

Abstract
Aims
The family of kynurenine pathway (KP) metabolites includes compounds produced along two arms of the path and acting in clearly opposite ways. The equilibrium between neurotoxic kynurenines, such as 3‐hydroxykynurenine (3‐HK) or quinolinic acid (QUIN), and neuroprotective kynurenic acid (KYNA) profoundly impacts the function and survival of neurons. This comprehensive review summarizes accumulated evidence on the role of KYNA in Alzheimer's, Parkinson's and Huntington's diseases, and discusses future directions of potential pharmacological manipulations aimed to modulate brain KYNA.
Discussion
The synthesis of specific KP metabolites is tightly regulated and may considerably vary under physiological and pathological conditions. Experimental data consistently imply that shift of the KP to neurotoxic branch producing 3‐HK and QUIN formation, with a relative or absolute deficiency of KYNA, is an important factor contributing to neurodegeneration. Targeting specific brain regions to maintain adequate KYNA levels seems vital; however, it requires the development of precise pharmacological tools, allowing to avoid the potential cognitive adverse effects.
Conclusions
Boosting KYNA levels, through interference with the KP enzymes or through application of prodrugs/analogs with high bioavailability and potency, is a promising clinical approach. The use of KYNA, alone or in combination with other compounds precisely influencing specific populations of neurons, is awaiting to become a significant therapy for neurodegenerative disorders.
Keywords: Alzheimer's disease, Huntington's disease, mitochondrial toxin, neurodegeneration, N‐methyl‐D‐aspartate, Parkinson's disease
Kynurenic acid (KYNA), metabolite of tryptophan along the kynurenine pathway, exhibits neuroprotective activity under in vitro and in vivo conditions. As novel KYNA targets were identified, distinctive biological role of the compound was recognized. Absolute or relative deficiency of KYNA in proportion to neurotoxic kynurenines in Huntington's, Parkinson's, and Alzheimer's diseases implicates potential therapeutic value of boosting brain KYNA levels.
![]()
1. INTRODUCTION
Tryptophan, an essential neutral amino acid, is a pivotal constituent of proteins and a source of numerous, biologically significant compounds. Only a small quantity of compound (1–2%) undergoes incorporation into peptides or proteins, whereas the remaining 98–99% follows two major metabolic routes. These include (a) the methoxyindole pathway yielding 5‐hydroxytryptamine (5‐HT; serotonin), and (b) the kynurenine pathway (KP) generating metabolites collectively called kynurenines (Figure 1). The KP converts 95% of tryptophan and ultimately leads to the formation of nicotinamide adenine dinucleotide (NAD+), with a number of neuroactive kynurenines en route. 1 , 2 The discoveries of last decades strongly support the concept of viewing the disturbed KP as an important link in the cycle of events leading to the development of brain pathologies. Various kynurenines are of substantial biological importance due to their ability to modify neurotransmission and to alter the immune response. 3 , 4
FIGURE 1.
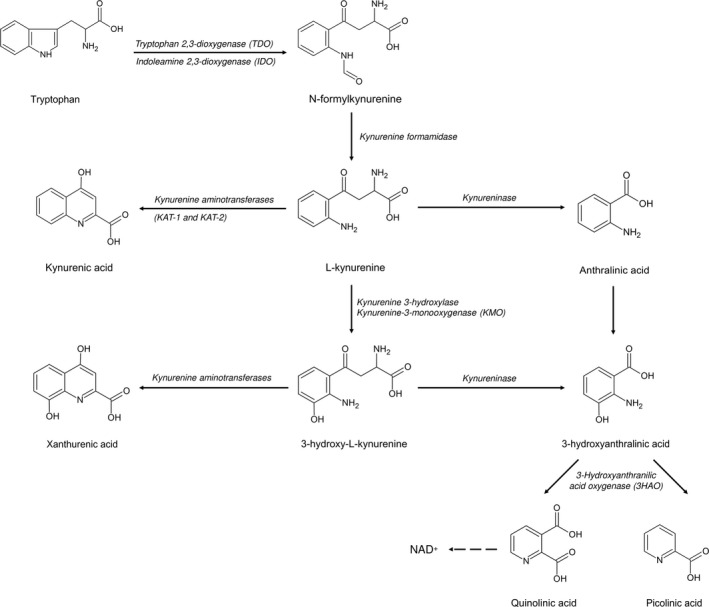
Scheme of kynurenine pathway
The family of KP metabolites comprises compounds acting in a divergent way and considered to be either neuroprotective or neurotoxic. 5 The synthesis of specific compounds is tightly regulated and may considerably vary under physiological and pathological conditions. 5 Kynurenic acid (KYNA), the main neuroprotective compound of the path, was discovered in the 19th century as a constituent of canine urine and initially regarded merely as a by‐product of tryptophan degradation. 6 The molecular structure of KYNA was unraveled at the beginning of the 20th century, 7 yet the particular steps of the KP leading to KYNA formation were determined much later. Discoveries of the 1980s revealed the ability of KYNA to block the excitatory amino acid receptors under in vitro and in vivo conditions. 8 , 9 Soon, abnormalities in cerebral KYNA synthesis have been implicated in the pathogenesis of neurodegeneration. 10 Intensified research during last four decades revolutionized our knowledge about KYNA and brought valuable data supporting the significant role of KYNA as an exceptional tryptophan metabolite in the mammalian brain. 11 This review aimed to discuss the involvement of altered KYNA metabolism in the development of neurodegenerative diseases, as well as the future of pharmacological manipulations aimed to boost brain KYNA as potential therapeutic agents.
The KP is functional in the brain and in the periphery. 12 The first step of tryptophan metabolism is catalyzed by the step‐limiting enzymes, indoleamine 2,3‐dioxygenases (IDO‐1 and 2) and tryptophan 2,3‐dioxygenase (TDO), yielding N‐formyl‐kynurenine (Figure 1). N‐formyl‐kynurenine is further converted to a direct precursor of KYNA, L‐kynurenine, by formamidase. In the periphery, the constitutive expression of IDO‐1 is restricted and was described mostly within endothelial, pancreatic, placental, or antigen‐presenting cells. Interestingly, IDO‐1 manifests pronounced susceptibility to the induction by proinflammatory molecules, such as interferon‐γ, tumor necrosis factor α (TNF‐α), interleukin‐6 (IL‐6), or IL‐10, in a variety of cells. 13 , 14
IDO‐2 and TDO show higher tissue specificity, mainly restricted to liver, and much lower expression level. 15 The enzymatic activity of TDO can be induced by estrogens, glucocorticoids, and tryptophan itself. 2 In the brain, striatal neurons and astrocytes express high levels of IDO‐1 mRNA. 16 TDO protein and its mRNA are also detectable in neurons and astrocytes. 17 , 18 , 19
The major central pool of KYNA is formed locally, from its precursor, L‐kynurenine. 20 , 21 L‐kynurenine, on the contrary, originates mostly from peripheral sources (60–70%), whereas the remaining 30–40% is produced in situ. 21 L‐kynurenine can be also converted along another arm of the KP to neurotoxic 3‐hydroxykynurenine (3‐HK), QUIN, and further down to NAD. 22 The fate of L‐kynurenine degradation and its availability for the synthesis of KYNA is determined by a number of factors, including tissue and cell type. Central KYNA production occurs mostly in astrocytes and endothelial cells and to a much lesser extent within neurons. 23 , 24 , 25 , 26 In contrast, neurotoxic QUIN is generated in the human brain mainly by the microglial cells and macrophages. 27
The principal route of KYNA synthesis is based on an irreversible transamination of L‐kynurenine catalyzed by kynurenine transaminases (KATs). 28 KYNA is produced by various tissues and organs, including liver, kidneys, intestines, or endothelium. 29 , 30 In the brain, KATs are expressed mainly in astrocytes and to a lesser degree in neuronal cells, for example, in hippocampus, substantia nigra, or striatum. 24 , 25 , 31 , 32 , 33 KATs are characterized by a different level of specific activity in various brain regions. 34 , 35
In humans and rodents, four isoforms of KATs, using L‐kynurenine as a donor for amino group, were characterized and include KAT I (glutamine transaminase K/cysteine conjugate beta‐lyase 1), KAT II (α‐aminoadipate aminotransferase), KAT III (glutamine transaminase L/cysteine conjugate beta‐lyase 2), and KAT IV (the mitochondrial aspartate aminotransferase/glutamic‐oxaloacetic transaminase 2). 36 Each KAT enzyme has an optimal pH range and a distinct substrate profile, despite sharing a number of amino acid and α‐keto acid substrates. 37 , 38 , 39 KATs manifest relatively low affinity for L‐kynurenine (Km approx. 1 mM). Under physiological conditions, KAT II is considered a major biosynthetic enzyme responsible for KYNA formation. 40
A targeted deletion of KAT II in mice leads to an early and transitory decrease in brain KAT activity and KYNA levels with commensurate behavioral and neuropathological changes. 41 In KAT II–deficient mice, striatal KYNA level was transiently reduced around the 2nd week of age and the degree of neuronal loss following the local administration of QUIN was strongly enhanced. 42 Later on, however, KYNA levels were normalized, possibly as a result of compensatory changes. 41 , 42
Indirectly, the activity of kynurenine monooxygenase (KMO), synthesizing 3‐HK and displaying a much lower Km value for L‐kynurenine, also impacts the synthesis of KYNA. Inhibition of KMO activity increases the pool of L‐kynurenine available for KATs. This, in turn, may easily shift the KP and direct it to the neuroprotective branch; conversely, an enhanced activity of KMO stimulates metabolism of tryptophan along the neurotoxic arm of the pathway. 43
As shown under in vitro and in vivo conditions, the composition of the extracellular milieu, the availability of oxygen and glucose, or level of ammonia and amino acids may influence the synthesis of KYNA. 44 , 45 , 46 , 47 Notably, neurotoxic compounds such as mitochondrial toxins or pyrethroid pesticides inhibit, whereas a number of therapeutic agents, including beta‐adrenoceptor agonists, nitric oxide donors, memantine, antidepressants, or some antiepileptics, stimulate KYNA production in the brain 48 , 49 , 50 , 51 , 52 , 53 .
