

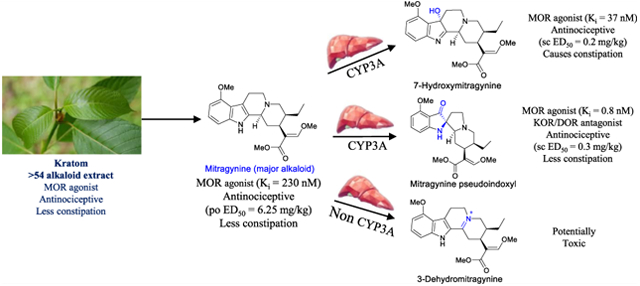
Abstract
The leaves of Mitragyna speciosa (kratom), a plant native to Southeast Asia, are increasingly used as a pain reliever and for attenuation of opioid withdrawal symptoms. Using the tools of natural products chemistry, chemical synthesis, and pharmacology, we provide a detailed in vitro and in vivo pharmacological characterization of the alkaloids in kratom. We report that metabolism of kratom’s major alkaloid, mitragynine, in mice leads to formation of (a) a potent mu opioid receptor agonist antinociceptive agent, 7-hydroxymitragynine, through a CYP3A-mediated pathway, which exhibits reinforcing properties, inhibition of gastrointestinal (GI) transit and reduced hyperlocomotion, (b) a multifunctional mu agonist/delta-kappa antagonist, mitragynine pseudoindoxyl, through a CYP3A-mediated skeletal rearrangement, displaying reduced hyperlocomotion, inhibition of GI transit and reinforcing properties, and (c) a potentially toxic metabolite, 3-dehydromitragynine, through a non-CYP oxidation pathway. Our results indicate that the oxidative metabolism of the mitragynine template beyond 7-hydroxymitragynine may have implications in its overall pharmacology in vivo.
Graphical Abstract
INTRODUCTION
Effective pain management remains one of the challenges of modern medicine. A recent study found that over 20 million adults in the United States (U.S.) are affected by chronic pain.1 Opioid agonists targeting the mu opioid receptor (MOR), such as morphine, are effective analgesics, but have high addiction potential and potentially lethal side effects in overdose attributed to profound respiratory suppression.2 Another lingering effect of MOR agonist use is opioid-induced constipation.3,4 As effective antinociceptives are essential to minimize pain and suffering, the identification of safer antinociceptive agents with diminished side effects and abuse potential is critical for breaking the vicious cycle fueling the opioid epidemic.
In the search for safer opioid agonists, we have focused on the alkaloids of Mitragyna speciosa (kratom),5 specifically mitragynine,6,7 7-hydroxymitragynine (7OH),6,7 and mitragynine pseudoindoxyl (MP),7,8 showing that these alkaloids are MOR partial agonists9 with reduced βarrestin-2 recruitment in vitro and mitigate lesser side effects compared to morphine and other clinical MOR agonists.7,10,11 The lower intrinsic efficacy12 of these alkaloids and negligible arrestin recruitment13 has been independently replicated and supported by a mechanistic conformational model for the activation of MOR by these alkaloids.14 Recent studies across numerous chemical templates suggest the possibility of designing safer opioids using the synthesis of low efficacy partial agonists.9,15–18
Kratom, a plant native to Southeast Asia, has been used for traditional treatment for various medical disorders, including pain, diarrhea, cough, alcoholism, and depression. Many aspects of the medicinal, pharmacological, and neurobiological potential of kratom-based natural products have been reviewed.19–23 Among the nearly 54 alkaloids found in kratom, mitragynine is the major alkaloid (approximately 1–2% mass of dry leaf matter, representing up to 66% of the total alkaloid content) thought to be largely responsible for the plant’s biological actions.
In 2016, the US Drug Enforcement Administration (DEA) released a notice of intent to place mitragynine and its oxidized analogue, 7OH, into Schedule I of the Controlled Substances Act (CSA). The notice was withdrawn two months later after outcry from the national media and public.24 However, kratom25 remains under regulatory scrutiny at the state and federal level. For example, the U.S. Food and Drug Administration (FDA) issued a statement on kratom in February 2018, describing its potential for abuse, addiction, and serious health consequences including death, concluding that kratom consumption has no health benefits.26 In contrast, an independent analysis that applied the FDA’s 7-point procedure to evaluate the risks and safety of drugs concluded that kratom presents a vastly safer profile compared to prescription and illicit opioids.27 Regardless, kratom-based products remain widely used in the U.S., with reported sales on an estimated 10,000 retail outlets and a market value of $207 million.27 At the same time, many aspects of kratom’s pharmacology remain understudied, although new findings are emerging. For example, recent reports have shown that mitragynine is metabolized into 7OH.28–30 The contribution of other metabolites to the various pharmacological effects of mitragynine remains unclear, as does a complete understanding how other component alkaloids might contribute to the effects of kratom as a whole. As part of the present study, we have examined the antinociceptive activity of the major individual alkaloids in kratom and a large variety of possible mitragynine metabolites in mice in order to better understand the mechanism of kratom’s biological action. We had previously used the 129S1 strain for antinociception and PK studies.29 In the present report, we used 129Sv6 mice for antinociception and peak effect PK assays to facilitate a direct comparison to our previous studies. In addition, we also used the inbred C57BL/6J strain which are well validated for pharmacokinetic investigations31 and the studies of opioid-induced antinociception32 and adverse effects such as reinforcement.33 We find that the antinociceptive actions of kratom are mediated by mitragynine through its first-pass metabolic product, namely, 7OH and MP. We also identify another previously unknown toxic metabolite, 3-dehydromitragynine (3DM), which seemed to be responsible for the lethality of mitragynine in mice. Further, we assess the locomotor effect, constipation, and conditioned-place preference induced by kratom, mitragynine, and its opioid-active metabolites, in order to better clarify the interaction of these compounds in mediating the side effect profile of the kratom plant.
RESULTS
Chemistry.
Extraction of Natural Products and Synthesis of Metabolites.
The natural products mitragynine, paynantheine, speciogynine, and speciociliatine (Figure 1A) were extracted from kratom leaves as described before by Varadi et al.7 The alkaloid derivatives, namely, 7-hydroxymitragynine (7OH),34 9-hydroxy corynantheidine (9OH), glucuronyl 9-hydroxycorynantheidine (9G), 3-dehydromitragynine (3DM),35 and mitragynine pseudoindoxyl (MP),7 were found as the major metabolites of mitragynine (Figure 1B, discussed in a later section) in mice dosed per os (po). The syntheses of 7-hydroxymitragynine, 9-hydroxycorynantheidine (9OH), and MP were accomplished by following our previously reported procedure.7 Briefly, the synthesis of 7OH was accomplished by the oxidation of mitragynine using oxone.7 The synthesis of 9G, 3DM, and mitragynine N-oxide (MNO) was completed by starting with mitragynine (Scheme 1). 3-dehydromitragynine could not be isolated in the pure form when mitragynine was heated with lead tetraacetate in acetic acid by following a previous report.35 Thus, the 3-dehydromitragynine (3DM) was prepared by an acid-catalyzed dehydration of the 7OH mitragynine using trifluoroacetic acid (TFA) in dichloromethane (DCM), isolated in quantitative yields as a TFA salt. The synthesis of glucuronyl 9-hydroxycorynantheidine (9G) was accomplished by the coupling of 9-hydroxycorynantheidine (9OH) with 2,3,4-tri-O-acetyl-α-d-glucuronic acid methyl ester and trichloroacetimidate in the presence of a mild Lewis acid BF3. OEt2.36 The crude glucuronide ester intermediate product was further subjected to hydrolysis directly since the ester intermediate was found not to be stable in column chromatography using silica or basic alumina. Hydrolysis of both the acetyl groups and methyl ester was performed in a one-pot reaction with a slight excess of NaOMe/MeOH in DCM for 10 min followed by alkaline hydrolysis of the methyl ester. Finally, the desired product 9G was purified by using the anion exchange resin (Amberlite(R) IRA-410 chloride, 20–25 mesh). Mitragynine N-oxide (MNO)37 has recently been reported to be a natural product present in kratom which was synthesized directly from mitragynine. The synthesis of MNO was achieved in three steps starting from mitragynine. Direct N-oxide formation38 in mitragynine using mCPBA resulted in various byproducts in our hands resulting in a complication during the purification of MNO. However, a clean reaction of Boc-mitragynine N-oxide formation was found using Boc-mitragynine with mCPBA in 79% yield. Finally, MNO was obtained from the Boc deprotection of Boc-mitragynine N-oxide using TFA/DCM.
Figure 1.
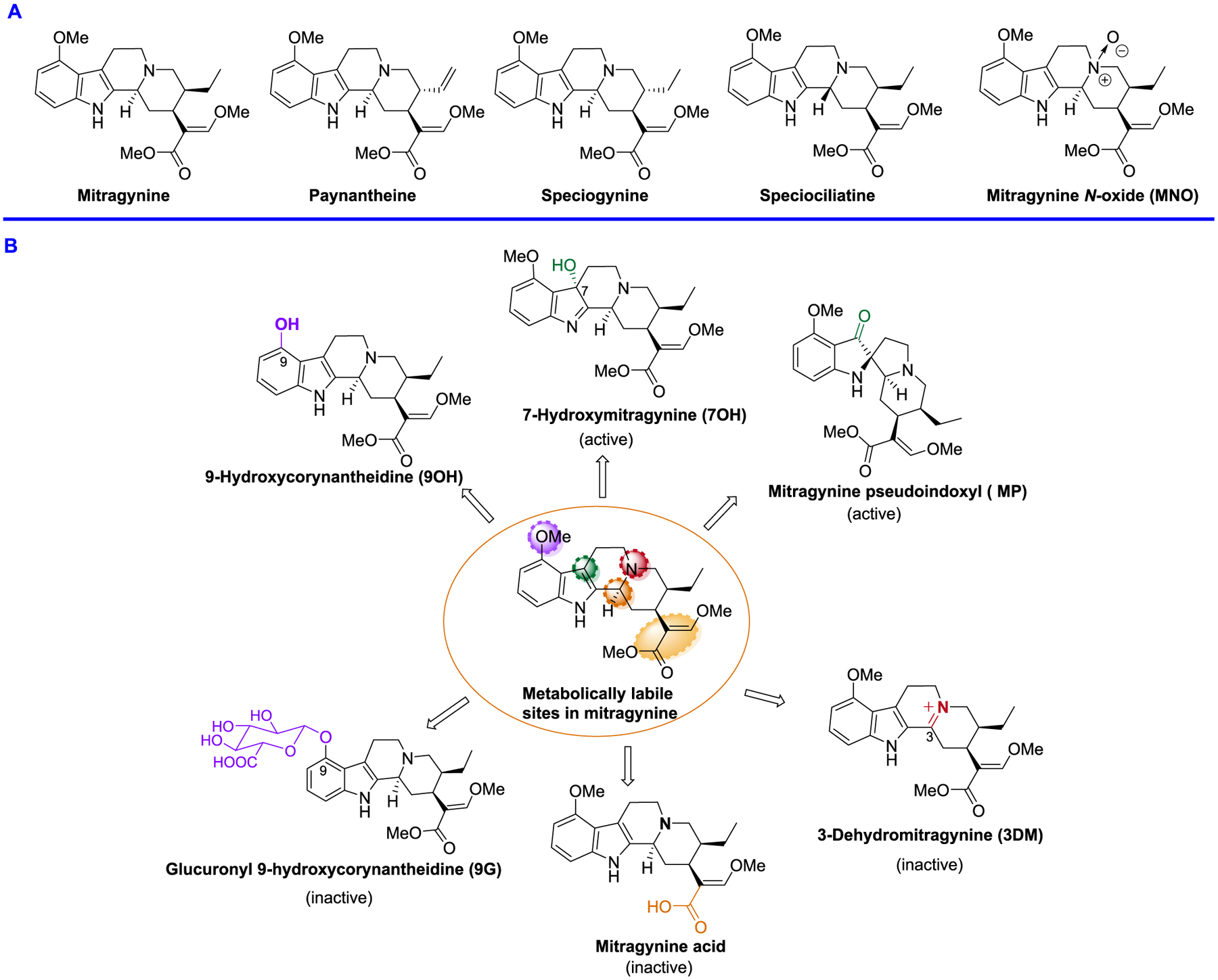
(A) Kratom alkaloids; major alkaloid natural product (B) metabolites of mitragynine in mice.
Scheme 1.
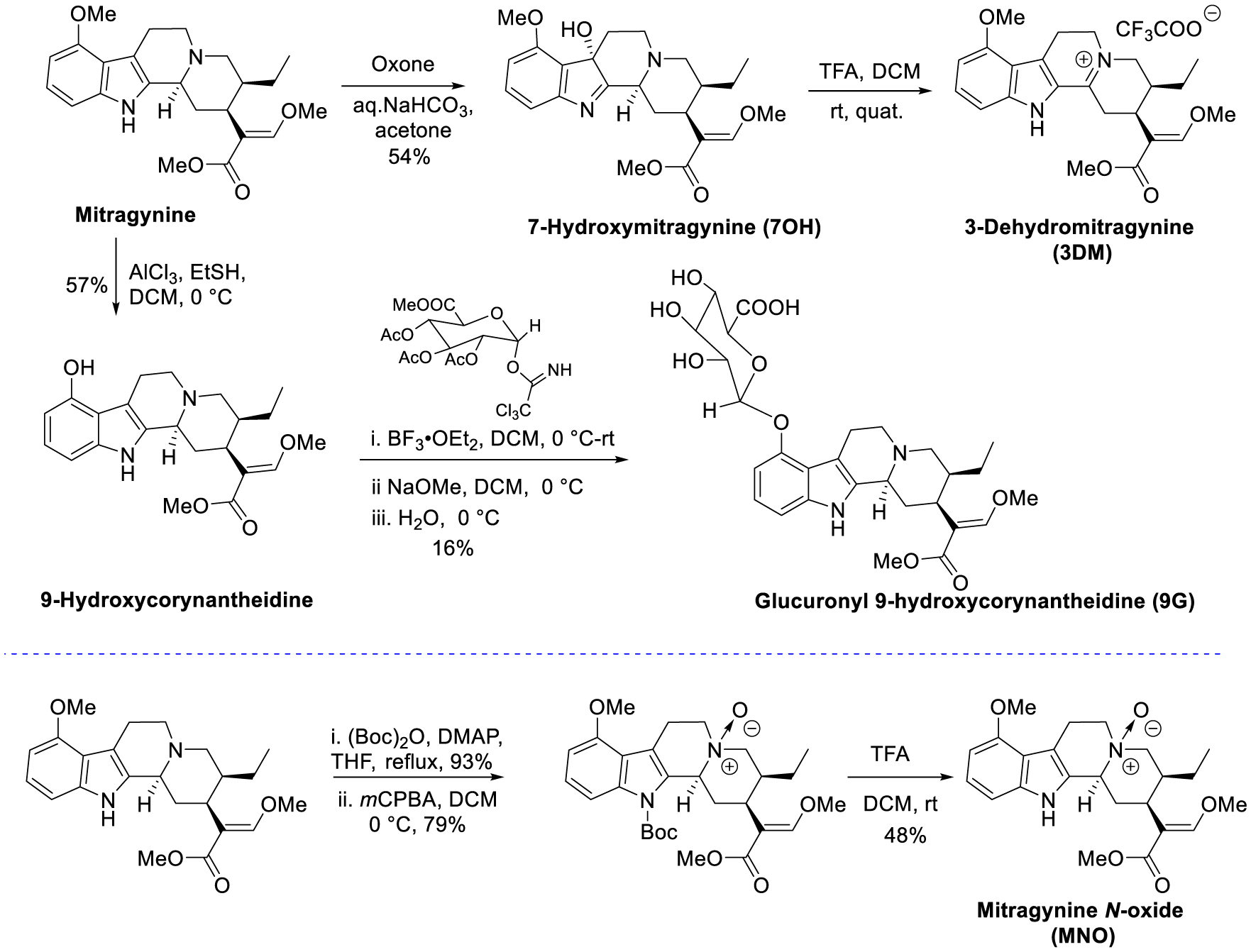
Synthesis of Mitragynine Metabolites and Mitragynine N-Oxide
Pharmacology of Kratom Alkaloid Extract.
To investigate the molecular actions of kratom, we utilized a robust extraction protocol developed to isolate kratom alkaloids from dry kratom leaf powder commercially available in the US.7 As it is well established that distribution of alkaloids in kratom is dependent on plant strain, growing climate, and when the leaves are harvested,37,39 our studies used the well-validated “Red Indonesian Micro Powder” from moon kratom, Austin, TX, USA. To begin, we studied the in vivo pharmacology of a kratom alkaloid extract (KAE) containing the major alkaloids, mitragynine (48.6%), paynantheine (5.8%), speciogynine (3.5%), speciocilliatine (13.7%), and MNO (not present in our kratom batches) (Figure 1A).
Since kratom is commonly consumed orally, we examined the effects of KAE in mice after oral (po) administration as our starting point to understand the pharmacology of kratom. Antinociception of the alkaloid fraction in rats following po administration has been reported previously using the hot plate assay at a single dose.40 In our hands, po KAE further showed potent, dose-dependent antinociception in a tail-flick assay in two strains of mice, C57BL/6J and 129Sv6. The peak antinociceptive effect of KAE was seen at 20 min in C57BL/6J mice (Figure 2A) and 15 min (Figure S2A) in 129Sv6 mice. The oral antinociceptive ED50 (and 95% C.I.) values of KAE were 17.1 (11.87–23.92) mg/kg in C57BL/6J mice (Figure 2A) and 35 (25.3–74.3) mg/kg in 129Sv6 (Figure S2A). The antinociception of KAE tested at an ED80 oral dose (45 mg/kg in C57BL/6J and 100 mg/kg in 129Sv6 mice) and was lost in MOR KO mice, while antinociception remained intact in delta opioid receptor (DOR) and kappa opioid receptor (KOR) KO mice in both C57BL/6J and 129Sv6 strains (Figure 2E and S2E, respectively). The antinociception was also attenuated by the opioid antagonist naloxone (Figure S2E), further confirming opioid-receptor mediation of KAE antinociception.
