

ABSTRACT
Trichoderma spp. represent one of the most important fungal genera to mankind and in natural environments. The genus harbors prolific producers of wood-decaying enzymes, biocontrol agents against plant pathogens, plant-growth-promoting biofertilizers, as well as model organisms for studying fungal-plant-plant pathogen interactions. Pursuing highly accurate, contiguous, and chromosome-level reference genomes has become a primary goal of fungal research communities. Here, we report the chromosome-level genomic sequences and whole-genome annotation data sets of four strains used as biocontrol agents or biofertilizers (Trichoderma virens Gv29-8, Trichoderma virens FT-333, Trichoderma asperellum FT-101, and Trichoderma atroviride P1). Our results provide comprehensive categorization, correct positioning, and evolutionary detail of both nuclear and mitochondrial genomes, including telomeres, AT-rich blocks, centromeres, transposons, mating-type loci, nuclear-encoded mitochondrial sequences, as well as many new secondary metabolic and carbohydrate-active enzyme gene clusters. We have also identified evolutionarily conserved core genes contributing to plant-fungal interactions, as well as variations potentially linked to key behavioral traits such as sex, genome defense, secondary metabolism, and mycoparasitism. The genomic resources we provide herein significantly extend our knowledge not only of this economically important fungal genus, but also fungal evolution and basic biology in general.
IMPORTANCE Telomere-to-telomere and gapless reference genome assemblies are necessary to ensure that all genomic variants are studied and discovered, including centromeres, telomeres, AT-rich blocks, mating type loci, biosynthetic, and metabolic gene clusters. Here, we applied long-range sequencing technologies to determine the near-completed genome sequences of four widely used biocontrol agents or biofertilizers: Trichoderma virens Gv29-8 and FT-333, Trichoderma asperellum FT-101, and Trichoderma atroviride P1. Like those of three Trichoderma reesei wild isolates [QM6a, CBS999.97(MAT1-1) and CBS999.97(MAT1-2)] we reported previously, these four biocontrol agent genomes each contain seven nuclear chromosomes and a circular mitochondrial genome. Substantial intraspecies and intragenus diversities are also discovered, including single nucleotide polymorphisms, chromosome shuffling, as well as genomic relics derived from historical transposition events and repeat-induced point (RIP) mutations.
KEYWORDS: biocontrol, CAZyme, comparative genomics, fungal-plant interaction, gene cluster, genome annotation, genome sequences, secondary metabolism biosynthesis, sexual development, Trichoderma
INTRODUCTION
Trichoderma (Hypocreales, Ascomycota) species are highly successful colonizers of the rhizosphere (as mycoparasites or versatile symbionts) or wherever decaying plant material is available. They are easy to culture, grow rapidly, and often outgrow or even attach to other microbes encountered in their natural habitats. Although some Trichoderma species are of clinical significance, many more are beneficial to humans and in natural processes. For example, the Trichoderma reesei QM6a wild isolate and its cellulase-producing mutants are widely used for commercial production of enzymes that degrade plant cell walls or to generate therapeutic proteins (1–3). Several Trichoderma spp. are also used in agriculture as biocontrol agents (or biopesticides) against plant pathogens or as plant-growth-promoting biofertilizers, including T. asperellum, T. atroviride, T. koningi, T. harzianum, T. rossicum, and T. virens. Trichoderma spp. also have long been model organisms for studying molecular mechanisms underlying industrial enzyme production, secondary metabolite (SM) biosynthesis, effector-like proteins, mycoparasitism, fungal-plant pathogen interactions and asexual sporulation (conidiation) in filamentous fungi (1–16). Recently, T. reesei has become an important emerging model filamentous fungus for studying ascomycete mating, meiosis, postmeiotic mitosis and repeat-induced point (RIP) mutation (17–27). RIP is a fungus-specific genome-level defense mechanism against mobile elements or transposons. RIP occurs premeiotically and targets duplicated sequences and causes frequent conversion of C:G base pairs to T:A within such duplicated sequences (28–30).
About 3 decades ago, the Trichoderma genus was divided into five taxonomic sections: Hypocreanum, Longibrachiatum, Pachybasium, Saturnisporum, and Trichoderma (31). Thanks to the rapid development of next-generation sequencing (NGS) and genome-wide annotation technology, researchers have gained more insights into the genomes of several Trichoderma species, including plant-cell-wall degradation enzymes, effector-like proteins and genes putatively involved in the biosynthesis of SMs (2, 3, 10, 32–37). Because the lengths of NGS reads are too short to exclude unidentified nucleotides and assembly errors, further validation is required before these NGS-based genome sequences and resulting annotation data sets can be used systematically. In a worst-case scenario, false-positive assemblies can result in incorrect assignments of gene gain or loss. NGS-based genome sequences also cannot reveal accurate information about genome synteny, diversity, or evolution (21, 38).
To obtain highly accurate and chromosome-level reference genomes to explore important biological questions, we previously applied third-generation sequencing (TGS) technology and the “Funannotate” gene-prediction tool to determine and annotate the nearly complete genome sequences of three T. reesei wild isolates, i.e., QM6a, CBS999.97(MAT1-1) and CBS999.97(MAT1-2) (21, 26, 27). Here, we further determined and annotated the nearly complete genome sequences of four Trichoderma biocontrol agents, Trichoderma virens Gv29-8 and FT-333, Trichoderma asperellum FT-101 and Trichoderma atroviride P1 (https://github.com/tfwangasimb/Trichoderma-biocontrol/releases; supplemental information [SI], Table S1). Hereafter, CBS999.97(MAT1-1) and CBS999.97(MAT1-2) and the four biocontrol agents are referred to as CBS1-1, CBS1-2, Gv29-8, FT-333, FT-101, and P1, respectively. We demonstrate in this study that these highest-quality genome resources represent powerful tools for comparative multiomics analyses of these economically important Trichoderma spp.
RESULTS
General properties of the nearly complete genome sequences of seven Trichoderma strains.
High-quality and chromosome-level genome sequences of Gv29-8, FT-333, FT-101, and P1 were determined by TGS technology, before performing genome-wide annotations (Table 1, Tables S2–S5 and supplemental data sets [SD], DS1–DS8). Like those of T. reesei wild isolates (21, 26, 38, 39), the nearly complete genomes of Gv29-8, FT-333, FT-101, and P1, each harbors seven telomere-to-telomere nuclear chromosomes (Fig. 1).
TABLE 1.
Summary of the annotated genes in the seven nearly complete Trichoderma genome sequences
| Species | T. reesei | T. reesei | T. reesei | T. virens | T. virens | T. asperellum | T. atroviride |
|---|---|---|---|---|---|---|---|
| Strain | CBS999.97 (MAT1-1) |
CBS999.97 (MAT1-2) |
QM6a | Gv29-8 | FT-333 | FT-101 | P1 |
| Sequencing technology | PacBio | PacBio | PacBio | Oxford Nanopore |
Oxford Nanopore |
PacBio | Oxford Nanopore |
| Locus_tag | TRC1 | TRC2 | TrQ | TrV | TrVFT-333 | TrA | TrAt |
| Genome size (base pairs; bps) | 34,319,199 | 34,324,311 | 34,922,528 | 40,979,523 | 41,418,917 | 37,545,380 | 37,300,646 |
| No. chromosomes | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| N50 (bps) | 5,258,134 | 5,262,578 | 5,311,445 | 6,490,838 | 6,644,895 | 5,512,738 | 5,658,044 |
| GC (%) | 51.63 | 51.63 | 51.08 | 47.35 | 47.07 | 47.06 | 48.72 |
| No. of AT-blocks (≥500 bps)a | 2259 | 2250 | 2249 | 3577 | 3367 | 4570 | 5510 |
| Length of AT-rich blocks (kbs) | 2,530 | 2,510 | 3,125 | 4,641 | 4,856 | 5,167 | 4,813 |
| Percentage of AT-rich blocks (%) | 7.37 | 7.77 | 8.95 | 11.33 | 11.72 | 13.76 | 12.90 |
| Mitogenome size (bps) | 38,995 | 39,005 | 42,130 | 27,943 | 31,081 | 30,285 | 29,981 |
| BUSCO genome metrics (%)b | 99.3 | 99.3 | 99.3 | 97.8 | 96.6 | 98.5 | 97.1 |
| Single complete (S) % Duplicated complete (D) % Fragment (F) % Missing (M) % |
S:99.3 D:0.0 F:0.0 M:0.7 |
S:99.3 D:0.0 F:0.0 M:0.7 |
S:99.3 D:0.0 F:0.0 M:0.7 |
S: 97.4 D:0.4 F:0.3 M:1.9 |
S:95.9 D:0.7 F:1.2 M:2.2 |
S:98.5 D:0.0 F:0.1 M:1.4 |
S:96.8 D:0.3 F:0.8 M:2.1 |
| BUSCO protein metrics (%)b | 99.2 | 98.9 | 98.4 | 95.5 | 94.2 | 98.6 | 90.8 |
| Single complete (S) % Duplicated complete (D) % Fragment (F) % Missing (M) % |
S:93.5 D:5.7 F:0.3 M:0.5 |
S:92.2 D:6.7 F:0.3 M:0.8 |
S:91.8 D:6.6 F:0.3 M:1.3 |
S: 95.1 D:0.4 F:1.6 M:2.9 |
S:93.7 D:0.5 F:2.5 M:3.3 |
S:96.2 D:2.4 F:0.8 M:0.6 |
S: 88.4 D:2.4 F:5.9 M:3.3 |
| Predicted protein-coding genes | 10,292 | 10,225 | 10,238 | 12,263 | 11,895 | 12,041 | 13,327 |
| Predicted proteins | 11,090 | 11,087 | 11,038 | 12,064 | 11,698 | 12,454 | 13,583 |
| tRNA genes | 150 | 144 | 159 | 202 | 200 | 184 | 185 |
| Predicted gene clusters | 32 | 32 | 31 | 57 | 57 | 54 | 45 |
| Transcription Factors (TF) | 691 | 710 | 680 | 739 | 641 | 882 | 843 |
| HET domains (PF06985) | 41 | 42 | 44 | 68 | 60 | 55 | 75 |
| Ankyrins (PF00023) | 79 | 81 | 75 | 117 | 137 | 105 | 114 |
| CAZymesc | |||||||
| Auxiliary activity (AA) | 48 | 51 | 49 | 66 | 58 | 54 | 58 |
| Carbohydrate-binding modules (CBM) | 11 | 11 | 12 | 16 | 14 | 13 | 13 |
| Carbohydrate esterases (CE) | 33 | 32 | 33 | 47 | 42 | 43 | 39 |
| Glycoside hydrolases (GH) | 199 | 198 | 199 | 230 | 222 | 240 | 229 |
| Glycosyl transferases (GT) | 89 | 91 | 88 | 71 | 72 | 77 | 77 |
| Polysaccharide lyases (PL) | 7 | 7 | 7 | 7 | 7 | 8 | 9 |
| CAZ-GCs | 31 | 31 | 29 | 35 | 26 | 31 | 33 |
| Predicted proteins with signal peptides (SP) | |||||||
| Total secretory signal peptidesd | 866 | 874 | 840 | 1,072 | 995 | 1,050 | 1,041 |
| Oxidoreductases (GO:0016491) | 43 | 42 | 45 | 61 | 60 | 50 | 54 |
| Hydrolases (GO:0016787) | 203 | 197 | 202 | 247 | 225 | 253 | 250 |
| Transferases (GO:0016740) | 10 | 11 | 11 | 9 | 8 | 11 | 9 |
| Catalytic activity (GO:0003824) | 284 | 280 | 282 | 357 | 328 | 347 | 350 |
| Lysases (GO:0016829) | 2 | 2 | 2 | 3 | 4 | 3 | 4 |
| Ligases (GO:0016874) | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Isomerases (GO:0016853) | 5 | 5 | 5 | 5 | 5 | 5 | 7 |
| Peptidoglycan muralytic activity (GO:0061783) | 2 | 2 | 2 | 2 | 2 | 0 | 1 |
| Secondary metabolite biosynthesis (SMB) | |||||||
| NRPS | 7 | 7 | 11 | 17 | 22 | 15 | 7 |
| PKS/NRPS-like proteins | 17 | 19 | 18 | 32 | 40 | 38 | 30 |
| Hybrid PKS-NRPS | 4 | 4 | 1 | 11 | 12 | 5 | 4 |
| NRPS-like proteins | 8 | 8 | 7 | 16 | 16 | 19 | 22 |
| Type I Iterative PKS | 10 | 10 | 10 | 16 | 15 | 12 | 16 |
| PKS-like proteins | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Cytochrome P450 (CYP) PF00067 | 79 | 80 | 71 | 104 | 87 | 72 | 67 |
| SM-BGCe | 32 | 32 | 32 | 58 | 57 | 52 | 46 |
As described previously (21), AT-rich blocks with GC contents ≥12% and ≥6% lower than the average GC content of all predicted genes (56.5%) and the entire QM6a genome (51.1%), respectively.
