

Abstract
Envelope phosphatidylserine (PtdSer) and phosphatidylethanolamine (PtdEtr) have been shown to mediate binding of enveloped viruses. However, commonly used PtdSer binding molecules such as Annexin V cannot block PtdSer-mediated viral infection. Lack of reagents that can conceal envelope PtdSer and PtdEtr and subsequently inhibit infection hinders elucidation of the roles of the envelope phospholipids in viral infection. Here, we developed sTIM1dMLDR801, a reagent capable of blocking PtdSer- and PtdEtr- dependent infection of enveloped viruses. Using sTIM1dMLDR801, we found that envelope PtdSer and/or PtdEtr can support ZIKV infection of not only human but also mosquito cells. In a mouse model for ZIKV infection, sTIM1dMLDR801 reduced ZIKV load in serum and the spleen, indicating envelope PtdSer and/or PtdEtr support in viral infection in vivo. sTIM1dMLDR801 will enable elucidation of the roles of envelope PtdSer and PtdEtr in infection of various virus species, thereby facilitating identification of their receptors and transmission mechanisms.
Keywords: Phosphatidylserine, Phosphatidylethanolamine, Zika virus, Dengue virus, Ebola virus, TIM-1, TIM-4, Axl, Gas6, CD300A
Introduction
Interactions between viral proteins and their respective receptors expressed on target cells are a common molecular mechanism of viral attachment and are thus intensively investigated. Additionally, enveloped viruses are known to utilize envelope lipid to facilitate the attachment step(Mercer and Helenius, 2008; Mercer et al., 2010; Schmidt et al., 2011) (Callahan et al., 2003; Laliberte and Moss, 2009; Puryear et al., 2013; Puryear et al., 2012). One such method involves viruses exposing PtdSer on their envelopes and exploiting various types of host PtdSer-recognizing molecular mechanisms to facilitate the binding step (Fig. 1A) (Amara and Mercer, 2015).
Fig 1. Proposed mechanisms of PtdSer- and/or PtdEtr-mediated viral infection and their inhibitors.
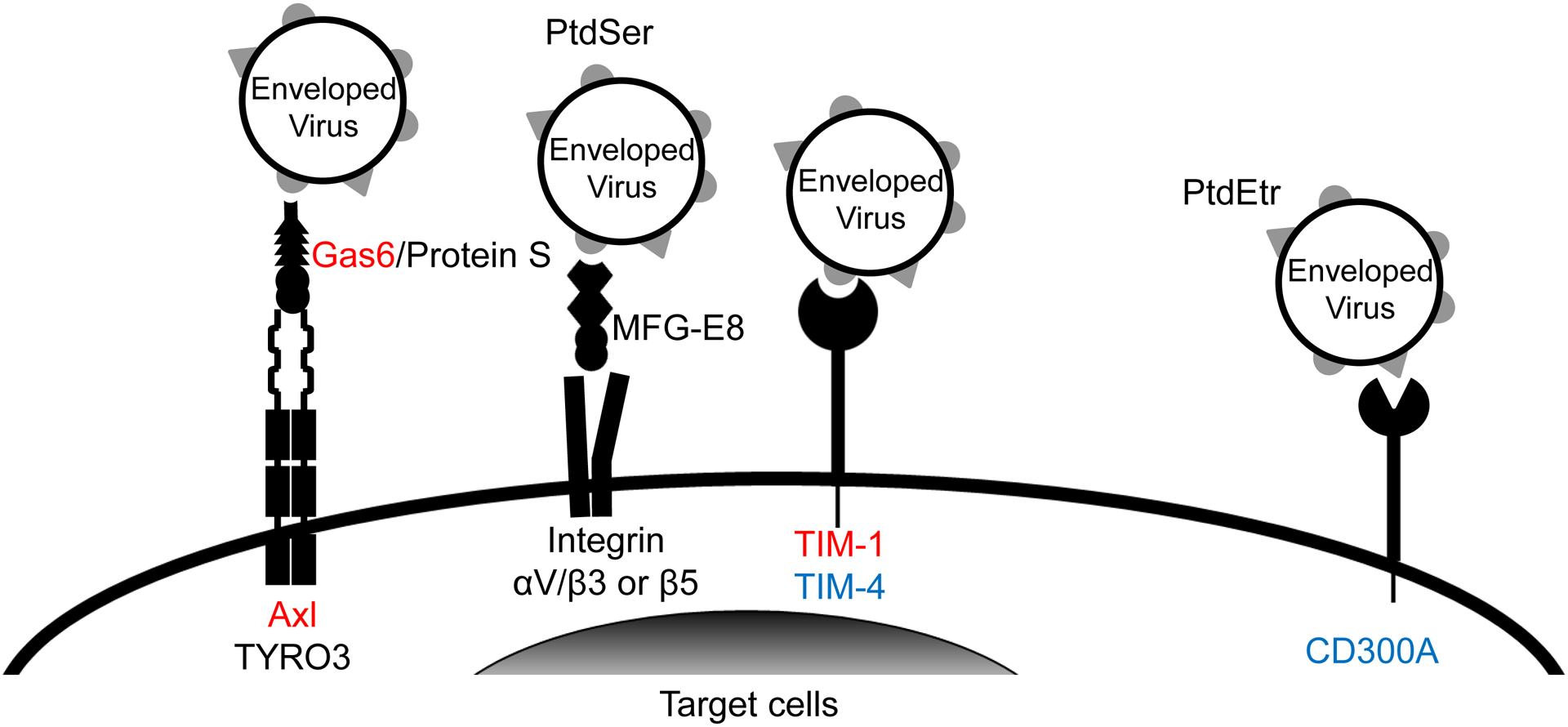
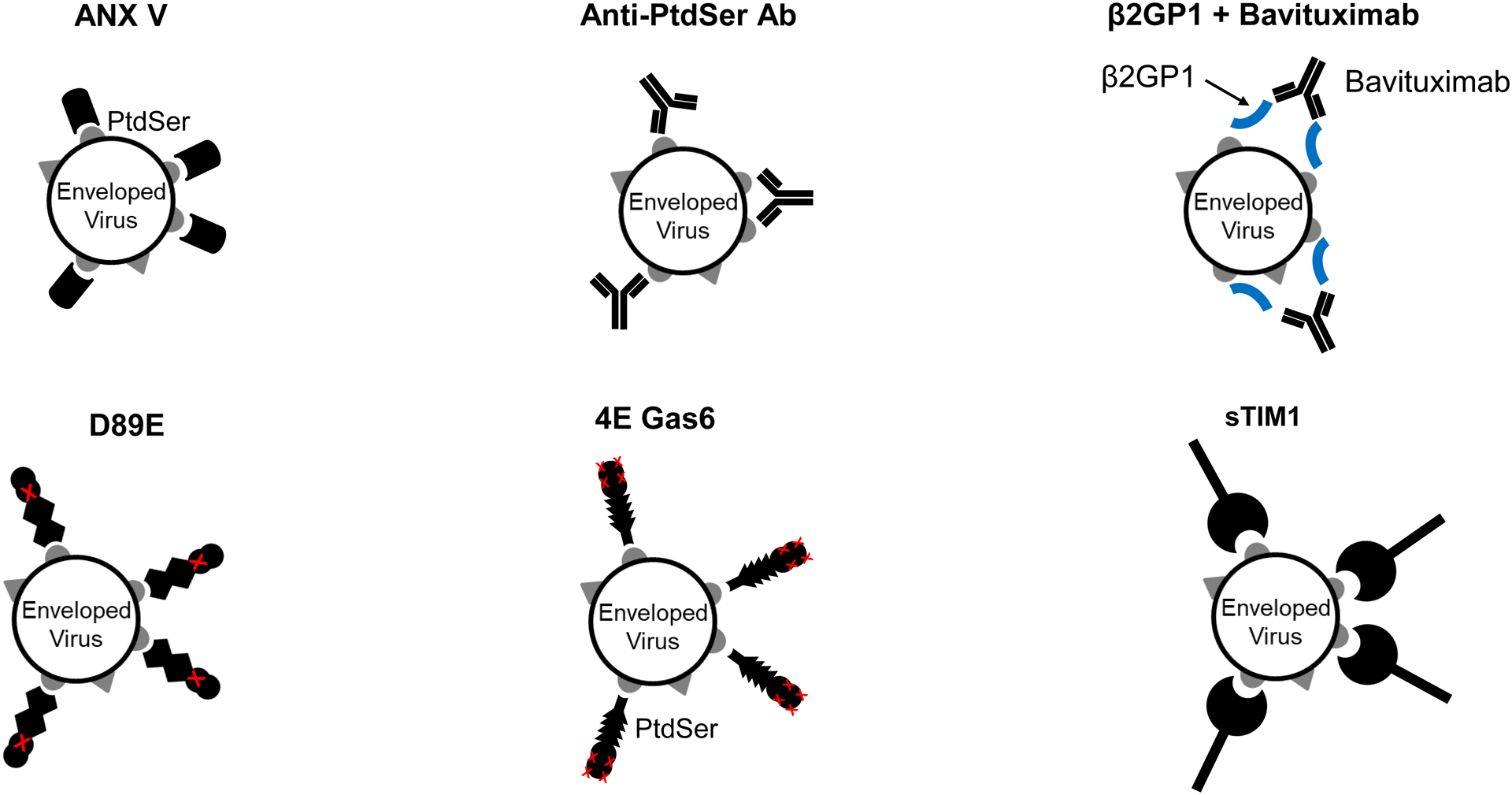
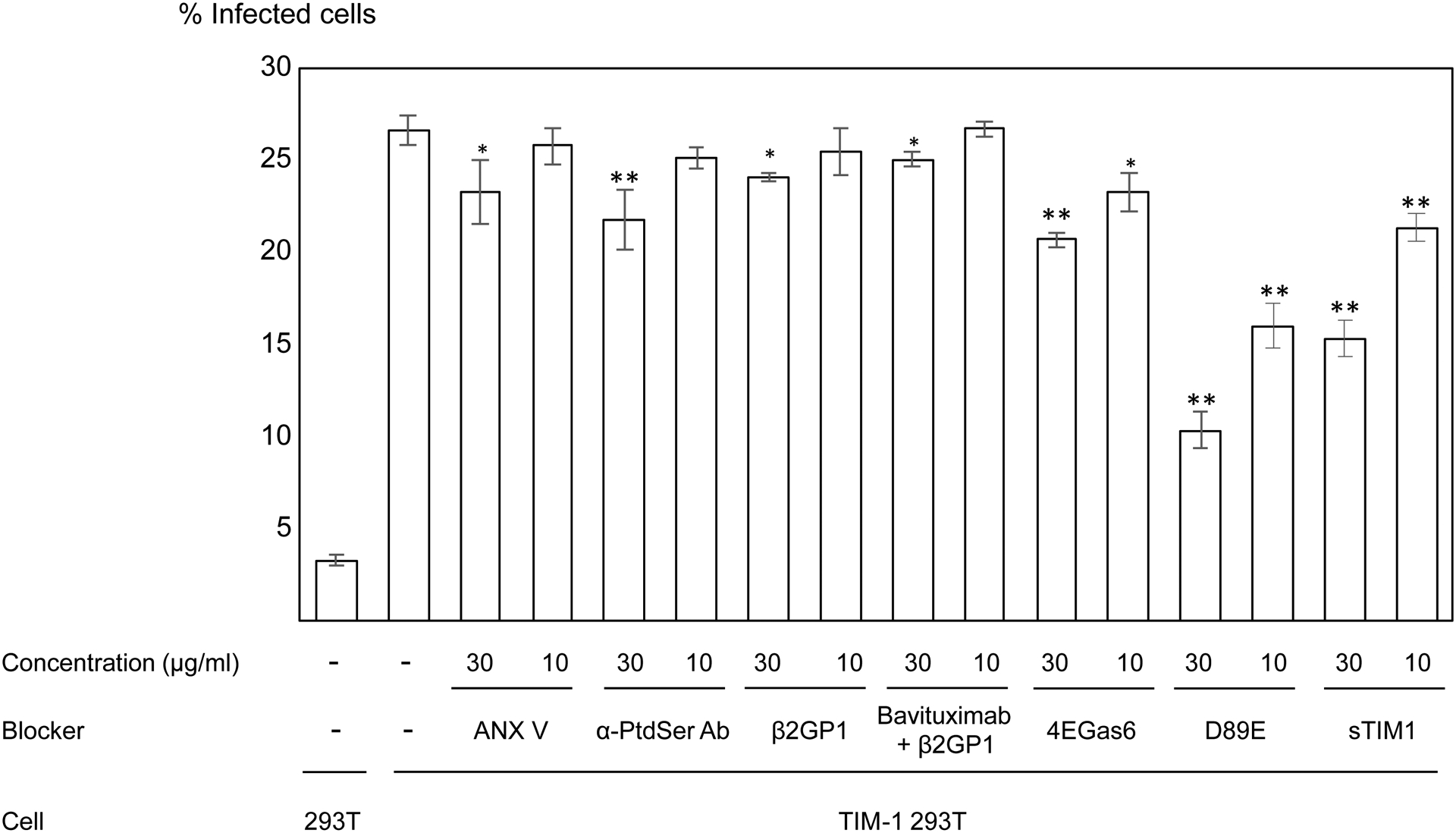
A) Gas6 and protein S bridge envelope PtdSer and Axl/TYRO3. MFG-E8 bridges envelope PtdSer and integrins αV/β3 and 5. TIM-1 and TIM-4 directly bind envelope PtdSer. CD300A directly binds to envelope PtdEtr. Some studies showed that TIM-1 and Gas6 can also bind envelope PtdEtr (Zhang et al., 2020) (Richard et al., 2015). Gas6/Axl and TIM-1 (red) mediate virus infection most efficiently. TIM-4 and CD300A (blue) mediate virus infection at moderate efficiencies. Protein S/Axl and TYRO3, Gas6/TYRO3, and MFG-E8/Integrins (black) mediate virus infection at the least efficiencies among the molecules shown (Chua et al., 2019). B) Inhibitory molecules of PtdSer-dependent infection. ANX V and anti-PtdSer Ab directly bind PtdSer. Serum protein β2GP1 binds PtdSer, and its binding is enhanced by crosslinking with the anti-β2GP1 antibody, Bavituximab. D89E was developed by mutating the integrin-binding domain of MFG-E8 and binds PtdSer by its PtdSer-binding discoidin domain (Hanayama et al., 2002). 4E Gas6 was developed by mutating TAM receptor-binding regions of Gas6 and binds PtdSer at its Gla domain (Chua et al., 2018). sTIM1 was developed by deleting the transmembrane and intracellular domains of TIM-1 and binds PtdSer by its IgV domain (Moller-Tank et al., 2014). 4EGas6 binds PtdSer at its Gla domain, D89E binds PtdSer by its discoidin domain, and sTIM1 binds PtdSer at its IgV domain. C) Blocking of TIM-1-mediated ZIKV infection by PtdSer-binding molecules. ZIKV replicon was incubated with 30 or 10 μg/ml of PtdSer-binding molecules or control medium for 1 hour at room temperature. 293T or TIM-1 293T cells were infected with the incubated virus for 2 hours, and infections were analyzed by flow cytometry 24 hours post-infection. The results shown are averages and standard deviations of the triplicate experiment. Significance was calculated by comparing TIM-1 293T cell infection without blocking reagents to those with blocking reagents, using a two-sample two-sided unpaired student t-test (*, p<0.05; **, p<0.01).
PtdSer-recognizing molecular mechanisms are originally known to mediate phagocytic removal of dead cells. Phagocytic cells recognize dead cells by binding to PtdSer exposed on the cell membrane (Fadok et al., 1992; Nagata et al., 2010). PtdSer usually resides in the inner leaflet of the cell membranes of live cells(van Meer et al., 2008) (Leventis and Grinstein, 2010). PtdSer is exposed on the cell surface upon cell death and subsequently recognized by phagocytes using various types of PtdSer-binding molecules(Fadok et al., 1992; Pietkiewicz et al., 2015; Wang et al., 2013).
Enveloped viruses can also expose PtdSer on their envelopes and exploit various types of host PtdSer-recognizing molecular mechanisms to facilitate the binding step (Fig. 1A) (Amara and Mercer, 2015). Soluble proteins Gas6 and protein S mediate phagocytic removal of dead cells by bridging PtdSer on dead cells to TAM receptors (TYRO3, Axl, and Mer) expressed on phagocytes(Ishimoto et al., 2000; Stitt et al., 1995). We, as well as other research groups, identified that Gas6/Protein S facilitates entry of enveloped viruses by bridging envelope PtdSer and Axl or Tyro3 expressed on target cells (Fig. 1A) (Bhattacharyya et al., 2013; Brindley et al., 2011; Fedeli et al., 2018; Hamel et al., 2015; Meertens et al., 2012; Morizono et al., 2011) (Meertens et al., 2017) (Zhang et al., 2020) (Richard et al., 2017). Additionally, we discovered that envelope viruses can bind cells by utilizing MFG-E8 to bridge envelope PtdSer and integrin αV/β3 or β5 (Fig. 1A) (Morizono and Chen, 2014). Other research groups have also established that enveloped viruses can utilize cell surface PtdSer-binding receptors TIM-1 and TIM-4 for virus attachment (Fig. 1A) (Jemielity et al., 2013; Meertens et al., 2012; Moller-Tank et al., 2013) (Brunton et al., 2019) (Rhein et al., 2016). Finally, recent studies demonstrated that several types of enveloped viruses can utilize another type of phospholipid, PtdEtr, to bind the PtdEtr receptor CD300A to facilitate their attachment (Carnec et al., 2016). Given that the receptors schematically displayed in Fig. 1A bind to PtdSer similar to the mechanism of phagocytosis-mediated dead cell removal, it is proposed that enveloped viruses utilize their own envelope PtdSer and PtdEtr as the primary host cell binding ligands.
However, our previous publications outline that widely used PtdSer-binding molecules such as Annexin V (ANX V) cannot efficiently block viral infections mediated by Gas6/protein S, MFG-E8, TIM-1, and TIM-4(Morizono and Chen, 2014; Morizono et al., 2011). Also, Duramycin, a well-known PtdEtr peptide, has been reported to be cytotoxic (Emoto et al., 1996), complicating its incorporation into PtdEtr-mediated virus infection experiments.
Current methods to investigate viral entry mediated by PtdSer- and PtdEtr- binding molecules include ectopic expression of the molecules shown in Fig. 1A, neutralization of their PtdSer/PtdEtr-binding activities by molecule-specific antibodies or liposomes containing PtdSer and/or PtdEtr, and knock out/down of these molecules using a variety of molecular biological reagents (Bhattacharyya et al., 2013; Brouillette et al., 2018; Dejarnac et al., 2018; Fedeli et al., 2018; Hamel et al., 2015; Hastings et al., 2017; Jemielity et al., 2013; Niu et al., 2018; Richard et al., 2015; Wang et al., 2017) (Brunton et al., 2019) (Das et al., 2017) (Jemielity et al., 2013; Moller-Tank et al., 2013; Zhang et al., 2020). Due to the lack of reagents that can conceal PtdSer and PtdEtr and block viral infection, previous studies could not directly examine the roles of envelope PtdSer and PtdEtr in infection. To investigate the role of envelope phospholipid in viral entry process of particular cells, it is necessary to knock out/down or neutralize all types of PtdSer and PtdEtr receptors on the cells. However, gene knockout/down may also affect the physiological functions of cells (Hastings et al., 2017). These approaches cannot be easily applied to non-human and mouse species since available antibodies, expression vectors, and gene knock out/down target sequences of PtdSer and PtdEtr receptors are limited to human and mouse systems. Non-human species play critical roles in the zoonotic transmission of several pathogenic enveloped viruses. For example, ZIKV infects mosquitoes, and mosquito bites transmit ZIKV to humans(Weissenbock et al., 2010). Bats have been shown to be the natural host of Ebola virus(Leroy et al., 2005), and bat-to-human transmission has been reported(Leroy et al., 2009). However, without sufficient information on PtdSer and PtdEr receptors in these species, we cannot investigate envelope PtdSer and PtdEr can mediate viral infection of mosquito and bat cells as with human and mouse cells. Blocking universal PtdSer/PtdEtr-mediated viral entry mechanisms by concealing envelope PtdSer and PtdEtr will demonstrate how envelope PtdSer and PtdEtr serve as viral ligands to bind various species and cell types.
In this study, we have developed a PtdSer and PtdEtr binding reagent, sTIM1dMLDR801, capable of specifically blocking the universal PtdSer- and PtdEtr- dependent virus entry mechanisms by multiple species of enveloped virus. Using sTIM1dMLDR801, we investigated the roles of envelope PtdSer and PtdEtr in ZIKV infection of human and mosquito cells in vitro as well as replication in vivo.
