

Abstract
Background
Janus kinase 2 (JAK2) is activated in diabetic mellitus (DM) conditions and may enhance oxidative stress, apoptosis and fibrosis in many tissues. Whether JAK2 activation is involved in the occurrence of diabetic erectile dysfunction (ED) is unknown.
Objectives
We performed this study to investigate the effect of JAK2 deficiency on diabetic ED.
Materials and methods
Conditional JAK2 gene knockout mice (Cre +/+‐JAK2fl/fl ) were used, in which JAK2 gene knockout could be induced by tamoxifen. Mice fell into four groups: control, JAK2 knockout (JAK2−/−), DM, and DM with JAK2−/−. DM was induced by intraperitoneal injection of streptozotocin. Two months later, JAK2 gene knockout was induced with tamoxifen in Cre+/+‐JAK2fl/fl mice. After another 2 months, erectile function was measured by electrical stimulation of the cavernous nerve, and penile tissues were harvested. Ratio of maximal intracavernosal pressure (MIP) to mean arterial blood pressure (MAP), expression and phosphorylation of JAK2, oxidative stress level, NO/Cyclic Guanosine Monophosphate (cGMP) pathway, apoptosis, fibrosis, and transforming growth factor beta 1 (TGF‐β1)/Smad/Collagen IV pathway in corpus cavernosum, were measured.
Results
JAK2 expression was remarkably decreased after induction with tamoxifen. JAK2 was activated in penile tissues of diabetic mice, and JAK2 deficiency could improve the impaired erectile function caused by DM. However, in mice without DM, JAK2 deficiency had no apparent influence on erectile function. Levels of oxidative stress, apoptosis, fibrosis, and TGF‐β1/Smad/Collagen IV pathway were all elevated by DM, whereas JAK2 deficiency lessened these alterations in diabetic mice. Moreover, JAK2 deficiency improved the expression of the down‐regulated NO/cGMP pathway in diabetic mice. In non‐diabetic mice, no apparent changes were found in aforementioned parameters after JAK2 gene knockout.
Discussion and conclusion
Our study showed that JAK2 deficiency could improve erectile function in diabetic mice, which might be mediated by reduction in oxidative stress, apoptosis, and fibrosis in corpus cavernosum.
Keywords: apoptosis, diabetic erectile dysfunction, fibrosis, Janus kinase 2, oxidative stress
1. INTRODUCTION
In diabetic patients, the prevalence of erectile dysfunction (ED) is about 3 times higher than that in patients without diabetes mellitus (DM). 1 Moreover, compared with non‐diabetic ED, diabetic ED is more severe and not sensitive to phosphodiesterase type 5 inhibitors. 2 Various pathological changes of corpus cavernosum have been reported to contribute to the development of diabetic ED, such as endothelial dysfunction, neural degeneration, apoptosis of smooth muscle cells, and cavernous fibrosis. 3 , 4 These deteriorated alterations in neurovascular function were caused by many factors, such as excessive production of reactive oxygen species (ROS) and advanced glycation endproducts (AGEs). 5 , 6
Janus kinase 2 (JAK2), a member of the Janus kinase family, is a non‐receptor tyrosine kinase and participates in various signaling pathways in mammals, such as cell differentiation, proliferation and apoptosis. 7 , 8 , 9 The main function of JAK2 is to deliver signals initiated by multiple cytokines and growth factors from extracellular to intracellular. Upon combination of ligands and receptors, two or more receptor‐associated JAK2 get close to each other and become auto‐phosphorylated or trans‐phosphorylated. Then the activated JAK2 phosphorylates downstream molecules to regulate gene transcription. 10 , 11
Numerous studies have shown that hyperglycemia could induce the phosphorylation of JAK2. In endothelial cells exposed to high glucose, the activation of JAK2 led to apoptosis. 12 Another study demonstrated that hyperglycemia‐induced activation of JAK2 was mediated through an angiotensin II (Ang II)‐dependent pathway in rat glomeruli. 13 Moreover, JAK2 activation was essential for hyperglycemia‐induced glomerular mesangial cell growth and collagen IV generation in rat kidney. 14 Increased production of transforming growth factor‐β (TGF‐β) in mesangial cells exposed to high glucose was also related to JAK2 activation. 15 In addition, activation of JAK2 by hydrogen peroxide was critical for the apoptosis of vascular smooth muscle cells. 16 Oxidative stress played an important role in the activation of JAK2, and in turn, JAK2 activation could exacerbate oxidative stress. 17 Our previous study showed that oxidative stress induced by high glucose in cavernous smooth muscle cells was significantly reduced upon treatment with the JAK2 inhibitor, AG490. 18 The above studies suggest that JAK2 is a major mediator of diabetic injuries in various tissues. We thereby hypothesize that JAK2 activation is a contributing factor to the development of diabetic ED.
AG490 is a common inhibitor of JAK2 and has been used in some studies. However, AG490 might non‐specifically target kinases other than JAK2, such as JAK3 and mitogen‐activated protein kinase (MAPK). 19 , 20 On the other hand, mice deficient in JAK2 tend to die in the embryonic period due to impaired hematopoiesis, limiting the use of traditional gene knockout technology to knockout JAK2 gene. 21 To address this issue, a conditional JAK2 gene knockout (Cre+/+‐JAK2fl/fl ) mouse model was generated with the Cre/LoxP recombination system. In this model, the knockout of JAK2 gene can be induced by tamoxifen in adult mice. 22
In this study, we used Cre+/+‐JAK2fl/fl mice to explore the effect of JAK2 deficiency on diabetic ED. We speculated that JAK2 deficiency was beneficial to the maintenance of ED, and the underlying mechanism might involve changes in oxidative stress, apoptosis, and fibrosis of corpus cavernosum.