Gene annotation completeness was evaluated in BUSCO (v4.1.4) using the database for fungi_odb10.
The CAZyme genes were determined by using the dbCAN2 meta server (http://bcb.unl.edu/dbCAN2) (96).
The SignalP server was used to predict the presence and location of signal peptide cleavage sites in amino acid sequences (56).
The gene clusters were determined by using the antiSMASH software tool.
FIG 1.

Diagrammatic representations of the seven chromosomes of CBS1-2 (A), QM6a (B), Gv29-8 (C), FT-333 (D), P1 (E), and FT-101 (F). For comparative genome analyses, we identified orthologous gene pairs using the annotation results generated by Funannotate v1.8 (91) (supplemental data set DS8). The colors of chromosome fragments represent orthologous proteins consistent with their colors in CBS1-2 to clearly show chromosomal rearrangements. Locations of predicted centromeres are shown by restricted width. White fragments in QM6a, Gv29-8, FT-333, P1, and FT-101 represent strain-specific sequences that do not exist in CBS1-2, respectively. Locations of AT-rich blocks are indicated by black bars in the lower chromosomal maps. Black, white and gray arrows indicate disruption of synteny occurring at AT-rich blocks of the subject genomes, the CBS1-2 genome, or both, respectively.
Telomeres, subtelomeres, AT-rich blocks, retrotransposons, and centromeres.
Using our assembled telomere-to-telomere genomes, we could assess and compare their structural components at fine-scale resolution. Telomeric repeats are evolutionarily conserved among these seven Trichoderma genomes, i.e., TTAGGG at the 3′-termini and the reverse complement CCCTAA at the 5′-termini (21, 32). Compared with those of CBS1-2, nearly all subtelomeres of the four Trichoderma biocontrol agent genomes are hypervariable and contain species-specific sequences (Fig. 1). New subtelomeric variants can be created by distinct mechanisms, including alternative lengthening of telomeres (ALT) and break-induced replication (BIR). ALT can lengthen telomeres without utilizing telomerase. It is a homologous recombination (HR)-based process that involves copying telomeric DNA template (40, 41). During BIR, homologous templates from either the same chromosome or even a nonallelic region can be used for template replication and to establish new subtelomeres (42, 43). A typical example of BIR in T. reesei is the reciprocal exchange between the right arm terminus of ChII in CBS1-1 or the right arm terminus of ChIV in CBS1-2 and QM6a, respectively (Fig. S1) (20, 26, 27). Another example is the subtelomeric fragment in the ChI left arm in FT-333. This subtelomeric fragment in Gv29-8 has relocated to the interior of ChIV, i.e., about a quarter of the chromosome’s length from the terminus of its right arm (Fig. S2B).
Compared with the genomes of the three T. reesei wild isolates, those of the four biocontrol agents contain more interspersed AT-rich blocks (≥500 bp). The respective overall numbers and genomic contents are 2,249 and 8.95% (QM6a), 2,259 and 7.37% (CBS1-1), 2,250 and 7.77% (CBS1-2) (26, 38), 3,577 and 11.33% (Gv29-8), 3,367 and 11.72% (FT-333), 4,570 and 13.76% (FT-101), and 5,510 and 12.90% (P1). These results are compatible with the average genomic GC contents of Gv29-8, FT-333, FT-101, and P1, all of which are lower than those of the three T. reesei strains (Table 1). The higher numbers of AT-rich blocks in these four Trichoderma species also partly account for their larger genome sizes. Because the numbers of AT-rich blocks of different lengths in Neurospora crassa (Sordarales, Ascomycota) and QM6a (21) are similar to those in CBS1-1, CBS1-2, and the four biocontrol agents (Fig. S3), all seven Trichoderma genomes have undergone extensive transposon invasions followed by RIP mutations.
The overall numbers of authentic transposons (i.e., which have not been extensively mutated or degenerated by RIP) in these seven genomes are 70 (QM6a), 62 (CBS1-1), 62 (CBS1-2) (27, 38), 93 (Gv29-8), 94 (FT-333), 92 (FT-101), and 78 (P1), respectively (Fig. 1 and Table S2). Because CBS1-1 and CBS1-2 were derived from two ascospores of a heterothallic CBS999.97 fruiting body, their genomes had undergone at least one round of RIP during sexual development (17, 27). In contrast, QM6a, Gv29-8, FT-333, FT-101, and P1 have been propagated asexually since they were isolated. Therefore, the two CBS999.97 strains contain fewer authentic transposable elements.
As in the filamentous fungal model organism Neurospora crassa (Sordariales, Ascomycota) (44–46) and QM6a (21), the putative centromeric loci in all seven Trichoderma genomes are the longest AT-rich blocks and the longest regions of each chromosome lacking an open-reading frame (ORF) or putative protein-encoding genes (Fig. S4–S8 and Table S3). Using BLASTN with an E value of 1e-8 (identity >80%) (47), all putative centromeric loci contain an array of repeats that are either short repetitive sequences or the relics derived from historical transposition and RIP events. Notably, there are no or very few authentic transposons in all putative centromeres (Fig. S4–S8 and Table S2). This scenario is consistent with our recent finding that all seven putative centromeres of QM6a and CBS1-1 generated no or only a few RIP mutations upon sexual crossing (26).
Extensive gross chromosome rearrangements between the genomes of different Trichoderma species.
Conserved synteny between the nearly complete Trichoderma genomes was revealed by ideogrammatic representations (Fig. 1 and Fig. S1) or by CIRCOS plots (48) (Fig. S2). Genomes of the same species (i.e., QM6a, CBS1-2, and CBS1-2 or Gv29-8 and FT-333) or of the same section/clade (i.e., P1 and FT-101) display a higher degree of chromosome synteny than when different Trichoderma species or sections are compared, respectively (Fig. S2). Disruption of synteny often (but not always) occurs at long AT-rich blocks or even within centromeres (Fig. 1) (see Discussion).
Highly divergent genomic contents of four Trichoderma species.
It was reported previously that the core genome derived from 14 of the most common Trichoderma species contains ∼7,000 genes (37). Our annotation results (SD, DS2–DS8) indicate that four representative Trichoderma species (CBS1-2, Gv29-8, P1, and FT-101) share 7,202 core protein-encoding genes. The four biocontrol agents (Gv29-8, FT-333, FT-101, and P1) possess an additional ∼800 conserved genes. The genomes of CBS1-2, Gv29-8, P1, and FT-101 each encodes 2,152, 2,439, 3,779, or 2,555 species-specific genes, equivalent to one-fifth to one-third of their overall protein-encoding genes (Fig. 2A). Gene ontology (GO) analyses further revealed that only 25% to 33% of the species-specific genes encode functionally annotated proteins (Fig. S9). Although the three T. reesei genomes have the smallest genome sizes and encode the lowest overall numbers of protein-encoding genes and tRNA genes, the Benchmarking Universal Single-Copy Ortholog (BUSCO) protein metrics of QM6a, CBS1-1, CBS1-2, and FT-101 are higher than those of Gv29-8, FT-333 and P1 (Table 1) (see Discussion).
FIG 2.