Figure 2.
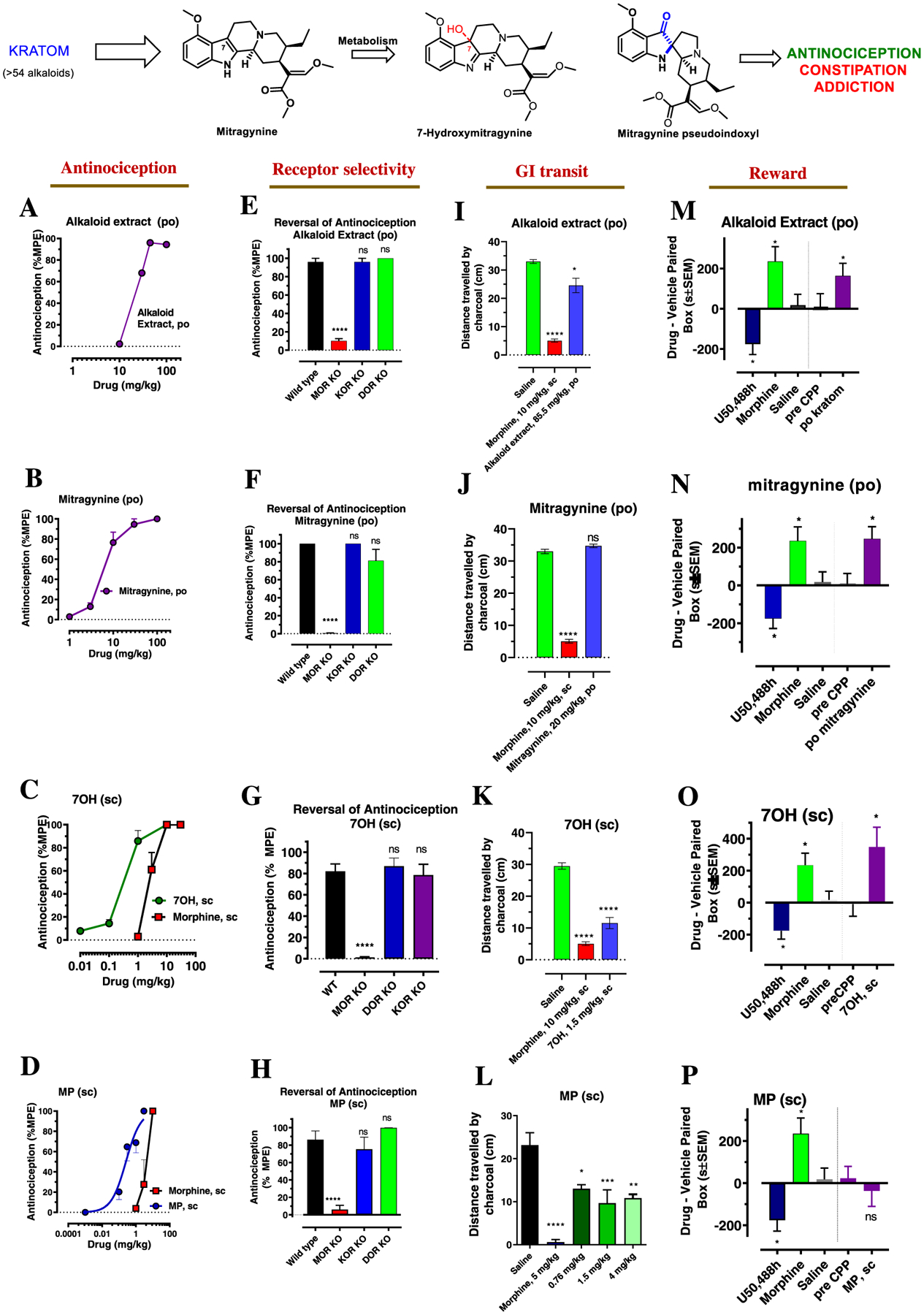
Kratom, mitragynine, and its metabolites show MOR-dependent antinociception with reduced constipation except for 7OH but retain reinforcing behavior except for MP in C57BL/6J mice. Oral tail flick dose–response curves (A,B): groups of mice (n = 8) per dose were given different doses of po kratom (A) or po mitragynine (B) by gavage and tested for the antinociceptive response at 20 min. ED50 (and 95% CI) was 17.1 (11.87–23.92) mg/kg for po kratom and 6.25 (4.49–8.68) mg/kg for po mitragynine. Subcutaneous tail flick dose–response curves (C,D): Groups of mice (n = 8) per dose were given different doses of sc 7OH and sc morphine (C) and sc MP and sc morphine (D) and tested for antinociceptive response at 20 min for 7OH and MP and 30 min for morphine. ED50 (and 95% CI) was 0.3 (0.19–0.48) mg/kg for sc 7OH, 0.26 (0.15–0.43) mg/kg for sc MP, and 2.48(1.57–3.87) mg/kg for sc morphine. The means of each point in each determination were calculated as percentage maximal possible effect (%MPE) [(observed latency − baseline latency)/(maximal latency − baseline latency)] × 100. Kratom alkaloid fraction, mitragynine showed antinociception given orally while the putative metabolites 7OH and MP showed antinociception given sc and about 10 fold more potent over sc morphine. Antinociception in KO mice (E–H): An ~ED80 antinociceptive dose of alkaloid extract (45 mg/kg, po), mitragynine (45 mg/kg, po), 7OH (1 mg/kg, sc), and MP (3 mg/kg, sc) was evaluated in group of (n = 8) of wildtype (WT), MOR KO, KOR KO, and DOR KO mice. Antinociceptive effects of all these drugs were found attenuated in MOR KO, whereas the effect was found intact in KOR KO and DOR KO mice (****p < 0.0001, one-way ANOVA followed by Dunnett’s multiple comparison comparisons test). Effects on GI transit in C57BL/6J mice (I–L): a group of C57BL/CJ mice (n = 8) were administered with either saline, morphine (10 mg/kg, sc), alkaloid extract (85.5 mg/kg, po), mitragynine (20 mg/kg, po) 7OH (1.5 mg/kg, sc), or MP (0.76, 1.5, 4 mg/kg, sc) and then were fed a charcoal meal. After 3 h, morphine significantly reduced the distance traveled by the charcoal through the intestines, consistent with the action of a MOR agonist (5.07 ± 0.57 cm), compared to 33 ± 0.68 cm for saline-treated mice (****p < 0.0001, one-way ANOVA followed by Dunnett’s multiple comparison test). In contrast, the alkaloid extract (24.6 ± 2.54 cm) showed constipation compared to saline (*p < 0.0019, one-way ANOVA followed by Dunnett’s multiple comparison test) but less than morphine, while mitragynine (34.7 ± 0.53 cm) showed no significant effect (I,J). 7OH (1.5 mg/kg, sc) showed significant reduction in the distance traveled by the charcoal (11.6 ± 1.8 cm) through the intestines (****p < 0.0001, one-way ANOVA followed by Dunnett’s multiple comparison test) (K). MP showed constipation at all doses (*p = 0.016, ***p = 0.001, **p = 0.005, one way ANOVA followed by Dunnett’s multiple comparison test) compared to saline but less than morphine. Conditioned place preference or aversion in C57BL/CJ mice (CPP/CPA) (M–P): A group of C57BL/CJ mice (n = 18–24) were habituated to freely explore both sides of a two-compartment apparatus for 3 h each for 2 days prior testing. The baseline preferences of 20 min were determined as the pre-condition test prior to the administration of a drug on the conditioning day. Mice were conditioned for 20 min on each session with either saline, morphine (10 mg/kg/d, sc), U50, 488H (30 mg/kg/d, sc), alkaloid extract (100 mg/kg/d), mitragynine (30 mg/kg/d) 7OH (1 mg/kg/d, sc), and MP (3.2 mg/kg/d, sc) after being habituated for 1 h. On the testing day, animals were placed in the side paired with saline and are allowed to freely explore both compartments for 20 min. The difference score was calculated to determine CPP/CPA. MP did not display significant CPP or CPA, whereas kratom alkaloid fraction, mitragynine, and 7OH displayed CPP (M–P); (p < 0.05) as determined by ANOVA followed by Tukey’s multiple-comparison test. Note: Figure 2F,G; data reprinted (adapted or reprinted in part) with permission from [Kruegel, A. C.; Uprety, R.; Grinnell, S. G.; Langreck, C.; Pekarskaya, E. A.; Le Rouzic, V.; Ansonoff, M.; Gassaway, M. M.; Pintar, J. E.; Pasternak, G. W.; Javitch, J. A.; Majumdar, S.; Sames, D. 7-Hydroxymitragynine Is an Active Metabolite of Mitragynine and a Key Mediator of Its Analgesic Effects. ACS Cent. Sci. 2019, 5 (6), 992–1001.] Copyright [2019/American Chemical Society]. Figure 2L,P data reprinted (adapted or reprinted in part) with permission from [Váradi, A.; Marrone, G. F.; Palmer, T. C.; Narayan, A.; Szabó, M. R.; Le Rouzic, V.; Grinnell, S. G.; Subrath, J. J.; Warner, E.; Kalra, S.; Hunkele, A.; Pagirsky, J.; Eans, S. O.; Medina, J. M.; Xu, J.; Pan, Y. X.; Borics, A.; Pasternak, G. W.; McLaughlin, J. P.; Majumdar, S. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016, 59 (18), 8381–8397.] Copyright [2016/American Chemical Society].
In addition to examining the antinociception induced by KAE and its molecular mechanism, we also compared this substance’s profile to that of established opioids in terms of three serious side effects: locomotor activity, inhibition of gastrointestinal (GI) transit, and addictive liability in C57BL/6J mice. Oral KAE (45 mg/kg) displayed significantly less inhibition of GI transit (Figure 2I) and no hyperlocomotion (Figure S7A) compared to morphine. Oral KAE did however show conditioned place preference (CPP) similar to morphine in C57BL/6J mice at doses 6-fold (100 mg/kg, po) higher than the antinociceptive ED50 (Figure 2M). Thus, in mice, KAE administered orally is antinociceptive (Figures 2A and S2A), and the antinociception is MOR-dependent (Figures 2E and S2E), yet associated with reduced constipation as well as the locomotor effect (Figures 2I and S7A). However, reward behavior as assessed by the CPP assay persists (Figure 2M), suggestive of addictive potential.
Pharmacology of Mitragynine.
We next evaluated in vitro binding and functional activity of the major kratom alkaloids in opioid-receptor-transfected CHO cells and antinociception in 129Sv6 mice after either subcutaneous (sc) or po administration, in order to identify the alkaloid responsible for the oral antinociceptive actions of KAE (Table 1). Briefly, among the natural products, mitragynine showed moderate binding affinity at mMOR (Ki = 230 nM), while speciocilliatine showed the highest affinity at mMOR (Ki = 79 nM). Both displayed partial agonism at mMOR (Emax = 63% and 45% for mitragynine and speciocilliatine, respectively) with potencies in line with the observed binding affinity. The other natural products had lower affinity as well as potency at opioid receptors. Among the six major natural individual alkaloids tested (Figure 1A) in 129Sv6 mice, only mitragynine showed antinociceptive activity and was more potent when given po (ED50 = 4 (2.21–7.16) mg/kg, Figure S3B) than when given sc (ED50 = 57.3 (44.6–73.5) mg/kg, Figure S3B). The po and parenteral antinociceptive actions of mitragynine in the hot plate assay in rats41,42 have been described by others, and our finding of higher potency by the oral route is consistent with previous work.43,44 Paynantheine, speciogynine, and speciocilliatine were inactive in the tail-flick assay at doses as high as 100 mg/kg (Table 1), suggesting that mitragynine is the major contributor to kratom’s antinociceptive effects. To further confirm that only mitragynine mediates antinociception, an alkaloid extract fraction without mitragynine (alkaloid 2) was administered (po and sc) to 129Sv6 mice. No antinociceptive activity was seen at doses as high as 100 mg/kg. The alkaloid extract fraction without mitragynine also showed no antagonism of morphine and/or mitragynine po antinociception, suggesting no functional opioid antagonist activity (Figure S4). The oral antinociceptive potency of mitragynine was similar to that of po morphine (ED50 = 3.3 (2.075–5.5) mg/kg, Figure S2B). However, unlike mitragynine, morphine’s antinociceptive potency was higher when administered sc (ED50 = 0.7 (0.47–0.99) mg/kg, Figure S3D). Similar to the 129Sv6 strain, in C57BL/6J mice, mitragynine was a more potent antinociceptive when given po (ED50 = 6.25 (4.49–8.68) mg/kg, Figures 2B–S3A) than when given sc (ED50 = 25.95 (15.43, 43.4) mg/kg, Figure S3A). In both strains and by both po and sc administration, the antinociceptive effect of mitragynine was rapid, reaching a peak in 15–20 min (Figure S1A–E). As with KAE, mitragynine po antinociception was mediated by MOR (but not DOR and KOR) in both C57BL/6J (Figure 2F) and 129Sv6 strains (Figure S2F), and could be reversed by naloxone (Figure S2F).
Table 1.
Receptor Affinities, [35S]GTPγS Functional Assays in Transfected Cell Lines, and Subcutaneous Antinociception in Mice
| affinity (Ki nM)a | [35S]GTPγS functional assaysb | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| mMOR | mKOR | mDOR | mMOR | mKOR | mDOR | analgesiac | |||||
| compound | EC50 | %Emax | IC50 | EC50 | %Emax | IC50 | IC50 | sc, mg/Kg, (CI) | |||
| mitragynine | 230.4 ± 47.2 | 231.1 ± 21.2 | 1011.2 ± 48.9 | 203.3 ± 13.19 | 63 ± 5 | no activity | >4 μM | >4 μM | 57.3 (44.6–73.5) | ||
| paynanthine | 665.7 ± 82.9 | 883.33 ± 293.72 | 3395 ± 962 | 9384 ± 801 | 54 ± 1 | 3586.3 ± 2621.4 | 57 ± 2 | >10 μM | >100 | ||
| speciogynine | 578.23 ± 63.46 | 2198 ± 764 | 7947 ± 823 | 6225.7 ± 3288 | 43 ± 0 | no activity | >100 | ||||
| speciociliatine | 78.6 ± 10.9 | 649 ± 168.8 | 1154.3 ± 200.1 | 72.7 ± 15 | 45 ± 1 | 10851 ± 2758 | 1355 ± 95.1 | 58 ± 2 | 12562.7 ± 1637 | >100 | |
| 7-hydroxymitragynine | 36.62 ± 4.12 | 131.67 ± 6.97 | 90.64 ± 8.49 | 52.99 ± 4.02 | 77 ± 5 | no activity | 2524 ± 551 | 691 ± 434 | 0.2 (0.07–0.6) | ||
| mitragynine pseudoindoxyl | 0.75 ± 0.18 | 24.23 ± 0.94 | 3.03 ± 1.32 | 1.66 ± 0.10 | 84 ± 5 | no activity | 30.9 ± 3.07 | 61.23 ± 6.34 | 0.3 (0.2—0.4) | ||
| 9-hydroxy corynanthedine | 216.2 ± 45.6 | 1177.2 ± 282.2 | 1882.4 ± 76 | 422.4 ± 134.5 | 62 ± 2 | 857.8 ± 33.5 | 87 ± 3 | no activity | >100 | ||
| 9-glucuronoyl mitragynine | 1674.7 ± 42.1 | 1274.3 ± 379.9 | 3239.3 ± 345.1 | 5778 ± 1357 | 102 ± 15 | 1143.5 ± 287.4 | 98 ± 2 | no activity | >30 | ||
| 3-dehydromitragynine | 34.1 ± 5.9 | 124.7 ± 14.9 | 49.1 ± 2.3 | 67.5 ± 8.3 | 166.1 ± 25.8 | 64 ± 9 | 1096 ± 64.7 | >100 | |||
| mitragynine N-oxide | 6902 ± 493 | 7180 ± 822 | 4143 ± 232 | ||||||||
| DAMGOd | 3.34 ± 0.43 | 1.39 ± 0.67 | |||||||||
| U50, 488Hd | 0.73 ± 0.32 | 9.5 ± 1.8 | |||||||||
| DPDPEd | 1.39 ± 0.67 | ||||||||||
| NTId | 0.46 ± 0.32 | 0.72 ±0.11 | |||||||||
| morphined | 4.6 ± 1.8 | 0.68 (0.47, 0.99) | |||||||||
Competition binding studies were performed for indicated compounds against 125IBNtxA (0.1 nM) in membranes from CHO cells stably expressing cloned mouse opioid receptors. Ki values were calculated from the IC50 values and represent the means ± SEM of at least three independent replications.
Efficacy data were determined using an agonist-induced stimulation of [35S]GTPγS binding assay. Efficacy is presented in EC50 (nM) and percent maximal stimulation (Emax) relative to standard agonists DAMGO (mMOR), DPDPE (mDOR), or U50, 488H (mKOR) at 1000 nM. To determine the antagonist properties of a compound, membranes were incubated with 100 nM of the appropriate agonist by varying its concentrations. Results are presented as nM ± SEM from three independent experiments performed as triplicate.
Cumulative dose–response curves were carried out on 129Sv6 mice (n = 10) using radiant heat tail flick assays with drugs at the indicated doses (sc), and antinociception was measured at 15 min (at peak effect). Results from two independent experiments are shown as mean (95% Cl).