2. MATERIALS AND METHODS
2.1. Treatment of animals
All animal procedures were conducted in accordance with the Animal Care and Use Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China), and ethical approval was obtained. The Cre+/+‐JAK2fl/fl mice were a generous gift from the Center for Biomedical Research, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). A Cre+/+‐JAK2fl/fl mouse carries LoxP sites at the first coding exon of JAK2 gene, and simultaneously, it carries a Cre‐ERT2 gene which can produce Cre recombinase. Cre recombinase can recognize LoxP sites and silence JAK2 gene. It is worth noting that Cre recombinase is inactivated without induction of tamoxifen. 22
The mice were housed in a pathogen‐free environment with a 12/12‐h light/dark cycle. 8‐week‐old male Cre+/+‐JAK2fl/fl mice were divided into four groups: control, JAK2 knockout (JAK2−/−), DM, and DM with JAK2−/− (n = 12 for each group).
DM was induced by intraperitoneal injection of streptozotocin (Sigma‐Aldrich, S0130, St. Louis, MO, USA) at a dose of 60 mg/kg body weight for 5 consecutive days in the latter two groups. 23 One week after the last injection, fasting blood glucose level was measured, with concentration ≥16.7 mmol/L considered to be DM. Mice with blood glucose < 16.7 mmol/L were excluded.
Two months after the establishment of DM model, mice in JAK2−/− group and DM with JAK2−/− group were intraperitoneally injected with tamoxifen (Sigma‐Aldrich, T5648) dissolved in corn oil at 20 mg/kg body weight for 5 consecutive days to induce JAK2 gene knockout. Mice in the other two groups were injected with an equal volume of corn oil. Mice were fed for 2 more months. The initial and final body weight and blood glucose level were recorded.
2.2. Evaluation of erectile function
The maximal intracavernosal pressure (MIP) and the corresponding mean arterial pressure (MAP) were measured with electrostimulation of cavernous nerve. The ratio of MIP to MAP (MIP/MAP) was calculated to reflect erectile function. 23 Briefly, after anesthesia with pentobarbital sodium (Sigma‐Aldrich, P3761) at 60 mg/kg body weight, mice were fixed on a warm pad in supine position. The carotid artery was catheterized with a PE‐10 tube to monitor the arterial pressure. A 29‐G needle was inserted into the corpus cavernosum to measure the intracavernosal pressure. A midline incision in the abdomen was made to expose the cavernous nerve. Cavernous nerve was stimulated using a bipolar electrode at 5 V with a frequency of 15 Hz and a pulse width of 1.2 ms for 60 s to induce erection. Data were recorded by a BL‐420F biological signal acquisition and processing system (Techman Soft, Chengdu, China). Each mouse was stimulated 3 times and data of the optimal erection were used to calculate MIP/MAP. Blood sample was then collected from the carotid artery and left to clot at room temperature for 1 h. The serum was collected and stored at −80°C. At last, mice were euthanized and the penis was cut into three parts: two parts were snap frozen in liquid nitrogen and stored at −80°C, another part was immersed in 4% paraformaldehyde to perform paraffin embedding.
2.2.1. Serum testosterone measurement
Serum T level was measured with an Elisa Kit (Abcam, ab108666, Cambridge, UK). Briefly, duplicate 25 μl of the standard, control or samples were added to the ELISA plate respectively, followed by the addition of 100 μl of testosterone‐HRP conjugate. After incubation at 37°C for 1 h, the plate was washed three times with 300 μl of wash buffer. 100 μl TMB substrate solution was added into the plate with a 15‐min incubation at 37°C in the dark. After adding 100 μl of Stop Solution, the absorbance was read at 450 nm on a microplate reader (Thermo Fisher, Waltham, MA, USA). Concentration of testosterone of was calculated according to the standard curve.
2.3. Western blot
Proteins were extracted from cryopreserved penile tissues and the concentration of proteins was measured with a BCA protein assay kit (Beyotime, P0012, Shanghai, China). 40 μg protein lysate was loaded on sodium dodecyl sulfate/polyacrylamide (SDS‐PAGE) gel for electrophoresis and then transferred to a polyvinylidene difluoride membrane. After blocking in 5% bovine serum albumin for 1 h, the membranes were incubated overnight with primary antibodies against the following proteins at 4°C: β‐actin (1:500; Boster, BM0627, Wuhan, China), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (NOX1, 1:500; Abcam, ab55831, Cambridge, UK), NOX2 (1:500; Abcam, ab80508), NOX4 (1:500; Abcam, ab154244), phospho‐JAK2 (Tyr1007/1008; 1:1000; CST, 3776, Danvers, MA, USA), JAK2 (1:1000; CST, 3230), caspase3 (1:1000; CST, 9665), cleaved caspase3 (1:1000; CST, 9664), TGF‐β1 (1:500; Abcam, ab64715), phospho‐Smad2/3 (1:1000; CST, 8828), Smad2/3 (1:1000; CST, 8685), and Collagen IV (1:500; Abcam, ab6586). Washed membranes were incubated with horseradish peroxidase‐conjugated secondary antibodies (1:1000; CST, 7074, 7076) at room temperature for 1 h. Finally, the protein bands were developed with Western ECL Substrate (Bio‐Rad Laboratories, 1705061, Hercules, CA, USA). Densitometry value of bands was analyzed with Image J software (National Institutes of Health, Bethesda, MD, USA).
2.4. Malondialdehyde (MDA) detection
Lipid peroxidation is an event caused by oxidative stress. MDA is an aldehydic metabolite of lipid peroxidation and can be used to indicate the level of oxidative stress. 24 We used an MDA assay kit (Beyotime, S0131) to measure the content of MDA in corpus cavernosum. Briefly, 0.1 ml supernatant of the tissue homogenate was mixed with 0.2 ml MDA testing reagent and incubated in boiling water for 15 min. After centrifugation, 0.2 ml supernatant of the mixture was removed to a 96‐well plate and the absorbance was measured at 532 nm with a microplate reader (Thermo). MDA level in each sample was calculated according to the standard curve and was normalized by protein concentration of the sample.