1.1. Other sources of brain KYNA
Apart from the canonical KAT‐related synthesis, alternative mechanisms were implicated in the synthesis of KYNA. 54 , 55 Indole‐3‐pyruvic acid, the keto‐analog of tryptophan, increases KYNA content in various organs, including brain. 56 Indole‐3‐pyruvic acid is effectively converted to KYNA in a non‐enzymatic reaction requiring ample presence of oxygen. Reactive oxygen species (ROS) target the enol form of indole‐3‐pyruvic acid, which undergoes pyrrole ring cleavage and subsequently forms KYNA. 57 , 58 L‐L‐kynurenine may also yield KYNA when incubated in the presence of H2O2, with or without peroxidases. 55 It is a pH‐dependent process, with the highest conversion of L‐kynurenine occurring at the pH between 7.4 and 8.0. 55 The contribution of alternative routes to the overall KYNA production still remains unclear. However, in the altered redox environment and when the antioxidant system is defective, as often is the case in neurodegenerative disorders, their significance may increase. Indeed, the lack of correlation between KATs activities and KYNA levels was reported in lead intoxication, Down syndrome, and disturbances of thyroid hormone levels. 59 , 60 , 61 , 62
Furthermore, although peripherally synthesized KYNA poorly passes through the blood‐brain barrier, certain conditions may facilitate its penetration into the brain. Systemic administration of KYNA prior to the cerebral ischemia potently increased its brain concentrations, 63 possibly as a result of passive diffusion. 21 In addition, KYNA was identified as a high‐affinity substrate for organic anion transporters, OAT1 and OAT3. 64 , 65 Experimental use of probenecid, a non‐selective inhibitor of Oat1, was shown to increase the brain level of KYNA. 66 , 67 Interestingly, thyroid hormones may enhance the removal of KYNA and modulate its brain level via diverse mechanisms, including the action of Oat. 59
Finally, apart from the de novo synthesis by the mammalian tissues, KYNA can be generated in the digestive system by microbiota and exogenously delivered with food products. 68 , 69
1.2. Neurotoxic branch of kynurenine pathway
Three major neurotoxic metabolites of the KP include 3‐HK, QUIN, and 3‐hydroxyanthranilic acid (3‐HANA). 3‐HK is an immediate product of L‐kynurenine conversion carried out by KMO. Metabolism of 3‐HK by kynureninase yields 3‐HANA, which, in two enzymatic steps, can be further converted to QUIN. The toxicity of 3‐HK has been attributed mainly to the formation of free radicals and an induction of apoptotic neuronal death. 70 , 71
The results of numerous research clearly indicate that QUIN is capable of acting as an endogenous excitotoxin. QUIN‐evoked neuronal loss is mostly associated with an excessive stimulation of NR2A and NR2B subunits of N‐methyl‐D‐aspartate (NMDA) receptor at agonist‐binding site. In the brain, physiological concentrations of QUIN are in nM range (~50–100 nM) and are approx. 20 times lower than in the periphery. 72 At low concentrations, QUIN induces proliferation of the stem cells and is an intermediate metabolite along the pathway yielding NAD+in human brain cells. 73 , 74 At high, close to millimolar concentrations, QUIN induces selective, axon‐sparing neuronal loss under various experimental conditions. The susceptibility of neurons to the QUIN‐induced damage depends on the brain area, with cortical, striatal, and hippocampal neurons being the most sensitive. 3 It was debated whether endogenous levels of QUIN are sufficient to cause neurotoxicity, yet, in the view of accumulated data, the compound undoubtedly may evoke neuronal death. In human brain, QUIN levels increase during inflammation or cerebral insults up to the micromolar values. 75 Locally, QUIN concentration may be much higher. 3 Notably, even low concentrations of QUIN may induce neuronal loss, providing that the exposure is prolonged. In organotypic corticostriatal, but not in caudate nucleus, cultures, exposed to low (100 nM) concentration of QUIN for up to 7 weeks, a clear focal neurodegeneration was developed. 76
QUIN was also demonstrated to enhance the glutamate release, to inhibit glutamate reuptake, and to stimulate lipid peroxidation. 77 Such local rise evokes depolarization of the postsynaptic membrane sufficient to remove the Mg2+ block of NMDA receptor–linked ion channel. Moreover, QUIN may impair the function of blood‐brain barrier, induce nitric oxide production, and cause hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons. 73 , 78 , 79 , 80 In the view of accumulated data, QUIN produces neurotoxicity through a link of events initiated by the excessive stimulation of the NMDA receptor, Ca2+ influx, energy deficit, and oxidative stress.
The proper balance between neurotoxic kynurenines and neuroprotective KYNA can be viewed, in fact, as the interplay between astrocytes, microglia, and neurons. Under physiological conditions, the astrocytic KP serves as a source of neuroprotective KYNA, whereas neuronal KP produces NAD+ improving cellular energy status. In diseased brain, the inflammatory signals stimulate the KP within macrophages, microglia, and dendritic cells to produce large quantities of QUIN. Astrocytes remove QUIN from synaptic cleft and catabolize it to NAD+; however, the enzyme responsible for catabolism is easily saturated. Thus, when the overall balance of the pathway is shifted toward QUIN and other neurotoxic kynurenines, with an absolute/relative deficiency of KYNA, neurodegenerative changes can follow.
2. BIOLOGICAL KYNA TARGETS IN THE BRAIN
The level of KYNA in the central nervous system (CNS) depends on the species, studied region, and the ontogenetic stage of development. 81 In human brain, KYNA occurs in low micromolar range (approx. 0.1–1.5 μM), which is 20–50 times higher than in rodent CNS (0.001–0.05 μM). 34 , 82 , 83 , 84 , 85 The content of KYNA was reported to be the lowest in cerebellum and medulla (0.1–0.3 pmol/mg), intermediate in cortical areas and substantia nigra (0.2–0.6 pmol/mg), and the highest in putamen and globus pallidus (0.7–1.4 pmol/mg). 34 , 82 In human CSF, KYNA concentration is low (0.001–0.01 μM), yet it steadily increases with age. 81 , 86 , 87 In other species, the brain content of KYNA also rises with age. 88 Over 50‐fold increase in brain KYNA between 1st week and 18th month of age was reported in rats. 89 Others demonstrated a threefold increase between the 3rd and 24th months of age. 90
KYNA is quickly liberated from the cell and is not a subject of enzymatic degradation or reuptake processes. 11 , 91 Extracellular KYNA interacts with a number of biological targets (Figure 2). KYNA was initially recognized as a broad‐spectrum antagonist of ionotropic excitatory amino acid receptors. KYNA displays the highest affinity for the co‐agonist glycine site of NMDA receptor complex (IC50 ~8–15 μM in the absence of glycine; ~50–200 μM in the presence of 10 μM glycine. 92 , 93 KYNA, in a competitive manner, blocks also agonist‐binding sites of NMDA, kainic acid, and alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptors, yet with lower affinity (IC50 of 100–500 μM). 3 , 94 Despite the discrepancy between physiologically occurring levels of KYNA and the levels needed to interfere with glutamatergic receptors, it is well established that KYNA synthesis may increase locally due to various factors and easily reach the concentration sufficient to interact with NMDA receptors. 95
FIGURE 2.
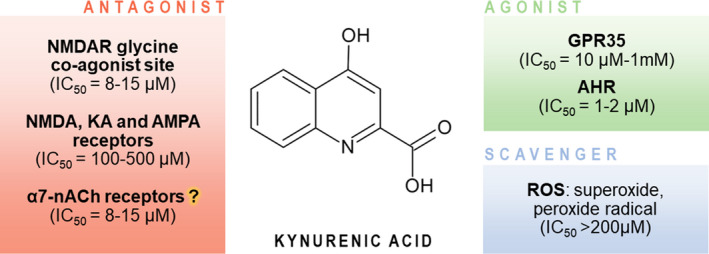
Targets of kynurenic acid. NMDA—N‐methyl‐D‐aspartate; KA—kainic acid; AMPA—alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid; AHR—aryl hydrocarbon receptor; GPR35—G protein–coupled orphan receptor 35; ROS—reactive oxygen species
Neuroprotective and anticonvulsant properties of KYNA are broadly documented under in vivo and in vitro conditions. 94 , 95 , 96 , 97 Notably, KYNA attenuates the morphological and behavioral consequences of experimental administration of its kynureninergic alter ego—excitotoxic QUIN.