The enhanced po versus sc potency of mitragynine (in two mice strains, Figures S3A,B) suggests the formation of a more potent active metabolite. The prototypic mu opioid agonist morphine also undergoes extensive first pass metabolism when given orally to yield both less active entities, like morphine 3-glucuronide and normorphine, and more active entities, like morphine 6-sulphate45 and morphine 6-gluronide.46,47 However, in contrast to mitragynine, morphine produces greater antinociception following parenteral versus oral administration; in humans, morphine given intravenously is ~3-fold more potent than when given orally.48 A similar trend was seen in mice. In the 129Sv6 strain, po morphine [ED50 = 3.32 (2.08–5.32) mg/kg] was less potent than sc morphine [ED50 = 0.68 (0.47–0.99) mg/kg] (Figure S3D), while in the C57BL/6J strain, morphine showed ED50 = 4.73 (3.24–6.85) mg/kg, po and 2.48 (1.57–3.87) mg/kg, sc (Figure S3C).
We also examined the locomotor, inhibition of GI transit, and addictive side effect profile of mitragynine in C57BL/6J mice. At doses 5-fold higher than its antinociceptive ED50, mitragynine (20 mg/kg, po in C57BL/6J mice) displayed attenuated inhibition of GI transit and locomotor compared to equi-antinociceptive doses of morphine (Figures 2J and S7B). However, po mitragynine produced reward behavior in C57BL/6J mice similar to morphine at a dose 5-fold higher than its antinociceptive ED50 dose (30 mg/kg, po) (Figure 2N). Mitragynine-conditioned place preference has been reported when administered parentally,49–51 but to the best of our knowledge, it has not been examined following po administration, despite the common oral consumption of kratom.
In light of these data, we conclude that mitragynine is responsible for kratom’s antinociceptive effects. When administered orally, it mediates MOR-dependent antinociception with reduced hyperlocomotion and constipation compared to morphine but displays rewarding behavior similar to morphine. The other tested alkaloids from kratom did not demonstrate antinociception nor did they antagonize mitragynine- or morphine-mediated antinociception.
Metabolic Mapping of Mitragynine Metabolites: PK Assays Show Formation of 7OH, MP, and 3DM as Important Entities.
Next, we sought to explore the potential contribution of active metabolites to the antinociceptive activity of mitragynine. To identify mitragynine metabolites, C57BL/6J and 129Sv6 mice were treated with mitragynine at doses of 30 mg/kg (po) and 10 mg/kg (po), respectively, at doses approximating the antinociceptive ED80 value. Plasma and brain samples were collected from C57BL/6J and samples analyzed at 20 min, 40 min, 60 min, 90 min, 2 h, 4 h, 8 h, and finally 24 h. In the case of 129Sv6 mice, samples were collected at peak antinociceptive effects of 15 min only. Liquid chromatography–tandem mass spectrometry (LC–MS/MS) was used to analyze and quantify parent drug and metabolites (See Table S3 and Figure 3). The identified mitragynine metabolites (shown in Figure 1B) were synthesized (see Scheme 1) and validated by LC–MS/MS using these authentic standards. Confirmed metabolites, namely 7OH,52 MP,7 3-dehydromitragynine (3DM), glucuronyl 9-hydroxycorynantheidine (9G),53 and 9-hydroxycorynantheidine (9OH),52 were evaluated for in vitro binding/functional activity in opioid-transfected cell lines and for antinociception in mice (after the sc route). Consistent with previous findings,6,7 in vitro, MP was a 200-fold more potent binder and 120-fold more potent agonist than mitragynine at MOR, while 7OH was a 6-fold more potent binder and 4-fold more potent agonist than mitragynine (Table 1). Among the other metabolites, only 3DM exhibited notable binding affinity for MOR (see the next section). 7OH and MP also showed potent antinociceptive activity in 129Sv6 mice [7OH ED50 (and 95% C.I.) value = 0.2 (0.07–0.6) mg/kg, sc; MP ED50 (and 95% C.I.) value = 0.3 (0.2–0.4) mg/kg, sc] (Figure S2C,D, respectively). Both metabolites displayed >100-fold more antinociceptive potency than mitragynine (sc),7 suggesting that these two metabolites might play a key role in mediating the oral antinociceptive effects of mitragynine.
Figure 3.
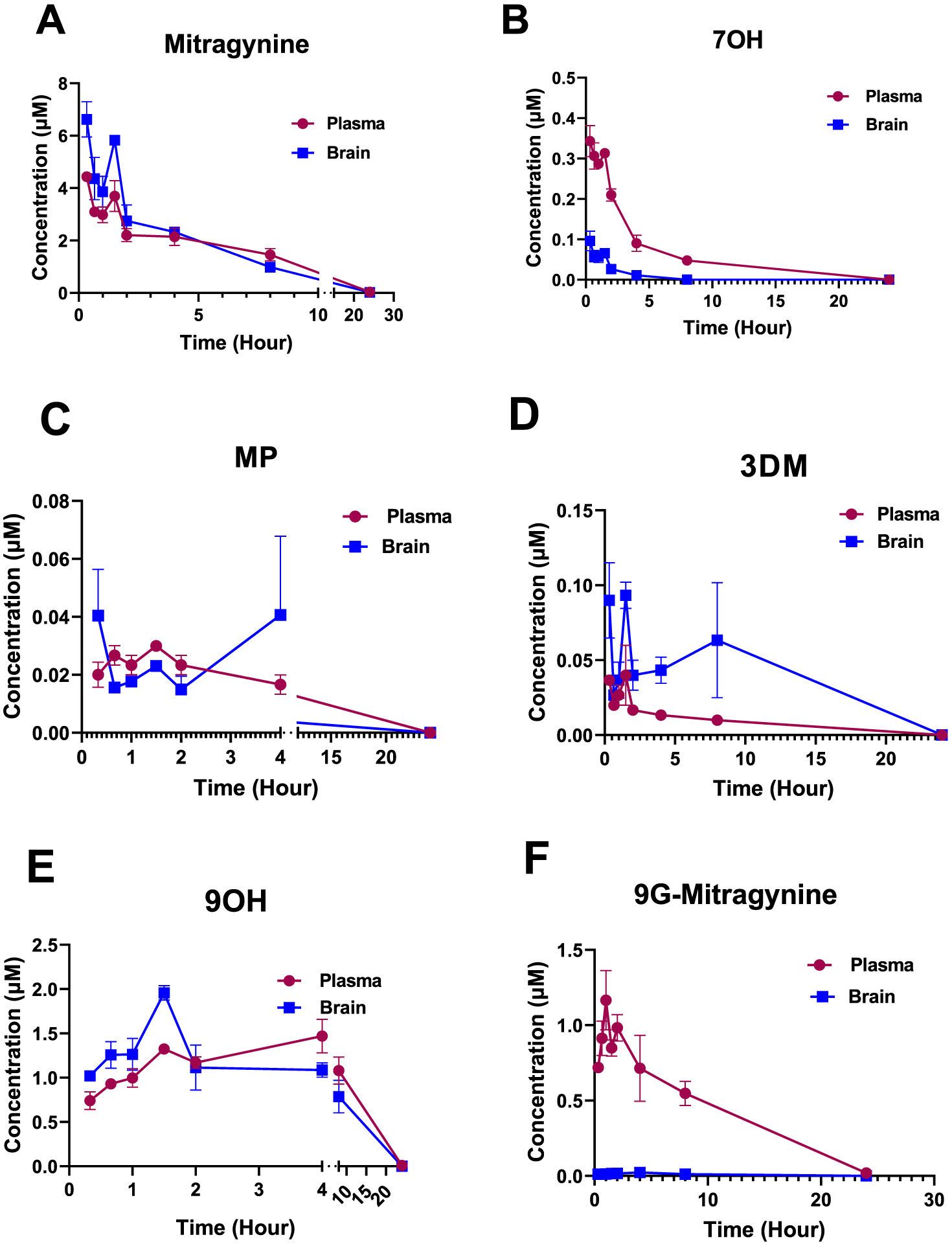
Concentration time profile for mitragynine to its metabolites was confirmed in vivo in C57BL/6 mice using LC–MS/MS. (A) Mitragynine was detected in both the plasma and brains of mice treated with mitragynine (30 mg/kg, po). n = 3 per time point for plasma; n = 3 per time point for brain. At the same time gradient, metabolites formed from mitragynine, namely, 7-OH (B), MP (C), 3DM (D), 9OH (E), and 9G-mitragynine (F) were also detected in the plasma and brains of the same animals, but at lower concentrations. 7OH was the major metabolite followed by MP and 3DM as other important metabolites.
The highest concentrations in both brain and plasma of mitragynine (administered po in C57BL/6J mice) 7OH and MP (metabolites formed from mitragynine po) matched the antinociceptive peak effect in C57BL/6J mice at 20 min generally (See Figure 3 for concentrations observed and Figure S1A,C for antinociception time course). The levels of parent drug mitragynine and metabolites 7OH and MP decreased with time with no quantifiable 7OH or MP found at 8 h. Mitragynine and 3DM levels reached below detection limits at the 24 h point. Moreover, at the time of peak antinociception, 129Sv6 mice possessed the same set of metabolites as seen in the C57BL/6J mice.
We next closely examined 7OH and MP administered alone in PK assays on plasma and brains collected from C57BL/6J and 129Sv6 mice at the time of peak antinociception. Plasma concentrations of 343 ± 80 and 830 ± 50 nM of 7OH and 20 ± 4 and 13 ± 0 nM of MP were detected, respectively, in C57BL/6J and 129Sv6 mice (Figure S5A,C). Similarly, 7OH at concentrations of 96 ± 24 and 1070 ± 12 nM and MP at concentrations of 40 ± 16 and 180 ± 0 nM were detected in the brains of C57BL/6J and 129Sv6 mice, respectively (Figure S5B,C). Next, we sought to compare the levels of 7OH and MP reached when formed as metabolites following mitragynine administration versus those achieved when administered directly at equi-antinociceptive dosing, in order to demonstrate that the metabolites were responsible for antinociception. 7OH and MP were administered alone in C57BL/6J mice at the ~ED80 antinociceptive dose sc. Brain and plasma samples were analyzed at the time point of the peak antinociceptive effect to determine concentrations of the intact drug. Concentrations of 270 ± 13 and 450 ± 30 nM were found for 7OH in plasma and brain, respectively, consistent with the mitragynine results. MP administered directly sc to C57BL/6J mice at its ED80 antinociceptive dose resulted in concentrations of 410 ± 120 and 70 ± 10 nM in plasma and brain, respectively (Figure S5A,B, Table S3).
These data suggest that po administration of mitragynine at antinociceptive doses yields concentrations of 7OH and MP as a metabolite that are 7–10-fold higher than the agonist potencies (EC50 = 53 ± 4 nM for 7OH and 1.7 ± 0.1 nM for MP) of these drugs at MOR, suggesting that these metabolites are present to activate MOR at relevant doses.
We also sought to explore the role that intact mitragynine plays in its antinociceptive activity. When mitragynine was administered po to mice at an antinociceptive dose equivalent to its ED80 value, 6.63 ± 0.67 μM and 12.22 ± 3.4 μM of intact mitragynine were found in the brains of C57BL/6J and 129Sv6 mice at concentrations 30-to-60-fold greater than the demonstrated MOR agonist potency. When mitragynine was administered subcutaneously in C57BL/6J or 129Sv6 mice at 30 and 10 mg/kg doses, respectively, brain concentrations of 9.74 ± 3.05 μM (in C57BL/6J mice) and 30.06 ± 8.12 μM (in 129Sv6 mice) were subsequently detected (Table S3). These mitragynine levels were approximately 45-to-150-fold greater than the demonstrated MOR agonist potency (EC50 = 203 nM) concentration in brain. Thus, these results suggest the parent mitragynine plays relatively a little role in the MOR-mediated antinociception associated with oral kratom or oral mitragynine.
Formation of Mitragynine Active Metabolites Is Dependent on CYP3A.
To determine if the oxidation of mitragynine to 7OH and MP is carried out by the liver, mitragynine was incubated with mouse liver microsomes from C57BL/6J or 129Sv6 mice or from human liver microsomes (HLM) and S9 fraction (Figure 4). Mitragynine was found to be labile in these assays (Figure 4A). The use of CYP3A inhibitor ketoconazole at 1 μM (Figure 4B) and liver microsomes from Cyp3a KO mice in the C57BL/6J strain (Figure 4C) decreased the decomposition of mitragynine. The half-life of mitragynine in microsomes was also increased by ~5 fold in the presence of ketoconazole and ~2 fold in microsomes from Cyp3a KO mice. The formation of 7OH and MP from mitragynine was also observed in mouse (129Sv6 and C57BL/6J) microsomes and HLM (Figure 4D,E). Lastly, the selective CYP3A inhibitor ketoconazole attenuated the formation of 7OH and MP from mitragynine in liver microsomes (Figures 4G,H).
Figure 4.
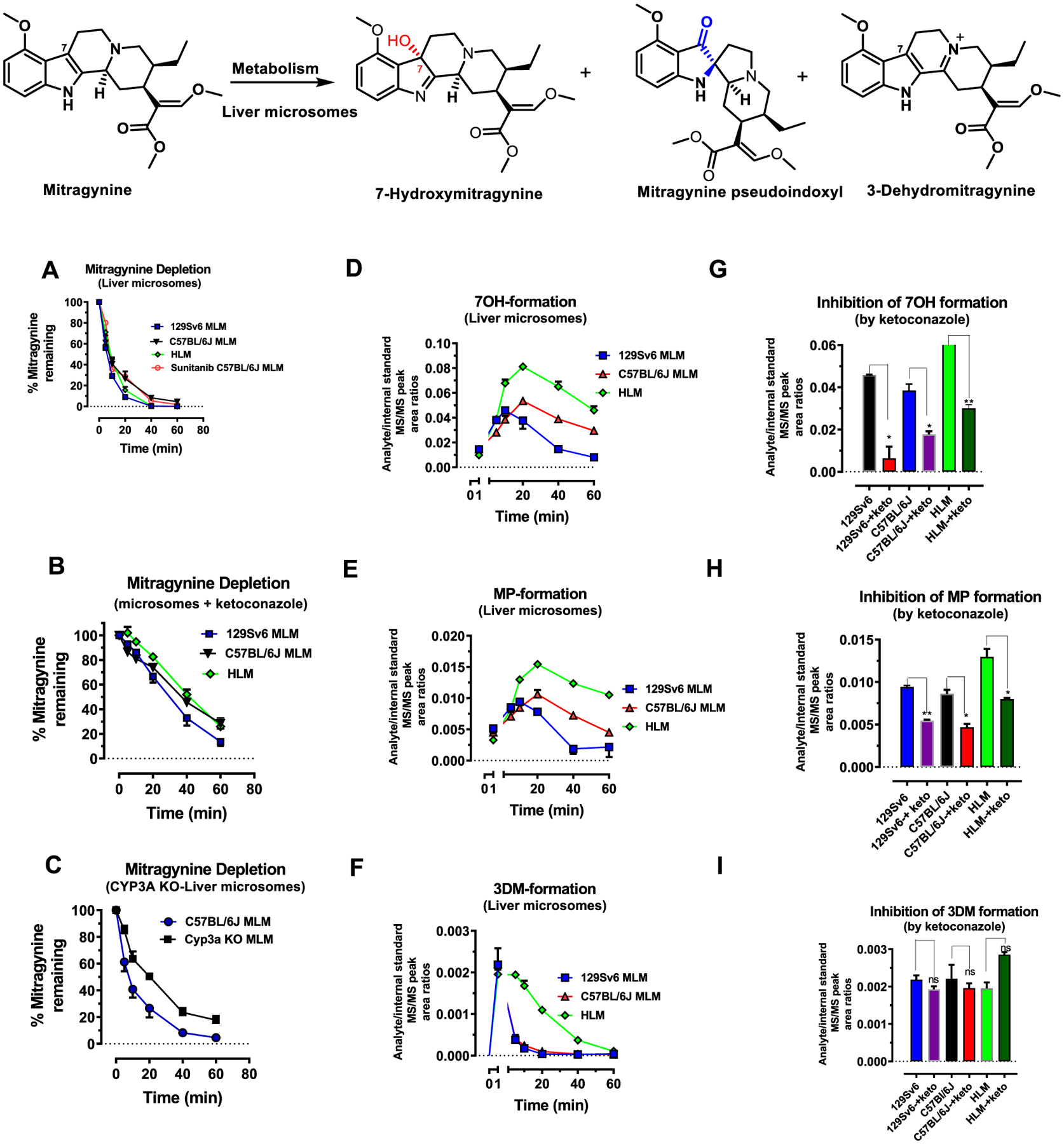
Metabolism of mitragynine to 7OH, MP, and 3DM in liver microsomes: mitragynine (1 μM) was incubated in the potassium phosphate buffer (0.1 M; pH 7.4) at 37 °C in the presence of liver microsomes of mice (129Sv6 and C57BL/6J) or human (HLM) for 60 min. Each point in the curve represents three independent determinations. Level of intact mitragynine decreases over time due to microsomal disintegration. The half-lives of mitragynine were 7.5 ± 0.82, 5.9 ± 0.3, and 13.5 ± 1.1 min in HLM, 129Sv6, and C57BL/6J mice liver microsomes. (A). Usage of a CYP3A blocker ketoconazole (1 μM). The half-lives of mitragynine in the presence of ketoconazole were 37.4 ± 7.5, 24.4 ± 4.9, and 35.8 ± 4 min in HLM, 129Sv6, and C57BL/6J mice liver microsomes (B). Liver microsomes from a Cyp3a KO male mice also inhibit the depletion of mitragynine. The half-life of mitragynine was 25.2 ± 2.05 min in Cyp3a KO mice microsomes (C). These liver microsomal assays indicated the formation of three major metabolites 7OH, MP, and 3DM (D–F). A decrease in the formation metabolites from mitragynine including 7OH and MP was observed in the presence of ketoconazole at the peak effect compared to control, where ketoconazole was not included in microsomal incubations (p < 0.05 as determined by unpaired t-test) (G–H). The formation of 3DM was found to be independent of CYP3A (I).
