

Abstract

The hexahydride complex OsH6(PiPr3)2 promotes the C–H bond activation of the 1,3-disubstituted phenyl group of the [BF4]− and [BPh4]− salts of the cations 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium and 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium. The reactions selectively afford neutral and cationic trihydride-osmium(IV) derivatives bearing κ2-C,N- or κ2-C,C-chelating ligands, a cationic dihydride-osmium(IV) complex stabilized by a κ3-C,C,N-pincer group, and a bimetallic hexahydride formed by two trihydride-osmium(IV) fragments. The metal centers of the hexahydride are separated by a bridging ligand, composed of κ2-C,N- and κ2-C,C-chelating moieties, which allows electronic communication between the metal centers. The wide variety of obtained compounds and the high selectivity observed in their formation is a consequence of the main role of the azolium group during the activation and of the existence of significant differences in behavior between the azolium groups. The azolium role is governed by the anion of the salt, whereas the azolium behavior depends upon its imidazolium or benzimidazolium nature. While [BF4]− inhibits the azolium reactions, [BPh4]− favors the azolium participation in the activation process. In contrast to benzimidazolylidene, the imidazolylidene resulting from the deprotonation of the imidazolium substituent coordinates in an abnormal fashion to direct the phenyl C–H bond activation to the 2-position. The hydride ligands of the cationic dihydride-osmium(IV) pincer complex display intense quantum mechanical exchange coupling. Furthermore, this salt is a red phosphorescent emitter upon photoexcitation and displays a noticeable catalytic activity for the dehydrogenation of 1-phenylethanol to acetophenone and of 1,2-phenylenedimethanol to 1-isobenzofuranone. The bimetallic hexahydride shows catalytic synergism between the metals, in the dehydrogenation of 1,2,3,4-tetrahydroisoquinoline and alcohols.
Introduction
The transition-metal-promoted activation of aromatic C–H bonds is one of the most relevant reactions in current chemistry,1 due to the wide range of fields with which it is connected, ranging from organic2 and organometallic3 synthesis to catalysis4 and materials science.5 The reaction is initiated by the coordination of the C–H bond to the unsaturated metal center of the promoter.6 The resulting σ-intermediate evolves by oxidative addition of the C–H bond or heterolytic C–H splitting. In the last case, the abstractor of the proton is a ligand of the metal coordination sphere or an external base.7 In accordance with this sequence of events, the activation energy for the C–H bond rupture depends upon two factors: the stability of the σ-intermediate and the C–H bond dissociation energy of the coordinated bond.8 Because in aromatic organic molecules the strengths of the different C(sp2)–H bonds are similar, the activation is mainly governed by the stability of the σ-intermediate, which is a function of the steric hindrance experienced by the coordinated C–H bond. As a consequence, the selectivity of C(sp2)–H bond activation in substituted aromatic arenes is kinetically controlled by steric factors.9
The presence of a substituent with coordinating ability in the arene selectively ties the activation at the ortho position.10 Although the latter is sterically hindered and therefore the last position being activated, the substituent thermodynamically abducts the ortho-activation product by coordination.11 This is of central importance for the comprehension of catalytic organic reactions of ortho-CH functionalization.12 Since a catalytic cycle represents the reaction pathway with the lowest activation energy and the ortho-metalation reaction has an activation energy higher than those of other C–H bond activations in the same ring, the o-CH bond activation should form part of the fast stage of the functionalization, the ortho-metalated intermediate being the resting state of the catalyst. An additional issue of selectivity appears when the arene bears several substituents with coordinating ability. Then, understanding the drivers of the selectivity in the activation process is especially relevant to control the products. In such a case, in addition to the steric hindrance of the C–H bonds, the coordinating ability of the different substituents should be also taken into account. The study of the selectivity is particularly challenging when the substituted arene is a part of an imidazolium salt because the imidazolylidene coordination can take place at different positions13 and has proved to be anion dependent.14 Furthermore, the necessary imidazolium C–H bond activation requires specific procedures for each case.15
The study of C–H bond activation reactions of aryl substrates asymmetrically 1,3-disubstituted with coordinating groups is particularly challenging. Three different activations can have a thermodynamic preference in this case, which give rise to four distinct stable situations (Chart 1). Activation at the congested 2-position lead to pincer-type derivatives (A),16 whereas separate activations at positions 4 and 6 provoke κ2-C,L and κ2-C,L’ coordinations of the activated substrate, which generate mononuclear derivatives bearing C–L and C–L′ chelating ligands (B and C, respectively).17 In contrast, the simultaneous or sequential C–H bond activations of both positions yield a bimetallic species (D).18 The 5-position is the most accessible. This kinetically favors its activation. However, the absence of a neighboring group with coordinating ability causes such a C–H bond activation to be inhibited from a thermodynamic point of view.
Chart 1. Possible Products of Thermodynamic Control for the C–H Bond Activation of an Aryl Substrate Asymmetrically Substituted with Coordinating Groups.

The chemistry of the polyhydrides of platinum-group metals is an area of great potential. Such a prospect is the consequence of the proven ability of these compounds to activate σ-bonds,19 which allows them to connect with fields such as organic synthesis,20 the preparation of new types of phosphorescent emitters for OLED devices,14e,21 and hydrogen storage and transport.22 Among the compounds of this class, the osmium-hexahydride OsH6(PiPr3)223 (1) occupies a prominent position due to its versatility for promoting C–H bond activation reactions.24 We are not strangers to the interest in the polyhydride chemistry nor to the aromatic C–H bond activations of substrates asymmetrically 1,3-disubstituted. Thus, in the search for understanding the factors that govern the challenging selectivity of these reactions, we have investigated the behavior of the [BF4]− and [BPh4]− salts of cations 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium and 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium (Chart 2) toward 1.
Chart 2. Azolium Salts Used in This Study.

This paper describes the selectivity of the osmium-promoted C–H bond activation of the salts shown in Chart 2, as a function of the anion and the azolium substituent and the catalytic performance of the isolated complexes for hydrogen generation by dehydrogenation of 1,2,3,4-tetrahydroisoquinoline and alcohols.
Results and Discussion
Complexes Resulting from [BF4]− and [BPh4]− Salts of 1-(3-(Isoquinolin-1-yl)phenyl)-3-methylimidazolium
The most clean, direct, and straightforward procedure to introduce an imidazolylidene ligand into the coordination sphere of a transition metal is generally direct metalation.15 The latter can take place by oxidative addition of an imidazolium C–H bond to an unsaturated metal fragment and by displacement of a coordinated Brønsted base, as a result of its protonation with the imidazolium salt. Complex 1 shows a marked tendency to undergo the reductive elimination of molecular hydrogen, at moderate temperatures (>50 °C), to afford the unsaturated tetrahydride OsH4(PiPr3)2 (E), which is the true species responsible for the proved ability of 1 to activate σ-bonds.11c,20d,20f,24 On the other hand, the hydrides of 1 are basic enough to promote the deprotonation of imidazolium salts. The addition of the proton initially leads to the known trihydride-bis(dihydrogen) derivative [OsH3(η2-H2)2(PiPr3)2]+, which loses molecular hydrogen and dimerizes to form the bimetallic cation [{OsH2(PiPr3)2}2(μ-H)3]+ in equilibrium with the deprotonated polyhydride (PiPr3)2H2Os(μ-H)3OsH(PiPr3)2.25 To prevent side products resulting from the formation of the OsH7 cation, the reactions of 1 with imidazolium salts are usually performed in the presence of triethylamine, including those where the imidazolylidene ligand acts as a chelating assistant.26
Treatment of toluene solutions of 1 with 1.0 equiv of the [BF4]− salt of 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium, in the presence of 15 equiv of triethylamine, under reflux leads to the cationic trihydride derivative 2 (Scheme 1) in 82% yield after 24 h, according to the 1H and 31P{1H} NMR spectra of the crude reaction product in dichloromethane-d2. The reaction can be rationalized as the isoquinolinyl-assisted activation of the C–H bond of the phenyl group at position 4 promoted by tetrahydride E. The imidazolium moiety of the salt does not interfere during the process. Consistently, in this case, the formation of the trihydride also takes place in the absence of the amine and in the same extension.
Scheme 1. Formation of Complexes 2–4.

Complex 2 was isolated as a red solid in 73% yield and characterized by X-ray diffraction analysis. The structure has two cations and two anions chemically equivalent, but these are crystallographically independent in the asymmetric unit. Figure 1 gives a view of a cation. The metal center displays a typical coordination for a d4 ion. Thus, the polyhedron can be idealized as a pentagonal bipyramid with axial phosphines (P(1)–Os(1)–P(2) = 164.64(4) and 166.91(4)°). The κ2-C,N-chelate group, which acts with bite angles of 75.51(14) and 75.06(14)° (C(1)–Os(1)–N(7)), and the hydride ligands, which are separated by more than 1.6 Å (X-ray and DFT calculations (B3LYP-D3(SMD)/6-31G**(SDD)), lie at the base. The 1H, 13C{1H}, and 31P{1H} NMR spectra in dichloromethane-d2 are consistent with the solid-state structure. In agreement with the presence of three inequivalent hydride ligands, the 1H spectrum at 193 K shows three high-field resonances at −6.19, −10.71, and −12.10 ppm. The most noticeable signal in the 13C{1H} spectrum is a triplet (2JC–P = 5.8 Hz) at 199.1 ppm, corresponding to the metalated carbon atom. The 31P{1H} spectrum displays a singlet at 22.1 ppm, as expected for equivalent phosphines.
Figure 1.

Molecular diagram of one of the two independent cations of complex 2 (ellipsoids shown at 50% probability) in the asymmetric units. All hydrogen atoms (except the hydrides) are omitted for clarity. Selected bond distances (Å) and angles (deg): Os–P(1) = 2.3527(10), 2.3368(11), Os–P(2) = 2.3367(10), 2.3423(11), Os–C(1) = 2.102(4), 2.098(4), Os–N(7) = 2.151(3), 2.151(3); P(1)–Os–P(2) = 164.64(4), 166.91(4), C(1)–Os–N(7) = 75.51(14), 75.06(14).
There are significant differences in behavior between the [BF4]− and [BPh4]− salts of the cation 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium. Tentatively, these differences may be associate with the distinct sizes of the anions, which influence the cation–anion association and the respective solvations. In contrast to the [BF4]− salt, the [BPh4]− counterpart allows the disinhibition of the reactivity of the imidazolium moiety. This favors the activations of the C–H bonds of the phenyl group at the 2- and 6-positions (Scheme 1). Thus, the treatment of a toluene solution of 1 with the [BPh4]− salt, in the presence of 15 equiv of triethylamine, under reflux affords a mixture of the cationic dihydride pincer complex 3 (73%) and the neutral trihydride 4 (27%). The major product, complex 3, results from the activations of C–H bonds of the imidazolium moiety and of the phenyl group at 5- and 2-positions, respectively, whereas the neutral trihydride 4 arises from similar ruptures at 2- and 6-positions of the respective rings. The formation of a pincer isomer of 3 involving the coordination of the carbon atom at the 2-position of the imidazolylidene instead of that at the 5-position was not observed. This suggests that the C–H ruptures leading to 3 are connected and take place in a sequential manner. Because the 2-position of the phenyl group is sterically more hindered than the 5-position of the imidazolium moiety, it seems reasonable to think that the latter is kinetically favored and therefore it is previous to the former. As expected from the imidazolium participation, the reaction is sensitive to triethylamine. In absence of the latter, in addition to the appearance of side products, the formation of 3 and 4 is slower.