Results
Blocking TIM-1-dependent ZIKV infection by various PtdSer-binding proteins
Previous studies that demonstrated that envelope PtdSer serves as a receptor-binding ligand required the inhibition of viral attachment and infection through the specific concealment of envelope PtdSer. We have previously used ANX V, which specifically binds PtdSer, for the specific concealment of envelope PtdSer. Despite expected concealment of envelope PtdSer, we found that ANX V cannot efficiently inhibit viral infection via TIM-1, TIM-4, Axl/Gas6, or MFG-E8(Morizono and Chen, 2014; Morizono et al., 2011). To study the roles of envelope PtdSer in virus infection, we prepared various types of PtdSer-binding molecules and investigated whether these molecules can inhibit TIM-1-dependent ZIKV infection (Fig. 1A). First, we obtained a monoclonal antibody against PtdSer from a commercial source (Fig. 1B). Second, we generated recombinant serum protein β2GP1,which binds PtdSer, and obtained a commercially available β2GP1-specific monoclonal antibody Bavituximab. The latter is known to crosslink β2GP1, further facilitates binding to PtdSer, and inhibits enveloped virus replication in vivo (Fig. 1B) (Luster et al., 2006). Third, we generated 4E Gas6, a mutant of Gas6 that can bind PtdSer but not Axl due to mutations in the Axl-binding domain (Fig 1B) (Chua et al., 2018); therefore, it is expected to conceal envelope PtdSer without bridging enveloped viruses. Fourth, we produced a mutant of MFG-E8 (D89E) that can bind PtdSer but not integrins due to mutations in its integrin-binding site (Fig. 1B) (Hanayama et al., 2002). Lastly, we generated soluble TIM-1 (sTIM-1) by deleting the transmembrane and cytoplasmic domains of TIM-1 to engineer PtdSer-binding molecule (Fig. 1B).
We ectopically expressed TIM-1 on 293T cells, designated as TIM-1 293T, and confirmed that expression of TIM-1 on 293T cells increased ZIKV infection (Fig. 1C). We then assessed the ability of each PtdSer-binding molecule to inhibit ZIKV infection of TIM-1 293T cells (Fig. 1C).
To quantitate the inhibitory effects of these molecules on a single cycle of ZIKV infection, we utilized a ZIKV replicon that only undergoes a single cycle of infection (Dowd et al., 2016; Pierson et al., 2006). As we have previously reported using pseudotyped lentiviral vectors (Morizono and Chen, 2014), ANX V cannot efficiently block TIM-1-mediated infection. We found that sTIM-1 and D89E can inhibit TIM-1-mediated ZIKV infection more efficiently than other tested molecules.
Improvement of antiviral activity of sTIM-1 and D89E by dimerization
Dimerization is known to increase the anti-HIV-1 effects of soluble CD4 by increasing their avidity for antigens(Capon et al., 1989). To enhance the inhibitory effects of D89E and sTIM1, we generated Fc-fusion proteins and dimerized the fusion proteins through their Fc domains. Fusion with the Fc domain has also been shown to increase the half-life of the fused molecules in vivo through the interactions between the Fc domain and FcRn (Capon et al., 1989) (Israel et al., 1996).
We investigated whether dimerization of D89E and sTIM1 resulted in increased inhibitory effects on ZIKV infection of TIM-1 293T cells by measuring the IC50 of the monomers and dimers.
Since PtdSer binding of D89E occurs at its C-terminus, there was a possibility that fusion of the Fc-domain to its C-terminus would hinder binding to PtdSer. Therefore, we dimerized D89E by fusing the Fc-domain to either its N-terminus end (D89E Fc) or C-teminus end (Fc D89E) (Fig. 2A). We then used D89E, D89D Fc, and Fc D89E at various concentrations to inhibit ZIKV infection of TIM-1 293T cells. We found that dimerization of D89E by N- and C- terminus fusion with the Fc-domain increased the IC50 from 3.7 μg/ml to greater than 30 μg/ml (Fig. 2B), indicating that dimerization of D89E hinders the binding of D89E to PtdSer.
Fig 2. Inhibition of TIM-1-mediated infection by dimers of D89E and sTIM-1.
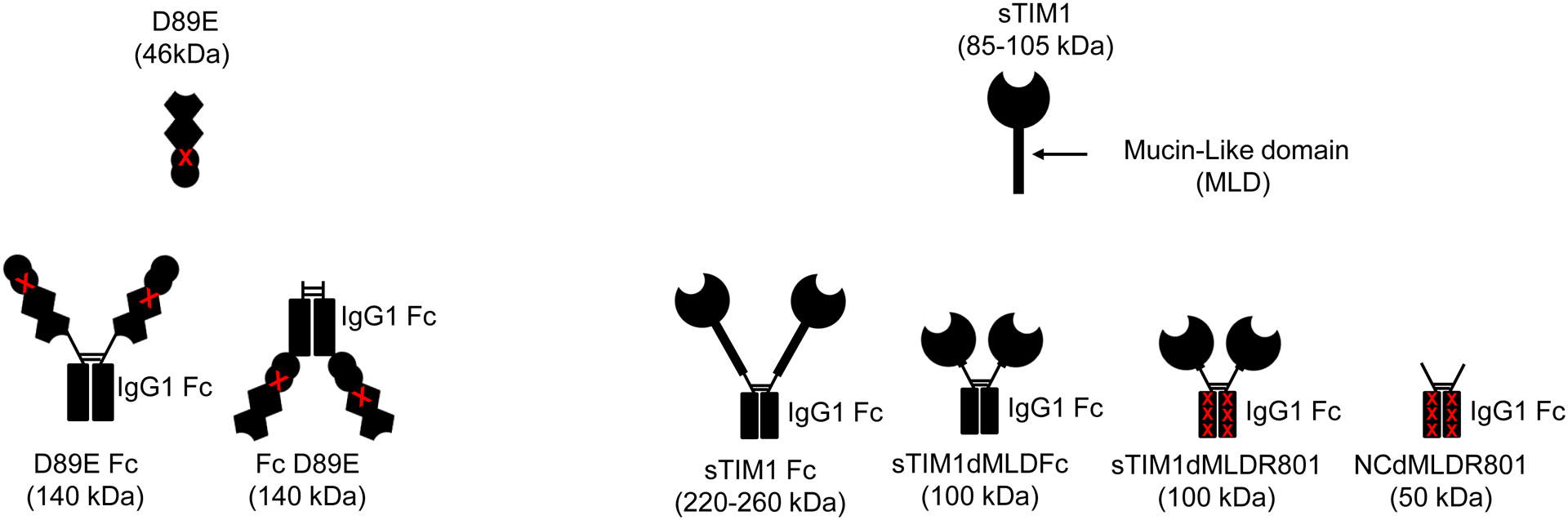
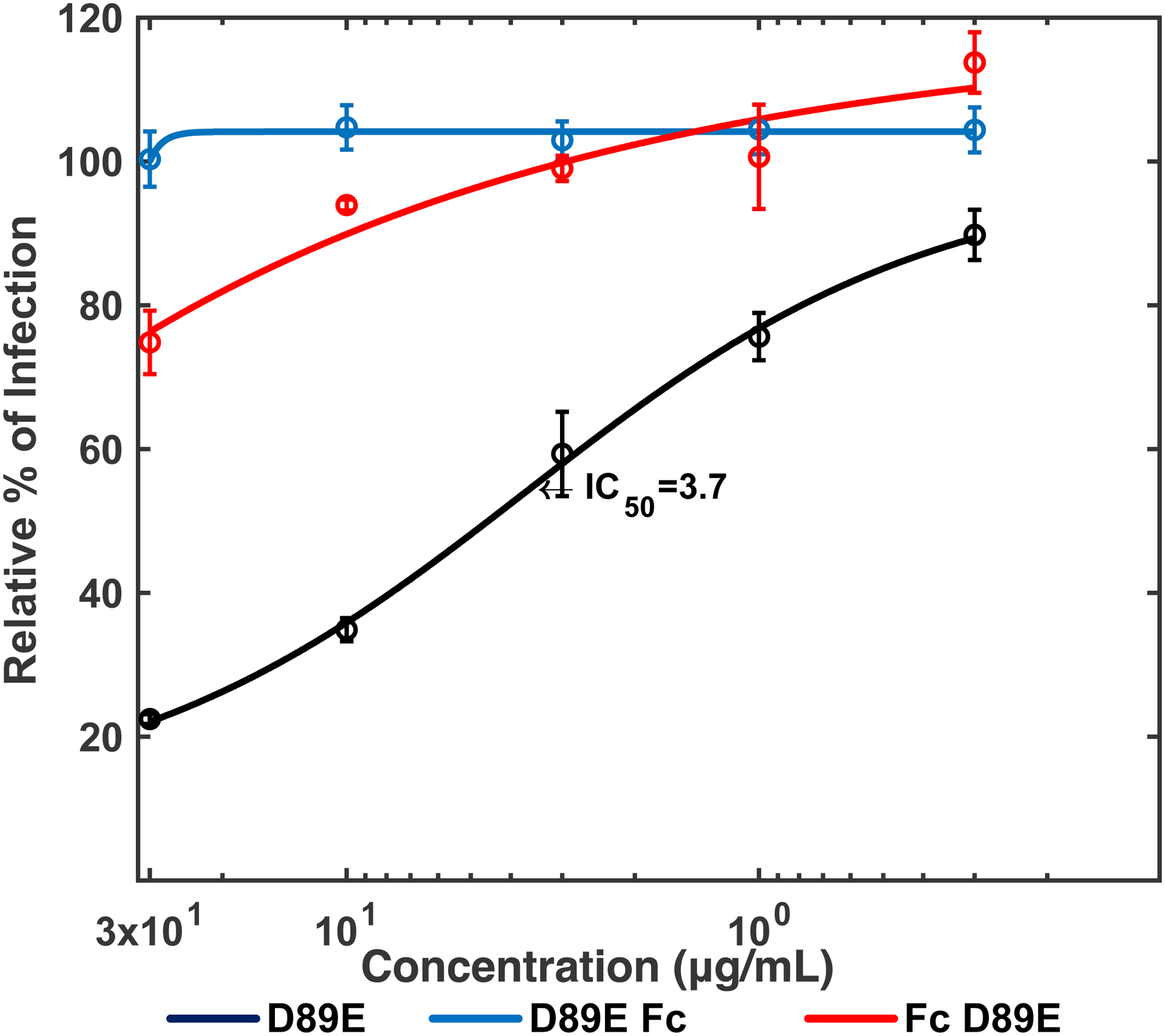
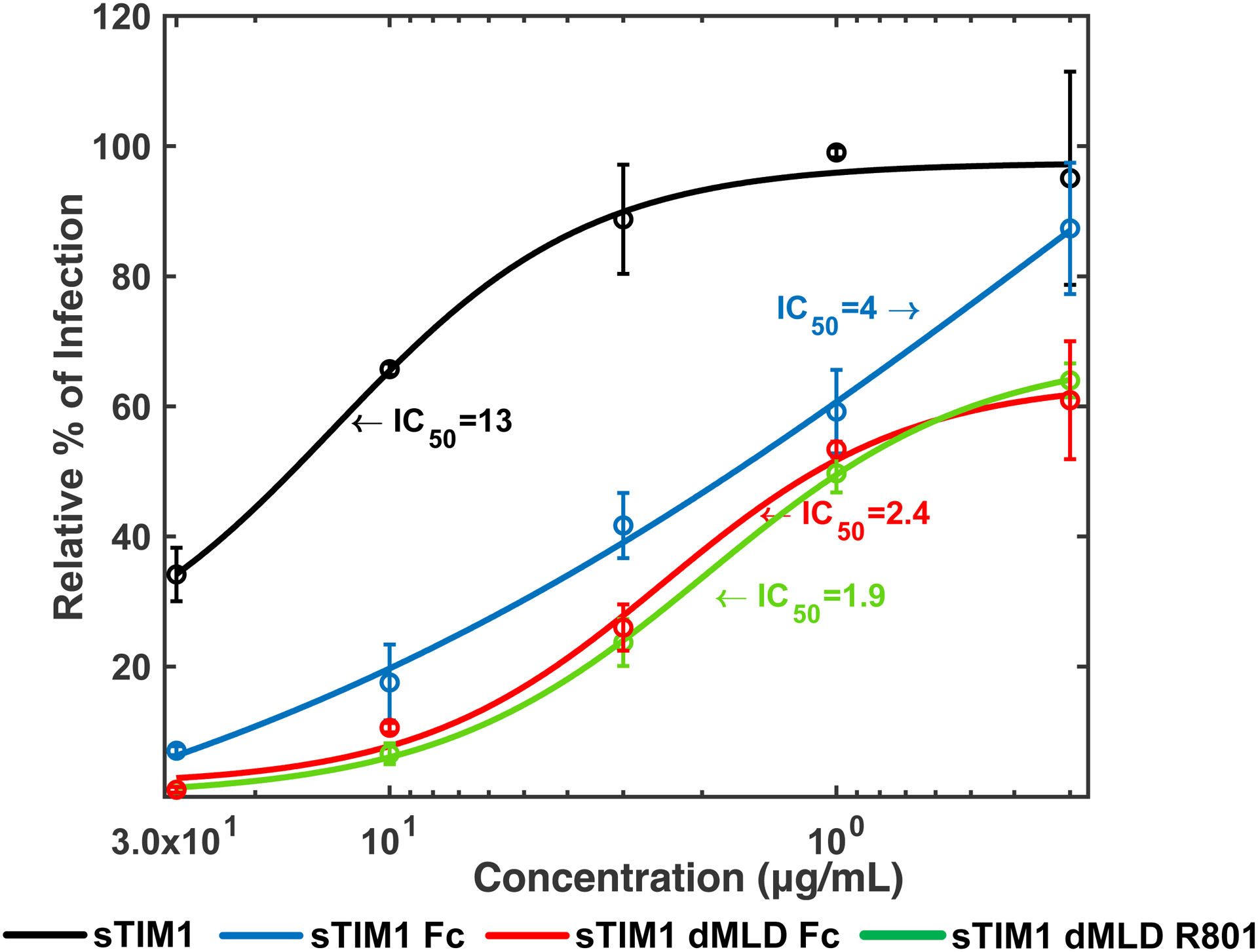
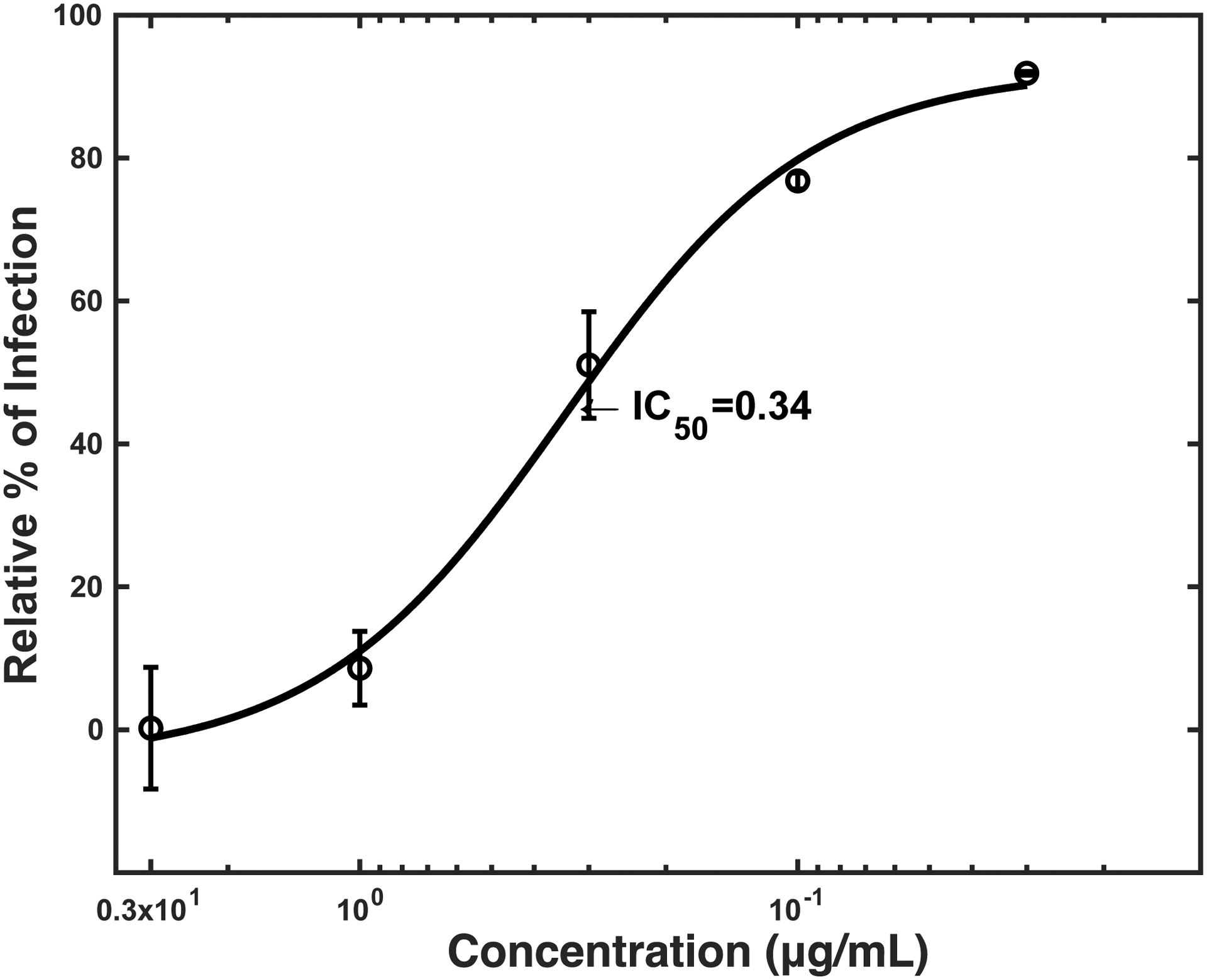
A) sTIM1 Fc was generated by fusion of sTIM1 with the human IgG1 Fc domain. sTIM1 dMDL Fc has deletions in the mucin-like domain of sTIM1 Fc. sTIM1 dMLD R801 contains four point mutations in its Fc domain to eliminate binding to Fc-receptors, except FcRn (Lee et al., 2017). NC dMLD R801 has deletions in the sTIM1 IgV domain of sTIM1dMLDR801. D89E Fc is D89E fused with the Fc domain at its C-terminus, and Fc D89E is D89E fused with the Fc-domain at its N-terminus. B-D) ZIKV replicon was incubated with various concentrations of D89E (B), D89E Fc (B), Fc linker D89E (B), sTIM1 (C), sTIM1 Fc (C), sTIM1dMLDFc (C), sTIM1dMLDR801 (C), or ZKA64 (D) for 1 hour at room temperature. 293T or TIM-1 293T cells were infected with the incubated virus for 2 hours, and the infections were analyzed by flow cytometry 24 hours post-infection. The MOI used for the experiments shown in Fig 2B, C, and D is 0.14. The results shown are averages and standard deviations of the triplicate experiment.
Since sTIM1 binds PtdSer at its N-terminal end, we fused the Fc-domain to its C terminus, and designated this Fc-fusion protein as sTIM1 Fc (Fig 2A). We found that the IC50 of sTIM1 and sTIM1 Fc are 13 and 4 μg/ml, respectively, when used for blocking ZIKV infection of TIM-1 293T cells, showing that the dimerization of sTIM-1 increased its antiviral activity (Fig. 2C). TIM-1 has a mucin-like domain (MLD) at its stalk region, which augments the size and weight of TIM-1 higher due to extensive glycosylation (Moller-Tank et al., 2014; Moller-Tank et al., 2013). The glycans at the MLD might interfere with movement and/or binding to PtdSer of the dimerized partners due to steric hindrance.
To decrease the molecular weight and size, we deleted the MLD of sTIM1 Fc, and designated this modified protein as sTIM1dMLDFc. Deletion of the MLD further reduced the IC50 from 4 μg/ml to 2.4 μg/ml, most likely due to its reduced molecular weight. We selected TIM1dMLDFc to further investigate the role of PtdSer in virus infection as it possessed the lowest IC50.