To evaluate NADPH dependency, we reevaluated metabolism in HLM and S9 fractions in presence and absence of NADPH (Figure 5). Depletion of mitragynine and formation of 7OH and MP was seen in HLM (Figure 5A) and S9 (Figure 5C) in the presence of NADPH, with 7OH being the major metabolite in both cases. As expected from a CYP-dependent mechanism, the formation of 7OH as well as MP was minimal in the absence of NADPH in both HLM (Figure 5B) and S9 incubations (Figure 5D). Together, these findings suggest that mitragynine is oxidized to 7OH and MP in the liver through a CYP3A-dependent mechanism.
Figure 5.
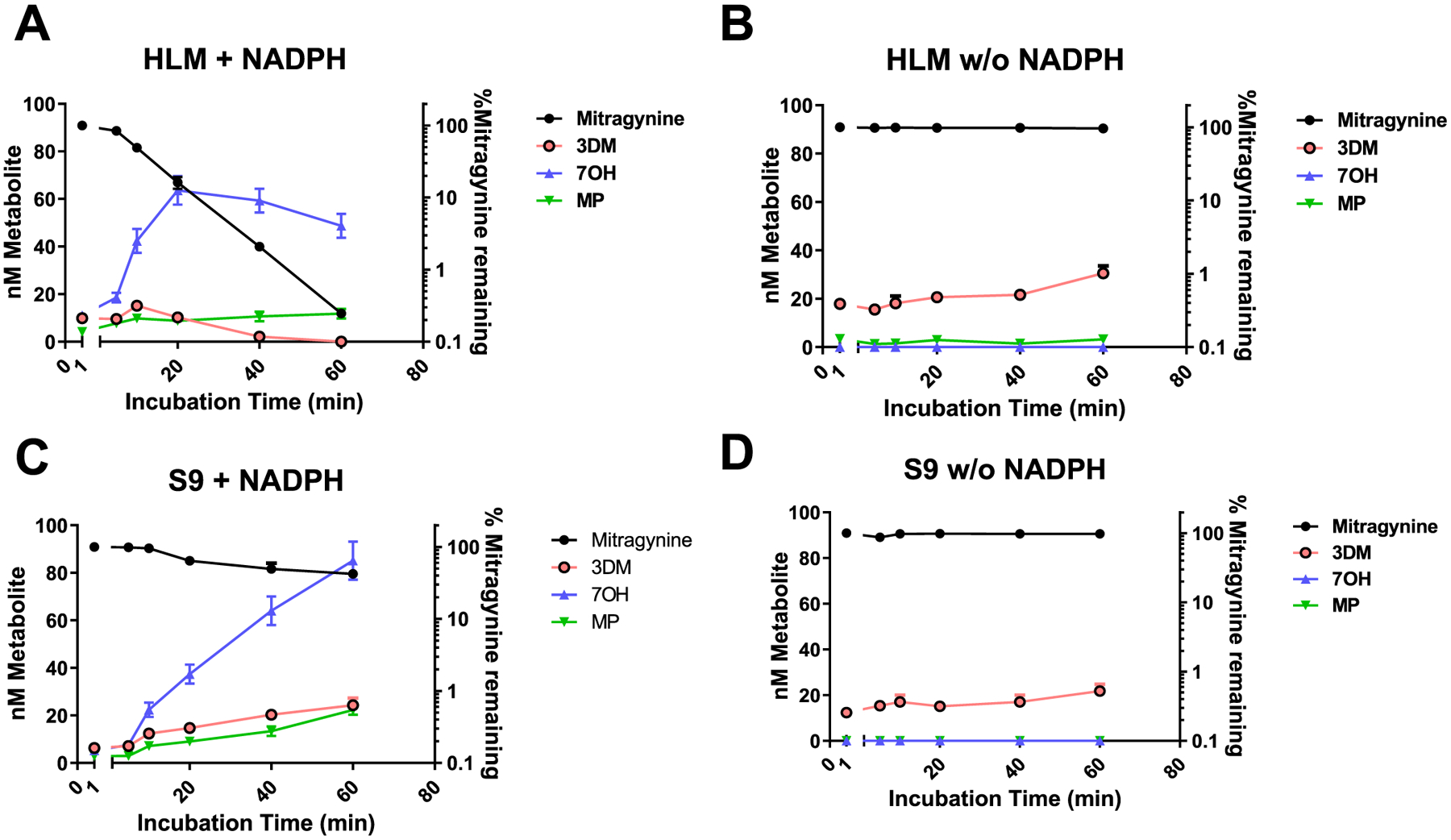
Metabolism of mitragynine to 7OH, MP, and 3DM in HLM and S9 fractions in the presence and absence of NADPH: mitragynine (1 μM) was incubated in the potassium phosphate buffer (0.1 M; pH 7.4) at 37 °C in the presence of HLM for 60 min. Level of intact mitragynine decreases over time due to microsomal disintegration. (A). In the presence of NADPH in HLM, concentration of mitragynine decreases with formation of 7OH, MP; 7OH being the major metabolite. (B) In the absence of NADPH in HLM, the depletion of mitragynine was inhibited with minimal formation of 7OH and MP, while formation of 3DM is less effected. (C) In the presence of NADPH in S9, the fraction concentration of mitragynine decreases with formation of 7OH, MP, and 3DM; 7OH being the major metabolite. (D) A decrease in the formation metabolites from mitragynine including 7OH and MP was observed in S9 fraction in the absence of NADPH, while 3DM levels were less effected.
In summary, among mitragynine’s known metabolites, only 7OH and MP are MOR agonists and antinociceptive. Further, formation of 7OH (major) and MP (minor) from mitragynine in mice occurs in the liver through a CYP3A-dependent mechanism, and these metabolites in turn play an important role in mediating mitragynine’s antinociceptive effects.
Antinociception of 7OH and MP is Mediated through MOR, but Associated with Reduced Locomotor Effects.
Opioids are associated with a myriad of physiological effects including (though not limited to) antinociception, hyperlocomotion (in mice), constipation, and reward. We quantified the potency of the active metabolites of mitragynine in multiple mouse behavioral assays in order to evaluate their activity and side effect profiles relative to morphine. Subcutaneous 7OH produced potent antinociception, with ED50 (and 95% C.I.) values of 0.3 (0.19–0.48) mg/kg, sc in C57BL/6J (Figure 2C) and 0.2 (0.07–0.6) mg/kg, sc in 129Sv6 mice (Figure S2C), approximately 3-to-10-fold more potent than morphine in these strains. The antinociceptive actions were mediated through MOR, independent of DOR or KOR, and antinociception was naloxone-sensitive (Figures 2G and S2G). Mitragynine pseudoindoxyl was 2–10-fold more potent than morphine [ED50 (and 95% C.I.) values of 0.26 (0.15–0.43) mg/kg, sc in C57BL6J, Figure 2D and 0.3 (0.2–0.4) mg/kg, sc in 129Sv6 mice (Figure S2D)]. The antinociception of both metabolites was mediated through MOR, independent of DOR/KOR, and was naloxone-sensitive (Figures 2G,H and S2G,H). To further evaluate MP-mediated antagonism of the KOR and DOR free of MOR agonist effects, we utilized MOR KO mice. In MOR KO mice, MP (30 mg/kg, sc) antagonized icv U50, 488H antinociception (100 nmol), as well as icv SNC 80 antinociception (100 nmol) (Figure S6). These results are in line with our in vitro [35S]-GTPγS assays, where MP was an antagonist at these receptors. It also further corroborates that MP is centrally active when administered systemically.
To determine whether other targets might contribute to the antinociceptive actions of these compounds, we also screened the major alkaloids and mitragynine metabolites for binding affinity at ~50 targets (Table S1) and βarrestin-2 recruitment (7OH and 3DM only, ~318 targets, Figure 6A,B) in a broad panel of CNS receptors. Mitragynine, 7OH, and MP selectively bound opioid receptors, consistent with the sensitivity of antinociception to naloxone and genetic disruption of MOR, and consistent with recent reports on the minimal activity of these alkaloids at non-opioid receptors.42,64 Above, we showed that po administration of either KAE or mitragynine induced less hyperlocomotion or constipation than did morphine when administered at doses 5-fold higher than their respective antinociceptive ED50 values. Similarly, 7OH (1 mg/kg, sc in C57BL/6J mice, Figure S7C) and MP (1.5 mg/kg, sc in C57BL/6J, Figure S7D) induced less hyperlocomotion than morphine at equi-antinociceptive doses. However, 7OH did inhibit GI transit (Figure 2K), while MP in the same assay was reported previously to have a ceiling effect in GI transit assays7 (Figure 2L).
Figure 6.
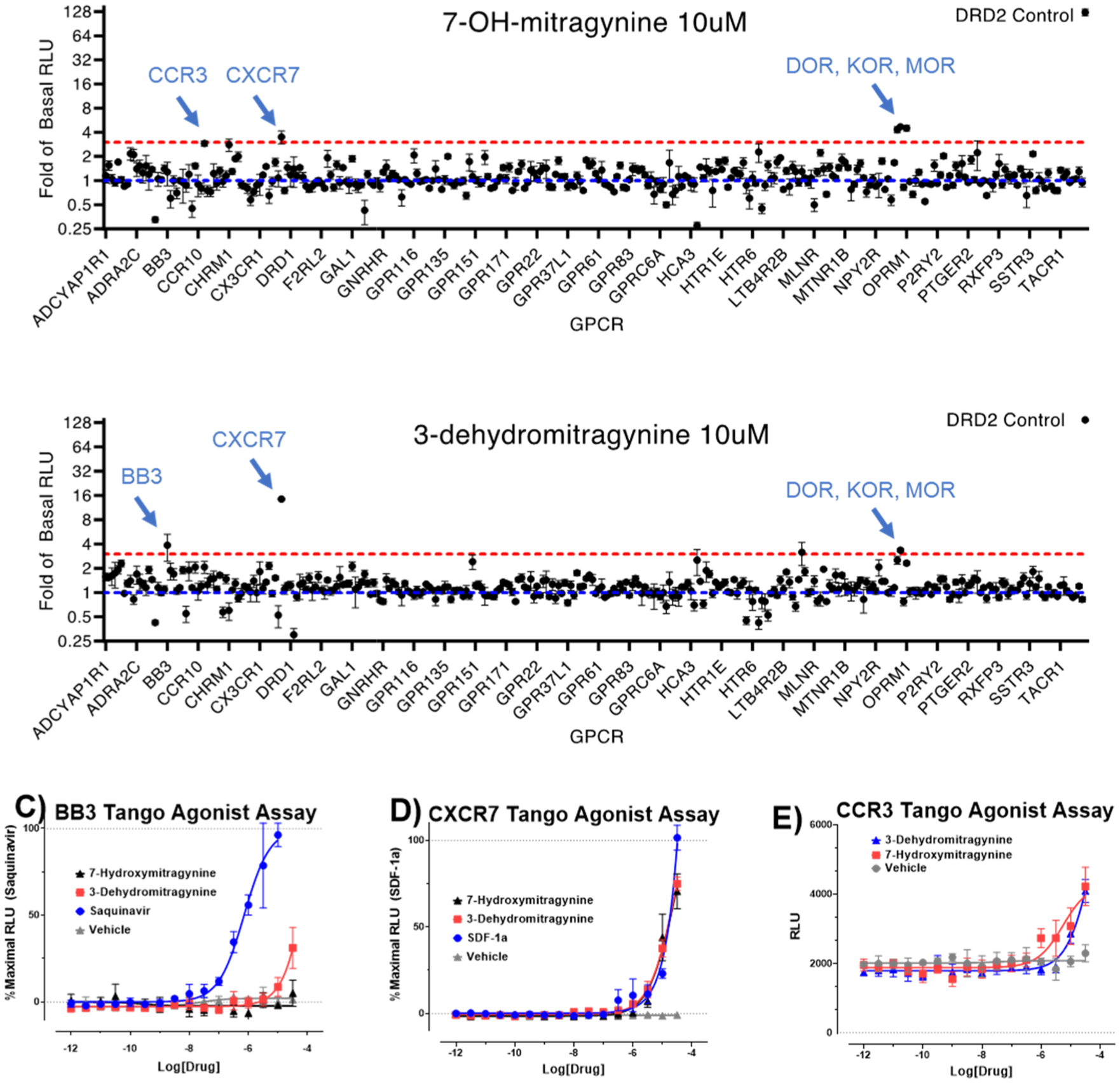
GPRome screening of 7OH and 3DM: 7OH and 3DM were screened against 318 non-olfactory GPCRs for agonism in the β-arrestin2 recruitment assay PRESTO-Tango. Each point shows change in the luminescence signal as fold-of-basal at a given GPCR with (A) 10 μM 7OH and (B) 10 μM 3DM. For clarity, GPCRs are listed alphabetically along the X-axis, and every 10th receptor is labeled. A full list of receptors is reported previously (Kroeze et al. PRESTO-Tango Nat. Struc. Mol. Bio. 201554). Both 7OH and 3DM induce increases in signal >3-fold of basal at opioid receptors. Non-opioid receptor screening hits (C) BB3, (D) CXCR7, and (E) CCR3 were tested against a concentration–response of 3DM, 7OH, and reference (if available) using PRESTO-Tango. Experiments were performed in triplicate and independently replicated three times.
Similar to po mitragynine, testing of the metabolite 7OH at 1 mg/kg, sc produced conditioned place preference (Figure 2O), while CPP was not reported in previous testing with MP7 in the C57BL/6J strain at doses 10-fold higher than its antinociceptive ED50 (3.2 mg/kg, sc in the C57BL/6J strain; Figure 2P). Notably, the finding of CPP following systemic administration of 7OH is consistent with previous reports examining this metabolite.56
Identification of 3-Dehydromitragynine as a Toxic Metabolite of Mitragynine.
We next closely evaluated the pharmacology of 3DM. The formation of 3DM35 from mitragynine was observed in vivo in mice of both strains (Figure 7D). Administration of the ED80 po antinociceptive dose of mitragynine resulted in detection of 0.094 ± 0.03 and 0.035 ± 0.01 μM concentrations of 3DM in the brains of C57BL/6J and 129Sv6 mice, respectively, coinciding with concentrations of 0.037 ± 0.01 and 0.028 ± 0.01 μM in plasma of C57BL/6J and 129Sv6 mice, respectively (Figure 7D). To further characterize the formation of 3DM, we looked at liver microsomes (Figure 4F), finding the formation of 3DM unlike 7OH or MP was not catalyzed by CYP3A (Figure 4I) and did not occur through 7OH as an intermediate, since incubation of 7OH with microsomes showed no formation of 3DM. The formation of 3DM from mitragynine was NADPH independent. 3DM formation was seen in both the presence and absence of NADPH in both HLM as well as S9 and in heat-inactivated HLM (Figure 5). Taken together, 3DM formation as a minor metabolite of mitragynine was found to be CYP3A independent. In vitro, 3DM had similar MOR affinity as 7OH in binding assays (Ki = 34.1 ± 5.9 nM), was a partial agonist at MOR (EC50 = 67.5 ± 8.3 nM; Emax = 44 ± 1%), and showed no other notable off–target activity beyond opioid receptors (Table 1 and Figure 6B). Poor (μM) activity at BB3, CXCR7, and CCR3 receptors (Figure 6C–E) in the PRESTO-Tango assay54 rule them out as possible targets for this molecule. Surprisingly, 3DM was not antinociceptive when administered directly to mice.
Figure 7.
