4°C, and then kept in 30% sucrose in 0.1 M phosphate buffer for 3 days to cryoprotect the tissues. The brains were embedded in O.C.T compound and cryosections were sliced at 16 μm. The sections were defrosted and air-dried for 2 h at RT. After washing with PBS, the sections were incubated in blocking buffer (10% goat serum and 0.1% Triton X-100 in PBS) for 1 h at RT followed by incubation overnight at 4°C with rabbit polyclonal anti-HIF-2α antibody (Novus Biologicals, LLC, Cat# NB100-122, 1:100), paired with mouse monoclonal Alexa Fluor488-conjugated anti-NeuN antibody (MilliporeSigma, Cat# MAB377X, RRID: AB_2149209, 1:100) for neurons; or mouse monoclonal coraLite488-conjugated anti-GFAP antibody (Proteintech Group Inc., Rosemont, IL, Cat# CL488-60190, RRID: AB_2883116, 1:100) for astrocytes. After washing with 0.025% Tween/PBS 3 times, the brain sections were incubated with goat anti-rabbit IgG (H+L) antibody - Alexa Fluor 568 (Invitrogen Corporation, Carlsbad, CA, Cat# A-11011, RRID: AB_143157, 1:500) for 1 h at RT, then with DAPI for 5 min, and cover-slipped with ProLong Diamond antifade reagent (Invitrogen). Images were captured using Leica TCS SP5 Spectral Confocal Microscope.
Histology and quantification of brain infarct volume
Six days or 3 months after HI, the mice were euthanized and brains processed for cresyl violet (CV) staining to evaluate brain injury. Following cryoprotection as above, 50-μm thickness of serial brain sections were collected using a vibratome and then stained with CV as we previously reported (Sheldon, Sedik, & Ferriero, 1998). Volumetric injury analysis was conducted using a series of 8 CV-stained sections with Image J software. The volumes of the ipsilateral and contralateral hemispheres of each brain were measured. Brain total infarct volume (%) was calculated and presented as [(volume of contralateral hemisphere – volume of ipsilateral hemisphere)/volume of contralateral hemisphere] × 100% (Lu et al., 2020). The infarct volumes of ipsilateral cortex, hippocampus and amygdala were also quantified using the same calculation. For histology study on 6 days, we used 8 WT (male=5, female=3) and 9 KO (male=4, female=5) HI animals. For evaluation at 3 months, 14 WT (male=8, female=6) and 17 KO (male=10, female=7) HI animals were used.
Behavioral testing
Three behavioral tests were performed between P90-P100, which included the rotarod (ROD) test, open field (OF) test and novel object recognition (NOR) test as we published previously (Lu et al., 2020).
Rotarod test:
The rotarod test examines the function of motor coordination, balance and muscle strength. After a 1-min adaptation period with a slow speed (4 rpm/min) on the rod (15 cm above the base), the rod was accelerated by 5 rpm every 15 sec, to a maximum speed of 32 rpm/min. The length of time (seconds, s) that the mouse remained on the rod (retention time or latency to fall) was recorded. The maximum test duration was 180 seconds.
Open field:
This test was conducted to assess anxiety-related behavior. The mouse was placed in a 40 x 40 cm plastic chamber that was divided into central (20 x 20 cm) and peripheral zones. The light intensity was 250 lux. The animal was allowed to explore freely in the entire arena for 5 minutes while being recorded by an overhead camera. The percentage of time each mouse spent in the center zone of the chamber was recorded to determine the level of anxiety.
2-object novel object recognition:
The novel object recognition test is a simple assessment of hippocampus- and cortex-dependent non-spatial learning and memory. The amount of time that the mouse spent exploring a novel object relative to a familiar one reflects the utilization of learning and recognition memory. On two consecutive days, the mouse was habituated to an open field arena with a light intensity of 15 lux for 5 min. On the third day, the mouse was exposed to the familiar arena with two identical objects and allowed to explore for 5 min. In the 3-minute test trial performed 24 h after the familiarization phase (the 4th day), one of the familiar objects was replaced by a novel object of the same size. The amount of time the mouse spent exploring each object was documented and the percentage of time spent with the novel object was determined as [time with novel object/(time with old + with novel object)] x 100.
For all tests, the animal numbers were as follows: n=10 for sham WT (male=3, female=7), n=6 for sham KO (male=2, female=4), n=16 for HI WT (male=9, female=7), n=19 for HI KO (male=11, female=8).
Statistical analysis
Statistical analyses were performed using Prism 5 (GraphPad Software, San Diego, CA). The immunoblotting, the infarct volume and the behavioral testing data were compared with the nonparametric Wilcoxon-Mann-Whitney test. Differences were considered significant at p < 0.05. The results are expressed as vertical scatter plot with median. On all graphs, each dot represents an individual mouse, and the horizontal bars represent the median.
Results
Neonatal HI enhanced HIF-2α protein expression
We examined the time course of HIF-2α protein expression after neonatal HI in the WT mice and confirmed its induction early after the insult at 30 min and sustained for at least 4 hours (Fig. 1 a). HIF-2α levels returned to the sham values by 24 h after HI. Cell death and brain damage following HI were validated by the significantly increased expression of cleaved caspase-3 at 4 h and 24 h after HI (Fig. 1 a).