Complex 3 was separated from the crude reaction mixture by silica column chromatography, isolated as a red solid in 57% yield, and subsequently fully characterized including an X-ray diffraction analysis. Figure 2 shows a view of the cation. The structure demonstrates the formation of the pincer, involving an abnormal coordination of the imidazolylidene moiety. The osmium–imidazolylidene bond length of 2.077(2) Å (Os–C(1)) compares well with those reported for osmium compounds displaying abnormal-NHC coordination.11c,27 The new monoanionic C,C,N-pincer ligand acts with C(1)–Os–N(1), C(1)–Os–C(10), and N(1)–Os–C(10) angles of 150.42(8), 76.18(8), and 74.24(8)°, respectively, which are close to the ideal values corresponding to three consecutive positions at the base of a pentagonal bipyramid (144, 72, and 72°), the observed coordination polyhedron in this case, and point out that this pincer should be particularly useful to stabilize compounds of d4 ions with such a disposition of donor atoms around the metal center. The ideal pentagonal bipyramid is completed with the phosphines, which lie at the apical positions (P(1)–Os–P(2) = 160.46(2)°), and the hydrides, which are situated at the pincer plane separated by 1.55(4) Å (1.647 Å in the DFT-optimized structure).
Figure 2.

Molecular diagram of the cation of complex 3 (ellipsoids shown at 50% probability). All hydrogen atoms (except the hydrides) are omitted for clarity. Selected bond distances (Å) and angles (deg): Os–P(1) = 2.3714(5), Os–P(2) = 2.3885(5), Os–C(1) = 2.077(2), Os–C(10) = 2.053(2), Os–N(1) = 2.1456(18); P(1)–Os–P(2) = 160.46(2), C(1)–Os–N(1) = 150.42(8), C(1)–Os–C(10) = 76.18(8), C(10)–Os–N(1) = 74.24(8).
The NMR spectra in dichloromethane-d2 are consistent with the solid-state structure. The 1H spectrum further reveals that the hydride ligands undergo quantum mechanical exchange coupling.19,28 As shown in Figure 3a, the observed H–H coupling constant (Jobs) in the AB part of the ABX2 (X = 31P) spin system corresponding to the dihydride resonance (−5.7 ppm) is temperature (T) dependent, increasing from 101 to 444 Hz as T increases from 183 to 243 K. For a given hydrogen–hydrogen separation (a), Jobs and T are related through eq 1, according to a two-dimensional harmonic oscillator model,29 where Jmag is the classical H–H coupling constant due to the Fermi contact interaction, λ represents the hard sphere radius of the hydrides, and ν describes the H–M–H vibrational wag mode that allows the movement along the H–H vector. As for a, these parameters are temperature independent. Using the hydrogen–hydrogen separation obtained by DFT calculations for the optimized structure, the fitting of the plot shown in Figure 3b yields values of Jmag = 9.5 Hz, λ = 1.0 Å, and ν = 497 cm–1, which compare well with those obtained for other osmium(IV)-hydride compounds.30 In the 13C{1H} spectrum, the most noticeable resonances are two triplets at 192.4 (2JC–P = 5.9 Hz) and 140.2 (2JC–P = 7.3 Hz) ppm, due to the metalated C(1) and C(10) atoms, respectively. The 31P{1H} spectrum shows a singlet at 3.4 ppm, in agreement with the equivalence of the phosphines.
| 1 |
Figure 3.

(a) 1H{31P} NMR spectra of complex 3 in the high-field region as a function of the temperature. (b) Plot of Jobs versus temperature for complex 3.
Complex 3 is a new case of a red phosphorescent emitter (601–644 nm) upon photoexcitation, in a 5 wt % doped poly(methyl methacrylate) (PMMA) film at room temperature and in 2-methyltetrahydrofuran (2-MeTHF) at room temperature and at 77 K (Table 1). The observed wavelengths are in accordance with those obtained by estimating the difference in energy between the optimized triplet state T1 and the singlet state S0 in THF (637 nm). Consistently, the emissions can be ascribed to this excited state. The emission spectra in the PMMA film and in 2-MeTHF at room temperature show broad structureless bands. In contrast, the spectrum in 2-MeTHF at 77 K displays a vibronic fine structure (Figure 4), which is consistent with a significant contribution of ligand-centered 3π–π* transitions to the excited state.31 The lifetimes are short and lie in a narrow range of 0.8–5.2 μs, whereas the quantum yields of about 0.20 are moderate. There is great interest in osmium(IV) emitters. In addition to being more difficult to oxidize than osmium(II) emitters, they should offer more flexibility for color tuning.32
Table 1. Emission Properties of Complex 3.
| HOMOcalc (eV) | LUMOcalc (eV) | HLGcalc (eV) | HOMOexp (eV)a | LUMOexp (eV)b | calc λem (nm)c | medium (T, K) | λem (nm) | τobs (μs) | Φ | kr (s–1)d | knr (s–1)d | kr/knr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PMMA (298) | 644 | 0.8 | 0.15 | 1.8 × 105 | 1.0 × 106 | 0.18 | ||||||
| –5.23 | –2.07 | 3.16 | –5.09 | –2.95 | 637 | MeTHF (298) | 642 | 1.1 | 0.19 | 1.7 × 105 | 7.3 × 105 | 0.23 |
| MeTHF (77) | 601 | 5.2 |
HOMO = −[Eox vs Fc/Fc+ + 4.8] eV.
LUMO = −[Ered vs Fc/Fc+ + 4.8] eV.
Predicted from TD-DFT calculations in THF at 298 K by estimating the energy difference between the optimized T1 and singlet S0 states.
Calculated according to the equations kr = Φ/τobs and knr = (1 – Φ)/τobs, where kr is the radiative rate constant, knr is the nonradiative rate constant, Φ is the quantum yield, and τobs is the excited-state lifetime.
Figure 4.

Emission spectra of complex 3.
The minor complex 4 was isolated as a yellow solid in 9% yield and characterized by X-ray diffraction analysis. Its structure (Figure 5) confirmed the molecular nature of the species and the coordination of the imidazolylidene moiety by the carbon atom at the 2-position of the ring. The polyhedron around the osmium atom resembles that of 2, with a P(1)–Os–P(2) angle of 163.47(3)°, the hydride ligands separated by more than 1.6 Å, and the imidazolylidene moiety, which forms a κ2-C,C-chelate with the ortho-metalated phenyl (C(1)–Os–C(5) = 75.83(12)°), occupying the isoquinolinyl group place. This ligand disposition is consistent with the NMR spectra of the molecule in dichloromethane-d2. In accordance with 2, the 1H spectrum at 183 K shows three hydride resonances at −8.90, −10.38, and −10.78 ppm. In the 13C{1H} spectrum, the signals due to the metalated carbon atoms appear at 190.1 (C(1)) and 162.8 (C(5)) ppm, as triplets with C–P coupling constants of 5.9 and 7.6 Hz, respectively. The 31P{1H} spectrum contains a singlet at 24.7 ppm for the equivalent phosphines.
Figure 5.

Molecular diagram of complex 4 (ellipsoids shown at 50% probability). All hydrogen atoms (except the hydrides) are omitted for clarity. Selected bond distances (Å) and angles (deg): Os–P(1) = 2.3481(8), Os–P(2) = 2.3511(8), Os–C(1) = 2.065(3), Os–C(5) = 2.128(3); P(1)–Os–P(2) = 163.47(3), C(1)–Os–C(5) = 75.83(12).
Complexes Resulting from [BF4]− and [BPh4]− Salts of 1-(3-(Isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium
The use of a benzimidazolium fragment instead of an imidazolium moiety should prevent the formation of a pincer species related to 3, confirming the linkage between the activation of the C–H bonds at the 5-position of the five-membered ring and the activation of the C–H bond at the 2-position of the phenyl group, while it would allow a better study of the C–H bond activation at the 6-position of the aryl group. This reasoning prompted us to study the reactions of 1 with the [BF4]− and [BPh4]− salts of the cation 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium, under the same conditions as those employed for the reactions summarized in Scheme 1. The results are consistent with those obtained for the cation 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium and confirm our previous conclusions (Scheme 2).
Scheme 2. Formation of Complexes 5 and 6.
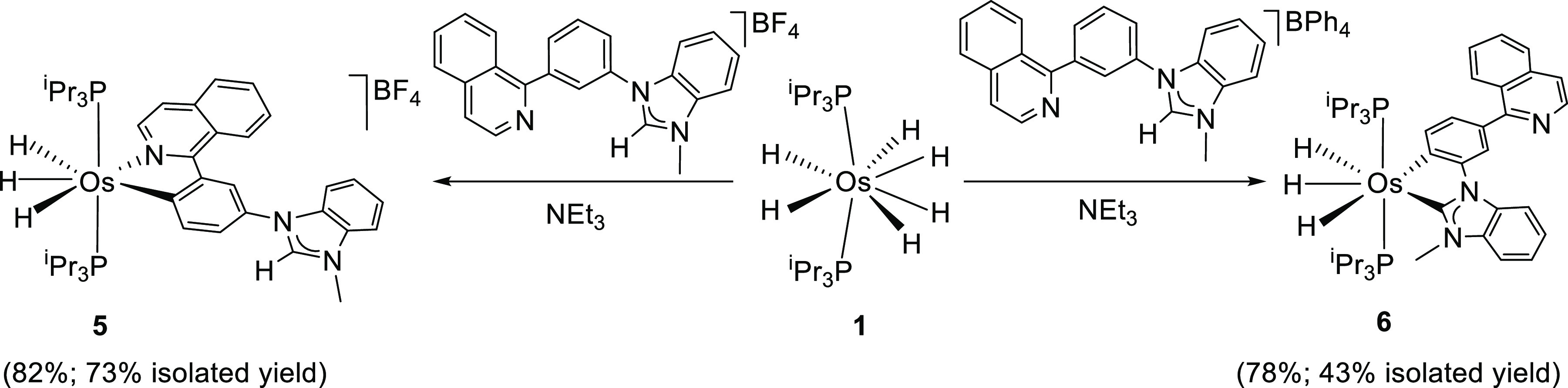
The [BF4]− anion inhibits the reactions of the benzimidazolium fragment, which favors the isoquinolinyl-assisted activation of the C–H bond of the phenyl group at the 4-position. Thus, the reaction of 1 with this salt leads to 5 (82%), the benzimidazolylidene counterpart of 2, while the reaction with the [BPh4]− salt selectively gives 6 (78%), the benzimidazolylidene counterpart of 4. Complex 6 results from the activation of the C–H bond at the 6-position of the phenyl group along with the activation of the C–H bond at the 2-position of the benzimidazolium fragment. No pincer complex resulting from C–H bond activation of the phenyl group at the 2-position was detected, which suggests that the rupture of the C–H bond of the phenyl group at the 6-position is a NHC-assisted reaction promoted by the tetrahydride E, the genesis of the Os–NHC bond being a heterolytic C–H activation mediated by the triethylamine external base. Complexes 5 and 6 were isolated as red and yellow solids in 73% and 43% yields, respectively, and fully characterized by NMR spectroscopy, in dichloromethane-d2. In agreement with the imidazolylidene counterparts 2 and 4, the 1H spectra at 203 K contain signals due to three inequivalent hydrides at −6.19, −10.54, and −11.99 ppm for 5 and at −8.43 and −10.02 (2H) ppm for 6. In the 13C{1H} spectra, the resonances corresponding to the metalated carbon atoms appear as triplets at 199.7 (2JC–P = 5.5 Hz) ppm for 5 and at 206.1 (2JC–P = 5.8 Hz) and 161.9 (2JC–P = 5.6 Hz) ppm for 6. The 31P{1H} spectra display a singlet at 21.9 ppm for 5 and at 26.1 ppm for 6.
