

Summary
CD4 T follicular helper (TFH) cells support B cells, which is critical for germinal center (GC) formation, but the importance of TFH-B cell interactions in cancer is unclear. We found enrichment of TFH cell transcriptional signature correlates with GC B cell signature and with prolonged survival in patients with lung adenocarcinoma (LUAD). We further developed a murine LUAD model in which tumor cells express B-cell- and T-cell-recognized neoantigens. Interactions between tumor-specific TFH and GC B cells, as well as IL-21 primarily produced by TFH cells, are necessary for tumor control and effector CD8 T cell function. Development of TFH cells requires B cells and B-cell-recognized neoantigens. Thus, tumor neoantigens can regulate the fate of tumor-specific CD4 T cells by facilitating their interactions with tumor-specific B cells, which in turn promote anti-tumor immunity by enhancing CD8 T cell effector functions.
Graphical Abstract
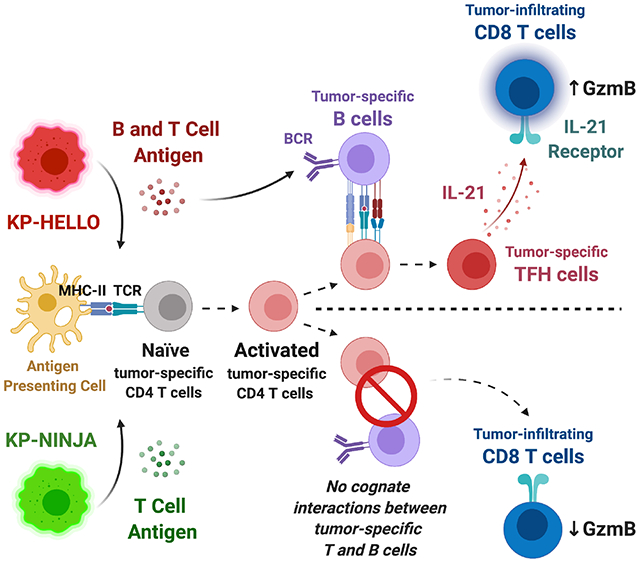
In brief
Germinal center B cells promote the development of tumor-specific CD4 T follicular helper cells in a neoantigen-dependent manner, which in turn enhance CD8 T cell effector functions by producing IL-21 and drive anti-tumor immunity in a murine model of lung adenocarcinoma.
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide, accounting for ~1.8 million deaths annually (Bray et al., 2018). Lung adenocarcinoma (LUAD) is the most common form of non-small cell lung cancer (NSCLC; 40% of cases), and is most frequently associated with mutations in KRAS proto-oncogene (30% of cases) (Schabath and Cote, 2019). Traditionally, therapeutic options for KRAS-mutant LUAD have been limited, but the successful application of immune checkpoint blockade (ICB) targeting the PD-1 pathway has changed the therapeutic landscape (Ettinger et al., 2019; Hanna et al., 2020). The success of PD-1-blockade therapy has also changed the perception that LUAD is a non-immunogenic cancer type because anti-PD-1/PD-L1 treatment potentiates the function of previously activated T cells, suggesting that therapy-responsive LUAD patients had potent, ongoing anti-tumor immune responses at the time of therapy administration (Forde et al., 2018; Gettinger et al., 2018; Lizotte et al., 2016; Topalian et al., 2012). Yet, only ~20% of LUAD patients benefit from ICB (Gandhi et al., 2018; Herbst et al., 2020; Reck et al., 2016) and improving response rates will require a better understanding of the anti-tumor immune response, including how different types of tumor-infiltrating T cells interact with other immune cells and contribute to anti-tumor immunity.
A growing body of evidence has highlighted the potential importance of CD4 T helper (Th) cells and B cells in cancer and immunotherapy (Alspach et al., 2019; Borst et al., 2018; Ferris et al., 2020; Helmink et al., 2020; Tran et al., 2014; Wieland et al., 2020). Much of the mechanistic research on CD4 T cells has focused on Th1, Th2, and Th17 cells (Galon et al., 2013; Goc et al., 2014; Guo et al., 2018; Liu et al., 2020; Martin-Orozco et al., 2009; Oh et al., 2020), and relatively little is known about the functional role of TFH cells in cancer. In non-cancer contexts, TFH cells provide necessary help for B cell maturation and function, and in a wide range of human cancers, the presence of B cells and TFH cells is correlated with prolonged survival and favorable therapeutic responses (Bindea et al., 2013; Cabrita et al., 2020; Cillo et al., 2020; Garaud et al., 2019; Germain et al., 2014; Griss et al., 2019; Gu-Trantien et al., 2013; Gu-Trantien et al., 2017; Helmink et al., 2020; Hennequin et al., 2016; Hollern et al., 2019; Kroeger et al., 2016; Lu et al., 2020; Petitprez et al., 2020; Truxova et al., 2018; Wieland et al., 2020). This correlation is especially strong when TFH cells and B cells are located in tumor-associated tertiary lymphoid structures (TLSs), as these structures could facilitate TFH-B cell interactions with subsequent effector functions in tumors (Rakaee et al., 2021; Ruffin et al., 2021; Sautes-Fridman et al., 2016; Sautes-Fridman et al., 2019; Sharonov et al., 2020). Yet, the functional importance of tumor-specific TFH cells remains uncertain, as does the importance of their interactions with B cells.
T cell-B cell interactions are necessary for the proper development and function of TFH cells and germinal center (GC) B cells in infection, vaccine responses, and autoimmune diseases (Crotty, 2019; Cyster and Allen, 2019). In canonical T-dependent immune responses, TFH cell differentiation requires interactions with two types of antigen-presenting cells. Dendritic cells (DCs) prime naïve CD4 T cells by providing antigen-MHC class II complexes in concert with co-stimulation, including that delivered by ICOS (inducible co-stimulator) via ICOS ligand (ICOSL) on DCs. These signals, plus cytokines, initiate a transcriptional program driven by the canonical TFH cell transcription factor B-cell lymphoma 6 (Bcl-6) and a second transcription factor Achaete-scute complex homolog 2 (Ascl2), leading to downregulation of P-selectin glycoprotein ligand 1 (PSGL1), and upregulation of PD1 and the chemokine receptor CXCR5, as cells acquire the TFH precursor cell phenotype (CD44hi PSGL1lo) (Choi et al., 2013; Nurieva et al., 2008; Poholek et al., 2010). Subsequent interactions with B cells provide additional antigen-MHC II and ICOSL signals and are required for further differentiation into mature TFH cells that are capable of migrating via CXCR5 to, and functioning within, the GC. (Crotty, 2011). Mature TFH cells (CD44hi ICOShi PSGL1lo PD1hi CXCR5hi) provide CD40 ligand (CD40L, CD154) and interleukin-21 (IL-21) to CD40+ B cells, signals necessary for the differentiation and maturation of GC B cells to memory B cells and long-lived plasma cells (Elgueta et al., 2009; Linterman et al., 2010; Zotos et al., 2010). Thus, antigen-engaged, bidirectional TFH-B cell collaboration plays a pivotal role in the initiation and maintenance of robust humoral immune responses (Mesin et al., 2016). Yet, the importance of interactions between tumor-specific TFH and B cells has not been studied in cancer. Nor is it clear whether anti-tumor TFH cells provide other functions in cancer beyond B cell help.
Studies in chronic viral infection and cancer have shown that CD4 T cells are critical to maintain effector CD8 T cell function (Battegay et al., 1994; Borst et al., 2018; Matloubian et al., 1994; Zajac et al., 1998; Zander et al., 2019), with the latter necessary for viral or tumor control and therapeutic efficacy after ICB (Fridman et al., 2017; Huang et al., 2017; van der Leun et al., 2020). The mechanisms of how CD4 T cells provide necessary help to CD8 T cells remain uncertain, but one proposal involves CD4 T cells providing IL-21, which drives cytolytic effector CD8 T cell differentiation, and prevents T cell exhaustion. (Elsaesser et al., 2009; Frohlich et al., 2009; Ren et al., 2020; Snell et al., 2018; Xin et al., 2015; Yi et al., 2009). IL-21 is the signature cytokine of TFH cells, although NKT and Th17 cells can also produce it under certain conditions (Chtanova et al., 2004; Coquet et al., 2007; Crotty, 2011; Korn et al., 2007; Nurieva et al., 2007; Suto et al., 2008; Wei et al., 2007). The cellular resources of IL-21 in chronic infection and cancer are still not well characterized.
Given their potential importance in anti-tumor immunity, we studied the function of tumor-specific TFH cells in lung cancer and determined how their interactions with tumor-specific B cells and CD8 T cells impacted the anti-tumor immune responses.
Results
GC B cells and TFH cells correlate with favorable clinical outcomes in LUAD patients
To investigate the potential importance of GC B cells and TFH cells in human LUAD, we first analyzed RNA sequencing data from 513 LUAD samples in the TCGA database using CIBERSORT and LM22-referenced single-cell gene expression profile matrix (Newman et al., 2015). We found that several immune cell types were enriched, including myeloid populations and CD8 T cells (Figure 1A), while B cell lineage and CD4 T cells were the first and third most abundant immune cell types, respectively (Figure 1A). Although the LM22 matrix does not distinguish GC B cells, the algorithm suggested that TFH cells and activated memory CD4 T cells comprised a large proportion of the CD4 T cells in tumors (Figure S1A). To more specifically delineate which types of CD4 T and B cells were present in LUAD, we analyzed published single cell RNA sequencing data from a study that included 44 patient samples of treatment-naïve LUAD from surgical resection or endobronchial ultrasound bronchoscopy biopsy (GSE131907) (Kim et al., 2020). We cataloged 49,901 T/NK cells and 22,592 B cells from primary tumors (tLung), metastatic lymph nodes (mLN), normal lung tissues (nLung) and normal lymph nodes (nLN), based on their previous annotations. T/NK cells were grouped into 15 clusters and visualized by dimension reduction method Uniform Manifold Approximation and Projection (UMAP) (Figure 1B). Eight clusters were distinguished as CD4 T cells, two of which displayed enrichment of TFH-signature genes. Notably, both TFH-like clusters (cluster 14 and 10) had higher expression of the TFH signature cytokine IL-21 (arrow) and were more prominent in tLung than nLung (Figure 1D). Likewise, from nine B cell clusters, we identified one GC B cell cluster (cluster 5) and two antibody-secreting cell (ASC) clusters (cluster 4 and 6), both of which were enriched in tLung, compared with nLung (Figure 1C and 1E; note, cluster numbers in Figure 1C and 1E do not correspond with those in Figure 1B and 1D). These data suggested that TFH and GC B cells are enriched in human LUAD tumor tissues.
Figure 1. GC B cells and TFH cells correlate with favorable clinical outcomes in LUAD patients.
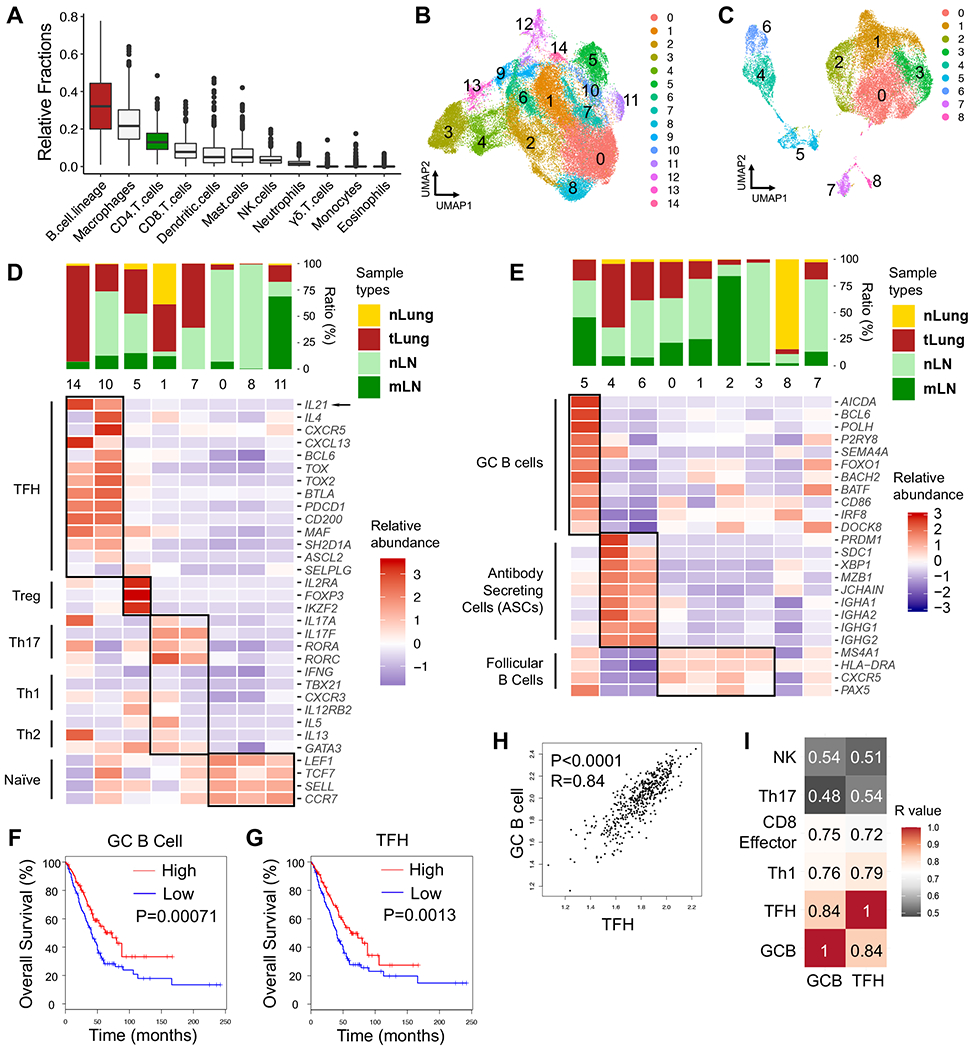
A, Estimated fractions of immune cell subsets from bulk expression data of primary tumor tissues in TCGA-LUAD patient cohort (n=513). CIBERSORT algorithm was used for deconvolution.
B – E, scRNA-seq analyses (GSE131907, Kim et.al.) of T/NK and B cell clusters in treatment-naïve human LUAD samples from primary sites (tLung), metastatic LNs (mLN), normal lung tissues (nLung) and normal LNs (nLN). B – C, UMAP displaying T/NK (B) and B cell clusters (C). D – E, Bar graphs displaying tissue origins (top) and heatmaps (bottom) showing relative abundance of signature genes in CD4 T cell (D) and B cell clusters (E).
F – G, Survival analyses based on GC B cell-signature (F) and TFH-signature (G) in TCGA-LUAD patient cohort (n=478).
H, Correlation analyses on GC B cell-signature and TFH-signature in TCGA-LUAD cohort.
I, Heatmap showing R values for the correlations among expression signatures of GC B cells, TFH, Th1, CD8 effector cells, Th17, NK cells in TCGA-LUAD cohort.
F – G, log-rank Mantel-Cox test. H – I, two-tailed Pearson correlation test.
To examine whether GC B cells and TFH cells associate with clinical outcomes of LUAD patients, we established gene expression signatures for LUAD GC B cells and TFH cells. Analysis of 478 LUAD cases from TCGA showed significant correlations between prolonged overall survival in LUAD patients and GC B cell- or TFH-signatures (Figure 1F and 1G, respectively). By contrast, Th1-, Th17- or NK cell-signatures did not correlate with overall survival, and effector CD8 T cell-signature correlated with slightly increased early survival benefits (Figure S1B). We also identified a significant correlation between the GC B cell- and TFH-signatures (R=0.84, p < 0.0001), which suggested the co-existence of these two cell types in tumors (Figure 1H). GC B cell- and TFH-signatures were also correlated with Th1- and CD8 effector T cell-signatures (Figure 1I and S1C). The correlation of TFH- and Th1- signatures was predicted as these cell types likely share a common developmental origin (Nakayamada et al., 2011; O’Shea and Paul, 2010). However, correlations between the signatures of GC B cells, TFH cells, and CD8 T cells were intriguing because they suggested potential connections between these three immune cell types. By contrast, neither the GC B cell- nor TFH-signatures were correlated with the signatures for Th17 cells or NK cells. Together, these data suggested that GC B cells and TFH cells are present in human lung cancer and contribute to anti-tumor immune responses.