As KYNA may impact the extracellular levels of glutamate, acetylcholine, GABA, and dopamine, neuromodulation is an important aspect of its role. 98 , 99 , 100 , 101 In striatal preparations, low nanomolar concentrations of KYNA reduced glutamate release in caudate nucleus and impaired the neurotransmitter release. 102 Experimental studies in vivo confirmed that fluctuations of KYNA level may alter glutamine, acetylcholine, and dopamine release. 98 , 99 , 101
KYNA has also been identified as a ligand of formerly orphan G protein–coupled receptor, GPR35, 103 broadly expressed in various immune cells. Apart from the regulation of immune response, KYNA‐GPR35 interaction may inhibit Ca2+ channels in sympathetic neurons and reduce synaptic activity in hippocampal neurons. 104 , 105 Therefore, KYNA capability to activate GPR35 might represent another way to reduce the excitatory transmission. 105 , 106 KYNA is also targeting xenobiotic receptor, the aryl hydrocarbon receptor (AHR). 107 KYNA‐related AHR stimulation increases the interleukin‐6 expression, which is often associated with promoting carcinogenesis and tumor outgrowth. 107 , 108 Moreover, KYNA displays the scavenging ability toward ROS. In the homogenates of rat brains, KYNA decreased the production of free radicals and lipid peroxidation. 109 It has been postulated that KYNA targets also α7‐nicotinic acetylcholine receptors (α7nAChR); however, this mechanism is still being controversial. 110 , 111 , 112
The inflammation emerged as one of the key factors contributing to the neuronal loss and compromised regeneration and thus was implicated in the pathogenesis of neurodegenerative disorders. An important link between proinflammatory status and the activation of KP is well substantiated. 11 , 86 Importantly, the metabolites of KP may act as pro‐ and antiinflammatory compounds. Genomic interventions aimed to eliminate IDO, TDO, or KMO were shown to alleviate the course of chronic inflammation, reduce viral replication, or change the expression of proinflammatory molecules. 113 On the contrary, a number of kynurenines, including KYNA, emerged as antiinflammatory compounds. 11 , 113 KYNA was demonstrated to attenuate inflammation by several ways including the reduction in TNF expression, diminished interleukin‐4 and α‐defensin secretion, or inhibition of Th17 cell differentiation, at least in part through activation of GPR35. 11 The interplay between immune activation and the KP activity results in a delicate balance, which may easily be shifted either to or away from neuroprotective KYNA (Figure 3).
FIGURE 3.
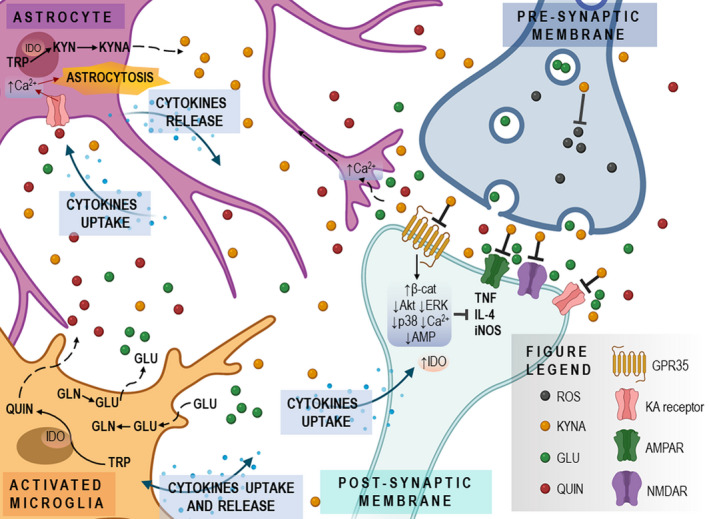
Role of kynurenic acid in neurodegeneration. The interplay between astrocytes, microglia, and neurons in terms of the quantities of produced KYNA and other kynurenines can be altered by various genetically determined and postnatal factors, including inflammation. Deficiency of KYNA may enhance the GLU‐mediated neurotransmission, reduce antioxidant capacity, and shift the kynurenine pathway toward neurotoxic metabolites, with ensuing neuronal loss
However, an excessive blockade of glutamate‐mediated neurotransmission may impair cognition and memory processes. 114 , 115 , 116 , 117 , 118 Thus, manipulations of the endogenous KYNA level may exert dual, conflicting effects—beneficial neuroprotection and unfavorable cognitive dysfunction. Considering the chronic nature of neurodegenerative disorders, neuroprotection seems to be essential, as it may slow the progress of disease. Maintaining adequate levels of brain KYNA seems vital to obtain neuroprotection without cognitive adverse effects. The optimal therapeutic intervention would include a region‐selective increase in KYNA; however, such pharmacological tools are not available yet.
3. KYNA ALTERATIONS IN NEURODEGENERATIVE DISEASES
3.1. Huntington's disease
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease. Clinically, it is characterized by a gradual deterioration of voluntary movements, appearance of chorea, cognitive decline, and complex psychiatric symptoms. 119 Symptoms begin slowly, usually in the fourth decade of life, and lead ultimately to death within 15–20 years. The genetic background of disease is linked with an expansion of CAG trinucleotide repeats within exon 1 on chromosome 4, following a single mutation within the IT15 gene encoding huntingtin. 120 Neurodegeneration affects primarily cerebral cortex and striatum, but as disease progresses, neuronal loss develops in multiple areas of the brain. Apart from the accumulation of huntingtin, the precise mechanisms leading to neurodegeneration and subsequent clinical symptoms are not fully elucidated. Aberrations in function of glial cells, inflammation, mitochondrial dysfunction, or oxidative stress were all implicated in the pathogenesis of HD. 121 , 122 , 123
The potential role of aberrant tryptophan metabolism in the pathogenesis of neurodegenerative disorders has been postulated by Schwarcz and co‐workers who discovered that intrastriatal application of excitotoxic KP metabolite, QUIN, results in neuropathological and behavioral alterations closely mimicking HD. 124 Further support to this concept was provided by numerous research data, including studies on the experimental KYNA deficiency caused by the pharmacological tools, aminooxyacetic acid (AOAA), and 3‐nitropropionic acid (3‐NPA). 96 , 125 , 126 AOAA, a non‐selective aminotransferase inhibitor, potently diminishes synthesis of KYNA in vitro, with very low, micromolar IC50 values. When administered intrastriatally, AOAA produces a pattern of neurodegenerative and behavioral changes modeling HD and astonishingly resembling the outcome of intrastriatal application of QUIN. 96 The axon‐sparing excitotoxic neuronal loss is age‐dependent, is susceptible to blockade with KYNA itself, and could be prevented by the ablation of corticostriatal glutamatergic input. 96
Similarly, 3‐NPA was shown to impair the synthesis of KYNA in rat cortical slices and to inhibit the activity of KAT I and KAT II. 126 In vivo, 3‐NPA decreased the number of KAT I immunopositive glial cells in the striatum (−3.57‐fold) and temporal cortex (‐twofold) of rats. 127 Behavioral consequences of 3‐NPA application in rodents are influenced by the mode of treatment. Acute application of 3‐NPA evokes seizures, whereas chronic administration of low doses of 3‐NPA results in a progressive locomotor deterioration and selective striatal degeneration resembling changes characteristic for HD. 128 , 129 , 130 Susceptibility to the effects of 3‐NPA increases with age; furthermore, 3‐NPA and mutated huntingtin seem to share certain mechanisms of toxicity. 131
Alterations in the metabolism of KP have also been demonstrated in genetic animal models of HD. In FVB/N mice with a mutation in the huntingtin gene, more than 10‐fold increase of 3‐HK level in the striatum and cortex was accompanied by a slight increase in KYNA levels and a considerable, 5.7‐fold, increase in the 3‐HK/KYNA ratio. 132 A study in R6/2 mice, modeling HD, also demonstrated the increased activity of KMO (1.65‐fold change in vmax value between 8‐week‐old wild‐type and R6/2 animals) and decreased activity of kynureninase (−1.5‐ to −1.67‐fold), resulting in an excessive enzymatic conversion of tryptophan to 3‐HK. 133
Data from human studies are in line with the experimental research, despite a clear limitation of postmortem analyses. Brain changes in KYNA content seem to be region‐selective. Neostriatal KYNA level was reported as either decreased or unaltered, whereas cortical KYNA levels seem to increase, especially during the late stage of disease. 34 , 83 , 134 , 135 , 136 Furthermore, an increase in frontocortical QUIN and 3‐HK (both c. 2.5‐fold) levels and a decrease in the KYNA/QUIN ratio (over −2.5‐fold in neostriatum and over ‐twofold in frontal cortex) were detectable at stage 1 of HD. Qualitatively similar changes were observed in mice transgenic for the full‐length mutant huntingtin. 132 , 137 In the advanced stages of HD, a reduction (−1.6‐fold) in KYNA CSF levels was observed. 136 In the periphery, the baseline L‐kynurenine levels were higher in HD and the difference remained obvious despite tryptophan depletion or loading. 138 Serum KYNA level in HD was not altered in comparison with control; however, the KYNA/L‐kynurenine ratio was lower. 138 In a cohort of patients at different stages of HD, the greatest increase in the L‐kynurenine/tryptophan ratio and their overall concentrations was observed among patients possessing more CAG repeats or in those in the later stages of HD. 139 These observations suggest that changes in the activity of KP, possibly leading to the excessive activity of the neurotoxic arm of the pathway, may have an impact on the development of HD.