C–H Bond Activation of 6
The hexahydride complex 1 also activates the C–H bond of the metalated phenyl group of 6 disposed in para position with regard to the benzimidazolylidene moiety and ortho to the isoquinolyl group, the 4-position in the starting cation, to give the bimetallic hexahydride 7 (Scheme 3). At first glance, one should expect that such a complex could be also prepared from 5, by activation of the C–H bond at 2-position of the benzimidazolium fragment along with the ortho metalation of the phenyl group: i.e., the activation of the C–H bond of the phenyl group disposed in a para position with respect to the isoquinolyl moiety, the 6-position of the original cation. However, the previously mentioned inhibition of the reactivity of the benzimidazolium moiety by the action of the [BF4]− anion prevents such a possibility, in the presence and in absence of triethylamine and in both toluene and tetrahydrofuran, as solvents, under reflux.
Scheme 3. Formation of Complex 7.

Complex 7 was isolated as a garnet solid in 80% yield and characterized by an X-ray diffraction analysis. Figure 6 shows its structure, which can be described as two OsH3(PiPr3)2 metal fragments linked by a bridging ligand resulting from activations at the 4- and 6-positions of a phenyl substrate asymmetrically 1,3-disubstituted with benzimidazolylidene and isoquinolyl groups. The polyhedron around Os(1) resembles that of 6 with P(1)–Os(1)–P(2) and C(1)–Os(1)–C(10) angles of 160.57(3) and 76.19(12)°, respectively, whereas the polyhedron around Os(2) resembles that of 5 with P(3)–Os(2)–P(4) and N(1)–Os(2)–C(12) angles of 164.30(3) and 76.20(12)°, respectively. The classical nature of the polyhydride is supported by both the X-ray structure and the optimized structure through DFT calculations, which display hydride–hydride separations longer than 1.6 Å. In agreement with the structure, the NMR spectra in dichloromethane-d2 are combinations of those of 5 and 6. The 1H spectrum at 203 K contains high-field signals for six inequivalent hydrides at −6.23, −8.05, −10.05 (2H), and −11.04 (2H) ppm. The 13C{1H} spectrum shows three triplets (2JC–P = 6.1–5.7 Hz) for the metalated carbon atoms at 204.9, 186.5, and 166.6 ppm. The two pairs of equivalent phosphines give rise to two singlets at 26.7 and 22.6 ppm in the 31P{1H} spectrum.
Figure 6.

Molecular diagram of complex 7 (ellipsoids shown at 50% probability). All hydrogen atoms (except the hydrides) are omitted for clarity. Selected bond distances (Å) and angles (deg): Os(1)–P(1) = 2.3411(9), Os(1)–P(2) = 2.3462(8), Os(2)–P(3) = 2.3319(9), Os(2)–P(4) = 2.3383(9), Os(1)–C(1) = 2.064(3), Os(1)–C(10) = 2.134(3), Os(2)–C(12) = 2.125(3), Os(2)–N(1) = 2.129(3); P(1)–Os(1)–P(2) = 160.57(3), P(3)–Os(2)–P(4) = 164.30(3); C(1)–Os(1)–C(10) = 76.19(12), C(12)–Os(2)–N(1) = 76.20(12).
The HOMO of the bimetallic complex 7 is delocalized between the metal centers and the bridge (Figure S38). Bimetallic complexes displaying frontier orbitals delocalized between the two metal centers connected by a π-linker can be viewed as being electronically coupled. Thus, one should expect that changes in the electron density at one site would perturb the electron density at the other.33 The redox potential separation between successive redox processes is frequently used as a first evaluation of the electronic coupling.34 To analyze this possibility in 7, we evaluated its redox properties. A cyclic voltammetry experiment was carried out under argon, in dichloromethane solution, with [Bu4N]PF6 as the supporting electrolyte (0.1 M). Four oxidation peaks at −0.60 ([Os2]/[Os2]+), −0.26 ([Os2]+/[Os2]2+), 0.01 ([Os2]2+/[Os2]3+), and 0.27 ([Os2]3+/[Os2]4+) V versus Fc/Fc+ were observed (Figure S39). The first oxidation is reversible, whereas the second and third oxidations are quasi-reversible and the fourth oxidation is irreversible. Reduction peaks were not observed in the range from −1.5 to +1.5 V. The consecutive separations between the three first oxidation peaks (ΔE) yield large values for the equilibrium constant Kc (Kc = e–nFΔE/RT)35 of the comproportionation reactions summarized by eq 2, of 5.6 × 10–5 and 3.7 × 10–4. These values point out the formation of class III radicals, with the odd electron being fully delocalized, according to the Robin–Day classification.36
| 2 |
The formation of mixed valence species was confirmed by an UV–vis–NIR spectroelectrochemical investigation on a 1 × 10–3 M dichloromethane solution of 7, in the presence of 0.1 M [Bu4N]PF6, under argon (Figures S50–S54). In agreement with the formation of species of this class, the spectra of [7]+ and [7]3+ contain broad absorptions centered at 927 and 1556 nm, respectively, which are ascribed to the respective intervalence charge transfer transitions (IVCTs). In accordance with the bandwidth at the half-height (Δν1/2 = 12500 cm–1 for [7]+ and 6436 cm–1 for [7]3+) and the maximum absorption (Δνmax = 657 cm–1 for [7]+ and 641 cm–1 for [7]3+) of the Gaussian-shaped ICTV band, the delocalization parameters Γ calculated according to eq 3(36b) are 0.87 for [7]+ and 0.83 for [7]3+. These values are characteristic of class III radicals.37
| 3 |
Catalytic Dehydrogenation of 1,2,3,4-Tetrahydroisoquinoline and Alcohols
The mononuclear complexes 2, 3, 5, and 6 and the bimetallic derivative 7 promote the dehydrogenation of 1,2,3,4-tetrahydroisoquinoline (Scheme 4). The reactions were carried out under argon, in p-xylene, at 140 °C, using a heterocycle concentration of 0.12 M and an osmium/heterocycle molar ratio of 1/14.6. Under these conditions, between 58% and 87% of all H2 capacity of the heterocycle, 1.50 × 10–2 mol g–1, is released after 48 h. The dehydrogenation is sequential, the release of the first hydrogen molecule being faster than the liberation of the second molecule. The behavior the bimetallic complex 7 should be pointed out, which reveals a nice example of catalytic synergism.38 This compound is significantly more active than complexes 5 and 6, the mononuclear units forming it. Although the metal centers are separated by the bridging ligand, the latter allows their electronic coupling, as was previously demonstrated. The catalysis can take place in an independent manner in each metal center, but the events in one metal center affect those in the other. The catalytic synergism is a consequence of the gain in the efficiency of each metal center by the action of its colleague.
Scheme 4. Dehydrogenation of 1,2,3,4-Tetrahydroisoquinoline.

Complexes 2, 3, and 5–7 also catalyze the dehydrogenation of primary and secondary alcohols, such as benzyl alcohol, 1-phenylethanol, and 1,2-phenylenedimethanol (Scheme 5). The reactions were performed under argon, in toluene, at 100 °C, using an alcohol concentration of 0.12 M and an osmium/substrate molar ratio of 1/14.6. The addition of a base to the catalytic solutions was not necessary. This finding is particularly notable in the case of 2, 3, and 5, given their cationic character.39 The hydride ligands of these compounds appear to have enough basic character to deprotonate the OH group of the substrates. The abstraction should release H2, generating the key metal-alkoxide intermediates. The organic product obtained and the amount of generated molecular hydrogen depend upon both the nature of the alcohol and the catalyst. The dehydrogenation reactions of benzyl alcohol (Scheme 5a) catalyzed by the mononuclear complexes 2, 3, 5, and 6 afford benzaldehyde in low to moderate yields, 6–55%, after 24 h. However, the alcohol is transformed in a mixture of benzylbenzoate (13%) and benzaldehyde (47%) in the presence of the bimetallic compound 7. The generation of esters in these reactions is not surprising. They are a consequence of a competitive dehydrogenative homocoupling and seem to result from the transitory formation of hemiacetals.40 The dehydrogenation of 1-phenylethanol (Scheme 5b) leads to the expected acetophenone also in moderate yields, with the exception of the pincer salt 3. The latter generates 73% of the ketone after 24 h. Complex 3 is more efficient than the bimetallic compound 7. Although the bimetallic species displays catalytic synergism, increasing its efficiency with regard to 5 and 6, its catalytic activity is still moderate. Thus, only 56% of the ketone is obtained with this catalyst, after 24 h. The dehydrogenation of 1,2-phenylenedimethanol (Scheme 5c) in the presence of 7 affords 1-isobenzofuranone and molecular hydrogen in a quantitative yield after 24 h, whereas 70% of the lactone is formed after 12 h. The reaction yield in the presence of the pincer complex 3 is also good, 86%.
Scheme 5. Dehydrogenation of Benzyl Alcohol, 1-Phenylethanol, and 1,2-Phenylenedimethanol.

Concluding Remarks
This study shows that the hexahydride complex OsH6(PiPr3)2 promotes the C–H bond activation of aryl compounds asymmetrically 1,3-disubstituted, with two coordinating groups. The rationalization of the products formed in the reactions with the [BF4]− and [BPh4]− salts of the cations 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium and 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium reveals that the azolium substituent establishes the position of the C–H bond activation. This is due to the main role of the azolium group during the activation, which can be governed through the election of the anion of the salt, and to the existence of significant differences in behavior between the azolium groups depending upon their imidazolium or benzimidazolium nature. The [BF4]− anion inhibits the reactions of the azolium groups. Consistently, both [BF4]− salts undergo the rupture of the aryl-CH bond at the 4-position, as a consequence of an isoquinolinyl-assisted C–H bond activation reaction. In contrast, the [BPh4]− anion disinhibits the azolium reactions. Then, the imidazolium substituent affords an imidazolylidene group. This moiety preferentially coordinates to the metal center in an abnormal fashion, to direct the C–H bond activation of the aryl at the 2-position and finally to yield a pincer ligand by the coordination of the isoquinolyl substituent. In contrast, the benzimidazolylidene, resulting from the deprotonation of the benzimidazolium substituent, assists the C–H bond activation at the 6-position.
The pincer complex resulting from the C–H activation of the central aryl group at the 2-position and the coordination of both substituents possesses interesting features. Its hydride ligands show an intense quantum mechanical exchange coupling, and it is a red phosphorescent emitter upon photoexcitation and displays a noticeable catalytic activity for the dehydrogenation of 1-phenylethanol to acetophenone and 1,2-phenylenedimethanol to 1-isobenzofuranone.
The sequential C–H bond activation of the 6- and 4-positions of the central aryl group of the 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium tetraphenylborate affords a bimetallic compound, which displays catalytic synergism between the metals, for the dehydrogenation of 1,2,3,4-tetrahydroisoquinoline and alcohols. The synergism seems to result from the electronic coupling between the metal centers and gives rise to a noticeable catalytic activity of this compound in the dehydrogenation of 1,2-phenylenedimethanol to 1-isobenzofuranone.
In summary, the introduction of an azolium substituent into a phenyl group previously bearing a coordinating group, to form an asymmetrically 1,3-disubstituted aryl salt, allows an efficient governing of the C–H bond activation of the aromatic ring. As a result, compounds with interesting physical properties and new catalysts can be prepared.