TFH cells are enriched in murine cancer model
Next, we studied whether TFH cells were enriched in the context of murine LUAD. For this, we used a LUAD cell line made from syngeneic KrasLSL-G12D; Trp53fl/fl (KP) mice (Damo et al., 2020). We subcutaneously (s.c.) implanted 2 × 105 KP cells into C57BL/6 (B6) recipients. As positive controls, we s.c. implanted MC38 or B16-F10, which have been previously shown to elicit TFH responses in vivo (Alvarez Arias et al., 2014; Bindea et al., 2013; Magen et al., 2019; Roberti et al., 2020). After 10 – 12 days, we analyzed TFH cells in tumor-draining LNs (dLNs) by flow cytometry. Consistent with a progressive development of CD4 T cell responses, we observed ~3-fold increases in CD44hi PSGL1lo CD4 T cells in MC38 and B16-F10 tumor-bearing mice, compared to non-tumor bearing controls, and ~11% of these PSGL1lo cells further displayed upregulated CXCR5 and PD-1, indicating development into mature TFH cells (Figure S2A) (Weinstein et al., 2018). By contrast, only a ~1.5-fold increase of CD44hi PSGL1lo CD4 T cells was observed in KP tumor-bearing mice, with TFH cells comprising ~4% of these (Figure 2D).
Figure 2. KP-HELLO tumor cells elicit tumor-specific CD4 T cell and B cell responses.
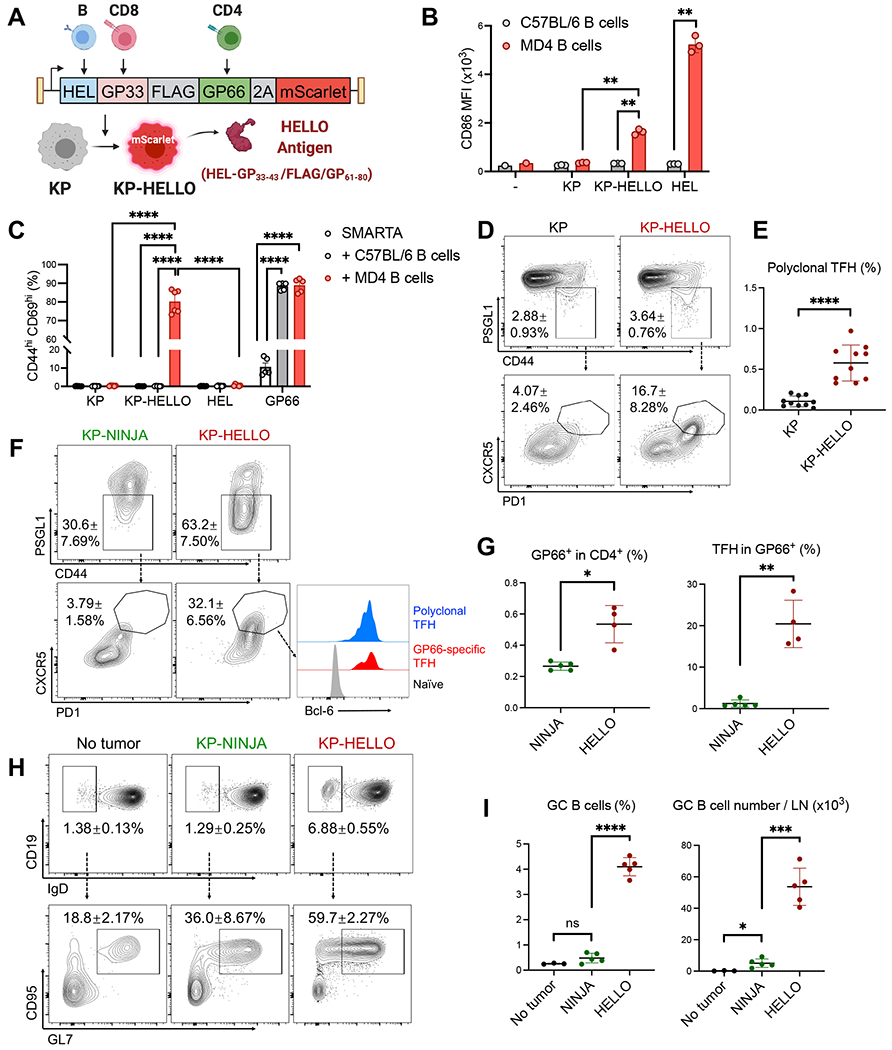
A, KP-HELLO model design.
B, CD86 expression (median fluorescence intensity, MFI) on MD4 or C57BL/6 B cells (CD19+ B220+ TCRb−) after 48-hour co-culture with KP or KP-HELLO supernatant, or 500ng/mL HEL.
C, CD44hi CD69hi frequency in SMARTA CD4 T cells, after 48-hour co-culture with MD4 or C57BL/6 B cells, and KP or KP-HELLO supernatant, 500ng/mL HEL or 5ug/mL GP61-80.
D – E, Representative flow plots (D) and frequency (E) of polyclonal CD4 TFH cells from tumor-draining LNs (dLNs) of KP or KP-HELLO tumor-bearing C57BL/6 mice.
F – G, Representative flow plots (F) and frequency (G) of I-Ab/GP66-77-specific (GP66-specific) CD4 TFH cells from dLNs of KP-NINJA or KP-HELLO tumor-bearing C57BL/6 mice.
H – I, Representative flow plots (H) and frequency (I) of GC B cells from dLNs of KP-NINJA or KP-HELLO tumor-bearing C57BL/6 mice. No tumor C57BL/6 mice were used as negative controls.
Data (mean ±SD) were pooled, from 2 – 5 mice/group/experiment, and representative of at least 2 independent experiments. LN tissues were harvested on day 10–12. B – C, E, G, I, unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Engineering KP cell lines to elicit antigen-specific T cell and B cell responses
KP cell lines are notable in that they have low numbers of somatic mutations (DuPage et al., 2011; McFadden et al., 2016; Yadav et al., 2014) and they do not elicit meaningful T or B cell responses, as evidenced by their unaffected growth in T and/or B cell deficient animals (Figure S2B). Taking advantage of this, the KP tumor cell line provided a system where we could assess how engineered neoantigens might drive anti-tumor T and B cell responses, and how these responses impact tumor growth.
We recently published the “NINJA” model and showed that the NINJA system could be used to program neoantigen-expressing KP lung tumors (Connolly et al., 2021; Damo et al., 2020; Fitzgerald et al., 2021). The cDNAs encoding NINJA neoantigens contain model antigens from the lymphocytic choriomeningitis virus (LCMV) glycoprotein: GP33-43 and GP61-80, which are recognized by GP33-specific CD8 (P14) and GP66-specific CD4 (SMARTA) T cell receptor (TCR) transgenic T cells, respectively, separated by the FLAG peptide sequence and embedded within a flexible loop region between β-strands 7 and 8 of GFP (called GFP-GP33-43/FLAG/GP61-80). The LCMV antigens were chosen because there is a wide array of immunologic tools that are available for studying T cell responses against these antigens (i.e., MHC class I and II tetramers and TCR transgenic mice). Moreover, we had previously engineered the sequence GP33-43/FLAG/GP61-80 into GFP (called GFP-GP33-43/FLAG/GP61-80 below) such that it allowed proper folding and fluorescence of GFP. Thus, we reasoned it would be an ideal starting point to design a neoantigen that was recognized by both T and B cells. Hen-Egg Lysozyme (HEL) is a widely used model B cell antigen, recognized by MD4 and SWHEL B cell receptor (BCR) transgenic B cells (Goodnow et al., 1988; Phan et al., 2003). Therefore, to facilitate the investigation of tumor-specific T cell and B cell responses, we created a fusion protein of HEL, LCMV GP33-43/FLAG/GP61-80, T2A, and codon-Optimized mScarlet (Figure 2A). This substrate was called HELLO and generated two polypeptides: the neoantigen fusion of HEL-GP33-43/FLAG/GP61-80, and mScarlet, a bright red fluorescent protein to distinguish neoantigen-expressing tumor cells.
We transduced the KP tumor cell line with HELLO-encoding lentiviral vectors, in which HELLO was the only protein product, and sorted mScarlet-positive KP-HELLO tumor cells with FACS. We placed a signal peptide sequence in the HELLO construct prior to the neoantigen fusion HEL-GP33-43/FLAG/GP61-80 so that it would be secreted by KP-HELLO cells, while mScarlet protein would remain in the cytoplasm (Figure 2A). To validate this, we isolated HEL-specific MD4 B cells, or polyclonal B6 B cells that are not enriched in HEL-specific BCRs (negative control), and added either HEL protein (positive control), KP or KP-HELLO supernatant into each condition. After 48 hours of culture, flow cytometric analyses showed activation of MD4 B cells, based on a significant increase in CD86 median fluorescence intensity (MFI), in samples where MD4 cells were incubated with KP-HELLO supernatant or HEL protein (Figure 2B). By contrast, B cell activation was not observed in negative control conditions. Next, we tested if HEL-specific B cells could present GP66-77 from HEL-GP33-43/FLAG/GP61-80 to GP66-specific CD4 T cells. For this, we cultured naïve SMARTA CD4 T cells with MD4 B cells, or as negative controls, SMARTA T cells alone or with polyclonal B cells from B6 mice. Into each co-culture, we added KP or KP-HELLO supernatant, HEL protein (negative control), or GP61-80 peptide (positive control). After 48 hours, there was a significant increase of CD44hi CD69hi (activated) CD4 T cells in GP61-80 containing samples, demonstrating that B6 and MD4 B cells were capable of presenting GP61-80 to SMARTA CD4 T cells (Figure 2C and S3A). By contrast, when cultured with KP-HELLO supernatant, only MD4 B cells could activate SMARTA cells, demonstrating that a HEL-specific BCR was required for B cells to uptake, process, and present HEL-GP33-43/FLAG/GP61-80 antigen to GP66-specific CD4 T cells. Of note, we could not detect robust GP33-specific CD8 T cell responses from KP-HELLO (Figure S3B – S3F). This was likely due to the biology of the GP33-43 epitope in HELLO system (i.e., antigen processing), rather than a technical breakdown, and HEL-GP33-43/FLAG/GP61-80 contained many other potential MHC class I restricted epitopes besides GP33-43 (Table S3). So we focused on endogenous polyclonal CD8 T cell responses in following analyses.
KP-HELLO tumors elicit tumor-specific TFH and GC B cell responses in vivo
To assess whether the introduction of HELLO neoantigens into KP tumors elicited TFH cell responses in vivo, we s.c. implanted 2 × 105 KP or KP-HELLO tumor cells into B6 mice and analyzed endogenous CD4 T cells in dLNs using flow cytometry. On day 10 – 12, the frequency of CD44hi PSGL1lo polyclonal CD4 T cells increased ~1.3 fold overall (KP-HELLO vs. KP), while there were ~4-fold increases in the frequency of these PSGL1lo cells that further upregulated PD-1 and CXCR5 (Figure 2D), resulting in a significant increase in the frequency of TFH cells (Figure 2E).
Next, we tested whether the linkage of GP61-80 to a B cell antigen (HEL) elicited an increase in TFH cells among tumor-specific (GP66-specific) CD4 T cells. The GFP-GP33-43/FLAG/GP61-80 in NINJA retains fluorescence and is recognized by conformation-specific anti-GFP antibodies. Thus, we reasoned that NINJA would be unlikely to elicit GFP-specific T or B cell responses in GFP-tolerant recipient mice (express GFP in an irrelevant location—we used Il4GFP [4get] mice), yet, GFP-tolerant recipients were still able to mount GP66-specific CD4 T cell responses contained in NINJA (Damo et al., 2020). Therefore, we could directly compare the differentiation of endogenous tumor-specific CD4 T cells under conditions where tumors expressed CD4 T cell-recognized neoantigens with different capabilities to elicit antigen-specific B cell responses (KP-NINJA versus KP-HELLO). We previously described KP-NINJA (KPN1.1) cell line, which was derived from an autochthonous KP-NINJA LUAD (Fitzgerald et al., 2021).
We s.c. implanted KP-NINJA or KP-HELLO tumors into Il4GFP mice and analyzed GP66-specific CD4 T cells for their expression of TFH markers. Endogenous GP66-specific CD4 T cells were identified in mice with KP-HELLO or KP-NINJA tumors using I-Ab/GP66-77-specific tetramers between days 10 – 12 after implant (Figure S4A and 2G, ~2-fold reduction in KP-NINJA). In KP-HELLO mice, ~50-60% of the GP66-specific CD4 T cells downregulated PSGL1, ~30% of which further upregulated PD-1 and CXCR5 (TFH cells; Figure 2F). These TFH cells were also marked by upregulation of Bcl-6, which is required for TFH cell differentiation (Johnston et al., 2009; Nurieva et al., 2009; Yu et al., 2009) (Figure 2F). Comparatively, the frequency of GP66-specific TFH cells was 17-fold higher in KP-HELLO tumor-bearing mice than KP-NINJA (Figure 2G). KP-HELLO tumors also potentiated GC B cell responses in vivo, as there was a ~5-fold increase in the frequency of IgDlo activated B cells compared with KP-NINJA, and ~50-60% of these cells were GL7+ CD95+, consistent with a GC B cell phenotype (Figure 2H). As a result, KP-HELLO tumors had ~8.5-fold and ~10.5-fold increases in the frequency and number, respectively, of GC B cells in dLN compared to KP-NINJA tumors (Figure 2I). Similarly, when B6 mice containing adoptively transferred allotype-marked naïve HEL-specific SWHEL B cells and SMARTA CD4 T cells were challenged with KP-HELLO tumor cells (Figure S4B), ~50% of SMARTA cells were PSGL1lo and ~25% of these were mature TFH cells (Figure S4C). Likewise, ~30% of the adoptively transferred SWHEL B cells were IgDlo, ~50% of which were GL7+ CD95+ Bcl-6hi (Figure S4D). Together, these data demonstrated that the B cell-containing neoantigens in KP-HELLO tumors were sufficient to elicit tumor-specific TFH and GC B cell responses.
Introduction of HELLO induces TFH- and B cell-dependent tumor control
KP-HELLO tumors displayed an increased growth rate in immunodeficient RAG1 KO mice compared with B6 mice (Figure S5A). This was not due to a cell intrinsic growth defect as parental KP and KP-HELLO tumors grew similarly in RAG1 KO mice (Figure S5B). Because KP tumor cells grow similarly in RAG1 KO and B6 hosts (Figure S2B), the reduced growth of KP-HELLO in B6 mice was due to the impact of adaptive immune cells responding to HELLO. To assess whether B cells, CD4 T cells, and specifically CD4 TFH cells were necessary for tumor control, we measured KP-HELLO tumor growth in uMT, CIITA KO mice (lacking mature B cells or CD4 T cells, respectively) and CD4-Cre Bcl6fl/fl, a commonly used TFH-deficient animal model (Crotty, 2019; Hollister et al., 2013). We observed that CD4 T cells, TFH cells and B cells were critical for optimal control over KP-HELLO tumor growth (Figure 3A – 3C). By contrast, KP-NINJA tumor growth was not significantly different in B6 vs. uMT hosts (Figure 3E). We further analyzed CD4 T cell phenotypes in KP-HELLO tumor-bearing uMT and CD4-Cre Bcl6fl/fl mice. Endogenous GP66-specific CD4 T cell responses were readily detectable in dLNs from KP-HELLO tumor-bearing B6, uMT and CD4-Cre Bcl6fl/fl mice (Figure S5C and S5F), but GP66-specific mature TFH cells were absent in uMT and CD4-Cre Bcl6fl/fl mice (Figure 3D, S5E and S5G). Notably, the latter also had significant reductions in GP66-specific CD44hi PSGL1lo cells, which comprise both TFH-precursors and TFH cells (Figure S5D and S5G). These data demonstrated that B cells and TFH cells were necessary to reduce the growth of KP-HELLO tumors.
Figure 3. CD4 T cells, TFH cells and B cells are necessary for optimal control over KP-HELLO tumors.