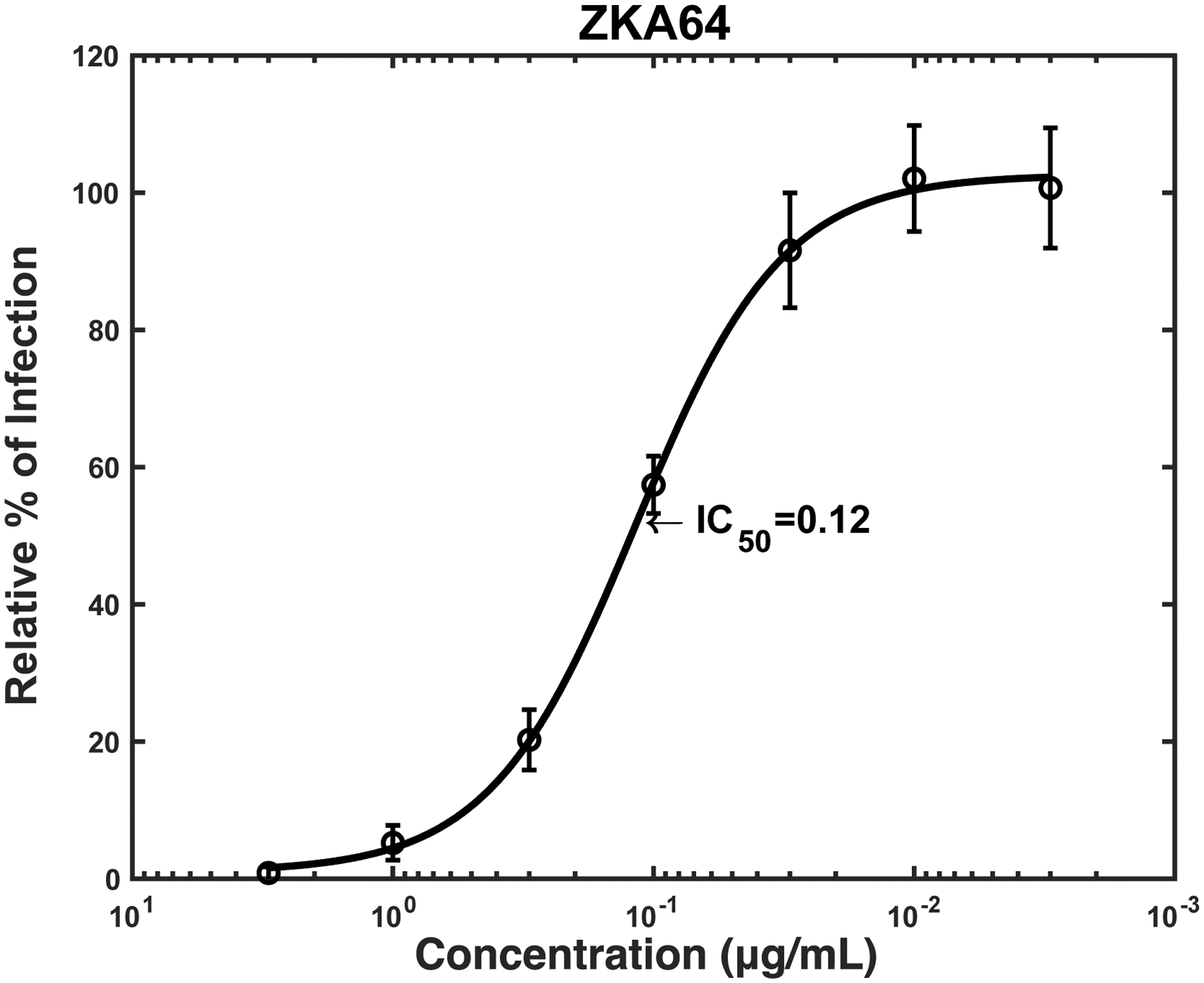
To establish a reference for an antibody-based antiviral reagent, we also measured the IC50 of ZKA64 (Stettler et al., 2016), as it is a well-studied antibody against the ZIKV E protein, in the infection of TIM-1 293T cells (IC50: 0.34 μg/ml) (Fig. 2D).
Elimination of FcR-mediated ZIKV infection by mutating the Fc-domain
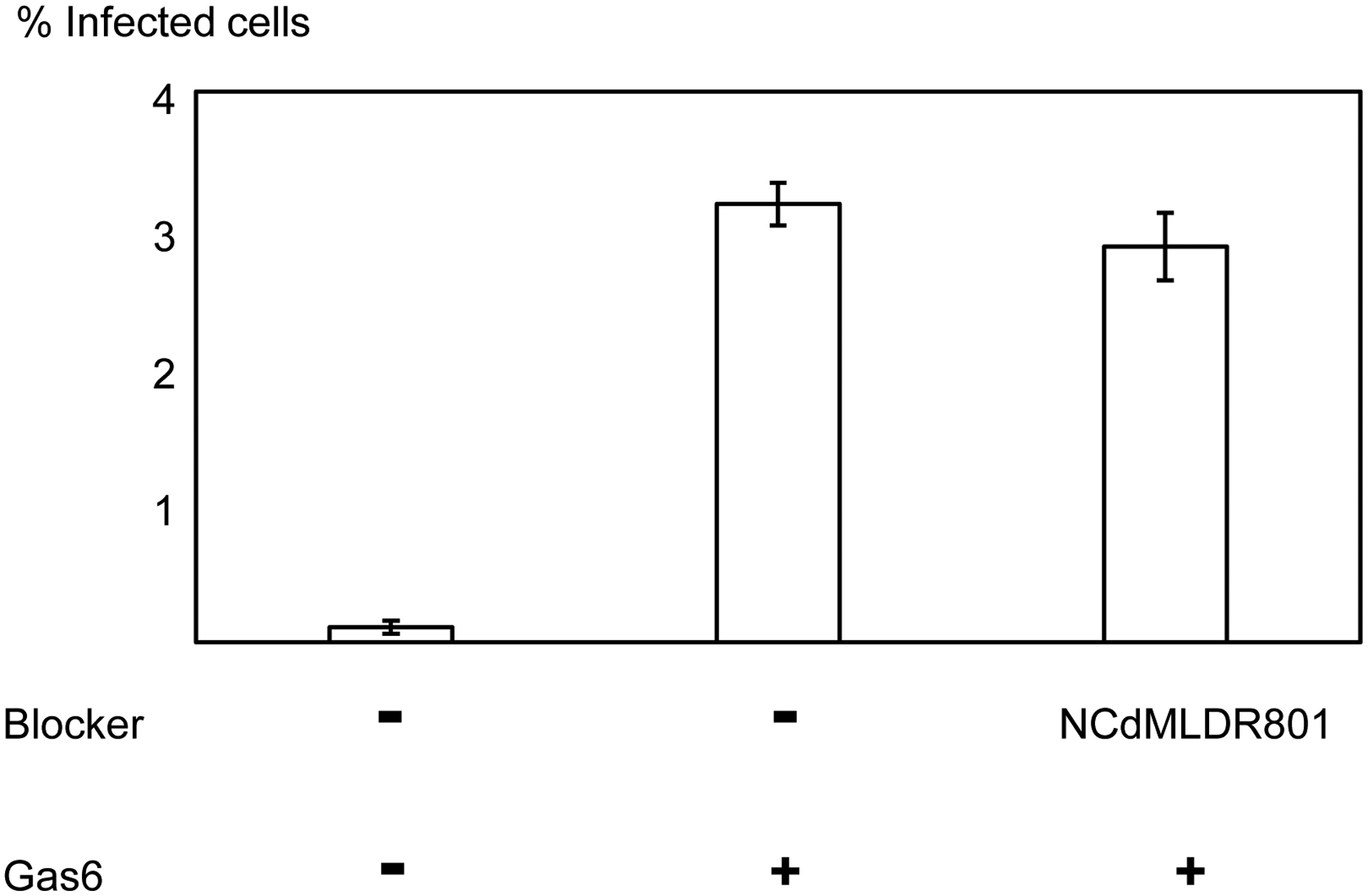
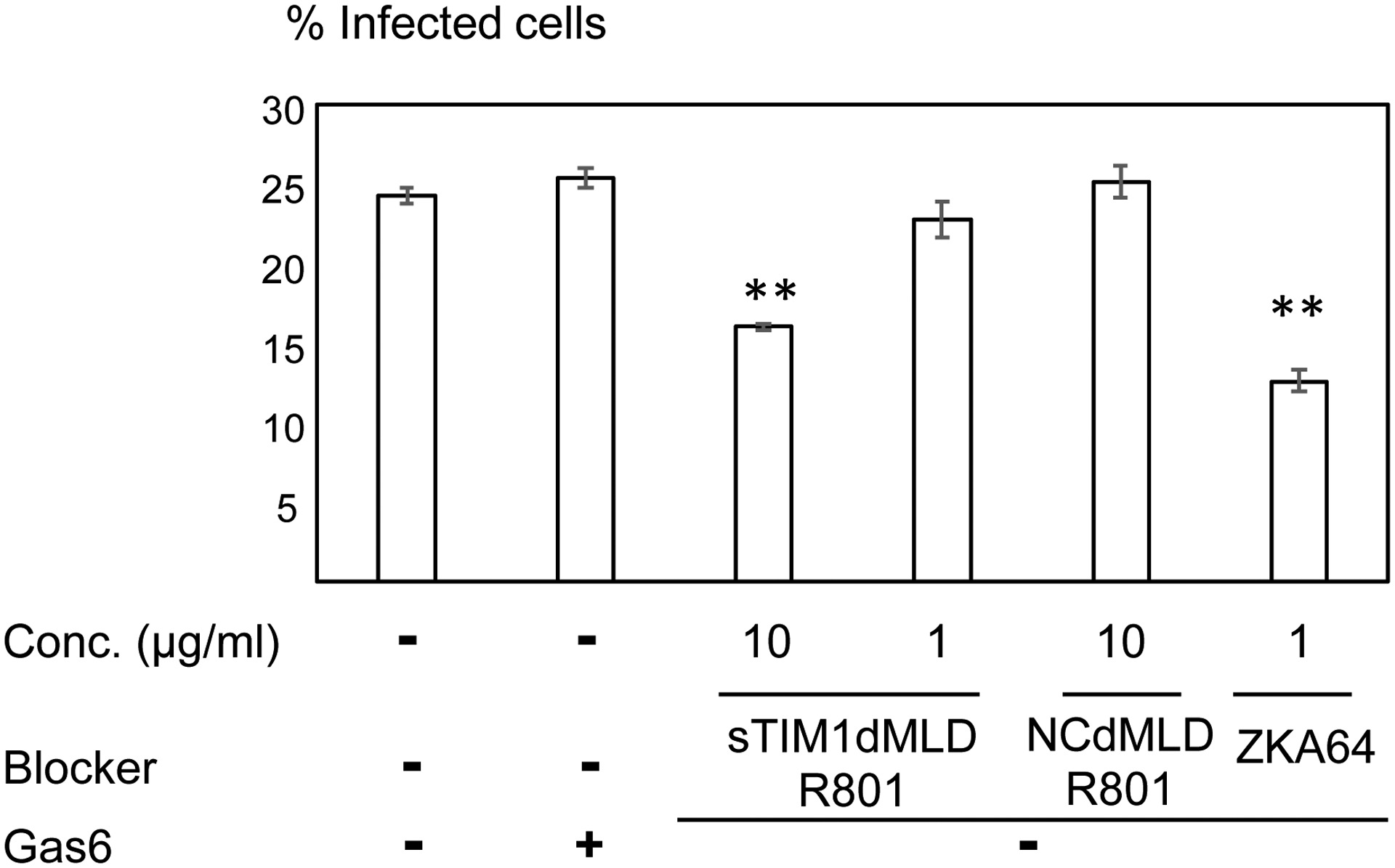
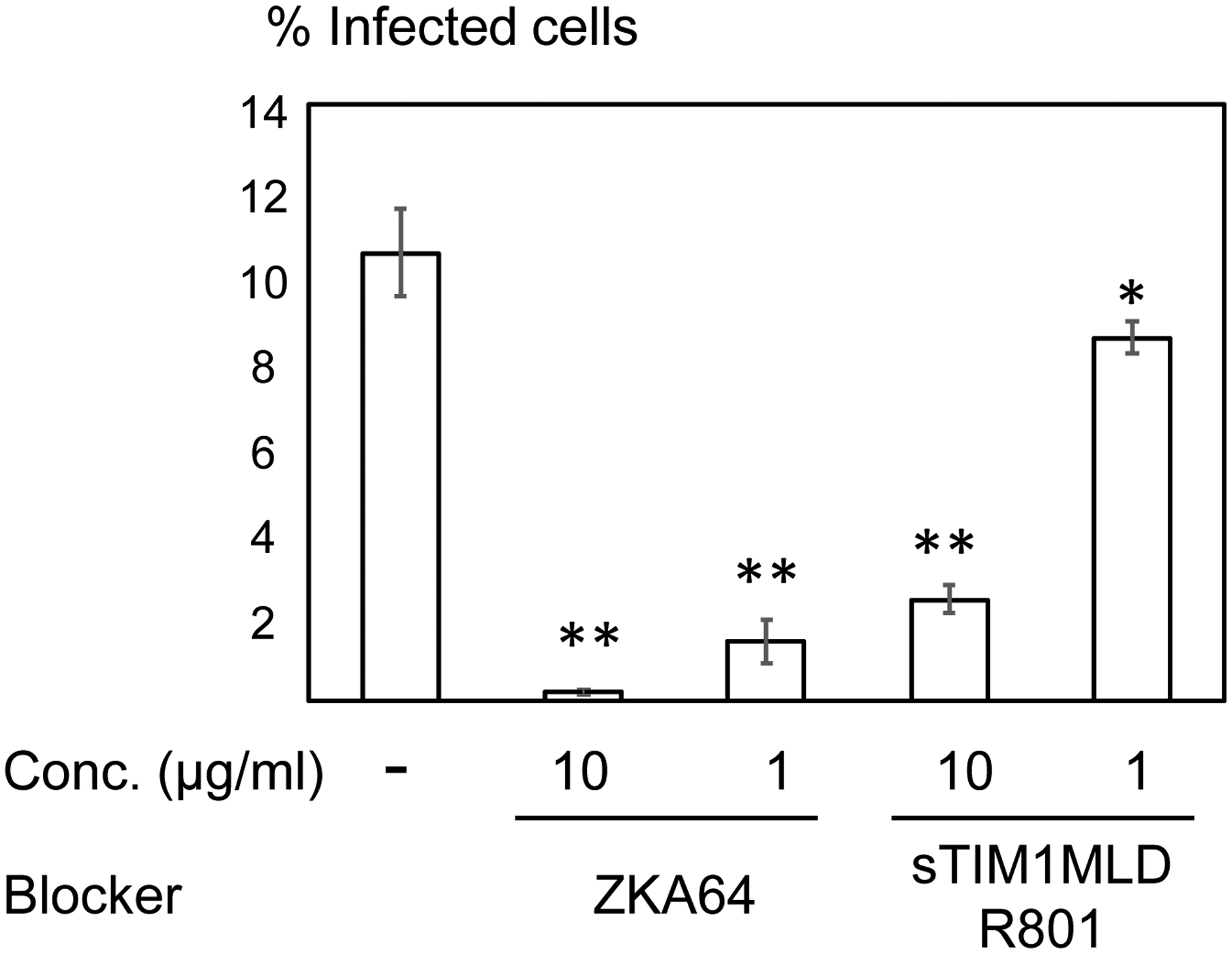
Antibodies against Flaviviruses, including ZIKV and dengue virus (DENV), are known to enhance Flavivirus infection of Fc-receptor-positive cells by bridging the viral E protein to Fc-receptors on target cells (Stettler et al., 2016). As previously reported, ZKA64 enhanced ZIKV infection of Fc-receptor (CD32)-positive K562 cells from 2- to 450- fold at concentrations ranging from 0.01 to 1 μg/ml (Fig. 3). To test the possibility that sTIM1dMLDFc may be similarly enhancing viral infection, we infected K562 cells with ZIKV in the presence of sTIM1dMLDFc and found that sTIM1dMLDFc increased infection 2.5-fold at 1 μg/ml. Although the extent of enhancement is not as high as with ZKA64, the Fc-receptor-mediated enhancement makes it nonetheless difficult to interpret the PtdSer-concealing effects of sTIM1 dMLD Fc on viral infection in various in vitro and in vivo experimental settings. Thus, we attempted to eliminate this Fc-dependent infection by introducing mutations at the Fc-domain of sTIM-1 dMLD Fc. The R801 mutation of the Fc domain has been shown to eliminate binding to Fc receptors (CD16, CD32, and CD64), while still maintaining interactions with FcRn with long half-lives in vivo (Lee et al., 2017). Therefore, we introduced R801 mutations into sTIM1dMLDFc, and designated the mutant as sTIM1dMLDR801 (Fig 2A). We found that sTIM1dMLDR801 did not enhance ZIKV infection of K562 cells at any of the concentrations tested (10–0.001 μg/ml) (Fig. 3). We confirmed that sTIM1dMLDR801 inhibits TIM-1-dependent ZIKV infection at an IC50 (1.9 μg/ml) similar to that of sTIM1dMLDFc (Fig. 2D). Of note, K562 cells express TYRO3, which might account for the inhibition of ZIKV infection by sTIM1dMLDR801 at 10 μg/ml. As a negative control protein, we generated NCdMLDR801, which lacks the PtdSer-binding domain of sTIM1dMLDR801 (Fig. 2A). NCdMLDR801 did not inhibit TIM-1 infection, even at 30 μg/ml (S1 Fig), indicating that PtdSer binding is necessary for the inhibitory effects of sTIM1dMLDR801 on ZIKV infection of TIM-1 cells.
Fig 3. Fc-dependent enhancement of ZIKV infection.
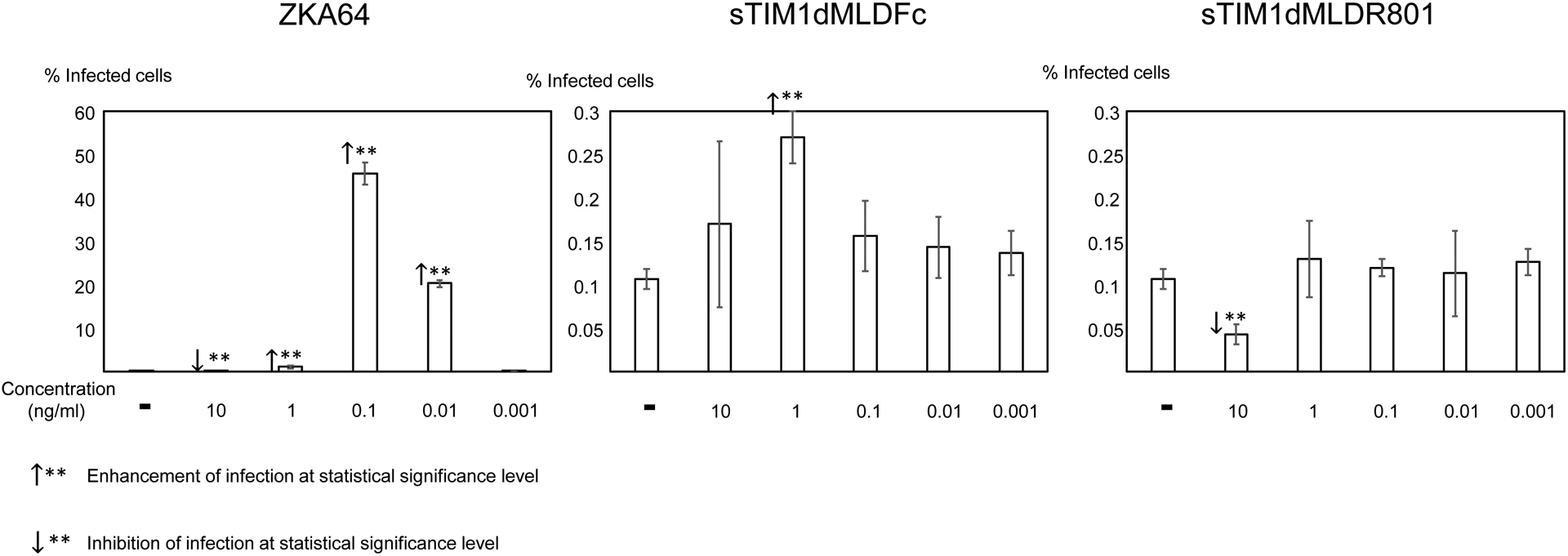
ZIKV replicon was incubated with various concentrations of ZKA64, sTIM1dMLDFc, or sTIM1dMLDR801 for 1 hour at room temperature. K562 cells were infected with the incubated virus for 2 hours, and the infections were analyzed by flow cytometry 24 hours post-infection. This experiment was repeated three times in singlicate and twice in triplicate, and the results shown are averages and standard deviations of one representative triplicate experiment. Significance was calculated by comparing K562 cell infection without reagents to those with reagents, using a two-sample two-sided unpaired student t-test (↑**: enhancement p<0.01; ↓**: inhibition p<0.01).
In order to focus on the effects of concealment of envelope PtdSer on viral infection and exclude Fc-dependent effects, we chose to use sTIM1dMLDR801 for further experiments.