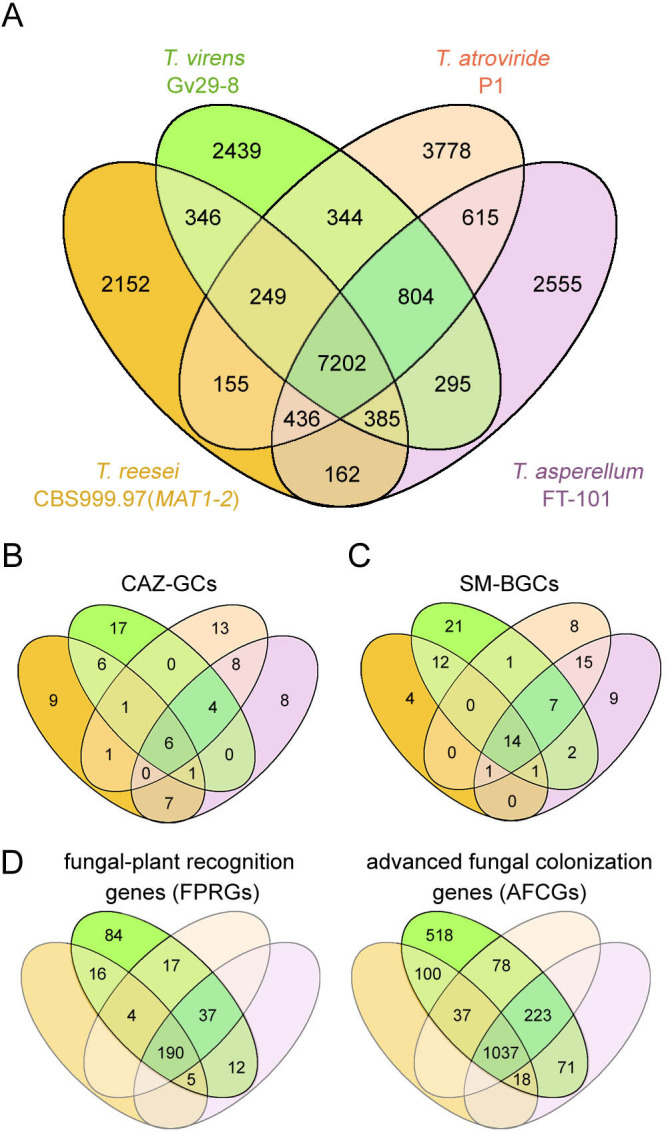
Comparison of the predicted proteomes encoded by the near complete genome sequences of CBS1-2, Gv29-8, P1, and FT-101. Venn diagram of the overall number of annotated protein-encoding genes (A), the number of CAZ-GCs (B), the number of SM-BGCs (C), and evolutionarily conserved FPRGs and AFCGs (D) (see SD, DS12-14).
Mating type loci and sexual development genes.
CBS1-1 and CBS1-2 are sexually competent. In contrast, like QM6a, Gv29-8, FT-333, FT-101, and P1 have been propagated asexually since they were isolated. To validate the accuracy of our genome annotation results, we first confirmed that only the ham5 gene in QM6a encodes a truncated protein (19) (Fig. S10). We then surveyed ∼160 gene orthologs in CBS1-2 and/or three other filamentous fungal model organisms [Neurospora crassa, Sordaria macrospora (Sphaeriales, Ascomycota), Saccharomyces cerevisiae (Saccharomycetales, Ascomycota)] (SD, DS9). All these gene orthologs have been implicated as being involved in or even essential to fungal sexual development (see reviews of 2, 3, 49–52), and their annotated functions are described in SD, DS9. The seven Trichoderma genomes encode nearly all of the normal protein homologs deemed to play roles in sexual mating signaling systems (e.g., pheromones, light, cell communication, and hyphal fusion), RIP, quelling, meiotic silencing by unpaired DNA (MSUD), meiotic DNA recombination, chromosome individualization or condensation, sister chromatid cohesion, chromosome synapsis, as well as the formation of fruiting bodies (SD, DS9). P1 and Gv29-8, like QM6a, each possesses a MAT1-2 locus with a normal mat1-2-1 gene. FT-101, like CBS1-1, has a normal MAT1-1 locus with three mating type genes: mat1-1-1, mat1-1-2, and mat1-1-3. The MAT1-1 locus of FT-333 only comprises mat1-1-1 and mat1-1-2, but not mat1-1-3 (Fig. 3). The genomes of QM6a, CBS1-1, and CBS1-2 all possess an ortholog of N. crassa male barren-3 (mb-3) (53). We were unable to identify or annotate this gene in the genomes of Gv29-8, FT-333, FT-101, or P1. Further investigations are needed to confirm whether Gv29-8, FT-333, FT-101, and P1 exhibit a male barren phenotype or if the mb-3-like gene in T. reesei is essential for male fertility. Interestingly, Gv29-8, FT-333, and P1, but not FT-101, also lack an ortholog of sad3, a gene essential for meiotic silencing by unpaired DNA (MUSD) and normal sexual development in N. crassa (54) (SD, DS9). MUSD, an RNA interference (RNAi)-related genome defense mechanism, occurs in prophase I of meiosis when unpaired DNA sequences are present and leads to the silencing of all homologous genes in the diploid ascus cell (54).
FIG 3.

Comparison of the nucleotide sequences within and around the mating-type loci in P1, FT-101, CBS1-1, CBS1-2, Gv29-8 and FT-333. The tracks between two strains are color-coded to indicate nucleotide sequence identity. The mating-type genes (mat1–1-1, mat1–1-2, mat1–1-3 and mat1–2-1) are dissimilar in sequence, but they are found at the same loci on the third chromosomes and are all flanked by two evolutionarily conserved genes, the DNA lyase apn2 and the complex I intermediate-associated protein 30 gene cia30.
Transcription factors.
In terms of overall numbers of transcription factors (TF) genes, the ranking is FT-101 > P1 > Gv29-8 > CBS1-2 > CBS1-1 > QM6a > FT-333 (Table 1). The main differences are due to specific gain or loss of two transcription factor subfamilies, i.e., the fungal Zn(2)-Cys(6) binuclear cluster domain subfamily (InterPro identity: 111138) and the fungal-specific TF domain family (InterPro identity: 007219). Compared with the genomes of Gv29-8, FT-333, FT-101, and P1, the three T. reesei genomes possess a species-specific PAS-fold protein, but lack a helix-turn-helix TF (Table S4). The physiological functions of these unique TFs need to be further explored. It is important to note that gene numbers of other TF families are nearly identical among the seven genomes. Thus, these TFs constitute the “core” transcriptional regulatory circuitry of the genus Trichoderma.
Predicted signal peptide proteins.
Signal peptides (SPs) are short amino acid sequences in the amino termini of many newly synthesized proteins that target to membranes or membrane-embedded export machines. The SignalP server has been widely used to predict the presence and location of SP cleavage sites in amino acid sequences of SP proteins (55, 56). The three T. reesei genomes each possesses 840 to 870 SP proteins. In contrast, those of Gv29-8, FT-333, FT-101, and P1 encode 1,072, 995, 1,050, and 1,041 SP proteins, respectively. GO indicates that all seven Trichoderma genomes are highly enriched with SP proteins having catalytic, oxidoreductive, or hydrolytic activities (Table 1). SP proteins revealed by the SignalP server are not necessarily cleaved or cleavable in vivo. Further experimental investigations are needed to validate if they are indeed secreted proteins.
Carbohydrate-active enzymes and CAZyme gene clusters.
Compared with the three T. reesei genomes, those of Gv29-8, FT-333, FT-101, and P1 encode more auxiliary activity (AA) proteins, carbohydrate esterases (CE) and glycoside hydrolases (GH). In contrast, all three T. reesei genomes possess more glycosyltransferase (GT) genes than those of Gv29-8, FT-333, FT-101 and P1 (Table 1). Further investigations will be needed to elucidate this unique property of T. reesei. There is considerable evidence showing that carbohydrate-active enzymes (CAZymes) cooperate with other CAZymes and signature proteins (e.g., transporters and transcription factors), and that the respective genes tend to form physically-linked CAZyme gene clusters (CAZ-GCs) in polysaccharide utilization loci (PUL) (57). To identify potential CAZ-GCs in these strains, we have developed a high-stringency predictive software that identified 31, 31, 29, 35, 26, 31, and 33 CAZ-GCs in QM6a, CBS1-1, CBS1-2, Gv29-8, FT-333, FT-101, and P1, respectively (Table 1, Table S5, and SD, DS10). These CAZ-GCs have been named according to their chromosomal location, e.g., CAZ-GC 3.2 indicates the second CAZ-GC from the left arm of ChIII. These results provide a comprehensive basis for further exploring CAZymes and PUL in Trichoderma spp. (Fig. 4). We identified 9, 17, 13, and eight species-specific CAZ-GCs in CBS1-2, Gv29-8, P1, and FT-101, respectively. Notably, consistent with their phylogenetic relationships, T. reesei and T. virens share 14 common CAZ-GCs, whereas T. atroviride and T. asperellum share 18 common CAZ-GCs (Fig. 2B).
FIG 4.

Locations of CAZyme genes (in gray), CAZ-GCs (in cyan) and SM-BGCs (in red) along chromosomal maps. Chromosome maps of CBS1-2, Gv29-8, P1 and FT-101 are shown. Locations of predicted centromeres are represented by narrowed segments.
Secondary metabolite biosynthetic genes and gene clusters.
Relative to the three T. reesei nuclear genomes, those of P1, FT-101, Gv29-8, and FT-333 have undergone expansions in almost all secondary metabolite biosynthetic genes and gene clusters (SM-BGC) subfamilies, including polyketide synthases (PKSs), nonribosomal peptide synthases (NRPSs) and cytochromes P450 (CYP450s) (Table 1). Using the Antibiotics and Secondary Metabolite Analysis Shell (antiSMASH) software tool (58), we have annotated a variety of SM-BGCs in the seven genomes: 32 in QM6a, 32 in CBS1-1, 32 in CBS1-2, 59 in Gv29-8, 57 in FT-333, 52 in FT-101, and 46 in P1 (Table 1 and Table S5). These SM-BGCs have also been named according to their chromosomal location; for example, SM-BGC 2.3 indicates the third SM-BGC from the left arm of ChII (Fig. 4). The four different Trichoderma species only share 14 common SM-BGCs. We identified four, 21, eight, and nine species-specific SM-BGCs in CBS1-2, Gv29-8, P1, and FT-101, respectively. Notably, consistent with their phylogenetic relationships (37), T. reesei and T. virens share 27 common SM-BGCs, whereas T. atroviride and T. asperellum share 37 common SM-BGCs (Fig. 2B).