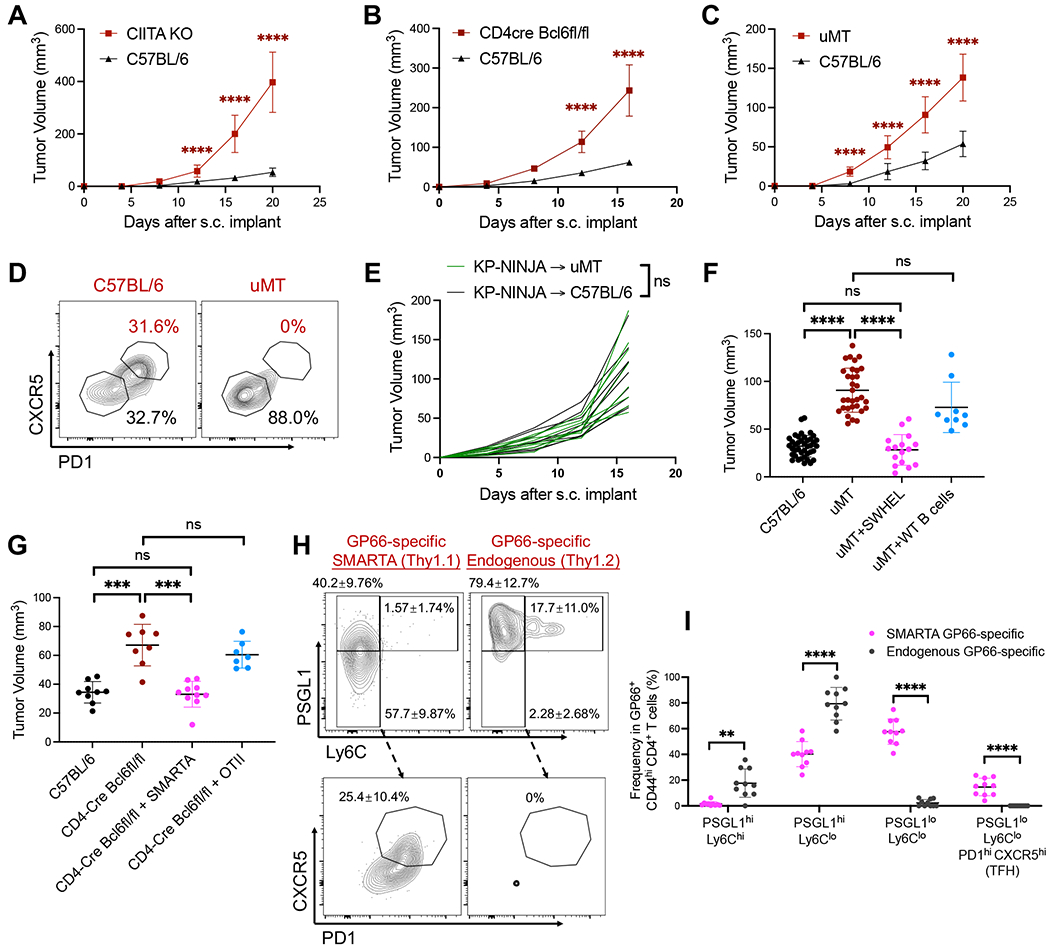
A and C, Tumor growth curves: C57BL/6, CIITA KO or uMT mice were subcutaneously (s.c.) implanted with 2 x 105 KP-HELLO. Growth curves of C57BL/6 (n = 40) and uMT (n = 32), pooled from 8 independent experiments, were set as the baseline of analyses.
B, Tumor growth curves: C57BL/6 or CD4-Cre Bcl6fl/fl mice were s.c. implanted with 5 x 105 KP-HELLO.
D, Representative flow plots of GP66-specific CD4 TFH cells from dLNs of KP-HELLO tumor-bearing C57BL/6 or uMT mice. Flow cytometric analyses were performed on day 10 – 12 and quantified in Figure S5. Flow plots were pre-gated on TCRb+ CD4+ GP66-tetramer+ CD44hi PSGL1lo.
E, Tumor growth curves: C57BL/6 or uMT mice were s.c. implanted with 5 x 105 KP-NINJA. Each line represented one mouse.
F, 1 x 106 SWHEL or WT C57BL/6 B cells were adoptively transferred into uMT recipients, 1 day prior to KP-HELLO s.c. implant. Graph shows tumor volumes on day 16.
G – I, 1 x 105 SMARTA CD4 T cells (Thy1.1/Thy1.1) or OT-II CD4 T cells (Thy1.1/Thy1.2) were adoptively transferred into CD4-Cre Bcl6fl/fl recipients (Thy1.2/Thy1.2), 1 day prior to KP-HELLO s.c. implant.
G, Tumor volumes on day 16 after s.c. implant.
H – I, Representative flow plots (H) and frequency (I) of GP66-specific CD4 T cell subsets from dLNs of [CD4-Cre Bcl6fl/fl + SMARTA] group, separated by transferred SMARTA and endogenous cells. Flow plots were pre-gated on TCRb+ CD4+ GP66-tetramer+ CD44hi.
Data (mean ±SD) were pooled, from 3–10 mice/group/experiment, and representative of at least 2 independent experiments. A – C and E, two-way ANOVA. F – G and I, unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
It remained possible that TFH and B cells acted in a non-antigen driven fashion to promote anti-tumor immune response. To formally test whether tumor-specific B cells were sufficient to control KP-HELLO tumors, we performed a rescue experiment by adoptively transferring naïve tumor-specific SWHEL or polyclonal WT B6 B cells into uMT recipients prior to KP-HELLO tumor initiation. Only adoptive transfer of the SWHEL B cells rescued tumor control in uMT mice back to the level observed in control mice (Figure 3F). Likewise, adoptive-transfer of naïve GP66-specific SMARTA CD4 T cells into CD4-Cre Bcl6fl/fl recipients, but not irrelevant-antigen-specific OT-II CD4 T cells, restored the ability of TFH-deficient animals to control KP-HELLO tumors (Figure 3G). ~50-60% of these transferred SMARTA cells downregulated PSGL1, with ~25% of these becoming mature TFH cells (Figure 3H). In contrast, endogenous GP66-specific T cells from the same CD4-Cre Bcl6fl/fl animal were incapable of developing into mature TFH cells or even becoming TFH precursors (note 96% decrease in PSGL1lo cells; Figure 3I). Thus, in the rescued CD4-Cre Bcl6fl/fl mice, the TFH compartment was solely comprised of SMARTA CD4 T cells. Lymphocyte antigen 6C (Ly6C) expression on CD44hi PSGL1hi has been widely used to discriminate TH1 from TFH cells, as Ly6C is regulated by TH1-specific transcription factor T-bet (Hale et al., 2013; Marshall et al., 2011; Weinstein et al., 2018; Yamanouchi et al., 1998; Zander et al., 2018). Compared to the transferred SMARTA CD4 T cells, the endogenous GP66-specific T cells in CD4-Cre Bcl6fl/fl mice had increased frequencies of both PSGL1hi Ly6Clo and PSGL1hi Ly6Chi TH1-like cells, the latter of which expressed higher amounts of T-bet than their TFH counterparts (Figures 3H–3I and S5G – S5H). Similar results were seen from endogenous GP66-specific T cells in CD4-Cre Bcl6fl/fl mice that had not received SMARTA CD4 T cells.
TFH cells, B cells and B-cell-recognized-neoantigens are critical for anti-tumor effector CD8 T cell responses
Given that differences in tumor growth were apparent between WT mice and B- or T-cell deficient mice at relatively early timepoints after tumor implant (8-12 days), we focused on whether CD8 T cells were impacted by B- and T-cell deficiency. First, we determined whether CD8 T cells were necessary for optimal tumor control in our model by examining KP-HELLO tumor growth in B6 mice with or without anti-CD8a antibody depletion. CD8 T cell depletion was efficient (data not shown) and led to impaired tumor control in mice (Figure 4A). We next examined the impact of TFH deficiency on CD8 T cells. In KP-HELLO tumor-bearing WT mice, ~40% of the tumor-infiltrating CD8 T cells were granzyme Bhi, consistent with an effector phenotype (Figure 4B). In line with this, most of these granzyme Bhi cells expressed PD-1. In comparison, TFH-deficient mice had a significant reduction in the frequency and number of tumor-infiltrating PD-1hi granzyme Bhi CD8 T cells (Figure 4B, see Figure 6O–6P for further quantification). Similarly, there was a significant reduction in the frequency and number of tumor-infiltrating PD-1hi granzyme Bhi CD8 T cells in uMT mice (Figure 4C – 4E). These data supported a model where TFH cells and B cells support the function of tumor-infiltrating CD8 T cells, but to test this further, we compared CD8 T cell function in WT and uMT KP-NINJA tumor-bearing mice, as these tumors did not elicit TFH or GC B cell responses. Critically, B cell-deficiency did not impact Granzyme B expression by tumor-infiltrating CD8 T cells in KP-NINJA tumors (Figure 4F–4H). These data demonstrated that B-cell antigens can drive effector CD8 T cell responses via a TFH- and B-cell dependent mechanism.
Figure 4. TFH and B cells are critical to drive anti-tumor effector CD8 T cell responses.
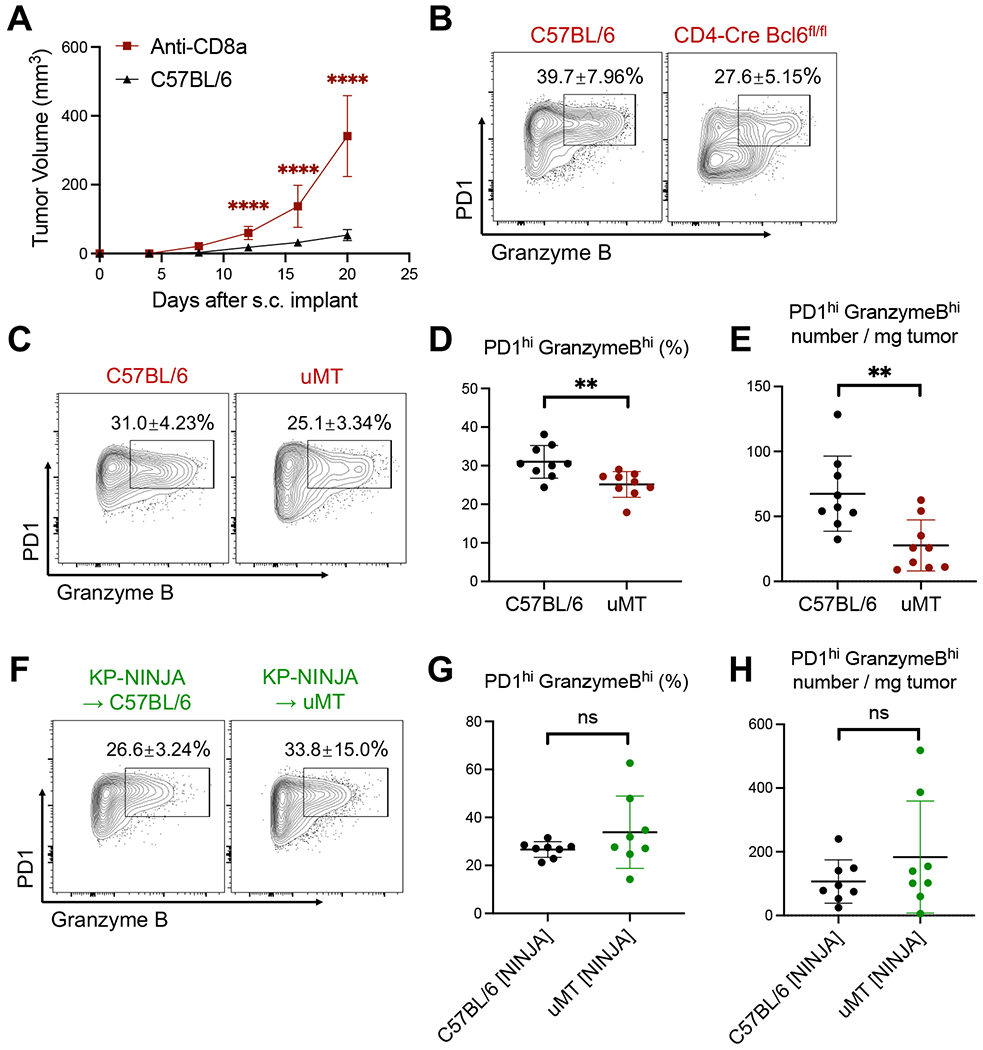
A, Control and anti-CD8a treated C57BL/6 mice were s.c. implanted with 2 x 105 KP-HELLO. Growth curve of C57BL/6 (n = 40), pooled from 8 independent experiments, was set as the baseline of analyses.
B, Representative flow plots of tumor-infiltrating PD1hi Granzyme Bhi CD8 T cells from KP-HELLO tumors in C57BL/6 or CD4-Cre Bcl6fl/fl mice, further quantified in Figure 6O–6P.
C – E, Representative flow plots (C), frequency (D) and number/mg tumor (E) of tumor-infiltrating PD1hi Granzyme Bhi CD8 T cells from KP-HELLO tumors in C57BL/6 or uMT mice.
F – H, Representative flow plots (F), frequency (G) and number/mg tumor (H) of tumor-infiltrating PD1hi Granzyme Bhi CD8 T cells from KP-NINJA tumors in C57BL/6 or uMT mice.
Data (mean ±SD) were pooled, from 3–5 mice/group/experiment, and representative of at least 2 independent experiments. Tumors were harvested on day 15–16. A, two-way ANOVA. D – E and G – H, unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 6. Collaborations between CD4 T cells and B cells are critical for anti-tumor immune responses.
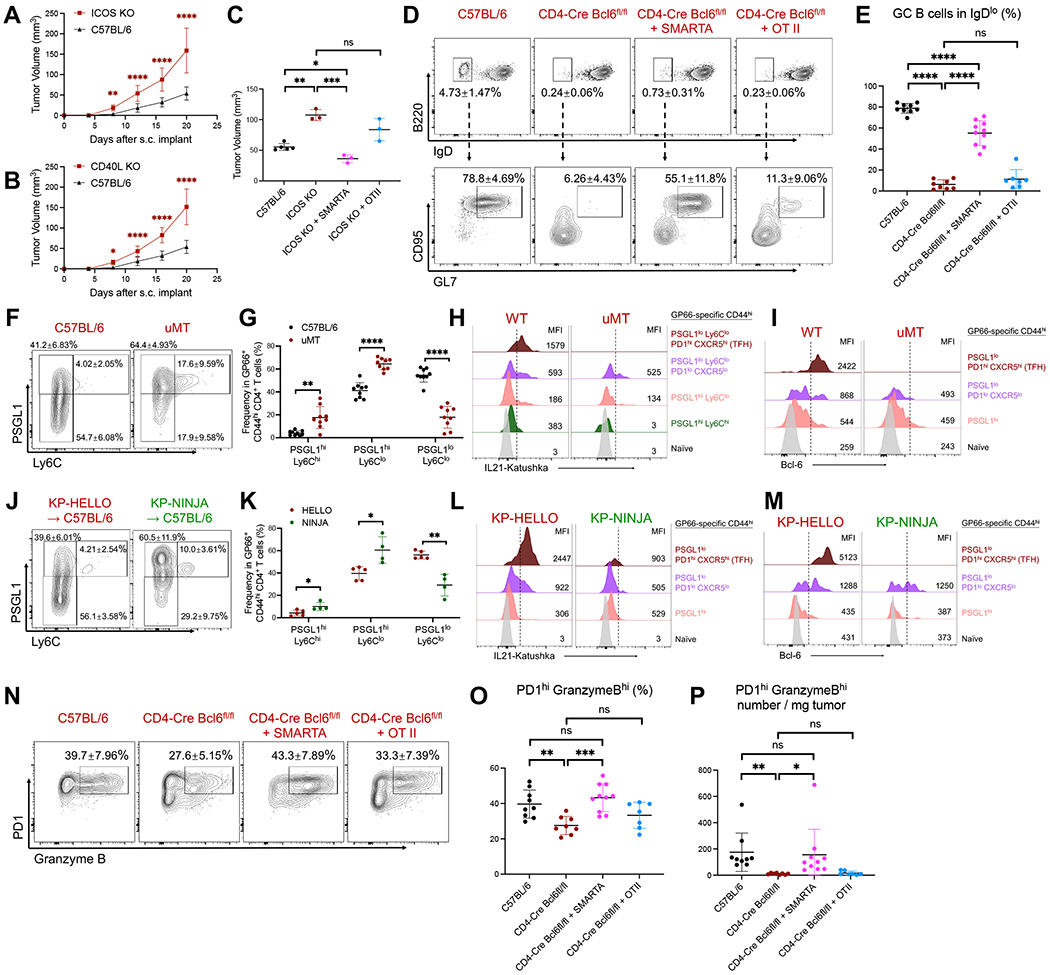
A – B, Tumor growth curves: C57BL/6, ICOS KO or CD40L KO mice were s.c. implanted with 2 x 105 KP-HELLO. Growth curve of C57BL/6 (n = 40), pooled from 8 independent experiments, was set as the baseline of analyses.
C, 1 x 105 SMARTA CD4 T cells or OT-II CD4 T cells were adoptively transferred to ICOS KO recipients prior to KP-HELLO s.c. implant. Graph shows tumor volumes on day 16.
D – E, Representative flow plots (D) and frequency (E) of GC B cells in dLNs of KP-HELLO tumors in C57BL/6, CD4-Cre Bcl6fl/fl mice, or CD4-Cre Bcl6fl/fl recipients following transfer of 1 x 105 SMARTA T cells or OT-II T cells (as described in Figure 3G).
F – G, Representative flow plots (F) and frequency (G) of GP66-specific CD4 T cell subsets from dLNs of KP-HELLO tumor-bearing WT or uMT mice.
J – K, Representative flow plots (J) and frequency (K) of GP66-specific CD4 T cell subsets from dLNs of KP-HELLO or KP-NINJA tumor-bearing WT mice.
H – I and L – M, Histograms displaying IL-21 or Bcl-6 expression on naïve and GP66-specific CD4 T cell subsets. Gating strategy was shown in Figure 3D, 6F and 6J.