2.4.1. NO content analysis
NO content in the penis was detected with a NO assay kit (Beyotime, S0023). Generally, NO is metabolized into nitrate and nitrite in vivo, and NO content can be reflected via the concentration of nitrate and nitrite. To measure the level of nitrate and nitrite, penis lysates were mixed with NADPH, flavin adenine dinucleotide and nitrate reductase at 30°C for 30 min. Then actate dehydrogenase was added into the mixture, followed by 30‐min incubation. Finally, Griess reagents were added and the absorbance was measured at 540 nm with a microplate reader (Thermo). NO level was calculated based on the standard curve and normalized by protein concentration of each sample.
2.4.2. Cyclic guanosine monophosphate (cGMP) measurement
Concentration of cGMP in corpus cavernosum was measured by an enzyme‐linked immunosorbent assay kit (R&D Systems, KGE003, Minneapolis, MN, USA) according to the manufacturer's instructions. The cGMP level was normalized by protein concentration of each sample.
2.4.3. Caspase3 activity evaluation
Caspase3 activity was detected with a caspase3 activity assay kit (Beyotime, C1116) following the manufacturer's instructions. Supernatant of penis lysates was briefly mixed with the detection buffer and Ac‐DEVD‐pNA. After a 2‐h incubation at 37°C, the absorbance of the mixture was measured at 405 nm with a microplate reader (Thermo). Caspase3 activity was calculated according to the standard curve and normalized by the protein concentration of each sample.
2.5. Histologic assessment
Immunohistochemistry was performed to detect the content of JAK2. Paraffin‐embedded tissues were cut into sections. After dewaxing and dehydration, the endogenous peroxidase was inactivated with 3% hydrogen peroxide, followed by antigen recovery. Then the sections were washed and blocked with goat serum. Sections were incubated with antibodies against JAK2 (1:200; CST) at room temperature overnight. They were then incubated successively with biotinylated secondary antibody, peroxidase‐conjugated streptavidin, diaminobenzidine and Harris's hematoxylin. Images were observed under microscope.
The content of endothelium and smooth muscle in the corpus cavernosum was assessed by immunofluorescence. Penile sections were prepared as above. After incubation with normal goat serum, sections were incubated with antibodies against α‐smooth muscle actin (α‐SMA; 1:100; Boster, A03744) or CD‐31 (1:200; Affinity, AF6191, Zhenjiang, China) at 4°C overnight. After that, sections were incubated with DyLight‐conjugated secondary antibodies (1:200; Abbkine, A23210, A23220, Redlands, CA, USA). Finally, nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; Beyotime, C1006). Images were observed with a fluorescence microscope (Olympus, Tokyo, Japan) and the percentage of staining‐positive area was calculated in three randomly chosen fields with Image‐Pro plus software (Media Cybernetics, Silver Spring, MD, USA).
Terminal deoxynucleotidyl transferase 2′‐deoxyuridine 5′‐triphosphate nick end labeling (TUNEL) staining was conducted on the sections with an In Situ Cell Death Detection Kit (Roche Applied Science, 06432344001, Indianapolis, IN, USA) following the manufacturer's instructions. Nuclei were stained blue in normal cells and brown in apoptotic cell. Apoptotic index, the ratio of apoptotic cells to all cells, was calculated in three randomly chosen fields of each sample.
Masson's trichrome staining was conducted to evaluate the level of cavernous fibrosis. The ratio of collagen to smooth muscle in corpus cavernosum was calculated in three randomly chosen fields of each sample.
2.6. Statistical analysis
Data were analyzed with GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA) and presented as mean ± SD. Kolmogoro–Smirnov test was used to determine normal distribution. Comparisons of normally distributed data were performed with t‐test or one‐way analysis of variance (anova) followed by Tukey test. Mann–Whitney test or Kruskal–Wallis test was used for non‐parametric data. Intergroup difference was considered to be significant with p < 0.05.
3. RESULTS
3.1. Changes in some metabolic indexes of mice
Initial body weight and fasting blood glucose level of mice had no apparent differences among groups (Table 1). After 4 months, the mean body weight of all groups increased remarkably (p < 0.05, compared with initial weight). However, compared with non‐diabetic mice, the mean body weight of diabetic mice was lower (p < 0.05). Furthermore, the final blood glucose levels of two diabetic groups increased a lot and were much higher than non‐diabetic groups (p < 0.05). Serum testosterone and MAP had no apparent difference among the 4 groups.
TABLE 1.
Metabolic parameters
| Control | JAK2−/− | DM | DM+JAK2−/− | |
|---|---|---|---|---|
| Initial body weight(g) | 20.73 ± 2.44 | 21.27 ± 1.82 | 20.67 ± 1.71 | 20.98 ± 2.32 |
| Final body weight (g) | 28.99 ± 2.98# | 29.08 ± 2.88# | 23.11 ± 2.09* , § , # | 23.26 ± 2.77* , § , # |
| Initial blood glucose (mmol/L) | 5.82 ± 1.92 | 5.55 ± 1.58 | 5.63 ± 1.53 | 5.64 ± 1.33 |
| Final blood glucose (mmol/L) | 5.68 ± 1.64 | 5.72 ± 1.63 | 27.29 ± 3.57* , § , # | 27.96 ± 2.64* , § , # |
| Serum testosterone (pg/mL) | 3.67 ± 0.72 | 3.25 ± 0.80 | 3.35 ± 0.86 | 3.29 ± 0.61 |
| Mean arterial pressure (mmHg) | 100.62 ± 6.39 | 98.33 ± 9.20 | 106.45 ± 6.4 | 102.68 ± 5.76 |
Data were expressed as mean ± SD. n = 10 for each group.
p < 0.05 compared with control group.
p < 0.05 compared with JAK2−/− group.
p < 0.05 compared with initial value.
3.2. JAK2 gene knockout reduced DM‐induced phosphorylation of JAK2
Compared with the control group, JAK2 phosphorylation was elevated in the DM group (p < 0.05), whereas total JAK2 expression did not increase notably (Figure 1A–C). After induction with tamoxifen, JAK2 expression and phosphorylation both decreased dramatically in JAK2−/− group and DM with JAK2−/− group (p < 0.05 for each). Immunohistochemistry showed that JAK2 was widely expressed in corpus cavernosum and the content of JAK2 was sparse after JAK2 gene knockout (Figure 1D).