To address the intriguing question of how SM-BGCs form and are regulated, we compared the biosynthetic genes of the five best-characterized SM-BGCs (see reviews of [2, 10]) to explore their commonalities and differences (Table S6). First, BGCs for siderophore (SID), ferrichrome (FRC), and conidial green pigment (CGP) are found in all four Trichoderma species we examined here. SID-BGCs and FRC-BGCs exhibit gene order conservation and contain none or only one AT-rich block (>500 bp) (Fig. S11 and S12). There are only three evolutionarily conserved genes in CGP-BGCs. Their neighboring sequences are quite variable and contain more AT-rich blocks. The neighboring genes of CGP-BGC in P1 are more conserved with those in FT-101, but they are completely different from those in Gv29-8 and CBS1-2 (Fig. S13). Second, the sorbicillinoid (SOR)-BGC is T. reesei-specific, as only T. reesei can secrete yellow sorbicillinoid compounds (59). The 5′ and 3′ termini of SOR-BGC are physically linked to usk1 and a well-characterized CAZ-GC harboring three CAZyme genes: axe1 (acetyl xylan esterase), cip1 (a CBM-containing auxiliary factor), and cel61a (previously named endoglucanase IV) (32) (Fig. S14). The entire chromosomal region (i.e., usk1-SOR-BGC-axe1-cip1-cel61a) contains 14 protein-encoding genes, but no AT-rich blocks. Although Gv29-8, P1, and FT-101 each possesses eight to 10 orthologs of those 14 protein-encoding genes, their orthologs are scattered across at least five different chromosomes (Fig. S14). Third, FT-101 possesses a terpene cyclase (TrA_010949) that is highly similar in amino acid sequence to the protein product of the T. brevicompactum tri5 trichothecene synthase gene (2). This putative tri5 gene and seven novel genes together constitute a potential trichothecene (TRI) or TRI-like BGC in FT-101 (i.e., SM-BGC-7.3) (Fig. S15). We were unable to identify or annotate this putative tri5 gene in P1, CBS1-2, or Gv29-8. It is also noteworthy that the protein products of these seven novel genes in FT-101 are different from the biosynthetic proteins encoded by the canonical TRI-BGCs in T. arundinaceum and T. brevicompactum (60, 61). Interestingly, six orthologs of these seven novel genes also cluster together in P1, whereas CBS1-2 and Gv29-8 each possesses three and five orthologs of these seven novel genes, respectively (Table S6). Fourth, T. virens has two species-specific BGCs: viridin (VIR)-BGC and gliotoxin (GTX)-BGC. These two SM-BGCs produce a group of furano-steroidal antibiotics and GTX or GTX-like compounds to compete with and restrict the growth of plant pathogenic fungi, respectively (36, 62). CBS1-2, P1, and FT-101 possess none or only two orthologs of the VIR biosynthetic genes, respectively (Table S6). Both GTX-BGCs in Gv29-8 and FT-333 contain 13 gli genes and are located at the far-right subtelomeres of their first chromosomes. Ten long AT-rich blocks separate these 13 gli genes. The order of these long AT-rich blocks is conserved in FT-333 and Gv29-8, but they exhibit highly variable lengths and sequences (Fig. S16A). Although T. reesei does not produce GTX during vegetative growth, QM6a and both CBS999.97 strains each possesses a gene cluster with only six and seven gli orthologs, respectively, i.e., SM-BGC-6.4 in CBS1-2 (Table S6 and Fig. S16B). gliH, an essential GTX biosynthetic gene, does not exist in any of the three T. reesei genomes. QM6a also lacks gliC, which encodes a CPY450 oxidoreductase that catalyzes the formation of C-S bonds. These C-S bonds constitute the internal disulfide bridge of GTX and other exopolysaccharide-type fungal toxins (63). The order of the gli genes in T. reesei is different from that in Gv29-8. There are no AT-rich blocks between the gli orthologs in T. reesei (Fig. S16B). These results suggest that the ancestral genomes of T. virens and T. reesei might have undergone multistep processes to acquire and/or reorganize their GTX or GTX-like BGCs. Further investigations will be needed to address the physiological roles of the incomplete GTX-BGCs in T. reesei.
T. reesei Sor7 and T. virens GliT are secreted proteins.
Most of our knowledge about the molecular mechanisms of GTX biosynthesis has come from studies of the GTX-BGC in Aspergillus fumigatus, an opportunistic fungal pathogen. A. fumigatus GliT is an intracellular FAD-dependent dithiol oxidase that converts the reduced GTX (i.e., dithio-GTX) to GTX, thus conferring host self-resistance toward highly reactive dithio-GTX and tolerance to exogeneous GTX (64, 65). We found that the GliT proteins in FT-333 hosts an SP, whereas that of A. fumigatus does not (Fig. S17). The GliC proteins in FT-333 and Gv29-8, like that of A. fumigatus, are predicted to possess signal peptides. In contrast, that of CBS1-2 does not (Fig. S18). Accordingly, the biosynthetic and regulatory mechanisms of GTX- or GTX-like BGCs in T. virens and T. reesei likely differ from those of A. fumigatus. Our annotation results also reveal that the Sor7 FAD-linked oxireductase of T. reesei hosts an SP (Fig. S19). Because the SP-hosting proteins predicted by the SignalP server are not necessarily cleaved or cleavable in vivo, we applied a proteomics approach to analyze them in the culture filtrate (CFs) of CBS1-2, FT-333, Gv29-8, FT-101, and P1, respectively, including by means of ammonium sulfate precipitation, in-solution trypsin digestion and LC-MS-MS analysis (SI, Materials and Methods). Although most of the secreted proteins we identified in CFs are functionally unannotated, our results confirm that T. virens GliT and T. reesei Sor7 are indeed secreted proteins (SD, DS11 and Table S7). Further investigations will be needed to reveal the functional roles of these two FAD-linked oxireductases in regulating biosynthesis and the functions of GTX and sorbicillinoids, respectively.
Our proteomics approach also identified two fungal secreted proteins, i.e., T. virens small protein 1 (Sml) and T. asperellum KatG2 catalase-peroxidase 2. T. virens Sm1 is a secreted elicitor-like protein that induces plant defense responses and systemic resistance by triggering production of reactive oxygen species. Sm1 lacks toxic activity against plants and microbes (66). The functional roles of KatG2 in T. asperellum remain unclear. KatG2 was originally identified as an extracellular protein in several phytopathogenic fungi (67). In Fusarium graminearum (Hypocreales, Ascomycota), KatG2 is exclusively located on the cell wall of invading hyphal cells and contributes to its pathogenicity by alleviating oxidative stress in the vicinity of invasion hyphae (68).
Genome-wide transcriptomic analyses reveal evolutionarily conserved and diverse genes contributing to fungal-plant interactions.
Trichoderma spp. can colonize plant roots, both externally and internally. Induction of plant defense via fungal-plant interactions is considered one of the most important mechanisms of Trichoderma-mediated biological control. Michael Kolomiets and colleagues performed RNA sequencing (RNA-seq) analysis on the roots of maize seedlings grown in hydroponic conditions and treated with T. virens Gv29-8 at 6 h and 30 h (69, 70). Notably, the time points represented fungal-plant recognition at 6 h and advanced fungal colonization at 30 h. Gv29-8 undergoes global repression of transcription upon recognition of maize roots and then induces expression of a broad spectrum of genes during root colonization (69). Using the complete genome sequence of Gv29-8 as a reference, we reanalyzed the previously acquired transcriptomic data sets (69, 70) to reveal 365 and 2,082 T. virens Gv29-8 genes that are transcriptionally upregulated (fold change [FC] ≥ 3 and P < 0.05) at the 6-h and 30-h time points, respectively (Table S8 and SD, DS12 and DS13). The protein products of 365 putative fungal-plant recognition genes (FPRGs) include 42 predicted SP proteins, 13 proteases, 14 CAZymes, and 75 membrane proteins. Three SM-BGCs and one CAZ-GC (1.4) were specifically upregulated at 6 h. The protein products of 2,082 putative advanced fungal colonization genes (AFCGs) include 349 predicted SP proteins, 100 proteases, 158 CAZymes, 433 membrane proteins, and 433 TFs. Almost 80% of SM-BGCs and CAZ-GCs in T. virens have at least one gene that was transcriptionally upregulated at 30 h. Notably, most genes in GTX-BGC-1.1 and CPG-BGC-10.1 are AFCGs, whereas all those in VIR-BGC-5.5 and SID-BGC-6.4 are not (Table S8). Further investigations will be needed to reveal whether and why CPG, GTX, or related SMs have nonantibiotic functions during advanced fungal colonization. Moreover, we found by genome-wide BLASTp searches that 190 (52%) FPRGs and 1,037 (50%) AFCGs are evolutionarily conserved in the genomes of CBS1-2, FT-101, P1, and FT-333, respectively (Fig. 2D, Table S9 and SD, DS12-DS14), prompting us to infer that different Trichoderma species might utilize both conserved and diverse SMs, proteins or signaling pathways to mediate fungal-plant interactions.
Mitogenomes and nuclear-encoded mitochondrial sequences.
Our results also reveal the circular mitogenomes of all seven Trichoderma strains (SD, DS9). We reported recently that T. reesei mitochondria are inherited maternally (27), so the mitogenomes of CBS1-1 and CBS1-2 should be identical. Indeed, we found that their sequence differences were attributable to sequencing read errors in 18 polyhomonucleotide runs. The lengths of the other six mitogenomes vary significantly, although both number and order of protein-encoding genes, tRNAs and rRNA are all well-conserved. We found that mobile genetic elements play key roles in shaping the mitogenomes of Trichoderma (Fig. S20–S22 and SD, DS15).
Although our results of Trichoderma mitogenomes are consistent with those of previous studies (2, 21, 71, 72), we have discovered that the mitogenomes of Gv29-8 and FT-333 each harbor three nuclear-encoded mitochondrial sequences (NUMTs). NUMTs, first detected in the mouse genome (73), have now been found in several other eukaryotes (74–76). NUMT integration has been implicated in increasing genetic diversity and facilitating genome evolution (77, 78). NUMTs have yet to be explicitly reported among filamentous fungi. Three lines of evidence suggest that the ancestral genome of T. virens underwent NUMT integration. First, FT-333 and Gv29-8 each contains three almost identical NUMTs in the subtelomeric regions of the left arm of their second chromosome (indicated by a black line in Fig. S2B). Their sequence lengths and coordinates in FT-333 are 139 bp (211,858 to 211,996), 166 bp (211,996 to 212,161) and 170 bp (115,955 to 116,124), whereas in Gv29-8 they are 146 bp (15,319 to 115,464), 168 bp (115,464 to 115,631), and 170 bp (115,955 to 116,124) (Fig. S23). The corresponding sequences of these three NUMTs localize within two mitochondrial NADH dehydrogenase subunit genes (nad5 and nad6) and a mitochondrial noncoding sequence, respectively (Fig. S21). Second, these three NUMTs are unlikely to represent mitochondrial DNA contamination arising from sequence assembly errors because the corresponding nucleotide sequences were observed in several long raw reads generated by the nanopore sequencer. Third, all three NUMTs in FT-333 and Gv29-8 are located within an AT-rich block (∼1,500 bp in length) that lacks protein-coding sequences (Fig. S24). It was reported previously that mammalian NUMTs tend to be inserted near retrotransposons and that the insertion sites often represent DNA sequences with high DNA “bendability” and lie immediately adjacent to AT-rich sequences (79). Therefore, filamentous fungi and mammalian cells may display an evolutionarily conserved origin for NUMTs (79).