Fig. 1:
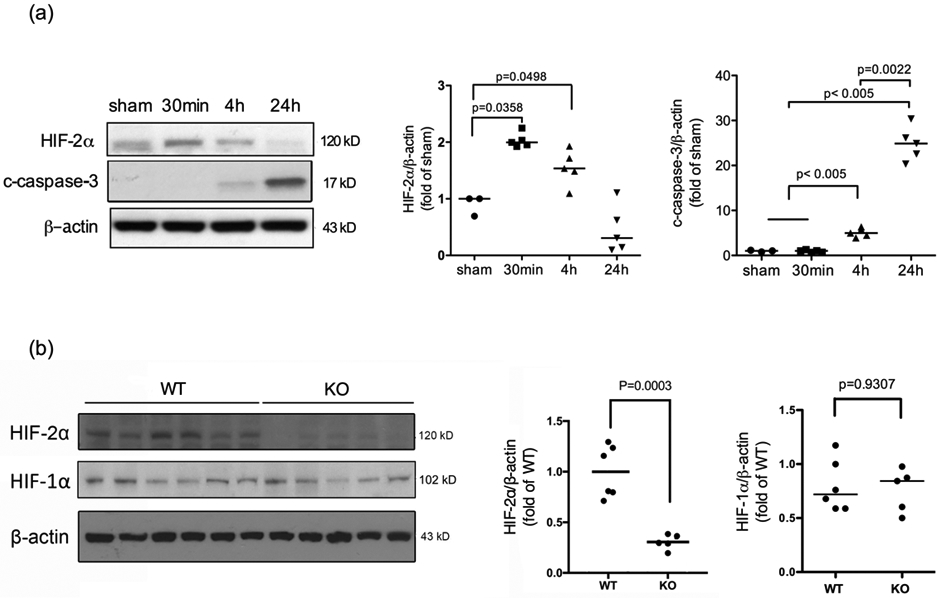
The time course of HIF-2α and cleaved caspase-3 protein expression after neonatal brain HI. (a) The nuclear fraction of cortical lysates was extracted at the indicated time points after HI and subjected to Western Blotting. The quantification of HIF-2α and cleaved caspase-3 (c-caspase-3) expression is shown on the right. The OD values were normalized to β-actin and expressed as fold change to the sham values (fold of sham). Expression of c-caspase-3 indicates brain injury at 4 h and 24 h (n=3 for sham, n=5 for HI mice). (b) Representative blots showing decreased HIF-2α expression in the KO mice compared to the WT animals at 4 h after HI. (n=6 for WT, n=5 for KO). The OD values were normalized to β-actin and expressed as fold change to the WT values (fold of WT).
Characterization of neuron-specific HIF-2α knockout mice
The protein expression of HIF-2α was compared between the WT and the neuron-specific KO mice at 4 h after HI. As shown in Fig.1b, HIF-2α protein levels were reduced to approximately 35% of that in the WT animals (Fig. 1b, p = 0.003 WT vs. KO). The remaining HIF-2α may be derived from other cell populations that also produce the protein. Alternatively, as CaMKIIα expression increases over time after birth, CaMKIIα-Cre-mediated recombination and HIF-2α deletion may remain incomplete at P9. The protein level of HIF-1α remained unchanged in the KO mice versus their WT littermates (Fig.1b). Double immunofluorescent staining confirmed a lack of HIF-2α expression in NeuN-positive neurons in HIF-2α KO brain at 4 h after HI compared to WT mice (Fig. 2). The images shown in Fig. 2 were captured from the similar ipsilateral cortical region, namely, the somatosensory cortical area. There was no localization of HIF-2α in GFAP-labeled astrocytes at 4 h (Fig. 2) and 24 h (data not shown).
Fig. 2:

HIF-2α localization in the WT and KO mice 4 h after HI. HIF-2α (red) was localized in NeuN-labeled neurons (green), and was diminished in the KO animals. HIF-2α was not co-localized with GFAP (green) in either WT or KO animals after HI. The images were captured from the similar ipsilateral cortical region, namely, the somatosensory cortical area. (Scale bar = 40μm)
Neuron-specific deletion of HIF-2α increased brain injury during the acute and subacute phases after HI
To elucidate the contribution of neuronal HIF-2α to HI brain damage, the WT and HIF-2α KO mice were subjected to HI at P9. Apoptotic and necrotic cell death were evaluated by the levels of spectrin breakdown products (SBDP) at 145/150KD and 120KD; and by cleaved caspase-3, indicators for activated calpain and caspase-3 (Z. Q. Zhang et al., 2009), at 24 h after HI. Fig. 3 showed that the levels of these proteins were higher in the HIF-2α KO mice than those in the WT littermates (p values and animal numbers are shown in the figure) suggesting that selective deletion of HIF-2α in forebrain neurons increased cell death at an acute time point post-HI. The histology outcome was determined at a subacute phase at 6 days after HI when cell death was complete and the full extent of injury was developed. The ipsilateral brain regions of cortex, hippocampus, striatum and thalamus were the primary lesion sites. Consistently, the HIF-2α KO mice had larger infarct volumes than the WT animals (Fig. 4, p = 0.023, KO vs. WT).