In a prospective single‐site controlled cohort study with standardized collection of CSF, blood, and phenotypic and imaging data, performed among 80 participants (20 healthy controls, 20 pre‐manifest HD, and 40 manifest HD), the KP metabolites in CSF and plasma were stable over 6 weeks of observation. 140 There were no differences regarding basal KYNA, L‐kynurenine, or tryptophan levels. However, an increase in 3‐HK/KYNA ratio was detected in the group of patients with evident HD compared with HD patients at early stage of disease. 140
Pathologically, high levels of neurotoxic L‐kynurenine metabolites accompanied by a relative lack of neuroprotective KYNA is a consistent finding in HD patients and in animal models of this disease. The deficiency of KYNA and malfunction of the neuroprotective arm of the KP may generate virtually identical consequences as an excessive production of QUIN and other neurotoxic kynurenines. In line with these observations, switching from the neurotoxic branch of the KP, yielding QUIN, to the neuroprotective branch producing KYNA was suggested to bring beneficial effects. Brain changes in KYNA and other KP metabolites can be considered the hallmarks of HD. The encouraging effects of KMO and TDO inhibition in HD models (vide paragraph 4.1.1) are the base for future clinical trials evaluating therapeutic potential of KMO inhibitors.
3.2. Parkinson's disease
Parkinson's disease (PD) is a common, progressive neurodegenerative disease, characterized by the gradual loss of dopaminergic brain neurons. Its most characteristic symptoms include resting tremor, limb rigidity, posture and gait instability, and bradykinesia. Loss of dopaminergic neurons in substantia nigra pars compacta and the appearance of intracytoplasmic proteinaceous inclusions, Lewy bodies, are characteristic morphological alterations. 141 While 10 to 15% of cases represent the familial form of PD, the idiopathic form of disorder prevails. 96 Disruptions in the ubiquitin‐proteasome and autophagolysosomal pathways, mitochondrial dysfunction, excessive oxidative stress, and enhanced apoptosis were all implicated in the pathogenesis of PD. 142 Furthermore, disturbed glutamate‐mediated transmission and KYNA deficiency are also among important factors contributing to the development of PD. Neuroprotective and antiparkinsonian effect of glutamate receptor antagonists was demonstrated already 3 decades ago, using various experimental models. 143 , 144 In line with these observations, an increase in brain KYNA level, either through the direct application or through increased availability of L‐kynurenine, effectively reduced neurodegeneration and behavioral symptoms in animal models of PD.
Canonical model of PD is based on the administration of lipophilic compound, 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP). In 1983, MPTP was discovered as contaminant of street heroin responsible for a rapid development of PD among young addicts. 145 , 146 This highly selective neurotoxin causing nigral degeneration, followed by a classical PD‐like behavioral pattern in various species, including rodents and primates, quickly became a valuable research tool. 146 The mechanisms underlying selective toxicity depend primarily on the glial conversion of MPTP to pyridinium metabolite (MPP+), which, upon release from astrocytes, inhibits neuronal mitochondrial respiratory chain and constitutes a source of free radicals. 147 Interestingly, MPP+ was discovered to inhibit the cortical KAT activity and to reduce KYNA formation in vitro in rat cortical slices. 126 The effect was confirmed in vivo, as MPTP decreased KYNA synthesis and the density of KAT I immunoreactive nigral neurons in mice. 148 , 149 Consistently, KYNA pretreatment was shown to reduce the apoptosis of neurons by downregulating Bax expression and maintaining mitochondrial function, in human neuroblastoma cell line exposed to MPP+. 150
Human studies mostly demonstrate that in the brain of PD victims, the metabolism of tryptophan is shifted toward neurotoxic kynurenines with ensuing deficiency of KYNA. Postmortem studies reported diminished KYNA and L‐kynurenine levels in frontal cortex, putamen, and substantia nigra, without change in tryptophan/L‐kynurenine and L‐kynurenine/KYNA ratios, in the brains of PD victims. 151 , 152 In caudate and precentral cortical gyrus, KYNA content did not differ from control values. 80
In the periphery, the results are not consistent. In erythrocytes obtained from PD patients, higher levels of KYNA and enhanced activity of KAT II, but not of KAT I, were detected. In serum, KYNA level remained unchanged, while KAT I and KAT II activities were lower. 153 Similarly, an increase in L‐kynurenine/tryptophan ratio, depletion of plasma tryptophan level, and increase in L‐kynurenine and KYNA were reported. 154 Increase in serum KYNA was also observed among patients without dyskinesia, but not in dyskinetic PD patients. 155
In contrast, the deficiency of KYNA was revealed in a metabolomic study performed on a larger cohort of PD patients. Findings included lower plasma KYNA/L‐kynurenine ratio, higher QUIN level, and increased QUIN/KYNA ratio. 156 Similarly, lower KYNA, higher QUIN, and an elevated QUIN/picolinic acid ratio in CSF, as well as high 3‐HK in plasma, were detected. 157
The above data suggest that deficient KYNA synthesis seems to be limited to the brain in the course of PD, whereas in the periphery, the direction of changes in the KP varies and may depend on the stage of disease and the presence of discrete inflammation. Future research should be aimed to analyze in detail the temporal dynamics of peripheral and central KYNA levels in PD. Prominent support to the concept of causal relationship between central KYNA deficiency and PD development comes from the studies utilizing pharmacological compounds to increase brain KYNA and indicating the beneficial effect of such approach in animal models (vide paragraph 4.1.2).
3.3. Alzheimer's disease
Alzheimer's disease (AD) is the major cause of age‐related dementia among elderly population. This progressive neurodegenerative disorder leads inevitably to a severe deterioration of cognitive functions and exerts dramatic negative impact on patients’ quality of life. The characteristic neuropathological features of AD include senile plaques composed of beta‐amyloid aggregates and neurofibrillary tangles built from hyperphosphorylated tau proteins. 158 , 159 Cholinergic neurons of the forebrain and hippocampal and cortical glutamatergic neurons are among the most affected areas. 160
In a transgenic mouse model of AD, a decrease in brain KYNA was confirmed. 161 However, the data on KYNA levels in AD patients are not consistent. 160 , 162 , 163 , 164 Up to our knowledge, the data from brains of AD patients are very limited. In a small study involving postmortem analyses of specimens obtained from 11 patients with an advanced stage of AD, KYNA concentration was not altered in cortical areas, and increased in putamen (1.92‐fold) and caudate nucleus (1.77‐fold). 160 In latter structures, elevated KYNA correlated with the KAT I activity. 160 Lower KYNA levels was also detected in 5 brain structures obtained from AD victims. 83
Analyses of KYNA content in CSF of AD patients is not conclusive. In mild (N = 41) and moderate‐severe (N = 20) AD patients, high KYNA and increased KYNA/tryptophan ratio (both c. 2.7‐fold) were detected. 165 Similar results were obtained by other groups, reporting 1.7‐fold increase (N = 20) vs controls (N = 18) 166 and 1.29‐fold increase (N = 40) vs cognitively healthy controls (N = 34). 167 Higher CSF KYNA levels in AD females and significant correlation in the AD group (N = 19) of CSF KYNA with sICAM‐1 and CSF P‐tau, but no association with T‐tau or Aβ1‐42, were found. 164 In contrast, a decrease in CSF KYNA levels among AD patients (−1.3‐fold) and reduction in KYNA content in erythrocytes (−1.54‐fold) and serum (−1.46‐fold) (N = 28) were detected by others. 60 , 168
In the periphery, patients with AD exhibited a profound (35%) decrease in KYNA in plasma and erythrocytes, although the activity of KAT I and KAT II was not altered. 60 A similar decrease (−1.48‐fold) of plasma KYNA level in AD patients (N = 34) accompanied by an enhanced tryptophan degradation (−1.35‐fold) was reported by others. 169 The same population of patients manifested higher L‐kynurenine/tryptophan ratio (1.61‐fold), whereas KYNA/L‐kynurenine and 3‐HK/L‐kynurenine ratios were decreased (−1.69‐fold and −1.25‐fold, respectively). 169 Furthermore, patients had elevated level of serum QUIN indicative of a shift in the peripheral KP toward the neurotoxic metabolites at the expense of KYNA. 169 In a recent large‐scale metabolic phenotyping study, analyzing urine (N = 560) and serum samples (N = 354) obtained from clinically diagnosed patients with AD and mild cognitive impairment, lower metabolite concentrations of L‐kynurenine (serum), kynurenic acid (urine), tryptophan (urine, serum), and L‐kynurenine/tryptophan ratio (urine) were reported. 170
The nature of a decreased KYNA in the periphery among AD patients requires consideration, especially in the view of a well‐established age dependency of serum and brain KYNA levels. AD and dementia affect, in a vast majority, elderly patients. Therefore, AD patients should manifest higher levels of KYNA, which, indeed, are observed in CSF. However, neither in peripheral blood nor in the brain tissue such increases occur. Let us hypothesize that the KP is defective among AD patients, leading to an enhanced formation of QUIN with concomitant decline in KYNA. In such scenario, KYNA levels should be low in periphery, brain, and CSF, which is not a universal finding. However, an important aspect of brain tissue obtained from patients with an advanced AD is evident and prominent widespread neurodegeneration. Neuronal loss results in inflammation and subsequent astrogliosis. Increased number of astrocytes is directly associated with an enhanced activity of KATs. 171 , 172 , 173 Thus, hyperactivity of KATs and subsequent overproduction of KYNA developing after the occurrence of neuronal loss would not be perceived as a cause, but rather as one of the consequences of neurodegeneration and reactive gliosis.
A support to the concept of deficient KYNA formation as one of the causative factors in AD comes from the elegant study in a transgenic mouse model of AD. The pharmacological manipulation aimed to increase KYNA level prevented a number of behavioral and neuropathological changes in this model 161 (vide paragraph 4.1.3).