Experimental Section
General Information
All reactions were carried out with exclusion of air using Schlenk-tube techniques or in a drybox. Instrumental methods and X-ray details are given in the Supporting Information. The chemical shifts (in ppm) in the NMR spectra (Figures S1–S36) are referenced to residual solvent peaks (1H, 13C{1H}) or external 85% H3PO4 (31P{1H}) or CFCl3 (19F{1H}), while the coupling constants J and N (N = JP–H + JP′–H for 1H and N = JP–C + JP′–C for 13C{1H}) are given in hertz.
Reaction of OsH6(PiPr3)2 (1) with 1-(3-(Isoquinolin-1-yl)phenyl)-3-methylimidazolium Tetrafluoroborate: Preparation of 2
A mixture of 1 (200 mg, 0.387 mmol), 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium tetrafluoroborate (145 mg, 0.387 mmol), and triethylamine (809 μL, 5.805 mmol) in toluene (8 mL) was refluxed for 24 h, giving a red suspension. After the mixture was cooled to room temperature, the solvent was removed in vacuo, affording a red residue. A small portion of the residue was dissolved in dichloromethane-d2, and its 1H and 31P{1H} NMR spectra showed the formation of 2 in 82% yield. Addition of pentane (3 mL) caused the precipitation of a pale red solid, which was washed with pentane (3 × 3 mL) and dried in vacuo. Yield: 250 mg (73%). Anal. Calcd for C37H60BF4N3OsP2: C, 50.16; H, 6.83; N, 4.74. Found: C, 49.78; H, 6.73; N, 4.97. HRMS (electrospray, m/z): calculated for C37H60N3OsP2 [M]+, 800.3951; found, 800.3874. IR (cm–1): ν(Os–H) 1975 (w), ν(BF4) 1016 (vs). 1H NMR (300.13 MHz, CD2Cl2, 298 K): δ 9.49–6.87 (12H, C19H15N3), 4.11 (s, 3H, CH3), 1.78 (m, 6H, PCH(CH3)2), 0.89 (dvt, 3JH–H = 7.0, N = 12.4, 36H, PCH(CH3)2), −8.25 (br, 2H, Os–H), −11.97 (br, 1H, Os–H). 1H NMR (300 MHz, CD2Cl2, high-field region, 193 K): δ −6.19 (br, 1H Os–H), −10.71 (br, 1H, Os–H), −12.10 (br, 1H, Os–H). 13C{1H}-apt NMR (75.48 MHz, CD2Cl2, 298 K): δ 199.1 (t, 3JC–P = 5.8, Os–C Ph), 164.6 (s, C), 152.9, 148.1 (both s, CH), 146.1 (s, C), 135.4 (s, CH), 134.6 (s, C), 129.6, 128.5 (both s, CH), 127.3 (s, C), 127.2, 126.7 (both s, CH), 125.6 (s, C), 124.3, 123.2, 122.1, 119.6, 119.4 (all s, CH), 37.0 (s, CH3), 26.6 (vt, N = 24.1, PCH(CH3)2), 20.0, 19.7 (both s, PCH(CH3)2). 31P{1H} NMR (121.50 MHz, CD2Cl2, 298 K): δ 22.1 (s). 19F{1H} NMR (282.38 MHz, CD2Cl2, 298 K): δ −153.7 (s). T1(min) (ms, OsH, 300 MHz, CD2Cl2, 213 K): 32 ± 3 (−6.19 ppm); 32 ± 3 (−10.71 ppm); 85 ± 8 (−12.10 ppm).
Reaction of OsH6(PiPr3)2 (1) with 1-(3-(Isoquinolin-1-yl)phenyl)-3-methylimidazolium Tetraphenylborate: Preparation of 3 and 4
A mixture of 1 (200 mg, 0.387 mmol), 1-(3-(isoquinolin-1-yl)phenyl)-3-methylimidazolium tetraphenylborate (330 mg, 0.503 mmol), and triethylamine (809 μL, 5.805 mmol) in toluene (8 mL) was refluxed for 24 h, giving a dark brown suspension. After the mixture was cooled to room temperature, the solvent was removed in vacuo, affording a brown residue. A small portion of the residue was dissolved in dichloromethane-d2, and its 1H and 31P{1H} NMR spectra showed the formation of 3 and 4 in a 73/27 molar ratio. The brown residue was purified by flash column chromatography (silica gel, toluene/CH2Cl2 100/0 to 0/100). Yellow and red bands were eluted, and after the solvents were removed, yellow and red residues were obtained. Addition of pentane (3 mL) to each of them afforded yellow and red solids, respectively, that were washed with pentane (2 × 4 mL) and dried in vacuo. Yield of 3 (red compound): 246 mg (57%). Yield of 4 (yellow compound): 28 mg (9%).
Data for 3
Anal. Calcd for C61H78BN3OsP2: C, 65.63; H, 7.04; N, 3.76. Found: C, 65.22; H, 6.92; N, 4.01. HRMS (electrospray, m/z): calculated for C37H58N3OsP2 [M]+, 798.3701; found, 798.3717. IR (cm–1): ν(Os–H) 2034 (w). 1H NMR (300.13 MHz, CD2Cl2, 298 K): δ 9.13–6.30 (31 H, 11H C19H14N3 plus 20H BPh4), 3.50 (s, 3H, CH3), 1.70 (m, 6H, PCH(CH3)2), 0.80 (dvt, 3JH–H = 6.7, N = 13.0, 18H, PCH(CH3)2), 0.69 (dvt, 3JH–H = 6.5, N = 13.0, 18H, PCH(CH3)2), −4.54 (br, 2H, Os–H). 1H{31P} NMR (300 MHz, CD2Cl2, high-field region, 193 K): δ −5.72 (AB spin system, Δν = 2678, JA-B = 135, 2H, OsH). 13C{1H}-apt NMR (75.48 MHz, CD2Cl2, 298 K): δ 192.4 (t, 2JC–P = 5.9, Os–C Ph), 167.2 (s, C), 164.5 (q, JC–B = 49.1, C BPh4), 153.2 (s, CH), 144.9 (s, C), 143.3 (s, C), 140.2 (t, 2JC–P = 7.3, Os–C im), 136.3 (s, CH BPh4), 135.6 (s, C), 135.5, 131.0, 128.7, 127.5, 127.3, 126.9 (all s, CH), 126.2 (q, JC–B = 2.1, CH BPh4), 124.1 (s, CH), 122.2 (s, CH BPh4), 121.3, 121.2, 111.8 (all s, CH), 35.8 (s, CH3), 26.2 (vt, N = 25.1, PCH(CH3)2), 19.0, 18.9 (both s, PCH(CH3)2). 31P{1H} NMR (121.50 MHz, CD2Cl2, 298 K): δ 3.4 (s). T1 (ms, OsH, 300 MHz, CD2Cl2, 183 K): 279 ± 28 (−5.72 ppm).
Data for 4
Anal. Calcd for C37H59N3OsP2: C, 55.68; H, 7.45; N, 5.27. Found: C, 55.47; H, 7.18; N, 5.32. HRMS (electrospray, m/z) calculated for C37H59N3OsP2 [M]+, 796.3604; found, 796.3561. IR (cm–1): ν(Os–H) 2033 (w). 1H NMR (300.13 MHz, CD2Cl2, 298 K): δ 8.54–6.94 (11H, C19H14N3), 3.96 (s, 3H, CH3), 1.82 (m, 6H, PCH(CH3)2), 0.97 (dvt, 3JH–H = 6.3, N = 12.4, 18H, PCH(CH3)2), 0.88 (dvt, 3JH–H = 6.4, N = 12.6, 18H, PCH(CH3)2), −8.77 (br, 1H, Os–H), −10.41 (br, 2H, Os–H). 1H NMR (300 MHz, CD2Cl2, high-field region, 183 K): δ −8.90 (t, 2JH–P = 15.7, 1H, Os–H), −10.38 (br, 1H, Os–H), −10.78 (br, 1H, Os–H). 13C{1H}-apt NMR (75.48 MHz, CD2Cl2, 298 K): δ 190.1 (t, 2JC–P = 5.9, Os–C im), 162.8 (t, 2JC–P = 7.6, Os–C Ph), 162.7, 147.7 (both s, C), 147.3, 142.6 (both s, CH), 137.5, 130.2 (both s, C), 129.9, 128.7 (both s, CH), 127.3 (s, C), 127.1, 126.9, 125.8, 120.9, 118.8, 114.7, 111.3 (all s, CH), 39.8 (s, CH3), 28.2 (vt, N = 24.4, PCH(CH3)2), 19.9, 19.8 (both s, PCH(CH3)2). 31P{1H} NMR (121.50 MHz, CD2Cl2, 298 K): δ 24.7 (s). T1(min) (ms, OsH, 300 MHz, CD2Cl2, 223 K): 70 ± 7 (−8.79 ppm); 75 ± 7 (−10.47 ppm).
Reaction of OsH6(PiPr3)2 (1) with 1-(3-(Isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium Tetrafluoroborate: Preparation of 5
A mixture of 1 (200 mg, 0.387 mmol), 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium tetrafluoroborate (164 mg, 0.387 mmol), and triethylamine (809 μL, 5.805 mmol) in toluene (8 mL) was refluxed for 24 h, giving a burgundy suspension. After the mixture was cooled to room temperature, the solvent was removed in vacuo, affording a burgundy residue. A small portion of the residue was dissolved in dichloromethane-d2, and its 1H and 31P{1H} NMR spectra showed the formation of 5 in 82% yield. Addition of diethyl ether (3 mL) to the residue caused the precipitation of a dark red solid, which was washed with diethyl ether (3 × 3 mL) and dried in vacuo. Yield: 265 mg (73%). Anal. Calcd for C41H62BF4N3OsP2: C, 52.61; H, 6.68; N, 4.49. Found: C, 52.72; H, 7.03; N, 4.72. HRMS (electrospray, m/z): calculated for C41H60N3OsP2 [M – 2H], 848.3865; found, 848.3874. IR (cm–1): ν(BF4) 1019 (vs). 1H NMR (300.13 MHz, CD2Cl2, 298 K): δ 9.54–6.94 (14H, C23H17N3), 4.32 (s, 3H, CH3), 1.83 (m, 6H, PCH(CH3)2), 0.93 (dvt, 3JH–H = 6.8, N = 12.3, 36H, PCH(CH3)2), −8.23 (br, 2H, Os–H), −11.90 (br, 1H, Os–H). 1H NMR (300 MHz, CD2Cl2, high-field region, 203 K): δ −6.19 (br, 1H, Os–H), −10.54 (br, 1H, Os–H), −11.99 (t, 2JH–P = 9.9, 1H, Os–H). 13C{1H}-apt NMR (75.48 MHz, CD2Cl2, 298 K): δ 199.7 (t, 3JC–P = 5.5,Os–C), 164.9 (s, C), 152.9 (s, CH), 148.2 (s, CH), 146.5 (s, C), 141.5 (s, CH), 135.4, 132.9, 132.5 (all s, C), 129.8, 128.5, 127.9, 127.8 (all s, CH), 127.4 (s, C), 127.1 (s, CH), 126.4 (s, CH), 122.9 (s, C), 122.2, 119.5, 114.2, 113.3 (all s, CH), 34.0 (s, CH3), 27.8 (vt, N = 24.3, PCH(CH3)2), 20.1, 19.8 (both s, PCH(CH3)2). 31P{1H} NMR (121.50 MHz, CD2Cl2, 298 K): δ 21.9 (s). 19F{1H} NMR (282.38 MHz, CD2Cl2, 298 K): δ −151.4 (s). T1(min) (ms, OsH, 300 MHz, CD2Cl2, 253 K): 61 ± 6 (−8.24 ppm); 33 ± 3 (−11.90 ppm).