Inhibition of various molecular mechanisms of ZIKV attachment by sTIM1dMLDR801
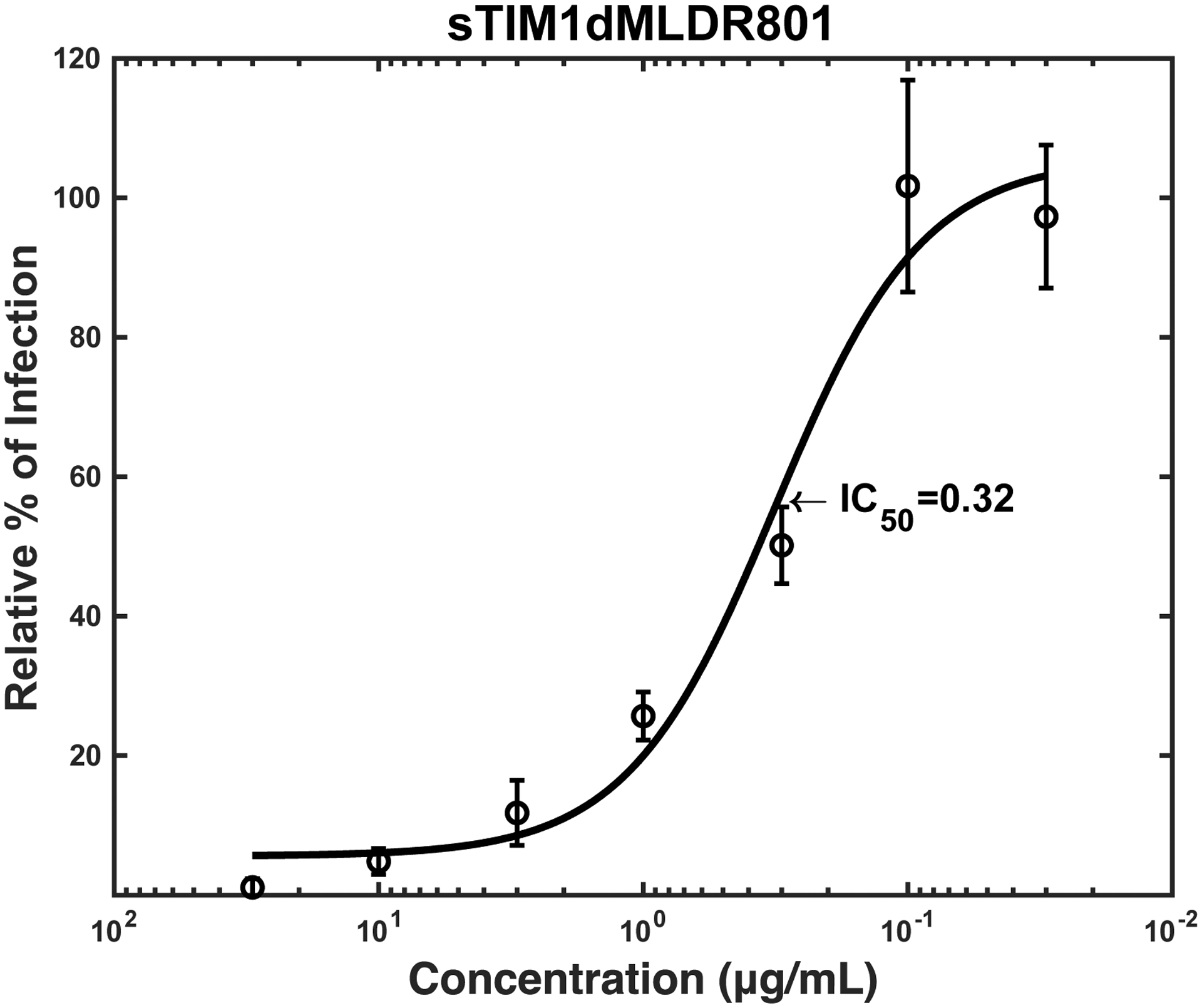
We then investigated whether sTIM1dMLDR801 can inhibit Gas6/Axl-dependent ZIKV infection, the other most efficient PtdSer-dependent virus infection mechanism. Infection of human microvascular endothelial cells (HMVEC) with ZIKV is enhanced 28-fold by addition of exogenous recombinant Gas6, which is consistent with our previous finding using vaccinia virus and pseudotyped lentiviral vectors (Fig. 4A) (Morizono et al., 2011). We incubated ZIKV with various concentrations of ZKA64 or sTIM1dMLD R801 before infection of HMVEC in the presence of Gas6. ZKA64 and sTIM1dMLDR801 inhibited Gas6-dependent ZIKV infection at IC50 0.12 and 0.32 μg/ml, respectively (Fig. 4B and C), while NCdMLDR801 did not exhibit any inhibition (Fig. 4A). These results indicated that sTIM1 can inhibit the two most efficient PtdSer-dependent virus infection mechanisms.
Fig 4. Inhibition of various mechanisms of ZIKV infection by sTIM1dMLDR801.
The results shown are averages and standard deviations of the triplicate experiment. A) HMVEC were infected with ZIKV replicon with or without Gas6 (1 μg/ml) and NC dMLD R801 (30 μg/ml). B) ZIKV replicon was incubated with various concentrations of ZKA64 or sTIM1dMLD R801, and HMVEC were infected with the incubated virus in the presence of Gas6 (1 μg/ml). C) ZIKV replicon was incubated with 1 or 10 μg/ml of ZKA64 or sTIM1dMLDR801, or control medium. 293T, TIM-4 293T, DC-SIGN 293T, or CD300A 293T cells were infected with the incubated virus for 2 hours, and infections were analyzed by flow cytometry 24 hours post-infection. The MOI used for the experiments shown in Fig 4B and C are 0.02 and 0.03, respectively. Significance was calculated by comparing TIM-4, CD300A, or DC-SIGN 293T cell infection without blocking reagents to those with blocking reagents, using a two-sample two-sided unpaired student t-test (**, p<0.01).
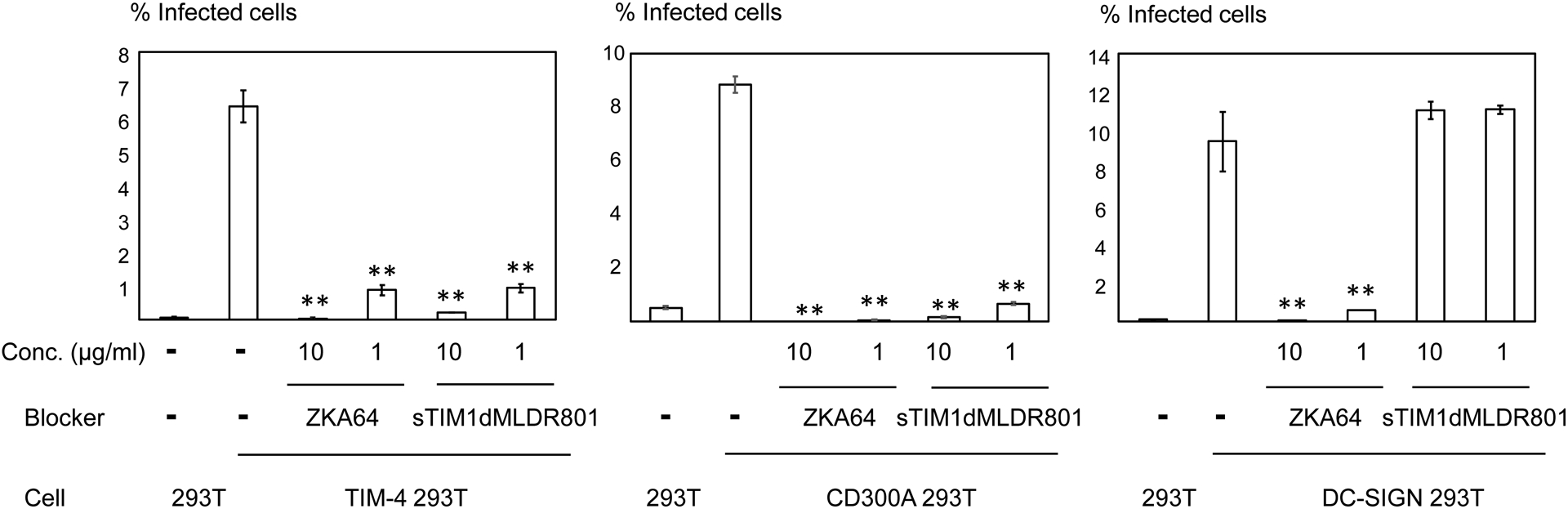
To further confirm the versatility of inhibitory effects of sTIM1dMLDR801, we investigated whether it could inhibit an additional PtdSer-dependent infection mechanism mediated by TIM-4, using 293T cells ectopically expressing TIM-4 (TIM-4 293T). sTIM1dMLDR801 inhibits TIM-4-dependent infection more efficiently than it does TIM-1-dependent infection (Fig. 4D), most likely because of the higher affinity of TIM-1 for PtdSer binding as compared to TIM-4 (Tietjen et al., 2014). Thus, both ZKA64 and sTIM1MLDR801 inhibited 85% of infection at the same concentration (1 μg/ml). Recent studies showed that TIM-1 binds to phosphatidylethanolamine (PtdEtr) (Richard et al., 2015), in addition to PtdSer. Therefore, we investigated whether sTIM1dMLDR801 inhibits ZIKV infection mediated by the interaction of envelope PtdEtr with CD300A. We found that it efficiently blocked ZIKV infection of 293T cells that ectopically express CD300A (CD300A 293T) (Fig. 4D). To confirm the phospholipid specificity of sTIM1dMLDR801, we investigated its effects on phospholipid-independent ZIKV infection mediated by the interaction between the N-glycan of the ZIKV E protein and DC-SIGN (Gong et al., 2018). While ectopic expression of DC-SIGN enhanced ZIKV infection of 293T (Fig. 4D), sTIM1dMLDR801 did not inhibit DC-SIGN-mediated ZIKV infection. On the other hand, ZKA64, which blocks ZIKV by binding to the ZIKV E protein, expectedly blocked both CD300A- and DC-SIGN- mediated ZIKV infection (Fig. 4D). These results conclude that sTIM1dMLDR801 specifically blocks PtdSer- and PtdEtr- dependent virus entry mechanisms.
Inhibition of PtdSer-dependent infection of replication-competent ZIKV and DENV and Ebola virus glycoprotein (EBOV GP)-pseudotyped lentiviral vector
In order to quantitatively analyze the effects of various molecules on single-cycle ZIKV infection, we used ZIKV replicons in our previous experiments. To examine whether sTIM1dMLDR801 can inhibit infection of replication-competent ZIKV, we next used replication-competent ZIKV. Since ZIKV produced from mosquito cells plays a critical role in transmission to humans, we generated replication-competent ZIKV produced from mammalian and insect cells. It is unknown whether the concentrations of PtdSer and PtdEtr exposed on the envelope differ between mammalian- and insect- derived ZIKV. So we investigated whether sTIM1dMLDR801 can inhibit TIM-1-dependent infection of replication-competent ZIKV produced from human cells and insect cells. TIM-1 enhanced infection of replication-competent ZIKV produced from both human and mosquito cells (Fig. 5A). We found that sTIM1dMLDR801 inhibited infection of TIM-1–293T cells with replication-competent ZIKV regardless of the producer cell (Fig. 5A).
Fig 5. Inhibition of infection by sTIM1dMLDR801 with various types of enveloped viruses.
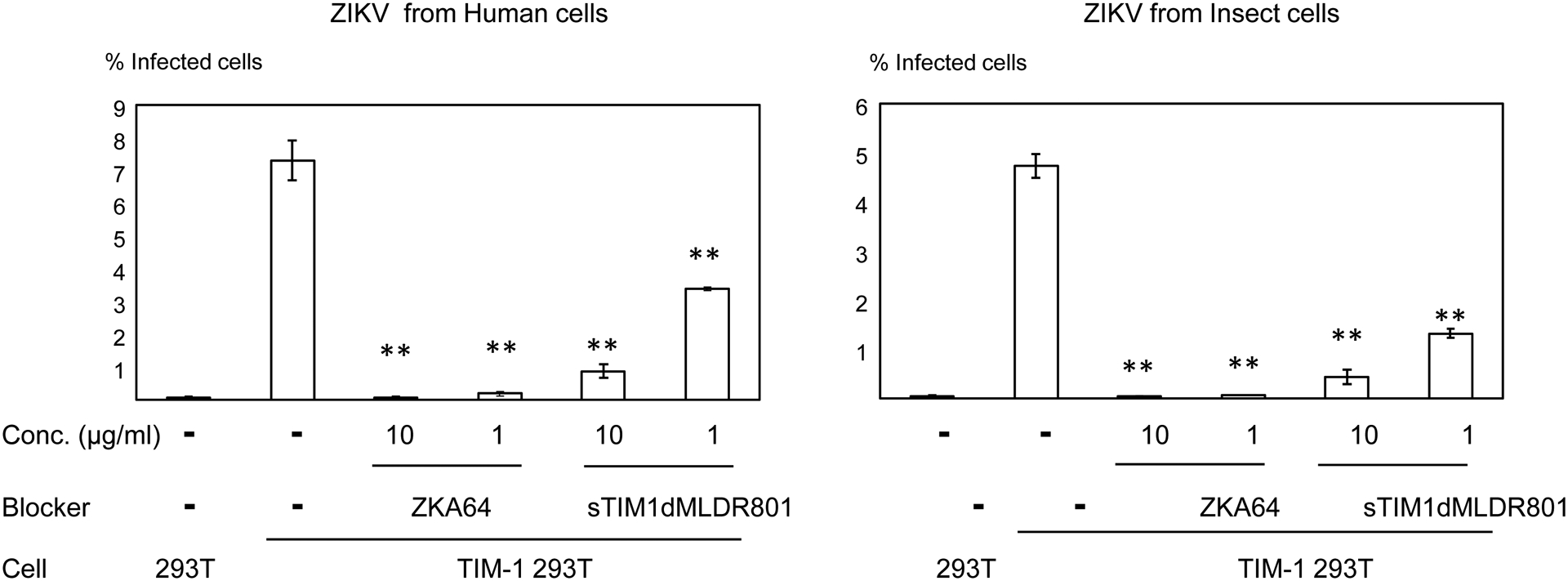
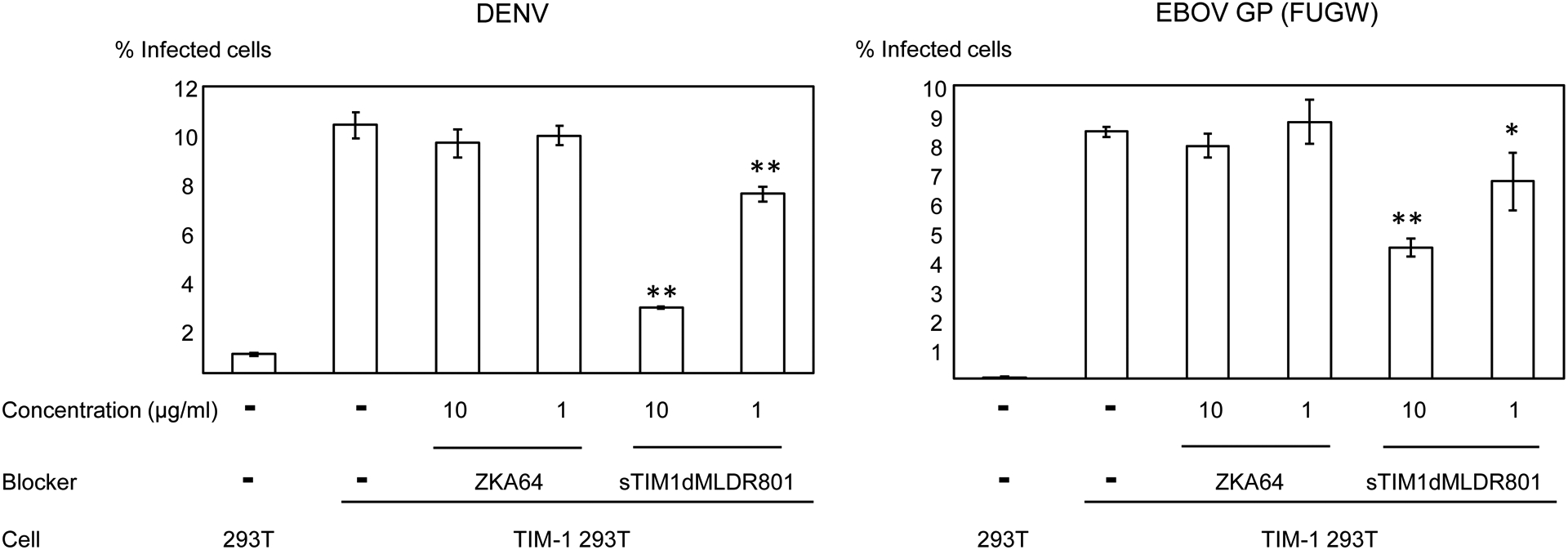
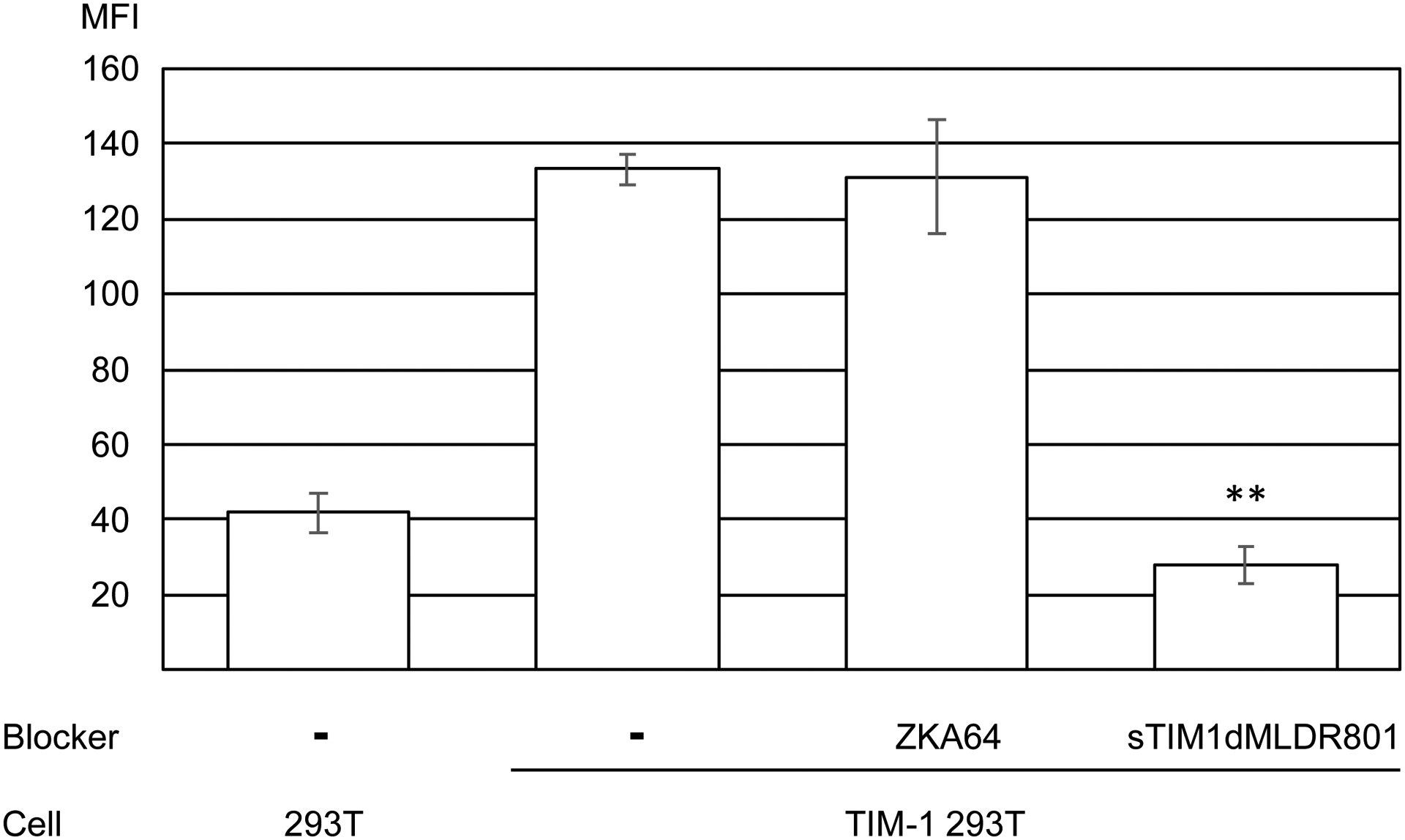
Replication-competent ZIKV produced by mammalian cells (A) or mosquito cells (A), DENV (B), and EBOV GP pseudotyped lentiviral vector [EBOV GP (FUGW)] (B) were incubated with 1 or 10 μg/ml of ZKA64 or sTIM1dMLDR801, or control medium for 1 hour at room temperature. 293T and TIM-1 293T cells were infected with the incubated virus for 2 hours, and the infections were analyzed by flow cytometry 24 hours (ZIKV and DENV) or 72 hours [EBOV GP (FUGW)] post-infection. The results shown are averages and standard deviations of the triplicate experiment. Significance was calculated by comparing TIM-1 293T cell infection without blocking reagents to those with blocking reagents, using a two-sample two-sided unpaired student t-test (*, p<0.05; **, p<0.01). (C) Quantitation of the EGFP-labeled EBOV GP pseudotype binding to 293T cells and TIM-1 293T cells. The values of the Y-axis correspond to the MFI of EGFP signals, which increase by binding of EGFP-labeled virus. This experiment was repeated once in singlicate and twice in triplicate, and the results shown are averages and standard deviations of one representative triplicate experiment. Significance was calculated by comparing binding of the EBOV GP pseudotype to TIM-1 293T cells with or without blocking reagents, using a two-sample two-sided unpaired student t-test (**, p<0.01).