The potential role of disturbed KYNA formation in the pathogenesis of dementias is not fully understood. It is important to note that disproportionately high KYNA production, in our opinion secondary to the neuronal loss, may be aimed to further prevent the death of neurons in AD. Unfortunately, at high concentrations KYNA may act as a double‐sword and actually impair working memory and contextual learning. 174 , 175 , 176 , 177 An increase in error frequency has been reported in rats treated intraperitoneally with L‐kynurenine and manifesting high levels of brain KYNA, produced de novo within the brain from its precursor. 116 Similarly, adult rats treated throughout their adolescence with L‐kynurenine exhibited deficits in contextual fear memory, a novel object recognition memory, but not cue‐specific fear memory. 175 Adult rats exposed pre‐ and postnatally (gestation day 15‐postnatal day 21) to L‐kynurenine manifested a threefold increase in forebrain KYNA levels, a 2.5‐fold increase in prefrontal cortex KYNA, and deficits in initial reversal learning and extra‐dimensional shift. 176
Hence, KYNA, a metabolite of KP with neuroprotective effects at physiological concentrations, may exacerbate cognitive dysfunction and memory impairment in AD. However, as discussed above, an increase of brain KYNA levels most probably results from and is not a cause of neurodegeneration. In order to clarify this issue, longitudinal studies assessing the level of KYNA prior to and during the occurrence of overt symptoms of AD and dementia should be performed.
4. THERAPEUTIC PERSPECTIVES OF INCREASING KYNA LEVELS IN NEURODEGENERATIVE DISORDERS
In the past, given the very limited penetration of KYNA through the BBB and its rapid removal from the brain and body, the use of KYNA in the treatment of neurodegenerative diseases seemed virtually impossible. 178 Various attempts aimed to refine the bioavailability of KYNA have brought promising results. The most successful approaches are based on the use of KYNA analogs penetrating through the blood‐brain barrier, or modulation of the KP aimed to increase the concentration of KYNA substrate, L‐kynurenine, in the periphery through an inhibition of selected key enzymes. The latter approach results in an enhanced availability of L‐kynurenine for brain KYNA synthesis, as this KP metabolite easily enters central compartment. As the current reports on the KYNA therapeutic abilities in vivo seem rather optimistic, bypassing the main obstacle by improving its bioavailability may be a milestone in introducing KYNA to the treatment of neurodegenerative diseases.
4.1. Inhibition of KMO
Modulation of the KP‐controlling enzymes is a crucial step toward the increased production of neuroprotective metabolites with a simultaneous reduction in neurotoxic QUIN and 3‐HK in the brain. Due to the fact that astrocytes do not express KMO, the major astrocytic product of tryptophan catabolism is KYNA. 179 In contrast, microglia and macrophages convert tryptophan along both arms of the KP—neuroprotective and neurodegenerative. 70 In such scenario, proinflammatory environment, consistently implied as one of the factors contributing to the development of neurodegeneration, leads to an ample production of neurotoxic kynurenines, such as QUIN or 3‐HK. 180 Thus, inhibition of KMO allows astrocytes to retain more of L‐kynurenine and to produce larger amounts of KYNA, sufficient to antagonize the glutamate and QUIN excitotoxicity. Indeed, it is broadly documented that KMO activity is crucial for directing the metabolic fate of L‐kynurenine, and thus influences the QUIN/KYNA ratio the most. 181 A number of KMO inhibitors were synthesized and tested in various experimental models. 182 , 183 The initial studies were carried out prior to the identification of crystal structure of KMO; thus, the design of earliest inhibitors was based on the structure of L‐kynurenine. 182 One of the first KMO inhibitors used experimentally was nicotinylalanine. 184 , 185 The development of selective KMO inhibitors started in the 1990s, with introduction of m‐nitrobenzoylalanine (mNBA) showing the IC50 = 0.9 μM. 186 Experimental administration of 400 mg/kg of mNBA to rats resulted in a substantial increase of L‐kynurenine and KYNA levels—13‐ and fivefold in the brain, fivefold and 2.4‐fold in the blood, and sixfold and 3.5‐fold in the liver, respectively. 183 mNBA served as a lead compound for the synthesis of novel inhibitors belonging to 4‐phenyl‐4‐oxobutanoic acids, such as (R, S)‐3,4‐dichlorobenzoylalanine (FCE 28833). 187 After intraperitoneal treatment with FCE 28833A in rats (400 mg/kg), extracellular brain KYNA levels remained significantly elevated (over 30‐fold) for at least 22 h. 187 Modifications of FCE 2883 have brought development of PNU‐168754 and UPF‐648, a potent and selective KMO inhibitors (IC50 = 40 and 20 nM, respectively). 188 In gerbils, administration of UPF‐648 evoked remarkable increase of L‐kynurenine and KYNA levels in brain and plasma. 189 Another class of potent inhibitors comprises sulfonamides, including Ro 61–8048 (3,4‐dimethoxy‐N‐[4‐(3‐nitrophenyl) thiazol‐2‐yl] benzenesulfonamide; IC50 = 37 nM). 188 Ro 61–8048 provided neuroprotection in the rat and gerbil ischemia models, displayed antiepileptic activity against electroshock‐induced seizures in mice and rats, and reduced the cerebral QUIN accumulation in mice subjected to immune activation. 66 , 190 , 191 , 192 Due to the relative instability of Ro 61–8048, its slow‐release prodrug form, 2‐(3,4‐dimethoxybenzenesulfonylamino)‐4‐(3‐nitrophenyl)‐5‐(piperidin‐1‐yl) methylthiazole (JM6), has been developed. 161 Next class of KMO inhibitors was developed after establishing the crystal structure of the Saccharomyces cerevisiae enzyme (ScKMO) and human enzyme (hKMO). 193 , 194 These compounds include aryl pyrimidine carboxylic acids, 3,4‐dichlorohippuric acid, or 5‐(3‐nitrobenzyl)‐1H‐tetrazole. 182
4.1.1. Huntington's disease
Reestablishment of the physiological ratios between KP metabolites and shifting of the KP toward neuroprotective compounds may yield a potentially therapeutic effect in HD. Indeed, protective action of KMO inhibition was demonstrated in various experimental models of HD. Both genetic (cinnabar and vermillion mutations) and chemical (UPF‐648) inhibition of KMO attenuated neuronal loss in Drosophila melanogaster HD models. 195 UPF‐648 ameliorated the QUIN‐induced excitotoxic neuronal damage in transgenic mKAT II −/− mice and prevented degeneration of the rhabdomeres (photoreceptor neurons) in fruit fly HD model. 195 In the transgenic model of HD, oral administration of KMO inhibitor was linked with the animal life span prolongation, neuroprotection, and reduced glial activation. 161 Inhibition of KMO activity with CHDI‐340246 diminished the brain formation of 3‐HK and QUIN, elevated L‐kynurenine and KYNA levels, and restored the electrophysiological alterations. However, chronic application of CHDI‐340246 did not modify behavioral phenotypes or natural progression in mouse models of HD. 196 Similar, favorable effects were obtained with an inhibitor of TDO in a fruit fly model of HD. Improved locomotor performance, extended life span, and reduced neurodegeneration in Alzheimer's model flies were linked with an increased KYNA/3‐HK ratio. 197
4.1.2. Parkinson's disease
Neuroprotective and antiparkinsonian effects of glutamate receptor antagonists are well documented. 143 , 198 In line with these data, either administration of KYNA itself or use of pharmacological tools increasing the availability of L‐kynurenine and its conversion to KYNA may reduce neuronal loss and behavioral symptoms in experimental PD models. 185 , 199 , 200 , 201 , 202 Modifications of tryptophan metabolism seem to exert dual therapeutic benefit in PD, neuroprotection, and prevention of L‐DOPA–induced motor side effects. 203
Direct application of KYNA into the medial segment of the globus pallidus reduced the behavioral symptoms in MPTP‐induced PD model. 199 Similarly, in monkeys with hemiparkinsonism induced by unilateral, intraarterial administration of MPTP, KYNA infusion into the contralateral globus pallidus internus alleviated the disease symptoms. 200 The intracerebroventricular infusion of nicotinylalanine, inhibiting kynureninase and L‐kynurenine hydroxylase activity, combined with L‐kynurenine and probenecid, an inhibitor of organic acid transport, substantially increases KYNA level in rodent brain. This approach was used to raise KYNA content in rat substantia nigra and appears to be sufficient to protect neurons from QUIN‐induced toxicity. 185 Furthermore, intraperitoneal administration of L‐kynurenine and probenecid was demonstrated to reduce the 6‐hydroxydopamine–evoked neuronal damage and behavioral alterations. 204 In a primate model of MPTP‐induced parkinsonism, orally administered KMO inhibitor, Ro 61–8048, increased central L‐kynurenine and KYNA levels. This treatment improved the L‐DOPA‐induced dyskinesias without alteration of drug's antiparkinsonian efficacy. 205 Ro 61–8048 also reduced L‐DOPA motor side effects without affecting PD exacerbation. 159 In QUIN‐lesioned striata, KMO inhibitor UPF‐648 decreased conversion of L‐kynurenine to downstream neurotoxic metabolites, 3‐HK and QUIN (by 77% and 66%, respectively) and moderately raised KYNA synthesis (by 27%). 95 On the contrary, intrastriatal application of UPF‐648 in naïve rats reduced 3‐HK synthesis (by 64%) without change in KYNA formation. 98
In a fly model of PD, TDO inhibition evoked dramatic reduction in the 3‐HK/KYNA ratio, mainly due to increased synthesis of KYNA, with ensuing amelioration of disease phenotypes. 197
Prolonged systemic administration of the KMO inhibitor Ro 61–8048 reduced L‐DOPA–induced dyskinesias in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)–treated monkeys with overt symptoms of parkinsonism. 206 Immunosuppressive drug, FK506, shown to exert neuroprotective effects in experimental PD models is also able to increase KYNA formation and to prevent the MPP+‐induced decline in KYNA synthesis. 207 , 208
Experimental data consistently imply that shift of the KP to neurotoxic branch producing 3‐HK and QUIN formation, with relative or absolute deficiency of KYNA, is an important factor contributing to the development and progress of PD. We are still awaiting the synthesis of more precise pharmacological tools, able to modulate the KP within basal ganglia in a selective manner, which may become a promising therapeutic option for PD.