Fig. 3:
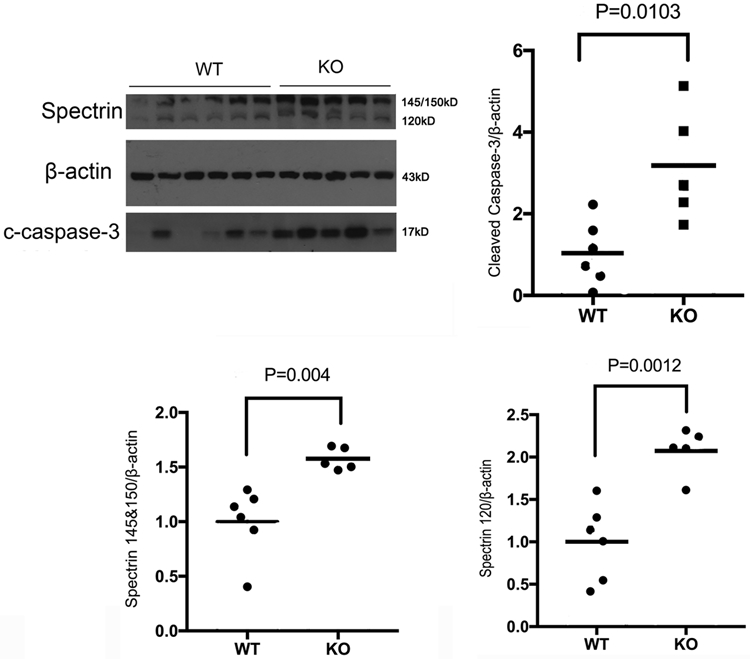
Neuronal HIF-2α deletion enhanced necrotic and apoptotic cell death 24 h after HI. The KO mice had increased levels of spectrin breakdown products at 145/150 kD and 120 kD, as well as cleaved caspase-3 compared to WT mice. (n = 6 for WT; n = 5 for KO). The OD values were normalized to β-actin and expressed as fold change to the WT values (fold of WT).
Fig. 4:

Brain infarct volume at 6 days after HI in the WT and KO mice. The percentage of total brain infarct volume (right) was quantified based on the cresyl violet-stained series of brain sections (left). (n=8 for WT; n=9 for KO animals).
Neuronal HIF-2α deletion did not provide long-term functional protection
HIF-2α-associated neuroprotection was assessed again at 3 months after HI when the mice reached adult age. Three behavioral tests were performed to evaluate motor and cognitive function. When compared to the sham animals, the HI-injured WT mice had reduced retention time on the rod (latency to fall) in the ROD test (Fig. 5a); spent less time in the center in the OF test (Fig. 5b); and spent less time exploring the novel object in the NOR test (Fig. 5c). These overall poorer behavioral performances indicate HI-induced long-term functional impairments. There were no differences between the WT and HIF-2α KO mice in the behavioral testing (Fig. 5), with the exception of the OF test, in which the HIF-2α KO mice spent more time in the center, indicating lower level of anxiety (Fig. 5b, p = 0.0382, WT vs. KO). The mice were sacrificed after the behavioral testing for histology staining to quantify brain volumes at this stage. The overall infarct volumes were similar in the WT and HIF-2α KO mice (Fig. 6, p = 0.3308, KO vs. WT). There were no gender differences, nor regional differences in the infarct volumes between these two genotypes (Fig. 6). There were fewer KO mice than the WT with small infarct while more KO mice showed moderate infarct (11.76% KO vs. 35.71% WT mice had small infarct with <5% infarct volume; and 76.47% KO vs. 50% of WT mice had moderate infarct volume ranged 5-40%). The HIF-2α KO mice had a trend towards more severe brain volume loss than the WT animals (Table 2).
