

Abstract
Somatic mutations in TET2 are common in myelodysplastic syndromes (MDS), myeloproliferative, and overlap syndromes. TET2 mutant (TET2MT) clones are also found in asymptomatic elderly individuals, a condition referred to as clonal hematopoiesis of indeterminate potential (CHIP). In various entities of TET2MT neoplasia, we examined the phenotype in relation to the strata of TET2 hits within the clonal hierarchy. Using deep sequencing, 1781 mutations were found in 1205 of 4930 patients; 40% of mutant cases were biallelic. Hierarchical analysis revealed that of TET2MT cases >40% were ancestral, e.g., representing 8% of MDS. Higher (earlier) TET2 lesion rank within the clonal hierarchy (greater clonal burden) was associated with impaired survival. Moreover, MDS driven by ancestral TET2MT is likely derived from TET2MT CHIP with a penetrance of ~1%. Following ancestral TET2 mutations, individual disease course is determined by secondary hits. Using multidimensional analyses, we demonstrate how hits following the TET2 founder defect induces phenotypic shifts toward dysplasia, myeloproliferation, or progression to AML. In summary, TET2MT CHIP-derived MDS is a subclass of MDS that is distinct from de novo disease.
Introduction
TET2 is one of the most commonly mutated genes in myeloid neoplasia [1–3] and also occurs at lower frequencies in some forms of T cell lymphoma [4]. TET2 mutations (TET2MT) have been detected in seemingly asymptomatic older controls, also referred as to having clonal hematopoiesis of indeterminate potential (CHIP). Their presence in these individuals is associated with a higher risk of developing a hematologic neoplasm [5–8]. TET2MT are also encountered in clonal cytopenias of undetermined significance (CCUS), a proportion of these cases likely representing early, subclinical MDS [9].
The TET2 gene product is a Fe2+-dependent dioxygenase, which uses electrons gained from vitamin C and α-ketoglutarate (aKG) decarboxylation to split O2 to hydroxylate 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). This reaction may lead to both passive indirect demethylation (whereby 5hmC prevents methylation of the newly synthesized strand during replication) and active demethylation via further TET2-mediated 5hmC oxidation followed by removal of the oxidized base by base excision repair [10, 11]. TET2MT are mainly loss-of-function mutations. The loss of hydroxymethylation skews myeloid differentiation toward monocytes, and, as expected, increases 5mC levels [12–15]. Tet2−/− mice develop fatal myeloproliferative disease and lymphomas. These conditions arise later in Tet2+/− mice suggesting a delayed impact of haploinsufficiency leading to acquisition of secondary hits [16, 17].
Prognostic impact of TET2MT has not been reproducibly demonstrated. Various groups have reported no impact on OS in MDS [18–20], while others have found TET2MT to be associated with an unfavorable prognosis [21–24]. An earlier study focused on coexisting JAK2MT and TET2MT in MPN has shown that the acquisition order of mutations influences not only the clinical phenotype, but also response to targeted therapy [25]. It is possible that the heterogeneity of TET2MT, their configuration, subclonal context, and co-associated variables result in biological heterogeneity that precludes the ability to establish precise phenotype–genotype associations.
We hypothesized that some of these aforementioned relationships can be clarified by studying cases in which TET2 hits are founder lesions. These cases appear to be derived from CHIP with TET2MT and differ from other CHIP-derived entities and de novo MDS. Specific mutations preceding or following TET2 in the clonal hierarchy can create signature phenotypic shifts that drive disease. Ancestral TET2 hits appear to predispose to a certain spectrum of secondary hits, which in turn imprint further clinical features, including myeloproliferative vs. dysplastic features or rates of progression. To address these questions, we analyzed a cohort of patients with myeloid neoplasms using whole exome and targeted deep sequencing of a panel of 22 genes most frequently affected by somatic mutations in myeloid neoplasms [26–28].
Methods
Patient samples
Blood and bone marrow samples were collected from patients at the Cleveland Clinic and the Munich Leukemia Laboratory with myeloid neoplasms (MN) according to protocols approved by the Cleveland Clinic IRB, and the Declaration of Helsinki. Germ line DNA was obtained from CD3+ lymphocytes. Sample which yielded low sequencing quality due to low depth were excluded from the study.
Next-generation sequencing (NGS)
Whole-exome libraries were prepared according to the Nextera Rapid Capture Exome protocol (Illumina, San Diego, CA) and subjected to massive parallel sequencing using the HiSeq 2000. Average coverage was ×115 and only variants with variant allele frequency (VAF) >5% were used. Multi-amplicon targeted deep sequencing was performed for a panel of 22 genes most commonly somatically mutated in MDS [26–28] (Supplementary Table 1). Paired-end libraries were generated and deep sequenced on MiSeq (Illumina, San Diego, CA) sequencers according to Illumina protocols. Average coverage was ×250. Variants were extracted using the GATK3.3 pipeline and best practices. TET2MT found in the CD3− fraction but not (or highly diminished frequencies) in the germ line CD3+ fraction were deemed somatic mutations and included in our analysis. We expect sequence alterations found in both the myeloid and lymphoid cells with equal VAF to be germ line and excluded from our study. Previously, usage of T cells as germ line [13, 29] resulted in similar frequencies of TET2MT compared to skin or buccal swabs [27, 30].
Distinction of ancestral and subclonal mutations
To identify the ancestral and secondary mutations for each patient, VAFs were serially analyzed in a subcohort of patients (N = 40; Supplementary Fig. 1). Mutations appearing during clinical course but absent at initial presentation were deemed subclonal, while ancestral mutations were detected at all time points. Evolution followed the expected order (including contractions or expansions) with the exception of ambivalent results for VAFs within ±5%. When serial samples were not available, VAFs of mutations (adjusted for copy number and zygosity) were ranked and assigned as ancestral for first or dominant hits, and secondary for any subsequent subclonal hit. Acknowledging resolution limitations we used a cutoff of at least a 5% difference between VAFs to identify ancestral mutations. If the difference in VAFs between two mutations was <5%, for the purpose of this study, we referred to them as codominant.