4.1.3. Alzheimer's disease
Only limited studies exploited KMO inhibitors as possible therapy for AD. Chronic oral therapy with JM6, inhibitor of KMO, was demonstrated to rise brain KYNA, due to de novo synthesis of the compound from L‐kynurenine, and to reduce extracellular glutamate in a transgenic mouse model of AD. 161 JM6 did not exert significant effects on Aβ plaque formation; however, it prevented spatial memory deficits. The compound also extended life span, prevented synaptic loss, and decreased microglial activation. 161
4.1.4. KMO inhibitors—limitations
Despite a large therapeutic potential, there are important drawbacks of some currently available KMO inhibitors. Certain compounds, such as mNBA and UPF‐648, were found to act as uncouplers of NADPH oxidation, which may actually potentiate neuronal loss via generation of cytotoxic hydrogen peroxide. 209 Therefore, precise design of novel compounds, effectively increasing brain KYNA levels, yet devoid of harmful production of free radicals, remains an important goal in the development of drugs against neurodegenerative disorders.
4.2. Analogs and prodrugs of KYNA
The goal of creating new KYNA analogs and prodrugs was to overcome the obstacle of poor BBB penetration by KYNA itself and to synthesize precursors that preferentially would not be metabolized to neurotoxic kynurenines. As a result, chlorokynurenines, including 4‐chlorokynurenine and 4,6‐dichlorokynurenine, emerged, meeting the above criteria, including fast delivery into the CNS and, once in the brain parenchyma, an easy conversion to potent NMDA antagonists acting at the glycine site, 7‐chlorokynurenic acid, or 5,7‐dichlorokynurenic acid. 210 Similar parameters characterize also esterified analogs and esterified 4‐amino analogs. 94 Another approach to improve the BBB penetration was based on utilizing D‐glucose ester of 7‐chlorokynurenic acid. The conjugate manifests improved BBB penetration as a result of an active transport by the glucose transporter GLUT1. 211 Indeed, systemic administration of the conjugates resulted in an anticonvulsant effect in mice affected by NMDA‐associated seizures.
4.2.1. Huntington's disease
Intraperitoneal injection of KYNA derivative, N‐(2‐N,N‐dimethylaminoethyl)‐4‐oxo‐1H‐quinoline‐2‐carboxamide hydrochloride (SzR72), diminished hypolocomotion, increased survival time, and provided striatal neuroprotection in transgenic N171‐82Q mice, without any major adverse effects. 212 Under in vivo conditions, both KYNA and SzR72 not only did not reduce but also actually enhanced the induction of long‐term potentiation (LTP). 213 The absence of memory impairment may result from the selective block of extrasynaptic NMDA and α7nACh receptors, while sparing the synaptic NMDA‐mediated currents. 213
4.2.2. Alzheimer's disease
Various KYNA analogs were synthesized and subsequently tested for their biological utility in the transgenic Caenorhabditis elegans line GMC101 with fully expressed Aβ42, AD model. 214 Three promising analogs representing a complex anti‐AD mechanism (free radical scavenger, AChE inhibitor, binding the mGluR5 and NMDA receptors and inhibiting the progression of Aβ fibrillation) include methyl 4‐hydroxy‐8‐methoxy‐5‐nitroquinoline‐2‐carboxylate, methyl 8‐amino‐4‐hydroxy‐6‐methoxyquinoline‐2‐carboxylate, and methyl 5‐amino‐4‐hydroxy‐8‐methoxyquinoline‐2‐carboxylate. The last two compounds exhibited fine permeability through the BBB model (cell‐based MDR1‐MDCKII), thus allowing to bypass the greatest limitation in KYNA bioavailability. The latter of the three analogs also showed a neuroprotective effect against Aβ‐related toxicity.
4.2.3. Parkinson's disease
Up to our knowledge, KYNA analogs were not studied in experimental models of Parkinson's disease. However, a number of compounds able to increase KYNA levels, through mechanisms distinct from interference with KP, successfully ameliorated L‐DOPA–induced dyskinesias. One of the interesting therapeutic options for PD seems to be the antiepileptic drug zonisamide, shown to reduce motor symptoms in patients with L‐DOPA–induced dyskinesias. 215 Zonisamide, apart from his broad pharmacological effects including inhibition of voltage‐gated sodium channels, T‐type calcium channels, and monoamine‐oxidase, has been shown to increase KYNA production. 216 Short and long exposure of the astrocytes to zonisamide increased the production of KYNA and other metabolites: xanthurenic acid and cinnabarinic acid, both with the properties of endogenous metabotropic glutamate receptor agonists (II and III groups, respectively). 216
FK506, a neuroimmunophilin ligand with immunosuppressive properties, used in the PD therapy, has been demonstrated both to enhance the cortical KYNA production and to restore the production of KYNA inhibited by the MPP+or 3‐NP. 207 Long‐term administration dose‐dependently increased dopaminergic neuron survival in an α‐synuclein–based rat model of Parkinson's disease. 217
5. CONCLUDING REMARKS
Alterations in central and peripheral KYNA synthesis have been demonstrated in the course of neurodegenerative diseases such as Alzheimer's, Parkinson's, or Huntington's disease, and deficiency of KYNA appears to contribute, at least in part, to the pathogenesis of neuronal loss. So far, accumulated data failed to show repeatedly the reliable correlation between peripheral and brain KYNA levels, as recently reviewed in a systematic review. 218 However, despite conflicting results, blood KYNA levels were linked with clinical symptoms and treatment response in psychiatric patients, as well as with observed neuroanatomical abnormalities and glial activity. 218 It would be optimal to combine the experiments involving simultaneous measurements of KYNA in blood and CSF, in future research involving experimental animal models, and in human studies aimed to elucidate KYNA contribution to neurodegeneration.
The current state of research is to develop promising experimental methods of manipulating the KP in humans without side effects, aimed to inhibit the KP part responsible for the synthesis of QUIN with concomitant stimulation of KYNA synthesis. Potential treatments include inhibitors of certain KP enzymes, as well as new prodrugs and analogs of KYNA penetrating via the blood‐brain barrier and thus able to enhance the KYNA‐induced block of the glycine site within the NMDA receptor complex. Emerging therapies may become an important route in the treatment of neurodegeneration, especially when the targeting of specific brain regions will be possible. Selectivity seems vital, especially considering that KYNA, apart from being neuroprotective, when produced in excessive quantities may also hamper cognition. The use of KYNA, alone or in combination with pharmacological tools precisely influencing specific populations of neurons, is awaiting to become a significant therapy for neurodegenerative disorders.
CONFLICT OF INTEREST
The authors declare that they do not have any conflicts of interest.
ACKNOWLEDGEMENTS
This study was supported by the Medical University in Lublin grants DS 450/20, 450/21.