Fig. 5:
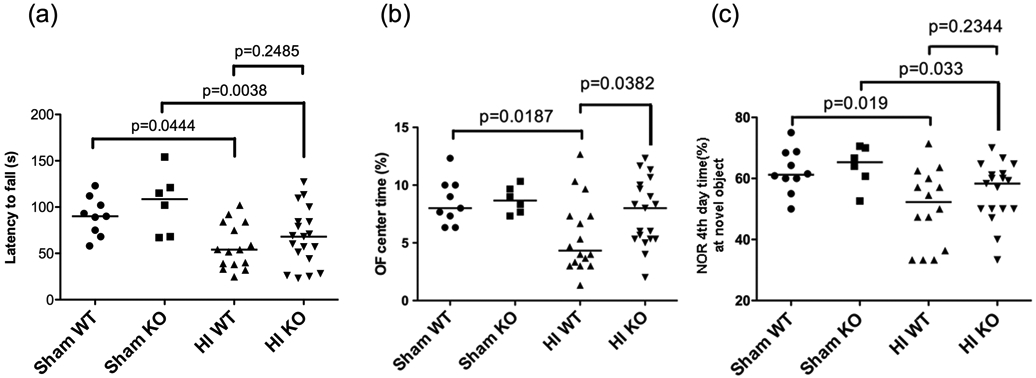
Brain functional outcomes at 3 months after HI evaluated by behavioral testing. (a) Compared to the sham animals, the HI-injured mice (both WT and KO) had less retention time on the rod or latency to fall (s). There was no difference between the HI WT and KO mice (p=0.2485). (b) In the OF test, the WT HI animals spent less time in the center compared to the WT sham (p=0.0187). The HI KO mice had significantly higher OF center time than the WT HI animals (p=00382). (c) Regardless of genotype, the HI animals spent less time exploring the novel object in the NOR test. There were no differences between HI-injured WT and KO mice (p=0.2344). In all tests, n=10 for sham WT (male=3, female=7), n=6 for sham KO (male=2, female=4), n=16 for HI WT (male=9, female=7), n=19 for HI KO (male=11, female=8).
Fig. 6:
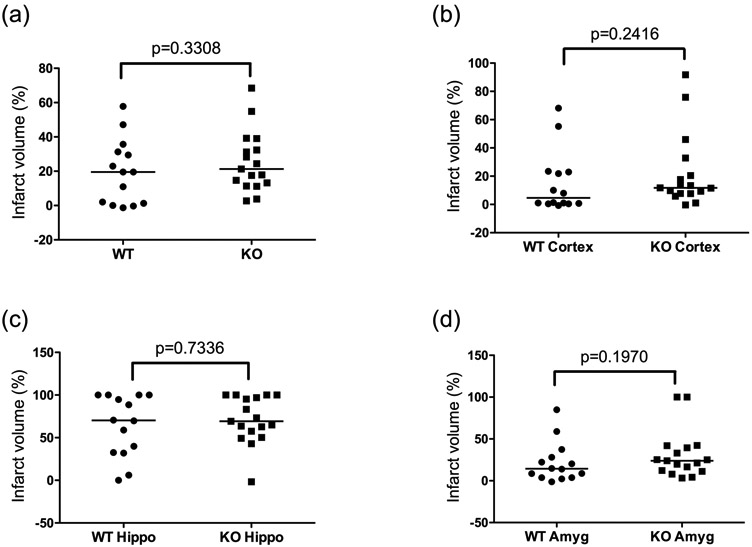
Brain infarct volume at 3 months after HI in the WT and KO mice. There were no differences in the overall (a) and regional (b: cortex; c: hippocampus; d: amygdala) infarct volume between the two genotypes. For all graphs, n=14 for WT (male=8, female=6), n=17 for KO (male=10, female=7). Hippo: hippocampus, Amyg: amygdala
Table 2.
Severity of brain volume loss by genotype at 3 months after HI
| Genotype | Animal number |
Infarct volume (%) |
||
|---|---|---|---|---|
| 0-5 | 5-40 | 40-60 | ||
| WT | 14 | 5 (35.71%) | 7 (50%) | 2 (14.29%) |
| KO | 17 | 2 (11.76%) | 13 (76.47%) | 2 (11.76%) |
Number and (percentage) of animals with small (0-5%), moderate (5-40%) or large (40-60%) infarct volume
Discussion
In this study, we demonstrate that conditional knockout of HIF-2α in mouse forebrain neurons results in more severe brain injury following neonatal HI at P9, suggesting that neuronal HIF-2α plays a role in promoting cell survival in our mouse model. The protection was prominent at acute (24 h) and subacute (6 days post-HI) phases. At adult age of 3-4 months, the HIF-2α KO mice showed a trend towards greater brain volume loss histologically; however, brain function for coordinated movement and recognition memory was not impacted.
The mechanisms underlying the protective effects of neuronal HIF-2α are unclear. We did not see any changes in HIF-1α expression in the HIF-2α KO mice compared to the WT control, which is consistent with the observation in adult neuronal HIF-2α knockout mice using the same mouse lines (Barteczek et al., 2017). This indicates that the increased injury was not attributed to altered activity of HIF-1α in these animals. HIF-1α did not compensate for the loss of HIF-2α; therefore neuronal HIF-2α should play unique roles for brain protection in neonatal HI. Our finding that deletion of neuronal HIF-2α worsens brain injury is in marked contrast to the results from an adult ischemic stroke (transient middle cerebral artery occlusion, MCAO) or global hypoxia model in which the same CaMKIIα-Cre mouse line was bred to floxed-HIF-1α or HIF-2α mice (Barteczek et al., 2017). That study showed that neuronal deficiency of either HIF-1α- or HIF-2α did not affect brain infarct and edema following MCAO or hypoxia, suggesting a mutual compensatory effect of both isoforms. Differential vulnerability to brain ischemia occurred only in neuronal HIF-1α/HIF-2α double knockout mice (Barteczek et al., 2017). One possible explanation for this age-related differences is that in the adult ischemic model, HIF-2α protein levels were only slightly decreased in the neuronal HIF-2α knockout mice, indicating that HIF-2α predominantly derives from non-neuronal cells (glial and endothelial cells), whereas in our neonatal HI condition, HIF-2α levels were markedly attenuated in the KO mice and there was robust HIF-2α expression in neurons, rather than astrocytes. Together, our previous work (Liang et al., 2019; Sheldon et al., 2009) and the present study show that in neonatal HI, deletion of neuronal HIF-1α or HIF-2α alone led to greater brain injury, which suggests that both isoforms participate in neuronal oxygen sensing and are indispensable for intrinsic neuroprotection. HIF-mediated adaptive and cellular responses differ in the context of brain maturity and injury paradigms.