FIGURE 1.
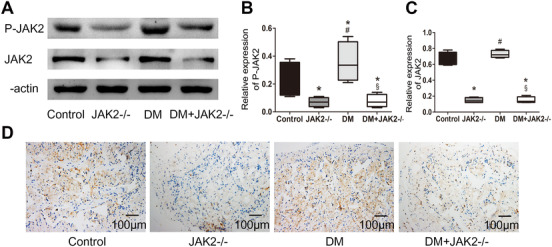
Expression and phosphorylation of JAK2 in corpus cavernosum. (A) Representative western blot results for JAK2 and phosphorylated JAK2. (B,C) Relative expression of phosphorylated JAK2 and JAK2 compared with β‐actin; data are shown as the fold changes over the control group; n = 4 for each group. (D) Representative images of immunohistochemistry detecting JAK2 expression (200×). *: p < 0.05 compared with control group. #: p < 0.05 compared with JAK2−/− group. §: p < 0.05 compared with DM group. DM: diabetes mellitus
3.3. JAK2 deficiency improved erectile function in diabetic mice
The erectile function of mice in the two diabetic groups was impaired severely and the MIP/MAP was lower than non‐diabetic groups (p < 0.05; Figure 2). In diabetic mice, JAK2 gene knockout could increase MIP/MAP (p < 0.05), whereas in non‐diabetic mice JAK2 gene knockout had no apparent influence on erectile function.
FIGURE 2.
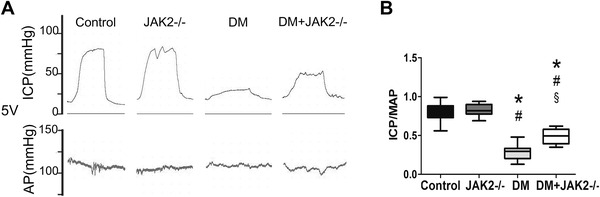
Erectile function of mice. (A) Representative recordings of intracavernosal pressure (ICP) and arterial pressure (AP) during 1‐min electrical stimulation at 5.0 V. (B) The ratio of maximal intracavernosal pressure (MIP) to mean systemic arterial blood pressure (MAP) for each group; n = 10 for each group. *: p < 0.05 compared with control group. #: p < 0.05 compared with JAK2−/− group. §: p < 0.05 compared with DM group. DM: diabetes mellitus
3.4. JAK2 deficiency ameliorated oxidative stress in corpus cavernosum of diabetic mice
It is reported that oxidative stress played an important role in the cellular damage caused by JAK2 activation. We found hyperglycemia triggered the over‐expression of three major NOXs: NOX1, NOX2 and NOX4. However, JAK2 deficiency reduced their expression in diabetic mice (p < 0.05 for each; Figure 3A–D). As an indicator of oxidative stress, the level of MDA in penile tissues was apparently higher in the two diabetic groups (p < 0.05 for each; Figure 3E). Compared with the DM group, MDA level in DM with JAK2−/− group was reduced (p < 0.05). In the non‐diabetic mice, JAK2 deficiency had no remarkable influence on abovementioned indexes regarding oxidative stress.
FIGURE 3.
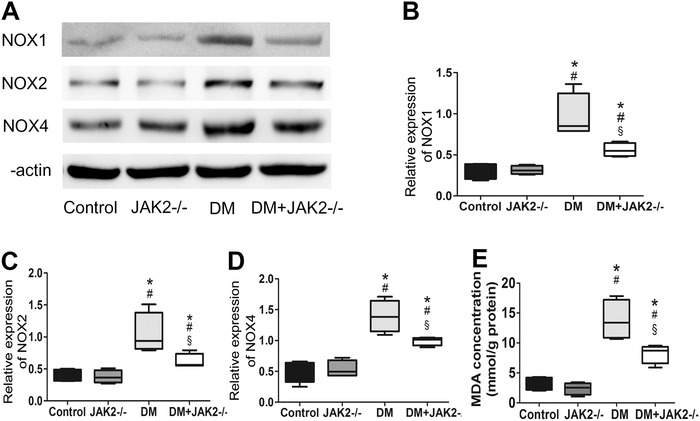
Oxidative stress level in corpus cavernosum. (A) Representative western blot results for NOX1, NOX2, and NOX4. (B–D) Relative expression of NOX1, NOX2, and NOX4 compared with β‐actin; data are shown as the fold changes over the control group; n = 4 for each group. (E) MDA concentration in corpus cavernosum was measured with an MDA assay kit; n = 4 for each group. *: p < 0.05 compared with control group. #: p < 0.05 compared with JAK2−/− group. §: p < 0.05 compared with DM group. DM: diabetes mellitus. NOX: NADPH oxidase. MDA: malondialdehyde
3.5. JAK2 deficiency protected NO/cGMP pathway in diabetic mice
NO/cGMP pathway is a main participator in the process of penile erection. We found that diabetes downregulated NO/cGMP pathway in corpus cavernosum (p < 0.05 for each; Figure 4). After JAK2 gene knockout, the level of NO and cGMP both increased in diabetic mice (p < 0.05 for each). In the non‐diabetic mice, JAK2 deficiency did not visibly change the NO/cGMP pathway.
FIGURE 4.