Reaction of OsH6(PiPr3)2 (1) with 1-(3-(Isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium Tetraphenylborate: Preparation of 6
A mixture of 1 (200 mg, 0.387 mmol), 1-(3-(isoquinolin-1-yl)phenyl)-3-methylbenzimidazolium tetraphenylborate (330 mg, 0.503 mmol), and triethylamine (809 μL, 5.805 mmol) in toluene (8 mL) was refluxed for 24 h, giving a dark brown suspension. After the mixture was cooled to room temperature, the solvent was removed in vacuo, affording a brown residue. A small portion of the residue was dissolved in dichloromethane-d2, and its 1H and 31P{1H} NMR spectra showed the formation of 6 in 78% yield. The brown residue was purified by flash column chromatography (silica gel, toluene), eluting a yellow band. Upon evaporation of toluene and addition of pentane (3 mL) a yellow solid was obtained, which was washed with pentane (2 × 4 mL) and dried in vacuo. Yield: 141 mg (43%). Anal. Calcd for C41H61N3OsP2: C, 58.06; H, 7.25; N, 4.95. Found: C, 58.31; H, 7.16; N, 5.35. HRMS (electrospray, m/z): calculated for C41H60N3OsP2 [M – H]+, 848.3879; found, 848.3874. IR (cm–1): ν(Os–H) 1904 (w). 1H NMR (300.13 MHz, CD2Cl2, 298 K): δ 8.61–7.05 (13H, C23H16N3), 4.24 (s, 3H, CH3), 1.90 (m, 6H, PCH(CH3)2), 0.99 (dvt, 3JH–H = 7.0, N = 12.6, 18H, PCH(CH3)2), 0.90 (dvt, 3JH–H = 7.0, N = 12.6, 18H, PCH(CH3)2), −8.40 (br, 1H, Os–H), −9.97 (br, 2H, Os–H). 1H NMR (300 MHz, CD2Cl2, high-field region, 233 K): δ −8.43 (tt, 2JH–H = 7.0, 2JH–P = 17.4,1H, Os–H), −10.02 (dt, 2JH–H = 7.0, 2JH–P = 14.0, 2H, Os–H). 13C{1H}-apt NMR (75.48 MHz, CD2Cl2, 298 K): δ 206.1 (t, 2JC–P = 5.8, Os–C), 162.5 (s, C), 161.9 (t, 2JC–P = 5.6, Os–C), 148.6 (s, C), 147.0 (s, CH), 142.3 (s, CH), 137.3, 137.1, 133.1, 130.2 (all s, C), 129.5, 128.2 (both s, CH), 126.9 (s, C), 126.7, 126.6, 125.1, 121.6, 121.1, 118.4, 113.1, 110.0, 109.1 (all s, CH), 36.7 (s, CH3), 27.9 (vt, N = 24.8, PCH(CH3)2), 19.4, 19.3 (both s, PCH(CH3)2). 31P{1H} NMR (121.50 MHz, CD2Cl2, 298 K): δ 26.1 (s). T1(min) (ms, OsH, 300 MHz, CD2Cl2, 223 K): 59 ± 6 (−8.45 ppm); 66 ± 7 (−10.02 ppm).
Reaction of Complex 6 with OsH6(PiPr3)2 (1): Preparation of 7
A solution of complexes 1 (79 mg, 0.153 mmol) and 6 (130 mg, 0.153 mmol) in toluene (5 mL) was refluxed for 24 h, giving a burgundy suspension. After the mixture was cooled to room temperature, the solvent was removed in vacuo, affording a burgundy residue. A small portion of the residue was dissolved in dichloromethane-d2, and its 1H and 31P{1H} NMR spectra showed the formation of 7 in 90% yield. Addition of methanol (3 mL) caused the precipitation of a garnet solid that was washed with methanol (3 × 4 mL) and dried in vacuo. Yield: 166 mg (80%). Anal. Calcd for C59H105N3Os2P4: C, 52.07; H, 7.78; N, 3.09. Found: C, 52.13; H, 7.56; N, 3.03. HRMS (electrospray, m/z): calculated for C59H105N3Os2P4 [M]+, 1360.5830; found, 1360.5691. IR (cm–1): ν(Os–H) 1989 (w), 1880 (m). 1H NMR (300.13 MHz, C6D6, 298 K): δ 9.57–6.67 (12H, C23H15N3), 3.98 (s, 3H, CH3), 1.99 (m, 12H, PCH(CH3)2), 1.14 (dvt, 3JH–H = 7.0, N = 12.6, 36H, PCH(CH3)2), 1.04 (dvt, 3JH–H = 7.0, N = 12.6, 18H, PCH(CH3)2), 0.96 (dvt, 3JH–H = 7.0, N = 12.6, 18H, PCH(CH3)2), −8.02 (br, 1H, Os–H), −10.13 (br, 3H, Os–H), −11.24 (br, 2H, Os–H). 1H NMR (300 MHz, toluene-d8, high-field region, 203 K): δ −6.23 (br, 1H, Os–H), −8.05 (br, 1H, Os–H), −10.05 (br, 2H, Os–H), −11.04 (br, 2H, Os–H). 13C{1H}-apt NMR (75.48 MHz, C6D6, 298 K): δ 204.9 (t, 2JC–P = 5.7, Os–C BzIm), 186.5 (t, 2JC–P = 6.1, Os–C Ph), 168.5 (s, C), 166.6 (t, 2JC–P = 5.9, Os–C Ph), 164.4 (s, CH), 153.2 (s, CH), 143.4 (s, C), 137.7 (s, C), 137.7 (s, C), 137.2 (s, CH), 136.2 (s, C), 135.9 (s, CH), 133.4 (s, C), 126.8, 125.9, 122.1, 120.9, 115.4, 115.1, 110.3, 109.2 (all s, CH), 36.8 (s, CH3), 28.1 (vt, N = 24.1, PCH(CH3)2), 26.4 (vt, N = 23.4, PCH(CH3)2), 21.0, 20.2, 20.0, 19.9 (all s, PCH(CH3)2). 31P{1H} NMR (121.50 MHz, C6D6, 298 K): δ 26.7 (s), 22.6 (s). T1(min) (ms, OsH, 300 MHz, toluene-d8, 248 K): 88 ± 8 (−6.23 ppm); 115 ± 11 (−8.05 ppm), 96 ± 10 (−10.05 ppm), 88 ± 8 (−11.04 ppm).
General Procedure for the Catalytic Dehydrogenation of 1,2,3,4-Tetrahydroisoquinoline
Under an argon atmosphere a solution of the catalyst (2, 3, 5, or 6, 0.0082 mmol; 7, 0.0041 mmol) and 1,2,3,4-tetrahydroisoquinoline (0.12 mmol) in p-xylene (1 mL) was placed in a Schlenk flask equipped with a condenser. The reaction mixture was heated at 140 °C for 48 h, and then it was cooled to room temperature. Gas chromatography was used to determine the extent of the reactions (Agilent Technologies 4890D gas chromatograph with a flame ionization detector, HP-5 column (30 m × 0.32 mm, with 0.25 μm film thickness), oven conditions 80 °C (hold 1 min) to 220 °C at 10 °C/min (hold 2 min)). The reactions were run in duplicate. The identity of the products was confirmed by comparison of their retention times with those of pure samples, as well as by GC-MS analyses.
General Procedure for the Catalytic Dehydrogenation of Alcohols
A toluene solution (1 mL) of the catalyst (2, 3, 5, or 6, 0.0082 mmol; 7, 0.0041 mmol) and the corresponding alcohol (0.12 mmol) was placed in a Schlenk flask equipped with a condenser. The reaction mixture was heated at 100 °C for 24 h. After this time the solution was cooled to room temperature, and the yield of the reaction was determined by different methods depending on the nature of the alcohol. In the case of 1-phenylethanol, the extent of the conversion to acetophenone was determined by GC on an Agilent Technologies 6890N gas chromatograph equipped with a flame ionization detector, using a HP-Innowax column (30 m × 0.25 mm, with 0.25 μm film thickness; oven conditions 80 °C (hold 5 min) to 200 °C at 15 °C/min (hold 7 min). The identity of the ketone was confirmed by a GC-MS analysis and by a comparison of its retention time with that of a pure sample. For benzyl alcohol and 1,2-phenylenedimethanol, the crude solution was concentrated under reduced pressure to obtain an oil. Then, 1,1,2,2-tetrachloroethane was added as an internal standard, and the mixture was dissolved in CDCl3 and analyzed by 1H NMR spectroscopy. Benzaldehyde and 1-isobenzofuranone were characterized by 1H NMR spectroscopy. The reactions were run in duplicate.
Acknowledgments
Financial support from the MICIN of Spain (PID2020-115286GB-I00/AEI/10.13039/501100011033 and RED2018-102387-T), Gobierno de Aragón (E06_20R), FEDER, and the European Social Fund is acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.1c00565.