In cases with multiple TET2MT, x–y plots of VAFs were generated to assess the probability that they are strictly biallelic or may also be biclonal or subclonal (Supplementary Fig. 2). In either case, higher VAFs indicate earlier events (including possibly an ancestral hit, if other higher hierarchal events are absent).
Calculation of TET2 mutant CHIP penetrance and TET2-derived CHIP fraction
A meta-analysis within large CHIP cohorts was performed to determine the prevalence of TET2MT CHIP [5, 6, 31–34]. For meta-analysis, cases were excluded when hematologic neoplasms were present before sampling, or clinical and molecular data were unavailable. The penetrance of TET2MT MDS derived from TET2MT CHIP was recapitulated according to the frequencies of ancestral TET2 hits in the MDS cohort. Supplementary Table 2 summarizes the meta-analysis. For the purpose of this study, CHIP was defined as the presence a somatic mutation with VAF ≥2% in an otherwise asymptomatic individual. Patients with unexplained persistent cytopenias, lack of dysplasia, and the absence of an MDS-associated somatic mutation are considered to be idiopathic cytopenias of undetermined significance (ICUS). Clonal cytopenias of undetermined significance (CCUS) describes a condition where somatic mutations are associated with cytopenias, but lack dysplasia [35].
Analysis of genotype/phenotype relationships
Wilcoxon tests were performed for pairwise continuous variable comparisons, Fisher’s exact test was used to compare proportions, and log-rank tests were used to compare survival times. All p values were two-sided and values less than 0.05 were considered statistically significant. All analyses were performed using the statistical computing environment R (3.4.3).
Three-dimensional plots
Odds ratios (OR) of phenotypes based on mutational status were calculated using 2 × 2 contingency tables, and plotted on an x–y–z plane to illustrate the trifold pheno–morphologic relationships. Confidence intervals of OR vs. the OR of patients with only a TET2 and no other mutations were deemed separate if they were non-overlapping. Phenotype definitions for the classification of dysplasia and cytopenia can be found in Supplementary Table 3.
Results
Patient characteristics
A total of 4930 patients with myeloid neoplasia were included (Supplementary Table 4) and underwent sequencing. TET2 was the most commonly mutated gene. Following sequencing of all-coding regions, we mapped and classified all somatic TET2MT according to their position and type. A total of 1781 somatic TET2MT were identified in 1205 patients (Table 1). Most were truncating: 47% were frame shifts, 34% were nonsense, and 19% were missense. Although the truncating mutations were widely dispersed across the gene, 89% of the missense mutations were located in the catalytic domain, which spans base pairs (bp) 1129–1936.
Table 1.
Distribution of TET2 mutations in myeloid neoplasms.
| TET2 MT | TET2 WT | ||
|---|---|---|---|
| Total | 1205 | 3725 | |
| DX | |||
| MDS | 247 | 1192 | p < .001 |
| MDS/MPN | 432 | 511 | p < .001 |
| MPN | 75 | 333 | p = .004 |
| pAML | 377 | 1436 | p < .001 |
| sAML | 58 | 188 | |
| t-MN | 16 | 65 | |
| Gender = M | 589 | 782 | |
| Age | |||
| Under 30 | 5 | 43 | p < .001 |
| 30–39 | 13 | 50 | p < .001 |
| 40–49 | 25 | 110 | p < .001 |
| 50–59 | 72 | 203 | p < .001 |
| 60–69 | 243 | 354 | |
| 70–79 | 409 | 377 | p < .001 |
| 80–89 | 156 | 133 | p < .001 |
| 90+ | 10 | 1 | p < .001 |
| Cytogenetics | |||
| Normal | 643 | 579 | p < .001 |
| Complex | 29 | 159 | p = .004 |
| 5q− | 26 | 70 | |
| Del 20 | 12 | 46 | |
| Del 7 | 22 | 45 | |
| Del Y | 31 | 21 | p < .001 |
| Trisomy 8 | 43 | 48 | p < .001 |
| Other | 123 | 229 | p < .001 |
The prevalence of TET2MT increased with patient age (Fig. 1a, R = 0.878, p < 0.0001), irrespective of histologic subtype and mutation type (missense, frame shift, and nonsense; Supplementary Fig. 3A). Among co-occurring mutations, JAK2 had the strongest correlation with increasing age (R2 = 0.924), followed by SRSF2 (R2 = 0.906) and ASXL1 (R2 = 0.867). Secondary (R2 = 0.876) and ancestral (R2 = 0.726) TET2MT correlated with an increase in age (Supplementary Fig. 3B). Of TET2MT patients, 43% harbored more than one TET2MT and 12% were either homo- or hemizygous (Fig. 1b). Of these, 65% had two truncating mutations, while the remaining cases showed a missense and truncation combination. Those with >1 TET2MT and low VAF could be either biallelic or biclonal, a distinction that is difficult to make using VAF. Plotting the VAFs of each TET2MT in patients with multiple TET2MT, on an x–y plot (individual VAF on each axis; Supplementary Fig. 2) yielded two populations, one clearly biallelic TET2MT (75% of double mutants) and the other either biallelic or biclonal TET2MT (25% of double mutants). In either case, the higher-ranked TET2MT may represent a founder event assuming that no earlier events exist in other genes.
Fig. 1.