N – P, Representative flow plots (N), frequency (O) and number/mg tumor (P) of tumor-infiltrating PD1hi Granzyme Bhi CD8 T cells from KP-HELLO tumors in C57BL/6, CD4-Cre Bcl6fl/fl mice, or CD4-Cre Bcl6fl/fl recipients following transfer of 1 x 105 SMARTA T cells or OT-II T cells (as described in Figure 3G).
Data (mean ±SD) were pooled, from 3–5 mice/group/experiment, and representative of at least 2 independent experiments. A – B, two-way ANOVA. C, E, G, K and O – P, unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
B cells are required to drive effective TFH and CD8 T cell responses in autochthonous tumors
In addition to s.c. implant tumor model, we also tested whether TFH and CD8 T cell responses required the presence of B cells in the autochthonous KrasLSL-G12D; Trp53fl/fl (KP) mouse LUAD model. KP tumors recapitulate cancers that lack neoantigens (McFadden et al., 2016), but have been programmed to express T cell neoantigens using lentiviral vectors (LV) (DuPage et al., 2011; Joshi et al., 2015; Pfirschke et al., 2016). Thus, we engineered non-replicating Lenti-HELLO-Cre LV, encoding both the HELLO fusion protein and Cre recombinase, and administered them intratracheally into KP and B-cell deficient uMT-KP mice (Figure S6A). This resulted in autochthonous HELLO-expressing lung tumors for analysis after 8 weeks. The lungs of mice are highly vascularized, so to distinguish lung tissue-infiltrating immune cells from circulating cells for flow cytometric analyses, we performed in vivo-labeling of circulating immune cells with PE-CF594 conjugated anti-CD45 (lung-tissue immune cells are CD45i.v.Neg; Figure S6B). In non-tumor-bearing mice, <1% of B, CD4 and CD8 T cells were in lung tissue, but in HELLO tumor-bearing mice, this increased to ~13% of B cells, ~60% of CD4 T cells, and ~ 40% of CD8 T cells (Figure S6C). Endogenous GP66-specific mature TFH cells were observed in the tumor lung tissues and dLNs of HELLO-tumor bearing KP mice, but not in uMT-KP mice (Figure S6D). Likewise, analysis of activated CD19+ B220+ IgDlo B cells showed ~2% and ~50% of these cells expressed GC B cell markers (IgDlo GL7+ CD95+ Bcl6hi) in lung and dLN, respectively (Figure S6E). Finally, the absence of B cells led to a significant drop in tumor-infiltrating CD44hi PD1hi effector CD8 T cells (Figure S6F). These data were consistent with our findings in s.c. implanted KP-HELLO tumors.
TFH and B cell-dependent IL-21 production is required for robust effector CD8 T cell responses in KP-HELLO tumors
We next investigated the mechanism for how TFH and B cells promote the effector functions of tumor-infiltrating CD8 T cells. We reasoned this could be via IL-21, a signature cytokine of TFH cells (Crotty, 2011) that can drive effector function of CD8 T cells (Zander et al., 2019). We first examined which cells in the dLN express IL-21 by s.c. implanting KP-HELLO into Il21-Katushka (Kat) reporter mice (Weinstein et al., 2016). IL-21 expression was only detected from mice with KP-HELLO tumors, but not KP tumors, demonstrating that IL-21 expression required the presence of the HELLO neoantigens (Figure 5A). >95% of the IL-21 expressing cells were CD4 T cells, and ~85% of these were PSGL1lo with ~70% expressing mature TFH cell markers (Figure 5A). We next addressed whether IL-21 expression on T cells was B-cell-dependent using Il21Kat/Kat x uMT mice. In the absence of B cells, we observed ~70% reduction in the frequency of IL-21-expressing GP66-specific CD44hi CD4T cells in dLNs and ~35% reduction in tumors (Figure 5B–5C). Likewise, B-cell-recognized neoantigens were required for IL-21 expression as KP-NINJA tumors exhibited ~70% reduction in the frequency of IL-21 expressing GP66-specific CD44hi CD4 T cells (Figure 5D–5E). Thus, the induction and/or maintenance of IL-21 in TFH cells by tumors required B cells and B-cell-recognized neoantigens.
Figure 5. IL-21 production is dependent on B cells and B-cell-recognized neoantigens and is required for effector CD8 T cell responses.
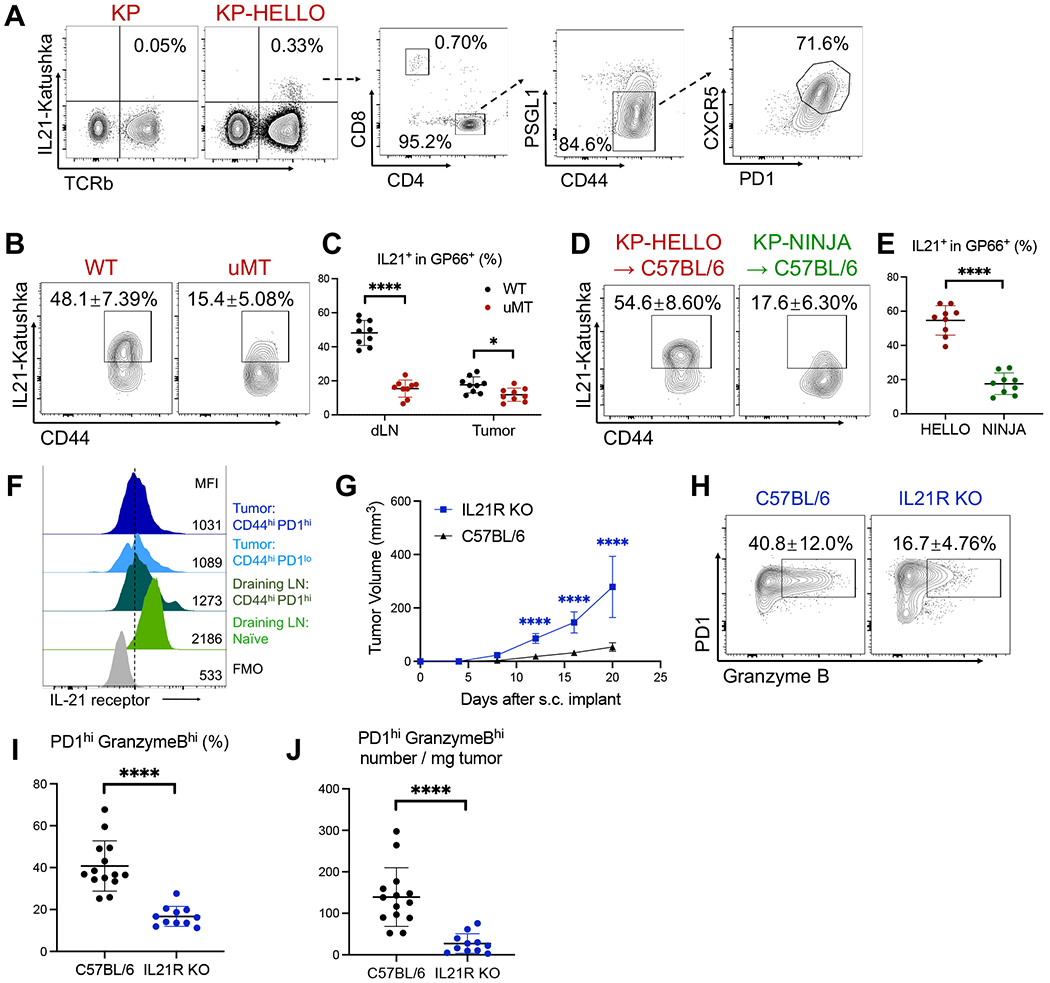
A, Representative flow plots showing IL-21-producing cells in dLNs of KP or KP-HELLO tumor-bearing mice.
B – C, Representative flow plots (B) and frequency (C) of IL-21 producers in GP66-specific CD44hi CD4 T cells from dLNs and tumors of KP-HELLO tumor-bearing WT or uMT mice.
D – E, Representative flow plots (D) and frequency (E) of IL-21 producers in GP66-specific CD44hi CD4 T cells from dLNs of KP-HELLO or KP-NINJA tumor-bearing WT mice
F, IL-21 receptor expression on CD8 T cell subsets from dLNs and tumors of KP-HELLO tumor-bearing mice. Gate was drawn based on FMO in CD44hi PD1hi tumor-infiltrating CD8 T cells.
G, Tumor growth curves: C57BL/6 and IL21R KO mice were s.c. implanted with 2 x 105 KP-HELLO. Growth curve of C57BL/6 (n = 40), pooled from 8 independent experiments, was set as the baseline of analyses.
H – J, Representative flow plots (H), frequency (I) and number/mg tumor (J) of tumor-infiltrating PD1hi Granzyme Bhi CD8 T cells from KP-HELLO tumors in C57BL/6 or IL21R KO mice.
Data (mean ±SD) were pooled, from 3–10 mice/group/experiment, and representative of at least 2 independent experiments. C, E, and I – J, unpaired t-test with Welch’s correction. G, two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The receptor for IL-21 (IL21R) was expressed on CD44hi PD1hi effector CD8 T cells in both tumors and dLNs (Figure 5F). To test if IL-21 was important for KP-HELLO tumor growth, we s.c. implanted KP-HELLO tumor cells into B6 or IL-21R knockout (IL21R KO) mice. KP-HELLO tumors grew more rapidly in IL-21R KO than B6 mice (Figure 5G). There was also a ~60% and ~80% reduction in the frequency and number of PD-1hi granzyme Bhi tumor-infiltrating CD8 T cells in IL-21R KO mice, respectively (Figure 5H–5J). These findings were in line with data from other groups showing that IL-21 acts on CD8 T cells to drive the effector functions (Elsaesser et al., 2009; Frohlich et al., 2009; Li et al., 2021; Snell et al., 2018; Yi et al., 2009; Zander et al., 2019) and confirmed the critical role of IL-21 in potentiating anti-tumor CD8 T cell responses in KP-HELLO tumors.
Interactions of TFH and B cells are required to induce anti-tumor immune response.
To close the loop between TFH, B cells, IL-21 signals and effector CD8 T cell responses, we first assessed whether T cell-B cell interactions were necessary for anti-tumor immunity. In T-dependent immune responses, CD40/CD40L-deficient and ICOS/ICOSL-deficient mice are not able to form GC B cells or mature TFH cells due to defects in the ability of T cells and B cells to provide mutual help (Choi et al., 2011; Ise et al., 2018; Liu et al., 2015; Nurieva et al., 2008; Renshaw et al., 1994; Zhang et al., 2020). Deficiencies in ICOS and CD40L led to more rapid growth of KP-HELLO tumors, on par with the changes observed in uMT mice (Figure 6A–6B). Mature TFH and GC B cell responses were impaired in ICOS deficient mice (data not shown). ICOS deficiency could impact many cell types, but adoptive transfer of WT SMARTA T cells into ICOS deficient hosts rescued KP-HELLO tumor control, while transfer of OT-II T cells did not (Figure 6C). These data are consistent with the idea that T cell and B cell interactions are necessary for control over the growth of KP-HELLO tumors.
Next, we assessed the impact of SMARTA T cell rescue on B cells and CD8 T cells in TFH-deficient mice, because under this experimental setting, the only functional TFH cells were transferred GP66-specific SMARTA cells (Figure 3G–3I). Polyclonal GC B cell responses in CD4-Cre Bcl6fl/fl mice were significantly reduced compared to B6 controls, and this was rescued by adoptive transfer of SMARTA but not OT-II T cells (Figure 6D–6E). This ~9-fold increase in polyclonal GC B cell frequency was particularly striking, as the rescue only restored TFH cells with a single specificity (GP66-specific) and was consistent with potent interactions between B cells and tumor-specific TFH cells in KP-HELLO tumor model. To assess the functional role of T cell-B cell interactions, we used flow cytometry to analyze how the differentiation programs of tumor-specific CD4 T cells were altered when tumor-bearing mice lacked B cells (B6 vs. uMT), or B-cell-recognized neoantigens (KP-HELLO vs. KP-NINJA). In both cases, tumor-specific TFH cell responses were significantly compromised (Figure 3D, S5E and 2F – 2G), but there was also a significant reduction of PSGL1lo Ly6Clo cells (Figure 6F–6G and 6J – 6K). These data are consistent with the hypothesis that B cells and B-cell-recognized neoantigens control the developmental fates of tumor-specific CD4 T cells by promoting the TFH differentiation program (beginning with downregulation of PSGL1). To assess the impact of T cell-B cell interactions over the course of TFH differentiation, we analyzed Bcl-6 and IL-21 expression at the different stages of CD4 T cell differentiation (based on markers) in B6 vs. uMT, or KP-HELLO vs. KP-NINJA. We found that both Bcl-6 and IL-21 expression were compromised at each stage in the absence of TFH-B cell interactions (Figure 6H–6I, 6L–6M, gating strategy in Figure 3D, 6F and 6J). Similar data were seen in mice with MC38 and B16-F10 tumors (Figure S7A–S7C). Finally, to assess the functional impact of TFH-B cell interactions on tumor-infiltrating CD8 T cells, we analyzed their phenotypes in the context of rescue of TFH-deficient mice adoptively transferred with WT SMARTA CD4 T cells. The significant decrease in PD-1hi granzyme Bhi amongst tumor-infiltrating CD8 T cells in TFH-deficient mice was rescued to WT levels by SMARTA T cell transfer, but not by OT-II T cell transfer, both in terms of frequency and number (Figure 6N–6P). Thus, the presence of tumor-specific TFH cells was sufficient to drive effector functions in tumor-infiltrating CD8 T cells.
Discussion
We have identified a mechanism by which interactions between tumor-specific CD4 T cells and B cells are necessary for the generation of IL-21-producing TFH cells, and this leads to increased Granzyme B expression by tumor-infiltrating CD8 T cells. We provided evidence for this B cell-TFH cell-IL21 axis through analyses of LUAD patients and animal models. We also demonstrated that this pathway can be driven by B-cell-recognized neoantigens, underscoring a mechanism by which tumor neoantigens can guide CD4 T cell differentiation.
Our findings on tumor-infiltrating TFH cells and GC B cells in human LUAD are in line with previous observations that NSCLC and other cancer types contain tumor-associated TFH-like cells with an enriched TFH signature (i.e., PDCD1, ICOS, CD200, MAF and IL21) (Bindea et al., 2013; Cillo et al., 2020; Gu-Trantien et al., 2013; Gu-Trantien et al., 2017; Guo et al., 2018; Li et al., 2019; Noel et al., 2021; Satpathy et al., 2019; Thommen et al., 2018; Zhang et al., 2021). In human studies including autoimmune diseases, TFH-like cells have been shown to express CXCL13, the ligand of CXCR5, which was associated with CXCR5+ B cell infiltration and tertiary lymphoid structure (TLS) formation (Bocharnikov et al., 2019; Li et al., 2015; Manzo et al., 2008; Morita et al., 2011; Rao et al., 2017). The presence of TLSs correlates with favorable clinical outcomes in many types of human cancers (Cabrita et al., 2020; Dieu-Nosjean et al., 2008; Germain et al., 2014; Goc et al., 2014; Helmink et al., 2020; Lu et al., 2020; Petitprez et al., 2020; Sautes-Fridman et al., 2016; Sautes-Fridman et al., 2019; Voabil et al., 2021; Wieland et al., 2020), and the TLSs containing mature GCs were further associated with more clinical benefits for many types of human cancers (Rakaee et al., 2021; Ruffin et al., 2021; Silina et al., 2018; Vanhersecke et al., 2021). These studies suggested tumor-specific antibody production as an important output of GC responses. However, our data suggested an alternative possibility that TLS could promote tumor-specific T cell-B cell collaborations that would support the effector functions of tumor-infiltrating CD8 T cells via IL-21.
Based on our data, we believe that TFH cells are a key physiological source of IL-21 for tumor-infiltrating effector CD8 T cells, and that this is one mechanism for how TFH cells promote better outcomes in LUAD patients. A critical question arising from our work is how and where TFH cells provide IL-21 to CD8 T cells. Several studies in chronic LCMV infection have focused on the critical role of CD4 T cell-derived IL-21 in promoting stem-like CXCR5+ TCF1+ CD8 T cells (TSL) to differentiate towards a granzyme Bhi effector cytolytic T cell fate (Chen et al., 2021; Elsaesser et al., 2009; Frohlich et al., 2009; Ren et al., 2020; Snell et al., 2018; Xin et al., 2015; Yi et al., 2009; Zander et al., 2019). Moreover, ~30% of splenic TSL cells are in B cell follicles while ~50-60% in T cell zones in LCMV infected mice (He et al., 2016; Im et al., 2016; Im et al., 2020; Leong et al., 2016). The infrastructure to promote interactions between CXCR5+ IL-21-producing TFH cells and CXCR5+ TCF1+ TSL cells likely resides in B cell follicles of secondary lymphoid organs (LNs and spleens) and/or in tumor-associated TLSs that have GCs. In line with this, CXCR5+ TCF1+ TSL cells are enriched in tumors from NSCLC patients, and this correlates with tumor infiltration of CXCR5+ TFH cells and CD19+ B cells (Brummelman et al., 2018; Noel et al., 2021). We and others have also recently demonstrated that draining LN associated with lung adenocarcinoma maintains a stable reservoir of tumor-specific TSLs throughout tumor development and that migration of these cells from dLNs to tumors is required to maintain TCF1+ CD8 T cells in tumors (Connolly et al., 2021; Schenkel et al., 2021). We have also found tumor-associated TLSs in our autochthonous tumor models [(Fitzgerald et al., 2021; Joshi et al., 2015), and data not shown], and TLSs are less likely to form in subcutaneously transplanted tumors given the spatial, temporal, chemokine and vasculature requirements of TLS formation/maintenance (Engelhard et al., 2018; Pitzalis et al., 2014; Ruddle, 2014; van de Pavert and Mebius, 2010). Thus, we hypothesize that TFH cell-CD8 T cell interactions are likely to occur in the dLNs of s.c. implanted KP-HELLO tumors, but these could also occur in tumor-associated TLS when GCs present. Further research will be necessary to elucidate the site of action for B cell-TFH cell-IL21 interactions in different tumor contexts.