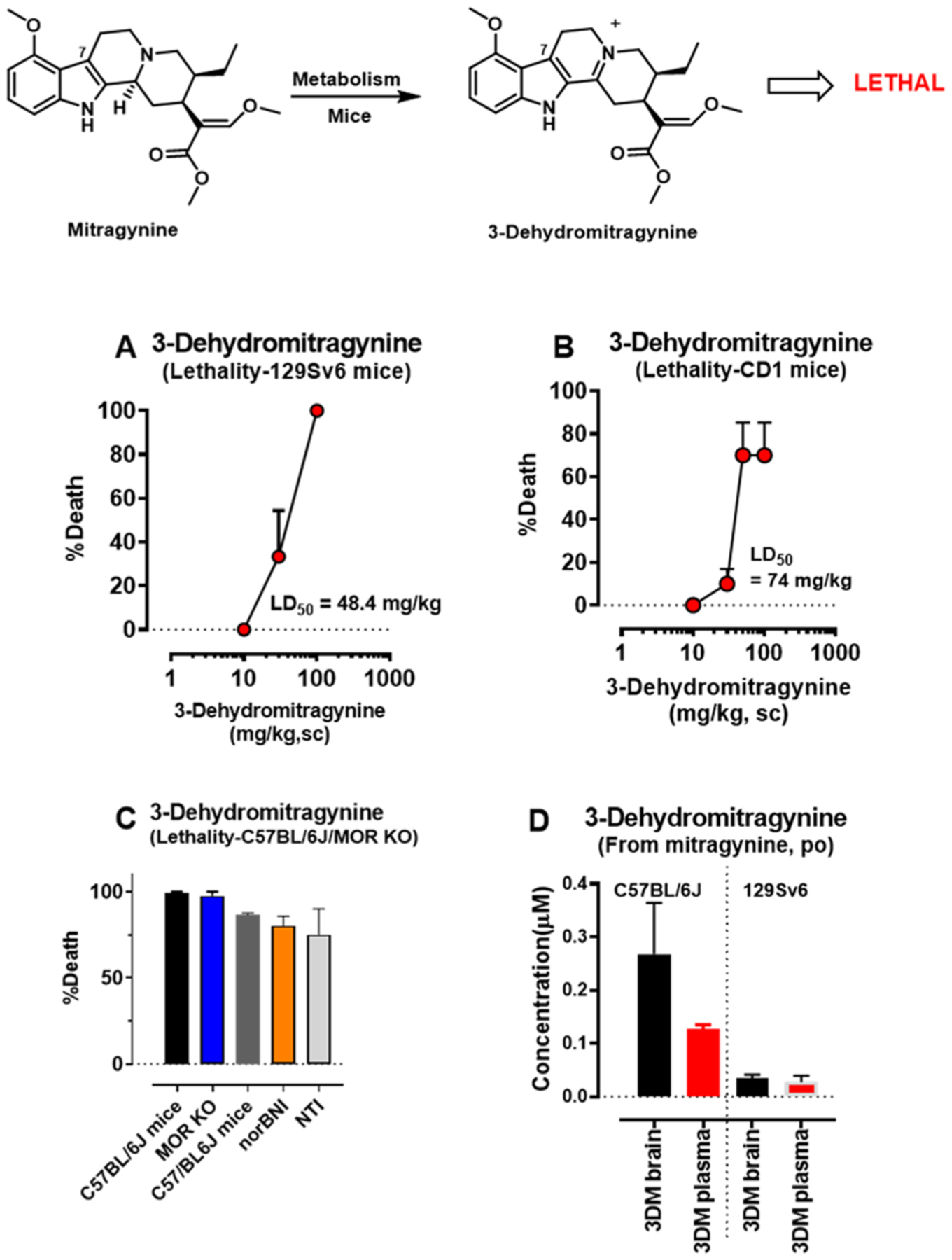
Detection of metabolic product 3-dehydromitragynine in brain and plasma in mice and its toxicity. Groups of mice (n = 6 per dose) were given different doses of sc 3DM to 129Sv6 mice (A) or CD1 mice (B) and tested for lethality. Similarly, C57BL/6J mice (n = 4) at 50 mg/kg, sc and MOR KO mice (n = 4) (C) at 50 mg/kg, sc and C57BL/6J mice pretreated with norBNI (10 mg/kg, sc; n = 15) for 24 h and NTI (20 mg/kg, sc; n = 12) for 15 min were tested for lethality. While 3-dehydromitragynine was not an antinociceptive agent, it was found to be toxic with a LD50 of 48.4 (20.85–129.8) mg/kg in 129Sv6 (A) and 74 (48.08–120.5) mg/kg in CD1 mice (B). It caused 100% lethality at 50 mg/kg, sc in C57BL/6J mice (C) and at 50 mg/kg, sc in MOR KO mice, suggesting that the toxicity is not MOR-mediated. Similarly, in the presence of KOR antagonist (nor-BNI) and DOR antagonist (NTI), the lethality was still observed. Results suggest lethality is opioid receptor-independent. The metabolite 3DM was detected at 0.035 ± 0.01 and 0.09 ± 0.025 μM in the brain of 129Sv6 and C57BL/6J mice when mice were dosed at ED80 po antinociceptive doses of mitragynine (D). Similarly, about 0.0275 ± 0.01 and 0.037 ± 0.01 μM were seen in plasma of 129Sv6 and C57BL/6J mice (D).
While not itself antinociceptive, 3DM was found to be toxic with 100% lethality after a 50 mg/kg, sc dose in C57BL/6J mice. Confirming this, 3DM demonstrated LD50 (and 95% C.I.) values of 48.4 (71.27–342.3) mg/kg sc in 129Sv6 and 74 (48.08–120.5) mg/kg, sc in CD1 mice (Figure 7A–C). However, the toxicity of this molecule seemed independent of mu, kappa, and delta opioid receptors, as either MOR KO mice or C57BL/6J mice treated with the kappa and delta opioid receptor antagonists, norbinaltorphimine (nor-BNI) and naltrindole (NTI), still demonstrated 3DM-mediated toxicity (Figure 7C). Supraspinal administration (10 μg) of 3DM to either CD1 or 129Sv6 mouse strains also led to fatality. At this time, the concentrations and half-life of 3DM achieved in plasma and brain following direct administration remain unknown, so interpretation of the toxicological relevance of its formation as a metabolite is admittedly difficult. Regardless, the present data demonstrates that mitragynine metabolizes to 3DM through a non-CYP mediated liver microsomal pathway and is lethal in multiple strains of mice.
DISCUSSION
The leaves of M. speciosa, also known as kratom, have been consumed by Southeast Asians for centuries. The pain-relieving properties of kratom have previously been attributed to the major indole alkaloid mitragynine. However, it has remained unclear how mitragynine acts as such a potent opioid antinociceptive despite having 50-fold weaker MOR binding affinity (230 nM) than morphine (4.6 nM) and only modest in vitro agonist potency (EC50 = 203 nM) and low efficacy at MOR (Table 1). Previous reports had shown that mitragynine administered by the oral route displayed more antinociceptive potency than when administered by parenteral routes,29,43 suggesting the potential involvement of active metabolites. Further, the opioid pharmacology of the other major alkaloids in kratom and the role they might play in the antinociceptive effects of the plant have remained less well understood. To better understand kratom’s pharmacological actions, we rigorously characterized in vitro and in vivo the opioid pharmacology of KAE, all the major natural kratom alkaloids (mitragynine, paynantheine, speciogynine, and speciociliatine) and minor (MNO) in pure form, and important metabolites of mitragynine. We show that mitragynine is converted to three biologically important metabolites (7OH, MP, and 3DM) in mice through two independent mechanisms. Formation of 7OH, as we have shown previously,29 was CYP3A-dependent and produced antinociception. A CYP3A-independent conversion of mitragynine led to 3DM, a newly identified and toxic metabolite. In this context, we would also like to state that mitragynine is very stable in pH 7.4 buffer. We recorded the 1H NMR spectroscopy (at 1, 5, 10, and 24 h time intervals) of mitragynine in D2O to confirm that it is stable in D2O and does not convert to 3DM at room temperature. We also confirmed this using LC/MS of mitragynine (at 1, 5, 10 and 24 h time intervals) incubated in pH 7.4 buffer. Again there was no formation of 3DM from mitragynine. We only see one peak that corresponds to mitragynine (1H NMR and LC/MS traces at different time intervals are provided in the SI).
The indole alkaloid mitragynine has at least five metabolically labile sites, including the C-9 methoxy group, C-7 unsaturated moiety (indole 2–3 double bond), C-3 axial hydrogen, and methyl (C-22), methoxy (C-17) acrylate functionalities (Figure 1B). Previous work suggested metabolism at these sites, and in particular, at the acrylate moiety.29,53 However, to our surprise, less than 1% of total plasma metabolites in mice involved the acrylate moiety. Further, previous literature has shown that hydrolysis of the acrylate ester to a carboxylic acid leads to a complete loss of opioid activity,19,21,29 suggesting that such metabolites do not play a major role in the pharmacological effects of mitragynine. In our PK studies in mice, we also observed demethylation of the 9-methoxy group to form 9OH, followed by glucuronidation to give 9G. Although 9OH showed an affinity similar to mitragynine at MOR (Ki = 216.2 ± 45.6 nM; Table 1), it did not produce antinociception when given subcutaneously at doses of up to 100 mg/kg and thus is not likely to contribute to mitragynine’s antinociception. Likewise, 9G was a weak MOR agonist (EC50 = 5.7 ± 1.36 μM) in vitro but not significantly antinociceptive due to lower potency; (Table 1).
The formation of 3DM35 from mitragynine, a natural product with unknown biosynthetic origins, was observed for the first time in both liver microsomes, S9 fractions, and in vivo in mice. 3DM has also been proposed to be a precursor which may lead to inhibition of CYP3A4 by Paine and co-workers.57 Though not antinociceptive in mice, 3DM was identified as a metabolite of mitragynine with lethal effects. These results with 3DM may mediate potential lethality of mitragynine and kratom, given the reported LD50 of po mitragynine in mice is 477 and 591 mg/kg for po kratom alkaloid fractions.58,59 In our hands, the LD50 of po mitragynine in the 129Sv6 strain was 750 mg/kg, while following administration of a 1 g/kg, po dose to C57BL/6J mice, 100% lethality was observed (data not shown). Importantly, administration of po mitragynine (1 g/kg) to C57BL/6J or MOR KO mice led to the death of all animals, confirming that the toxicity of mitragynine, similar to that of 3DM, does not seem to depend on MOR. While further study is warranted, this mechanistic similarity further suggests that 3DM may mediate the toxicity of mitragynine in rodents. Notably, the dose discrepancy between antinociception and fatality has precedence. In our previous work, the LD50 of morphine in CD1 mice was 750 mg/kg, sc while the antinociceptive ED50 value was 5 mg/kg.60 The therapeutic indices, calculated as the ratio of antinociceptive and lethal ED50 doses, are ~150 for both morphine sc and mitragynine po. Given the difference in metabolism between humans and rodents, it is premature to determine whether 3DM has toxic effects in humans. Further, when corrected by standard allometric scaling, the oral LD50 value of mitragynine observed in rodents corresponds to >2.5 g of mitragynine in an adult human. At typical concentrations of mitragynine found in dry leaf powder available in the US (~1%), such an exposure would require consumption of >250 g of plant matter, a situation that seems unlikely given that most human users report consumption of <10 g per dose. Nevertheless, considering a number of reported deaths associated with kratom use26,61 mediated by unknown causes, caution in the human usage of kratom is advisable until further studies of 3DM are available.
Our mouse PK studies also identified 7OH (major) and MP (minor) as metabolites of mitragynine. Inconsistent with its affinity and functional activity at MOR, po mitragynine in both mouse strains produced potent antinociception, with an ED50 value comparable to that of morphine. However, the higher potency of 7OH and MP in functional assays and antinociception in mice suggest that they are likely contributors to the analgesic effects of mitragynine. The brain/plasma ratio of 7OH was found to be 1.5:1, while MP appeared less brain permeable in our studies with ratios of 1:6. The higher brain permeability of 7OH might explain why despite its moderate binding (37 nM) and MOR agonistic potency (EC50 = 53 nM), 7OH displays a 10-fold greater analgesic potency over morphine and comparable potency to MP in the C57BL/6J strain.
Detailed PK studies with multiple time points and doses are required to conclusively prove this hypothesis, which is beyond the scope of this paper. The present results corroborate previous findings identifying 7OH as the key element in the antinociceptive actions of mitragynine.29
KAE, mitragynine, 7OH, and MP showed reduced hyperlocomotion compared to morphine while KAE, mitragynine, and 7OH all induced CPP in mice, suggesting that these substances are likely to be habit forming (Figure 2M–O). Recent reports have shown that mitragynine is not self-administered in rats given by the intravenous (iv) route.62,63 Our results contrast with these studies, perhaps because iv administration of mitragynine enables less first pass metabolism of the drug, and in turn, attenuates formation of the 7OH concomitant to MOR-mediated reward. It is important to note that this discrepancy may also be due to differences in species (rats instead of mice), assay performed (self-administration vs CPP), or low doses tested in the self-administration assay (0.5 mg/kg/injection in Hemby et al. and 3 mg/kg/injection in Yue et al.), doses at which the drug does not show antinociception in mice in our hands.
Differences between the KAE, mitragynine and 7OH were seen in their ability to cause constipation. KAE and mitragynine showed reduced constipation while 7OH was similar to morphine in this assay. MP in the same assay behaved like a partial agonist. It is possible that the non-opioid actions42,64,65 of mitragynine may be responsible for reduced constipation over 7OH which exclusively labels MOR. More studies are required to test this observation.
As an indole alkaloid, the metabolism of mitragynine may provide broader insight into the metabolism of other indole-based drugs, which is important given that over 41% of all drugs approved by the FDA are natural products or derived analogues.66,67 The oxidative lability of the C-3 position of an indole ring (C-7 position in the case of mitragynine) has been previously reported62,68 and conversion of mitragynine to mitragynine pseudoindoxyl via the transformation of an indole ring to a spirocyclic core through microbial fermentation is known.69 However, we believe that the importance of metabolic oxidation at this position is still underappreciated. Here, we report for the first time a CYP-mediated transformation of indole to a spirocyclic pseudoindoxyl core. Incubation of 7OH with liver microsomes showed no conversion to MP (though a recent report shows formation in human plasma),70 suggestive of a distinct CYP3A-mediated conversion of an indole (mitragynine) to a spirocyclic pseudoindoxyl (MP), which does not involve oxidation of the C-3 position (7OH) as a precursor. In addition, we present the first insight into a potential biosynthetic route to 3DM via C-3 oxidation of mitragynine.
CONCLUSIONS
To summarize, we present a comprehensive evaluation of kratom’s pharmacological actions in vitro and in vivo. We show that KAE, mitragynine, and the mitragynine metabolites 7OH and MP are potent MOR antinociceptive agents. Oral administration of mitragynine displays antinociceptive activity similar to sc morphine. The antinociception of po mitragynine results from its bioactivation to potent 7OH (major) and MP (minor) metabolites, while the parent compound mitragynine is not itself antinociceptive. We also report for the first time that metabolism of mitragynine leads to a highly toxic metabolite, 3DM. Further study is undoubtedly required to elucidate the relevance, if any, of this toxic metabolite to reported kratom toxicities and fatalities in man, but caution among kratom consumers in the interim is advised. Kratom, mitragynine, and the metabolite 7OH may be safer antinociceptive agents in view of their attenuated locomotor activity and reduced ability to cause GI transit inhibition for mitragynine and kratom. However, they still retain at least some degree of abuse potential, as evidenced by our findings in the CPP paradigm.
EXPERIMENTAL SECTION
Chemistry.
General Methods.
Reagents purchased from Sigma-Aldrich Chemicals, Fisher scientific, Alfa Aesar, were used without further purification. Reaction mixtures were purified by silica gel flash chromatography on E. Merck 230–400 mesh silica gel 60 using a Teledyne ISCO CombiFlash Rf instrument with UV detection at 280 and 254 nm. RediSep Rf silica gel normal phase columns were used with a gradient range of 0–10% MeOH in DCM. The yields reported are isolated yields (UPLC purity ≥95%). NMR spectra were recorded on Bruker Avance III 500, Avance III 600 with DCH CryoProbe instruments. Chemical shifts are reported in parts per million (ppm) relative to residual solvent peaks at the nearest 0.01 for proton and 0.1 for carbon (CDCl3 1H: 7.26, 13C: 77.1; and CD3OD 1H: 3.31, 13C: 49.0). Peak multiplicity is reported as the NMR spectra were processed with MestreNova software, namely, s—singlet, d—doublet, t—triplet, q—quartet, and m—multiplet for examples. Coupling constant (J) values are expressed in Hz. Mass spectra were obtained at the MSKCC Analytical Core Facility using The Waters Acuity SQD LC MS by electrospray ionization (ESI). High-resolution mass spectra were obtained on a Waters Acuity Premiere XE TOF LC–MS by ESI, and the accurate masses are reported for the molecular ion [M + H]+. All compounds are >95% pure by UPLC analysis. Purity of the products (≥95%) was confirmed by Waters Acquity UPLC: equipped with a binary solvent manager system, Waters XBridge C18 column (1.7 μm × 2.1 × 100 mm), PDA, ELS, and QDa mass detectors; mobile phase: solvent A: water with 0.1% TFA; solvent B: acetonitrile with 0.1% TFA. The flow rate was 1 mL/min in a reversed phase; total run time 8 min.
Isolation of Mitragynine from M. speciosa (Kratom).
Mitragynine was extracted from the powdered leaves by following our previously reported methods,7 which is a modified method with an extra step of petroleum ether wash of the acidic aqueous phase reported by Ponglux et al.71 Kratom powder (500 g) was heated to reflux in MeOH 700 mL for 40 min. The suspension was filtered, and the methanolic extraction process was repeated (3 × 500 mL). The solvent of combined methanolic extract was removed under reduced pressure and the content was dried using high vacuum. The dry residue was resuspended in 20% acetic acid solution (1 L) and washed with petroleum ether (4 × 500 mL). The aqueous layer was then cooled on ice bath and basified (pH ~ 9) with aqueous NaOH solution (3.5 M. ~ 1 L) slowly. Alkaloids were extracted in DCM (4 × 400 mL) from the aqueous layer. The combined DCM layer was washed with brine 300 mL and dried over anhydrous Na2SO4 and filtered. The solvent was removed under reduced pressure, and the residue was dried under high vacuum to obtain kratom alkaloid extract (11 g). Then, 200 mg of the kratom extract was subjected to silica gel column chromatography, using 0–40% EtOAc in hexanes to isolate mitragynine (97.2 mg); paynantheine (11.6 mg), speciogynine (7 mg), and 0–15% MeOH in DCM to isolate speciociliatine (27.4 mg), which correspond to approximately 48.6, 5.8, 3.5, and 13.7% of each alkaloids in the kratom extract. After removing the mitragynine fraction out, the total alkaloid content in the enriched alkaloid extract 2 (alkaloid 2; devoid of mitragynine), when isolated as a combined fraction using the above protocol, approximately contains paynen-theine (24%), speciogynine (14%), and speciociliatine (57%) as the major alkaloids.
Synthesis and Characterization of New Compounds.
(2S,3S)-2-((E)-1,3-Dimethoxy-3-oxoprop-1-en-2-yl)-3-ethyl-8-methoxy-2,3,4,6,7,12-hexahydro-1H-indolo[2,3-a]quinolizin-5-ium TFA Salt (3DM TFA).