TIM-1 has been shown to mediate infection of various types of enveloped viruses, including Ebola virus and DENV(Kondratowicz et al., 2011; Moller-Tank et al., 2013) (Moller-Tank et al., 2014) (Dejarnac et al., 2018) (Kuroda et al., 2015). We investigated whether sTIM1dMLDR801 blocks TIM-1-dependent infection regardless of the species of enveloped virus. We found that TIM-1 enhanced infection of replication-competent DENV (Fig. 5B). sTIM1dMLDR801 inhibited DENV infection of TIM-1 293T cells, but ZKA64 did not because of its specificity to the ZIKV E protein. A lentiviral vector pseudotyped with EBOV GP has been used to investigate Ebola virus entry mechanisms(Diehl et al., 2016; Kobinger et al., 2001; Sinn et al., 2017). Using EBOV GP pseudotyped lentiviral vectors, we investigated whether sTIM1dMLDR801 can block PtdSer-dependent Ebola virus entry. We observed that TIM-1 enhanced EBOV GP pseudotype infection, and sTIM1dMLDR801 inhibited the TIM-1-mediated EBOV GP pseudotype, while ZKA64 did not (Fig. 5B). These results confirmed that sTIM1dMLDR801 blocks infection of PtdSer-dependent enveloped virus regardless of the species of the enveloped virus.
We next examined the ability of sTIM1dMLDR801 to block viral infection/transduction at the virus-binding step. We used EBOV GP pseudotyped lentiviral vectors containing the EGFP-gag fusion protein (Hermida-Matsumoto and Resh, 2000) (Morizono and Chen, 2014; Morizono et al., 2011). Due to the incorporation of the EGFP fusion protein into virions, binding of this virus to target cells can be quantitatively analyzed by flow cytometry. Using the fluorescently labelled virus, we observed whether sTIM1dMLDR801 blocks virus binding (Fig. 5C). Expression of TIM-1 enhanced binding of the EBOV GP pseudotype. sTIM1dMLDR801 inhibited virus binding to TIM-1 293T cells, while ZKA64 did not. These results confirmed that sTIM1dMLDR801 blocks PtdSer-dependent virus binding.
The role of envelope PtdSer and PtdEtr in ZIKV infection of various types of human cells and mosquito cells.
We next used sTIM1dMLDR801 to investigate the roles of envelope PtdSer and PtdEtr in infection of various human cell types significant to the pathogenesis of ZIKV, including glial cells (U87), adult normal human dermal fibroblasts (NHDF), newborn human skin fibroblasts (CCD1064sk), fetal retinal pigment epithelial cells (FRPE), and dendritic cells(Garcia et al., 2020; Gong et al., 2018) (Hamel et al., 2015) (Meertens et al., 2017). To anticipate the roles of PtdSer and PtdEtr in enveloped virus infection in these cells, we initially analyzed the expression of the PtdSer receptors shown in Fig. 1A as well as DC-SIGN on these cells. None of these cells expressed TYRO3, TIM-1, or TIM-4 whereas all cell types except dendritic cells expressed Axl. DC-SIGN and CD300A are expressed only on dendritic cells.
Consistent with Axl expression, ZIKV infection of U87, NHDF, CCD1064sk, and FRPE, but not dendritic cells, was enhanced by addition of Gas6 (Fig. 6A–E). sTIM1dMLDR801 at 10 μg/ml inhibited 77–91% of ZIKV infection of these cells, regardless of the presence or absence of Gas6. These results demonstrated that ZIKV attachment to these cell types can be largely attributed to envelope PtdSer and/or PtdEtr. In contrast, sTIM1dMLDR801 at 10 μg/ml weakly inhibited ZIKV infection of dendritic cells (35%) (Fig 6E), indicating that attachment of the virus is mainly mediated in a PtdSer- and PtdEtr- independent manner, most likely by the interaction between the N-glycan of the ZIKV E protein and DC-SIGN. These results showed that the roles of PtdSer and PtdEtr shown by blocking infection of sTIM1dMLDR801 are consistent with the prediction made by expression levels of all known PtdSer and PtdEtr receptors and other viral receptors.
Fig. 6. Inhibition of ZIKV infection of various cells types.
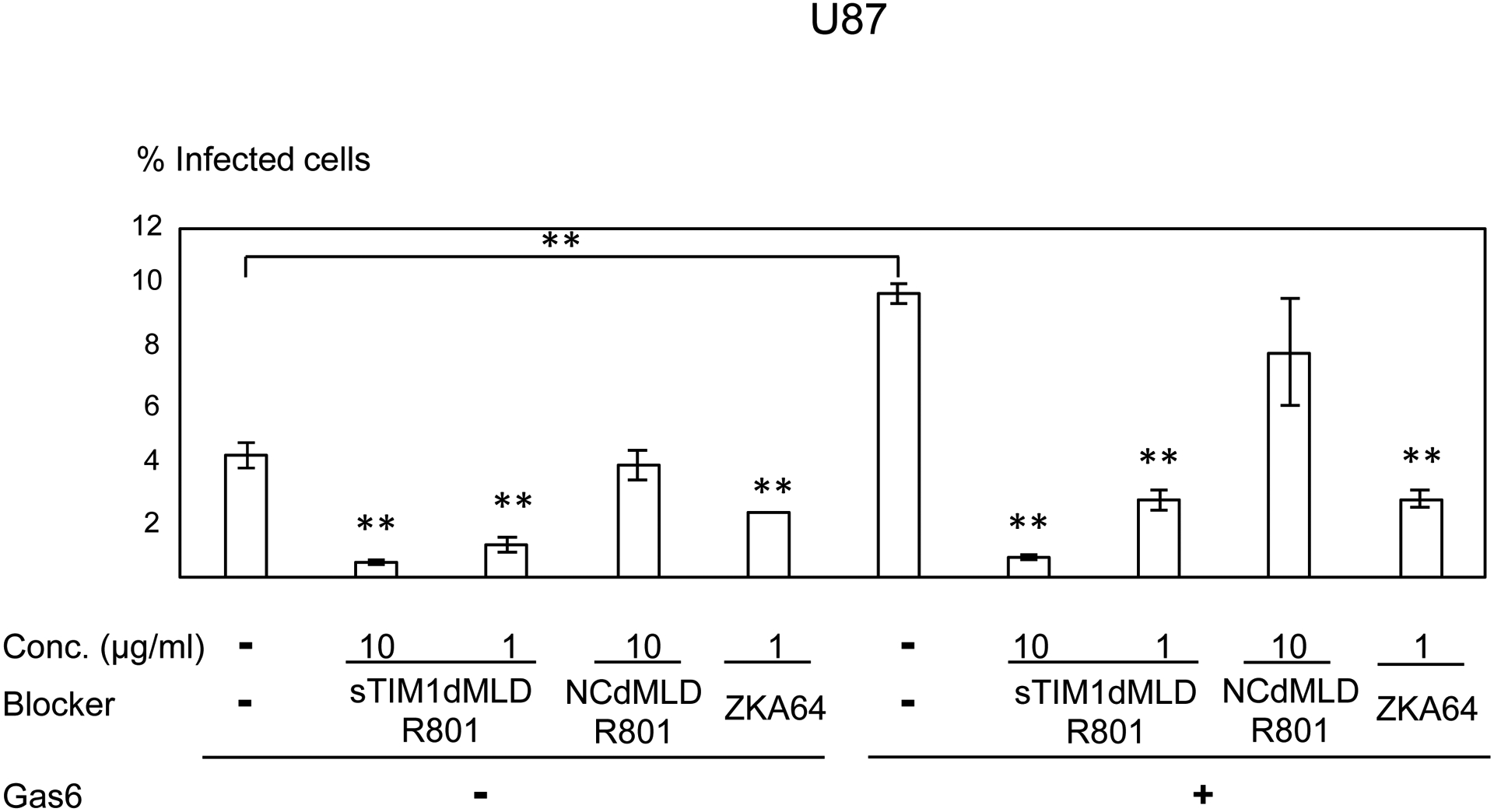
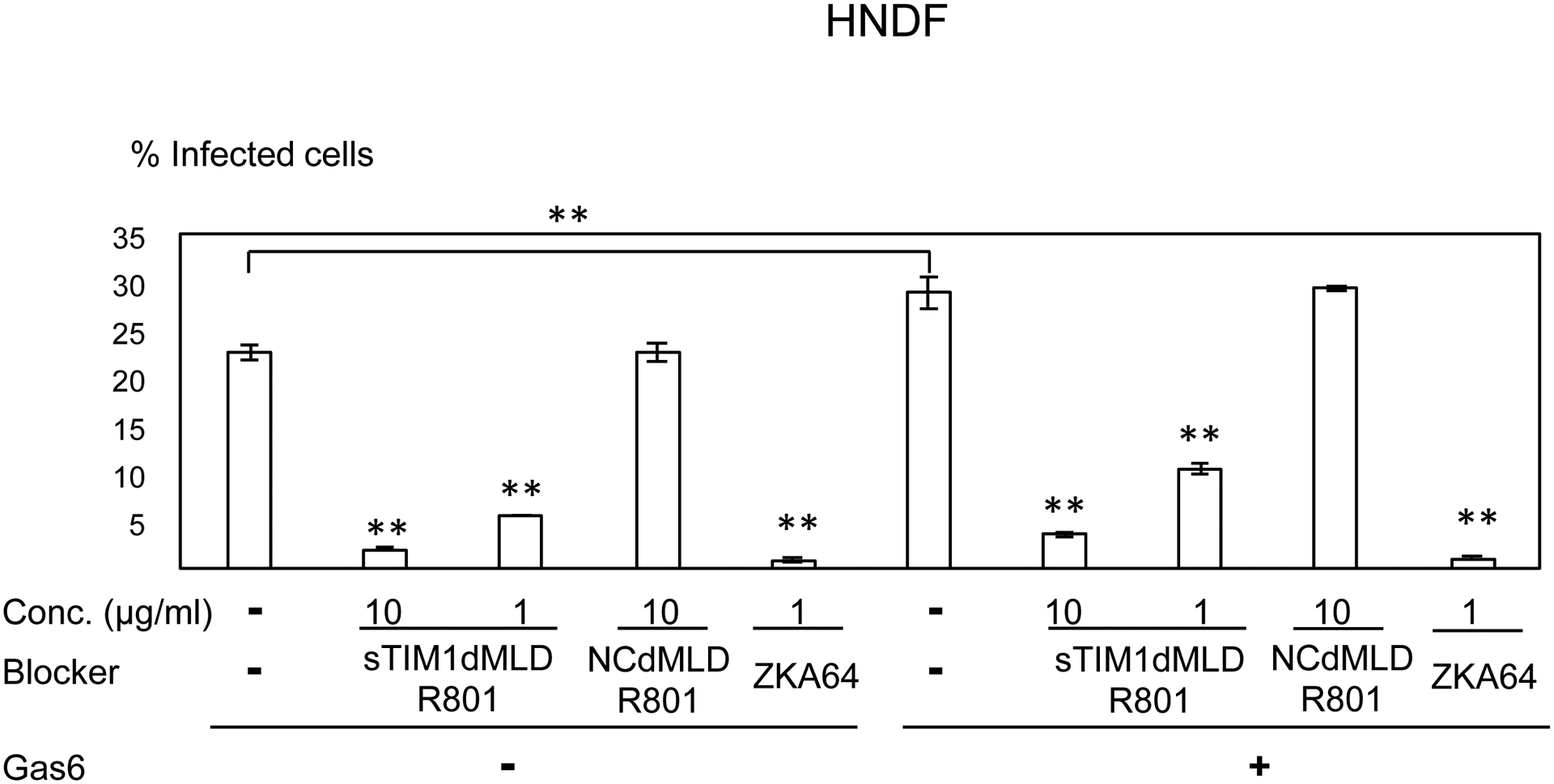
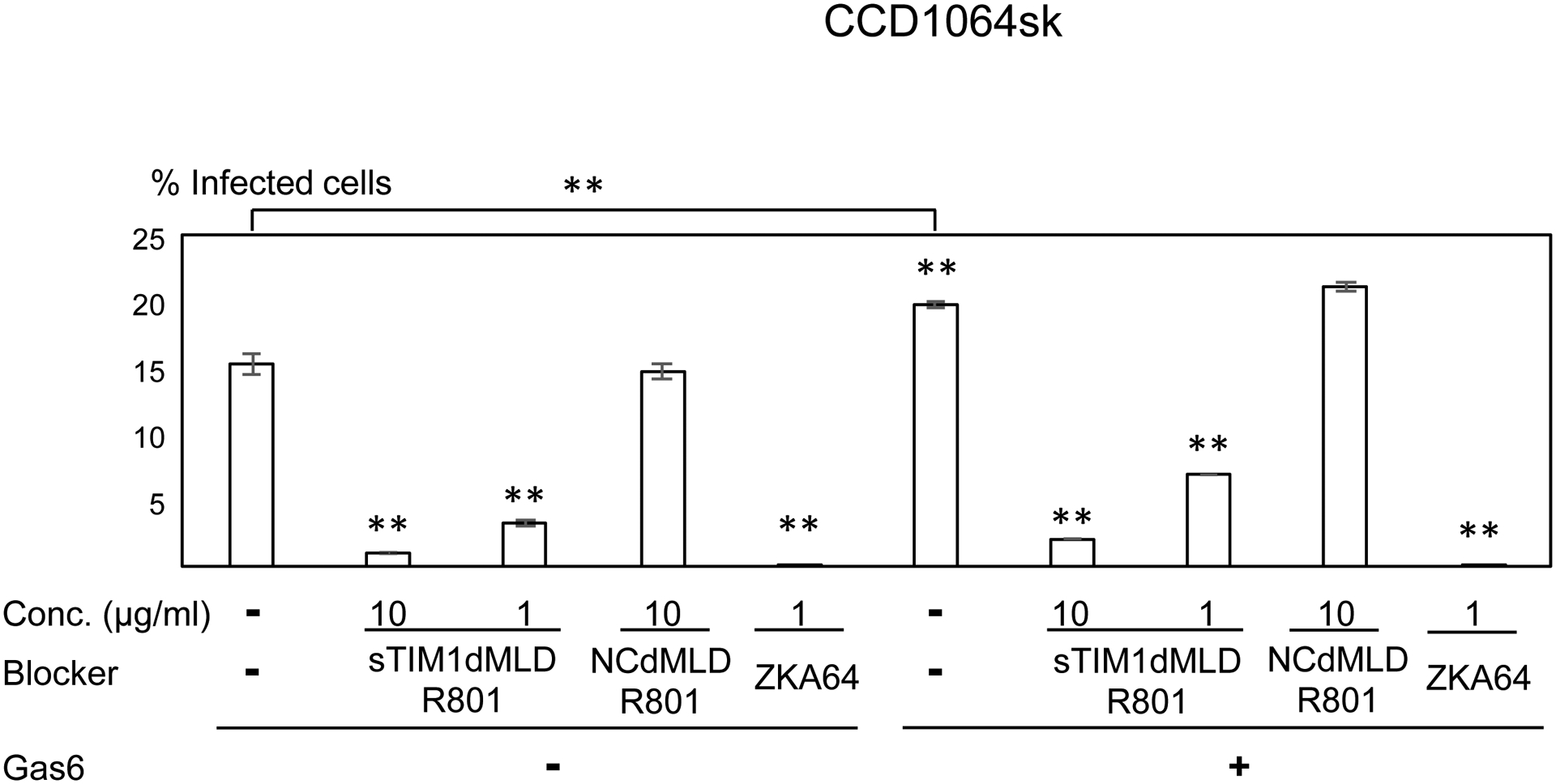
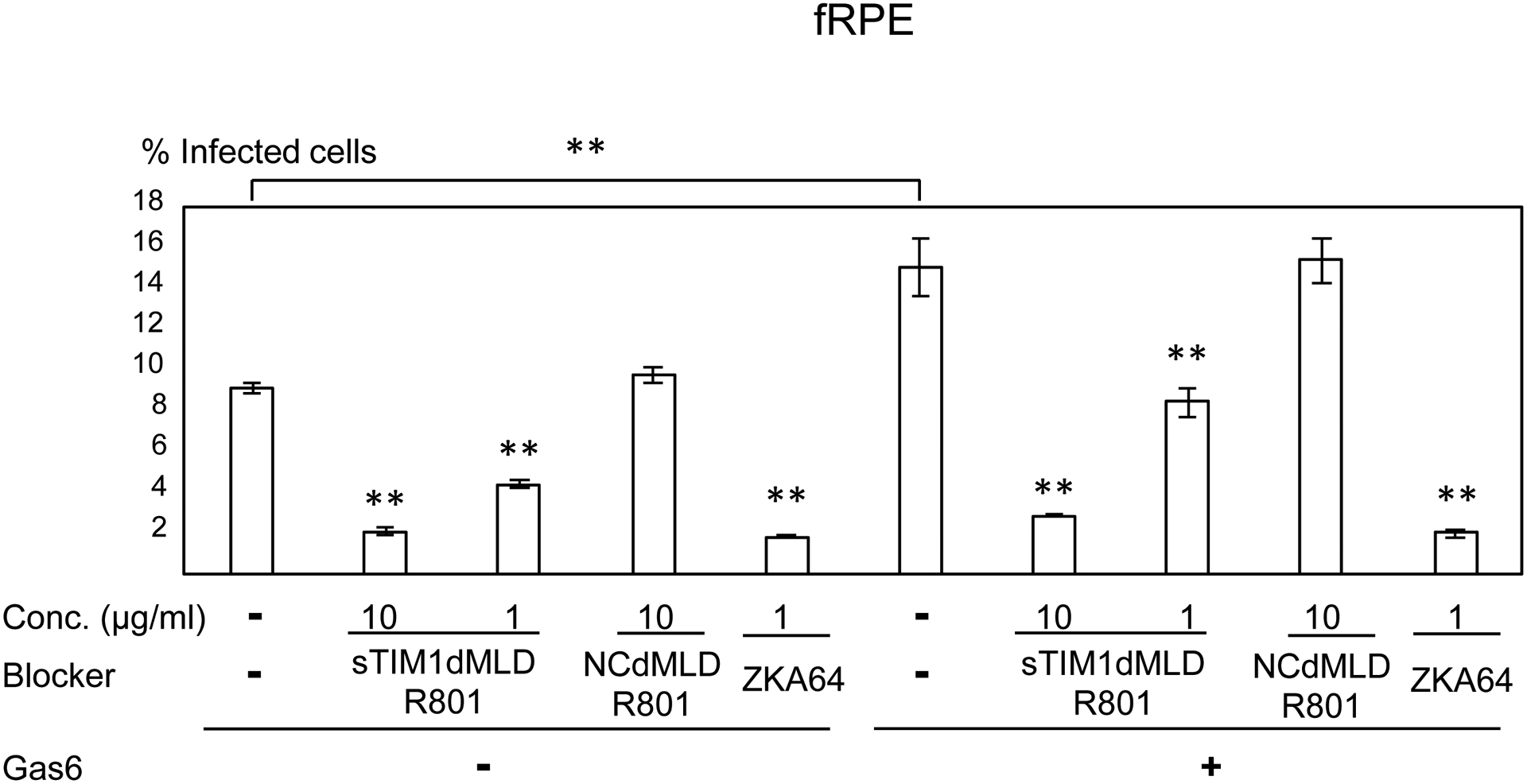
ZIKV replicon was incubated with 1 or 10 μg/ml of ZKA64, sTIM1dMLDR801, or NCFcR801, or control medium for 1 hour at room temperature. U87 (A), NHDF (B), CCD1064sk (C), FRPE (D), dendritic cells (E), and C6/36 cells were infected with the incubated virus for 2 hours, and the infections were analyzed by flow cytometry 24 hours post-infection. The results shown are averages and standard deviations of the triplicate experiment. Significance was first calculated by comparing infections of each cell type without Gas6 to those with Gas6, using a two-sample two-sided unpaired student t-test. Significance was next calculated by comparing infection of each cell types without blocking reagents in the absence of Gas6 to those with blocking reagents in the absence of Gas6, using a two-sample two-sided unpaired student t-test. Significance was then calculated by comparing infection of each cell types without blocking reagents in the presence of Gas6 to those with blocking reagents in the presence of Gas6, using a two-sample two-sided unpaired student t-test (*, p<0.05; **, p<0.01).
In the life-cycle of ZIKV, ZIKV infection of mosquitoes has a critical role in transmission to humans(Weissenbock et al., 2010). It is not known whether ZIKV uses its envelope PtdSer and/or PtdEtr in its infection of mosquito cells, so we investigated whether sTIM1dMLDR801 inhibits ZIKV infection of a mosquito cell line, C6/36 (Fig. 6F) (Weissenbock et al., 2010). We found that sTIM1dMLDR801 inhibited ZIKV infection of C6/36 cells, demonstrating that ZIKV can use its envelope PtdSer and/or PtdEtr in its infection of mosquito cells.