In our HI model, HIF-2α may act through pathways that overlap with or are distinct from those of HIF-1α to protect brain. They share certain protein targets due to their capacity to bind to the same HRE motif in the promoter or enhancer regions of the target DNA. Despite that, there is a preference for induction and transcriptional activation of their common genes depending on cell type, severity of insults and injury evolvement. For example, Epo, which is initially considered as a HIF-1α target that is involved in cell survival and neuroregeneration, is preferentially controlled by HIF-2α, but not HIF-1α, in both neurons and astrocytes (Chavez et al., 2006; Ruscher et al., 2002; Yeo et al., 2008). VEGF-A, another HIF-1α target implicated in angiogenesis and cerebral microvasculature reconstruction during brain recovery, is also more significantly regulated by HIF-2α in human umbilical vein endothelial cells (Downes et al., 2018) and primary cortical neurons (Ralph et al., 2004). This is in line with the function of HIF-2α in neurovascular development (Cristante et al., 2018). Chromatin immunoprecipitation (ChIP) studies using cancer (Mole et al., 2009; Smythies et al., 2019) or endothelial cell lines (Bartoszewski et al., 2019) revealed that the two HIFα isoforms have differential but complementary transcriptional regulation (Downes et al., 2018). HIF-1α participates in the initial acute adaptation to hypoxia by regulating glycolysis and processes related to anaerobic metabolic reprogramming. HIF-2α, however, is induced by more prolonged hypoxia and regulates many more genes in response to chronic stresses, for example angiogenic signaling, vascular function and extracellular matrix reorganization (Bartoszewski et al., 2019; Downes et al., 2018; Koh & Powis, 2012). To address the distinct mechanisms by which neuronal HIF-1α and HIF-2α benefit neonatal HI, a genome-wide profiling of HIF DNA binding is required to identify their common and unique transcriptional targets. This approach will help us understand the specific molecular pathways triggered by HIF-1α or HIF-2α respectively, and how the HIF system orchestrates the diverse responses along the processes of brain injury, recovery and repair in newborn brain injury.
Similar to HIF-1α, HIF-2α may directly regulate cell survival and death machineries. HIF-1α is known to induce pro-apoptotic members of the Bcl-2 family genes such as Bnip3 (Bcl-2 interacting protein 3) and Pmaip1 (Phorbol-12-myristate-13-acetate-induced protein 1, Noxa) (Althaus et al., 2006; Bruick, 2000; Kim, Ahn, Ryu, Suk, & Park, 2004). Bnip3 is also implicated in autophagy and mitophagy (Shi et al., 2014; J. Zhang & Ney, 2009) that occur in neonatal HI (Carloni, Buonocore, & Balduini, 2008; Thornton et al., 2018). Neuronal Bnip3 expression is regulated by HIF-1α rather than HIF-2α (Barteczek et al., 2017). In addition, HIF-1α is required in p53-mediated apoptosis in tumor cells (C. Zhang et al., 2021), whereas HIF-2α inhibits p53 pathway activation by limiting accumulation of reactive oxygen species (ROS), thereby reducing cell death (Bertout et al., 2009; Das et al., 2012). In fact, studies with a global HIF-2α deficient mouse line reported that HIF-2α enhances the expression of multiple antioxidant genes including superoxide dismutase 1 (SOD1), SOD2, glutathione peroxidase 1 (GPx1) and catalase in developing embryos and neonates; and reduces the expression of genes associated with oxidative stress (Scortegagna et al., 2003; Semenza, 2012). HIF-2α particularly upregulates SOD2, which scavenges superoxide in chronic intermittent hypoxia (Nanduri et al., 2009; Semenza, 2012). By contrast, HIF-1α mediates increased expression of NADPH oxidase-2 and the resultant superoxide formation (Yuan et al., 2011). These findings suggest that HIF-2α is beneficial in maintaining ROS and mitochondrial homeostasis.