An ice-cold solution of trifluoroacetic acid (150 μL, 4.2 mmol) dissolved in DCM (1.0 mL) was added to the stirred solution of 7 hydroxymitragynine (330 mg, 0.79 mmol) in DCM (6 mL) at 0 °C under inert atmosphere. LC/MS indicated that the dehydration reaction was clean and completed in 2 h at 0 °C. Solvent and the excess of reagent were removed under reduced pressure to yield 3-dehyromitragynine TFA salt. The salt was redissolved in a mixture of acetonitrile in water (1:3, 15 mL) and was lyophilized to get 3DM TFA salt as a yellow solid, (405 mg, quantitatively). 1H NMR (600 MHz, chloroform-d): δ 12.24 (s, 1H), 7.49 (s, 1H), 7.28−7.25 (m, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.41 (d, J = 7.7 Hz, 1H), 3.90 (s, 3H), 3.88−3.80 (m, 2H), 3.75 (s, 3H), 3.61 (s, 3H), 3.59−3.51 (m, 3H), 3.50−3.40 (m, 3H), 3.28 (t, J = 12.9 Hz, 1H), 2.04 (ddt, J = 10.3, 7.7, 5.3 Hz, 1H), 1.46 (ddd, J = 13.7, 7.5, 5.9 Hz, 1H), 1.19−1.13 (m, 1H), 0.97 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 168.3, 166.9, 162.4, 155.9, 143.3, 130.1, 125.2, 122.4, 107.7, 107.0, 100.0, 62.1, 55.3, 54.3, 52.9, 51.5, 38.5, 31.5, 28.0, 23.0, 21.3, 11.6. HRMS calcd for C23H29N2O4 [M]+, 397.2127; found, 397.2114.
(2S,3S,5R,6S)-6-(((2S,3S)-2-((E)-1,3-Dimethoxy-3-oxoprop-1-en-2-yl)-3-ethyl-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-8-yl)oxy)-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic Acid (9G).
The 9G was prepared by following a previously reported protocol with the modification.72 2,3,4-Tri-O-acetyl-α-d-glucuronic acid methyl ester and trichloroacetimidate (120.4 mg, 0.10 mmol) were added to the solution of 9 hydroxy corynantheidine (50 mg, 0.31 mmol) stirred in DCM (6 mL), over a molecular sieve, at rt under argon. The mixture was cooled to −78 °C bath for 5 min, BF3·OEt2 (42.1 μL, 0.15 mmol) was added, and the reaction was continued for 10 min at the temperature. The cold bath was removed, and the reaction was continued over ice bath (at 0 °C) for 5 h. The reaction mixture was quenched with MeOH (30 μL) at the temperature, and the volume was concentrated down under reduced pressure. The concentrate was redissolved in DCM (15 mL), and the organic layer was washed with saturated aqueous NaHCO3 solution (2 × 15 mL), brine (15 mL), dried over anhydrous Na2SO4, filtered, concentrated, and dried under reduced pressure. The crude material was redissolved in DCM (6 mL) and cooled to 0 °C. A solution of NaOMe (62 μL, 0.31 mmol; 0.5 M) was added at 0 °C, and the reaction was continued for 10 min; MS indicated that deacetylation was completed. Water (30 μL) was added, and the reaction was continued for 10 min to complete the hydrolysis of methyl ester. The reaction mixture was diluted with water (3 mL), acidified to pH 6 using acetic acid (20 μL), and the volatilities were removed under reduced pressure. The pH of the aqueous layer (pH ~ 6) was adjusted to slightly basic (pH ~ 8) using saturated aqueous NaHCO3 solution, and the aqueous layer was washed with diethyl ether (3 × 5 mL) to ensure the removal of unreacted 9-hydroxy corynantheidine and the small amount of 9-acetyl corynantheidine byproduct; MS indicated the 9G as the major peak. Then, the aqueous layer was loaded into the anion exchange resin, (Amberlite IRA-410 chloride, 20–25 mesh, 5 g pre-incubated in MeOH for 30 min and washed) and incubated in 10 min. The flow through the aqueous layer was collected with argon positive pressure and reloaded to the resin; and the process was repeated 5–6 times to optimize loading, MS of the final flow through indicated only a trace amount the product. The resin was washed with acetonitrile, EtOAc, and water (5 mL of each) to remove trace amount of impurities. Finally, the product was eluted with aqueous acetic acid (0.5%, 14 mL), and the aqueous layer was lyophilized to obtained the desired product 9G as a white solid; (yield: 9.2 mg, 16%).1H NMR (600 MHz, methanol-d4): δ = 7.63 (d, J = 3.4 Hz, 1H), 7.19−7.00 (m, 2H), 6.77 (dd, J = 6.9, 1.7 Hz, 1H), 5.14 (d, J = 7.5 Hz, 1H), 4.23 (d, J = 12.3 Hz, 1H), 3.99 (d, J = 9.7 Hz, 1H), 3.86 (s, 3H), 3.72 (s, 5H), 3.68−3.61 (m, 2H), 3.60−3.47 (m, 3H), 3.46−3.32 (m, 2H), 2.80−2.60 (m, 1H), 2.40 (d, J = 14.5 Hz, 1H), 1.99 (s, 1H), 1.65 (td, J = 13.5, 12.8, 7.0 Hz, 1H), 1.51−1.42 (m, 1H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, MeOD): δ 172.2, 169.8, 163.2, 153.0, 140.0, 128.3, 124.3, 118.86, 110.0, 107.6, 106.6, 105.7, 102.9, 77.7, 76.6, 74.9, 73.1, 64.2, 62.6, 56.9, 55.0, 52.0, 40.3, 38.6, 28.4, 21.8, 19.3, 12.6. HRMS calcd for C28H35N2O10 [M + H]+, 561.2448; found, 561.2443.
(2S,3S)-12-(tert-Butoxycarbonyl)-2-(€-1,3-dimethoxy-3-oxoprop-1-en-2-yl)-3-ethyl-8-methoxy-1,2,3,4,6,7,12,12b-octahydro-5H-indolo[2,3-a]quinolizine 5-Oxide (Boc-MNO).
Di-tert-butyl dicar-bonate (41.0 mg, 0.18 mmol) and DMAP (5 mg, 0.04 mmol) were added to the stirred solution of mitragynine (50 mg, 0.12 mmol) dissolved in THF (5.0 mL) at rt. under argon. The reaction mixture was heated to reflux at 80 °C for 4 h. The reaction mixture was concentrated under reduced pressure, the residue was redissolved in EtOAc (20 mL), and the organic layer was washed with saturated aqueous NaHCO3 (20 mL), brine (20 mL), dried over anhydrous Na2SO4, filtered, concentrated, and the crude product was purified by SiO2 column chromatography (ISCO, 4 g) using 1–20% MeOH in DCM to get Boc-mitragynine as a white solid, (yield: 58 mg, 93%); 1H NMR (500 MHz, chloroform-d): δ 7.72 (d, J = 8.4 Hz, 1H), 7.39 (s, 1H), 7.13 (t, J = 8.2 Hz, 1H), 6.62 (d, J = 8.0 Hz, 1H), 3.86 (s, 3H), 3.69 (s, 3H), 3.68 (s, 3H), 3.60−3.54 (m, 1H), 3.10 (dt, J = 13.2, 3.6 Hz, 1H), 3.07−3.01 (m, 2H), 2.91−2.83 (m, 2H), 2.72−2.61 (m, 2H), 2.23 (td, J = 13.0, 10.4 Hz, 1H), 1.95 (d, J = 12.8 Hz, 1H), 1.72 (ddd, J = 13.8, 11.0, 7.1 Hz, 1H), 1.59 (s, 9H), 1.56 (d, J = 16.1 Hz, 1H), 1.21 (qd, J = 7.4, 2.9 Hz, 1H), 0.86 (t, J = 7.4 Hz, 3H). ESI-MS m/z: 499.2 [M + H]+.Then, the meta-Chloroperoxybenzoic acid (41.8 mg, 0.24 mmol) was added to the stirred solution of Boc-mitragynine (110 mg, 0.22 mmol) dissolved in DCM (3.0 mL) at 0 °C under argon and the reaction was continued overnight. The solvent was removed under reduced pressure, and the crude product was purified by alumina basic column chromatography (ISCO, 8 g) using MeOH (3–5%) in DCM to get Boc-mitragynine N-oxide (Boc-MNO) as a white solid, (yield: 90 mg, 79%); 1H NMR (600 MHz, chloroform-d): δ 7.70 (d, J = 8.4 Hz, 1H), 7.42 (s, 1H), 7.15 (t, J = 8.2 Hz, 1H), 6.63 (d, J = 8.0 Hz, 1H), 4.93−4.87 (m, 1H), 4.05−3.99 (m, 1H), 3.87 (s, 3H), 3.82 (ddd, J = 15.5, 9.8, 4.6 Hz, 2H), 3.78 (s, 3H), 3.68 (s, 3H), 3.65 (dd, J = 11.4, 5.7 Hz, 1H), 3.51 (dt, J = 12.8, 5.0 Hz, 1H), 3.34 (dd, J = 11.6, 6.0 Hz, 1H), 3.17 (dd, J = 16.8, 4.5 Hz, 1H), 2.40−2.35 (m, 1H), 2.29 (dt, J = 15.2, 3.4 Hz, 1H), 2.22 (s, 1H), 1.63 (s, 9H), 1.46 (ddq, J = 21.5, 14.3, 7.2 Hz, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ 168.4, 161.1, 154.2, 149.7, 137.7, 130.3, 125.0, 118.3, 112.1, 109.6, 109.0, 103.64, 84.2, 74.3, 73.8, 61.9, 57.9, 55.4, 53.5, 51.5, 32.8, 28.2, 21.2, 20.4, 12.5. ESI-MS m/z: 515.2 [M + H]+.
(2S,3S)-2-((E)-1,3-Dimethoxy-3-oxoprop-1-en-2-yl)-3-ethyl-8-methoxy-1,2,3,4,6,7,12,12b-octahydro-5H-indolo[2,3-a]quinolizine 5-Oxide (MNO).
An ice-cold solution of trifluoroacetic acid in DCM (1:1, 1.0 mL) was added to the stirred solution of Boc-MNO (15 mg, 0.03 mmol) dissolved in DCM (0.5 mL) at 0 °C under an argon. After 10 min, the ice bath was removed, and the reaction was continued for 60 min. Volatilities were removed under reduced pressure, and the crude product was purified by HPLC using acetonitrile/water with 0.1% TFA to get MNO as a white solid, (yield: 5.8 mg, 48%); 1H NMR (600 MHz, methylene chloride-d2): δ 8.92 (s, 1H), 7.28 (s, 1H), 7.06 (t, J = 8.0 Hz, 1H), 6.92 (d, J = 8.2 Hz, 1H), 6.45 (d, J = 7.8 Hz, 1H), 5.09 (dd, J = 9.3, 3.6 Hz, 1H), 4.44 (s, 1H), 3.98 (dt, J = 12.2, 5.3 Hz, 1H), 3.87 (ddd, J = 12.2, 7.5, 4.2 Hz, 1H), 3.82 (s, 3H), 3.75−3.69 (m, 1H), 3.66−3.53 (m, 3H), 3.50 (s, 3H), 3.45 (t, J = 7.4 Hz, 1H), 3.31−3.17 (m, 2H), 2.55−2.35 (m, 3H), 1.29 (dq, J = 14.5, 7.2 Hz, 1H), 1.21 (dq, J = 14.4, 7.2 Hz, 1H), 0.88 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CD2Cl2): δ 168.6, 161.5, 154.7, 138.3, 125.6, 123.9, 117.4, 116.2, 115.5, 108.4, 105.2, 104.9, 100.1, 68.9, 62.6, 62.0, 55.4, 51.6, 34.6, 30.9, 22.0, 20.3, 12.0. HRMS calcd for C23H30N2O5 [M + H]+, 415.2233; found, 415.2228.
Biological Methods.
Drugs and Materials.
Opiates were provided by the Research Technology Branch of the National Institute on Drug Abuse (Rockville, MD). IBNtxA and [125I]BNtxA were synthesized in our laboratory as previously described.73,74 Na125I and [35S]GTPγS were purchased from PerkinElmer (Waltham, MA). Selective opioid antagonists were purchased from Tocris Bioscience or Sigma-Aldrich. Miscellaneous chemicals and buffers were purchased from Sigma-Aldrich. Moon Kratom “Red Indonesian Micro Powder was purchased from Moon Kratom (Austin, TX).
Mice.
Mice including CD1, 129Sv6, and C57BL/6J (24–38 g) were purchased from Jackson Laboratories (Bar Harbor, ME). Exon-1/Exon-11 MOR KO mice were bred in the Pasternak/Pan laboratory at Sloan Kettering, while DOR and KOR KO mice in 129Sv6 strain were bred in the Pintar laboratory at Rutgers University. Exon 2 MOR KO mice on the C57BL/6J background were bred in the McLaughlin laboratory at University of Florida, from progenitors obtained from commercial sources [Jackson Laboratories (Bar Harbor, ME)]. All mice used throughout the manuscript were opioid naïve. All mice were maintained on a 12-h light/dark cycle with Purina rodent chow and water available ad libitum and housed in groups of five until testing. All animal studies were preapproved by the Institutional Animal Care and Use Committees of the Memorial Sloan Kettering Cancer Center or University of Florida or Rutgers University, in accordance with the 2002 National Institutes of Health Guide for the Care and Use of Laboratory Animals. The formulation used for all mice experiments was 1:1:8 of DMSO/solutol/saline. The volume of injection varied with the weight of mice, with 250 μL for a 25 g mouse typically used.
Power analysis to justify minimal numbers of animals was used. Mice are a valuable resource, prompting the application of power analysis to determine the minimal significant subject number. On the basis of analysis aiming for 95% power with an alpha significance of p = 0.05 and an ANOVA analysis with repeated measures within-between interaction, G*Power 3.19.2 was used to calculate a minimum number mice per antinociceptive of place conditioning testing condition. For the tail-withdrawal (“tail-flick”), a moderate effect size (f = 0.7) and 10 measurements assumed over time, resulting in an estimated sample size of eight mice, with a predicted critical F-value of 1.80 and actual power = 0.996. Assuming a large effect size (f = 2.6) and four treatment groups, eight mice were estimated needed for the GI transit testing, with a predicted critical F-value of 6.59 and actual power = 0.960. Assuming a small effect size (f = 0.5) and two measures, G*Power estimated 24 mice per condition for conditioned place preference/aversion were needed to achieve statistical significance in this work, defined with a critical F-value of 3.098, with actual power = 0.973.
Radioligand Competition Binding Assays.
[125I]IBNtxA binding was carried out in membranes prepared from Chinese Hamster Ovary (CHO) cells stably expressing murine clones of MOR, DOR, and KOR, as previously described.73,74 Binding in MOR/CHO was carried out in 50 mM potassium phosphate buffer with 5 mM MgSO4 and 20 μg/mL protein, while binding in KOR/CHO and DOR/CHO was carried out in 50 mM potassium phosphate pH = 7.0 buffer and 40 μg/mL protein at 25 °C for 90 min. After the incubation, the reaction was filtered through glass-fiber filters (Whatman Schleicher & Schuell, Keene, NH) and washed (3 × 3 mL of ice-cold 50 mM Tris–HCl, pH 7.4) on a semiautomatic cell harvester. Nonspecific binding was determined by the addition of levallorphan (8 μM) to matching samples and was subtracted from total binding to yield specific binding. Ki values were calculated by nonlinear regression analysis (GraphPad Prism, San Diego, CA). Protein concentrations were determined using the Lowry method75 with BSA as the standard.
[35S]GTPγS Functional Assay.
[35S]GTPγS binding was performed on membranes prepared from transfected cells stably expressing opioid receptors in the presence and absence of the indicated compound for 60 min at 30 °C in the assay buffer (50 mM Tris–HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 10 mM NaCl) containing 0.05 nM [35S]GTPγS; 2 μg/mL each leupeptin, pepstatin, aprotinin, and bestatin; and 30 μM GDP, as previously described.7,76,77 After the incubation, the reactions were filtered through glass fiber filters (Whatman Schleicher & Schuell, Keene, NH) and washed (3 × 3 mL of ice-cold buffer, 50 mM Tris–HCl, pH 7.4) on a semi-automatic cell harvester. Filters were transferred into vials with 3 mL of Liquiscint (National Diagnostics, Atlanta, GA), and the radioactivity in vials was determined by scintillation spectroscopy in a Tri-Carb 2900TR counter (PerkinElmer Life and Analytical Sciences). Basal binding was determined in the presence of GDP and the absence of drug. Data were normalized to 100 nM DAMGO, DPDPE, and U50, 488H for MOR, DOR, and KOR binding, respectively. EC50, IC50, and %Emax values were calculated by nonlinear regression analysis (GraphPad Prism, San Diego, CA).
Antinociception. Pasternak/Pan Labs.
Tail flick antinociception in 129Sv6 mice was determined using the radiant heat tail flick technique using an Ugo Basile model 37360 instrument as previously described.7,76,78 The intensity was set to achieve a baseline between 2 and 3 s. Baseline latencies were determined before experimental treatments for all mice. Tail flick antinociception was assessed quantally as a doubling or greater of the baseline latency, with a maximal 10 s latency to minimize damage to the tail. Data were analyzed as the percent maximal effect, %MPE, and was calculated according to the formula: % MPE [(observed latency − baseline latency)/(maximal latency − baseline latency)] × 100. Compounds were administered either orally (po) or subcutaneously (sc) or intracerebroventricularly (icv), and antinociception was assessed at the peak effect. Intracerebroventricular dosing (icv) was carried out as described previously. In brief, mice were anesthetized using isoflurane. A small incision was made, and the drug (2 μL/mouse) was injected (using a 10 μL Hamilton syringe fitted to a 27-gauge needle) into the right lateral ventricle at the following coordinates: 2 mm caudal to bregma, 2 mm lateral to sagittal suture, and 2 mm in depth. Mice were tested for antinociception at the peak effect post injection. For the antagonism studies, naloxone (1 mg/kg, sc) was administered 15 min prior to the drug administration.
McLaughlin Lab.