Inhibition of multiple-cycle of ZIKV replication by sTIM1dMLDR801
We next investigated how sTIM1dMLDR801 inhibits multiple cycles of viral replication in cells that endogenously express PtdSer receptors. We examined TIM1dMLDR801 inhibition of ZIKV replication in Vero cells which endogenously express both TIM-1 and Axl and produce high titers of ZIKV after infection(Brouillette et al., 2018; Hamel et al., 2015). We first determined how ZKA64 and sTIM1 inhibit single-cycle ZIKV infection of Vero cells, using a ZIKV replicon, and found that sTIM1 inhibits Vero cell infection more efficiently than ZKA64 at concentrations lower than 700 ng/ml, but less efficiently at concentrations higher than 700 ng/ml. While increasing concentrations of ZKA64 from 300 ng/ml to 3 μg/ml increased its inhibitory effect from 57% to 98%, the same increases in sTIM1dMLDR801 concentrations increased its inhibitory effects only from 64% to 90% (Fig. 7A). These results indicated that infection of Vero cells is mainly mediated by PtdSer- and/or PtdEtr- dependent mechanisms; however, infection is partially mediated by a PtdSer/PtdEtr-independent mechanism(s) that is resistant to inhibition by sTIM1dMLDR801. On the other hand, ZKA64 can inhibit ZIKV infection regardless of the viral receptors used as displayed in Fig. 4C.
Fig. 7. Inhibition of multiple-cycle replication by sTIM1dMLDR801.
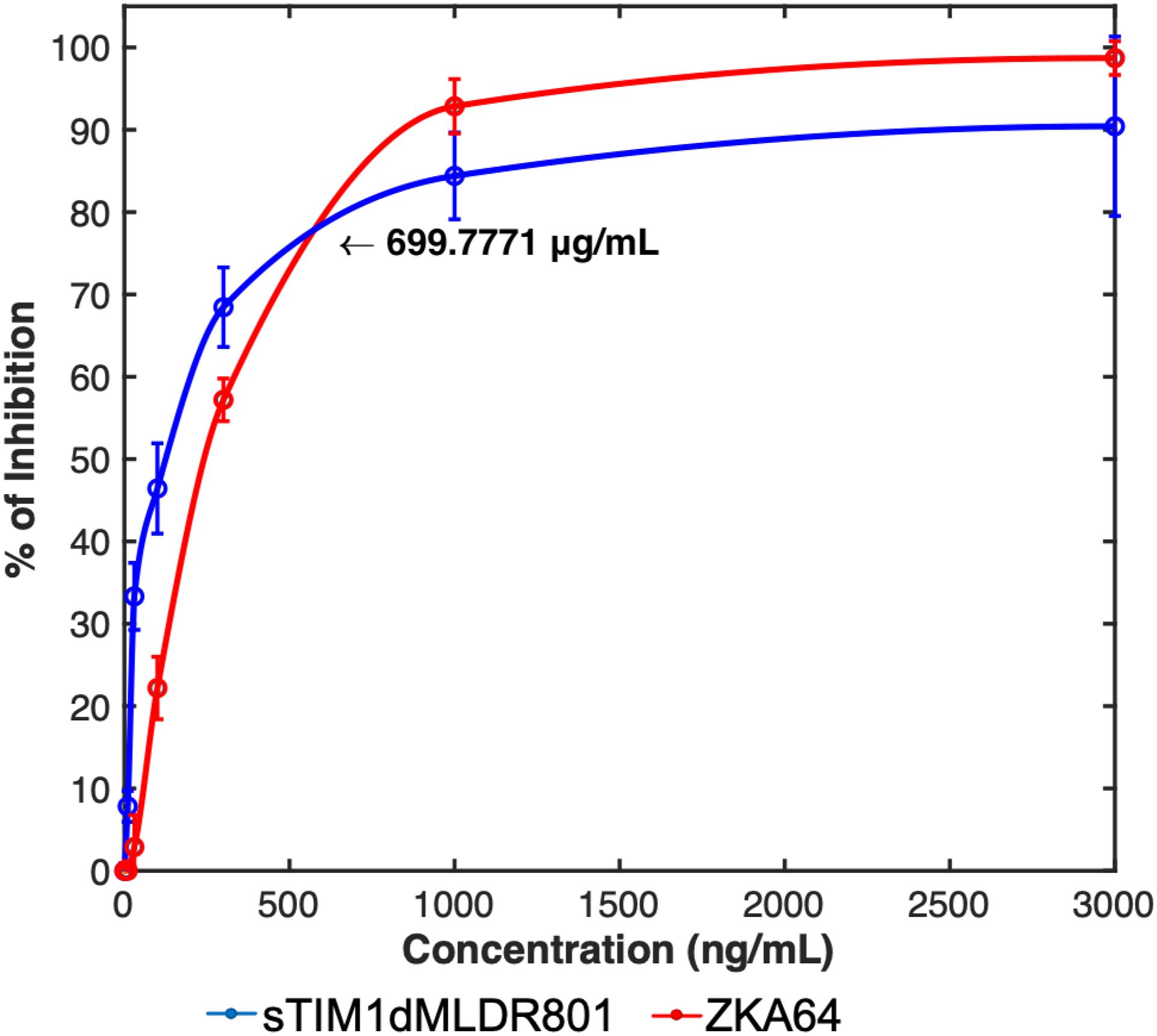
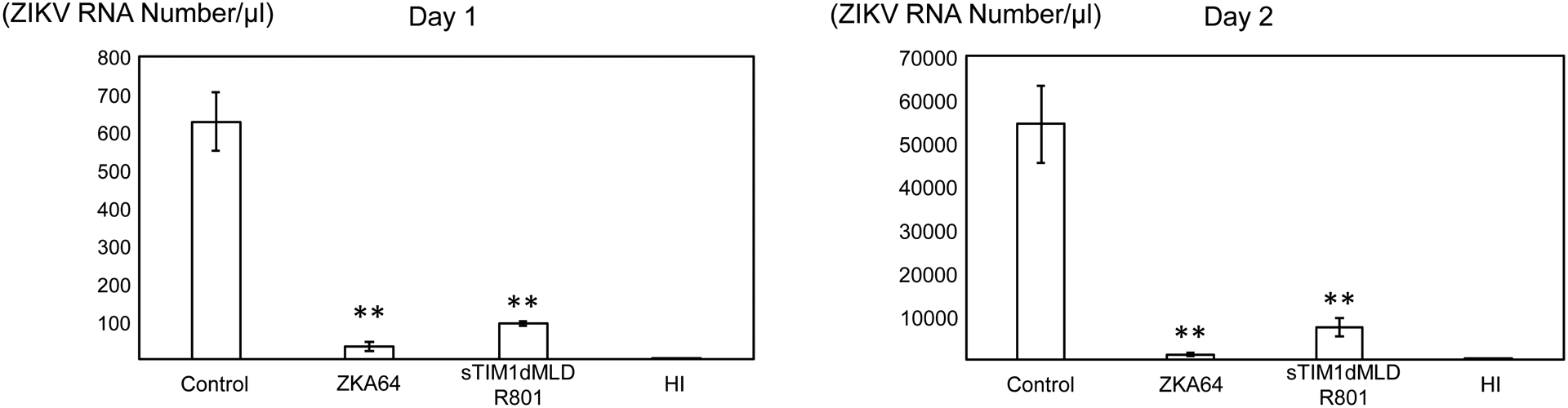
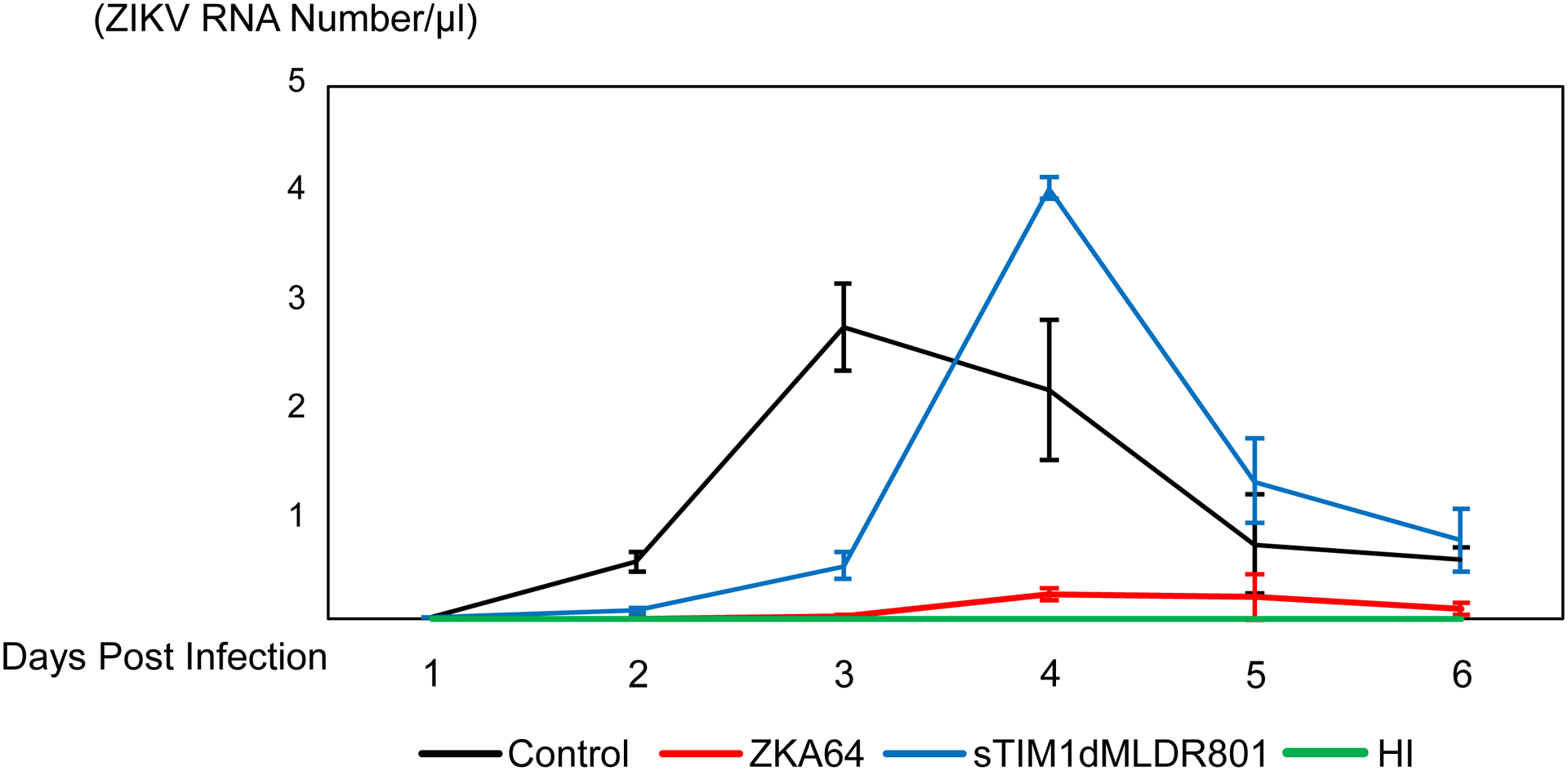
The results shown are averages and standard deviations of the triplicate experiment. A) Inhibition of single-cycle infection of Vero cells with ZIKV replicon by various concentrations of sTIM1dMLDR801 or ZKA64. B) Production of replication-competent ZIKV production at 1 and 2 days post-infection. Replication-competent ZIKV was incubated with 700 ng/ml of ZKA64 or sTIM1dMLDR801 for 30 min at 37°C. Vero cells were infected with the virus (MOI 0.01) preincubated with or without ZKA64, sTIM1dMLD R801, or heat-inactivated virus for 2 hours at 37°C. The cells were then washed 3 times with culture medium and cultured in fresh culture medium containing the same reagents (700 ng/ml) used for incubation with the virus. ZKA64 and sTIM1dMLDR801 were present days 0–6 post-infection at 700 ng/ml. Significance was calculated by comparing Vero cell infection without blocking reagents to those with blocking reagents, using a two-sample two-sided unpaired student t-test (**, p<0.01). C) Production of replication-competent ZIKV production from day 1 to day 6 post-infection.
We studied the effects of ZKA64 and sTIM1dMLDR801 on multiple cycles of ZIKV replication in Vero cells at the concentration of 700 ng/ml, which is the concentration at which both reagents inhibit 70% of single-cycle of ZIKV infection. To rigorously quantify the amount of virus produced in the supernatants, we used digital droplet PCR (ddPCR). One day post-infection, ZKA64 inhibited 90% of virus production and sTIM1dMLDR801 inhibited 80% (Fig. 7B). Two and three days post-infection, ZKA64 inhibited 99.9% of virus production (Fig. 7B and C) due to its inhibitory effects being amplified when the virus undergoes multiple cycles of replication. Overall, ZKA64 efficiently inhibited ZIKV replication for 6 days (Fig. 7C). In contrast, the inhibitory effects of sTIM1dMLDR801 remained at 80% for both 2 and 3 days post-infection, suggesting that there is (an) effects of sTIM1dMLDR801 on production of replication-competent ZIKV, which were not evident in the assays using single-cycle infection by ZIKV replicon. At three days post-infection, ZIKV production in untreated cells reached its maximum level and subsequently decreased due to cell death (Fig. 7C). Although sTIM1dMLDR801 can reduce virus production for up to three days post-infection, treated cells produced virus at higher levels than untreated cells at four days post-infection. Consistent with our virus production data, sTIM1dMLDR801 inhibited cell death for the first 3 days, but we observed massive cell death at 4 days post-infection (data not shown). These results suggest that sTIM1dMLDR801 can inhibit and slow ZIKV replication most notably during the early cycles of ZIKV replication. However, during later cycles of viral replication, sTIM1dMLDR801 shows minimal inhibitory effects and may even enhance viral production.
Inhibition of ZIKV replication by sTIM1dMLDR801 in vivo
We next used sTIM1dMLDR801 to investigate the roles of envelope PtdSer and PtdEtr in in vivo virus replication. Intraperitoneal injection of ZKA64 (15 μg/g) either before or after subcutaneous ZIKV infection has been shown to inhibit more than 99% of ZIKV production in mouse tissues compared to untreated mice at 13 days post-infection (Stettler et al., 2016). We anticipated that sTIM1dMLDR801 would not inhibit ZIKV replication in vivo as efficiently as ZKA64 due to its inability to block PtdSer- and/or PtdEtr- independent ZIKV infection, as well as its incomplete inhibition of multiple-cycle infection in Vero cells. However, we expected the viral load to decrease, particularly during the early cycles of viral replication, if PtdSer and/or PtdEtr do support ZIKV replication in such organs and tissues. To that end, we examined whether sTIM1 dMLDR801 can decrease viral load when used in the ZIKV infection of mice. As the IC50 of sTIM1dMLDR801 is 2.7- and 5.5- fold higher than that of ZKA64 for either Axl/Gas6-dependent or TIM-1-dependent infection mechanisms, respectively, we increased the administered concentration four-fold (60 μg/g). To ensure that sTIM1dMLDR801 is present when the virus begins to infect and replicate, we injected sTIM1dMLDR801 both one day before and after ZIKV injection (Fig 8A). We limited administration of sTIM1dMLDR801 to these two time points to avoid its potential enhancing effects on the later ZIKV replication cycle observed in vitro. To compare viral replication in live mice, we sacrificed all mice at 7 days after virus injection before we observed death of any of infected mice and isolated serum, brain, and spleen to quantitate viral RNA. sTIM1dMLDR801 did not prevent weight loss of infected mice (S2 Fig). Quantification of serum viral load by ddPCR showed that administration of sTIM1dMLDR801 decreased the serum viral load by 60% with statistical significance (Fig 8B). We next investigated viral load in the brain and spleen. Large molecules such as antibodies cannot migrate into the brain due to the blood-brain barrier(Pardridge, 2002), but the spleen has sinusoid capillaries that allows passage of large molecules (Eigenmann et al., 2017). To rigorously assess viral production in these two organs, we measured the copy numbers of both GAPDH mRNA and viral mRNA in the same reverse-transcribed cDNA samples and showed the results as viral RNA copy numbers/GAPDH RNA. We found that sTIM1dMLDR801 did not decrease the average viral RNA copy numbers/GAPDH in the brain at a statistically significant level (Fig. 8B). On the other hand, sTIM1dMLDR801 decreased viral RNA copy numbers/GAPDH in the spleen by 60% with statistical significance, suggesting that envelope PtdSer and/or PtdEtr can support viral replication in vivo (Fig. 8B).
Fig 8. Effects of sTIM1dMLDR801 on ZIKV replication in vivo.
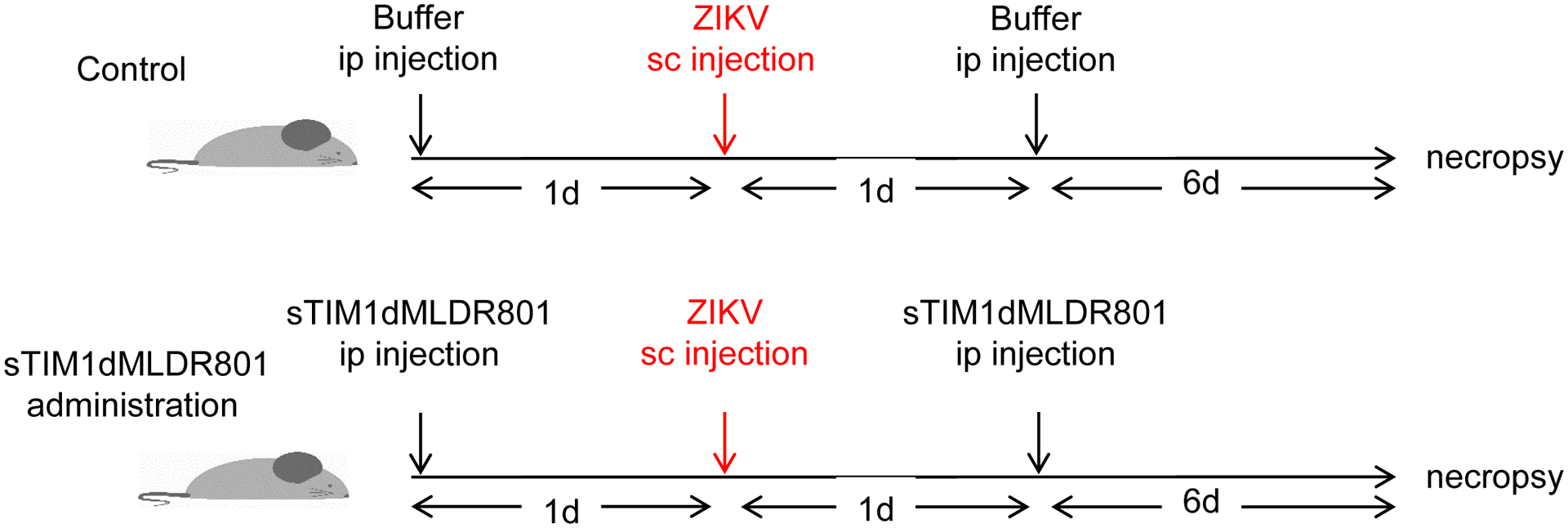
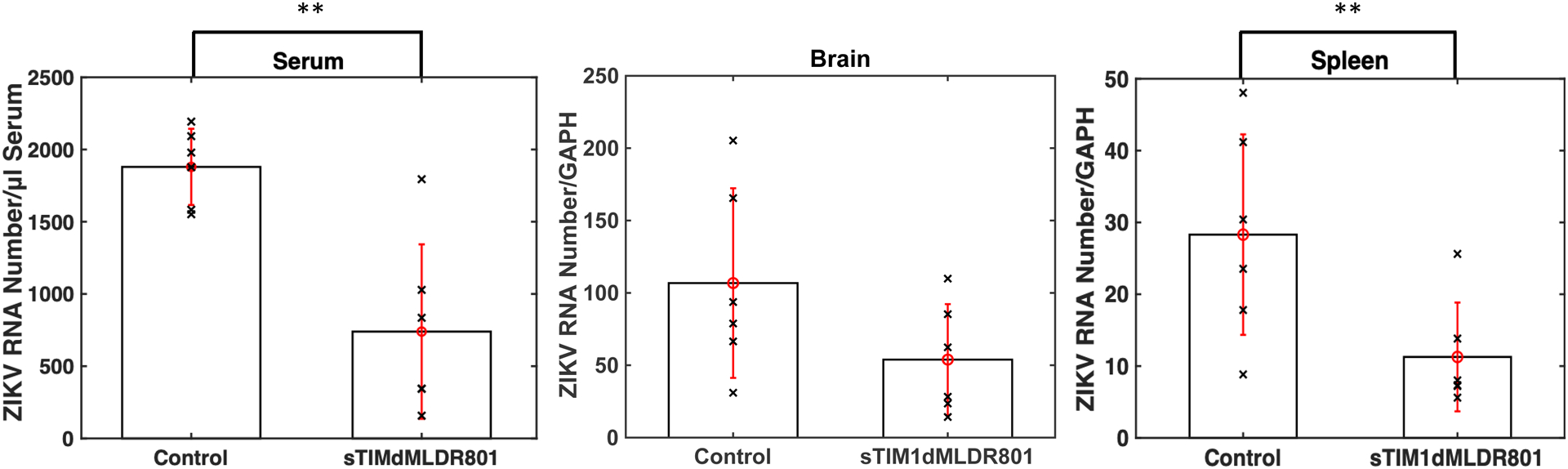
A) Schematic representation of the experimental design. Ifnar1−/− mice received intraperitoneal injections of control buffer or sTIM1dMLDR801 before and after subcutaneous injection of replication-competent ZIKV. Seven days post-injection, the mice were euthanized and viral RNA was quantitated in serum, brain, and spleen. B) Viral RNA in serum, brain, and spleen. The serum viral RNA are shown as ZIKV copy numbers/μL of serum. Brain and spleen viral RNA amount is shown as ZIKV RNA copy numbers/GAPDH RNA copy number. P values were calculated using a two-sided Wilcoxon Rank Sum Exact Test (*, p<0.05; **, p<0.01).