DISCUSSION
Although >50 different Trichoderma genomes have been determined by NGS technology, almost all of the NGS genomic drafts are far from nearly complete or chromosome-level assemblies due to the shortcomings of short NGS reads and the exceedingly low-complexity nature of Trichoderma genomes. In contrast, the seven nearly complete Trichoderma genomes we have determined by TGS technology represent the highest quality yet achieved. The greatest benefits of precise genome assemblies are that they enable accurate determination of structural components in each genome (e.g., telomeres, centromeres, interspersed AT-rich blocks and authentic transposable elements), as well as the chromosome synteny between different Trichoderma species. First, the results of our comparative genomic analysis reveal that all seven genomes likely underwent extensive transposon invasions followed by RIP mutations (Fig. S3), explaining why the overall numbers of authentic transposons (i.e., which have not been extensively mutated or degenerated by RIP) in these seven genomes are relatively low (Table S2). Second, we reported previously that the longest AT-rich blocks in all seven QM6a chromosomes are likely their centromeres, as they collectively harbor 24 centromere-specific and conserved satellite repeats (21). Here, we further demonstrate that the putative centromeric loci in all seven Trichoderma genomes are not only the longest AT-rich blocks but also the longest regions of each chromosome lacking an open-reading frame (ORF) or putative protein-encoding genes (Fig. S4–S8 and Table S3). Third, the genomes of different Trichoderma species exhibit extensive chromosome shuffling. Disruption of chromosome synteny often but not always occurs near or within AT-rich blocks or even the predicted centromeres (Fig. 1). One possibility is that mobilization of ancient retrotransposons might induce chromosome rearrangements. Alternatively, chromosome translocations via recombination of AT-rich blocks or centromere scission (80) may drive chromosome shuffling to reshape the genomes of different species. In this regard, retrotransposition and RIP might act as a critical driver of genome reorganization, eventually leading to genome diversification within a species (microevolution) and between species (macroevolution). Consequently, the newly generated strains harboring chromosome translocations may fail to undergo successful sexual reproduction with the parental wild-type strain because extensive chromosomal heterozygosity results in recombination suppression during meiosis, i.e., via nonhomologous pairing, synaptic adjustments or shifts in recombination (or chiasma) positions (81, 82).
Locally, the order of biosynthetic genes, AT-rich blocks, and retrotransposons in SM-BGCs provides detailed insights into the mechanisms of genome evolution. Our data indicate that evolutionarily conserved SM-BGCs often lack or only contain a few longer AT-rich blocks (≥500 bp). In contrast, the GTX-BGCs in FT-333 and Gv29-8 have 10 such blocks. This unique property of GTX-BGCs had not been reported before because NGS sequence reads are too short to assemble chromosomal regions of low sequence complexity (36). We suggest that GTX-BGCs represent versatile systems for future experimental evolution studies to reveal the functional impacts of AT-rich blocks for the expression and regulation of GTX biosynthesis. Also noteworthy is that orthologs of GTX biosynthetic genes are randomly allocated across the genomes of T. atroviride and T. asperellum. The gene order of the putative GTX-BGC in T. reesei also differs from that of the authentic GTX-BGCs in T. virens (Fig. S16). We conclude that diverse and/or multistep mechanisms were involved in the formation of different SM-BGCs during evolution (83), such as lateral gene transfer, unequal crossing over, or transposon-mediated gene transfer. Lateral gene transfer (also called horizontal gene transfer) represents the acquisition of genetic material from another organism (e.g., bacteria), whereas unequal crossing over involves the exchange of sequences between chromosomes. Because the GTX-BGCs of both FT-333 and Gv29-8 are located in the subtelomeres of their first chromosomes, it would be interesting to determine if BIR (42, 43) or other molecular mechanisms might play critical roles in generating or maintaining this T. virens-specific gene cluster.
High-quality and near complete genome sequences also ensure accurate and comprehensive genome annotation. Based on NGS-based genome sequences, it was reported previously that T. reesei and its ancestor might have undergone rapid gene loss relative to several other of the most common Trichoderma species (37). Notably, a smaller gene inventory might result from less intensive expansion of certain gene families rather than profound loss of distinct protein-encoding genes. In contrast, our genome-wide annotation data indicate that QM6a, CBS1-1, CBS1-2, and FT-101 encode more near-universal single-copy orthologs (NUSCOs) than the genomes of P1, Gv29-8, and FT-333, with the BUSCO protein metrics of QM6a, CBS1-1, CBS1-2, and FT-101 being higher respectively than those of P1, Gv29-8, and FT-333 (Table 1). BUSCOs are ideal for quantifying genome and proteome completeness, as the assumptions for these NUSCOs are evolutionarily sound (84). The genome sizes of QM6a, CBS1-1, and CBS1-2 are much smaller than those of FT-101, P1, Gv29-8, and FT-333. Thus, the genomes of T. reesei and its ancestor might not have experienced rapid gene loss. Instead, compared with the genomes of QM6a and the two CBS999.97 strains, the genomes of P1, Gv29-8, and FT-333 appear to have experienced both gene loss and extensive gene family expansions during evolution. In contrast, the genome of FT-101 only underwent extensive gene family expansions and not gene loss (Table 1). These findings represent a striking example of why TGS-based genomes are superior to NGS-based genomes for comparative and functional studies. Because the overall numbers of interspersed AT-rich blocks (≥500 bp) in QM6a, CBS1-1, and CBS1-2 are much lower (50% to 70%) than those in FT-101, P1, Gv29-8, and FT-333, we further infer that the larger genome sizes of these latter and their more extensive gene family expansions might have arisen from more profound mobilization and RIP of ancient retrotransposons in their ancestral genomes.
In this study, we have shown by comparative genomic and transcriptomic analyses that different Trichoderma species might utilize both conserved and diverse proteins or signaling pathways to mediate fungal-plant interactions (Fig. 2D). Our genome-wide annotation results also have revealed a comprehensive array of SM-BGCs and CAZ-BGCs in each of the four Trichoderma species we considered herein (Fig. 4). The majority of SM-BGCs, CAZ-BGCs, and effector-like SP proteins are poorly understood and, consequently, their corresponding products and physiological functions are largely unknown. Because a variety of genome mining methods and tools have been developed to guide characterization and activation of gene clusters (85–87), the nearly complete genome resources provided in this study will help discover and/or functionally characterize new SMs, CAZymes, and effector-like SP proteins, as well as their roles in and underlying evolutionary mechanisms of mycoparasitism, fungal-plant interactions and biocontrol activities.
Conclusion.
Our study highlights that high-quality and nearly complete genome sequences are necessary tools for accurate comparative and functional genomic analyses. The data we have presented herein provide numerous insights into fundamental biology and industrial biotechnology.
MATERIALS AND METHODS
Fungal strains.
All Trichoderma wild-isolate strains described in this study are listed in Table S1. FT-101 and FT-333 were isolated by Ruey-Shyang Chen (National Chia-Yi University). CBS1-1, CBS1-2, Gv29-8, and P1 were provided by Monika Schmoll (Austrian Institute of Technology, Austria).
Whole-genome DNA sequencing, RNA sequencing, and whole-genome gene prediction.
Isolation of genomic DNA and RNA, as well as PacBio single-molecular real-time (SMRT) genome sequencing and assembly, were carried out as described previously (21, 38, 88). Oxford Nanopore direct DNA library preparation and sequencing were performed at the Genomic Core Lab of the Institute of Molecular Biology, Academia Sinica. In brief, small fragments of purified genomic DNA were removed by using the Ampure cleanup kit (Beckman Coulter). High-quality DNA quantification was conducted using a Qubit fluorometer (Thermo Fisher Scientific). The average length of genomic DNA was scaled using Femto Pulse (Agilent). We used 1 μg of genomic DNA for library construction. The library contained three or four DNA samples barcoded using the EXP-NBD104 Rapid barcoding kit (Oxford Nanopore Technologies), and adaptors were added with the SQK-LSK109 ligation sequencing kit (Oxford Nanopore Technologies). The library was loaded onto a R9.4.1 Flow Cell system (FLO-MIN106) and processed for 48 h on the MinION platform. The sequencing process was controlled using the MinKNOW software (version 3.6.5; Oxford Nanopore Technologies). All experimental procedures were carried out according to the manufacturer’s instructions. FAST5 data files were generated upon completion of sequencing. These files were converted into FASTQ files in Guppy (v3.6.0). All reads were split into separate FASTQ files based on their barcode by means of the qcat tool (v1.1.0), and then Canu (v2.1.1) (89) was used to perform whole-genome assembly on the corresponding FASTQ files for each sample. The parameter of corOutCoverage was set to 60 based on sequencing read depth. Finally, we used medaka (v1.2.0) to polish the assemblies with the raw reads. Because Canu generated expected numbers of contigs for each sample, and a comparison of the genome to wild-type QM6a indicated no broken contigs, we did not perform any manual finishing or validation. The completeness of genome assemblies was evaluated in BUSCO (v4.0.6) (90) against the database of fungi_odb10 (OrthoDB; https://www.orthodb.org). All other experimental procedures have been described previously in detail (38), including PacBio SMRT genome sequencing, NGS-based RNA sequencing, as well as genome-wide gene prediction in Funannotate (91). BUSCO was also used to quantitatively measure the completeness of genome assemblies and gene predictions. We selected evolutionarily informed expectations of gene content based on the Ascomycota odb9 database from OrthoDB (https://www.orthodb.org). Genome or protein matrix scores >95% for model organisms are generally deemed complete reference genomes (92).
Analysis of genome-wide synteny.
For comparative genome analyses and to identify duplicated regions, we identified orthologous gene pairs using the annotation results generated in Funannotate v1.8 (91). Alignments were performed using BLASTP with an expect value (E) ≤ 1e-20. CIRCOS (48) was used to display sequence similarity and conservation. The inner ribbon track of CIRCOS outputs is used to show synteny, whereas the exterior tracks quantify the degree of sequence conservation between the T. reesei CBS1-2 genome and those of the three other Trichoderma species.
Comparative genomic analysis.
Funannotate (91) not only parses the protein-coding models from the annotation, but also identifies numbers and classes of carbohydrate-active enzymes (CAZymes), proteases, transcriptional factors, secreted proteins, polyketide synthases (PKSs), and nonribosomal peptide synthetases (NRPSs). To predict secreted proteins, we downloaded the SignalP 4.1 (93), TMHMM 2.0 (94), and big-PI Fungal Predictor (95) programs into “Funannotate” and then applied them with default settings. In this study, effector candidate proteins were defined as predicted secreted proteins (with a signal peptide present, but no transmembrane domains or glycosylphosphatidylinositol anchors) having lengths of <300 amino acids. To assess if effector candidates presented similarity to known proteins, we performed BLASTP analysis (cutoff E-value 10−5) using the Swiss-Prot database (downloaded October 22, 2016).