NO‐cGMP pathway in corpus cavernosum. (A) NO concentration in corpus cavernosum measured with a NO assay kit; n = 4 for each group. (B) Concentration of cGMP in corpus cavernosum measured with an enzyme‐linked immunosorbent assay kit; n = 4 for each group. *: p < 0.05 compared with control group. #: p < 0.05 compared with JAK2−/− group. §: p < 0.05 compared with DM group. DM: diabetes mellitus. cGMP: cyclic guanosine monophosphate
3.6. JAK2 deficiency improved cell survival in diabetic mice
We detected apoptosis in corpus cavernosum with multiple methods. Immunofluorescence results showed that hyperglycemia decreased the number of both endothelial cells and smooth muscle cells, whereas JAK2 deficiency had a protective effect on their survival in diabetic mice (p < 0.05 for each; Figure 5A–D). Western blot showed that the levels of caspase3 and cleaved caspase3 were increased by hyperglycemia (p < 0.05 for each; Figure 5E–G). JAK2 deficiency reduced caspase3 cleavage in diabetic mice (p < 0.05). Cleaved caspase3 is the activated form of caspase3 and can reflect the activity of caspase3. In consistence with this finding, caspase3 activity measured with a kit was enhanced by DM and decreased by JAK2 deficiency in diabetic mice (p < 0.05 for each; Figure 5H). TUNEL staining suggested that the elevated apoptotic index in corpus cavernosum of diabetic mice was lowered with JAK2 deficiency (p < 0.05 for each; Figure 5I,J). In the non‐diabetic mice, JAK2 deficiency had no influence on apoptosis.
FIGURE 5.
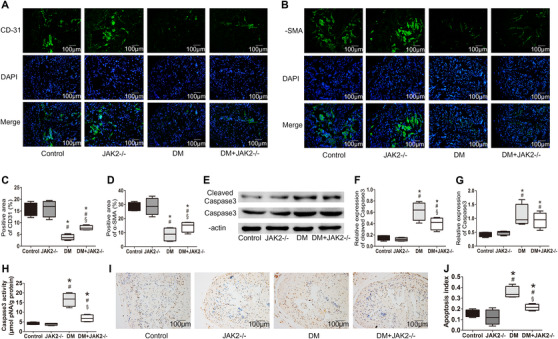
Apoptosis in corpus cavernosum. (A,B) Representative images of immunofluorescence detecting endothelium and smooth muscle with antibodies against CD‐31 or α‐SMA (green, 200×); nuclei were stained with DAPI (blue). (C,D) Percentage of endothelium and smooth muscle content detected with immunofluorescence; n = 4 for each group. (E) Representative western blot results for cleaved caspase‐3 and caspase‐3. (F,G) Relative expression of cleaved caspase‐3 and caspase‐3 compared with β‐actin detected with western blot; data are shown as the fold changes over the control group; n = 4 for each group. (H) Caspase‐3 activity detected with a caspase‐3 activity assay kit; n = 4 for each group. (I) Representative images of terminal deoxynucleotidyl transferase 2′‐deoxyuridine 5′‐triphosphate nick end labeling (TUNEL) staining (200×); apoptotic cells were stained brown and non‐apoptotic cells were blue. (J) Apoptosis index in the corpus cavernosum detected with TUNEL staining; n = 4 for each group. *: p < 0.05 compared with control group. #: p < 0.05 compared with JAK2−/− group. §: p < 0.05 compared with DM group. DM: diabetes mellitus
3.7. JAK2 deficiency attenuated cavernous fibrosis in diabetic mice
Cavernous fibrosis caused by TGF‐β1/Smad/Collagen IV signaling pathway is involved in the development of diabetic ED. Our study showed that TGF‐β1/Smad/Collagen IV pathway was upregulated by hyperglycemia, while JAK2 deficiency could downregulate this pathway in diabetic mice (p < 0.05 for each; Figure 6A–E). Moreover, Masson's staining indicated that the ratio of collagen to smooth muscle in corpus cavernosum was elevated by hyperglycemia and JAK2 deficiency could relieve this change up to a point (p < 0.05 for each; Figure 6F,G). In the non‐diabetic mice, TGF‐β1/Smad/Collagen IV pathway and cavernous fibrosis were not influenced by JAK2 deficiency.
FIGURE 6.
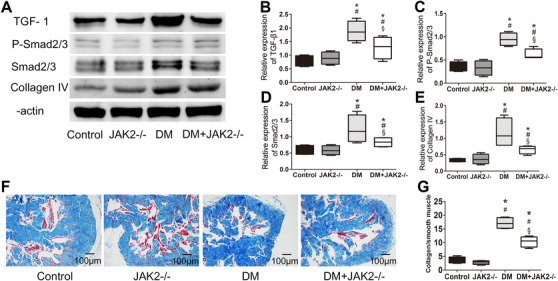
Fibrosis in corpus cavernosum. (A) Representative western blot results for TGF‐β1, phosphorylated Smad2/3, Smad2/3, and Collagen IV. (B–E) Relative expression of TGF‐β1, phosphorylated Smad2/3, Smad2/3, and Collagen IV compared with β‐actin detected with western blot; data are shown as the fold changes over the control group; n = 4 for each group. (F) Representative images of Masson's trichrome staining (200×); smooth muscle was stained red and collagen was blue. (G) Ratio of collagen to smooth muscle detected with Masson's trichrome staining; n = 4 for each group. *: p < 0.05 compared with control group. #: p < 0.05 compared with JAK2−/− group. §: p < 0.05 compared with DM group. DM: diabetes mellitus. TGF‐β1: transforming growth factor beta 1
4. DISCUSSION
Diabetes is a major risk factor of ED. We innovatively utilized Cre+/+‐JAK2fl/fl mice to explore the role of JAK2 in the occurrence of diabetic ED. Our work found that JAK2 was activated in penile tissues of diabetic mice and JAK2 deficiency could improve the impaired erectile function caused by diabetes. Oxidative stress, apoptosis, and fibrosis in corpus cavernosum were alleviated subsequent to JAK2 gene knockout. However, no aforementioned changes appeared after JAK2 gene knockout in the non‐diabetic mice.