General information for the Experimental Section, structural analysis, NMR spectra, computational details and energies of optimized structures, experimental and computed UV–vis and UV–vis–NIR spectra, cyclic voltammogram of complex 7, and frontier orbitals (PDF)
Cartesian coordinates of the optimized structures (XYZ)
Accession Codes
CCDC 2112116–2112119 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Jones W. D.; Feher F. J. Comparative Reactivities of Hydrocarbon C-H Bonds with a Transition-Metal Complex. Acc. Chem. Res. 1989, 22, 91–100. 10.1021/ar00159a002. [DOI] [Google Scholar]; b Shilov A. E.; Shul’pin G. B. Activation of C-H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]; c Eisenstein O.; Milani J.; Perutz R. N. Selectivity of C-H Activation and Competition between C-H and C-F Bond Activation at Fluorocarbons. Chem. Rev. 2017, 117, 8710–8753. 10.1021/acs.chemrev.7b00163. [DOI] [PubMed] [Google Scholar]; d Pabst T. P.; Chirik P. J. A Tutorial on Selectivity Determination in C(sp2)-H Oxidative Addition of Arenes by Transition Metal Complexes. Organometallics 2021, 40, 813–831. 10.1021/acs.organomet.1c00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. C-H Activation for the Construction of C-B Bonds. Chem. Rev. 2010, 110, 890–931. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; b Davies H. M. L.; Morton D. Recent Advances in C-H Functionalization. J. Org. Chem. 2016, 81, 343–350. 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]; c Hartwig J. F.; Larsen M. A. Undirected, Homogeneous C-H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Esteruelas M. A.; López A. M. C-C Coupling and C-H Bond Activation Reactions of Cyclopentadienyl-Osmium Compounds: The Rich and Varied Chemistry of Os(η5-C5H5)Cl(PiPr3)2 and Its Major Derivatives. Organometallics 2005, 24, 3584–3613. 10.1021/om050272g. [DOI] [Google Scholar]; b Werner H. Carbene-Transition Metal Complexes Formed by Double C-H Bond Activation. Angew. Chem., Int. Ed. 2010, 49, 4714–4728. 10.1002/anie.201000306. [DOI] [PubMed] [Google Scholar]
- a Hummel J. R.; Boerth J. A.; Ellman J. A. Transition-Metal-Catalyzed C-H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev. 2017, 117, 9163–9227. 10.1021/acs.chemrev.6b00661. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Park Y.; Kim Y.; Chang S. Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]; c Roudesly F.; Oble J.; Poli G. Metal-catalyzed CH activation/functionalization: The fundamentals. J. Mol. Catal. A: Chem. 2017, 426, 275–296. 10.1016/j.molcata.2016.06.020. [DOI] [Google Scholar]; d Dalton T.; Faber T.; Glorius F. C-H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 7, 245–261. 10.1021/acscentsci.0c01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chi Y.; Chou P.-T. Transition-metal phosphors with cyclometalating ligands: fundamentals and applications. Chem. Soc. Rev. 2010, 39, 638–655. 10.1039/B916237B. [DOI] [PubMed] [Google Scholar]; b Caporale C.; Massi M. Cyclometalated iridium(III) complexes for life science. Coord. Chem. Rev. 2018, 363, 71–91. 10.1016/j.ccr.2018.02.006. [DOI] [Google Scholar]; c Yoon S.; Teets T. S. Red to near-infrared phosphorescent Ir(III) complexes with electron-rich chelating ligands. Chem. Commun. 2021, 57, 1975–1988. 10.1039/D0CC08067E. [DOI] [PubMed] [Google Scholar]
- a Kubas G. J.Metal Dihydrogen and σ-Bond Complexes: Structure, Theory and Reactivity; Kluwer Academic/Plenum: 2001. [Google Scholar]; b Kubas G. J. Metal-dihydrogen and σ-bond coordination: the consummate extension of the Dewar-Chatt-Duncanson model for metal-olefin π bonding. J. Organomet. Chem. 2001, 635, 37–68. 10.1016/S0022-328X(01)01066-X. [DOI] [Google Scholar]
- a Esteruelas M. A.; Oliván M.. C-H Activation Coupling Reactions. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook, 3rd ed.; Cornils B., Herrmann W. A., Beller M., Paciello R., Eds.; Wiley: 2017; Chapter 23, pp 1307–1332. [Google Scholar]; b Esteruelas M. A.; Oliván M.; Oñate E. Sigma-bond activation reactions induced by unsaturated Os(IV)-hydride complexes. Adv. Organomet. Chem. 2020, 74, 53–104. 10.1016/bs.adomc.2020.04.002. [DOI] [Google Scholar]
- a Eguillor B.; Esteruelas M. A.; García-Raboso J.; Oliván M.; Oñate E. Stoichiometric and Catalytic Deuteration of Pyridine and Methylpyridines by H/D Exchange with Benzene-d6 Promoted by an Unsaturated Osmium Tetrahydride Species. Organometallics 2009, 28, 3700–3709. 10.1021/om900335b. [DOI] [Google Scholar]; b Boutadla Y.; Davies D. L.; Macgregor S. A.; Poblador-Bahamonde A. I. Mechanisms of C-H bond activation: rich synergy between computation and experiment. Dalton Trans. 2009, 5820–5831. 10.1039/b904967c. [DOI] [PubMed] [Google Scholar]; c Balcells D.; Clot E.; Eisenstein O. C-H Bond Activation in Transition Metal Species from a Computational Perspective. Chem. Rev. 2010, 110, 749–823. 10.1021/cr900315k. [DOI] [PubMed] [Google Scholar]; d Xue X.-S.; Ji P.; Zhou B.; Cheng J.-P. The Essential Role of Bond Energetics in C-H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. 10.1021/acs.chemrev.6b00664. [DOI] [PubMed] [Google Scholar]
- a Hartwig J. F. Regioselectivity of the borylation of alkanes and arenes. Chem. Soc. Rev. 2011, 40, 1992–2002. 10.1039/c0cs00156b. [DOI] [PubMed] [Google Scholar]; b Esteruelas M. A.; Martínez A.; Oliván M.; Oñate E. Direct C-H Borylation of Arenes Catalyzed by Saturated Hydride-Boryl-Iridium-POP Complexes: Kinetic Analysis of the Elemental Steps. Chem. - Eur. J. 2020, 26, 12632–12644. 10.1002/chem.202001838. [DOI] [PubMed] [Google Scholar]; c Esteruelas M. A.; Martínez A.; Oliván M.; Oñate E. Kinetic Analysis and Sequencing of Si-H and C-H Bond Activation Reactions: Direct Silylation of Arenes Catalyzed by an Iridium-Polyhydride. J. Am. Chem. Soc. 2020, 142, 19119–19131. 10.1021/jacs.0c07578. [DOI] [PubMed] [Google Scholar]
- a Albrecht M. Cyclometalation Using d-Block Transition Metals: Fundamental Aspects and Recent Trends. Chem. Rev. 2010, 110, 576–623. 10.1021/cr900279a. [DOI] [PubMed] [Google Scholar]; b Cerón-Camacho R.; Roque-Ramires M. A.; Ryabov A. D.; Le Lagadec R. Cyclometalated Osmium Compounds and beyond: Synthesis, Properties, Applications. Molecules 2021, 26, 1563. 10.3390/molecules26061563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang X.; Kanzelberger M.; Emge T. J.; Goldman A. S. Selective Addition to Iridium of Aryl C-H Bonds Ortho to Coordinating Groups. Not Chelation-Assisted. J. Am. Chem. Soc. 2004, 126, 13192–13193. 10.1021/ja046476q. [DOI] [PubMed] [Google Scholar]; b Alós J.; Esteruelas M. A.; Oliván M.; Oñate E.; Puylaert P. C-H Bond Activation Reactions in Ketones and Aldehydes Promoted by POP-Pincer Osmium and Ruthenium Complexes. Organometallics 2015, 34, 4908–4921. 10.1021/acs.organomet.5b00416. [DOI] [Google Scholar]; c Cancela L.; Esteruelas M. A.; López A. M.; Oliván M.; Oñate E.; San-Torcuato A.; Vélez A. Osmium- and Iridium-Promoted C-H Bond Activation of 2,2’-Bipyridines and Related Heterocycles: Kinetic and Thermodynamic Preferences. Organometallics 2020, 39, 2102–2115. 10.1021/acs.organomet.0c00156. [DOI] [Google Scholar]
- a Ros A.; Fernández R.; Lassaletta J. M. Functional group directed C-H borylation. Chem. Soc. Rev. 2014, 43, 3229–3243. 10.1039/C3CS60418G. [DOI] [PubMed] [Google Scholar]; b De Sarkar S.; Liu W.; Kozhushkov S. I.; Ackermann L. Weakly Coordinating Directing Groups for Ruthenium(II)-Catalyzed C-H Activation. Adv. Synth. Catal. 2014, 356, 1461–1479. 10.1002/adsc.201400110. [DOI] [Google Scholar]; c Kakiuchi F.; Kochi T.; Murai S. Chelation-Assisted Regioselective Catalytic Functionalization of C-H, C-O, C-N and C-F Bonds. Synlett 2014, 25, 2390–2414. 10.1055/s-0034-1379210. [DOI] [Google Scholar]; d Sambiagio C.; Schönbauer D.; Blieck R.; Dao-Huy T.; Pototschnig G.; Schaaf P.; Wiesinger T.; Zia M. F.; Wencel-Delord J.; Besset T.; Maes B. U. W.; Schnürch M. A comprehensive overview of directing groups applied in metal-catalysed C-H functionalization chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. 10.1039/C8CS00201K. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Rej S.; Chatani N. Rhodium-Catalyzed C(sp2)- or C(sp3)-H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem., Int. Ed. 2019, 58, 8304–8329. 10.1002/anie.201808159. [DOI] [PubMed] [Google Scholar]
- a Schuster O.; Yang L.; Raubenheimer H. G.; Albrecht M. Beyond Conventional N-Heterocyclic Carbenes: Abnormal, Remote, and Other Classes of NHC Ligands with Reduced Heteroatom Stabilization. Chem. Rev. 2009, 109, 3445–3478. 10.1021/cr8005087. [DOI] [PubMed] [Google Scholar]; b Crabtree R. H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257, 755–766. 10.1016/j.ccr.2012.09.006. [DOI] [Google Scholar]; c Vivancos Á.; Segarra C.; Albrecht M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018, 118, 9493–9586. 10.1021/acs.chemrev.8b00148. [DOI] [PubMed] [Google Scholar]
- a Kovacevic A.; Gründemann S.; Miecznikowski J. R.; Clot E.; Eisenstein O.; Crabtree R. H. Counter-ion effects switch ligand binding from C-2 to C-5 in kinetic carbenes formed from an imidazolium salt and IrH5(PPh3)2. Chem. Commun. 2002, 2580–2581. 10.1039/B207588C. [DOI] [Google Scholar]; b Gründemann S.; Kovacevic A.; Albrecht M.; Faller J. W.; Crabtree R. H. Abnormal Ligand Binding and Reversible Ring Hydrogenation in the Reaction of Imidazolium Salts with IrH5(PPh3)2. J. Am. Chem. Soc. 2002, 124, 10473–10481. 10.1021/ja026735g. [DOI] [PubMed] [Google Scholar]; c Appelhans L. N.; Zuccaccia D.; Kovacevic A.; Chianese A. R.; Miecznikowski J. R.; Macchioni A.; Clot E.; Eisenstein O.; Crabtree R. H. An Anion-Dependent Switch in Selectivity Results from a Change of C-H Activation Mechanism in the Reaction of an Imidazolium Salt with IrH5(PPh3)2. J. Am. Chem. Soc. 2005, 127, 16299–16311. 10.1021/ja055317j. [DOI] [PubMed] [Google Scholar]; d Baya M.; Eguillor B.; Esteruelas M. A.; Oliván M.; Oñate E. Influence of the Anion of the Salt Used on the Coordination Mode of an N-Heterocyclic Carbene Ligand to Osmium. Organometallics 2007, 26, 6556–6563. 10.1021/om700746h. [DOI] [Google Scholar]; e Alabau R. G.; Eguillor B.; Esler J.; Esteruelas M. A.; Oliván M.; Oñate E.; Tsai J.-Y.; Xia C. CCC-Pincer-NHC Osmium Complexes: New Types of Blue-Green Emissive Neutral Compounds for Organic Light-Emitting Devices (OLEDs). Organometallics 2014, 33, 5582–5596. 10.1021/om500905t. [DOI] [Google Scholar]