CAZymes were reannotated using the dbCAN2 meta server (http://bcb.unl.edu/dbCAN2) (96). We have developed a software tool “IBM-CAZ” to predict potential CAZyme gene clusters (CAZ-GCs) with the following requirements: a potential CAZ-GC must contain ≥3 CAZyme genes or ≥2 CAZyme genes with ≥1 other specific signature genes (namely, transporters or transcription factors), and be present within ≤2 intergenic distances. Although the prediction requirements in IMB-CAZ are more stringent than those of dbCAN2 (96) and dbCAN-PUL (97), IBM-CAZ was able to identify all four previously identified CAZ-GCs in Trichoderma reesei QM6a, the ancestor of all currently used cellulase-producing mutants (32).
Proteomics analysis of secreted proteins.
To identify proteins secreted by different Trichoderma strains, we germinated 1 mL of conidia (OD600nm = 0.3) and cultured it at 25°C in 250 mL potato dextrose broth (PDB) medium in shake flasks at 120 rpm for 24 h. The vegetative mycelia and conidia were removed from culture medium by filtering through a 0.22-μm filter (Merck Millipore, Darmstadt, Germany). The proteins in the culture filtrate (CF) were precipitated by adding ammonium sulfate (AS) to 80% saturation at 4°C. After centrifugation at 10,000 × g for 30 min at 4°C, the AS precipitates were recovered and then resuspended in 500 μL of 50 mM Tris-HCl (pH 7.5). Protein concentration was determined using Bradford reagent (Sigma-Aldrich).
For in-solution trypsin digestion, the target proteins (20 μg) were reconstituted in 100 mM Tris-HCl (pH 8.0) and 5% acetonitrile, mixed with 1/10 volume of 100 mM 1,4-dithiothreitol in 100 mM Tris-HCl (pH 8.0), and incubated at 37°C for 1 h. Then, 1/10 volume of 550 mM iodoacetamide in 100 mM Tris-HCl (pH 8.0) was added to the mixture, gently vortexed, and incubated at room temperature for 1 h. Promega Sequencing Grade Modified Trypsin was added to give a final substrate:trypsin ratio of 50:1. The digestion reaction was carried out overnight at 37°C and then the reaction was stopped by adding 5% formic acid to adjust the pH of the solution to below pH 6.0. We determined pH by placing 1-μL aliquots onto pH paper. The peptide digestion products were desalted using C18 Zip-Tips (Millipore, ZTC 18M 096). The peptide solutions were dried down in a SpeedVac vacuum concentrator (Thermo Fisher Scientific) and stored at −20°C before undergoing mass spectrometric analysis.
Liquid chromatography-mass spectrometry (LC-MS-MS) was applied to analyze trypsin-digested peptides. LC-MS analysis was performed by Shu-Yu Lin (Institute of Biological Chemistry, Academia Sinica) using an EASY-nLC 1200 system linked to a Thermo Orbitrap Fusion Lumos mass spectrometer equipped with a Nanospray Flex ion source (Thermo Fisher Scientific) located at the Academia Sinica Common Mass Spectrometry Facilities (https://www.ibc.sinica.edu.tw/facilities/mass-spectrometry-facilities/). All related experimental procedures were described previously in detail (98). Proteomic data were searched against our whole-genome annotation data sets (see below) using the Mascot search engine (v.2.6.2; Matrix Science, Boston, MA, USA) in Proteome Discoverer (v 2.2.0.388; Thermo Fisher Scientific, Waltham, MA, USA). We used the search criterion trypsin digestion. The fixed modification was set as carbamidomethyl (C), and variable modifications were set as oxidation (M), acetylation (protein N-terminal) allowing up to two missed cleavages, and mass accuracy of 10 ppm for the parent ion and 0.6 Da for the fragment ions. The false discovery rate (FDR) was calculated with the Proteome Discoverer Percolator function, and identifications with an FDR > 1% were rejected.
Data availability.
The complete genome sequences, annotation, and raw data sets have been deposited in the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA700774 (Table S1). All study data are included in the article as well as in SI and SD appendixes. All genomic databases data and 15 supplemental data sets are also available at the following websites: https://github.com/tfwangasimb/Trichoderma-biocontrol/releases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Institute of Molecular Biology, Academia Sinica, Taiwan, Republic of China. We thank Shu-Yun Tung (IMB Genomics Core) for the NGS sequencing service, Kun-Hai Ye (IMB Bioinformatics Core) for statistical assistance and bioinformatics consultancy, John O’Brien for English editing, and Yu-Tang Huang (IMB Computer Room) for maintaining the computer workstation. This work was supported by Academia Sinica, Taipei, Taiwan, Republic of China.
H.F.N. and R.S.C. provided the two biocontrol strains, T. asperellum FT-101 and T. virens FT-333. W.C.L. and C.L.C. performed HMW gDNA isolation and Funannotate gene prediction. J.L.C. performed nanopore sequencing. W.C.L. and H.N.L. assembled genome sequences. T.C.L. performed comparative proteomic analyses. C.H.H. performed in-solution trypsin digestion. W.C.L. and T.F.W. performed comparative genomic analyses. T.F.W. conceived and designed the experiments. T.F.W. and W.C.L. wrote the original and revised manuscripts. All authors read and approved the manuscript.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Ruey-Shyang Chen, Email: rschen@mail.ncyu.edu.tw.
Ting-Fang Wang, Email: tfwang@gate.sinica.edu.tw.
Christina A. Cuomo, Broad Institute
REFERENCES
- 1.Peterson R, Nevalainen H. 2012. Trichoderma reesei RUT-C30–thirty years of strain improvement. Microbiology (Reading) 158:58–68. doi: 10.1099/mic.0.054031-0. [DOI] [PubMed] [Google Scholar]
- 2.Schmoll M, Dattenböck C, Carreras-Villaseñor N, Mendoza-Mendoza A, Tisch D, Alemán MI, Baker SE, Brown C, Cervantes-Badillo MG, Cetz-Chel J, Cristobal-Mondragon GR, Delaye L, Esquivel-Naranjo EU, Frischmann A, Gallardo-Negrete J.dJ, García-Esquivel M, Gomez-Rodriguez EY, Greenwood DR, Hernández-Oñate M, Kruszewska JS, Lawry R, Mora-Montes HM, Muñoz-Centeno T, Nieto-Jacobo MF, Nogueira Lopez G, Olmedo-Monfil V, Osorio-Concepcion M, Piłsyk S, Pomraning KR, Rodriguez-Iglesias A, Rosales-Saavedra MT, Sánchez-Arreguín JA, Seidl-Seiboth V, Stewart A, Uresti-Rivera EE, Wang C-L, Wang T-F, Zeilinger S, Casas-Flores S, Herrera-Estrella A. 2016. The genomes of three uneven siblings: footprints of the lifestyles of three Trichoderma species. Microbiol Mol Biol Rev 80:205–327. doi: 10.1128/MMBR.00040-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Druzhinina IS, Kubicek CP. 2016. Familiar stranger: ecological genomics of the model saprotroph and industrial enzyme producer Trichoderma reesei breaks the stereotypes. Adv Appl Microbiol 95:69–147. doi: 10.1016/bs.aambs.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Montenecourt BS, Eveleigh DE. 1977. Preparation of mutants of Trichoderma reesei with enhanced cellulase production. Appl Environ Microbiol 34:777–782. doi: 10.1128/aem.34.6.777-782.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harman GE, Howell CR, Viterbo A, Chet I, Lorito M. 2004. Trichoderma species–opportunistic, avirulent plant symbionts. Nat Rev Microbiol 2:43–56. doi: 10.1038/nrmicro797. [DOI] [PubMed] [Google Scholar]
- 6.Druzhinina I, Kubicek CP. 2005. Species concepts and biodiversity in Trichoderma and Hypocrea: from aggregate species to species clusters? J Zhejiang Univ Sci B 6:100–112. doi: 10.1631/jzus.2005.B0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster A, Schmoll M. 2010. Biology and biotechnology of Trichoderma. Appl Microbiol Biotechnol 87:787–799. doi: 10.1007/s00253-010-2632-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steyaert JM, Weld RJ, Mendoza-Mendoza A, Stewart A. 2010. Reproduction without sex: conidiation in the filamentous fungus Trichoderma. Microbiology (Reading) 156:2887–2900. doi: 10.1099/mic.0.041715-0. [DOI] [PubMed] [Google Scholar]
- 9.Masunaka A, Hyakumachi M, Takenaka S. 2011. Plant growth-promoting fungus, Trichoderma koningi suppresses isoflavonoid phytoalexin vestitol production for colonization on/in the roots of Lotus japonicus. Microbes Environ 26:128–134. doi: 10.1264/jsme2.me10176. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee PK, Horwitz BA, Kenerley CM. 2012. Secondary metabolism in Trichoderma–a genomic perspective. Microbiology (Reading) 158:35–45. doi: 10.1099/mic.0.053629-0. [DOI] [PubMed] [Google Scholar]
- 11.Vinale F, Sivasithamparam K, Ghisalberti EL, Ruocco M, Wood S, Lorito M. 2012. Trichoderma secondary metabolites that affect plant metabolism. Nat Prod Commun 7:1545–1550. [PubMed] [Google Scholar]
- 12.Yao L, Tan C, Song J, Yang Q, Yu L, Li X. 2016. Isolation and expression of two polyketide synthase genes from Trichoderma harzianum 88 during mycoparasitism. Braz J Microbiol 47:468–479. doi: 10.1016/j.bjm.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzman-Guzman P, Porras-Troncoso MD, Olmedo-Monfil V, Herrera-Estrella A. 2019. Trichoderma species: versatile plant symbionts. Phytopathology 109:6–16. doi: 10.1094/PHYTO-07-18-0218-RVW. [DOI] [PubMed] [Google Scholar]
- 14.Ramirez-Valdespino CA, Casas-Flores S, Olmedo-Monfil V. 2019. Trichoderma as a model to study effector-like molecules. Front Microbiol 10:1030. doi: 10.3389/fmicb.2019.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan RAA, Najeeb S, Mao Z, Ling J, Yang Y, Li Y, Xie B. 2020. Bioactive secondary metabolites from Trichoderma spp. against phytopathogenic bacteria and root-knot Nematode. Microorganisms 8:401. doi: 10.3390/microorganisms8030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras-Cornejo HA, Macías-Rodríguez L, del-Val E, Larsen J. 2020. p 263–290. In Mérillon J.M, and Ramawat K.G. (ed), Interactions of Trichoderma with plants, insects, and plant pathogen microorganisms: chemical and molecular bases. Co-evolution of secondary metabolites. Springer International Publishing, Switzerland. [Google Scholar]