Studies of neoantigen immunogenicity generally focus on metrics such as neoantigen abundance, MHC binding, and T cell recognition to identify candidate neoantigens for therapeutic efforts (Schumacher and Hacohen, 2016; Schumacher et al., 2019; Wells et al., 2020). Our findings, however, raise the possibility that other aspects of neoantigens should also be considered. These include the assessment of mutation sites and whether they would result in structural alterations that are able to change BCR binding and B cell recognition. B-cell-recognized neoantigens were unique in that they were sufficient to potentiate TFH cell differentiation pathways, which provided non-redundant help for anti-tumor CD8 T cell responses via IL-21 in our model. This is in line with multiple studies showing that the presence of both cognate B cells and antigens with B-cell- and CD4 T cell-recognized epitopes was required for IL-21 production by PD1hi CXCR5hi TFH cells (Kerfoot et al., 2011; Shulman et al., 2014). Moreover, the principle of intermolecular help, in which antigen-engaged B cells are able to internalize and process whole antigenic proteins and subsequently present different MHCII epitopes to CD4 T cells, ensures that rare, cognate interactions between tumor-specific B and CD4 T cells are enabled and augmented by B-cell-recognized neoantigens (Milich et al., 1987; Mitchison, 1971a; b; Russell and Liew, 1979; Sette et al., 2008). MHC-II restricted neoantigens, recognized by CD4 T cells, have been shown to be indispensable for successful anti-tumor responses (Alspach et al., 2019; Kreiter et al., 2015; Linnemann et al., 2015), but less is known about how individual neoantigens, or their structures, could impact fate decisions by anti-tumor CD4 T cells. We found B cell-recognized epitopes were necessary for TFH cell development, and concurrently, co-existence of GC B cells and TFH cells in many human cancers strongly suggests that many tumors could express B-cell-recognized neoantigens. Here, it is possible that the B-cell-recognized portions are neoepitopes or are non-mutated epitopes connected to neoantigens recognized by CD4 T cells. Thus, tumors not only shape T cell responses via their production of chemokines, cytokines, and innate immune signaling modulators (i.e., STING/RIG-I), but also via structural features of their neoantigens. Gaining a better understanding of how various features of neoantigens drive different CD4 T cell fates could help to design therapies that leverage a wider variety of immune cell functions. Our approach provides an easily adaptable platform to investigate different aspects of B and T cell neoantigens, such as secreted/membrane-bound/intracellular features, and BCR binding affinity, of which available tools have already been established from many studies (Burnett et al., 2018; Paus et al., 2006).
Given the pivotal functional role of T cell-B cell interactions in controlling tumors, demonstrated by our work and others, the B cell-TFH cell-IL21 axis could provide an entry point for therapeutic manipulation. Preclinical studies and phase I clinical trials of personalized neoantigen vaccines, utilizing MHC class I binding epitopes, have successfully demonstrated the induction of de novo neoantigen-specific CD4 and CD8 T cell responses in patients with NSCLC, bladder cancer, melanoma and glioblastoma (Keskin et al., 2019; Ott et al., 2017; Ott et al., 2020). Here, one possibility could be to modify these personalized neoantigen vaccines to include epitopes recognized by both B and T cells. We predict that this would boost responses by neoantigen-specific TFH cells, facilitate their IL-21 production, and promote anti-tumor responses by effector CD8 T cells. Alternatively, one study found that B cell-based neoantigen vaccines could promote checkpoint blockade efficacy by activating autologous CD8 T cells in animal models of glioblastoma (Lee-Chang et al., 2021). Beyond vaccines, agonist antibodies or cytokines targeting the B cell-TFH cell-IL21 axis could also provide therapeutic benefits. Anti-ICOS agonist antibody (GSK3359609) is currently in phase II/III clinical trials in combination with PD-1 or PD-L1 blockade. Moreover, combining engineered IL-21 with existing immunotherapies can elicit robust anti-tumor immune responses (Deng et al., 2020; Li et al., 2021; Singh et al., 2011; Sondergaard and Skak, 2009; Wang et al., 2019; Štach et al., 2018). Taken together, these data suggested that therapeutic designs leveraging the B cell-TFH cell-IL21 axis could advance existing immunotherapies and lead to improved response rates for patients with lung and other cancers.
Limitations of the study
We showed the importance of T and B cell collaborations on driving IL-21-producing CD4 T cells in detail, but relied on previous work demonstrating how IL-21 helps to sustain cytolytic effector functions of CD8 T cells. (Chen et al., 2021; Elsaesser et al., 2009; Frohlich et al., 2009; Ren et al., 2020; Snell et al., 2018; Xin et al., 2015; Yi et al., 2009; Zander et al., 2019). Our studies did not delve into the location where IL-21 is provided to CD8 T cells or which CD8 T cell types receive the IL-21. We discuss the idea that this may differ depending on the presence of tumor-associated TLSs. Further studies will be necessary to dissect the spatial and temporal dynamics of TFH-IL21-CD8 T cell interactions.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nikhil Joshi (nikhil.joshi@yale.edu).
Materials availability
Cell lines and plasmids generated in this study will be deposited to ATCC and Addgene for public requests. See http://www.nikjoshilab.org/reagents for details.
Data and code availability
This paper analyzed existing, publicly available scRNA-seq data (GSE131907). Code is available at https://github.com/wangjw17/TfhInLungCancer (DOI: https://doi.org/10.5281/zenodo.5590959). All data reported in this paper will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were housed in pathogen-free conditions at Yale School of Medicine (New Haven, CT). CD45.2 WT C57BL/6 mice, and CD45.1 allotype-marked mice were purchased from the Jackson Laboratory and Charles River Laboratory. uMT, ICOS KO, IL21R KO, and MD4 mice were obtained from the Jackson Laboratory. Class II major histocompatibility complex (MHCII) transactivator (CIITA) KO, CD40 ligand KO, CD4-cre Bcl6fl/fl mice were provided by Stephanie Eisenbarth (Yale University), and RAG1 KO by David Schatz (Yale University). SMARTA, OT-II, P14 and KP mice were described previously (Damo et al., 2020; DuPage et al., 2011; Joshi et al., 2015; Weinstein et al., 2016). SWHEL mice were provided by Robert Brink (Garvan Institute of Medical Research). Il21Kat/Kat and Il4GFP [4get] reporter was described previously (Weinstein et al., 2016). uMT-IL21Kat/Kat reporter and uMT-KP were bred in-house. All studies were carried out in male and female recipients to account for sex as a biological variable and no differences were noted. All mice were used in accordance with protocols approved by the Institutional Animal Care and Use Committees of Yale University.
In vitro T cell and B cell co-culture
In vitro co-cultures were modeled after previously reported systems (Bruno et al., 2017; Kolenbrander et al., 2018; Lapointe et al., 2003). KP and KP-HELLO cells had been cultured at 37°C and 5% CO2, in complete RPMI-1640 (10% HI-FBS + 1% Penicillin & Streptomycin + 1x L-Glutamine + 55μM β-mercaptoethanol) for 2 days. Supernatant of KP and KP-HELLO were filtered with 0.45μm filters. Naïve CD4+ T cells and B cells were separated from spleens by negative selection with EasySep Mouse CD4+ T Cell Isolation Kit and Mouse B Cell Isolation Kit, respectively (STEMCELL technologies), according to the manufacturer’s protocol. 1 × 106/mL B cells were plated with CD4+ T cells in a ratio of B:T=1:2, and co-cultured with the supernatant of KP or KP-HELLO, or complete RPMI-1640 supplemented with LCMV GP61-80 peptide GLKGPDIYKGVYQFKSVEFD (5μg/mL, Anaspec) or HEL (0.5μg/mL, Sigma-Aldrich). BAFF (10ng/mL, R&D Systems) were added in all the co-culture experimental conditions. Flow cytometric analysis were performed after 48 hours of co-culture.
Tumor cell line and growth studies
The KP cell line had been described previously (Damo et al., 2020) and was derived from an Ad-CRE-infected KrasG12D, p53fl/fl mouse (DuPage et al., 2009). KP-NINJA (KPN1.1) has been described previously (Fitzgerald et al., 2021). MC38 and B16-F10 cells were obtained from Akiko Iwasaki laboratory (Jiang et al., 2019) and Aaron M. Ring laboratory (Zhou et al., 2020), respectively. KP, KP-HELLO, KP-NINJA, MC38 and B16-F10 cells were cultured at 37°C and 5% CO2, in complete DMEM (4.5g/L D-glucose + L-glutamine + 110mg/L sodium pyruvate + 10% HI-FBS + 1% Penicillin & Streptomycin). For tumor growth measurements and FACS analysis, mice were subcutaneously (s.c.) implanted with 2 x 105or 5 x 105 KP, KP-HELLO, KP-NINJA cells (individual numbers in figure legends), or 5 x 105 MC38, B16-F10 cells. Tumor volume was measured as [(Width2 × Length)/2], every four days with a digital caliper. Growth curves of C57BL/6 (n = 40) and uMT (n = 32) mice that were challenged with 2 x 105 KP-HELLO cells, pooled from 8 independent experiments, were set as the baseline of tumor growth analyses. Mice with ≧ 1cm3 or ulcerated tumors were euthanized in accordance with the protocols approved by the Institutional Animal Care and Use Committees of Yale University.
METHOD DETAILS
Generation of KP-HELLO cell line
KP was transduced with HELLO Lentiviral vector (LV). The LV backbone for HELLO has been described previously (Akama-Garren et al., 2016). HELLO-LV contains HELLO driven by phosphoglycerate kinase (pGK) promoter. HELLO was generated as a protein fusion of codon-optimized hen egg lysozyme (HEL), LCMVgp33-43 (KAVYNFATCGI), LCMVgp61-80 (GLNGPDIYKGVYQFKSVEFD), and 2A peptide-linked mScarlet. The DNA and amino acid sequences of HELLO construct are available in Table S2. NET MHC4.0 web server was used to predict MHC class I binding peptides (H-2Db and H-2Kb) in HELLO, with results summarized in Table S3. KP cells were transduced with HELLO-LV produced by co-transfection of 293FS* cells with HELLO, PSPax2 (gag/pol) and VSV-G (vesicular stomatitis virus glycoprotein) vectors as described previously (Damo et al., 2020). Transduced LGKP cells were FACS-sorted, yielding a pure mScarlet population of KP-HELLO cells. 25 frozen aliquots of KP-HELLO were generated after 2 passages and were named as “G0”. Based on 1 aliquot of “G0”, 20 frozen aliquots of “G1” were generated after 2 passages, and so as “G2” based on “G1”. For each experiment, one fresh aliquot of “G2” was thawed and used within 2 passages. After 6-7 passages described above, >99% of KP-HELLO “G2” cells were still mScarlet positive with flow cytometric analysis, which suggested the stable expression of HELLO protein in KP-HELLO cell line.
KP-HELLO cell line proviral sequence validation
Genomic DNA was extracted from two separate cultures of KP-HELLO (2 x 106 cells for each lysis) using the Takara Bio NucleoSpin Tissue kit (Cat#: 740952.50). PCR (Takara Bio High Fidelity PCR EcoDry Premix, Cat. # 639280) was performed to amplify the proviral DNA sequences encoding the GP33-43/FLAG/GP61-80 from the KP-HELLO cell genomic DNA. Amplified products were purified using the Takara Bio NucleoSpin Gel and PCR Clean-up kit (Cat. #740609.50) and were subsequently sequenced with Sanger sequencing using 2 different sets of specific primers.
Lentiviral vector production and administration
“Lenti-HELLO-Cre” plasmids were generated by cloning Cre into HELLO-LV. Lentiviral vectors were produced by transfecting 293FS* cells with Lenti-HELLO-Cre, PSPax2 and VSV-G vectors at a 4:3:1 ratio, of which supernatant was harvested at 100,000g for 2 hours (20°C), after 32-hour incubation at 37°C, as described previously (Joshi et al., 2015). LV titer was quantified by transducing GreenGo cell line (105 cells/well) in serial dilutions to 1mL of final volume and calculated with the formula: (% fluorescent cells x 105 x Dilution Factor)/100 (pfu/mL). For autochthonous KP-HELLO tumor initiation, 5 x 104 pfu Lenti-HELLO-Cre were administered intratracheally to KP mice.
Antibody depletion and adoptive transfer
To deplete CD8 T Cells, 200ug/mouse anti-CD8a antibodies (clone: 53-6.7; InVivoMab) were diluted in PBS, and injected intraperitoneally every three days, 10 days prior to tumor initiation until the end point of experiments. For adoptive transfer, naïve CD8+ T cells, CD4+ T cells and/or B cells were separated from spleens by negative selection with EasySep Mouse CD8+ T Cell Isolation Kit, Mouse CD4+ T Cell Isolation Kit and/or Mouse B Cell Isolation Kit, respectively (STEMCELL technologies). For rescue experiments, 1 x 106 SWHEL or WT C57BL/6 B cells were transferred into age- and sex-matched uMT mice, and 1 x 105 SMARTA or OT-II T cells were transferred into age- and sex-matched CD4-cre Bcl6fl/fl mice, with 2 x 105 KP-HELLO s.c. implanted one day later. For co-transfer experiments, CD45.1/CD45.2 or Thy1.1/Thy1.2 allotype-marked P14 (5 x 105/mouse), SMARTA (1 x 105/mouse) and SWHEL (1 x 105/mouse) cells were co-transferred into naïve CD45.2/CD45.2 Thy1,2/Thy1.2 mice one day prior to tumor initiation.
Immune cell isolation from implanted or lung tumors and lymphoid organs
Subcutaneously implanted solid tumors were dissected, cut into small pieces and digested with 5mL Collagenase IV buffer (1x HEPES buffer, 0.5mg/mL Collagenase IV, 20μg/mL DNase in 1x HBSS with MgCl2 and CaCl2) for 30-40 minutes at 37°C, as described previously (Connolly et al., 2021; Fitzgerald et al., 2021; Joshi et al., 2015). Samples were then run on the default lung_02 program on a gentleMACS Dissociator instrument (Miltenyi Biotec). Digestion was neutralized by adding 5mL 1% HI-FBS RPMI-1640. Tumor samples were filtered with 70 μm cell strainers and washed with 1% HI-FBS RPMI-1640. Cell suspensions were then processed with Mouse CD45 (TIL) MicroBeads (Miltenyi Biotec) to enrich for CD45+ TILs. For autochthonous lung tumors, mice were injected intravenously with CD45-PE-CF594 (1:100 diluted in PBS, clone: 30-F11), 2-3 minutes before sacrifice as described previously (Joshi et al., 2015). Lungs were harvested, run on lung_01 program with gentleMACS Dissociator and digested with Collagenase IV buffer as described above, for 30-40 minutes at 37°C. Samples were then run on the lung_02 program, neutralized by 1% HI-FBS RPMI-1640, filtered with 70 μm cell strainers, washed with 1% HI-FBS RPMI-1640 and treated with RBC lysis buffer (eBioscience) to remove red blood cells. Tumor-draining lymph nodes were harvested, filtered with 70 μm cell strainers and processed as described previously (Damo et al., 2020; Joshi et al., 2015).
Flow cytometry
Single cell suspensions were stained with antibodies for surface markers. For I-Ab/GP66-77− specific tetramer (NIH Tetramer Core Facility) staining, cells were incubated 2-3 hours at 37°C prior to surface staining. For intracellular staining, cells were processed using either Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for transcription factors, or Fixation/Permeabilization Solution Kit (BD Cytofix/Cytoperm) for cytokines. Cells were washed and resuspended in FACS Buffers (PBS + 0.5% HI-FBS) until data collection. Flow cytometry was performed with LSR II flow cytometer (BD Bioscience), and analyzed by FlowJo software (version 10, TreeStar). For MFI quantification, median was applied to logarithmic histogram for specific molecules.