Ostapiuk A, Urbanska EM. Kynurenic acid in neurodegenerative disorders—unique neuroprotection or double‐edged sword? CNS Neurosci Ther.2022;28:19–35. doi: 10.1111/cns.13768
REFERENCES
- 1. Massudi H, Grant R, Guillemin GJ, Braidy N. NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep. 2012;17(1):28‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cervenka I, Agudelo LZ, Kynurenines RJL. Tryptophan’s metabolites in exercise, inflammation, and mental health. Science. 2017;357(6349):378–398.e5. [DOI] [PubMed] [Google Scholar]
- 3. Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci. 2013;34(2):136‐143. [DOI] [PubMed] [Google Scholar]
- 4. Bohár Z, Toldi J, Fülöp F, Vécsei L. Changing the face of kynurenines and neurotoxicity: therapeutic considerations. Int J Mol Sci. 2015;16(12):9772‐9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen Y, Guillemin GJ. Kynurenine pathway metabolites in humans : disease and healthy states. Int J Tryptophan Resear. 2009;12:1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liebig J. Uber kynurensaeure. Justus Liebigs Ann Chem. 1853;86:125‐126. [Google Scholar]
- 7. Homer A. The constitution of kynurenic acid. J Biol Chem. 1914;17:509‐518. [Google Scholar]
- 8. Perkins MN, Stone TW. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982;247(1):184‐187. [DOI] [PubMed] [Google Scholar]
- 9. Foster AC, Vezzani A, French ED, Schwarcz R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci Lett. 1984;48(3):273‐278. [DOI] [PubMed] [Google Scholar]
- 10. Schwarcz R, Du F, Schmidt W, et al. Kynurenic acid: a potential pathogen in brain disorders. Ann N Y Acad Sci. 1992;648:140‐153. [DOI] [PubMed] [Google Scholar]
- 11. Wirthgen E, Hoeflich A, Rebl A, Günther J. Kynurenic acid: the janus‐faced role of an immunomodulatory tryptophan metabolite and its link to pathological conditions. Front Immunol. 2017;8:1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badawy AA‐B. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res. 2017;10:1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asp L, Johansson A‐S, Mann A, et al. Effects of pro‐inflammatory cytokines on expression of kynurenine pathway enzymes in human dermal fibroblasts. J Inflamm. 2011;8(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Myint A‐M, Kim Y‐K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog Neuro‐Psychopharmacol Biol Psychiatry. 2014;48:304‐313. [DOI] [PubMed] [Google Scholar]
- 15. Hornyák L, Dobos N, Koncz G, et al. The role of indoleamine‐2,3‐dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. 2018;9:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mazarei G, Leavitt BR. Indoleamine 2,3 dioxygenase as a potential therapeutic target in Huntington’s disease. J Huntingtons Dis. 2015;4(2):109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayaishi O. Properties and function of indoleamine 2,3‐dioxygenase. J Biochem. 1976;79(4):13‐21. [DOI] [PubMed] [Google Scholar]
- 18. Gál EM, Sherman AD. L‐kynurenine: its synthesis and possible regulatory function in brain. Neurochem Res. 1980;5(3):223‐239. [DOI] [PubMed] [Google Scholar]
- 19. Biernacki T, Sandi D, Bencsik K, Vécsei L. Kynurenines in the pathogenesis of multiple sclerosis: therapeutic perspectives. Cells. 2020;9(6):1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Németh H, Toldi J, Vécsei L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl. 2006;70:285‐304. [DOI] [PubMed] [Google Scholar]
- 21. Fukui S, Schwarcz R, Rapoport SI, Takada Y, Smith QR. Blood‐brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991;56(6):2007‐2017. [DOI] [PubMed] [Google Scholar]
- 22. Gál EM, Sherman AD. Synthesis and metabolism of L‐kynurenine in rat brain. J Neurochem. 1978;30(3):607‐613. [DOI] [PubMed] [Google Scholar]
- 23. Tamburin M, Mostardini M, Benatti L. Kynurenine aminotransferase I (KATI) isoform gene expression in the rat brain: an in situ hybridization study. NeuroReport. 1999;10(1):61‐65. [DOI] [PubMed] [Google Scholar]
- 24. Roberts RC, Du F, McCarthy KE, Okuno E, Schwarcz R. Immunocytochemical localization of kynurenine aminotransferase in the rat striatum: a light and electron microscopic study. J Comp Neurol. 1992;326(1):82‐90. [DOI] [PubMed] [Google Scholar]
- 25. Rzeski W, Kocki T, Dybel A, et al. Demonstration of kynurenine aminotransferases I and II and characterization of kynurenic acid synthesis in cultured cerebral cortical neurons. J Neurosci Res. 2005;80(5):677‐682. [DOI] [PubMed] [Google Scholar]
- 26. Stazka J, Luchowski P, Urbanska EM. Homocysteine, a risk factor for atherosclerosis, biphasically changes the endothelial production of kynurenic acid. Eur J Pharmacol. 2005;517(3):217‐223. [DOI] [PubMed] [Google Scholar]
- 27. Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3‐dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. 2005;49(1):15‐23. [DOI] [PubMed] [Google Scholar]
- 28. Guillemin GJ, Kerr SJ, Smythe GA, et al. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem. 2001;78(4):842‐853. [DOI] [PubMed] [Google Scholar]
- 29. Fujigaki H, Yamamoto Y, Saito K. L‐Tryptophan‐kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology. 2017;112(Pt B):264‐274. [DOI] [PubMed] [Google Scholar]
- 30. Stazka J, Luchowski P, Wielosz M, Kleinrok Z, Urbanska EM. Endothelium‐dependent production and liberation of kynurenic acid by rat aortic rings exposed to L‐kynurenine. Eur J Pharmacol. 2002;448(2–3):133‐137. [DOI] [PubMed] [Google Scholar]
- 31. Gramsbergen JB, Hodgkins PS, Rassoulpour A, Turski WA, Guidetti P, Schwarcz R. Brain‐specific modulation of kynurenic acid synthesis in the rat. J Neurochem. 1997;69(1):290‐298. [DOI] [PubMed] [Google Scholar]
- 32. Guidetti P, Hoffman GE, Melendez‐Ferro M, Albuquerque EX, Schwarcz R. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia. 2007;55(1):78‐92. [DOI] [PubMed] [Google Scholar]
- 33. Herédi J, Berkó AM, Jankovics F, et al. Astrocytic and neuronal localization of kynurenine aminotransferase‐2 in the adult mouse brain. Brain Struct Funct. 2017;222(4):1663‐1672. [DOI] [PubMed] [Google Scholar]
- 34. Jauch D, Urbańska EM, Guidetti P, et al. Dysfunction of brain kynurenic acid metabolism in Huntington’s disease: focus on kynurenine aminotransferases. J Neurol Sci. 1995;130(1):39‐47. [DOI] [PubMed] [Google Scholar]
- 35. Eastman CL, Urbanska EM, Chapman AG, Schwarcz R. Differential expression of the astrocytic enzymes 3‐hydroxyanthranilic acid oxygenase, kynurenine aminotransferase and glutamine synthetase in seizure‐prone and non‐epileptic mice. Epilepsy Res. 1994;18(3):185‐194. [DOI] [PubMed] [Google Scholar]
- 36. Okada K, Angkawidjaja C, Koga Y, Kanaya S. Structural and mechanistic insights into the kynurenine aminotransferase‐mediated excretion of kynurenic acid. J Struct Biol. 2014;185(3):257‐266. [DOI] [PubMed] [Google Scholar]
- 37. Guidetti P, Amori L, Sapko MT, Okuno E, Schwarcz R. Mitochondrial aspartate aminotransferase: a third kynurenate‐producing enzyme in the mammalian brain. J Neurochem. 2007;102(1):103‐111. [DOI] [PubMed] [Google Scholar]
- 38. Okuno E, Schmidt W, Parks DA, Nakamura M, Schwarcz R. Measurement of rat brain kynurenine aminotransferase at physiological kynurenine concentrations. J Neurochem. 1991;57(2):533‐540. [DOI] [PubMed] [Google Scholar]
- 39. Bellocchi D, Macchiarulo A, Carotti A, Pellicciari R. Quantum mechanics/molecular mechanics (QM/MM) modeling of the irreversible transamination of l‐kynurenine to kynurenic acid: the round dance of kynurenine aminotransferase II. Biochim Biophys Acta ‐ Proteins Proteomics. 2009;1794(12):1802‐1812. [DOI] [PubMed] [Google Scholar]
- 40. Guidetti P, Okuno E, Schwarcz R. Characterization of rat brain kynurenine aminotransferases I and II. J Neurosci Res. 1997;50(3):457‐465. [DOI] [PubMed] [Google Scholar]
- 41. Yu P, Di Prospero NA, Sapko MT, et al. Biochemical and phenotypic abnormalities in kynurenine aminotransferase II‐deficient mice. Mol Cell Biol. 2004;24(16):6919‐6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sapko M, Guidetti P, Yu P, et al. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: implications for Huntington’s disease. Exp Neurol. 2006;197:31‐40. [DOI] [PubMed] [Google Scholar]
- 43. Smith JR, Jamie JF, Guillemin GJ. Kynurenine‐3‐monooxygenase: a review of structure, mechanism, and inhibitors. Drug Discov Today. 2016;21(2):315‐324. [DOI] [PubMed] [Google Scholar]
- 44. Kocki T, Luchowski P, Luchowska E, Wielosz M, Turski WA, Urbanska EM. L‐cysteine sulphinate, endogenous sulphur‐containing amino acid, inhibits rat brain kynurenic acid production via selective interference with kynurenine aminotransferase II. Neurosci Lett. 2003;346(1):97‐100. [DOI] [PubMed] [Google Scholar]
- 45. Saran T, Hilgier W, Urbanska EM, Turski WA, Albrecht J. Kynurenic acid synthesis in cerebral cortical slices of rats with progressing symptoms of thioacetamide‐induced hepatic encephalopathy. J Neurosci Res. 2004;75(3):436‐440. [DOI] [PubMed] [Google Scholar]