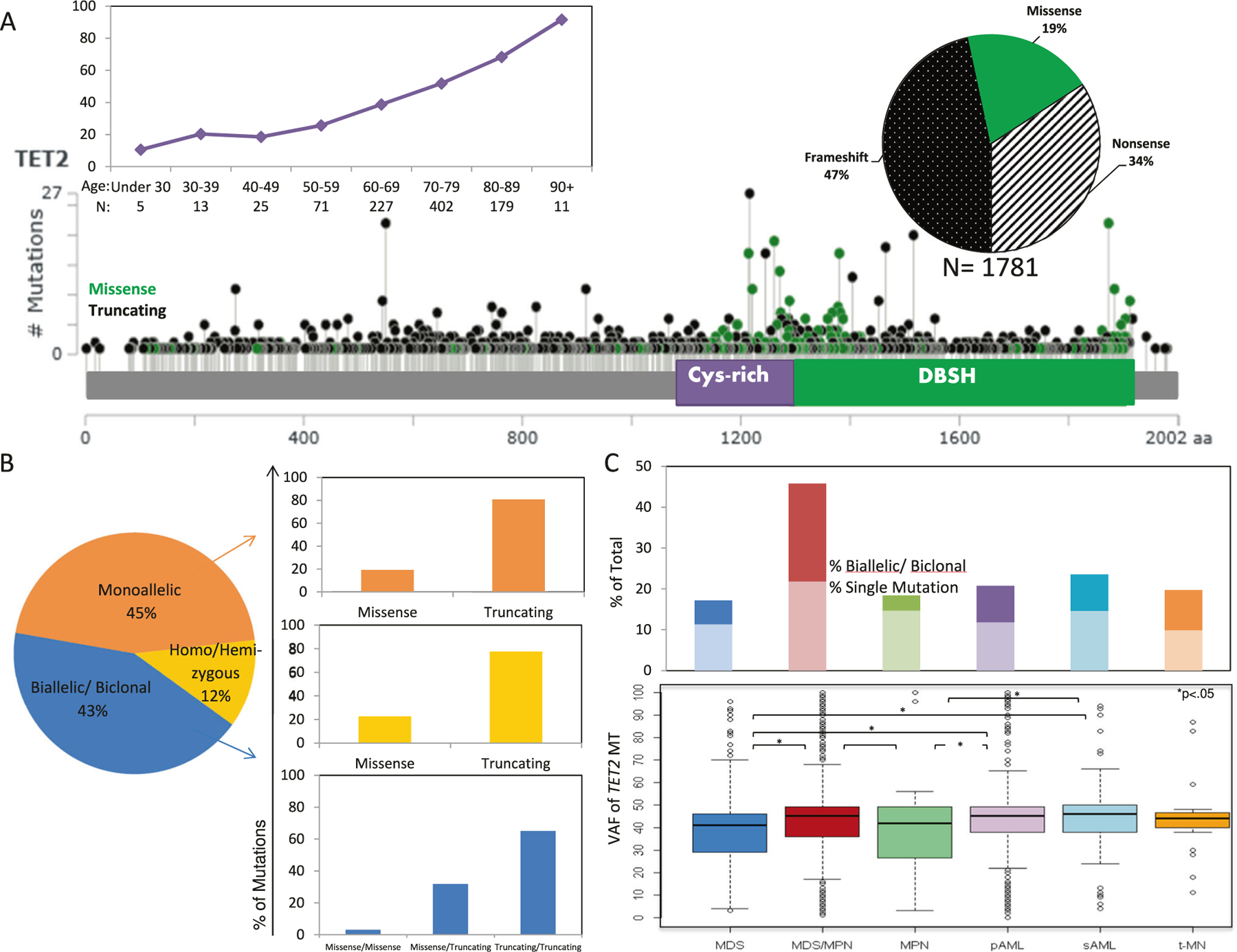
Topology and demographics of TET2 mutations in myeloid neoplasms. a Schematic drawing of TET2 gene showing location, distribution, types of mutations, and age-related increases in the number of mutations. For details of mutation and disease subtypes, see Supplementary Fig. 1. b Distribution of number and type of TET2MT across the spectrum TET2MT. c The frequencies of single and multiple mutations in each disease subtype and the distribution of mutant VAF by MDS subtype
TET2MT were found in 17% of patients with MDS, 46% of MDS/myeloproliferative neoplasms (MDS/MPN), 19% of MPN, 21% of primary acute myeloid leukemia (pAML), 24% of secondary AML (sAML), and 20% of treatment-related MN (t-MN) patients. In general, single mutations were more common than multiple mutations, except in MDS/MPN, where there was a similar proportion of single and multiple mutant cases. No differences in TET2MT clonal burden were found among disease subtypes (Fig. 1c).
Genotypic context of TET2 mutations
TET2MT may occur in association with distinct mutational spectra (Fig. 2a). Overall, TET2MT most often co-occurred with another mutation in TET2 (43%) and with mutations in ASXL1 (21%), SRSF2 (18%), and NPM1 (13%, Fig. 2b). Patients with multiple TET2MT harbored, on average, more alterations than those with one or TET2 wild type (TET2WT; Fig. 2c). By disease subtype, in MDS patients, TET2MT most often co-occurred with another TET2MT, ASXL1, SF3B1, and SRSF2. MDS patients with TET2MT more frequently had mutations in ASXL1 (22% vs. 10%, p < .001), SRSF2 (20% vs. 9%, p < .001), and RUNX1 (14%, vs. 6%, p = .003), compared to those with TET2WT (Fig. 2d). In MDS/MPN, TET2MT most often coincided with another TET2, or SRSF2, ASXL1, RUNX1, and CBL. SRSF2 mutations were more common in TET2MT vs. TET2WT MDS/ MPN (52% vs. 14%, p < .001) as were ASXL1 mutations (36% vs. 14%, p < .001). In MPN, the most common coexisting mutations were JAK2, ASXL1, and SRSF2; while double TET2MT were less frequent. Subclonal lesions of NPM1 and DNMT3A were predominant in pAML, while the lesions seen in sAML were more similar to that of MDS. TET2MT were significantly associated with normal cytogenetics, deletion Y, and trisomy 8, while TETWT were associated with more complex karyotypes (Table 1).
Fig. 2.