It is surprising that there were no long-term behavioral differences in the motor coordination and recognition memory between the WT and KO animals despite the fact that there were differences histologically after HI in the subacute phase. At 3 months of age, the overall and regional brain infarct volumes were similar between the two genotypes. As cre-mediated HIF-2α deletion is driven by CaMKIIα promoter, whose expression starts after birth and increases with age (Bayer, Lohler, Schulman, & Harbers, 1999; Colbran, 1992), perhaps persistent deficiency of HIF-2α in neurons differentially affects the chronic reparative process and regeneration after the full extent of injury is developed at 6 days after HI. Another possibility is that our HI paradigm produced mainly mild to moderate brain injuries with wide variation, in which cases the behavioral differences may be difficult to discern. Unexpectedly, the HIF-2α KO mice showed less anxiety in the open field test. At least one more valid anxiety-related behavioral test is needed to ascertain the role of neuronal HIF-2α in regulating anxiety in response to neonatal HI. It has been reported that deletion of astrocyte HIF-1α or HIF-2α does not alter anxiety levels in adult rats (Leiton et al., 2018), by contrast, endothelial cell-specific inactivation of HIF-1α, which also express a lower level of HIF-2α, showed gender-dependent effects on the average center times in the open field test when evaluated at P20, even under normoxic conditions (Li et al., 2017). These findings suggest the implication of HIF signaling in anxiety-like behavior, in an isoform-, cell type-, and age-dependent manner.
In summary, we show that elimination of neuronal HIF-2α augments destructive responses of HI brain injury early in neonatal mice. HIF-2α is also expressed by non-neuronal cells in the brain. In models of in vitro cortical neurons and astrocytes undergoing hypoxia or oxygen-glucose deprivation, as well as in adult hypoxic mouse brain, HIF-2α expression is localized in astrocytes rather than in neurons where HIF-1α is expressed at a higher level (Chavez et al., 2006; Leiton et al., 2018). Surprisingly, we did not find HIF-2α expression in astrocytes in our model in the first 24 h after HI. It could be upregulated at later time points in astrocytes, similar to what we have observed that Epo is expressed in astrocytes at one week following neonatal stroke (Mu et al., 2005). The cell type- and isoform-based differences in HIF signaling pathways have not been fully characterized in vivo in the developing brain. The current study demonstrates that HIF-2α is present in neurons and contributes to cell survival in neonatal brain hypoxia-ischemia. Future studies will reveal the underlying mechanisms for this neuroprotection.
Supplementary Material
Significance Statement.
Brain hypoxia-ischemia is a significant cause of perinatal death and life-long neurological disabilities. There is a critical need to develop strategies that promote endogenous brain protection. In this study, we demonstrated that selective deletion of hypoxia-inducible factor 2α in forebrain neurons increased acute cell death and subacute brain injury in a mouse model of neonatal hypoxia-ischemia, suggesting its role in neuroprotection. Although the underlying mechanisms are not clear, our findings indicate that induction of neuronal HIF-2α is a potential strategy for developing treatment for hypoxic-ischemic encephalopathy.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke (awards R35NS097299 to Dr. Ferriero and R56NS114563 to Dr. Jiang); and China Scholarship Council (No. 201806230241 to Dr. Sun).
Footnotes
Conflict of Interest Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Accessibility
The original data that support the findings of this study are available from the corresponding author upon request.
References:
- Althaus J, Bernaudin M, Petit E, Toutain J, Touzani O, & Rami A (2006). Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1 alpha (HIF-1 alpha) protein following focal cerebral ischernia in rats. Neurochemistry International, 48(8), 687–695. doi: 10.1016/j.neuint.2005.12.008 [DOI] [PubMed] [Google Scholar]
- Barteczek P, Li LX, Ernst AS, Bohler LI, Marti HH, & Kunze R (2017). Neuronal HIF-1 alpha and HIF-2 alpha deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. Journal of Cerebral Blood Flow and Metabolism, 37(1), 291–306. doi: 10.1177/0271678x15624933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoszewski R, Moszynska A, Serocki M, Cabaj A, Polten A, Ochocka R, … Collawn JF (2019). Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J, 33(7), 7929–7941. doi: 10.1096/fj.201802650RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, Lohler J, Schulman H, & Harbers K (1999). Developmental expression of the CaM kinase II isoforms: ubiquitous gamma- and delta-CaM kinase II are the early isoforms and most abundant in the developing nervous system. Brain Res Mol Brain Res, 70(1), 147–154. doi: 10.1016/s0169-328x(99)00131-x [DOI] [PubMed] [Google Scholar]
- Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B, … Simon MC (2009). HIF2 alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proceedings of the National Academy of Sciences of the United States of America, 106(34), 14391–14396. doi: 10.1073/pnas.0907357106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK (2000). Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proceedings of the National Academy of Sciences of the United States of America, 97(16), 9082–9087. doi:DOI 10.1073/pnas.97.16.9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni S, Buonocore G, & Balduini W (2008). Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiology of Disease, 32(3), 329–339. doi: 10.1016/j.nbd.2008.07.022 [DOI] [PubMed] [Google Scholar]