The exact role of JAK2 is not clear at the moment; however, the overexpression or activation of JAK2 is linked to the occurrence of some diseases. Previously, AG490 was used as an inhibitor of JAK2 to explore the function of JAK2 in organisms. Nevertheless, the poor specificity of AG490 limited its wide application. At present, more studies have adopted conditional gene knockout technology to inhibit the expression of JAK2. 22 , 25
In our study, we found JAK2 expressed widely in corpus cavernosum and the content of JAK2 declined notably after tamoxifen‐induced JAK2 gene knockout in Cre+/+‐JAK2fl/fl mice. In diabetic mice, phosphorylation of JAK2 in corpus cavernosum was increased, although the expression of JAK2 itself was not elevated apparently. JAK2 gene knockout lowered both the expression and phosphorylation of JAK2, and further improved the erectile function of diabetic mice. This finding indicated that excess phosphorylation of JAK2 might play a role in the development of diabetic ED. Although JAK2 deficiency was not specific to penile tissues in our study, the changes of JAK2 expression did not lead to apparent systematic influence, such as blood glucose level and arterial pressure. Moreover, some alterations indeed occurred in corpus cavernosum after JAK2 gene knockout. This indicated that the protective effects of JAK2 gene knockout on erectile function in diabetic mice was unlikely secondary to its systemic effects. In the non‐diabetic mice, JAK2 gene knockout did not change MIP/MAP notably, signifying that JAK2 deficiency did not influence erectile function in non‐diabetic conditions.
Tamoxifen, usually used as an oestrogen antagonist, acted as an inducer of JAK2 gene knockout in our study. It is reported that administration of tamoxifen might increase testosterone production mildly. However, the testosterone level decreased to baseline after stopping tamoxifen. 26 In our study, the short‐time tamoxifen treatment had no apparent influence on the final serum testosterone level of mice. Some studies have reported impaired erectile function after long‐term use of tamoxifen in men with breast cancer. 27 , 28 To explore the influence of tamoxifen on erectile function, we treated non‐transgenic B6 mice with intraperitoneal injection of tamoxifen at 20 mg/kg body weight for 5 consecutive days. Two months later, no apparent difference in MIP/MAP was found between tamoxifen‐treated mice and control mice (Figure S1). This finding eliminated the interference of tamoxifen on the effect of JAK2 deficiency.
The mechanism of hyperglycemia‐induced activation of JAK2 might implicate multiple factors. A study showed that AGEs elevated tyrosin phosphorylation of JAK2 in rat kidney fibroblast. 11 Furthermore, JAK2 was activated by Ang II in diabetic rat glomeruli. Ang II‐receptor antagonist hindered hyperglycemia‐triggered phosphorylation of JAK2. 13 Another important inducing factor of JAK2 activation is oxidative stress. In rat aortic vascular smooth muscle cells cultured under high glucose conditions, endothelin‐1(ET‐1) activated JAK2 via NOXs. 29 Additionally, many studies have verified that cell apoptosis caused by oxidative stress was directly dependent on JAK2 activation. 30 , 31 All aforementioned findings suggested that JAK2 might be a mediator of the occurrence of diabetic complications.
JAK2 was proven to be associated with hyperglycemia‐induced apoptosis in several types of cells. In endothelial cells cultured with high glucose medium, tyrosin phosphorylation of JAK2 was triggered, leading to apoptosis. Inhibiting JAK2 with AG490 could protect endothelial cells from apoptosis. 12 Additionally, JAK2 activation by oxidative stress mediated the apoptosis of vascular smooth muscle cells and caspase3 might play a role in this process. Pre‐treatment with AG490 decreased the number of apoptotic cells remarkably. 16 Similarly, cell apoptosis was enhanced in corpus cavernosum of mice with diabetic ED in our study. Caspase3 is a major cysteine protease involved in apoptosis and the cleavage of caspase3 leads to caspase3 activation. 32 , 33 We found elevated expression of cleaved caspase3 in diabetic penile tissues. The activation of caspase3 was partly related to JAK2 phosphorylation as JAK2 deficiency decreased caspase3 activity.
Oxidative stress could activate JAK2 and trigger JAK2‐mediated cell apoptosis. Hence JAK2 was considered to be an ROS‐activated kinase. 30 In turn, JAK2 could also promote oxidative stress. 17 Consistent with this finding, our study showed that JAK2 deficiency could ameliorate the oxidative stress level in corpus cavernosum of diabetic mice. NOX1, NOX2 and NOX4 are three members of the NOX family that are expressed abundantly in rodents and are main sources of ROS production in diabetic environments. 34 , 35 We found that the expression of these three NOX isoforms were all increased in the penile tissues of diabetic mice. After tamoxifen‐induced JAK2 deficiency, their expressions were decreased together with reduction in MDA content, indicating that JAK2 might cause oxidative stress via NOXs.
It was reported that ROS excess induced the endothelial nitric oxide synthase (eNOS) uncoupling, resulting in the generation of superoxide and reduction of NO. 36 Furthermore, ROS could react with NO directly and attenuate NO bioavailability. 37 NO/cGMP pathway is essential for penile erection, and its downregulation impaired erectile function heavily. 38 In this study we found that excessive JAK2 phosphorylation reduced NO and cGMP expression, while JAK2 deficiency could increase the content of NO and its downstream cGMP in diabetic mice.
In addition to oxidative stress, JAK2 activation by hyperglycemia could also promote synthesis of extracellular matrix. Amiri et al. found that JAK2 phosphorylation is essential for hyperglycemia‐induced Collagen IV generation. 14 TGF‐β is a major cytokine responsible for matrix protein production and diabetes can increase TGF‐β expression. 39 TGF‐β phosphorylates Smad2 and Smad3, and the latter can regulate the transcription of some fibrosis‐related genes. 40 AG490 treatment abolished hyperglycemia‐induced increase of TGF‐β synthesis, suggesting that JAK2 activation might be a cause of TGF‐β overexpression. 15 In corpus cavernosum of diabetic mice, TGF‐β1 can induce cavernous fibrosis via Smad‐signaling pathway, impairing erectile function. 41 , 42 Our study showed that TGF‐β1/Smad/Collagen IV pathway was upregulated in penile tissue of diabetic mice, whereas JAK2 deficiency lowered the expression of TGF‐β1 and its downstream molecules. As reflected by the ratio of collagen to smooth muscle in Masson's staining, cavernous fibrosis was ameliorated by JAK2 deficiency.