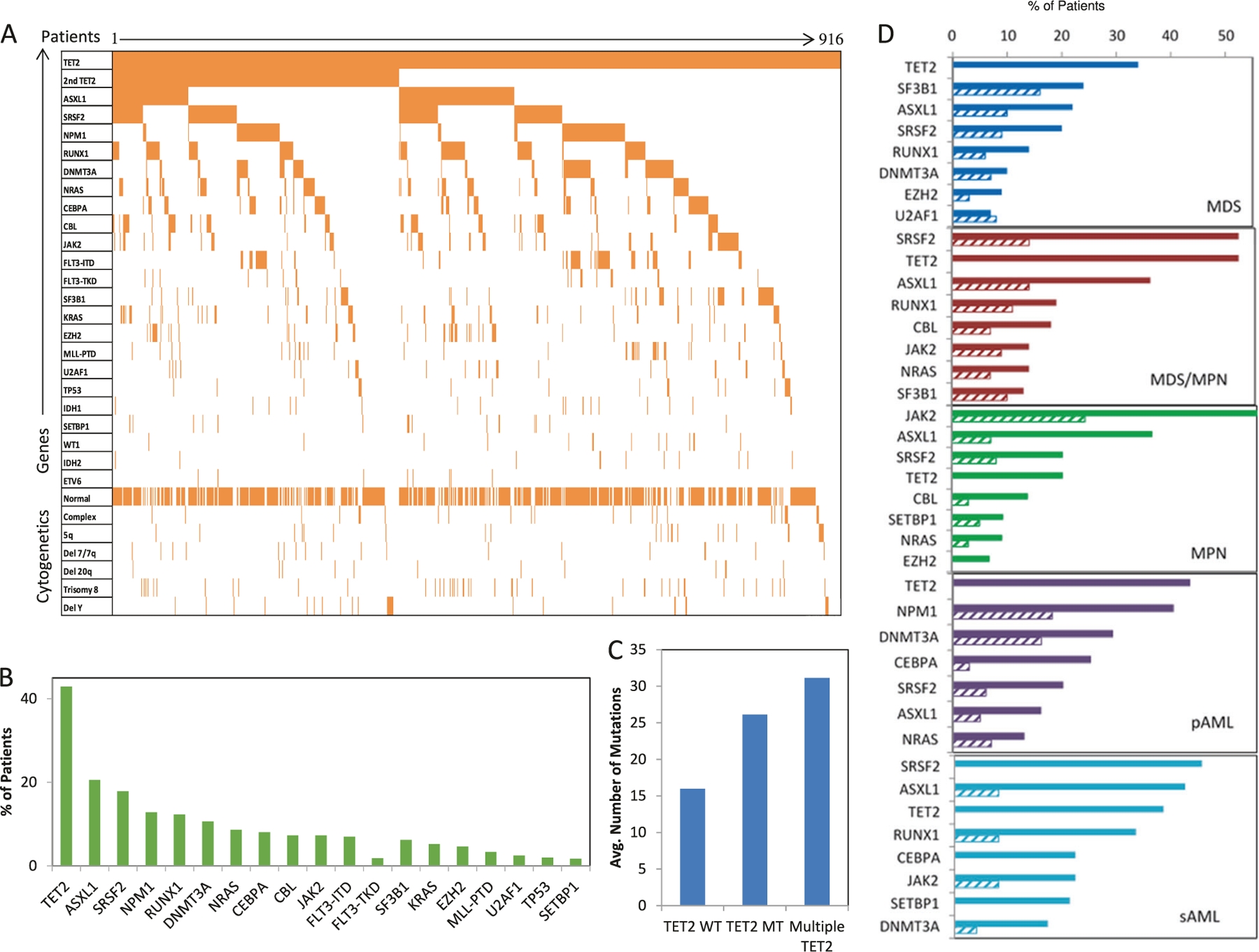
Clonal architecture of TET2 mutants. a Co-occurring mutations in TET2MT patients. b Frequency of somatic mutations co-occurring with TET2MT. c The average number of mutations of patients without a TET2MT, a single TET2MT, or double TET2MT. d Mutational profiles of TET2MT (solid bars) and TET2WT (hashed bars) within disease subtype
Molecular implications from clonal architecture
The position of TET2MT in the clonal hierarchy can be inferred by ranking heterogeneous somatic events. Where serial samples are not available, the position can be inferred from a cross-sectional analysis based on VAF (Fig. 3a). The results indicate that TET2MT are first hits (dominant clones) in 40% of TET2MT cases and later hits (subclonal events) in other cases. Furthermore, a subclonal TET2MT can follow an ancestral TET2 hit or be subclonal to other mutations in a linear or branching fashion. When TET2 is the first hit, the most common second mutation is another TET2 lesion, followed by SRSF2, ASXL1, DNMT3A, and SF3B1 mutations. When TET2 is subclonal, the dominant antecedent clone is defined by the presence of SRSF2, EZH2, ASXL1, DNMT3A, or CEBPA mutations (Fig. 3b). A significantly greater number of pAML patients had ancestral TET2 lesions than sAML and were also associated with abnormal cytogenetics and NPM1 mutations (Supplementary Fig. 4). This observation could be explained by the older age of our cohort as compared to TCGA AML patients (69 vs. 55 years in TCGA). There was no significant difference in the frequency of FLT3ITD between primary and secondary AML. Secondary TET2 hits coincided with ASXL1, DNMT3A, EZH2, JAK2, and RUNX1 mutations. When subgrouped according to “class-defining” ancestral hits (TET2, SRSF2, SF3B1, etc.), patients with ancestral frame shift TET2MT harbored the highest numbers of additional subclonal alterations (Fig. 3c).
Fig. 3.