- a Peris E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. 10.1021/acs.chemrev.6b00695. [DOI] [PubMed] [Google Scholar]; b Scattolin T.; Nolan S. P. Synthetic Routes to Late Transition Metal-NHC Complexes. Trends Chem. 2020, 2, 721–736. 10.1016/j.trechm.2020.06.001. [DOI] [Google Scholar]
- a Bailey W. D.; Luconi L.; Rossin A.; Yakhvarov D.; Flowers S. E.; Kaminsky W.; Kemp R. A.; Giambastiani G.; Goldberg K. I. Pyrazole-Based PCN Pincer Complexes of Palladium(II): Mono- and Dinuclear Hydroxide Complexes and Ligand Rollover C-H Activation. Organometallics 2015, 34, 3998–4010. 10.1021/acs.organomet.5b00355. [DOI] [Google Scholar]; b Ito J.; Sugino K.; Matsushima S.; Sakaguchi H.; Iwata H.; Ishihara T.; Nishiyama H. Synthesis of NHC-Oxazoline Pincer Complexes of Rh and Ru and Their Catalytic Activity for Hydrogenation and Conjugate Reduction. Organometallics 2016, 35, 1885–1894. 10.1021/acs.organomet.6b00239. [DOI] [Google Scholar]; c Yan J.; Wang Y.-B.; Zhu Z.-H.; Li Y.; Zhu X.; Hao X.-Q.; Song M.-P. Synthesis, Characterization, and Catalytic Studies of Unsymmetrical Chiral NCC Pincer Pd(II) and Ni(II) Complexes Bearing (Imidazolinyl)aryl NHC Ligands. Organometallics 2018, 37, 2325–2334. 10.1021/acs.organomet.8b00300. [DOI] [Google Scholar]; d Luconi L.; Osipova E. S.; Giambastiani G.; Peruzzini M.; Rossin A.; Belkova N. V.; Filippov O. A.; Titova E. M.; Pavlov A. A.; Shubina E. S. Amine Boranes Dehydrogenation Mediated by an Unsymmetrical Iridium Pincer Hydride: (PCN) vs (PCP) Improved Catalytic Performance. Organometallics 2018, 37, 3142–3153. 10.1021/acs.organomet.8b00488. [DOI] [Google Scholar]; e Mousa A. H.; Polukeev A. V.; Hansson J.; Wendt O. F. Carboxylation of the Ni-Me Bond in an Electron-Rich Unsymmetrical PCN Pincer Nickel Complex. Organometallics 2020, 39, 1553–1560. 10.1021/acs.organomet.9b00817. [DOI] [Google Scholar]
- a Ares R.; López-Torres M.; Fernández A.; Pereira M. T.; Alberdi G.; Vázquez-García D.; Fernández J. J.; Vila J. M. Functionalized cyclopalladated compounds with bidentate Group 15 donor atom ligands: the crystal and molecular structures of [{Pd[5-(COH)C6H3C(H)=NCy-C2,N](Cl)}2(μ-Ph2PRPPh2)] (R = CH2CH2, Fe(C5H4)2], [Pd{5-(COH)C6H3C(H)=NCy-C2,N}(Ph2PCH2PPh2-P,P)][PF6] and [Pd{5-(COH)C6H3C(H)=N(Cy)-C2,N}(Ph2PCH2CH2AsPh2-P,As)][PF6]. J. Organomet. Chem. 2003, 665, 76–86. 10.1016/S0022-328X(02)02065-X. [DOI] [Google Scholar]; b Wilkinson A. J.; Goeta A. E.; Foster C. E.; Williams J. A. G. Synthesis and Luminescence of a Charge-Neutral, Cyclometalated Iridium(III) Complex Containing N̂ĈN- and ĈN̂C-Coordinating Terdentate Ligands. Inorg. Chem. 2004, 43, 6513–6515. 10.1021/ic049272j. [DOI] [PubMed] [Google Scholar]; c Blanck S.; Geisselbrecht Y.; Kräling K.; Middel S.; Mietke T.; Harms K.; Essen L.-O.; Meggers E. Bioactive cyclometalated phthalimides: design, synthesis and kinase inhibition. Dalton Trans. 2012, 41, 9337–9348. 10.1039/c2dt30940h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Okamura N.; Maeda T.; Fujiwara H.; Soman A.; Unni K. N. N.; Ajayaghosh A.; Yagi S. Photokinetic study on remarkable excimer phosphorescence from heteroleptic cyclometalated platinum(II) complexes bearing a benzoylated 2-phenylpyridinate ligand. Phys. Chem. Chem. Phys. 2018, 20, 542–552. 10.1039/C7CP06944H. [DOI] [PubMed] [Google Scholar]
- a Meijer M. D.; Kleij A. W.; Lutz M.; Spek A. L.; van Koten G. A new homobimetallic arenediyl diplatinum(II) unit as building block for macromolecular synthesis. X-ray crystal structure of [C6H2{CH2NMe2}2-1,5-{PtCl(PPh3)}2-2,4]. J. Organomet. Chem. 2001, 640, 166–169. 10.1016/S0022-328X(01)01176-7. [DOI] [Google Scholar]; b Esteruelas M. A.; Masamunt A. B.; Oliván M.; Oñate E.; Valencia M. Aromatic Diosmatricyclic Nitrogen-Containing Compounds. J. Am. Chem. Soc. 2008, 130, 11612–11613. 10.1021/ja8048892. [DOI] [PubMed] [Google Scholar]; c Williams J. A. G. The coordination chemistry of dipyridylbenzene: N-deficient terpyridine or panacea for brightly luminescent metal complexes?. Chem. Soc. Rev. 2009, 38, 1783–1801. 10.1039/b804434c. [DOI] [PubMed] [Google Scholar]; d Hu Y. X.; Zhang J.; Zhang F.; Wang X.; Yin J.; Hartl F.; Liu S. H. Electronic Properties of Oxidized Cyclometalated Diiridium Complexes: Spin Delocalization Controlled by the Mutual Position of the Iridium Centers. Chem. - Eur. J. 2020, 26, 4567–4575. 10.1002/chem.201904894. [DOI] [PubMed] [Google Scholar]
- Esteruelas M. A.; López A. M.; Oliván M. Polyhydrides of Platinum Group Metals: Nonclassical Interactions and σ-Bond Activation Reactions. Chem. Rev. 2016, 116, 8770–8847. 10.1021/acs.chemrev.6b00080. [DOI] [PubMed] [Google Scholar]
- a Takaya H.; Naota T.; Murahashi S.-I. Iridium Hydride Complex Catalyzed Addition of Nitriles to Carbon-Nitrogen Triple Bonds of Nitriles. J. Am. Chem. Soc. 1998, 120, 4244–4245. 10.1021/ja974106e. [DOI] [Google Scholar]; b Takaya H.; Yoshida K.; Isozaki K.; Terai H.; Murahashi S.-I. Transition-Metal-Based Lewis Acid and Base Ambiphilic Catalysts of Iridium Hydride Complexes: Multicomponent Synthesis of Glutarimides. Angew. Chem., Int. Ed. 2003, 42, 3302–3304. 10.1002/anie.200351689. [DOI] [PubMed] [Google Scholar]; c Esteruelas M. A.; Honczek N.; Oliván M.; Oñate E.; Valencia M. Direct Access to POP-Type Osmium(II) and Osmium(IV) Complexes: Osmium a Promising Alternative to Ruthenium for the Synthesis of Imines from Alcohols and Amines. Organometallics 2011, 30, 2468–2471. 10.1021/om200290u. [DOI] [Google Scholar]; d Babón J. C.; Esteruelas M. A.; López A. M.; Oñate E. Osmium-Promoted Transformation of Alkyl Nitriles to Secondary Aliphatic Amines: Scope and Mechanism. Organometallics 2020, 39, 2177–2188. 10.1021/acs.organomet.0c00236. [DOI] [Google Scholar]; e Babón J. C.; Esteruelas M. A.; Fernández I.; López A. M.; Oñate E. Dihydroboration of Alkyl Nitriles Catalyzed by an Osmium-Polyhydride: Scope, Kinetics, and Mechanism. Organometallics 2020, 39, 3864–3872. 10.1021/acs.organomet.0c00582. [DOI] [Google Scholar]; f Babón J. C.; Esteruelas M. A.; López A. M.; Oñate E. Hydration of Aliphatic Nitriles Catalyzed by an Osmium Polyhydride: Evidence for an Alternative Mechanism. Inorg. Chem. 2021, 60, 7284–7296. 10.1021/acs.inorgchem.1c00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Crespo O.; Eguillor B.; Esteruelas M. A.; Fernández I.; García-Raboso J.; Gómez-Gallego M.; Martín-Ortiz M.; Oliván M.; Sierra M. A. Synthesis and characterisation of [6]-azaosmahelicenes: the first d4-heterometallahelicenes. Chem. Commun. 2012, 48, 5328–5330. 10.1039/c2cc30356f. [DOI] [PubMed] [Google Scholar]; b Eguillor B.; Esteruelas M. A.; Fernández I.; Gómez-Gallego M.; Lledós A.; Martín-Ortiz M.; Oliván M.; Oñate E.; Sierra M. A. Azole Assisted C-H Bond Activation Promoted by an Osmium-Polyhydride: Discerning between N and NH. Organometallics 2015, 34, 1898–1910. 10.1021/acs.organomet.5b00174. [DOI] [Google Scholar]; c Alabau R. G.; Esteruelas M. A.; Oliván M.; Oñate E.; Palacios A. U.; Tsai J.-Y.; Xia C. Osmium(II) Complexes Containing a Dianionic CCCC-Donor Tetradentate Ligand. Organometallics 2016, 35, 3981–3995. 10.1021/acs.organomet.6b00776. [DOI] [Google Scholar]; d Esteruelas M. A.; Oñate E.; Palacios A. U. Selective Synthesis and Photophysical Properties of Phosphorescent Heteroleptic Iridium(III) Complexes with Two Different Bidentate Groups and Two Different Monodentate Ligands. Organometallics 2017, 36, 1743–1755. 10.1021/acs.organomet.7b00108. [DOI] [Google Scholar]; e Alabau R. G.; Esteruelas M. A.; Oliván M.; Oñate E. Preparation of Phosphorescent Osmium(IV) Complexes with N,N’,C and C,N,C’-Pincer Ligands. Organometallics 2017, 36, 1848–1859. 10.1021/acs.organomet.7b00193. [DOI] [Google Scholar]; f Castro-Rodrigo R.; Esteruelas M. A.; Gómez-Bautista D.; Lezáun V.; López A. M.; Oliván M.; Oñate E. Influence of the Bite Angle of Dianionic C,N,C-Pincer Ligands on the Chemical and Photophysical Properties of Iridium(III) and Osmium(IV) Hydride Complexes. Organometallics 2019, 38, 3707–3718. 10.1021/acs.organomet.9b00466. [DOI] [Google Scholar]
- a Liu F.; Goldman A. S. Efficient thermochemical alkane dehydrogenation and isomerization catalyzed by an iridium pincer complex. Chem. Commun. 1999, 655–656. 10.1039/a900631a. [DOI] [Google Scholar]; b Bertoli M.; Choualeb A.; Gusev D. G.; Lough A. J.; Major Q.; Moore B. PNP pincer osmium polyhydrides for catalytic dehydrogenation of primary alcohols. Dalton Trans. 2011, 40, 8941–8949. 10.1039/c1dt10342c. [DOI] [PubMed] [Google Scholar]; c Esteruelas M. A.; Lezáun V.; Martínez A.; Oliván M.; Oñate E. Osmium Hydride Acetylacetonate Complexes and Their Application in Acceptorless Dehydrogenative Coupling of Alcohols and Amines and for the Dehydrogenation of Cyclic Amines. Organometallics 2017, 36, 2996–3004. 10.1021/acs.organomet.7b00521. [DOI] [Google Scholar]; d Buil M. L.; Esteruelas M. A.; Gay M. P.; Gómez-Gallego M.; Nicasio A. I.; Oñate E.; Santiago A.; Sierra M. A. Osmium Catalysts for Acceptorless and Base-Free Dehydrogenation of Alcohols and Amines: Unusual Coordination Modes of a BPI Anion. Organometallics 2018, 37, 603–617. 10.1021/acs.organomet.7b00906. [DOI] [Google Scholar]; e Buil M. L.; Esteruelas M. A.; Izquierdo S.; Nicasio A. I.; Oñate E. N-H and C-H Bond Activations of an Isoindoline Promoted by Iridium- and Osmium-Polyhydride Complexes: A Noninnocent Bridge Ligand for Acceptorless and Base-Free Dehydrogenation of Secondary Alcohols. Organometallics 2020, 39, 2719–2731. 10.1021/acs.organomet.0c00316. [DOI] [Google Scholar]