In vitro P14 T cell and antigen-loaded DC co-culture
P14 CD8+ T cells were isolated with EasySep™ Mouse CD8 T Cell Isolation Kit (StemCell). After isolation, P14 cells were resuspend in 5 mL warm PBS at 2 × 106 cells/mL. 5 mL of 2 uM CFSE (ThermoFisher) staining buffer (2×) was freshly prepared in warm PBS and then added to P14 suspension drop by drop during vortex. CFSE labeling was allowed at 37 C for 5 minutes and quenched with 5 mL of 100% FBS at 37 C for another minute. CFSE-labeled P14 cells were washed and resuspended in complete RPMI and ready to be used for DC-P14 coculture. DCs were isolated from pooled spleens and LNs using the EasySep™ Mouse Pan-DC Enrichment Kit (StemCell) per manufacturer’s instruction. 2 x 104 DCs were stimulated by 100 ng/mL LPS in complete RPMI for 16 hours in a 96-well format. LPS-stimulated DCs were then incubated with 5 ug/mL GP33 peptide or HELLO cell culture supernatant for 2 hours (unless indicated otherwise). Following LPS stimulation and antigen loading, 2 x 105 CFSE-labeled P14 cells were added to each well and cocultured with DC in complete RPMI media. Media was replaced once 2 days following coculture. On day 3, cell culture was collected to assess P14 cell proliferation (by CFSE dilution) and activation (by CD44/CD69 upregulation or CD62L downregulation) by flow cytometry. Note, we could not detect robust GP33-specific CD8+ T cell responses with either in vitro (Figure S3C) or in vivo (Figure S3D–S3F) assays, which was unlike what we observed in NINJA-expressing tumors (Connolly et al., 2021; Damo et al., 2020; Fitzgerald et al., 2021). This was not a technical breakdown, as the proviral sequence from KP-HELLO tumor cell line was correct (Figure S3B) and GP61-80 was presented to SMARTA CD4 T cells (Figure 2C).
CIBERSORT analysis
Fractions of different cell types were estimated with CIBERSORT (Newman et al., 2015), a cell type deconvolution program that derives cell type-specific abundances from bulk RNA-seq data. Here bulk RNA-seq data were downloaded from the whole TCGA LUAD cohort. Among the 594 samples with FPKM data, the first sample was used for each series of primary tumor samples. In total, 513 samples of primary tumor from different cases were selected for deconvolution. Reference marker gene expression profile (GEP) matrix was assigned with a widely used leukocyte signature matrix LM22 (Newman et al., 2015) provided by CIBERSORT program.
Quantile normalization was disabled, and 100 permutations were used as default to calculate P values. From 22 cell subsets of CIBERSORT output using LM22 matrix, “B cells naïve”, “B cells memory” and “plasma cells” were combined as “B cell lineage”; “T cells CD4 memory resting” and “T cells CD4 memory activated” were combined as “CD4 memory T cells”; “NK cells resting” and “NK cells activated” were combined as “NK cells”; “macrophages M0”, “macrophages M1”, and “macrophages M2” were combined as “macrophages”; “dendritic cells resting” and “dendritic cells activated” were combined as “dendritic cells”; “mast cells resting” and “mast cells activated” were combined as “mast cells”.
Single-cell RNA sequencing analysis
Data was downloaded from GEO with accession code GSE131907 (Kim et al., 2020). The raw UMI count matrix and annotations were used. Data normalization and analysis were performed with R package Seurat (version 3.1.0) and only cells from lungs or lymph nodes were used with original annotations “nLung”, “tLung”, “nLN”, or “mLN”. For the analysis of CD4+ T cells and B cells, 49,901 cells with original annotation “T/NK cells” and 22,592 cells with original annotations “B lymphocytes” were selected for the following analysis. UMI counts were log-normalized using function NormalizeData with parameter normalization.method set to “LogNormalize”. PCA was run based on the normalized data and clusters were called with FindNeighbors and FindClusters using the top 20 PCs and resolution set to 0.5 or 0.25 for T/NK cells or B cells, respectively. For both T/NK cells and B cells, dimension reduction and visualization were performed with UMAP using the top 15 PCs. Relative abundances for marker genes were calculated as Z-scaled average of log2(RC+1), where RC is relative counts that is calculated with NormalizeData and normalization.method set to “RC”.
Survival analysis and correlation analysis
Survival analysis and pairwise correlation analysis of gene expression signatures were performed using web server GEPIA2 (Tang et al., 2019), based on TCGA and GTEx databases. 478 LUAD tumor samples were included in survival analysis. We established gene expression signatures for GC B cells and TFH cells based on previous human studies in lymphoid tissues and cancers (Cabrita et al., 2020; Cillo et al., 2020; Milpied et al., 2018; Vella et al., 2019), and refined them based on the data in Figures 1D–1E to reflect the gene expression of GC B cells and TFH cells in LUAD. For each signature gene set, LUAD cohort was divided into high and low expression groups by median value (50% cut-off). GC B cell: AICDA, BATF, BACH2, BCL6, CD79A, CD79B, CD86, DOCK8, IRF4, IRF8, MYC. TFH: ASCL2, BCL6, CD4, CD200, CXCR5, IL4, IL21, IL6ST, MAF, PDCD1, SH2D1A, TOX2. Th1: BHLHE40, CXCR3, CD4, IFNG, IL2, IL12RB2, STAT4, TBX21. CD8 effector cell: CD8A, CD8B, CX3CR1, GZMA, GZMB, KLRG1, PRF1, ZEB2. Th17: CD4, IL6R, IL17A, IL17F, IL23R, RORA, RORC, STAT3. NK cell: CD160, CD244, GNLY, GZMB, KLRC3, KLRF1, NKG2A, NKG7. Survival analyses were performed with log-rank Mantel-Cox test. Pairwise correlation analyses of gene expression signature were performed with two-tailed Pearson correlation test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism 9 (GraphPad software) was used for quantification and statistical analysis. Methods of statistical analysis, number of mice used per experiment per group, number of experimental repeats and P values were indicated in figure legends. P < 0.05 was considered statistically significant.
Supplementary Material
Figure S1. Analyses of CD4 T cells and B cells in LUAD samples from TCGA; Related to Figure 1
A, Estimated fractions of subtypes in CD4 T cells and B cell lineage described in Figure 1A, from CIBERSORT analyses of TCGA-LUAD samples (n=513).
B, Survival analyses based on expression signatures of Th1, effector CD8 T cells, Th17, NK cells in TCGA-LUAD patient cohort (n=478).
C, Correlation analyses among expression signatures of GC B cells, TFH, Th1, CD8 effector cells, Th17, NK cells in lung adenocarcinoma patients.
B, log-rank Mantel-Cox test. C, two-tailed Pearson correlation test.
Figure S2. TFH cell responses in murine tumor models; Related to Figure 2
A, Representative flow plots showing TFH gating in tumor-draining LNs (dLNs) of MC38 or B16-F10 tumor-bearing C57BL/6 mice (pre-gated on TCRb+ CD4). Flow cytometric analyses were performed on day 10 – 12. Data (mean ±SD) represented 4 – 5 mice/group.
B, Tumor growth curves: C57BL/6, RAG1 KO, CIITA KO and uMT mice were s.c. implanted with 2 x 105 KP. Data (mean ±SD) were pooled, from 3 – 10 mice/group/experiment, and representative of at least 2 independent experiments. Two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure S3. KP-HELLO LUAD cell line elicits GP66-specific CD4 T cell responses in vitro, while unable to elicit robust GP33-specific CD8 T responses; Related to Figure 2
A, Representative flow plots showing CD69 and CD44 expression on SMARTA CD4 T cells, after 48-hour co-culture with MD4 or C57BL/6 B cells, and KP or KP-HELLO supernatant, 500ng/mL HEL or 5ug/mL GP61-80.
B, Chromatogram displaying DNA sequence of GP33-43/FLAG/GP61-80 from the proviral insertion in the KP-HELLO tumor cell line. This demonstrated that the integrated provirus in KP-HELLO cells encodes the correct sequence for GP33-43/FLAG/GP61-80. The amino acid sequence is indicated above the chromatogram as a reference for translation. Results were repeated in 4 independent experiments.
C, In vitro co-culture of P14 T cells and antigen loaded DCs. Bar graphs displaying division and activation marker expression on P14 T cells under different experimental conditions. CFSE-labeled P14 cells were co-cultured with LPS-stimulated and antigen-loaded DCs. On day 3, cell culture was collected to assess P14 cell proliferation (by CFSE dilution) and activation (by CD44 upregulation or CD62L downregulation) by flow cytometry.
D – F, GP33-specific CD8+ T cell responses in KP-HELLO models. (D) Frequency and representative flow plots displaying GP33-specific CD8+ T cells in tumors (top) and dLNs (bottom). C57BL/6 and uMT mice were s.c. implanted with 2 x 105 KP-HELLO. Tumors and dLNs were harvested on day 14 – 16. GP33 gating (red: pre-gated on singlets, TCRb+ CD8+) was drawn based on FMO control (blue). (E) Frequency and representative flow plots displaying P14 tumor-specific CD8+ T cells in tumors (top) and dLNs (bottom). 5 x 105 Thy1.1 P14 T cells were transferred (i.v.) into C57BL/6 and uMT mice, one day prior to 5 x 105 KP-HELLO s.c. implant. Tumors and dLNs were harvested on day 14. (F) Frequency and representative flow plots displaying GP33-specific CD8+ T cells in tumor tissues (top) and dLNs (bottom). KP and uMT-KP mice were infected intratracheally with 5 x 104 PFU Lenti-HELLO-Cre. Lung tumors were harvested 8 weeks post-infection. GP33 gating (red: pre-gated on singlets, TCRb+ ivCD45Neg, CD8+) was drawn based on FMO control (blue). Here, Y-axis of the bar graphs were adjusted based on figures and analyses of previous publications (Connolly et al., 2021; Fitzgerald et al., 2021) to display the differences of GP33-specific responses in KP-HELLO vs. KP-NINJA.
Figure S4. KP-HELLO LUAD tumors elicit antigen-specific TFH and GC B cell responses in vivo; Related to Figure 2
A, Representative flow plots of GP66-specific CD4 T cells from dLNs of KP-HELLO tumor-bearing C57BL/6 mice.
B, Experimental design of Figure S4C–S4D.
C – D, Representative flow plots showing tumor-specific SMARTA TFH cells (C) and SWHEL GC B cells (D) in dLNs of KP-HELLO bearing mice. As shown in Figure S4B, CD45.2/CD45.2 Thy1.2/Thy1.2 C57BL/6 recipient, transferred with 1 x 105 CD45.1/CD45.2 HEL-specific SWHEL B cells and 1 x 105 Thy1.1/Thy1.2 GP66-specific SMARTA CD4 T cells, were s.c. implanted with 5 x 105 KP-HELLO. Flow cytometric analyses were performed on day 10 – 12. Data (mean ± SD) represented at least 2 independent experiments.
Figure S5. B cells are required to drive tumor-specific TFH cell responses; Related to Figure 3
A – B, Tumor growth curves: RAG1 KO or C57BL/6 mice were s.c. implanted with 2 × 105 KP or KP-HELLO. Growth curve of KP-HELLO in C57BL/6 (n = 40), pooled from 8 independent experiments, was set as the baseline of analyses.
C – G, Frequency and number of GP66-specific total CD4 T cells (C and F), TFH cells (CD44hi PSGL1lo Ly6Clo PD1hi CXCR5hi) (E and G), and other CD4+ CD44hi T cell subsets (D and G) from dLNs of C57BL/6, uMT or CD4-Cre Bcl6fl/fl mice with KP-HELLO tumors.
H, Representative histogram displaying T-bet expression on naïve, GP66-specific PSGL1hi Ly6Chi (TH1-like) and PSGL1lo Ly6Clo PD1hi CXCR5hi (TFH) T cells from dLNs of KP-HELLO tumor bearing mice.
Data (mean ±SD) were pooled, from 3–10 mice/group/experiment, and representative of at least 2 independent experiments. A – B, Two-way ANOVA. C – G, Unpaired t-test with Welch’s correction. **P < 0.05, **P < 0.01, ***P < 0.001, ****p < 0.0001.
Figure S6. B cells are required to drive tumor-specific TFH cell responses and tumor-infiltrating effector CD8 T cell responses in KP-HELLO autochthonous lung tumor model; Related to Figure 2–4 and STAR Methods
A, Design of KP-HELLO autochthonous lung tumor model. KP or uMT-KP mice were intratracheally infected with 5 × 104 PFU Lenti-HELLO-Cre. Tumor lung tissues and dLNs were harvested at 8 weeks post-infection.
B, Diagram showing the technique to label circulating immune cells in lung tissues for flow cytometric analyses. 2–3 minutes prior to sacrifice, PE-CF594-conjugated CD45 antibodies were injected intravenously. Immune cells in circulation were labelled, while the time was not enough for antibodies to diffuse into tissue and bind to tumor-infiltrating immune cells.
C, Gating strategy and representative histogram of CD45-PE-CF594 (in vivo labeling) showing tissue-infiltrating B cells, CD4 T cells and CD8 T cells in non-tumor-bearing mice, or tumor-bearing KP and uMT-KP mice.
D, Representative flow plots and frequency of GP66-specific CD4 TFH cells (CD44hi PSGL1lo PD1hi CXCR5hi) from tumor lung tissues and dLNs of KP or uMT-KP mice. Flow plots were pre-gated on CD45i.v.Neg TCRb+ CD4+ GP66-tetramer+ CD44hi PSGL1lo.
E, Representative flow plots showing GC B cells in tumor lung tissues and dLNs of KP mice (pre-gated on CD45i.v.Neg CD19+ B220+ IgDlo).
F, Representative flow plots and frequency of tumor-infiltrating CD44hi PD1hi CD8+ T cells in KP or uMT-KP mice.
Data (mean ±SD) were pooled, from 3–10 mice/group/experiment, and representative of at least 2 independent experiments. D – F, Unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure S7. TFH-derived IL-21 in murine tumor models; Related to Figure 6
A – B, Representative flow plots showing IL-21-expressing TFH cells (A) and histogram displaying IL-21 expression through different stages of TFH differentiation (B) in dLNs of MC38 and B16-F10 tumor-bearing mice. Flow cytometric analyses were performed on day 10 – 12.
C, IL-21 expression by TFH cells (CD44hi PSGL1lo PD1hi CXCR5hi) in dLNs of KP, MC38, B16-F10 and KP-HELLO tumors.
Data (mean ±SD) were pooled, from 3 – 5 mice/group/experiment, and representative of 2 independent experiments. C, Unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Table S1. Differentially expressed genes in T/NK cell clusters and B cell clusters (GSE131907). Related to Figure 1.
Table S2. DNA and amino acid sequences of HELLO construct. Related to Figure 2 and STAR Methods.
Table S3. Prediction of CD8 T cell epitopes in HELLO neoantigen. Related to Figure 2 and STAR Methods.
Highlights.