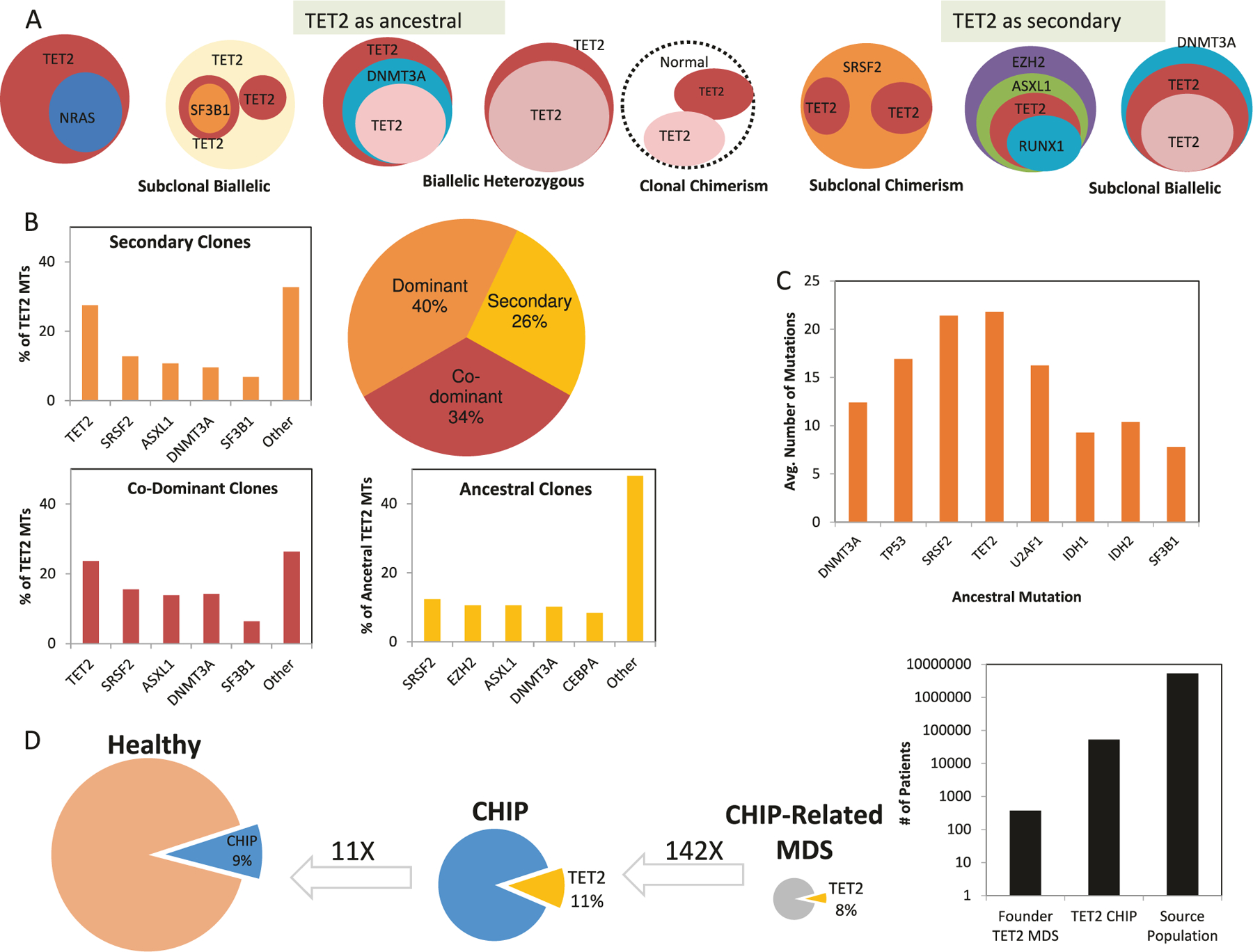
Clonal hierarchy of TET2 mutations. a Cross-sectional analysis of patient samples to identify clonal hierarchy of TET2 (second sphere to the left top row represents a patient with three TET2 mutations, including an ancestral and two different subclonal hits). b Distribution of TET2MT patients based on the position of TET2MT within the clonal hierarchy, and the frequency of other mutations throughout the clonal hierarchy. c The average number of mutations of MDS, MPN, and AML patients with ancestral hits of various genes. d Meta-analysis to show the frequency of TET2MT CHIP and CHIP-related MDS. Values above arrows are reverse direction multipliers of percentages of individuals
Demographic and pathogenic relationship between TET2MT CHIP and MDS
Meta-analysis of six major CHIP studies revealed that 9% of healthy individuals have CHIP (4470/49290). It was found that 11–15% of CHIP is due to TET2 hits (513/4470, Fig. 3d and Supplementary Table 2). We estimate that in turn <1% of TET2MT CHIP evolves to MDS (0.7%, 2/277), but the low number of events available precludes precise estimates. Conversely, we show 8% of MDS are initiated by TET2 hits and thus likely derive from antecedent TET2-mutated CHIP. From the 373 ancestral TET2MT MDS cases in our cohort, we can estimate that we “need” 53,285 individuals with TET2MT CHIP to account for the cases diagnosed and calculate the evolution rate. These calculations allowed us to conclude that while most of TET2MT CHIP is not “productive,” all of the ancestral TET2MT MDS cases are likely CHIP derived rather than de novo cases. The mean age of our cohort was 67, while it was found to be 59 for the individuals of the meta-analysis. Consequently, it is more likely that we are underestimating the number of CHIP cases because the population is younger than ours, providing further evidence that nearly all MDS with ancestral TET2MT are CHIP derived. Of note, is that our study primarily focused on TET2MT CHIP and other CHIP-associated mutations were not further investigated.
Impact of subclonal lesions on phenotype and disease progression in TET2MT-initiated cases
The odds of a molecular lesion being associated with proliferation vs. dysplasia, and low vs. high-risk disease (based on blasts >5% and IPSS) can be used to separate secondary mutations acquired after a founder TET2MT in MDS and MDS/MPN patients (Fig. 4a). Such three-dimensional plots illustrate how hits secondary to those in TET2 bias the rate of progression (high- vs. low-risk disease) and phenotype (dysplastic vs. proliferative features). Patients with only an ancestral TET2MT tend to be low-risk MPN. Secondary hits following an ancestral TET2 lesion alter disease trajectories in distinct manners, pushing them toward different phenotypes and progression rates; e.g., a second (biallelic) TET2 hit increases the “MDS-like” character of the disease. A secondary SRSF2 mutation greatly increases a patient’s progression risk, and retained its impact in a multivariate analysis that adjusted for IPSS (Supplementary Table 5). When mutational profiles of TET2MT were correlated with cytopenias, patients with exclusive TET2MT tended to be thrombocytopenic, while the addition of a U2AF1 mutation increased odds of the anemia phenotype (Fig. 4b). Anemia and leukopenia were associated with secondary SRSF2 and KRAS mutations. A similar analysis was done for lineage dysplasia; we found subclonal ASXL1 mutations to be associated with myeloid, rather than erythroid or megakaryocytic dysplasia (Fig. 4c). We also investigated how phenotypic features change when TET2 is secondary to ancestral hits affecting other genes. The latter originated from different starting points within the phenotypic continuum, and subclonal TET2 mutations may further redirect the founder phenotype. For example, when we examined this scenario with ancestral ASXL1 and secondary TET2 hit (Supplementary Fig. 5A vs. Fig. 4a), we found that sub-clonal TET2 hits increase the propensity to progression. The opposite succession (TET2 preceding ASXL1) was not associated with the risk of evolution. Similarly, when we studied the effects of subclonal TET2 hits on SRSF2-initiated disease, we found a shift toward anemia and less prominent leukopenia more than in TET2-initated disease with a secondary SRSF2 mutation (Supplementary Fig. 5B vs. Fig. 4b). SRSF2-initiated disease with a secondary TET2MT had greater myeloid and less erythroid dysplasia than the reciprocal scenario (Supplementary Fig. 5C vs. Fig. 4c).