This work demonstrated that JAK2 phosphorylation mediated the occurrence and development of diabetic ED. JAK2 deficiency improved diabetic ED by reducing oxidative stress, apoptosis, and fibrosis. However, some limitations exist in our study. First, we did not explore how JAK2 was phosphorylated in diabetic environments. It was verified by other studies that hyperglycemia could activate JAK2 through ROS, AGEs, Ang II, ET‐1, and other cytokines. Whether these factors mediated JAK2 phosphorylation in corpus cavernosum of diabetic mice needs further investigation. Second, how JAK2 activation led to high level of oxidative stress, apoptosis, and cavernous fibrosis was not clear. STATs family, Src kinase cascade, PI3K‐AKT pathway, and Ras‐MAPK pathway were all downstream targets of JAK2. 43 More studies are required to investigate the role of these pathways in JAK2‐induced alterations in corpus cavernosum.
5. CONCLUSIONS
JAK2 phosphorylation was increased in corpus cavernosum of diabetic mice. Adopting conditional JAK2 gene knockout mice to reduce the expression and activation of JAK2, we found JAK2 deficiency could improve diabetic ED. The underlying mechanism might involve changes in oxidative stress, apoptosis, fibrosis, and the NO/cGMP pathway. JAK2 might be a novel mediator of diabetes‐induced ED and its role deserves further investigation.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTION
Hao Li participated in the design of the study, conducted the experiments, and drafted the manuscript. Wenchao Xu and Hongyang Jiang conducted the data acquisition. Xiaming Liu and Hao Li interpreted and analyzed the data. Tao Wang and Shaogang Wang polished and revised the manuscript. Jihong Liu guided the experiment direction and contributed to the study materials. All authors read and approved the final manuscript.
Supporting information
Supporting Information
ACKNOWLEDGMENT
The conditional JAK2 knockout mice were a generous gift from Prof. Congyi Wang, Center for Biomedical Research, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China).
Li H, Xu W, Liu X, et al. JAK2 Deficiency improves erectile function in diabetic mice through attenuation of oxidative stress, apoptosis and fibrosis. Andrology. 2021;9:1662–1671. 10.1111/andr.13061
Funding information National Natural Science Foundation of China; NSFC #81471451 and NSFC #82001536
Contributor Information
Jihong Liu, Email: jhliu@tjh.tjmu.edu.cn.
Hongyang Jiang, Email: jiang.hongyang@163.com.
REFERENCES
- 1. Feldman HA, Goldstein I, Hatzichristou DG, Krane RJ, McKinlay JB. Impotence and its medical and psychosocial correlates: results of the Massachusetts Male Aging Study. J Urol. 1994;151:54–61. [DOI] [PubMed] [Google Scholar]
- 2. Vickers MA, Satyanarayana R. Phosphodiesterase type 5 inhibitors for the treatment of erectile dysfunction in patients with diabetes mellitus. Int J Impot Res. 2002;14:466–471. [DOI] [PubMed] [Google Scholar]
- 3. Li M, Li H, Ruan Y, Wang T, Liu J. Stem cell therapy for diabetic erectile dysfunction in rats: a meta‐analysis. PLoS One. 2016;11:e0154341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cameron NE, Cotter MA. Erectile dysfunction and diabetes mellitus: mechanistic considerations from studies in experimental models. Curr Diabetes Rev. 2007;3:149–158. [DOI] [PubMed] [Google Scholar]
- 5. Srivastava AK. High glucose‐induced activation of protein kinase signaling pathways in vascular smooth muscle cells: a potential role in the pathogenesis of vascular dysfunction in diabetes [review]. Int J Mol Med. 2002;9:85–89. [PubMed] [Google Scholar]
- 6. Rask‐Madsen C, King GL. Proatherosclerotic mechanisms involving protein kinase C in diabetes and insulin resistance. Arterioscler Thromb Vasc Biol. 2005;25:487–496. [DOI] [PubMed] [Google Scholar]
- 7. Liu KD, Gaffen SL, Goldsmith MA. JAK/STAT signaling by cytokine receptors. Curr Opin Immunol. 1998;10:271–278. [DOI] [PubMed] [Google Scholar]
- 8. Sandberg EM, Wallace TA, Godeny MD, VonDerLinden D, Sayeski PP. Jak2 tyrosine kinase: a true jak of all trades? Cell Biochem Biophys. 2004;41:207–232. [DOI] [PubMed] [Google Scholar]
- 9. Ihle JN, Gilliland DG. Jak2: normal function and role in hematopoietic disorders. Curr Opin Genet Dev. 2007;17:8–14. [DOI] [PubMed] [Google Scholar]
- 10. Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang JS, Guh JY, Hung WC, et al. Role of the Janus kinase (JAK)/signal transducters and activators of transcription (STAT) cascade in advanced glycation end‐product‐induced cellular mitogenesis in NRK‐49F cells. Biochem J. 1999;342:231–238. [PMC free article] [PubMed] [Google Scholar]
- 12. Tawfik A, Jin L, Banes‐Berceli AK, et al. Hyperglycemia and reactive oxygen species mediate apoptosis in aortic endothelial cells through Janus kinase 2. Vascul Pharmacol. 2005;43:320–326. [DOI] [PubMed] [Google Scholar]
- 13. Banes AK, Shaw S, Jenkins J, et al. Angiotensin II blockade prevents hyperglycemia‐induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol. 2004;286:F653–F659. [DOI] [PubMed] [Google Scholar]
- 14. Amiri F, Shaw S, Wang X, et al. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002;61:1605–1616. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, Shaw S, Amiri F, Eaton DC, Marrero MB. Inhibition of the Jak/STAT signaling pathway prevents the high glucose‐induced increase in tgf‐beta and fibronectin synthesis in mesangial cells. Diabetes. 2002;51:3505–3509. [DOI] [PubMed] [Google Scholar]
- 16. Sandberg EM, Sayeski PP. Jak2 tyrosine kinase mediates oxidative stress‐induced apoptosis in vascular smooth muscle cells. J Biol Chem. 2004;279:34547–34552. [DOI] [PubMed] [Google Scholar]