Enrichment of TFH and GC B cells correlates with favorable clinical outcomes of LUAD
B-cell-recognized neoantigens drive tumor specific B cell and TFH cell responses
Tumor-specific TFH cells produce IL-21
IL-21 is critical for tumor control and tumor-infiltrating CD8 T cell effector function
Acknowledgements
This work was supported by grants from the NCI K22CA200912 (N.S.J.), NCI 1RO1CA237037-01A1 (N.S.J.), NIH R37AR40072 (J.C.), NIH AR074545 (J.C.), Yale SPORE in Lung Cancer 1P50CA196530 (N.S.J. and J.C.), and a Pilot Grant from Yale Cancer Center (N.S.J. and J.C.). C.C. was supported by the Gruber Science Fellowship and Richard K. Gershon Research Fellowship. J.F.C, K.A.C and B.F. were supported by the Yale Immunobiology NIH T32 training grant (5T32AI007019). We thank N.S.J. and J.C. laboratory members for reviewing the manuscript, Z. Chen, C. Yang, and N. Ruddle for helpful discussions and W. Wei for important guidance on statistical analysis. We also thank the Yale Flow Cytometry Core, Yale Keck DNA Sequencing Facility, Yale School of Medicine Histology Facility, and NIH Tetramer Core Facility. SWHEL mice were kindly provided by Dr. Robert Brink. Figures 2A, S4B, S6A–S6B and graphical abstract were created with www.biorender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Akama-Garren EH, Joshi NS, Tammela T, Chang GP, Wagner BL, Lee DY, Rideout WM 3rd, Papagiannakopoulos T, Xue W, and Jacks T (2016). A Modular Assembly Platform for Rapid Generation of DNA Constructs. Sci Rep 6, 16836. 10.1038/srep16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, Meng W, Lichti CF, Esaulova E, Vomund AN, et al. (2019). MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701. 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Arias DA, Kim HJ, Zhou P, Holderried TA, Wang X, Dranoff G, and Cantor H (2014). Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol Res 2, 207–216. 10.1158/2326-6066.CIR-13-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay M, Moskophidis D, Rahemtulla A, Hengartner H, Mak TW, and Zinkernagel RM (1994). Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J Virol 68, 4700–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, Muise ES, Zhang KX, Arazi A, Keras G, et al. (2019). PD-1hiCXCR5- T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight 4. 10.1172/jci.insight.130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst J, Ahrends T, Babala N, Melief CJM, and Kastenmuller W (2018). CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 18, 635–647. 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, and Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, et al. (2018). High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med 215, 2520–2535. 10.1084/jem.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, Singhal S, Mitchell JD, Franklin WA, Merrick DT, et al. (2017). Antigen-Presenting Intratumoral B Cells Affect CD4(+) TIL Phenotypes in Non-Small Cell Lung Cancer Patients. Cancer Immunol Res 5, 898–907. 10.1158/2326-6066.CIR-17-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, Bourne K, Reed JH, Patterson K, Porebski BT, et al. (2018). Germinal center antibody mutation trajectories are determined by rapid self/foreign discrimination. Science 360, 223–226. 10.1126/science.aao3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. (2020). Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, Zheng S, Burns R, Taparowsky EJ, and Cui W (2021). BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nat Immunol 22, 996–1007. 10.1038/s41590-021-00965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, and Crotty S (2011). ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34, 932–946. 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Yang JA, and Crotty S (2013). Dynamic regulation of Bcl6 in follicular helper CD4 T (Tfh) cells. Curr Opin Immunol 25, 366–372. 10.1016/j.coi.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, and Mackay CR (2004). T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol 173, 68–78. 10.4049/jimmunol.173.1.68. [DOI] [PubMed] [Google Scholar]
- Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, Liu A, Duvvuri U, Kim S, Soose RJ, et al. (2020). Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 52, 183–199 e189. 10.1016/j.immuni.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, Hornick NI, Fitzgerald BL, Damo M, Kasmani MY, et al. (2021). A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol 6, eabg7836. 10.1126/sciimmunol.abg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, and Godfrey DI (2007). IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol 178, 2827–2834. 10.4049/jimmunol.178.5.2827. [DOI] [PubMed] [Google Scholar]
- Crotty S (2011). Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29, 621–663. 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- Crotty S (2019). T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 50, 1132–1148. 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, and Allen CDC (2019). B Cell Responses: Cell Interaction Dynamics and Decisions. Cell 177, 524–540. 10.1016/j.cell.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damo M, Fitzgerald B, Lu Y, Nader M, William I, Cheung JF, Connolly KA, Foster GG, Akama-Garren E, Lee DY, et al. (2020). Inducible de novo expression of neoantigens in tumor cells and mice. Nat Biotechnol. 10.1038/s41587-020-0613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Sun Z, Qiao J, Liang Y, Liu L, Dong C, Shen A, Wang Y, Tang H, Fu YX, and Peng H (2020). Targeting tumors with IL-21 reshapes the tumor microenvironment by proliferating PD-1intTim-3-CD8+ T cells. JCI Insight 5. 10.1172/jci.insight.132000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L, et al. (2008). Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol 26, 4410–4417. 10.1200/JCO.2007.15.0284. [DOI] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85. 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4, 1064–1072. 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, and Noelle RJ (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229, 152–172. 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser H, Sauer K, and Brooks DG (2009). IL-21 is required to control chronic viral infection. Science 324, 1569–1572. 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, and Slingluff CL Jr. (2018). Immune Cell Infiltration and Tertiary Lymphoid Structures as Determinants of Antitumor Immunity. J Immunol 200, 432–442. 10.4049/jimmunol.1701269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger DS, Wood DE, Aggarwal C, Aisner DL, Akerley W, Bauman JR, Bharat A, Bruno DS, Chang JY, Chirieac LR, et al. (2019). NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020. J Natl Compr Canc Netw 17, 1464–1472. 10.6004/jnccn.2019.0059. [DOI] [PubMed] [Google Scholar]
- Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson J.T.t., Bagadia P, Liu T, Briseno CG, et al. (2020). cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584, 624–629. 10.1038/s41586-020-2611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald B, Connolly KA, Cui C, Fagerberg E, Mariuzza DL, Hornick NI, Foster GG, William I, Cheung JF, and Joshi NS (2021). A mouse model for the study of anti-tumor T cell responses in Kras-driven lung adenocarcinoma. Cell Rep Methods 1. 10.1016/j.crmeth.2021.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, et al. (2018). Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med 378, 1976–1986. 10.1056/NEJMoa1716078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Zitvogel L, Sautes-Fridman C, and Kroemer G (2017). The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 14, 717–734. 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, and Kopf M (2009). IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 324, 1576–1580. 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, and Marincola FM (2013). The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39, 11–26. 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. (2018). Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 378, 2078–2092. 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- Garaud S, Buisseret L, Solinas C, Gu-Trantien C, de Wind A, Van den Eynden G, Naveaux C, Lodewyckx JN, Boisson A, Duvillier H, et al. (2019). Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight 5. 10.1172/jci.insight.129641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Lepelley A, Becht E, Katsahian S, Bizouard G, et al. (2014). Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 189, 832–844. 10.1164/rccm.201309-1611OC. [DOI] [PubMed] [Google Scholar]
- Gettinger SN, Choi J, Mani N, Sanmamed MF, Datar I, Sowell R, Du VY, Kaftan E, Goldberg S, Dong W, et al. (2018). A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune checkpoint blockers. Nat Commun 9, 3196. 10.1038/s41467-018-05032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, de Chaisemartin L, Ouakrim H, Becht E, Alifano M, et al. (2014). Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res 74, 705–715. 10.1158/0008-5472.CAN-13-1342. [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, and et al. (1988). Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676–682. 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, Maurer-Granofszky M, Roka F, Penz T, Bock C, et al. (2019). B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun 10, 4186. 10.1038/s41467-019-12160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, Ravoet M, Le Buanec H, Sibille C, Manfouo-Foutsop G, et al. (2013). CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest 123, 2873–2892. 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu-Trantien C, Migliori E, Buisseret L, de Wind A, Brohee S, Garaud S, Noel G, Dang Chi VL, Lodewyckx JN, Naveaux C, et al. (2017). CXCL13-producing TFH cells link immune suppression and adaptive memory in human breast cancer. JCI Insight 2. 10.1172/jci.insight.91487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R , et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24, 978–985. 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, Wu T, Iyer SS, and Ahmed R (2013). Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity 38, 805–817. 10.1016/j.immuni.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna NH, Schneider BJ, Temin S, Baker S Jr., Brahmer J, Ellis PM, Gaspar LE, Haddad RY, Hesketh PJ, Jain D, et al. (2020). Therapy for Stage IV Non-Small-Cell Lung Cancer Without Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. J Clin Oncol 38, 1608–1632. 10.1200/JCO.19.03022. [DOI] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5-expressing CD8(+) T cells curtail chronic viral infection. Nature 537, 412–428. 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennequin A, Derangere V, Boidot R, Apetoh L, Vincent J, Orry D, Fraisse J, Causeret S, Martin F, Arnould L, et al. (2016). Tumor infiltration by Tbet+ effector T cells and CD20+ B cells is associated with survival in gastric cancer patients. Oncoimmunology 5, e1054598. 10.1080/2162402X.2015.1054598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, Morise M, Felip E, Andric Z, Geater S, et al. (2020). Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. N Engl J Med 383, 1328–1339. 10.1056/NEJMoa1917346. [DOI] [PubMed] [Google Scholar]
- Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, He X, Garay JP, Carey-Ewend K, Marron D, et al. (2019). B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell 179, 1191–1206 e1121. 10.1016/j.cell.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, and Dent AL (2013). Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J Immunol 191, 3705–3711. 10.4049/jimmunol.1300378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Konieczny BT, Hudson WH, Masopust D, and Ahmed R (2020). PD-1+ stemlike CD8 T cells are resident in lymphoid tissues during persistent LCMV infection. Proc Natl Acad Sci U S A 117, 4292–4299. 10.1073/pnas.1917298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T, and Kurosaki T (2018). T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity 48, 702–715 e704. 10.1016/j.immuni.2018.03.027. [DOI] [PubMed] [Google Scholar]
- Jiang X, Muthusamy V, Fedorova O, Kong Y, Kim DJ, Bosenberg M, Pyle AM, and Iwasaki A (2019). Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J Exp Med 216, 2854–2868. 10.1084/jem.20190801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, and Crotty S (2009). Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010. 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43, 579–590. 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, and Haberman AM (2011). Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34, 947–960. 10.1016/j.immuni.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, Lee JI, Suh YL, Ku BM, Eum HH, et al. (2020). Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11, 2285. 10.1038/s41467-020-16164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolenbrander A, Grewe B, Nemazee D, Uberla K, and Temchura V (2018). Generation of T follicular helper cells in vitro: requirement for B-cell receptor cross-linking and cognate B- and T-cell interaction. Immunology 153, 214–224. 10.1111/imm.12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, and Kuchroo VK (2007). IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448, 484–487. 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, et al. (2015). Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696. 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger DR, Milne K, and Nelson BH (2016). Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res 22, 3005–3015. 10.1158/1078-0432.CCR-15-2762. [DOI] [PubMed] [Google Scholar]
- Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, and Hwu P (2003). CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res 63, 2836–2843. [PubMed] [Google Scholar]
- Lee-Chang C, Miska J, Hou D, Rashidi A, Zhang P, Burga RA, Jusue-Torres I, Xiao T, Arrieta VA, Zhang DY, et al. (2021). Activation of 4-1BBL+ B cells with CD40 agonism and IFNgamma elicits potent immunity against glioblastoma. J Exp Med 218. 10.1084/jem.20200913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, Minnich M, Meckiff BJ, Wei Y, Hou Z, et al. (2016). CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol 17, 1187–1196. 10.1038/ni.3543. [DOI] [PubMed] [Google Scholar]
- Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen J, Blank CU, et al. (2019). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 176, 775–789 e718. 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cong Y, Jia M, He Q, Zhong H, Zhao Y, Li H, Yan M, You J, Liu J, et al. (2021). Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat Commun 12, 951. 10.1038/s41467-021-21241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YJ, Zhang F, Qi Y, Chang GQ, Fu Y, Su L, Shen Y, Sun N, Borazanci A, Yang C, et al. (2015). Association of circulating follicular helper T cells with disease course of NMO spectrum disorders. J Neuroimmunol 278, 239–246. 10.1016/j.jneuroim.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. (2015). High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 21, 81–85. 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, and Vinuesa CG (2010). IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med 207, 353–363. 10.1084/jem.20091738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, Luo D, and Qi H (2015). T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature 517, 214–218. 10.1038/nature13803. [DOI] [PubMed] [Google Scholar]
- Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, Chou C, Do MH, Stamatiades EG, Gao S, et al. (2020). TGF-beta suppresses type 2 immunity to cancer. Nature 587, 115–120. 10.1038/s41586-020-2836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, Dries R, Almonte C, Herter-Sprie GS, Santos A, et al. (2016). Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 1, e89014. 10.1172/jci.insight.89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, Li Y, Li J, Zhou B, Ye Y, et al. (2020). Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell 180, 1081–1097 e1024. 10.1016/j.cell.2020.02.015. [DOI] [PubMed] [Google Scholar]
- Magen A, Nie J, Ciucci T, Tamoutounour S, Zhao Y, Mehta M, Tran B, McGavern B, Hannenhalli S, and Bosselut R (2019). Single-Cell Profiling Defines Transcriptomic Signatures Specific to Tumor-Reactive versus Virus-Responsive CD4(+) T Cells. Cell Rep 29, 3019–3032 e3016. 10.1016/j.celrep.2019.10.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo A, Vitolo B, Humby F, Caporali R, Jarrossay D, Dell’accio F, Ciardelli L, Uguccioni M, Montecucco C, and Pitzalis C (2008). Mature antigen-experienced T helper cells synthesize and secrete the B cell chemoattractant CXCL13 in the inflammatory environment of the rheumatoid joint. Arthritis Rheum 58, 3377–3387. 10.1002/art.23966. [DOI] [PubMed] [Google Scholar]
- Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, and Kaech SM (2011). Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35, 633–646. 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, and Dong C (2009). T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31, 787–798. 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Concepcion RJ, and Ahmed R (1994). CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68, 8056–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, et al. (2016). Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A 113, E6409–E6417. 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, and Victora GD (2016). Germinal Center B Cell Dynamics. Immunity 45, 471–482. 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milich DR, McLachlan A, Thornton GB, and Hughes JL (1987). Antibody production to the nucleocapsid and envelope of the hepatitis B virus primed by a single synthetic T cell site. Nature 329, 547–549. 10.1038/329547a0. [DOI] [PubMed] [Google Scholar]
- Milpied P, Cervera-Marzal I, Mollichella ML, Tesson B, Brisou G, Traverse-Glehen A, Salles G, Spinelli L, and Nadel B (2018). Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol 19, 1013–1024. 10.1038/s41590-018-0181-4. [DOI] [PubMed] [Google Scholar]
- Mitchison NA (1971a). The carrier effect in the secondary response to hapten-protein conjugates. I. Measurement of the effect with transferred cells and objections to the local environment hypothesis. Eur J Immunol 1, 10–17. 10.1002/eji.1830010103. [DOI] [PubMed] [Google Scholar]
- Mitchison NA (1971b). The carrier effect in the secondary response to hapten-protein conjugates. II. Cellular cooperation. Eur J Immunol 1, 18–27. 10.1002/eji.1830010104. [DOI] [PubMed] [Google Scholar]
- Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, Foucat E, Dullaers M, Oh S, Sabzghabaei N, et al. (2011). Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34, 108–121. 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, Sun HW, Vahedi G, Hakim O, Handon R, et al. (2011). Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity 35, 919–931. 10.1016/j.immuni.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel G, Fontsa ML, Garaud S, De Silva P, de Wind A, Van den Eynden GG, Salgado R , Boisson A, Locy H, Thomas N, et al. (2021). Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J Clin Invest 131. 10.1172/JCI139905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, and Dong C (2007). Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448, 480–483. 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q, and Dong C (2008). Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 29, 138–149. 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, and Dong C (2009). Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005. 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, and Paul WE (2010). Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102. 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, Aran D, Ilano A, Pai CS, Rancan C, et al. (2020). Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 181, 1612–1625 e1613. 10.1016/j.cell.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ, et al. (2020). A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 183, 347–362 e324. 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- Paus D, Phan TG, Chan TD, Gardam S, Basten A, and Brink R (2006). Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med 203, 1081–1091. 10.1084/jem.20060087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougouin A, et al. (2020). B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560. 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TG, Amesbury M, Gardam S, Crosbie J, Hasbold J, Hodgkin PD, Basten A, and Brink R (2003). B cell receptor-independent stimuli trigger immunoglobulin (Ig) class switch recombination and production of IgG autoantibodies by anergic self-reactive B cells. J Exp Med 197, 845–860. 10.1084/jem.20022144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzalis C, Jones GW, Bombardieri M, and Jones SA (2014). Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol 14, 447–462. 10.1038/nri3700. [DOI] [PubMed] [Google Scholar]