The 55 °C warm-water tail-withdrawal assay was conducted in C57BL/6J mice as a measure of acute thermal antinociception as described previously.79 Briefly, each mouse was tested for baseline tail-withdrawal latency prior to drug administration. Following drug administration, the latency for each mouse to withdraw the tail was measured every 10 min until latency returned to the baseline value. A maximum response time of 15 s was utilized to prevent tissue damage. If the mouse failed to display a tail-withdrawal response within 15 s, the tail was removed from the water and the animal was assigned a maximal antinociceptive score of 100%. Data are reported as percent antinociception, calculated by the equation: % antinociception = 100 × [(test latency − baseline latency)/(15 − baseline latency)]. This was utilized to account for innate variability between mice. Compounds were administered subcutaneously (sc), and the antinociceptive action of compounds was assessed at the peak effect.
Conditioned Place Preference and Aversion.
Mice were conditioned with a counterbalanced place conditioning paradigm using similar timing as detailed previously.7,10 Groups of mice (n = 18–24) were habituated to freely explore both sides of a three-compartment apparatus for 3 h each for 2 days prior to testing. The amount of time subjects spent in each of three compartments was measured over a 20 min testing period. Prior to place conditioning, the animals did not demonstrate significant differences in their time spent exploring the left versus right compartments. During each of the next 2 days, mice were administered vehicle (0.9% saline) and consistently confined in a randomly assigned outer compartment for 20 min, half of each group in the right chamber, half in the left chamber. Four hours later, mice were administered drugs morphine (10 mg/kg/d, sc), U50, 488H (30 mg/kg/d, sc), Kratom alkaloid extract (100 mg/kg/d, po), mitragynine (30 mg/kg/d, po), or 7-OH (1 mg/kg/d; 3.2 mg/kg/d, sc) and were confined to the opposite compartment for 20 min. Conditioned place preference data are presented as the difference in time spent in drug- and vehicle-associated chambers and were analyzed via repeated measures two-way ANOVA with the difference in time spent on the treatment-versus vehicle-associated side as the dependent measure and conditioning status as the between-groups factor. Where appropriate, Tukey’s HSD or Sidak’s multiple comparison post hoc tests were used to assess group differences. Effects were considered significant when p < 0.05. All effects are expressed as mean ± SEM.
Assessment of GI Transit.
C57BL/6J mice (8 per drug treatment) were administered saline (0.9%, s.c.), morphine (10 mg/kg, s.c.), KAE (85.5 mg/kg, p.o.), mitragynine (25 mg/kg, p.o.), 7OH (1.5 mg/kg, s.c.), or mitragynine pseudoindoxyl (0.75–4 mg/kg, s.c.) 20 min prior to oral gavage with 0.3 mL of a 5% aqueous solution of charcoal meal. After 3 h, mice were euthanized, and the intestines were removed. The progression of charcoal through the intestines was measured as distance traveled from the jejunum to the cecum as utilized elsewhere.80,81
Pharmacokinetic Study.
Mitragynine was administered to mice either orally or subcutaneously at appropriate doses in C57BL/6J and 129Sv6 mice.82 At peak effects, after 15 or 20 min post administration of the drug, mice were anesthetized under isoflurane, blood was removed, and animals were sacrificed for brain removal. Blood was separated to plasma in K2EDTA tubes by centrifugation and then plasma was extracted with 50% volume of methanol, vortexed, pelleted, and supernatants dried down. Brains were quickly rinsed off with PBS, blot-dried, homogenized 50% methanol in water, pelleted, and supernatants dried down to analyze metabolites. Each brain sample reflects material recovered from half a brain. The dried-down metabolites content was resuspended in the acetonitrile/water mixture (100 μL) prior to the LC–MS/MS analysis. In the case of direct plasma analysis, 5 μL of plasma was mixed with 100 μL of 80/20 acetonitrile/water (containing 5 ng/mL carbamazepine, an internal standard), centrifuged, and passed through a filter plate (Millipore Multiscreen Solvinter 0.45 μm, low binding PTFE hydrophilic filter plate). Samples were analyzed by LC–MS/MS.
Liver Microsomal Assays.
Liver microsomal assays were performed by following the previously reported protocols.82 Briefly, microsome stability of the kratom alkaloid was evaluated by incubating mitragynine(1 μM) with 1 mg/mL hepatic microsomes (human, rat, or mouse) in a 100 mM potassium phosphate buffer, pH 7.4. The reactions were held at 37 °C with continuous shaking. The reaction was initiated by adding NADPH, 1 mM final concentration. The final incubation volume was 300, and 40 μL aliquots were removed at 0, 5, 10, 20, 40, and 60 min. The removed aliquot was added to 160 μL acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction was evaluated in parallel incubations without NADPH. Mitragynine metabolism by CYP3A4 was verified using a validated 3A4 inhibitor assay using 1 μM mitragynine in the presence of 1 μM ketoconazole for selective 3A4 blockade. At the end of the assay, the samples are centrifuged through a 0.45-μm filter plate (Millipore Solventer low binding hydrophilic plates, cat# MSRLN0450) and analyzed by LC–MS/MS. The data were log-transformed, and the results are reported as half-life. Note: Protein concentration for both liver micrsomes ans well as S9 was 1 mg/mL. For S9 fraction experiments, we simply swapped microsomes with S9. S9 was bought from XenoTech. Varapamil was used as control for S9 experiemnts. The half-life in min for verapamil was 24.14 min (data not shown).
Assessment of Off–target Activity of Kratom alkaloids and Metabolite Using PRESTO-Tango GPCR-ome.
To identify potential off–target activity of 7OH and 3DM, we used the National Institutes of Mental Health Psychoactive Drug Screen Program. 3DM and 7OH was first tested for activity against 330 non-olfactory GPCRs using the PRESTO-Tango GPCRome screening β-arrestin2 recruitment assay at 10 μM. The activity at each receptor was measured in quadruplicate. Screening of compounds was accomplished using previously described methods with several modifications (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4424118/).54 First, HTLA cells were plated in DMEM with 2% dialyzed FBS and 10 U/mL penicillin–streptomycin. Next, the cells were transfected using an in-plate PEI method (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4012321/).55 PRESTO-Tango receptor DNAs were resuspended in OptiMEM and hybridized with PEI prior to dilution and distribution into 384-well plates and subsequent addition to cells. After overnight incubation, drugs diluted in DMEM with 1% dialyzed FBS were added to cells without replacement of the medium. The remaining steps of the PRESTO-Tango protocol were followed as previously described.
Supplementary Material
ACKNOWLEDGMENTS
S.M. and D.S. are supported by the National Institute on Drug Addiction (NIDA) of the National Institute of Health (NIH), grants R21DA045884 and R01DA046487. S.M. is further supported by AA026949 and W81XWH-17-1-0256 (Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program) and start-up funds from Center for Clinical Pharmacology, St. Louis College of Pharmacy and Washington University to S.M. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. Work done by the lab of JPM was funded through funds from the University of Florida. The authors wish to thank George Sukenick and Rong Wang of the NMR Analytical Core Facility at MSKCC for their assistance with NMR and MS instruments and experiments. This manuscript is dedicated to the memory of Professor Gavril W Pasternak who passed away on Feb 22nd, 2019.
ABBREVIATIONS
- C.I.
confidence interval
- CLAMS
comprehensive lab animal monitoring system
- DCM
dichloromethane
- 3-DM
3-dehydromitragynine
- DOR
delta opioid receptor
- 9G
9-glucuronyl corynantheidine
- HLM
human liver microsomes
- KAE
kratom alkaloid extract
- KOR
kappa opioid receptor
- MNO
mitragynine N-oxide
- MOR
mu opioid receptor
- MP
mitragynine pseudoindoxyl
- MPE
maximum possible effect
- 7OH
7-hydroxymitragynine
- po
per os
- Rt
room temperature
- sc
subcutaneous
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01111.
Time course of antinociception of oral and systemic kratom, mitragynine and metabolites, kratom, mitragynine, and its metabolites show MOR-dependent antinociception, oral mitragynine is more potent as an antinociceptive agent over systemically administered mitragynine in mice, mitragynine is the principal component found in kratom for producing antinociception in mice, metabolism of mitragynine to 7OH and MP, MP kappa/delta antagonism, ambulation effect in C57BL/6J mice, binding affinity (Ki ± SEM nM/pKi ± SEM) of kratom alkaloids and mitragynine metabolites at select CNS receptors, tables of binding assay conditions, concentration of parent compounds and metabolites detected from mitragynine-dosed mice (μM), 1H–13C NMR spectra, and UPLC chromato-grams of new compounds (PDF)
Molecular formula strings for compounds (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c01111
The authors declare the following competing financial interest(s): SM, GWP, YXP are co-founders of Sparian biosciences. DS and ACK are co-founders of Kures, Inc. GWP, ACK, DS, AK, AV and SM have filed a provisional patents on mitragynine and related molecules. The other authors declare no other competing interests.
Contributor Information
Soumen Chakraborty, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy and Washington University School of Medicine, St. Louis, Missouri 63110, United States.
Rajendra Uprety, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Samuel T. Slocum, Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Takeshi Irie, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Valerie Le Rouzic, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Xiaohai Li, Department of Molecular Therapeutics, Scripps Research Institute, Jupiter, Florida 33458, United States.
Lisa L. Wilson, Department of Pharmacodynamics, University of Florida, Gainesville, Florida 32610, United States;.
Brittany Scouller, Centre for Biodiscovery, School of Biological Science, Victoria University of Wellington, Wellington 6012, New Zealand.
Amy F. Alder, Centre for Biodiscovery, School of Biological Science, Victoria University of Wellington, Wellington 6012, New Zealand
Andrew C. Kruegel, Department of Chemistry, Columbia University, New York 10027, United States
Michael Ansonoff, Department of Neuroscience and Cell Biology, Rutgers Robert Wood Johnson Medical School, Piscataway, New Jersey 08854-8021, United States.
Andras Varadi, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States;.
Shainnel O. Eans, Department of Pharmacodynamics, University of Florida, Gainesville, Florida 32610, United States
Amanda Hunkele, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Abdullah Allaoa, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Sanjay Kalra, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Jin Xu, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Ying Xian Pan, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
John Pintar, Department of Neuroscience and Cell Biology, Rutgers Robert Wood Johnson Medical School, Piscataway, New Jersey 08854-8021, United States.
Bronwyn M. Kivell, Centre for Biodiscovery, School of Biological Science, Victoria University of Wellington, Wellington 6012, New Zealand
Gavril W. Pasternak, Department of Neurology and Molecular Pharmacology, Memorial Sloan Kettering Cancer Center, New York 10065, United States
Michael D. Cameron, Department of Molecular Therapeutics, Scripps Research Institute, Jupiter, Florida 33458, United States;.
Jay P. McLaughlin, Department of Pharmacodynamics, University of Florida, Gainesville, Florida 32610, United States;.
Dalibor Sames, Department of Chemistry, Columbia University, New York 10027, United States;.
Susruta Majumdar, Center for Clinical Pharmacology, University of Health Sciences & Pharmacy and Washington University School of Medicine, St. Louis, Missouri 63110, United States;.
REFERENCES
- (1).Institute of Medicine (US) Committee on Advancing Pain Research, C. and E. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research; National Academies Press, 2011. [PubMed] [Google Scholar]
- (2).Pasternak GW; Pan Y-X Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev 2013, 65, 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Szigethy E; Knisely M; Drossman D Opioid Misuse in Gastroenterology and Non-Opioid Management of Abdominal Pain. Nat. Rev. Gastroenterol. Hepatol 2018, 15, 168–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Imam MZ; Kuo A; Ghassabian S; Smith MT Progress in Understanding Mechanisms of Opioid-Induced Gastrointestinal Adverse Effects and Respiratory Depression. Neuropharmacology 2018, 131, 238–255. [DOI] [PubMed] [Google Scholar]
- (5).Gutridge AM; Robins MT; Cassell RJ; Uprety R; Mores KL; Ko MJ; Pasternak GW; Majumdar S; Rijn RM G protein-biased kratom-alkaloids and synthetic carfentanil-amide opioids as potential treatments for alcohol use disorder. Br. J. Pharmacol 2020, 177, 1497–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kruegel AC; Gassaway MM; Kapoor A; Váradi A; Majumdar S; Filizola M; Javitch JA; Sames D Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc 2016, 138, 6754–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Váradi A; Marrone GF; Palmer TC; Narayan A; Szabó MR; Le Rouzic V; Grinnell SG; Subrath JJ; Warner E; Kalra S; Hunkele A; Pagirsky J; Eans SO; Medina JM; Xu J; Pan Y-X; Borics A; Pasternak GW; McLaughlin JP; Majumdar S Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem 2016, 59, 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stoeber M; Jullié D; Li J; Chakraborty S; Majumdar S; Lambert NA; Manglik A; von Zastrow M Agonist-Selective Recruitment of Engineered Protein Probes and of GRK2 by Opioid Receptors in Living Cells. Elife 2020, 9, No. e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bhowmik S; Galeta J; Havel V; Nelson M; Faouzi A; Bechand B; Ansonoff M; Fiala T; Hunkele A; Kruegel AC; Pintar JE; Majumdar S; Javitch JA; Sames D Site selective C-H functionalization of Mitragyna alkaloids reveals a molecular switch for tuning opioid receptor signaling efficacy. Nat. Commun 2021, 12, 3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wilson LL; Harris HM; Eans SO; Brice-Tutt AC; Cirino TJ; Stacy HM; Simons CA; León F; Sharma A; Boyer EW; Avery BA; McLaughlin JP; McCurdy CR Lyophilized Kratom Tea as a Therapeutic Option for Opioid Dependence. Drug Alcohol Depend. 2020, 216, 108310. [DOI] [PubMed] [Google Scholar]
- (11).Wilson LL; Chakraborty S; Eans SO; Cirino TJ; Stacy HM; Simons CA; Uprety R; Majumdar S; McLaughlin JP Kratom Alkaloids, Natural and Semi-Synthetic, Show Less Physical Dependence and Ameliorate Opioid Withdrawal. Cell. Mol. Neurobiol 2021, 41, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Obeng S; Wilkerson JL; León F; Reeves ME; Restrepo LF; Gamez-Jimenez LR; Patel A; Pennington AE; Taylor VA; Ho NP; Braun T; Fortner JD; Crowley ML; Williamson MR; Pallares VLC; Mottinelli M; Lopera-Londoño C; McCurdy CR; McMahon LR; Hiranita T Pharmacological Comparison of Mitragynine and 7-Hydroxymitragynine: In Vitro Affinity and Efficacy for μ-Opioid Receptor and Opioid-Like Behavioral Effects in Rats. J. Pharmacol. Exp. Ther 2021, 376, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Todd DA; Kellogg JJ; Wallace ED; Khin M; Flores-Bocanegra L; Tanna RS; McIntosh S; Raja HA; Graf TN; Hemby SE; Paine MF; Oberlies NH; Cech NB Chemical Composition and Biological Effects of Kratom (Mitragyna Speciosa): In Vitro Studies with Implications for Efficacy and Drug Interactions. Sci. Rep 2020, 10, 19158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhou Y; Ramsey S; Provasi D; El Daibani A; Appourchaux K; Chakraborty S; Kapoor A; Che T; Majumdar S; Filizola M Predicted Mode of Binding to and Allosteric Modulation of the μ-Opioid Receptor by Kratom’s Alkaloids with Reported Antinociception In Vivo. Biochemistry 2020, 60, 1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gillis A; Sreenivasan V; Christie MJ Intrinsic Efficacy of Opioid Ligands and Its Importance for Apparent Bias, Operational Analysis, and Therapeutic Window. Mol. Pharmacol 2020, 98, 410–424. [DOI] [PubMed] [Google Scholar]
- (16).Feinberg EN; Farimani AB; Uprety R; Hunkele A; Pasternak GW; Majumdar S; Pande VS Machine Learning Harnesses Molecular Dynamics to Discover New μ Opioid Chemo-types. 2018. arXiv:1803.04479 [q-bio.BM].
- (17).Faouzi A; Uprety R; Gomes I; Massaly N; Keresztes AI; Le Rouzic V; Gupta A; Zhang T; Yoon HJ; Ansonoff M; Allaoa A; Pan YX; Pintar J; Morón JA; Streicher JM; Devi LA; Majumdar S Synthesis and Pharmacology of a Novel μ-δ Opioid Receptor Heteromer-Selective Agonist Based on the Carfentanyl Template. J. Med. Chem 2020, 63, 13618–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Faouzi A; Varga BR; Majumdar S Biased Opioid Ligands. Molecules 2020, 25, 4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Takayama H Chemistry and Pharmacology of Analgesic Indole Alkaloids from the Rubiaceous Plant, Mitragyna Speciosa. Chem. Pharm. Bull 2004, 52, 916–928. [DOI] [PubMed] [Google Scholar]
- (20).Brown PN; Lund JA; Murch SJ A Botanical, Phytochemical and Ethnomedicinal Review of the Genus Mitragyna Korth: Implications for Products Sold as Kratom. J. Ethnopharmacol 2017, 202, 302–325. [DOI] [PubMed] [Google Scholar]
- (21).Kruegel AC; Grundmann O The Medicinal Chemistry and Neuropharmacology of Kratom: A Preliminary Discussion of a Promising Medicinal Plant and Analysis of Its Potential for Abuse. Neuropharmacology 2018, 134, 108–120. [DOI] [PubMed] [Google Scholar]
- (22).Hassan Z; Muzaimi M; Navaratnam V; Yusoff NHM; Suhaimi FW; Vadivelu R; Vicknasingam BK; Amato D; von Hörsten S; Ismail NIW; Jayabalan N; Hazim AI; Mansor SM; Müller CP From Kratom to Mitragynine and Its Derivatives: Physiological and Behavioural Effects Related to Use, Abuse, and Addiction. Neurosci. Biobehav. Rev 2013, 37, 138–151. [DOI] [PubMed] [Google Scholar]
- (23).Chakraborty S; Majumdar S Natural Products for the Treatment of Pain: Chemistry and Pharmacology of Salvinorin A, Mitragynine, and Collybolide. Biochemistry 2020, 60, 1381–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen A Kratom Drug Ban May Cripple Promising Painkiller Research - Scientific American. https://www.scientificamerican.com/article/kratom-drug-ban-may-cripple-promising-painkiller-research/ (accessed Feb 4, 2020).