- 17.Seidl V, Seibel C, Kubicek CP, Schmoll M. 2009. Sexual development in the industrial workhorse Trichoderma reesei. Proc Natl Acad Sci USA 106:13909–13914. doi: 10.1073/pnas.0904936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CL, Kuo HC, Tung SY, Hsu PW, Wang CL, Seibel C, Schmoll M, Chen RS, Wang TF. 2012. Blue light acts as a double-edged sword in regulating sexual development of Hypocrea jecorina (Trichoderma reesei). PLoS One 7:e44969. doi: 10.1371/journal.pone.0044969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linke R, Thallinger GG, Haarmann T, Eidner J, Schreiter M, Lorenz P, Seiboth B, Kubicek CP. 2015. Restoration of female fertility in Trichoderma reesei QM6a provides the basis for inbreeding in this industrial cellulase producing fungus. Biotechnol Biofuels 8:155. doi: 10.1186/s13068-015-0311-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuang YC, Li WC, Chen CL, Hsu PW, Tung SY, Kuo HC, Schmoll M, Wang TF. 2015. Trichoderma reesei meiosis generates segmentally aneuploid progeny with higher xylanase-producing capability. Biotechnol Biofuels 8:30. doi: 10.1186/s13068-015-0202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li WC, Huang CH, Chen CL, Chuang YC, Tung SY, Wang TF. 2017. Trichoderma reesei complete genome sequence, repeat-induced point mutation, and partitioning of CAZyme gene clusters. Biotechnol Biofuels 10:170. doi: 10.1186/s13068-017-0825-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W-C, Chuang Y-C, Chen C-L, Wang T-F. 2016. Hybrid infertility: The dilemma or opportunity of applying sexual development to improve Trichoderma reesei industrial strains, p 351–359. In Schmoll M. and Dattenböck C. (ed), Gene expression systems in fungi: advancements and applications. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 23.Dattenbock C, Tisch D, Schuster A, Monroy AA, Hinterdobler W, Schmoll M. 2018. Gene regulation associated with sexual development and female fertility in different isolates of Trichoderma reesei. Fungal Biol Biotechnol 5:9. doi: 10.1186/s40694-018-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li WC, Chen CL, Wang TF. 2018. Repeat-induced point (RIP) mutation in the industrial workhorse fungus Trichoderma reesei. Appl Microbiol Biotechnol 102:1567–1574. doi: 10.1007/s00253-017-8731-5. [DOI] [PubMed] [Google Scholar]
- 25.Li WC, Chuang YC, Chen CL, Timofejeva L, Pong WL, Chen YJ, Wang CL, Wang TF. 2019. Two different pathways for initiation of Trichoderma reeseiRad51-only meiotic recombination. BioRxiv. doi: 10.1101/644443. [DOI]
- 26.Li WC, Liu HC, Lin YJ, Tung SY, Wang TF. 2020. Third-generation sequencing-based mapping and visualization of single nucleotide polymorphism, meiotic recombination, illegitimate mutation and repeat-induced point mutation. NAR Genom Bioinform 2:lqaa056. doi: 10.1093/nargab/lqaa056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W-C, Lee C-Y, Lan W-H, Woo T-T, Liu H-C, Yeh H-Y, Chang H-Y, Chuang Y-C, Chen C-Y, Chuang C-N, Chen C-L, Hsueh Y-P, Li H-W, Chi P, Wang T-F. 2021. Trichoderma reesei Rad51 tolerates mismatches in hybrid meiosis with diverse genome sequences. Proc Natl Acad Sci USA 118:e2007192118. doi: 10.1073/pnas.2007192118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selker EU. 1990. Premeiotic instability of repeated sequences in Neurospora crassa. Annu Rev Genet 24:579–613. doi: 10.1146/annurev.ge.24.120190.003051. [DOI] [PubMed] [Google Scholar]
- 29.Aramayo R, Selker EU. 2013. Neurospora crassa, a model system for epigenetics research. Cold Spring Harb Perspect Biol 5:a017921. doi: 10.1101/cshperspect.a017921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gladyshev E. 2017. Repeat-induced point mutation and other genome defense mechanisms in fungi. Microbiol Spectr 5. doi: 10.1128/microbiolspec.FUNK-0042-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bissett J. 1991. A revision of the genus Trichoderma. II. Infrageneric classification. Can J Bot 69:2357–2373. doi: 10.1139/b91-297. [DOI] [Google Scholar]
- 32.Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EGJ, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, Schoch CL, Yao J, Barabote R, Barbote R, Nelson MA, Detter C, Bruce D, Kuske CR, Xie G, Richardson P, Rokhsar DS, Lucas SM, Rubin EM, Dunn-Coleman N, Ward M, Brettin TS. 2008. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26:553–560. doi: 10.1038/nbt1403. [DOI] [PubMed] [Google Scholar]
- 33.Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK, Mukherjee M, Kredics L, Alcaraz LD, Aerts A, Antal Z, Atanasova L, Cervantes-Badillo MG, Challacombe J, Chertkov O, McCluskey K, Coulpier F, Deshpande N, von Döhren H, Ebbole DJ, Esquivel-Naranjo EU, Fekete E, Flipphi M, Glaser F, Gómez-Rodríguez EY, Gruber S, Han C, Henrissat B, Hermosa R, Hernández-Oñate M, Karaffa L, Kosti I, Le Crom S, Lindquist E, Lucas S, Lübeck M, Lübeck PS, Margeot A, Metz B, Misra M, Nevalainen H, Omann M, Packer N, Perrone G, Uresti-Rivera EE, Salamov A, et al. -. 2011. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol 12:R40. doi: 10.1186/gb-2011-12-4-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atanasova L, Knox BP, Kubicek CP, Druzhinina IS, Baker SE. 2013. The polyketide synthase gene pks4 of Trichoderma reesei provides pigmentation and stress resistance. Eukaryot Cell 12:1499–1508. doi: 10.1128/EC.00103-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derntl C, Guzman-Chavez F, Mello-de-Sousa TM, Busse HJ, Driessen AJM, Mach RL, Mach-Aigner AR. 2017. In vivo study of the sorbicillinoid gene cluster in Trichoderma reesei. Front Microbiol 8:2037. doi: 10.3389/fmicb.2017.02037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulgari D, Fiorini L, Gianoncelli A, Bertuzzi M, Gobbi E. 2020. Enlightening gliotoxin biological system in agriculturally relevant Trichoderma spp. Front Microbiol 11:200. doi: 10.3389/fmicb.2020.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kubicek CP, Steindorff AS, Chenthamara K, Manganiello G, Henrissat B, Zhang J, Cai F, Kopchinskiy AG, Kubicek EM, Kuo A, Baroncelli R, Sarrocco S, Noronha EF, Vannacci G, Shen Q, Grigoriev IV, Druzhinina IS. 2019. Evolution and comparative genomics of the most common Trichoderma species. BMC Genomics 20:485. doi: 10.1186/s12864-019-5680-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li WC, Wang TF. 2021. PacBio long-read sequencing, assembly, and funannotate reannotation of the complete genome of Trichoderma reesei QM6a. Methods Mol Biol 2234:311–329. doi: 10.1007/978-1-0716-1048-0_21. [DOI] [PubMed] [Google Scholar]
- 39.Liu HC, Li WC, Wang TF. 2021. TSETA: A third-generation sequencing-based computational tool for mapping and visualization of SNPs, meiotic recombination products, and RIP mutations. Methods Mol Biol 2234:331–361. doi: 10.1007/978-1-0716-1048-0_22. [DOI] [PubMed] [Google Scholar]
- 40.Lundblad V, Blackburn EH. 1993. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 73:347–360. doi: 10.1016/0092-8674(93)90234-H. [DOI] [PubMed] [Google Scholar]
- 41.Teng SC, Zakian VA. 1999. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19:8083–8093. doi: 10.1128/MCB.19.12.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malkova A, Ivanov EL, Haber JE. 1996. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci USA 93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lydeard JR, Jain S, Yamaguchi M, Haber JE. 2007. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 44.Borkovich KA, Alex LA, Yarden O, Freitag M, Turner GE, Read ND, Seiler S, Bell-Pedersen D, Paietta J, Plesofsky N, Plamann M, Goodrich-Tanrikulu M, Schulte U, Mannhaupt G, Nargang FE, Radford A, Selitrennikoff C, Galagan JE, Dunlap JC, Loros JJ, Catcheside D, Inoue H, Aramayo R, Polymenis M, Selker EU, Sachs MS, Marzluf GA, Paulsen I, Davis R, Ebbole DJ, Zelter A, Kalkman ER, O’Rourke R, Bowring F, Yeadon J, Ishii C, Suzuki K, Sakai W, Pratt R. 2004. Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol Mol Biol Rev 68:1–108. doi: 10.1128/MMBR.68.1.1-108.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith KM, Galazka JM, Phatale PA, Connolly LR, Freitag M. 2012. Centromeres of filamentous fungi. Chromosome Res 20:635–656. doi: 10.1007/s10577-012-9290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim KE, Peluso P, Babayan P, Yeadon PJ, Yu C, Fisher WW, Chin C-S, Rapicavoli NA, Rank DR, Li J, Catcheside DEA, Celniker SE, Phillippy AM, Bergman CM, Landolin JM. 2014. Long-read, whole-genome shotgun sequence data for five model organisms. Sci Data 1:140045. doi: 10.1038/sdata.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang Y, Ding W, Zhang M, Han J, Jing Y, Yao W, Hasterok R, Wang Z, Wang K. 2021. The formation and evolution of centromeric satellite repeats in Saccharum species. Plant J 106:616–629. doi: 10.1111/tpj.15186. [DOI] [PubMed] [Google Scholar]
- 48.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang SS, Zhang Z, Liu Y. 2012. RNA interference pathways in fungi: mechanisms and functions. Annu Rev Microbiol 66:305–323. doi: 10.1146/annurev-micro-092611-150138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunter N. 2015. Meiotic Recombination: The essence of heredity. Cold Spring Harb Perspect Biol 7:a016618. doi: 10.1101/cshperspect.a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Zickler D, Kleckner N, Zhang L. 2015. Meiotic crossover patterns: obligatory crossover, interference and homeostasis in a single process. Cell Cycle 14:305–314. doi: 10.4161/15384101.2014.991185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fischer MS, Glass NL. 2019. Communicate and fuse: how filamentous fungi establish and maintain an interconnected mycelial. Front Microbiol 10:619. doi: 10.3389/fmicb.2019.00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCluskey K, Wiest AE, Grigoriev IV, Lipzen A, Martin J, Schackwitz W, Baker SE. 2011. Rediscovery by whole genome sequencing: classical mutations and genome polymorphisms in Neurospora crassa. G3 (Bethesda) 1:303–316. doi: 10.1534/g3.111.000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hammond TM, Xiao H, Boone EC, Perdue TD, Pukkila PJ, Shiu PK. 2011. SAD-3, a putative helicase required for meiotic silencing by unpaired DNA, interacts with other components of the silencing machinery. G3 (Bethesda) 1:369–376. doi: 10.1534/g3.111.000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nielsen H. 2017. Predicting secretory proteins with SignalP. Methods Mol Biol 1611:59–73. doi: 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