Fig. 4.
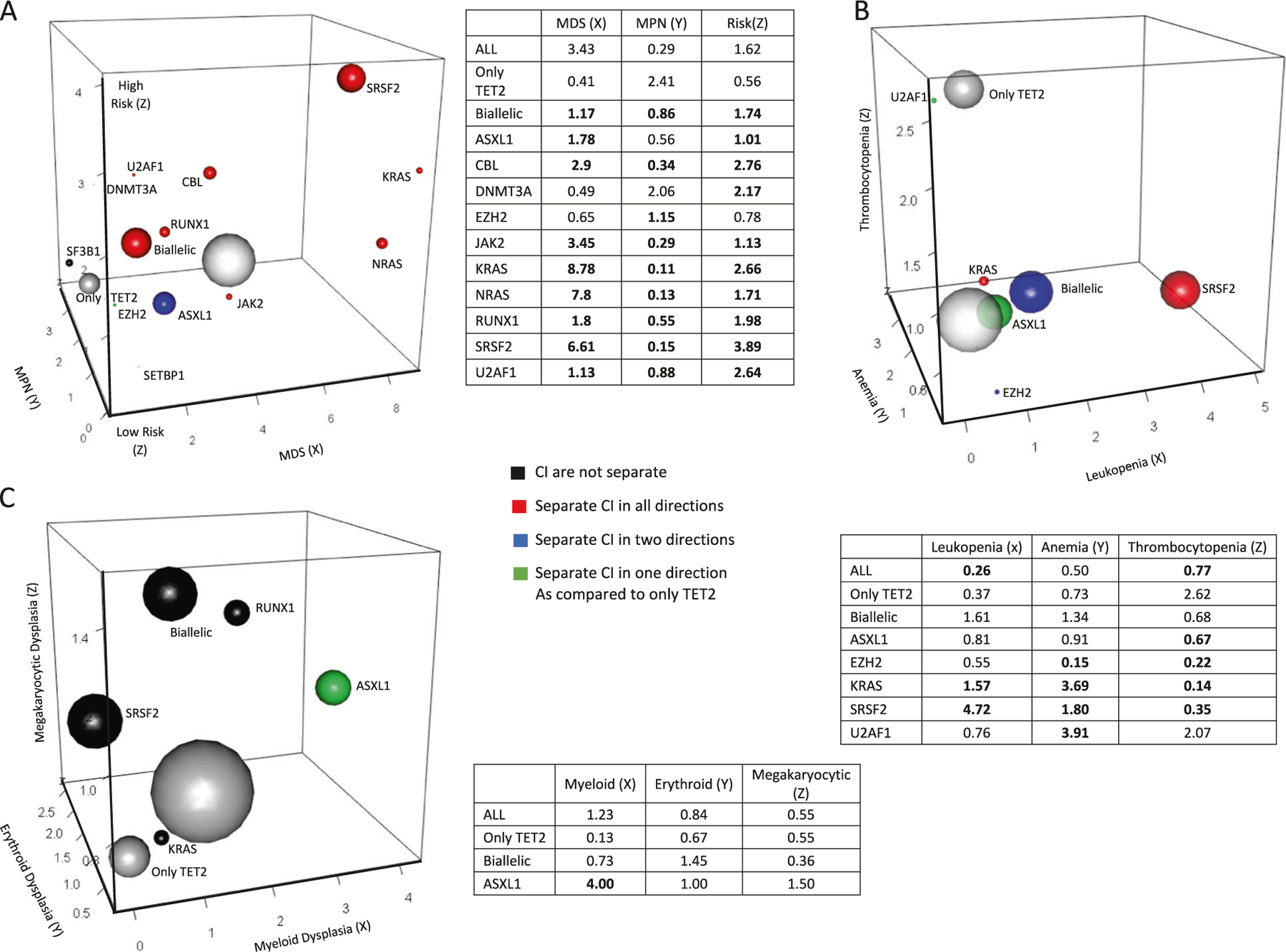
Secondary hits of TET2 mutants. a Associations between disease phenotypes and mutation rates are quantified by the odds ratios, MDS (X) vs. MPN (Y), and high risk vs. low risk (Z). Mutations showing significant enrichment in comparison to patients with only a TET2MT (shown in smaller gray ball) are indicated by color according to OR 95% CI limits. Red color indicates separation of CI in all directions, blue indicates separation in two of the three directions, green indicates separation in a single direction, black indicates no separation. The sizes of the spheres are proportional to the frequency of the mutation in our cohort. The largest white ball is the total cohort of all ancestral TET2MT carriers combined. Tables shown provide odds ratio point estimates for each associated plot. b. Associations between phenotypes and mutation rates are quantified by odds ratios, leukopenia (X), anemia (Y), and thrombocytopenia (Z). c. Associations between phenotypes and mutation rates are quantified by odds ratios, myeloid dysplasia (X), erythroid dysplasia (Y), and megakaryoctye dysplasia (Z). Definitions for classification of dysplasia types and cytopenias are provided in Supplementary Table 2
Overall, TET2MT had no impact on survival (Supplementary Fig. 6A–D). When the size of the TET2MT clone was considered, survival was worse in patients with larger clones (p = .014; Supplementary Fig. 7A), yet there was no statistical survival difference between patients with ancestral vs. secondary TET2MT. Focusing on disease subgroups, MPN and sAML patients with ancestral TET2 hits showed a trend toward worse survival (not shown). When patients were grouped by genetic configuration of TET2MT, those with hemizygous and homozygous mutations had significantly poorer survival (p = .008; Supplementary Fig. 7B).