- Chavez JC, Baranova O, Lin J, & Pichiule P (2006). The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J Neurosci, 26(37), 9471–9481. doi: 10.1523/JNEUROSCI.2838-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ (1992). Regulation and Role of Brain Calcium Calmodulin-Dependent Protein Kinase-Ii. Neurochemistry International, 21(4), 469–497. doi:Doi 10.1016/0197-0186(92)90080-B [DOI] [PubMed] [Google Scholar]
- Cristante E, Liyanage SE, Sampson RD, Kalargyrou A, De Rossi G, Rizzi M, … Bainbridge JWB (2018). Late neuroprogenitors contribute to normal retinal vascular development in a Hif2a-dependent manner. Development, 145(8). doi:ARTN dev157511 10.1242/dev.157511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Bayat-Mokhtari R, Tsui M, Lotfi S, Tsuchida R, Felsher DW, & Yeger H (2012). HIF-2 alpha Suppresses p53 to Enhance the Stemness and Regenerative Potential of Human Embryonic Stem Cells. Stem Cells, 30(8), 1685–1695. doi: 10.1002/stem.1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas-Escobar M, & Weiss MD (2015). Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA Pediatr, 169(4), 397–403. doi: 10.1001/jamapediatrics.2014.3269 [DOI] [PubMed] [Google Scholar]
- Downes NL, Laham-Karam N, Kaikkonen MU, & Yla-Herttuala S (2018). Differential but Complementary HIF1alpha and HIF2alpha Transcriptional Regulation. Mol Ther, 26(7), 1735–1745. doi: 10.1016/j.ymthe.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, & Zeitlin S (2000). CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis, 26(2), 133–135. [DOI] [PubMed] [Google Scholar]
- Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, & Simon MC (2007). Acute postnatal ablation of Hif-2 alpha results in anemia. Proceedings of the National Academy of Sciences of the United States of America, 104(7), 2301–2306. doi: 10.1073/pnas.0608382104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamrick SE, McQuillen PS, Jiang X, Mu D, Madan A, & Ferriero DM (2005). A role for hypoxia-inducible factor-1alpha in desferoxamine neuroprotection. Neurosci Lett, 379(2), 96–100. doi: 10.1016/j.neulet.2004.12.080 [DOI] [PubMed] [Google Scholar]
- Hu CJ, Wang LY, Chodosh LA, Keith B, & Simon MC (2003). Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol, 23(24), 9361–9374. doi: 10.1128/mcb.23.24.9361-9374.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke QD, & Costa M (2006). Hypoxia-inducible factor-1 (HIF-1). Molecular Pharmacology, 70(5), 1469–1480. doi: 10.1124/mol.106.027029 [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, & Simon MC (2011). HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer, 12(1), 9–22. doi: 10.1038/nrc3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Ahn HJ, Ryu JH, Suk K, & Park JH (2004). BH3-only protein noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1 alpha. Journal of Experimental Medicine, 199(1), 113–123. doi: 10.1084/jem.20030613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh MY, & Powis G (2012). Passing the baton: the HIF switch. Trends Biochem Sci, 37(9), 364–372. doi: 10.1016/j.tibs.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyasu S, Kobayashi M, Goto Y, Hiraoka M, & Harada H (2018). Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Science, 109(3), 560–571. doi: 10.1111/cas.13483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiton CV, Chen E, Cutrone A, Conn K, Mellanson K, Malik DM, … Floyd TF (2018). Astrocyte HIF-2 alpha supports learning in a passive avoidance paradigm under hypoxic stress. Hypoxia, 6, 35–56. doi: 10.2147/Hp.S173589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Michaud M, Park C, Huang Y, Couture R, Girodano F, … Madri JA (2017). The role of endothelial HIF-1 alpha in the response to sublethal hypoxia in C57BL/6 mouse pups. Laboratory Investigation, 97(4), 356–369. doi: 10.1038/labinvest.2016.154 [DOI] [PubMed] [Google Scholar]
- Liang X, Liu X, Lu F, Zhang Y, Jiang X, & Ferriero DM (2019). HIF1alpha Signaling in the Endogenous Protective Responses after Neonatal Brain Hypoxia-Ischemia. Dev Neurosci, 1–10. doi: 10.1159/000495879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Fan S, Romo AR, Xu D, Ferriero DM, & Jiang X (2020). Serum 24S-hydroxycholesterol predicts long-term brain structural and functional outcomes after hypoxia-ischemia in neonatal mice. J Cereb Blood Flow Metab, 271678X20911910. doi: 10.1177/0271678X20911910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar LJ, Shi L, Hoerder-Suabedissen A, & Molnar Z (2017). Neonatal Hypoxia Ischaemia: Mechanisms, Models, and Therapeutic Challenges. Front Cell Neurosci, 11, 78. doi: 10.3389/fncel.2017.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, & Ratcliffe PJ (2009). Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem, 284(25), 16767–16775. doi: 10.1074/jbc.M901790200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Chang YS, Vexler ZS, & Ferriero DM (2005). Hypoxia-inducible factor 1alpha and erythropoietin upregulation with deferoxamine salvage after neonatal stroke. Exp Neurol, 195(2), 407–415. doi: 10.1016/j.expneurol.2005.06.001 [DOI] [PubMed] [Google Scholar]
- Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, & Ferriero DM (2003). Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis, 14(3), 524–534. [DOI] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan GX, Khan SA, Souvannakitti D, Peng YJ, … Prabhakar NR (2009). Intermittent hypoxia degrades HIF-2 alpha via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proceedings of the National Academy of Sciences of the United States of America, 106(4), 1199–1204. doi: 10.1073/pnas.0811018106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan G, Pappas A, & Shankaran S (2016). Outcomes in childhood following therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy (HIE). Seminars in Perinatology, 40(8), 549–555. doi: 10.1053/j.semperi.2016.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph GS, Parham S, Lee SR, Beard GL, Craigon MH, Ward N, … Krige D (2004). Identification of potential stroke targets by lentiviral vector mediated overexpression of HIF-1 alpha and HIF-2 alpha in a primary neuronal model of hypoxia. Journal of Cerebral Blood Flow and Metabolism, 24(2), 245–258. doi: 10.1097/01.Wcb.0000110532.48786.46 [DOI] [PubMed] [Google Scholar]
- Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, … Meisel A (2002). Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci, 22(23), 10291–10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, … Garcia JA (2003). Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1(−/−) mice. Nature Genetics, 35(4), 331–340. doi: 10.1038/ng1266 [DOI] [PubMed] [Google Scholar]
- Semenza GL (1998). Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev, 8(5), 588–594. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2001a). HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol, 13(2), 167–171. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2001b). Hypoxia-inducible factor 1: control of oxygen homeostasis in health and disease. Pediatr Res, 49(5), 614–617. doi: 10.1203/00006450-200105000-00002 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2007). Life with oxygen. Science, 318(5847), 62–64. doi: 10.1126/science.1147949 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2012). Hypoxia-inducible factors in physiology and medicine. Cell, 148(3), 399–408. doi: 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran S, Natarajan G, Chalak L, Pappas A, McDonald SA, & Laptook AR (2016). Hypothermia for neonatal hypoxic-ischemic encephalopathy: NICHD Neonatal Research Network contribution to the field. Seminars in Perinatology, 40(6), 385–390. doi: 10.1053/j.semperi.2016.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Lee CL, Jiang X, Knox RN, & Ferriero DM (2014). Hypoxic preconditioning protection is eliminated in HIF-1alpha knockout mice subjected to neonatal hypoxia-ischemia. Pediatr Res, 76(1), 46–53. doi: 10.1038/pr.2014.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Osredkar D, Lee CL, Jiang X, Mu D, & Ferriero DM (2009). HIF-1 alpha-deficient mice have increased brain injury after neonatal hypoxia-ischemia. Dev Neurosci, 31(5), 452–458. doi: 10.1159/000232563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Sedik C, & Ferriero DM (1998). Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res, 810(1-2), 114–122. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Windsor C, & Ferriero DM (2019). Strain-Related Differences in Mouse Neonatal Hypoxia-Ischemia. Developmental Neuroscience, 40(5-6), 490–496. doi: 10.1159/000495880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, & Kong JM (2014). BNIP3 Interacting with LC3 Triggers Excessive Mitophagy in Delayed Neuronal Death in Stroke. Cns Neuroscience & Therapeutics, 20(12), 1045–1055. doi: 10.1111/cns.12325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythies JA, Sun M, Masson N, Salama R, Simpson PD, Murray E, … Mole DR (2019). Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep, 20(1). doi: 10.15252/embr.201846401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C, Jones A, Nair S, Aabdien A, Mallard C, & Hagberg H (2018). Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. Febs Letters, 592(5), 812–830. doi: 10.1002/1873-3468.12943 [DOI] [PubMed] [Google Scholar]
- Tian H, McKnight SL, & Russell DW (1997). Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev, 11(1), 72–82. doi: 10.1101/gad.11.1.72 [DOI] [PubMed] [Google Scholar]
- Yeo EJ, Cho YS, Kim MS, & Park JW (2008). Contribution of HIF-1alpha or HIF-2alpha to erythropoietin expression: in vivo evidence based on chromatin immunoprecipitation. Ann Hematol, 87(1), 11–17. doi: 10.1007/s00277-007-0359-6 [DOI] [PubMed] [Google Scholar]
- Yuan GX, Khan SA, Luo WB, Nanduri J, Semenza GL, & Prabhakar NR (2011). Hypoxia-Inducible Factor 1 Mediates Increased Expression of NADPH Oxidase-2 in Response to Intermittent Hypoxia. Journal of Cellular Physiology, 226(11), 2925–2933. doi: 10.1002/jcp.22640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu JA, Wang JM, Zhang TL, Xu DD, Hu WW, & Feng ZH (2021). The Interplay Between Tumor Suppressor p53 and Hypoxia Signaling Pathways in Cancer. Frontiers in Cell and Developmental Biology, 9. doi:ARTN 648808 10.3389/fcell.2021.648808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, & Ney PA (2009). Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death and Differentiation, 16(7), 939–946. doi: 10.1038/cdd.2009.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZQ, Larner SF, Liu MC, Zheng WR, Hayes RL, & Wang KKW (2009). Multiple alphaII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis, 14(11), 1289–1298. doi: 10.1007/s10495-009-0405-z [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.