- 57.Terrapon N, Lombard V, Gilbert HJ, Henrissat B. 2015. Automatic prediction of polysaccharide utilization loci in Bacteroidetes species. Bioinformatics 31:647–655. doi: 10.1093/bioinformatics/btu716. [DOI] [PubMed] [Google Scholar]
- 58.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antisMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abe N, Sugimoto O, Arakawa T, Tanji K, Hirota A. 2001. Sorbicillinol, a key intermediate of bisorbicillinoid biosynthesis in Trichoderma sp. USF-2690. Biosci Biotechnol Biochem 65:2271–2279. doi: 10.1271/bbb.65.2271. [DOI] [PubMed] [Google Scholar]
- 60.Cardoza RE, Malmierca MG, Hermosa MR, Alexander NJ, McCormick SP, Proctor RH, Tijerino AM, Rumbero A, Monte E, Gutierrez S. 2011. Identification of loci and functional characterization of trichothecene biosynthesis genes in filamentous fungi of the genus Trichoderma. Appl Environ Microbiol 77:4867–4877. doi: 10.1128/AEM.00595-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mousa WK, Raizada MN. 2015. Biodiversity of genes encoding anti-microbial traits within plant associated microbes. Front Plant Sci 6:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bansal R, Sherkhane PD, Oulkar D, Khan Z, Banerjee K, Mukherjee PK. 2018. The Viridin biosynthesis gene cluster of Trichoderma virens and its conservancy in the bat white-nose fungus Pseudogymnoascus destructans. Chemistryselect 3:1289–1293. doi: 10.1002/slct.201703035. [DOI] [Google Scholar]
- 63.Scharf DH, Heinekamp T, Remme N, Hortschansky P, Brakhage AA, Hertweck C. 2012. Biosynthesis and function of gliotoxin in Aspergillus fumigatus. Appl Microbiol Biotechnol 93:467–472. doi: 10.1007/s00253-011-3689-1. [DOI] [PubMed] [Google Scholar]
- 64.Scharf DH, Remme N, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. 2010. Transannular disulfide formation in gliotoxin biosynthesis and its role in self-resistance of the human pathogen Aspergillus fumigatus. J Am Chem Soc 132:10136–10141. doi: 10.1021/ja103262m. [DOI] [PubMed] [Google Scholar]
- 65.Schrettl M, Carberry S, Kavanagh K, Haas H, Jones GW, O'Brien J, Nolan A, Stephens J, Fenelon O, Doyle S. 2010. Self-protection against gliotoxin–a component of the gliotoxin biosynthetic cluster, GliT, completely protects Aspergillus fumigatus against exogenous gliotoxin. PLoS Pathog 6:e1000952. doi: 10.1371/journal.ppat.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Djonovic S, Pozo MJ, Dangott LJ, Howell CR, Kenerley CM. 2006. Sm1, a proteinaceous elicitor secreted by the biocontrol fungus Trichoderma virens induces plant defense responses and systemic resistance. Mol Plant Microbe Interact 19:838–853. doi: 10.1094/MPMI-19-0838. [DOI] [PubMed] [Google Scholar]
- 67.Zamocky M, Gasselhuber B, Furtmuller PG, Obinger C. 2012. Molecular evolution of hydrogen peroxide degrading enzymes. Arch Biochem Biophys 525:131–144. doi: 10.1016/j.abb.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo Y, Yao S, Yuan T, Wang Y, Zhang D, Tang W. 2019. The spatiotemporal control of KatG2 catalase-peroxidase contributes to the invasiveness of Fusarium graminearum in host plants. Mol Plant Pathol 20:685–700. doi: 10.1111/mpp.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malinich EA, Wang K, Mukherjee PK, Kolomiets M, Kenerley CM. 2019. Differential expression analysis of Trichoderma virens RNA reveals a dynamic transcriptome during colonization of Zea mays roots. BMC Genomics 20:280. doi: 10.1186/s12864-019-5651-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang KD, Gorman Z, Huang PC, Kenerley CM, Kolomiets MV. 2020. Trichoderma virens colonization of maize roots triggers rapid accumulation of 12-oxophytodienoate and two -ketols in leaves as priming agents of induced systemic resistance. Plant Signal Behav 15:1792187. doi: 10.1080/15592324.2020.1792187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mukherjee PK, Horwitz BA, Herrera-Estrella A, Schmoll M, Kenerley CM. 2013. Trichoderma research in the genome era. Annu Rev Phytopathol 51:105–129. doi: 10.1146/annurev-phyto-082712-102353. [DOI] [PubMed] [Google Scholar]
- 72.Kwak Y. 2020. Complete mitochondrial genome of the fungal biocontrol agent Trichoderma atroviride: Genomic features, comparative analysis and insight into the mitochondrial evolution in Trichoderma. Front Microbiol 11:785. doi: 10.3389/fmicb.2020.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Du Buy HG, Riley FL. 1967. Hybridization between the nuclear and kinetoplase DNA’s of Leishmania enriettii and between nuclear and mitochondrila DNA’s of mouse liver. Proc Natl Acad Sci USA 57:790–797. doi: 10.1073/pnas.57.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blanchard JL, Schmidt GW. 1996. Mitochondrial DNA migration events in yeast and humans: integration by a common end-joining mechanism and alternative perspectives on nucleotide substitution patterns. Mol Biol Evol 13:537–548. doi: 10.1093/oxfordjournals.molbev.a025614. [DOI] [PubMed] [Google Scholar]
- 75.Ricchetti M, Fairhead C, Dujon B. 1999. Mitochondrial DNA repairs double-strand breaks in yeast chromosomes. Nature 402:96–100. doi: 10.1038/47076. [DOI] [PubMed] [Google Scholar]
- 76.Yu X, Gabriel A. 1999. Patching broken chromosomes with extranuclear cellular DNA. Mol Cell 4:873–881. doi: 10.1016/S1097-2765(00)80397-4. [DOI] [PubMed] [Google Scholar]
- 77.Ricchetti M, Tekaia F, Dujon B. 2004. Continued colonization of the human genome by mitochondrial DNA. PLoS Biol 2:E273. doi: 10.1371/journal.pbio.0020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang GJ, Dong R, Lan LN, Li SF, Gao WJ, Niu HX. 2020. Nuclear integrants of organellar DNA contribute to genome structure and evolution in plants. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsuji J, Frith MC, Tomii K, Horton P. 2012. Mammalian NUMT insertion is non-random. Nucleic Acids Res 40:9073–9088. doi: 10.1093/nar/gks424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yadav V, Sun S, Coelho MA, Heitman J. 2020. Centromere scission drives chromosome shuffling and reproductive isolation. Proc Natl Acad Sci USA 117:7917–7928. doi: 10.1073/pnas.1918659117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.White M. 1978. Modes of speciation. W. H. Freeman, San Francisco, CA. [Google Scholar]
- 82.Potter S, Bragg JG, Blom MP, Deakin JE, Kirkpatrick M, Eldridge MD, Moritz C. 2017. Chromosomal speciation in the genomics era: disentangling phylogenetic evolution of Rock-wallabies. Front Genet 8:10. doi: 10.3389/fgene.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nutzmann HW, Scazzocchio C, Osbourn A. 2018. Metabolic gene clusters in eukaryotes. Annu Rev Genet 52:159–183. doi: 10.1146/annurev-genet-120417-031237. [DOI] [PubMed] [Google Scholar]
- 84.Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. 2013. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res 41:D358–365. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim FY, Sanchez JF, Wang CC, Keller NP. 2012. Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Methods Enzymol 517:303–324. doi: 10.1016/B978-0-12-404634-4.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ziemert N, Alanjary M, Weber T. 2016. The evolution of genome mining in microbes - a review. Nat Prod Rep 33:988–1005. doi: 10.1039/c6np00025h. [DOI] [PubMed] [Google Scholar]
- 87.Hindra Zhang X, Elliot MA. 2019. Unlocking the trove of metabolic treasures: activating silent biosynthetic gene clusters in bacteria and fungi. Curr Opin Microbiol 51:9–15. doi: 10.1016/j.mib.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 88.Woo TT, Chuang CN, Wang TF. 2021. Budding yeast Rad51: a paradigm for how phosphorylation and intrinsic structural disorder regulate homologous recombination and protein homeostasis. Curr Genet 67:389–396. doi: 10.1007/s00294-020-01151-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. CANU: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 91.Palmer J. 2017. Funannotate: Fungal genome annotation scripts. https://github.com/nextgenusfs/funannotate
- 92.Seppey M, Manni M, Zdobnov EM. 2019. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol Biol 1962:227–245. doi: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- 93.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 94.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 95.Eisenhaber B, Schneider G, Wildpaner M, Eisenhaber F. 2004. A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. J Mol Biol 337:243–253. doi: 10.1016/j.jmb.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 96.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46:W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ausland C, Zheng J, Yi H, Yang B, Li T, Feng X, Zheng B, Yin Y. 2021. dbCAN-PUL: a database of experimentally characterized CAZyme gene clusters and their substrates. Nucleic Acids Res 49:D523–D528. doi: 10.1093/nar/gkaa742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shih PY, Hsieh BY, Lin MH, Huang TN, Tsai CY, Pong WL, Lee SP, Hsueh YP. 2020. CTTNBP2 controls synaptic expression of zinc-related autism-associated proteins and regulates synapse formation and autism-like behaviors. Cell Rep 31:107700. doi: 10.1016/j.celrep.2020.107700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM00663-21_Supp_1_seq7.pdf, PDF file, 3.5 MB (3.6MB, pdf)
Data Availability Statement
The complete genome sequences, annotation, and raw data sets have been deposited in the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA700774 (Table S1). All study data are included in the article as well as in SI and SD appendixes. All genomic databases data and 15 supplemental data sets are also available at the following websites: https://github.com/tfwangasimb/Trichoderma-biocontrol/releases.