We also examined the impact of additional mutations on survival in TET2MT cases (Supplementary Fig. 8). U2AF1, TP53, and SRSF2 mutants have significantly higher hazard ratios, but in combinations with a TET2MT, their hazard ratio decreases. TET2MT with EZH2 mutations had a significantly higher hazard ratio; the same was seen in TET2WT patients.
Discussion
While the immediate biochemical consequences of TET2MT are known [4, 10, 12, 36–39] their downstream proleukemogenic impact on disease evolution remains elusive. Apart from its biochemical function and role in passive demethylation, our speculative view is that other TET2 functions (and their deficiency) may be important, such as its oxygen-sensing function, modification of double-stranded RNA, and DNA repair. Irrespective of these activities, TET2 is a tumor suppressor gene because it limits both the number of hematopoietic stem cells (HSC, i.e., target cells of MDS and MDS/MPN) and also likely the rate at which they mutate per cell [17]. Nevertheless, the shared prevalence of TET2 defects implies their “general” pathogenic importance and propensity for leukemogenesis. The lack of mutations in highly homologous TET1/TET3 indicates a distinct pathophysiologic role of this specific gene likely due to differences in tissue-specific expression [2, 40].
To date, only a modest impact of TET2MT on clinical outcomes has been described, and its effect on AML progression in MDS has been found to be rather neutral [18–23, 40, 41]. We and others have demonstrated a favorable association of TET2MT with responsiveness to hypomethylating agents; however, this finding has not been uniformly reproduced, likely due to the diverse impact of co-associated events [22, 42, 43]. Larger treatment groups will be needed to appropriately taxonomize such heterogeneity.
The diversity of TET2-associated phenotypes is high. This might be due to the heterogeneity of TET2 lesions, including their intragenic topology, allelic configuration, and position within the clonal hierarchy and combinations of other mutations with which they tend to coincide. For instance, it might reflect a generic role of TET2 loss in the expansion of HSC, perhaps enabling them to exist in a more oxic/DNA-damaging environment prone to acquisition of secondary hits.
Our study of TET2MT included enough patients to perform subset analyses and therefore answers some outstanding questions regarding the role of TET2 in correlation to morphologic phenotypes. Although using VAF rankings to decipher clonal architecture is not ideal, we have various lines of computational and analytic confirmation of our method allowing it to apply in principle to the large number of samples included in our study. Our results indicate that most TET2MT represent phenotype-neutral ubiquitous ancestral hits, which seem to create a leukemogenic pre-disposition (mutator phenotype) rather than leukemic drive as evidenced by the lack of impact on progression and a higher total number of subclonal mutational events. The presumed mutator phenotype is consistent with results of murine studies showing accumulation of secondary hits during evolution of Tet2KD-mediated disease [44]. We also show that phenotypic features in mutant cases are determined by pairings with secondary events that are not entirely random. Analysis of mutational events in cases with ancestral TET2MT indicate a higher number of subsequent subclonal events than with other ancestral events, including hits in SF3B1, SRSF2, DNMT3A, TP53, U2AF1, or IDH1/2 as previously described [18, 45]. This effect might arise by increases of absolute numbers of target cells, by increases in mutation rates per cell, or both. In Tet2KD/KO mouse models, increases in numbers of HSC (target cells) [14] and elevated mutation rates have been described [17]; the latter was confirmed in human samples using whole-exome sequencing [44].
TET2MT also increase with age in CHIP [5] and CHIP is associated with an increased risk of developing hematologic neoplasms [6]. To that end myeloid neoplasia, especially MDS that are characterized by ancestral TET2 hits are likely to have developed from previous CHIP. A higher frequency of CHIP mutations, such as TET2, are seen in people over the age of 60 years old explaining that more ancestral TET2MT were found in pAML than sAML likely due to underrepresentation of CBF AML occurring in younger age [34]. Indeed, our pAML cohort is older than previously reported (69 vs. 55 years, p < 0.001) [46].
We demonstrated that TET2MT can also arise, albeit less frequently, as secondary events. TET2 hits tend to accumulate, with second hits resulting in biallelic mutations, hemizygous deletions, or uniparental disomy with homozygous mutations. Patients with secondary TET2MT, with the exception of those with ancestral events observed in CHIP (chiefly, DNMT3A or ASXL1), are not CHIP-related and have founder hits with higher pathogenicity and likely faster progression.
The secondary hits following ancestral TET2MT are not entirely random; the probabilities of different secondary hits differ following TET2 vs. other founder mutations and they result in differences in the phenotype they generate (dysplastic vs. proliferative disease phenotypes) and their propensity to progress. For instance, TET2MT show predilection for biallelic mutations, hemizygous deletions involving the TET2 locus, or somatic uniparental disomy [47]. Other secondary mutations also show a pattern with certain secondary hits overrepresented compared to neoplasia that was initiated by other founder mutations. Such secondary hits affect phenotypic features. For example, SRSF2 and K/NRAS secondary hits are common in CMML and thus lead to development of MDS/MPN overlap syndromes. Similarly, ASXL1, EZH2, and SF3B1 secondary hits are common in MDS, and JAK2 V617F secondary hits are common in MPN. Other TET2MT associations include RUNX1 and EZH2 and its mutual exclusivity with del5q in MDS or DNMT3A and NPM1 mutations negatively correlating with IDH1/2 mutations in AML. Some secondary hits (e.g., in SRSF2 and CBL) impact the pace of progression and therefore survival [48, 49]. Some secondary hits such as ASXL1, EZH2, and TP53 following ancestral TET2MT were surprisingly not associated with advanced disease, but, as expected showed a negative impact on survival [27, 50–56]. This finding indicates that high-risk lesions can be found early in the disease course.