- (25).Prozialeck WC; Avery BA; Boyer EW; Grundmann O; Henningfield JE; Kruegel AC; McMahon LR; McCurdy CR; Swogger MT; Veltri CA; Singh D Kratom Policy: The Challenge of Balancing Therapeutic Potential with Public Safety. Int. J. Drug Policy 2019, 70, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).FDA. Statement from FDA Commissioner Scott Gottlieb, M.D., on the agency’s scientific evidence on the presence of opioid compounds in kratom, underscoring its potential for abuse. https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-agencys-scientific-evidence-presence-opioid-compounds (accessed Jul 20, 2020).
- (27).Henningfield JE; Fant RV; Wang DW The Abuse Potential of Kratom According the 8 Factors of the Controlled Substances Act: Implications for Regulation and Research. Psychopharmacology 2018, 235, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kamble SH; Sharma A; King TI; León F; McCurdy CR; Avery BA Metabolite Profiling and Identification of Enzymes Responsible for the Metabolism of Mitragynine, the Major Alkaloid of Mitragyna Speciosa (Kratom). Xenobiotica 2019, 49, 1279. [DOI] [PubMed] [Google Scholar]
- (29).Kruegel AC; Uprety R; Grinnell SG; Langreck C; Pekarskaya EA; Le Rouzic V; Ansonoff M; Gassaway MM; Pintar JE; Pasternak GW; Javitch JA; Majumdar S; Sames D 7-Hydroxymitragynine Is an Active Metabolite of Mitragynine and a Key Mediator of Its Analgesic Effects. ACS Cent. Sci 2019, 5, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Maxwell EA; King TI; Kamble SH; Raju KSR; Berthold EC; León F; Avery BA; McMahon LR; McCurdy CR; Sharma A Pharmacokinetics and Safety of Mitragynine in Beagle Dogs. Planta Med. 2020, 86, 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang D; Luo G; Ding X; Lu C Preclinical Experimental Models of Drug Metabolism and Disposition in Drug Discovery and Development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar]
- (32).Mogil JS; Kest B; Sadowski B; Belknap JK Differential Genetic Mediation of Sensitivity to Morphine in Genetic Models of Opiate Antinociception: Influence of Nociceptive Assay. J. Pharmacol. Exp. Ther 1996, 276, 532. [PubMed] [Google Scholar]
- (33).Orsini C; Bonito-Oliva A; Conversi D; Cabib S Susceptibility to Conditioned Place Preference Induced by Addictive Drugs in Mice of the C57BL/6 and DBA/2 Inbred Strains. Psychopharmacology 2005, 181, 327–336. [DOI] [PubMed] [Google Scholar]
- (34).Jansen KLR; Prast CJ Ethnopharmacology of Kratom and the Mitragyna Alkaloids. J. Ethnopharmacol 1988, 23, 115–119. [DOI] [PubMed] [Google Scholar]
- (35).Houghton PJ; Said IM 3-Dehydromitragynine: An Alkaloid from Mitragyna Speciosa. Phytochemistry 1986, 25, 2910–2912. [Google Scholar]
- (36).Hayes JA; Eccles KS; Lawrence SE; Moynihan HA Preparation and Characterisation of Solid State Forms of Paracetamol-O-Glucuronide. Carbohydr. Res 2012, 349, 108–112. [DOI] [PubMed] [Google Scholar]
- (37).Flores-Bocanegra L; Raja HA; Graf TN; Augustinović M; Wallace ED; Hematian S; Kellogg JJ; Todd DA; Cech NB; Oberlies NH The Chemistry of Kratom [Mitragyna speciosa]: Updated Characterization Data and Methods to Elucidate Indole and Oxindole Alkaloids. J. Nat. Prod 2020, 83, 2165–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Takayama H; Ishikawa H; Kurihara M; Kitajima M; Aimi N; Ponglux D; Koyama F; Matsumoto K; Moriyama T; Yamamoto LT; Watanabe K; Murayama T; Horie S Studies on the Synthesis and Opioid Agonistic Activities of Mitragynine-Related Indole Alkaloids: Discovery of Opioid Agonists Structurally Different from Other Opioid Ligands. J. Med. Chem 2002, 45, 1949–1956. [DOI] [PubMed] [Google Scholar]
- (39).Chear NJ-Y; León F; Sharma A; Kanumuri SRR; Zwolinski G; Abboud KA; Singh D; Restrepo LF; Patel A; Hiranita T; Ramanathan S; Hampson AJ; McMahon LR; McCurdy CR Exploring the Chemistry of Alkaloids from Malaysian Mitragyna Speciosa (Kratom) and the Role of Oxindoles on Human Opioid Receptors. J. Nat. Prod 2021, 84, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sabetghadam A; Ramanathan S; Mansor SM The Evaluation of Antinociceptive Activity of Alkaloid, Methanolic, and Aqueous Extracts of Malaysian Mitragyna Speciosa Korth Leaves in Rats. Pharmacogn. Res 2010, 2, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hiranita T; Leon F; Felix JS; Restrepo LF; Reeves ME; Pennington AE; Obeng S; Avery BA; McCurdy CR; McMahon LR; Wilkerson JL The Effects of Mitragynine and Morphine on Schedule-Controlled Responding and Antinociception in Rats. Psychopharmacology 2019, 236, 2725–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Obeng S; Kamble SH; Reeves ME; Restrepo LF; Patel A; Behnke M; Chear NJ-Y; Ramanathan S; Sharma A; León F; Hiranita T; Avery BA; McMahon LR; McCurdy CR Investigation of the Adrenergic and Opioid Binding Affinities, Metabolic Stability, Plasma Protein Binding Properties, and Functional Effects of Selected Indole-Based Kratom Alkaloids. J. Med. Chem 2020, 63, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Macko E; Weisbach JA; Douglas B Some Observations on the Pharmacology of Mitragynine. Arch. Int. Pharmacodyn. Ther 1972, 198, 145–161. [PubMed] [Google Scholar]
- (44).Carpenter JM; Criddle CA; Craig HK; Ali Z; Zhang Z; Khan IA; Sufka KJ Comparative Effects of Mitragyna Speciosa Extract, Mitragynine, and Opioid Agonists on Thermal Nociception in Rats. Fitoterapia 2016, 109, 87–90. [DOI] [PubMed] [Google Scholar]
- (45).Zuckerman A; Bolan E; De Paulis T; Schmidt D; Spector S; Pasternak GW Pharmacological characterization of morphine-6-sulfate and codeine-6-sulfate. Brain Res. 1999, 842, 1–5. [DOI] [PubMed] [Google Scholar]
- (46).Pasternak GW; Bodnar RJ; Clark JA; Inturrisi CE Morphine-6-Glucuronide, a Potent Mu Agonist. Life Sci. 1987, 41, 2845–2849. [DOI] [PubMed] [Google Scholar]
- (47).De Gregori S; Minella CE; De Gregori M; Tinelli C; Ranzani GN; Govoni S; Allegri M; Regazzi M Clinical Pharmacokinetics of Morphine and Its Metabolites During Morphine Dose Titration for Chronic Cancer Pain. Ther. Drug Monit 2014, 36, 335–344. [DOI] [PubMed] [Google Scholar]
- (48).Gammaitoni AR; Fine P; Alvarez N; McPherson ML; Bergmark S Clinical Application of Opioid Equianalgesic Data. Clin. J. Pain 2003, 19, 286–297. [DOI] [PubMed] [Google Scholar]
- (49).Yusoff NHM; Suhaimi FW; Vadivelu RK; Hassan Z; Rümler A; Rotter A; Amato D; Dringenberg HC; Mansor SM; Navaratnam V; Müller CP Abuse Potential and Adverse Cognitive Effects of Mitragynine (Kratom). Addict. Biol 2016, 21, 98–110. [DOI] [PubMed] [Google Scholar]
- (50).Yusoff NHM; Mansor SM; Müller CP; Hassan Z Opioid Receptors Mediate the Acquisition, but Not the Expression of Mitragynine-Induced Conditioned Place Preference in Rats. Behav. Brain Res 2017, 332, 1–6. [DOI] [PubMed] [Google Scholar]
- (51).Sufka KJ; Loria MJ; Lewellyn K; Zjawiony JK; Ali Z; Abe N; Khan IA The Effect of Salvia Divinorum and Mitragyna Speciosa Extracts, Fraction and Major Constituents on Place Aversion and Place Preference in Rats. J. Ethnopharmacol 2014, 151, 361–364. [DOI] [PubMed] [Google Scholar]
- (52).Le D; Goggin MM; Janis GC Analysis of Mitragynine and Metabolites in Human Urine for Detecting the Use of the Psychoactive Plant Kratom. J. Anal. Toxicol 2012, 36, 616–625. [DOI] [PubMed] [Google Scholar]
- (53).Philipp AA; Wissenbach DK; Zoerntlein SW; Klein ON; Kanogsunthornrat J; Maurer HH Studies on the Metabolism of Mitragynine, the Main Alkaloid of the Herbal Drug Kratom, in Rat and Human Urine Using Liquid Chromatography-Linear Ion Trap Mass Spectrometry. J. Mass Spectrom 2009, 44, 1249–1261. [DOI] [PubMed] [Google Scholar]
- (54).Kroeze WK; Sassano MF; Huang X-P; Lansu K; McCorvy JD; Giguère PM; Sciaky N; Roth BL PRESTO-Tango as an Open-Source Resource for Interrogation of the Druggable Human GPCRome. Nat. Struct. Mol. Biol 2015, 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Longo PA; Kavran JM; Kim M-S; Leahy DJ Transient Mammalian Cell Transfection with Polyethylenimine (PEI). Methods in Enzymology; Academic Press Inc., 2013; Vol. 529; pp 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Matsumoto K; Takayama H; Narita M; Nakamura A; Suzuki M; Suzuki T; Murayama T; Wongseripipatana S; Misawa K; Kitajima M; Tashima K; Horie S MGM-9 [(E)-methyl 2-(3-ethyl-7a,12a-(epoxyethanoxy)-9-fluoro-1,2,3,4,6,7,12,12b-octahydro-8-methoxyindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate], a derivative of the indole alkaloid mitragynine: A novel dual-acting μ- and κ-opioid agonist with potent antinociceptive and weak rewarding effects in mice. Neuropharmacology 2008, 55, 154–165. [DOI] [PubMed] [Google Scholar]
- (57).Tanna RS; Tian D-D; Cech NB; Oberlies NH; Rettie AE; Thummel KE; Paine MF Refined Prediction of Pharmacokinetic Kratom-Drug Interactions: Time-Dependent Inhibition Considerations. J. Pharmacol. Exp. Ther 2021, 376, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sabetghadam A; Navaratnam V; Mansor SM Dose-Response Relationship, Acute Toxicity, and Therapeutic Index between the Alkaloid Extract ofMitragyna speciosaand Its Main Active Compound Mitragynine in Mice. Drug Dev. Res 2013, 74, 23–30. [Google Scholar]
- (59).Smith LC; Lin L; Hwang CS; Zhou B; Kubitz DM; Wang H; Janda KD Lateral Flow Assessment and Unanticipated Toxicity of Kratom. Chem. Res. Toxicol 2019, 32, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Gistrak MA; Paul D; Hahn EF; Pasternak GW Pharmacological actions of a novel mixed opiate agonist/antagonist: naloxone benzoylhydrazone. J. Pharmacol. Exp. Ther 1989, 251, 469–476. [PubMed] [Google Scholar]
- (61).Gershman K; Timm K; Frank M; Lampi L; Melamed J; Gerona R; Monte AA Deaths in Colorado Attributed to Kratom. N. Engl. J. Med 2019, 380, 97–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Hemby SE; McIntosh S; Leon F; Cutler SJ; McCurdy CR Abuse Liability and Therapeutic Potential of the Mitragyna Speciosa (Kratom) Alkaloids Mitragynine and 7-Hydroxymitragynine. Addict. Biol 2019, 24, 874–885. [DOI] [PubMed] [Google Scholar]
- (63).Yue K; Kopajtic TA; Katz JL Abuse Liability of Mitragynine Assessed with a Self-Administration Procedure in Rats. Psychopharmacology 2018, 235, 2823–2829. [DOI] [PubMed] [Google Scholar]
- (64).Ellis CR; Racz R; Kruhlak NL; Kim MT; Zakharov AV; Southall N; Hawkins EG; Burkhart K; Strauss DG; Stavitskaya L Evaluating Kratom Alkaloids Using PHASE. PLoS One 2020, 15, No. e0229646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Matsumoto K; Mizowaki M; Suchitra T; Murakami Y; Takayama H; Sakai S.-i.; Aimi N; Watanabe H Central Antinociceptive Effects of Mitragynine in Mice: Contribution of Descending Noradrenergic and Serotonergic Systems. Eur. J. Pharmacol 1996, 317, 75–81. [DOI] [PubMed] [Google Scholar]
- (66).Newman DJ; Cragg GM Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- (67).Harvey AL; Edrada-Ebel R; Quinn RJ The Re-Emergence of Natural Products for Drug Discovery in the Genomics Era. Nat. Rev. Drug Discovery 2015, 14, 111–129. [DOI] [PubMed] [Google Scholar]
- (68).Gillam EMJ; Notley LM; Cai H; De Voss JJ; Guengerich FP Oxidation of Indole by Cytochrome P450 Enzymes. Biochemistry 2000, 39, 13817–13824. [DOI] [PubMed] [Google Scholar]
- (69).Zarembo JE; Douglas B; Valenta J; Weisbach JA Metabolites of Mitragynine. J. Pharm. Sci 1974, 63, 1407–1415. [DOI] [PubMed] [Google Scholar]
- (70).Kamble SH; León F; King TI; Berthold EC; Lopera-Londoño C; Siva Rama Raju K; Hampson AJ; Sharma A; Avery BA; McMahon LR; McCurdy CR Metabolism of a Kratom Alkaloid Metabolite in Human Plasma Increases Its Opioid Potency and Efficacy. ACS Pharmacol. Transl. Sci 2020, 3, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Ponglux D; Wongseripipatana S; Takayama H; Kikuchi M; Kurihara M; Kitajima M; Aimi N; Sakai S.-i. A New Indole Alkaloid, 7 α-Hydroxy-7H-mitragynine, fromMitragyna speciosain Thailand. Planta Med. 1994, 60, 580–581. [DOI] [PubMed] [Google Scholar]
- (72).Martin R; Schürenkamp J; Pfeiffer H; Lehr M; Köhler H Synthesis, Hydrolysis and Stability of Psilocin Glucuronide. Forensic Sci. Int 2014, 237, 1–6. [DOI] [PubMed] [Google Scholar]
- (73).Grinnell SG; Uprety R; Varadi A; Subrath J; Hunkele A; Pan YX; Pasternak GW; Majumdar S Synthesis and Characterization of Azido Aryl Analogs of IBNtxA for Radio-Photoaffinity Labeling Opioid Receptors in Cell Lines and in Mouse Brain. Cell. Mol. Neurobiol 2020, 41, 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Majumdar S; Burgman M; Haselton N; Grinnell S; Ocampo J; Pasternak AR; Pasternak GW Generation of Novel Radiolabeled Opiates through Site-Selective Iodination. Bioorg. Med. Chem. Lett 2011, 21, 4001–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).LOWRY O; ROSEBROUGH N; FARR AL; RANDALL R Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem 1951, 193, 265–275. [PubMed] [Google Scholar]
- (76).Váradi A; Marrone GF; Eans SO; Ganno ML; Subrath JJ; Le Rouzic V; Hunkele A; Pasternak GW; McLaughlin JP; Majumdar S Synthesis and Characterization of a Dual Kappa-Delta Opioid Receptor Agonist Analgesic Blocking Cocaine Reward Behavior. ACS Chem. Neurosci 2015, 6, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Váradi A; Palmer TC; Haselton N; Afonin D; Subrath JJ; Le Rouzic V; Hunkele A; Pasternak GW; Marrone GF; Borics A; Majumdar S Synthesis of Carfentanil Amide Opioids Using the Ugi Multicomponent Reaction. ACS Chem. Neurosci 2015, 6, 1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Urai Á; Váradi A; Szőcs L; Komjáti B; Le Rouzic V; Hunkele A; Pasternak GW; Majumdar S; Hosztafi S Synthesis and pharmacological evaluation of novel selective MOR agonist 6β-pyridinyl amidomorphines exhibiting long-lasting antinociception. Medchemcomm 2017, 8, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Reilley KJ; Giulianotti M; Dooley CT; Nefzi A; McLaughlin JP; Houghten RA Identification of Two Novel, Potent, Low-Liability Antinociceptive Compounds from the Direct in Vivo Screening of a Large Mixture-Based Combinatorial Library. AAPS J. 2010, 12, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Eans SO; Ganno ML; Mizrachi E; Houghten RA; Dooley CT; McLaughlin JP; Nefzi A Parallel Synthesis of Hexahydrodiimidazodiazepines Heterocyclic Peptidomimetics and Their in Vitro and in Vivo Activities at μ (MOR), δ (DOR), and κ (KOR) Opioid Receptors. J. Med. Chem 2015, 58, 4905–4917. [DOI] [PubMed] [Google Scholar]
- (81).Raehal KM; Walker JKL; Bohn LM Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther 2005, 314, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (82).Kennedy NM; Schmid CL; Ross NC; Lovell KM; Yue Z; Chen YT; Cameron MD; Bohn LM; Bannister TD Optimization of a Series of Mu Opioid Receptor (MOR) Agonists with High G Protein Signaling Bias. J. Med. Chem 2018, 61, 8895–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.