- Poholek AC, Hansen K, Hernandez SG, Eto D, Chandele A, Weinstein JS, Dong X, Odegard JM, Kaech SM, Dent AL, et al. (2010). In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol 185, 313–326. 10.4049/jimmunol.0904023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakaee M, Kilvaer TK, Jamaly S, Berg T, Paulsen EE, Berglund M, Richardsen E, Andersen S, Al-Saad S, Poehl M, et al. (2021). Tertiary lymphoid structure score: a promising approach to refine the TNM staging in resected non-small cell lung cancer. Br J Cancer. 10.1038/s41416-021-01307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, et al. (2017). Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114. 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. (2016). Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 375, 1823–1833. 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- Ren HM, Kolawole EM, Ren M, Jin G, Netherby-Winslow CS, Wade Q, Shwetank Rahman ZSM, Evavold BD, and Lukacher AE (2020). IL-21 from high-affinity CD4 T cells drives differentiation of brain-resident CD8 T cells during persistent viral infection. Sci Immunol 5. 10.1126/sciimmunol.abb5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw BR, Fanslow WC 3rd, Armitage RJ, Campbell KA, Liggitt D, Wright B, Davison BL, and Maliszewski CR (1994). Humoral immune responses in CD40 ligand-deficient mice. J Exp Med 180, 1889–1900. 10.1084/jem.180.5.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberti MP, Yonekura S, Duong CPM, Picard M, Ferrere G, Tidjani Alou M, Rauber C, Iebba V, Lehmann CHK, Amon L, et al. (2020). Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat Med 26, 919–931. 10.1038/s41591-020-0882-8. [DOI] [PubMed] [Google Scholar]
- Ruddle NH (2014). Lymphatic vessels and tertiary lymphoid organs. J Clin Invest 124, 953–959. 10.1172/JCI71611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffin AT, Cillo AR, Tabib T, Liu A, Onkar S, Kunning SR, Lampenfeld C, Atiya HI, Abecassis I, Kurten CHL, et al. (2021). B cell signatures and tertiary lymphoid structures contribute to outcome in head and neck squamous cell carcinoma. Nat Commun 12, 3349. 10.1038/s41467-021-23355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SM, and Liew FY (1979). T cells primed by influenza virion internal components can cooperate in the antibody response to haemagglutinin. Nature 280, 147–148. 10.1038/280147a0. [DOI] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019). Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936. 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautes-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, and Dieu-Nosjean MC (2016). Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front Immunol 7, 407. 10.3389/fimmu.2016.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautes-Fridman C, Petitprez F, Calderaro J, and Fridman WH (2019). Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 19, 307–325. 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- Schabath MB, and Cote ML (2019). Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol Biomarkers Prev 28, 1563–1579. 10.1158/1055-9965.EPI-19-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan SL, Gibbons G, Smith OC, Kim JY, Westcott P, et al. (2021). Conventional type I dendric cells maintain a reservoir of proliferative tumor-antigen specific TCF-1(+) CD8(+) T cells in tumor-draining lymph nodes. Immunity. 10.1016/j.immuni.2021.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, and Hacohen N (2016). Neoantigens encoded in the cancer genome. Curr Opin Immunol 41, 98–103. 10.1016/j.coi.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Scheper W, and Kvistborg P (2019). Cancer Neoantigens. Annu Rev Immunol 37, 173–200. 10.1146/annurev-immunol-042617-053402. [DOI] [PubMed] [Google Scholar]
- Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ, Peters B, Rafii-El-Idrissi Benhnia M, Hoffmann J, Su HP, et al. (2008). Selective Cd4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity 28, 847–858. 10.1016/j.immuni.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, and Chudakov DM (2020). B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol 20, 294–307. 10.1038/s41577-019-0257-x. [DOI] [PubMed] [Google Scholar]
- Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, Craft JE, and Nussenzweig MC (2014). Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345, 1058–1062. 10.1126/science.1257861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silina K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H, Wandres M, Isajevs S, Cheng P, Curioni-Fontecedro A, et al. (2018). Germinal Centers Determine the Prognostic Relevance of Tertiary Lymphoid Structures and Are Impaired by Corticosteroids in Lung Squamous Cell Carcinoma. Cancer Res 78, 1308–1320. 10.1158/0008-5472.CAN-17-1987. [DOI] [PubMed] [Google Scholar]
- Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, Mi T, Maiti S, Kebriaei P, Lee DA, et al. (2011). Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res 71, 3516–3527. 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell LM, MacLeod BL, Law JC, Osokine I, Elsaesser HJ, Hezaveh K, Dickson RJ, Gavin MA, Guidos CJ, McGaha TL, and Brooks DG (2018). CD8(+) T Cell Priming in Established Chronic Viral Infection Preferentially Directs Differentiation of Memory-like Cells for Sustained Immunity. Immunity 49, 678–694 e675. 10.1016/j.immuni.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondergaard H, and Skak K (2009). IL-21: roles in immunopathology and cancer therapy. Tissue Antigens 74, 467–479. 10.1111/j.1399-0039.2009.01382.x. [DOI] [PubMed] [Google Scholar]
- Suto A, Kashiwakuma D, Kagami S, Hirose K, Watanabe N, Yokote K, Saito Y, Nakayama T, Grusby MJ, Iwamoto I, and Nakajima H (2008). Development and characterization of IL-21-producing CD4+ T cells. J Exp Med 205, 1369–1379. 10.1084/jem.20072057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Kang B, Li C, Chen T, and Zhang Z (2019). GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 47, W556–W560. 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. (2018). A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 24, 994–1004. 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443–2454. 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645. 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L, Belicova L, Praznovec I, Halaska MJ, Brtnicky T, et al. (2018). Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J Immunother Cancer 6, 139. 10.1186/s40425-018-0446-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, and Mebius RE (2010). New insights into the development of lymphoid tissues. Nat Rev Immunol 10, 664–674. 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 20, 218–232. 10.1038/s41568-019-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhersecke L, Brunet M, Guégan J-P, Rey C, Bougouin A, Cousin S, Le Moulec S, Besse B, Loriot Y, Larroquette M, et al. (2021). Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nature Cancer 2, 794–802. 10.1038/s43018-021-00232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella LA, Buggert M, Manne S, Herati RS, Sayin I, Kuri-Cervantes L, Bukh Brody I, O’Boyle KC, Kaprielian H, Giles JR, et al. (2019). T follicular helper cells in human efferent lymph retain lymphoid characteristics. J Clin Invest 129, 3185–3200. 10.1172/JCI125628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, Broeks A, Sanders J, Herzig P, Zippelius A, et al. (2021). An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat Med. 10.1038/s41591-021-01398-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jiang H, Luo H, Sun Y, Shi B, Sun R, and Li Z (2019). An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front Immunol 10, 1691. 10.3389/fimmu.2019.01691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Laurence A, Elias KM, and O’Shea JJ (2007). IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem 282, 34605–34610. 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, and Craft J (2016). TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol 17, 1197–1205. 10.1038/ni.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JS, Laidlaw BJ, Lu Y, Wang JK, Schulz VP, Li N, Herman EI, Kaech SM, Gallagher PG, and Craft J (2018). STAT4 and T-bet control follicular helper T cell development in viral infections. J Exp Med 215, 337–355. 10.1084/jem.20170457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. (2020). Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818–834 e813. 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland A, Patel MR, Cardenas MA, Eberhardt CS, Hudson WH, Obeng RC, Griffith CC, Wang X, Chen ZG, Kissick HT, et al. (2020). Defining HPV-specific B cell responses in patients with head and neck cancer. Nature. 10.1038/s41586-020-2931-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, Esplugues E, Wen R, Wang D, Parish IA, et al. (2015). A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Rep 13, 1118–1124. 10.1016/j.celrep.2015.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- Yamanouchi S, Kuwahara K, Sakata A, Ezaki T, Matsuoka S, Miyazaki J, Hirose S, Tamura T, Nariuchi H, and Sakaguchi N (1998). A T cell activation antigen, Ly6C, induced on CD4+ Th1 cells mediates an inhibitory signal for secretion of IL-2 and proliferation in peripheral immune responses. Eur J Immunol 28, 696–707. . [DOI] [PubMed] [Google Scholar]
- Yi JS, Du M, and Zajac AJ (2009). A vital role for interleukin-21 in the control of a chronic viral infection. Science 324, 1572–1576. 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, et al. (2009). The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468. 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, and Ahmed R (1998). Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188, 2205–2213. 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W (2019). CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51, 1028–1042 e1024. 10.1016/j.immuni.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander RA, Vijay R, Pack AD, Guthmiller JJ, Graham AC, Lindner SE, Vaughan AM, Kappe SHI, and Butler NS (2018). Th1-like Plasmodium-Specific Memory CD4(+) T Cells Support Humoral Immunity. Cell Rep 23, 1230–1237. 10.1016/j.celrep.2018.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Liu E, Gertie JA, Joseph J, Xu L, Pinker EY, Waizman DA, Catanzaro J, Hamza KH, Lahl K, et al. (2020). Divergent T follicular helper cell requirement for IgA and IgE production to peanut during allergic sensitization. Sci Immunol 5. 10.1126/sciimmunol.aay2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, Liu B, Niu L, Sun X, Yu X, et al. (2021). Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell. 10.1016/j.ccell.2021.09.010. [DOI] [PubMed] [Google Scholar]
- Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, Fischer S, Jackson R, Flavell RA, Wang J, et al. (2020). IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614. 10.1038/s41586-020-2422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner KM, Smyth MJ, et al. (2010). IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med 207, 365–378. 10.1084/jem.20091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štach M, Musil J, Cetkovsky P, and Otahal P (2018). Interleukin 21 Enhances Survival and Expansion of CAR T Cells Via Inhibition of Their Terminal Differentiation during Interaction with Tumor Target Cells. Blood 132, 4545–4545. 10.1182/blood-2018-99-116294. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analyses of CD4 T cells and B cells in LUAD samples from TCGA; Related to Figure 1
A, Estimated fractions of subtypes in CD4 T cells and B cell lineage described in Figure 1A, from CIBERSORT analyses of TCGA-LUAD samples (n=513).
B, Survival analyses based on expression signatures of Th1, effector CD8 T cells, Th17, NK cells in TCGA-LUAD patient cohort (n=478).
C, Correlation analyses among expression signatures of GC B cells, TFH, Th1, CD8 effector cells, Th17, NK cells in lung adenocarcinoma patients.
B, log-rank Mantel-Cox test. C, two-tailed Pearson correlation test.
Figure S2. TFH cell responses in murine tumor models; Related to Figure 2
A, Representative flow plots showing TFH gating in tumor-draining LNs (dLNs) of MC38 or B16-F10 tumor-bearing C57BL/6 mice (pre-gated on TCRb+ CD4). Flow cytometric analyses were performed on day 10 – 12. Data (mean ±SD) represented 4 – 5 mice/group.
B, Tumor growth curves: C57BL/6, RAG1 KO, CIITA KO and uMT mice were s.c. implanted with 2 x 105 KP. Data (mean ±SD) were pooled, from 3 – 10 mice/group/experiment, and representative of at least 2 independent experiments. Two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure S3. KP-HELLO LUAD cell line elicits GP66-specific CD4 T cell responses in vitro, while unable to elicit robust GP33-specific CD8 T responses; Related to Figure 2
A, Representative flow plots showing CD69 and CD44 expression on SMARTA CD4 T cells, after 48-hour co-culture with MD4 or C57BL/6 B cells, and KP or KP-HELLO supernatant, 500ng/mL HEL or 5ug/mL GP61-80.
B, Chromatogram displaying DNA sequence of GP33-43/FLAG/GP61-80 from the proviral insertion in the KP-HELLO tumor cell line. This demonstrated that the integrated provirus in KP-HELLO cells encodes the correct sequence for GP33-43/FLAG/GP61-80. The amino acid sequence is indicated above the chromatogram as a reference for translation. Results were repeated in 4 independent experiments.
C, In vitro co-culture of P14 T cells and antigen loaded DCs. Bar graphs displaying division and activation marker expression on P14 T cells under different experimental conditions. CFSE-labeled P14 cells were co-cultured with LPS-stimulated and antigen-loaded DCs. On day 3, cell culture was collected to assess P14 cell proliferation (by CFSE dilution) and activation (by CD44 upregulation or CD62L downregulation) by flow cytometry.
D – F, GP33-specific CD8+ T cell responses in KP-HELLO models. (D) Frequency and representative flow plots displaying GP33-specific CD8+ T cells in tumors (top) and dLNs (bottom). C57BL/6 and uMT mice were s.c. implanted with 2 x 105 KP-HELLO. Tumors and dLNs were harvested on day 14 – 16. GP33 gating (red: pre-gated on singlets, TCRb+ CD8+) was drawn based on FMO control (blue). (E) Frequency and representative flow plots displaying P14 tumor-specific CD8+ T cells in tumors (top) and dLNs (bottom). 5 x 105 Thy1.1 P14 T cells were transferred (i.v.) into C57BL/6 and uMT mice, one day prior to 5 x 105 KP-HELLO s.c. implant. Tumors and dLNs were harvested on day 14. (F) Frequency and representative flow plots displaying GP33-specific CD8+ T cells in tumor tissues (top) and dLNs (bottom). KP and uMT-KP mice were infected intratracheally with 5 x 104 PFU Lenti-HELLO-Cre. Lung tumors were harvested 8 weeks post-infection. GP33 gating (red: pre-gated on singlets, TCRb+ ivCD45Neg, CD8+) was drawn based on FMO control (blue). Here, Y-axis of the bar graphs were adjusted based on figures and analyses of previous publications (Connolly et al., 2021; Fitzgerald et al., 2021) to display the differences of GP33-specific responses in KP-HELLO vs. KP-NINJA.
Figure S4. KP-HELLO LUAD tumors elicit antigen-specific TFH and GC B cell responses in vivo; Related to Figure 2
A, Representative flow plots of GP66-specific CD4 T cells from dLNs of KP-HELLO tumor-bearing C57BL/6 mice.
B, Experimental design of Figure S4C–S4D.
C – D, Representative flow plots showing tumor-specific SMARTA TFH cells (C) and SWHEL GC B cells (D) in dLNs of KP-HELLO bearing mice. As shown in Figure S4B, CD45.2/CD45.2 Thy1.2/Thy1.2 C57BL/6 recipient, transferred with 1 x 105 CD45.1/CD45.2 HEL-specific SWHEL B cells and 1 x 105 Thy1.1/Thy1.2 GP66-specific SMARTA CD4 T cells, were s.c. implanted with 5 x 105 KP-HELLO. Flow cytometric analyses were performed on day 10 – 12. Data (mean ± SD) represented at least 2 independent experiments.
Figure S5. B cells are required to drive tumor-specific TFH cell responses; Related to Figure 3
A – B, Tumor growth curves: RAG1 KO or C57BL/6 mice were s.c. implanted with 2 × 105 KP or KP-HELLO. Growth curve of KP-HELLO in C57BL/6 (n = 40), pooled from 8 independent experiments, was set as the baseline of analyses.
C – G, Frequency and number of GP66-specific total CD4 T cells (C and F), TFH cells (CD44hi PSGL1lo Ly6Clo PD1hi CXCR5hi) (E and G), and other CD4+ CD44hi T cell subsets (D and G) from dLNs of C57BL/6, uMT or CD4-Cre Bcl6fl/fl mice with KP-HELLO tumors.
H, Representative histogram displaying T-bet expression on naïve, GP66-specific PSGL1hi Ly6Chi (TH1-like) and PSGL1lo Ly6Clo PD1hi CXCR5hi (TFH) T cells from dLNs of KP-HELLO tumor bearing mice.
Data (mean ±SD) were pooled, from 3–10 mice/group/experiment, and representative of at least 2 independent experiments. A – B, Two-way ANOVA. C – G, Unpaired t-test with Welch’s correction. **P < 0.05, **P < 0.01, ***P < 0.001, ****p < 0.0001.
Figure S6. B cells are required to drive tumor-specific TFH cell responses and tumor-infiltrating effector CD8 T cell responses in KP-HELLO autochthonous lung tumor model; Related to Figure 2–4 and STAR Methods
A, Design of KP-HELLO autochthonous lung tumor model. KP or uMT-KP mice were intratracheally infected with 5 × 104 PFU Lenti-HELLO-Cre. Tumor lung tissues and dLNs were harvested at 8 weeks post-infection.
B, Diagram showing the technique to label circulating immune cells in lung tissues for flow cytometric analyses. 2–3 minutes prior to sacrifice, PE-CF594-conjugated CD45 antibodies were injected intravenously. Immune cells in circulation were labelled, while the time was not enough for antibodies to diffuse into tissue and bind to tumor-infiltrating immune cells.
C, Gating strategy and representative histogram of CD45-PE-CF594 (in vivo labeling) showing tissue-infiltrating B cells, CD4 T cells and CD8 T cells in non-tumor-bearing mice, or tumor-bearing KP and uMT-KP mice.
D, Representative flow plots and frequency of GP66-specific CD4 TFH cells (CD44hi PSGL1lo PD1hi CXCR5hi) from tumor lung tissues and dLNs of KP or uMT-KP mice. Flow plots were pre-gated on CD45i.v.Neg TCRb+ CD4+ GP66-tetramer+ CD44hi PSGL1lo.
E, Representative flow plots showing GC B cells in tumor lung tissues and dLNs of KP mice (pre-gated on CD45i.v.Neg CD19+ B220+ IgDlo).
F, Representative flow plots and frequency of tumor-infiltrating CD44hi PD1hi CD8+ T cells in KP or uMT-KP mice.
Data (mean ±SD) were pooled, from 3–10 mice/group/experiment, and representative of at least 2 independent experiments. D – F, Unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure S7. TFH-derived IL-21 in murine tumor models; Related to Figure 6
A – B, Representative flow plots showing IL-21-expressing TFH cells (A) and histogram displaying IL-21 expression through different stages of TFH differentiation (B) in dLNs of MC38 and B16-F10 tumor-bearing mice. Flow cytometric analyses were performed on day 10 – 12.
C, IL-21 expression by TFH cells (CD44hi PSGL1lo PD1hi CXCR5hi) in dLNs of KP, MC38, B16-F10 and KP-HELLO tumors.
Data (mean ±SD) were pooled, from 3 – 5 mice/group/experiment, and representative of 2 independent experiments. C, Unpaired t-test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Table S1. Differentially expressed genes in T/NK cell clusters and B cell clusters (GSE131907). Related to Figure 1.
Table S2. DNA and amino acid sequences of HELLO construct. Related to Figure 2 and STAR Methods.
Table S3. Prediction of CD8 T cell epitopes in HELLO neoantigen. Related to Figure 2 and STAR Methods.
Data Availability Statement
This paper analyzed existing, publicly available scRNA-seq data (GSE131907). Code is available at https://github.com/wangjw17/TfhInLungCancer (DOI: https://doi.org/10.5281/zenodo.5590959). All data reported in this paper will be shared by the lead contact upon request.