In summary, our results demonstrate how originally phenotype-neutral founder TET2MT are followed by hits resulting in modifications of clone morphology, type of lineage production defect, myeloproliferative features, or rates of progression to AML.
Supplementary Material
Acknowledgements
This work was supported by grants R01HL118281, R01HL123904, R01HL132071, R35HL135795, and Edward P. Evans Foundation.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1038/s41375–018-0150–9) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Eng J Med 2009;360:2289–301. [DOI] [PubMed] [Google Scholar]
- 2.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 2009;41:838–42. [DOI] [PubMed] [Google Scholar]
- 3.Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013; 122:4021–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011;20:25–38. [DOI] [PubMed] [Google Scholar]
- 5.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012;44:1179–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Eng J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mason CC, Khorashad JS, Tantravahi SK, Kelley TW, Zabriskie MS, Yan D, et al. Age-related mutations and chronic myelomonocytic leukemia. Leukemia 2016;30:906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep 2015; 10:1239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwok B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015;126:2355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahfoudhi E, Talhaoui I, Cabagnols X, Della Valle V, Secardin L, Rameau P, et al. TET2-mediated 5-hydroxymethylcytosine induces genetic instability and mutagenesis. DNA Repair 2016;43:78–88. [DOI] [PubMed] [Google Scholar]
- 11.An J, Gonzalez-Avalos E, Chawla A, Jeong M, Lopez-Moyado IF, Li W, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun 2015;6:10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010;468:839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009;113:6403–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci 2014;105: 1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011;118:4509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan F, Wingo TS, Zhao Z, Gao R, Makishima H, Qu G, et al. Tet2 loss leads to hypermutagenicity in haematopoietic stem/progenitor cells. Nat Commun 2017;8:15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin TL, Nagata Y, Kao HW, Sanada M, Okuno Y, Huang CF, et al. Clonal leukemic evolution in myelodysplastic syndromes with TET2 and IDH1/2 mutations. Haematologica 2014; 99:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 2009;114:3285–91. [DOI] [PubMed] [Google Scholar]
- 20.Smith AE, Mohamedali AM, Kulasekararaj A, Lim Z, Gaken J, Lea NC, et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 2010;116:3923–32. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Zhang G, Yi Y, Xiao L, Pei M, Liu S, et al. Decreased 5-hydroxymethylcytosine levels are associated with TET2 mutation and unfavorable overall survival in myelodysplastic syndromes. Leuk Lymphoma 2013;54:2466–73. [DOI] [PubMed] [Google Scholar]
- 22.Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014;124:2705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009;94: 1676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim M, Yahng SA, Kwon A, Park J, Jeon YW, Yoon JH, et al. Mutation in TET2 or TP53 predicts poor survival in patients with myelodysplastic syndrome receiving hypomethylating treatment or stem cell transplantation. Bone Marrow Transplant 2015; 50:1132–4. [DOI] [PubMed] [Google Scholar]
- 25.Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of mutation order on myeloproliferative neoplasms. N Eng J Med 2015;372:601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014;28:241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Eng J Med 2011;364:2496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013;122: 3616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langemeijer SM, Aslanyan MG, Jansen JH. TET proteins in malignant hematopoiesis. Cell Cycle 2009;8:4044–8. [DOI] [PubMed] [Google Scholar]
- 30.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 2013;27:1275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lepine G, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017;130:753–62. [DOI] [PubMed] [Google Scholar]
- 32.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017;130:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell 2017;21:374–82 e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Eng J Med 2014;371: 2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahn JS, Kim HJ, Kim YK, Jung SH, Yang DH, Lee JJ, et al. Adverse prognostic effect of homozygous TET2 mutation on the relapse risk of acute myeloid leukemia in patients of normal karyotype. Haematologica 2015;100:e351–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011;8:200–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 2011;118:2551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev 2015;29:910–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet 2017;49:204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol 2014;32:2691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel BJ, Przychodzen B, Thota S, Radivoyevitch T, Visconte V, Kuzmanovic T, et al. Genomic determinants of chronic myelomonocytic leukemia. Leukemia 2017;31:2815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014;28:78–87. [DOI] [PubMed] [Google Scholar]
- 44.Hirsch CM, Przychodzen BP, Radivoyevitch T, Patel B, Thota S, Clemente MJ, et al. Molecular features of early onset adult myelodysplastic syndrome. Haematologica 2017;102:1028–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012;120:3080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Eng J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patnaik MM, Zahid MF, Lasho TL, Finke C, Ketterling RL, Gangat N, et al. Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J 2016;6:e472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kao HW, Sanada M, Liang DC, Lai CL, Lee EH, Kuo MC, et al. A high occurrence of acquisition and/or expansion of C-CBL mutant clones in the progression of high-risk myelodysplastic syndrome to acute myeloid leukemia. Neoplasia 2011;13:1035–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makishima H, Jankowska AM, McDevitt MA, O’Keefe C, Dujardin S, Cazzolli H, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood 2011;117:e198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou WC, Huang HH, Hou HA, Chen CY, Tang JL, Yao M, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010;116:4086–94. [DOI] [PubMed] [Google Scholar]
- 51.Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica 2012;97:388–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin I, Such E, Navarro B, Villamon E, Vicente A, Mora E, et al. Prognostic impact of gene mutations in myelodysplastic syndromes with ring sideroblasts. Blood Cancer J 2017;7:630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyl-transferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010;42:665–7. [DOI] [PubMed] [Google Scholar]
- 54.Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Gohring G, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol 2011;29:1971–9. [DOI] [PubMed] [Google Scholar]
- 55.Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia 2017;31:705–11. [DOI] [PubMed] [Google Scholar]
- 56.Nazha A, Narkhede M, Radivoyevitch T, Seastone DJ, Patel BJ, Gerds AT, et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016;30:2214–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.