- Aracama M.; Esteruelas M. A.; Lahoz F. J.; Lopez J. A.; Meyer U.; Oro L. A.; Werner H. Synthesis, Reactivity, Molecular Structure, and Catalytic Activity of the Novel Dichlorodihydridoosmium(IV) Complexes OsH2Cl2(PR3)2 (PR3 = P-i-Pr3, PMe-t-Bu2). Inorg. Chem. 1991, 30, 288–293. 10.1021/ic00002a025. [DOI] [Google Scholar]
- a Esteruelas M. A.; García-Raboso J.; Oliván M. Preparation of Half-Sandwich Osmium Complexes by Deprotonation of Aromatic and Pro-aromatic Acids with a Hexahydride Brønsted Base. Organometallics 2011, 30, 3844–3852. 10.1021/om200371r. [DOI] [Google Scholar]; b Eguillor B.; Esteruelas M. A.; Lezáun V.; Oliván M.; Oñate E.; Tsai J.-Y.; Xia C. A Capped Octahedral MHC6 Compound of a Platinum Group Metal. Chem. - Eur. J. 2016, 22, 9106–9110. 10.1002/chem.201601729. [DOI] [PubMed] [Google Scholar]; c Eguillor B.; Esteruelas M. A.; Lezáun V.; Oliván M.; Oñate E. Elongated Dihydrogen versus Compressed Dihydride in Osmium Complexes. Chem. - Eur. J. 2017, 23, 1526–1530. 10.1002/chem.201605843. [DOI] [PubMed] [Google Scholar]; d Valencia M.; Merinero A. D.; Lorenzo-Aparicio C.; Gómez-Gallego M.; Sierra M. A.; Eguillor B.; Esteruelas M. A.; Oliván M.; Oñate E. Osmium-Promoted σ-Bond Activation Reactions on Nucleosides. Organometallics 2020, 39, 312–323. 10.1021/acs.organomet.9b00693. [DOI] [Google Scholar]; e Cancela L.; Esteruelas M. A.; Galbán J.; Oliván M.; Oñate E.; Vélez A.; Vidal J. C. Electronic Communication in Binuclear Osmium- and Iridium-Polyhydrides. Inorg. Chem. 2021, 60, 2783–2796. 10.1021/acs.inorgchem.0c03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Smith K.-T.; Tilset M.; Kuhlman R.; Caulton K. G. Reactions of (PiPr3)2OsH6 Involving Addition of Protons and Removal of Electrons. Characterization of (PiPr3)2Os(NCMe)xHyz+ (x = 0, 2, 3; y = 1, 2, 3, 4, 7; z = 1, 2), Including Dicationic >2-H2 Complexes. J. Am. Chem. Soc. 1995, 117, 9473–9480. 10.1021/ja00142a013. [DOI] [Google Scholar]; b Buil M. L.; Esteruelas M. A.; Garcés K.; García-Raboso J.; Oliván M. Trapping of a 12-Valence-Electron Osmium Intermediate. Organometallics 2009, 28, 4606–4609. 10.1021/om9002544. [DOI] [Google Scholar]; c Anderson B. G.; Hoyte S. A.; Spencer J. L. Synthesis and Characterization of the Dinuclear Polyhydrides [Os2H7(PPhiPr2)4]+ and [Os2H6(PPhiPr2)4]. Inorg. Chem. 2009, 48, 7977–7983. 10.1021/ic900111s. [DOI] [PubMed] [Google Scholar]
- Bolaño T.; Esteruelas M. A.; Fernández I.; Oñate E.; Palacios A.; Tsai J.-Y.; Xia C. Osmium(II)-Bis(dihydrogen) Complexes Containing Caryl,CNHC-Chelate Ligands: Preparation, Bonding Situation, and Acidity. Organometallics 2015, 34, 778–789. 10.1021/om501275c. [DOI] [Google Scholar]
- Eguillor B.; Esteruelas M. A.; Oliván M.; Puerta M. Abnormal and Normal N-Heterocyclic Carbene Osmium Polyhydride Complexes Obtained by Directed Metalation of Imidazolium Salts. Organometallics 2008, 27, 445–450. 10.1021/om7010722. [DOI] [Google Scholar]
- Castillo A.; Esteruelas M. A.; Oñate E.; Ruiz N. Dihydrido and Trihydrido Diolefin Complexes Stabilized by the Os(PiPr3)2 Unit: New Examples of Quantum Mechanical Exchange Coupling in Trihydrido Osmium Compounds. J. Am. Chem. Soc. 1997, 119, 9691–9698. 10.1021/ja9700212. [DOI] [Google Scholar]
- Heinekey D. M.; Hinkle A. S.; Close J. D. Quantum Mechanical Exchange Coupling in Iridium Trihydride Complexes. J. Am. Chem. Soc. 1996, 118, 5353–5361. 10.1021/ja952142c. [DOI] [Google Scholar]
- a Castillo A.; Barea G.; Esteruelas M. A.; Lahoz F. J.; Lledós A.; Maseras F.; Modrego J.; Oñate E.; Oro L. A.; Ruiz N.; Sola E. Thermally Activated Site Exchange and Quantum Exchange Coupling Processes in Unsymmetrical Trihydride Osmium Compounds. Inorg. Chem. 1999, 38, 1814–1824. 10.1021/ic9804061. [DOI] [PubMed] [Google Scholar]; b Casarrubios L.; Esteruelas M. A.; Larramona C.; Muntaner J. G.; Oñate E.; Sierra M. A. 2-Azetidinones as Precursors of Pincer Properties of CC’N-Osmium Complexes. Inorg. Chem. 2015, 54, 10998–11006. 10.1021/acs.inorgchem.5b02138. [DOI] [PubMed] [Google Scholar]
- a Hsieh C.-H.; Wu F.-I.; Fan C.-H.; Huang M.-J.; Lu K.-Y.; Chou P.-Y.; Yang Y.-H. O.; Wu S.-H.; Chen I.-C.; Chou S.-H.; Wong K.-T.; Cheng C.-H. Design and Synthesis of Iridium Bis(carbene) Complexes for Efficient Blue Electrophosphorescence. Chem. - Eur. J. 2011, 17, 9180–9187. 10.1002/chem.201100317. [DOI] [PubMed] [Google Scholar]; b Zanoni K. P. S.; Kariyazaki B. K.; Ito A.; Brennaman M. K.; Meyer T. J.; Iha N. Y. M. Blue-Green Iridium(III) Emitter and Comprehensive Photophysical Elucidation of Heteroleptic Cyclometalated Iridium(III) Complexes. Inorg. Chem. 2014, 53, 4089–4099. 10.1021/ic500070s. [DOI] [PubMed] [Google Scholar]
- Buil M. L.; Esteruelas M. A.; López A. M. Recent Advances in Synthesis of Molecular Heteroleptic Osmium and Iridium Phosphorescent Emitters. Eur. J. Inorg. Chem. 2021, 10.1002/ejic.202100663. [DOI] [Google Scholar]
- Kaim W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. 10.1021/ic2003832. [DOI] [PubMed] [Google Scholar]
- Winter R. F. Half-Wave Potential Splittings ΔE1/2 as a Measure of Electronic Coupling in Mixed-Valent Systems: Triumphs and Defeats. Organometallics 2014, 33, 4517–4536. 10.1021/om500029x. [DOI] [Google Scholar]
- D’Alessandro D. M.; Keene F. R. A cautionary warning on the use of electrochemical measurements to calculate comproportionation constants for mixed-valence compounds. Dalton Trans. 2004, 3950–3954. 10.1039/b413980a. [DOI] [PubMed] [Google Scholar]
- a Robin M. B.; Day P. Mixed Valence Chemistry: A Survey and Classification. Adv. Inorg. Chem. Radiochem. 1968, 10, 247–422. 10.1016/S0065-2792(08)60179-X. [DOI] [Google Scholar]; b Aguirre-Etcheverry P.; O’Hare D. Electronic Communication through Unsaturated Hydrocarbon Bridges in Homobimetallic Organometallic Complexes. Chem. Rev. 2010, 110, 4839–4864. 10.1021/cr9003852. [DOI] [PubMed] [Google Scholar]
- Brunschwig B. S.; Creutz C.; Sutin N. Optical transitions of symmetrical mixed-valence systems in the Class II-III transition regime. Chem. Soc. Rev. 2002, 31, 168–184. 10.1039/b008034i. [DOI] [PubMed] [Google Scholar]
- Buchwalter P.; Rosé J.; Braunstein P. Multimetallic Catalysis Based on Heterometallic Complexes and Clusters. Chem. Rev. 2015, 115, 28–126. 10.1021/cr500208k. [DOI] [PubMed] [Google Scholar]
- The base deprotonates the alcohol to generate an alkoxide, which coordinates to the metal and subsequently yields the carbonyl compound by a β-hydrogen elimination reaction. See for example:; a Fujita K.; Yoshida T.; Imori Y.; Yamaguchi R. Dehydrogenative Oxidation of Primary and Secondary Alcohols Catalyzed by a Cp*Ir Complex Having a Functional C,N-Chelate Ligand. Org. Lett. 2011, 13, 2278–2281. 10.1021/ol2005424. [DOI] [PubMed] [Google Scholar]; b Kamitani M.; Ito M.; Itazaki M.; Nakazawa H. Effective dehydrogenation of 2-pyridylmethanol derivatives catalyzed by an iron complex. Chem. Commun. 2014, 50, 7941–7944. 10.1039/C4CC01498G. [DOI] [PubMed] [Google Scholar]; c Toyomura K.; Fujita K. Synthesis of Coordinatively Unsaturated Iridium Complexes Having Functional 8-Quinolinolato Ligands: New Catalysts for Dehydrogenative Oxidation of Alcohols in Aqueous Media. Chem. Lett. 2017, 46, 808–810. 10.1246/cl.170166. [DOI] [Google Scholar]; d Wang Z.; Pan B.; Liu Q.; Yue E.; Solan G. A.; Ma Y.; Sun W.-H. Efficient acceptorless dehydrogenation of secondary alcohols to ketones mediated by a PNN-Ru(II) catalyst. Catal. Sci. Technol. 2017, 7, 1654–1661. 10.1039/C7CY00342K. [DOI] [Google Scholar]
- a Murahashi S.-I.; Naota T.; Ito K.; Maeda Y.; Taki H. Ruthenium-Catalyzed Oxidative Transformation of Alcohols and Aldehydes to Esters and Lactones. J. Org. Chem. 1987, 52, 4319–4327. 10.1021/jo00228a032. [DOI] [Google Scholar]; b Zhang J.; Balaraman E.; Leitus G.; Milstein D. Electron-Rich PNP- and PNN-Type Ruthenium(II) Hydrido Borohydride Pincer Complexes. Synthesis, Structure, and Catalytic Dehydrogenation of Alcohols and Hydrogenation of Esters. Organometallics 2011, 30, 5716–5724. 10.1021/om200595m. [DOI] [Google Scholar]; c Langer R.; Fuchs I.; Vogt M.; Balaraman E.; Diskin-Posner Y.; Shimon L. J. W.; Ben-David Y.; Milstein D. Stepwise Metal-Ligand Cooperation by a Reversible Aromatization/Deconjugation Sequence in Ruthenium Complexes with a Tetradentate Phenanthroline-Based Ligand. Chem. - Eur. J. 2013, 19, 3407–3414. 10.1002/chem.201204003. [DOI] [PubMed] [Google Scholar]; d Nguyen D. H.; Trivelli X.; Capet F.; Swesi Y.; Favre-Réguillon A.; Vanoye L.; Dumeignil F.; Gauvin R. M. Deeper Mechanistic Insight into Ru Pincer-Mediated Acceptorless Dehydrogenative Coupling of Alcohols: Exchanges, Intermediates, and Deactivation Species. ACS Catal. 2018, 8, 4719–4734. 10.1021/acscatal.8b00995. [DOI] [Google Scholar]; e Alabau R. G.; Esteruelas M. A.; Martínez A.; Oliván M.; Oñate E. Base-Free and Acceptorless Dehydrogenation of Alcohols Catalyzed by an Iridium Complex Stabilized by a N,N,N-Osmaligand. Organometallics 2018, 37, 2732–2740. 10.1021/acs.organomet.8b00380. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.