Discussion
We developed sTIM1dMLDR801 that inhibits PtdSer-dependent infection of enveloped viruses most efficiently among the PtdSer-binding molecules tested. Using sTIM1dMLDR801, we showed that ZIKV can utilize envelope PtdSer and/or PtdEtr when infecting not only human cells but also insect cells. sTIM1dMLDR801 inhibited ZIKV replication in mice, suggesting that envelope PtdSer and/or PtdEtr support enveloped virus replication in vivo.
Using comprehensive immunostaining of all known PtdSer receptors, we will be able to predict whether the virus can utilize PtdSer- and/or PtdEtr- mediated viral entry mechanisms on target cells of interest. However, PtdSer and PtdEtr receptors of many species other than those of humans and mice have yet to be fully identified. Our results demonstrated that ZIKV can infect mosquito cells, using envelope PtdSer and/or PtdEtr. sTIM1dMLDR801 will be applicable to further study the roles of envelope PtdSer and PtdEtr in infection of various types of enveloped viruses on the cells of various species, especially those with roles in zoonosis.
The results of blocking infection by sTIM1dMLDR801 will be informative for identification of receptors of enveloped viruses. Viral receptors have been identified by seeking host molecules binding to viral envelope proteins. However, if sTIM1dMLDR801 completely blocks infection of particular target cells with the virus of interest, investigation of PtdSer- and PtdEtr- binding molecules instead of envelope protein-binding molecules will be more appropriate for identification of viral receptors.
We found that the mutants of MFG-E8 and TIM-1 could inhibit TIM-1-mediated virus infection, while ANX V, an anti-PtdSer monoclonal antibody, as well as the PtdSer-binding host protein, β2GP1, with or without its cross-linking antibody, Bavituximab, could not efficiently block TIM-1-mediated infection. The underlying causes for such observed differences in inhibitory capacities have yet to be determined. ANX V was previously reported to bind to PtdSer with high affinities (Kd: 5×10−10); therefore, it is unlikely that the affinity of ANX V accounts for its inability to inhibit viral infection(Andree et al., 1990). One possible explanation is that the affinity of sTIM1 and D89E for PtdEtr is important for inhibiting the binding of PtdSer-binding viral receptors to envelope PtdSer. However, in our previous studies, viral infection mediated by TIM-4, which bind PtdSer but not PtdEtr, cannot be efficiently blocked by ANX V(Morizono and Chen, 2014). Therefore, we are not certain that the lack of affinity for PtdEtr can generally explain why some of the PtdSer-binding molecules cannot block PtdSer-dependent viral infection. A second possible explanation is that ANX V, anti-PtdSer antibody, and/or β2Gp1 bind PtdSer at sites different than those used by TIM-1, TIM-4, MFG-E8, and Gas6. However, all these PtdSer-binding molecules should bind the head of PtdSer, which is less than 1 nm in length(Pan et al., 2014). It is unlikely that these proteins can bind different sites of such a small portion of the phospholipid. A third possible explanation is that ANX V, anti-PtdSer-antibody, and/or β2GP1 cannot completely conceal all PtdSer exposed on the viral envelope, due to the steric hindrance between themselves. Thus, TIM-1, TIM-4, Gas6, and MFG-E8 can reach and bind envelope PtdSer even if ANX V, anti-PtdSer antibody, and β2GP1 maximally bind envelope PtdSer. On the other hand, sTIM-1 and D89E might conceal envelope PtdSer in a manner that inhibits the access of TIM-1, TIM-4, Gas6, and MFG-E8 to envelope PtdSer. Investigation of these possibilities will require analytical methods that can analyze the interactions between PtdSer-binding molecules and PtdSer exposed on the virus but not on cells or coated ELISA plates, since curvature of the membrane and the polarity of PtdSer was shown to play important roles for binding to PtdSer-binding molecules (Shi et al., 2004) (Otzen et al., 2012) (Iwamoto et al., 2007).
Fusion with the IgG Fc-domain has been used to increase the avidity and half-lives of many molecules in vivo (Duivelshof et al., 2021). While dimerization by fusion with the Fc-domain reduced the IC50 of sTIM-1 from 13 μg/ml to 4 μg/ml, fusion of D89E with the Fc-domain increased its IC50 regardless of whether Fc was fused to its C- or N-terminus. Dimerization and/or fusion with the Fc-domain is likely to cause steric hindrance for binding of the PtdSer-binding domain (discoid domains) of D89E to PtdSer.
When TIM-1 was identified as the receptor that mediates phagocytosis of dead cells, it was reported not to bind PtdEtr (Miyanishi et al., 2007). However, recent virological research showed that TIM-1 binds PtdEtr on the viral envelope (Richard et al., 2015). Our results confirmed the binding of TIM-1 to envelope PtdEtr by showing that sTIM1dMLDR801 can inhibit PtdEtr-dependent infection mediated by CD300A. It is possible that PtdEtr exposed on the viral envelope behaves differently than PtdEtr coated on plates and/or exposed on dead cells.
Inhibition of multiple cycles of ZIKV replication by sTIM1dMLDR801 showed very different kinetics compared to inhibition by ZKA64. We adjusted the concentrations of both reagents to inhibit 75% of single-cycle replication. While ZKA64 inhibited virus replication for 6 days, sTIM1dMLDR801 inhibited viral replications only for 3 days. At four days post infection, the cells treated sTIM1dMLDR801 produced greater amount of ZIKV than maximum amounts produced from the untreated cells at 3 days post infection. The difference between the effects of sTIM1dMLDR801 on single-cycle infection and multiple-cycles replication of ZIKV is likely due to the effects of sTIM1dMLDR801 on virus budding. It has been reported that the interaction between TIM-1 and envelope PtdSer traps HIV-1 and lentiviral vectors on cell surface, thereby inhibiting budding of the virus (Li et al., 2014; Li et al., 2019). Thus, it is possible that ZIKV is trapped on the surface of Vero cells by TIM-1. sTIM1dMLDR801 could potentially release trapped ZIKV by inhibiting the interaction between TIM-1 and envelope PtdSer, thereby resulting in an increased amount of virus released. Because of the potential effects of sTIM1dMLDR801 on virus budding, it will be important to carefully design the experimental settings to focus its effects on the specific viral replication step of interest (Li et al., 2014; Li et al., 2019).
We observed that sTIM1dMLDR801 decreased viral load in serum and spleen. Since sTIM1dMLDR801 cannot inhibit PtdSer-independent virus entry, it is possible that sTIM1dMLDR801 reduces viral load but cannot completely suppress virus replication in the mouse model experiments. sTIM1dMLDR801 did not decrease the viral mRNA copy numbers in the brain at a statistically significant level, which is likely due to its inability to cross the blood-brain barrier. However, sTIM1dMLDR801 was shown to decrease serum viral loads and the copy numbers of mRNA in the spleen, suggesting that envelope PtdSer and PtdEtr can support ZIKV infection in vivo. Our in vitro data showed that sTIM1dMLDR801 can also inhibit PtdSer-dependent DENV and EBOV GP pseudotype infection. sTIM1dMLDR801 might be able to slow their replication in vivo.
Bavituximab was previously used as a PtdSer-targeting antiviral reagent (Luster et al., 2006; Ran et al., 2003; Soares et al., 2008). Bavituximab has been shown to inhibit the replication of various enveloped viruses, including Pichinde virus and cytomegalovirus in vivo (Soares et al., 2008). Animals that received Bavituximab showed significant improvement in survival rates and decrease in viral load. However, our data showed that Bavituximab does not inhibit PtdSer-dependent infection of TIM-1-expressing cells, thus it is unclear how Bavituximab inhibits viral replication in vivo. It is possible that the binding of β2GP1 and Bavituximab to viruses enables Fc-receptor-positive cells to capture viruses and activate antiviral immunity against the virus. Since we wanted to focus on the roles of PtdSer and PtdEtr in in vivo replication of enveloped viruses, we eliminated the affinity for Fc-receptors to improve interpretation of the data. It will be of future interest to determine whether reverting the Fc-domain of sTIM1dMLDR801 back to wild-type changes its effects on viral replication in vivo through effector functions of Fc-receptor-expressing cells.
Materials and Methods
Plasmids
The expression vector of 4E Gas6 was generated as previously described (Chua et al., 2018), and the vector of D89E was obtained from Dr. Shigekazu Nagata (Hanayama et al., 2002). The expression vector of sTIM1 was generated by replacing the VSV-g of pCMV VSV-g (Burns et al., 1993) with the extra cellular domain of human TIM-1 (a.a. 1–297) fused with a 3-teminus Histidine tag. The expression vectors of sTIM1Fc and sTIM1dMLDFc were generated by replacing the VSV-g of pCMV VSV-g (Burns et al., 1993) with the human IgG1 Fc region fused with sTIM1 with or without MLD (a.a 145–297), respectively. sTIM1dMLDR801 was generated by introducing three mutations into the Fc-domain as previously reported (Lee et al., 2017), and NCdMLDR801 was generated by deleting the IgV domain of TIM-1 from sTIM1dMLDR801. D89E Fc and Fc D89E were generated by replacing the VSV-g cDNA of pCMV VSV-g with D89E fused with the human IgG1 Fc-region at its C- and N- terminus, respectively. Human β2GP1 expression was generated by replacing the VSV-g cDNA of pCMV VSV-g with synthesized human β2GP1 cDNA (GENWIZ, La Jolla, CA) fused with a 3-terminus Flag tag sequence. ZIKV packaging plasmid ZIKV H/PF CprME and EGFP-expressing WNV-based replicon plasmid WNVII-Rep-G/Z were kindly provided by Dr. Benjamin Hurley’s lab (Dowd et al., 2016; Pierson et al., 2006). The EGFP-expressing ZIKV-based replicon, C1P9, was generated by Dr. Vaithi Arumugaswami’s lab (manuscript in preparation).
Viruses, replicons, and viral vectors
The PRVABC59 (GenBank accession number KU501215) ZIKV strain of Asian genotype was used as the replication-competent ZIKV (Lanciotti et al., 2016). Working viral stock for the specified experiments was generated by subjecting the original ZIKV strain (passage=3) to two additional passages in A549 or C6/36 cells. DENV Type 1, UIS 998, NR-49713 was acquired from BEI Resources, NIAID, NIH, as part of the WRCEVA program. DENV stock for the specified experiments was generated by subjecting the original dengue virus strain (passage=3) to one additional passage in Vero cells.
The ZIKV replicon was generated as previously described (Dowd et al., 2016; Pierson et al., 2006). Briefly, 293T cells were transfected with the packaging plasmid, ZIKV H/PF CprME, together with either WNVII-Rep-G/Z or C1P9, using TransIT LT1 (Mirus Bio, Madison, WI) according to the manufacturer’s protocol. Twenty-four hours post-transfection, medium was changed to AIM-V, and cells were cultured at 30°C. Supernatant was collected, filtered, and frozen daily from 5 to 9 days post-transfection daily. For infection of most cell types used, we used replicon derived from WNVII-Rep-G/Z as ZIKV replicon. For infection of HMVEC, the replicon derived from WNVII-Rep-G/Z does not express the EGFP transgene in HMVEC so we instead used the replicon derived from C1P9.
EBOV GP pseudotype was produced in 293T cells using a previously described transfection method (Situ et al., 2018). The EGFP-expressing lentiviral vector was produced by transfecting 293T cells with an expression vector of EBOV GP protein without the mucin-like domain (Addgene, Watertown, MA) (Diehl et al., 2016), the packaging plasmid PAX2, and a lentiviral vector containing EGFP (FUGW) as its transgene (Lois et al., 2002), as previously described.
The EBOV GP pseudotype labeled by EGFP was generated by transfecting 293T cells with the EBOV GP expression vector, PAX2, and p-gagEGFP (NIH AIDS Research & Reference Reagent Program, Germantown, MD) (Hermida-Matsumoto and Resh, 2000). The supernatant was harvested, filtered, and concentrated by ultracentrifugation 72 hours post-transfection, as previously described.
Proteins
Recombinant human Gas6 and 4E Gas6 were produced as previously described (Chua et al., 2018; Morizono and Chen, 2014). Β2GP1, sTIM1, sTIM1 Fc, sTIM1dMLDR801, NCdMLDR801, D89E Fc, and Fc D89E were produced by transfecting 293F cells with their expression vector plasmids, using TransIT LT1 according to the manufacturer’s protocol. One day post-transfection, cells were cultured with Pro293s CDM medium supplemented with glutamine, penicillin, and streptomycin, and were rotated in roller bottles at 8 RPM. The supernatants were collected and filtered 5 days post-transfection. The supernatant of sTIM1 was first concentrated by ammonium sulfate purification, followed by purification by TALON column (Takarabio, Mountain View, CA) according to the manufacturer’s protocol. The supernatant of β2GP1 was incubated with anti-Flag tag antibody-conjugated beads (Sigma-Aldrich, St. Louis, MO) for 1 hour at room temperature. The supernatants of sTIM1 Fc, sTIM1dMLDR801, NCdMLDR801, D89E Fc, and Fc D89E were incubated with MabCapture A (Thermo Fisher Scientific, Canoga Park, CA) beads for 1 hour at room temperature. The beads were pelleted by centrifugation, then washed in columns, and recombinant proteins were eluted according to the manufacturer’s protocols. The buffers of all purified proteins were changed to Hanks buffered solution (Thermo Fisher Scientific), using Zeba spin columns (Thermo Fisher Scientific).
Annexin V was purchased from eBioscience (San Diego, CA). ZKA64 anti-ZIKV E protein antibody was purchased from Absolute Antibody (Boston, MA). Bavituximab was purchased from Creative Biolab (Shirley, NY), and Anti-PtdSer monoclonal antibody was purchased from Millipore (Burlington, MA).
Cells
Human microvascular endothelial cells, normal human dermal fibroblasts (PromoCell, Heidelberg, Germany), and CCD1064sk (ATCC, Manassas, VA) cells were cultured in the EGM-2 bullet kit (Lonza, Walkersville, MD). Human peripheral blood mononuclear cells (PBMC) were obtained from the UCLA Virology Core Laboratory. Monocytes were isolated from PBMC using the Pan Monocyte Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s protocol. Dendritic cells were prepared by culturing monocyte populations with 800 IU/ml of GM-CSF (Bayer Healthcare, Seattle, WA) and 63 ng/ml IL-4 (R&D Systems, Minneapolis, MN) for 7 days at 37°C, as previously described. DC were recovered on day 7, and the surface markers of isolated cells were confirmed by flow cytometry after staining the cells with antibodies against various dendritic cell markers, including HLA DR, CD86, CD14, CD11c, and the DC-SIGN antibody conjugated with APC (Morizono et al., 2010). K562 and 293T cells were cultured in IMDM (Thermo Fisher Scientific) containing 10% FCS (Sigma-Aldrich) and 1% penicillin/streptomycin (Thermo Fisher Scientific). 293T cells stably expressing TIM-1, TIM-4, CD300a, or DC-SIGN were designated as TIM-1 293T, TIM-4 293T, CD300a 293T, and DC-SIGN 293T, respectively. TIM-1, TIM-4, CD300a, and DC-SIGN were expressed by transducing 293T cells with lentiviral vectors expressing the corresponding cDNAs at multiplicity of infection (MOI) of 0.1 (Morizono and Chen, 2014). The lentiviral vectors expressing TIM-4, CD300A, and DC-SIGN co-expressed a puromycin-resistant gene, and the lentiviral vectors expressing TIM-1 co-expressed a Blasticidine-resistance gene. The transduced cells were cultured for 1–2 weeks in IMDM containing 10% FCS, 1% penicillin/streptomycin, and Blasticidin (Invivogen, San Diego, CA) or Puromycin (Invivogen) according to the type of drug-resistant genes present and the manufacturer’s protocol. Since the expression of TIM-1 decreases during long-term passages, even after sorting and culturing under Blasticidine, we cloned cells that stably expressed TIM-1 at high levels by limiting dilution. FRPE cells were kindly provided by Dr. Guoping Fan at UCLA, with required informed consent described in a previous publication (Liao et al., 2010; Liu et al., 2014). FRPEs were originally isolated from fetal retina (~20-week-old) in Guoping Fan’s laboratory, using a previously published protocol. U87 (ATCC) and Vero cells (ATCC) were grown in DMEM high-glucose medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Infection of cells with virus, replicon, and vector
Target cells were seeded into 24-well plates one day before transduction for all infection experiments. The volumes of reagents and viruses used for all transduction experiments of this study were 250 μl/well. Replication-competent ZIKV and ZIKV replicons were titrated on target cells, and the virus amount (MOI) used for infection was adjusted for each cell type to infect less than 30% of total cells to precisely investigate the effects of various inhibitors on virus infection in the linear range of MOI and percentage of infection. IMDM supplemented with 0.3% BSA was used for dilution of virus stocks. The virus, replicons, or lentiviral vector was incubated in the presence or absence of various concentrations of ZKA64, PtdSer-targeting reagents, or a control protein for 30 min at room temperature, followed by infection of the cells at 37°C for 2 hours. After infection, infected/transduced cells were cultured in fresh medium. The percentages of cells infected with the ZIKV replicon or EBOV GP-pseudotyped lentiviral vector were analyzed by measuring EGFP expression, using flow cytometry 24 or 72 hours post-infection, respectively. The cells infected with replication-competent ZIKV or DENV were harvested 24 hours post-infection. The cells were then fixed by 4% paraformaldehyde, followed by staining with Alexa 647-conjugated Flavivirus group antigen Antibody (Novus Biologicals, Littleton, CO) in cell permeabilization buffer (Thermo Fisher Scientific). The percentages of cells were analyzed by measuring Alexa 647-positive cells using flow cytometry.
Antibody-dependent enhancement of ZIKV infection
ZIKV replicon was incubated with various concentrations of ZKA64, sTIM1dMLDFc, or sTIM1dMLDR801 for 30 min at room temperature prior to infection. K562 cells were infected with the ZIKV replicon in the presence or absence of the reagents for 2 hours at 37°C. The cells were cultured in 24-well plates with IMDM containing 10% FCS and 1% penicillin/streptomycin. Twenty-four hours post-infection, infections were analyzed by measuring EGFP expression, using flow cytometry.
Virus binding assay
EGFP-labeled EBOV GP pseudotype (200 ng p24/ml) was incubated with or without ZKA64 or sTIM1dMLDR801 (30 μg/ml) for 30 min at RT. 293T cells and TIM-1 293T cells (4×105) were then incubated for 2 hours with 200 μl of the preincubated EGFP-labeled EBOV GP pseudotype, followed by fixation with the 4% paraformaldehyde. Viral binding was analyzed by flow cytometry.
Analysis of multiple cycles of ZIKV replication
Replication-competent ZIKV produced by A549 cells was used for infection. The virus heat-inactivated for 2 hours at 65°C was used as a negative control. Vero cells were seeded into 24-well plates one day prior to infection. Replication-competent ZIKV were incubated with 700 ng/ml of ZKA64 or sTIM1dMLDR801 for 30 min at 37°C. Vero cells were infected with the virus pre-incubated with or without ZKA64, sTIM1dMLD R801, or heat-inactivated virus for 2 hours at 37°C. The cells were then washed 3 times with culture medium and cultured in fresh culture medium containing the same reagents (700 ng/ml) used for incubation with the virus. The supernatants of the infected cells were harvested daily and replaced with fresh medium. The harvested supernatant was filtered, and viral RNA was isolated by the QIAamp viral RNA kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. The viral RNA was reverse-transcribed to DNA using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) according to the manufacturer’s protocol. The viral DNA was quantitated by ddPCR, using QuantStudio 3D Digital PCR 20K Chip Kit v2 and Master Mix (Thermo Fisher Scientific) according to the manufacturers’ protocols. The primers and probe for reverse transcription and ddPCR were previously published (Enlow et al., 2020). ZIKV RNA levels are expressed as ZIKV genome copies per microliter of serum.
Animal experiments
Ifnar1−/− (IFN-αβR-KO) mice (B6.129S2-Ifnar1tm1Agt/Mmjax) were congenic C57BL/6 genetic background and purchased from the Jackson Laboratory (derived by Mutant Mouse Resource and Research Centers (MMRRC) supported by NIH) for infection studies (Garcia et al., 2020). One day prior to infection of 4–6-week old Ifnar1−/− male or female mice, they received intraperitoneal injection of sTIM1dMLDR801 (60 μg/g) (n=6 mice) or the same volume of HBS. The mice were then inoculated with PRVABC59 ZIKV (1 ×105 pfu per mouse in 40 μl volume) by the subcutaneous route in the hind limb region while under isoflurane anesthesia. One day post-infection the mice received intraperitoneal injection of the same reagents (sTIM1dMLDR801) at the same dose. Tissues and sera were collected at 7 days post-infection. The study was approved by the Institutional Animal Care and Use Committee (IACUC) at UCLA.