- 17. Neria F, Castilla MA, Sanchez RF, et al. Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin A toxicity. Kidney Int. 2009;75:227–234. [DOI] [PubMed] [Google Scholar]
- 18. Song J, Tang Z, Li H, et al. Role of JAK2 in the pathogenesis of diabetic erectile dysfunction and an intervention with berberine. J Sex Med. 2019;16:1708–1720. [DOI] [PubMed] [Google Scholar]
- 19. Zhong J, Yang P, Muta K, et al. Loss of Jak2 selectively suppresses DC‐mediated innate immune response and protects mice from lethal dose of LPS‐induced septic shock. PLoS One. 2010;5:e9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meydan N, Grunberger T, Dadi H, et al. Inhibition of acute lymphoblastic leukaemia by a Jak‐2 inhibitor. Nature. 1996;379:645–648. [DOI] [PubMed] [Google Scholar]
- 21. Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. [DOI] [PubMed] [Google Scholar]
- 22. Yang P, Zhang Y, Pang J, et al. Loss of Jak2 impairs endothelial function by attenuating Raf‐1/MEK1/Sp‐1 signaling along with altered eNOS activities. Am J Pathol. 2013;183:617–625. [DOI] [PubMed] [Google Scholar]
- 23. Li H, Chen LP, Wang T, Wang SG, Liu JH. Calpain inhibition improves erectile function in diabetic mice via upregulating endothelial nitric oxide synthase expression and reducing apoptosis. Asian J Androl. 2018;20:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang H, Wang D, Yang L, et al. Compact bone‐derived mesenchymal stem cells attenuate nonalcoholic steatohepatitis in a mouse model by modulation of CD4 cells differentiation. Int Immunopharmacol. 2017;42:67–73. [DOI] [PubMed] [Google Scholar]
- 25. Kirabo A, Kearns PN, Jarajapu YP, et al. Vascular smooth muscle Jak2 mediates angiotensin II‐induced hypertension via increased levels of reactive oxygen species. Cardiovasc Res. 2011;91:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smit DL, de Ronde W. Outpatient clinic for users of anabolic androgenic steroids: an overview. Neth J Med. 2018;76:167. [PubMed] [Google Scholar]
- 27. Pemmaraju N, Munsell MF, Hortobagyi GN, Giordano SH. Retrospective review of male breast cancer patients: analysis of tamoxifen‐related side‐effects. Ann Oncol. 2012;23:1471–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Motofei IG, Rowland DL, Popa F, et al. A pilot study on tamoxifen sexual side effects and hand preference in male breast cancer. Arch Sex Behav. 2015;44:1589–1594. [DOI] [PubMed] [Google Scholar]
- 29. Banes‐Berceli AK, Ogobi S, Tawfik A, et al. Endothelin‐1 activation of JAK2 in vascular smooth muscle cells involves NAD(P)H oxidase‐derived reactive oxygen species. Vascul Pharmacol. 2005;43:310–319. [DOI] [PubMed] [Google Scholar]
- 30. Yu HM, Zhi JL, Cui Y, et al. Role of the JAK‐STAT pathway in protection of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress in PC12 cells. Apoptosis. 2006;11:931–941. [DOI] [PubMed] [Google Scholar]
- 31. Ponnusamy M, Pang M, Annamaraju PK, et al. Transglutaminase‐1 protects renal epithelial cells from hydrogen peroxide‐induced apoptosis through activation of STAT3 and AKT signaling pathways. Am J Physiol Renal Physiol. 2009;297:F1361–F1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Xu W, Ni P, Li A, Zhou J, Xu S. MiR‐99a and MiR‐491 regulate cisplatin resistance in human gastric cancer cells by targeting CAPNS1. Int J Biol Sci. 2016;12:1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu B, Li F, Shi J, Yang D, Deng Y, Gong Q. Gastrodin ameliorates subacute phase cerebral ischemia‐reperfusion injury by inhibiting inflammation and apoptosis in rats. Mol Med Rep. 2016;14:4144–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sonta T, Inoguchi T, Tsubouchi H, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–123. [DOI] [PubMed] [Google Scholar]
- 35. Manea SA, Antonescu ML, Fenyo IM, Raicu M, Simionescu M, Manea A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase‐dependent mechanisms in experimental diabetes. Redox Biol. 2018;16:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Virdis A, Santini F, Colucci R, et al. Vascular generation of tumor necrosis factor‐α reduces nitric oxide availability in small arteries from visceral fat of obese patients. J Am Coll Cardiol. 2011;58:238–247. [DOI] [PubMed] [Google Scholar]
- 37. Francis SH, Corbin JD. PDE5 inhibitors: targeting erectile dysfunction in diabetics. Curr Opin Pharmacol. 2011;11:683–688. [DOI] [PubMed] [Google Scholar]
- 38. Ruan Y, Li M, Wang T, et al. Taurine supplementation improves erectile function in rats with streptozotocin‐induced type 1 diabetes via amelioration of penile fibrosis and endothelial dysfunction. J Sex Med. 2016;13:778–785. [DOI] [PubMed] [Google Scholar]
- 39. Kim SI, Han DC, Lee HB. Lovastatin inhibits transforming growth factor‐beta1 expression in diabetic rat glomeruli and cultured rat mesangial cells. J Am Soc Nephrol. 2000;11:80–87. [DOI] [PubMed] [Google Scholar]
- 40. Watabe T, Nishihara A, Mishima K, et al. TGF‐beta receptor kinase inhibitor enhances growth and integrity of embryonic stem cell‐derived endothelial cells. J Cell Biol. 2003;163:1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gonzalez‐Cadavid NF. Mechanisms of penile fibrosis. J Sex Med. 2009;6:353–362. [DOI] [PubMed] [Google Scholar]
- 42. Cui K, Ruan Y, Wang T, et al. FTY720 supplementation partially improves erectile dysfunction in rats with streptozotocin‐induced type 1 diabetes through inhibition of endothelial dysfunction and corporal fibrosis. J Sex Med. 2017;14:323–335. [DOI] [PubMed] [Google Scholar]
- 43. Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19:5662–5679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information