Viral RNA was isolated from sera and quantitated as described previously. To isolate RNA from the spleen and brain, the tissues were placed in RNAlater (Thermo Fisher Scientific) immediately after harvest and stored at −80°C. Tissues were homogenized and RNA was extracted using the gentleMACS Octo Dissociator (Miltenyi Biotec ) and PureLink RNA Mini kit (Thermo Fisher Scientific). Tissue RNA was quantified using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). cDNAs of ZIKV and GAPDH were prepared from 1 μg of RNA, using ZIKV or mouse GAPDH-specific primers (Warren et al., 2006) and the SuperScript IV Reverse Transcriptase Kit. The viral and GAPDH cDNA was quantitated by ddPCR as previously described. The probes for ZIKV and GAPDH were labeled by FAM and VIC, respectively, which enables quantitation of both genes from the same cDNA sample. The sequences of the primers and probe for reverse transcription and ddPCR were previously published. ZIKV RNA levels were expressed as ZIKV genome copies per microliter of serum or per one copy of GAPDH in the spleen or brain.
Statistical analysis
For in vitro experiments, p-values were calculated by a two-sample two-sided unpaired student t-test. For animal experiments, p-values were calculated using a two sided Wilcoxon Rank Sum exact Test. Tests were done using R studio Version 1.2.5033.
Supplementary Material
S1 Fig. The effects of NCdMLDR801 on TIM-1-mediated ZIKV infection. ZIKV replicon was incubated with 30 μg/ml of NCdMLDR801 or control medium for 1 hour at room temperature. 293T or TIM-1 293T cells were infected with the incubated virus for 2 hours, and the infections were analyzed by flow cytometry 24 hours post-infection. The results shown are averages and standard deviations of the triplicate experiment.
S2 Fig. Change of body weight after ZIKV infection.
Acknowledgements
We thank Drs. Melody Li, Samuel French, Danyang Gong, Ren Sun, and Gene-Errol Ringpis for discussion and Ms. Wendy Aft for proof-reading of the manuscript. We thank Dr. Benjamin Hurley, Theodore Pierson, and David Gordon for providing ZIKV H/PF CprME and WNVII-Rep-G/Z and Drs. Jeremy Luban and Paris Sabeti for providing pGL4.23 WT 2014 EBOV Delta. Contemporary Dengue Virus Panel, NR-51131, was obtained through BEI Resources, NIAID, NIH. We thank support from the UCLA AIDS Institute, the James B. Pendleton Trust and McCarthy Foundation. This work was supported by the UCLA AIDS Institute and the UCLA Center for AIDS Research, the James B. Pendleton Trust and McCarthy Foundation, and U.S. National Institute of Health grants R21AI095004 (K.M.), R01AI108400 (K. M.), R01AI145044 (K.M.), U19AI149504 (K.M.), and R01EY032149 (V.A.) and the California Institute for Regenerative Medicine Discovery Award DISC2-10188 (V.A.).
Footnotes
We have no financial conflicts of interest to declare.
References
- Amara A, Mercer J, 2015. Viral apoptotic mimicry. Nat Rev Microbiol 13, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andree HA, Reutelingsperger CP, Hauptmann R, Hemker HC, Hermens WT, Willems GM, 1990. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. The Journal of biological chemistry 265, 4923–4928. [PubMed] [Google Scholar]
- Bhattacharyya S, Zagorska A, Lew ED, Shrestha B, Rothlin CV, Naughton J, Diamond MS, Lemke G, Young JA, 2013. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell host & microbe 14, 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley MA, Hunt CL, Kondratowicz AS, Bowman J, Sinn PL, McCray PB Jr., Quinn K, Weller ML, Chiorini JA, Maury W, 2011. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 415, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillette RB, Phillips EK, Patel R, Mahauad-Fernandez W, Moller-Tank S, Rogers KJ, Dillard JA, Cooney AL, Martinez-Sobrido L, Okeoma C, Maury W, 2018. TIM-1 Mediates Dystroglycan-Independent Entry of Lassa Virus. Journal of virology 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunton B, Rogers K, Phillips EK, Brouillette RB, Bouls R, Butler NS, Maury W, 2019. TIM-1 serves as a receptor for Ebola virus in vivo, enhancing viremia and pathogenesis. PLoS neglected tropical diseases 13, e0006983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK, 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci U S A 90, 8033–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ, 2003. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J Immunol 170, 4840–4845. [DOI] [PubMed] [Google Scholar]
- Capon DJ, Chamow SM, Mordenti J, Marsters SA, Gregory T, Mitsuya H, Byrn RA, Lucas C, Wurm FM, Groopman JE, et al. , 1989. Designing CD4 immunoadhesins for AIDS therapy. Nature 337, 525–531. [DOI] [PubMed] [Google Scholar]
- Carnec X, Meertens L, Dejarnac O, Perera-Lecoin M, Hafirassou ML, Kitaura J, Ramdasi R, Schwartz O, Amara A, 2016. The Phosphatidylserine and Phosphatidylethanolamine Receptor CD300a Binds Dengue Virus and Enhances Infection. Journal of virology 90, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua BA, Ngo JA, Situ K, Morizono K, 2019. Roles of phosphatidylserine exposed on the viral envelope and cell membrane in HIV-1 replication. Cell Commun Signal 17, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua BA, Ngo JA, Situ K, Ramirez CM, Nakano H, Morizono K, 2018. Protein S and Gas6 induce efferocytosis of HIV-1-infected cells. Virology 515, 176–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Hirai-Yuki A, Gonzalez-Lopez O, Rhein B, Moller-Tank S, Brouillette R, Hensley L, Misumi I, Lovell W, Cullen JM, Whitmire JK, Maury W, Lemon SM, 2017. TIM1 (HAVCR1) Is Not Essential for Cellular Entry of Either Quasi-enveloped or Naked Hepatitis A Virions. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejarnac O, Hafirassou ML, Chazal M, Versapuech M, Gaillard J, Perera-Lecoin M, Umana-Diaz C, Bonnet-Madin L, Carnec X, Tinevez JY, Delaugerre C, Schwartz O, Roingeard P, Jouvenet N, Berlioz-Torrent C, Meertens L, Amara A, 2018. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell reports 23, 1779–1793. [DOI] [PubMed] [Google Scholar]
- Diehl WE, Lin AE, Grubaugh ND, Carvalho LM, Kim K, Kyawe PP, McCauley SM, Donnard E, Kucukural A, McDonel P, Schaffner SF, Garber M, Rambaut A, Andersen KG, Sabeti PC, Luban J, 2016. Ebola Virus Glycoprotein with Increased Infectivity Dominated the 2013–2016 Epidemic. Cell 167, 1088–1098 e1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd KA, DeMaso CR, Pelc RS, Speer SD, Smith ARY, Goo L, Platt DJ, Mascola JR, Graham BS, Mulligan MJ, Diamond MS, Ledgerwood JE, Pierson TC, 2016. Broadly Neutralizing Activity of Zika Virus-Immune Sera Identifies a Single Viral Serotype. Cell reports 16, 1485–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duivelshof BL, Murisier A, Camperi J, Fekete S, Beck A, Guillarme D, D’Atri V, 2021. Therapeutic Fc-fusion proteins: Current analytical strategies. J Sep Sci 44, 35–62. [DOI] [PubMed] [Google Scholar]
- Eigenmann MJ, Karlsen TV, Krippendorff BF, Tenstad O, Fronton L, Otteneder MB, Wiig H, 2017. Interstitial IgG antibody pharmacokinetics assessed by combined in vivo- and physiologically-based pharmacokinetic modelling approaches. The Journal of physiology 595, 7311–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto K, Kobayashi T, Yamaji A, Aizawa H, Yahara I, Inoue K, Umeda M, 1996. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proceedings of the National Academy of Sciences of the United States of America 93, 12867–12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enlow W, Piret J, Boivin G, 2020. Droplet Digital PCR and Immunohistochemistry Techniques to Detect Zika Virus in the Central Nervous System of Mice. Methods in molecular biology 2142, 41–57. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM, 1992. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148, 2207–2216. [PubMed] [Google Scholar]
- Fedeli C, Torriani G, Galan-Navarro C, Moraz ML, Moreno H, Gerold G, Kunz S, 2018. Axl Can Serve as Entry Factor for Lassa Virus Depending on the Functional Glycosylation of Dystroglycan. Journal of virology 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G Jr., Paul S, Beshara S, Ramanujan VK, Ramaiah A, Nielsen-Saines K, Li MMH, French SW, Morizono K, Kumar A, Arumugaswami V, 2020. Hippo Signaling Pathway Has a Critical Role in Zika Virus Replication and in the Pathogenesis of Neuroinflammation. The American journal of pathology 190, 844–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong D, Zhang TH, Zhao D, Du Y, Chapa TJ, Shi Y, Wang L, Contreras D, Zeng G, Shi PY, Wu TT, Arumugaswami V, Sun R, 2018. High-Throughput Fitness Profiling of Zika Virus E Protein Reveals Different Roles for Glycosylation during nfection of Mammalian and Mosquito Cells. iScience 1, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Despres P, Amara A, Yssel H, Misse D, 2015. Biology of Zika Virus Infection in Human Skin Cells. Journal of virology 89, 8880–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S, 2002. Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187. [DOI] [PubMed] [Google Scholar]
- Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, Gaitsch HF, Parnell LA, Cao B, Mysorekar IU, Rothlin CV, Fikrig E, Diamond MS, Iwasaki A, 2017. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep 19, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermida-Matsumoto L, Resh MD, 2000. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imaging. J Virol 74, 8670–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto Y, Ohashi K, Mizuno K, Nakano T, 2000. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem 127, 411–417. [DOI] [PubMed] [Google Scholar]
- Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE, 1996. Increased clearance of IgG in mice that lack beta 2-microglobulin: possible protective role of FcRn. Immunology 89, 573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Hayakawa T, Murate M, Makino A, Ito K, Fujisawa T, Kobayashi T, 2007. Curvature-dependent recognition of ethanolamine phospholipids by duramycin and cinnamycin. Biophys J 93, 1608–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, Bu X, Farzan M, Freeman GJ, Umetsu DT, Dekruyff RH, Choe H, 2013. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog 9, e1003232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobinger GP, Weiner DJ, Yu QC, Wilson JM, 2001. Filovirus-pseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nature biotechnology 19, 225–230. [DOI] [PubMed] [Google Scholar]
- Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, Meyerholz DK, Rennert P, Mullins RF, Brindley M, Sandersfeld LM, Quinn K, Weller M, McCray PB Jr., Chiorini J, Maury W, 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proceedings of the National Academy of Sciences of the United States of America 108, 8426–8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Fujikura D, Nanbo A, Marzi A, Noyori O, Kajihara M, Maruyama J, Matsuno K, Miyamoto H, Yoshida R, Feldmann H, Takada A, 2015. Interaction between TIM-1 and NPC1 Is Important for Cellular Entry of Ebola Virus. Journal of virology 89, 6481–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laliberte JP, Moss B, 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc Natl Acad Sci U S A 106, 17517–17521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti RS, Lambert AJ, Holodniy M, Saavedra S, Signor Ldel C, 2016. Phylogeny of Zika Virus in Western Hemisphere, 2015. Emerging infectious diseases 22, 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Romain G, Yan W, Watanabe M, Charab W, Todorova B, Lee J, Triplett K, Donkor M, Lungu OI, Lux A, Marshall N, Lindorfer MA, Goff OR, Balbino B, Kang TH, Tanno H, Delidakis G, Alford C, Taylor RP, Nimmerjahn F, Varadarajan N, Bruhns P, Zhang YJ, Georgiou G, 2017. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat Immunol 18, 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy EM, Epelboin A, Mondonge V, Pourrut X, Gonzalez JP, Muyembe-Tamfum JJ, Formenty P, 2009. Human Ebola outbreak resulting from direct exposure to fruit bats in Luebo, Democratic Republic of Congo, 2007. Vector Borne Zoonotic Dis 9, 723–728. [DOI] [PubMed] [Google Scholar]
- Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Delicat A, Paweska JT, Gonzalez JP, Swanepoel R, 2005. Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576. [DOI] [PubMed] [Google Scholar]
- Leventis PA, Grinstein S, 2010. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39, 407–427. [DOI] [PubMed] [Google Scholar]
- Li M, Ablan SD, Miao C, Zheng YM, Fuller MS, Rennert PD, Maury W, Johnson MC, Freed EO, Liu SL, 2014. TIM-family proteins inhibit HIV-1 release. Proceedings of the National Academy of Sciences of the United States of America 111, E3699–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Waheed AA, Yu J, Zeng C, Chen HY, Zheng YM, Feizpour A, Reinhard BM, Gummuluru S, Lin S, Freed EO, Liu SL, 2019. TIM-mediated inhibition of HIV-1 release is antagonized by Nef but potentiated by SERINC proteins. Proceedings of the National Academy of Sciences of the United States of America 116, 5705–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JL, Yu J, Huang K, Hu J, Diemer T, Ma Z, Dvash T, Yang XJ, Travis GH, Williams DS, Bok D, Fan G, 2010. Molecular signature of primary retinal pigment epithelium and stem-cell-derived RPE cells. Human molecular genetics 19, 4229–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Jiang R, Yuan S, Wang N, Feng Y, Hu G, Zhu X, Huang K, Ma J, Xu G, Liu Q, Xue Z, Fan G, 2014. Integrated analysis of DNA methylation and RNA transcriptome during in vitro differentiation of human pluripotent stem cells into retinal pigment epithelial cells. PLoS One 9, e91416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D, 2002. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872. [DOI] [PubMed] [Google Scholar]
- Luster TA, He J, Huang X, Maiti SN, Schroit AJ, de Groot PG, Thorpe PE, 2006. Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti-tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. The Journal of biological chemistry 281, 29863–29871. [DOI] [PubMed] [Google Scholar]
- Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, Lemke G, Schwartz O, Amara A, 2012. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell host & microbe 12, 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, Le Charpentier T, Hafirassou ML, Zamborlini A, Cao-Lormeau VM, Coulpier M, Misse D, Jouvenet N, Tabibiazar R, Gressens P, Schwartz O, Amara A, 2017. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep 18, 324–333. [DOI] [PubMed] [Google Scholar]
- Mercer J, Helenius A, 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320, 531–535. [DOI] [PubMed] [Google Scholar]
- Mercer J, Knebel S, Schmidt FI, Crouse J, Burkard C, Helenius A, 2010. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc Natl Acad Sci U S A 107, 9346–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S, 2007. Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439. [DOI] [PubMed] [Google Scholar]
- Moller-Tank S, Albritton LM, Rennert PD, Maury W, 2014. Characterizing functional domains for TIM-mediated enveloped virus entry. Journal of virology 88, 6702–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller-Tank S, Kondratowicz AS, Davey RA, Rennert PD, Maury W, 2013. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. Journal of virology 87, 8327–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizono K, Chen IS, 2014. Role of phosphatidylserine receptors in enveloped virus infection. Journal of virology 88, 4275–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizono K, Ku A, Xie Y, Harui A, Kung SK, Roth MD, Lee B, Chen IS, 2010. Redirecting lentiviral vectors pseudotyped with Sindbis virus-derived envelope proteins to DC-SIGN by modification of N-linked glycans of envelope proteins. J Virol 84, 6923–6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, Chen IS, 2011. The Soluble Serum Protein Gas6 Bridges Virion Envelope Phosphatidylserine to the TAM Receptor Tyrosine Kinase Axl to Mediate Viral Entry. Cell Host Microbe 9, 286–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S, Hanayama R, Kawane K, 2010. Autoimmunity and the clearance of dead cells. Cell 140, 619–630. [DOI] [PubMed] [Google Scholar]
- Niu J, Jiang Y, Xu H, Zhao C, Zhou G, Chen P, Cao R, 2018. TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otzen DE, Blans K, Wang H, Gilbert GE, Rasmussen JT, 2012. Lactadherin binds to phosphatidylserine-containing vesicles in a two-step mechanism sensitive to vesicle size and composition. Biochim Biophys Acta 1818, 1019–1027. [DOI] [PubMed] [Google Scholar]
- Pan J, Cheng X, Monticelli L, Heberle FA, Kucerka N, Tieleman DP, Katsaras J, 2014. The molecular structure of a phosphatidylserine bilayer determined by scattering and molecular dynamics simulations. Soft Matter 10, 3716–3725. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, 2002. Drug and gene delivery to the brain: the vascular route. Neuron 36, 555–558. [DOI] [PubMed] [Google Scholar]
- Pierson TC, Sanchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW, 2006. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346, 53–65. [DOI] [PubMed] [Google Scholar]
- Pietkiewicz S, Schmidt JH, Lavrik IN, 2015. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical Annexin V/propidium iodide staining. J Immunol Methods 423, 99–103. [DOI] [PubMed] [Google Scholar]
- Puryear WB, Akiyama H, Geer SD, Ramirez NP, Yu X, Reinhard BM, Gummuluru S, 2013. Interferon-inducible mechanism of dendritic cell-mediated HIV-1 dissemination is dependent on Siglec-1/CD169. PLoS Pathog 9, e1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puryear WB, Yu X, Ramirez NP, Reinhard BM, Gummuluru S, 2012. HIV-1 incorporation of host-cell-derived glycosphingolipid GM3 allows for capture by mature dendritic cells. Proc Natl Acad Sci U S A 109, 7475–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran S, Huang X, Downes A, Thorpe PE, 2003. Evaluation of novel antimouse VEGFR2 antibodies as potential antiangiogenic or vascular targeting agents for tumor therapy. Neoplasia 5, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein BA, Brouillette RB, Schaack GA, Chiorini JA, Maury W, 2016. Characterization of Human and Murine T-Cell Immunoglobulin Mucin Domain 4 (TIM-4) IgV Domain Residues Critical for Ebola Virus Entry. Journal of virology 90, 6097–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, Choe H, 2017. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proceedings of the National Academy of Sciences of the United States of America 114, 2024–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard AS, Zhang A, Park SJ, Farzan M, Zong M, Choe H, 2015. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proceedings of the National Academy of Sciences of the United States of America 112, 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FI, Bleck CK, Helenius A, Mercer J, 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. Embo J 30, 3647–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Heegaard CW, Rasmussen JT, Gilbert GE, 2004. Lactadherin binds selectively to membranes containing phosphatidyl-L-serine and increased curvature. Biochim Biophys Acta 1667, 82–90. [DOI] [PubMed] [Google Scholar]
- Sinn PL, Coffin JE, Ayithan N, Holt KH, Maury W, 2017. Lentiviral Vectors Pseudotyped with Filoviral Glycoproteins. Methods in molecular biology 1628, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Situ K, Chua BA, Bae SY, Meyer AS, Morizono K, 2018. Versatile targeting system for lentiviral vectors involving biotinylated targeting molecules. Virology 525, 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MM, King SW, Thorpe PE, 2008. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat Med 14, 1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, Vanzetta F, Minola A, Jaconi S, Mele F, Foglierini M, Pedotti M, Simonelli L, Dowall S, Atkinson B, Percivalle E, Simmons CP, Varani L, Blum J, Baldanti F, Cameroni E, Hewson R, Harris E, Lanzavecchia A, Sallusto F, Corti D, 2016. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826. [DOI] [PubMed] [Google Scholar]
- Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al. , 1995. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80, 661–670. [DOI] [PubMed] [Google Scholar]
- Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD, Heffern CT, Henderson JM, Meron M, Lin B, Roux B, Schlossman ML, Steck TL, Lee KY, Adams EJ, 2014. Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proceedings of the National Academy of Sciences of the United States of America 111, E1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW, 2008. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Qiao L, Hou Z, Luo G, 2017. TIM-1 Promotes Hepatitis C Virus Cell Attachment and Infection. Journal of virology 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T, 2013. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol 25, 363–372. [DOI] [PubMed] [Google Scholar]
- Warren L, Bryder D, Weissman IL, Quake SR, 2006. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proceedings of the National Academy of Sciences of the United States of America 103, 17807–17812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenbock H, Hubalek Z, Bakonyi T, Nowotny N, 2010. Zoonotic mosquito-borne flaviviruses: worldwide presence of agents with proven pathogenicity and potential candidates of future emerging diseases. Veterinary microbiology 140, 271–280. [DOI] [PubMed] [Google Scholar]
- Zhang L, Richard AS, Jackson CB, Ojha A, Choe H, 2020. Phosphatidylethanolamine and Phosphatidylserine Synergize To Enhance GAS6/AXL-Mediated Virus Infection and Efferocytosis. Journal of virology 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Fig. The effects of NCdMLDR801 on TIM-1-mediated ZIKV infection. ZIKV replicon was incubated with 30 μg/ml of NCdMLDR801 or control medium for 1 hour at room temperature. 293T or TIM-1 293T cells were infected with the incubated virus for 2 hours, and the infections were analyzed by flow cytometry 24 hours post-infection. The results shown are averages and standard deviations of the triplicate experiment.
S2 Fig. Change of body weight after ZIKV infection.