- 46. Luchowska E, Luchowski P, Paczek R, et al. Dual effect of DL‐homocysteine and S‐adenosylhomocysteine on brain synthesis of the glutamate receptor antagonist, kynurenic acid. J Neurosci Res. 2005;79(3):375‐382. [DOI] [PubMed] [Google Scholar]
- 47. Luchowska E, Kloc R, Olajossy B, et al. β‐adrenergic enhancement of brain kynurenic acid production mediated via cAMP‐related protein kinase A signaling. Prog Neuro‐Psychopharmacol Biol Psychiatry. 2009;33(3):519‐529. [DOI] [PubMed] [Google Scholar]
- 48. Urbanska EM, Kocki T, Saran T, Kleinrok Z, Turski WA. Impairment of brain kynurenic acid production by glutamate metabotropic receptor agonists. NeuroReport. 1997;8(16):3501‐3505. [DOI] [PubMed] [Google Scholar]
- 49. Luchowski P, Urbanska EM. SNAP and SIN‐1 increase brain production of kynurenic acid. Eur J Pharmacol. 2007;563(1):130‐133. [DOI] [PubMed] [Google Scholar]
- 50. Kloc R, Luchowska E, Wielosz M, Owe‐Larsson B, Urbanska EM. Memantine increases brain production of kynurenic acid via protein kinase A‐dependent mechanism. Neurosci Lett. 2008;435(2):169‐173. [DOI] [PubMed] [Google Scholar]
- 51. Kocki T, Wnuk S, Kloc R, Kocki J, Owe‐Larsson B, Urbanska EM. New insight into the antidepressants action: modulation of kynurenine pathway by increasing the kynurenic acid/3‐hydroxykynurenine ratio. J Neural Transm. 2012;119(2):235‐243. [DOI] [PubMed] [Google Scholar]
- 52. Kocki T, Wielosz M, Turski WA, Urbanska EM. Enhancement of brain kynurenic acid production by anticonvulsants–novel mechanism of antiepileptic activity? Eur J Pharmacol. 2006;541(3):147‐151. [DOI] [PubMed] [Google Scholar]
- 53. Chmiel‐Perzyńska I, Perzyński A, Olajossy B, Gil‐Kulik P, Kocki J, Urbanska EM. Losartan reverses hippocampal increase of kynurenic acid in type 1 diabetic rats: a novel procognitive aspect of sartan action. J Diabetes Res. 2019;2019:4957879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blanco Ayala T, Lugo Huitrón R, Carmona Aparicio L, et al. Alternative kynurenic acid synthesis routes studied in the rat cerebellum. Front Cell Neurosci. 2015;9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ramos‐Chávez LA, Lugo Huitrón R, González Esquivel D, et al. Relevance of alternative routes of kynurenic acid production in the brain. Oxid Med Cell Longev. 2018;2018:5272741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Russi P, Carlà V, Moroni F. Indolpyruvic acid administration increases the brain content of kynurenic acid. Is this a new avenue to modulate excitatory amino acid receptors in vivo? Biochem Pharmacol. 1989;38(15):2405‐2409. [DOI] [PubMed] [Google Scholar]
- 57. Bartolini B, Corniello C, Sella A, Somma F, Politi V. The Enol Tautomer of Indole‐3‐Pyruvic Acid as A Biological Switch in Stress Responses BT ‐ Developments in Tryptophan and Serotonin Metabolism. Comparative aspects of circadian rhythms, Kerala, India: Transworld Research Network. Springer US; 2003; 601‐608. [DOI] [PubMed] [Google Scholar]
- 58. Politi V, Lavaggi MV, Di Stazio G, Margonelli A. Indole‐3‐pyruvic acid as a direct precursor of kynurenic acid. Adv Exp Med Biol. 1991;294:515‐518. [DOI] [PubMed] [Google Scholar]
- 59. Tomczyk T, Urbanska EM. Experimental hypothyroidism raises brain kynurenic acid – Novel aspect of thyroid dysfunction. Eur J Pharmacol. 2020;883:1‐8. [DOI] [PubMed] [Google Scholar]
- 60. Ramos‐Chávez LA, Lugo Huitrón R, González Esquivel D, et al. Decreased serum and red blood cell kynurenic acid levels in Alzheimer’s disease. Neurochem Int. 2007;50(2):308‐313. [DOI] [PubMed] [Google Scholar]
- 61. Baran H, Cairns N, Lubec B, Lubec G. Increased kynurenic acid levels and decreased brain kynurenine aminotransferasei in patients with down syndrome. Life Sci. 1996;58(21):1891‐1899. [DOI] [PubMed] [Google Scholar]
- 62. Ramirez Ortega D, Ovalle Rodríguez P, Pineda B, et al. Kynurenine pathway as a new target of cognitive impairment induced by lead toxicity during the lactation. Sci Rep. 2020;10(1):3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salvati P, Ukmar G, Dho L, et al. Brain concentrations of kynurenic acid after a systemic neuroprotective dose in the gerbil model of global ischemia. Prog Neuro‐Psychopharmacol Biol Psychiatry. 1999;23(4):741‐752. [DOI] [PubMed] [Google Scholar]
- 64. Uwai Y, Honjo H, Iwamoto K. Interaction and transport of kynurenic acid via human organic anion transporters hOAT1 and hOAT3. Pharmacol Res. 2012;65(2):254‐260. [DOI] [PubMed] [Google Scholar]
- 65. Uwai Y, Honjo E. Transport of xanthurenic acid by rat/human organic anion transporters OAT1 and OAT3. Biosci Biotechnol Biochem. 2013;77(7):1517‐1521. [DOI] [PubMed] [Google Scholar]
- 66. Russi P, Alesiani M, Lombardi G, Davolio P, Pellicciari R, Moroni F. Nicotinylalanine increases the formation of kynurenic acid in the brain and antagonizes convulsions. J Neurochem. 1992;59(6):2076‐2080. [DOI] [PubMed] [Google Scholar]
- 67. Shepard PD, Joy B, Clerkin L, Schwarcz R. Micromolar brain levels of kynurenic acid are associated with a disruption of auditory sensory gating in the rat. Neuropsychopharmacology. 2003;28(8):1454‐1462. [DOI] [PubMed] [Google Scholar]
- 68. Turski MP, Turska M, Zgrajka W, Kuc D, Turski WA. Presence of kynurenic acid in food and honeybee products. Amino Acids. 2009;36(1):75‐80. [DOI] [PubMed] [Google Scholar]
- 69. Kuc D, Zgrajka W, Parada‐Turska J, Urbanik‐Sypniewska T, Turski WA. Micromolar concentration of kynurenic acid in rat small intestine. Amino Acids. 2008;35(2):503‐505. [DOI] [PubMed] [Google Scholar]
- 70. Chiarugi A, Calvani M, Meli E, Traggiai E, Moroni F. Synthesis and release of neurotoxic kynurenine metabolites by human monocyte‐derived macrophages. J Neuroimmunol. 2001;120(1–2):190‐198. [DOI] [PubMed] [Google Scholar]
- 71. Guidetti P, Schwarcz R. 3‐Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur J Neurosci. 1999;11(11):3857‐3863. [DOI] [PubMed] [Google Scholar]
- 72. Morrison PF, Morishige GM, Beagles KE, Heyes MP. Quinolinic acid is extruded from the brain by a probenecid‐sensitive carrier system: a quantitative analysis. J Neurochem. 1999;72(5):2135‐2144. [DOI] [PubMed] [Google Scholar]
- 73. Braidy N, Grant R, Adams S, Brew BJ, Guillemin GJ. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res. 2009;16(1):77‐86. [DOI] [PubMed] [Google Scholar]
- 74. Jones SP, Guillemin GJ, Brew BJ. The kynurenine pathway in stem cell biology. Int J Tryptophan Res. 2013;6:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lugo‐Huitrón R, Ugalde Muñiz P, Pineda B, Pedraza‐Chaverrí J, Ríos C, Pérez‐de la Cruz V. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longev. 2013;2013:104024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Whetsell WO, Schwarcz R. Prolonged exposure to submicromolar concentrations of quinolinic acid causes excitotoxic damage in organotypic cultures of rat corticostriatal system. Neurosci Lett. 1989;97(3):271‐275. [DOI] [PubMed] [Google Scholar]
- 77. Guillemin GJ. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012;279(8):1356‐1365. [DOI] [PubMed] [Google Scholar]
- 78. Schurr A, Rigor BM. Quinolinate potentiates the neurotoxicity of excitatory amino acids in hypoxic neuronal tissue in vitro. Brain Res. 1993;617(1):76‐80. [DOI] [PubMed] [Google Scholar]
- 79. Pierozan P, Zamoner A, Soska AK, et al. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp Neurol. 2010;224(1):188‐196. [DOI] [PubMed] [Google Scholar]
- 80. Steiner J, Bogerts B, Sarnyai Z, et al. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: potential role of glial NMDA receptor modulators and impaired blood–brain barrier integrity. World J Biol Psychiatry. 2012;13(7):482‐492. [DOI] [PubMed] [Google Scholar]
- 81. Kepplinger B, Baran H, Kainz A, Ferraz‐Leite H, Newcombe J, Kalina P. Age‐related increase of kynurenic acid in human cerebrospinal fluid ‐ IgG and beta2‐microglobulin changes. Neurosignals. 2005;14(3):126‐135. [DOI] [PubMed] [Google Scholar]
- 82. Turski WA, Nakamura M, Todd WP, Carpenter BK, Whetsell WOJ, Schwarcz R. Identification and quantification of kynurenic acid in human brain tissue. Brain Res. 1988;454(1–2):164‐169. [DOI] [PubMed] [Google Scholar]
- 83. Beal MF, Matson WR, Storey E, et al. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J Neurol Sci. 1992;108(1):80‐87. [DOI] [PubMed] [Google Scholar]
- 84. Swartz KJ, Matson WR, MacGarvey U, Ryan EA, Beal MF. Measurement of kynurenic acid in mammalian brain extracts and cerebrospinal fluid by high‐performance liquid chromatography with fluorometric and coulometric electrode array detection. Anal Biochem. 1990;185(2):363‐376. [DOI] [PubMed] [Google Scholar]
- 85. Carlá V, Lombardi G, Beni M, Russi P, Moneti G, Moroni F. Identification and measurement of kynurenic acid in the rat brain and other organs. Anal Biochem. 1988;169(1):89‐94. [DOI] [PubMed] [Google Scholar]
- 86. Sorgdrager FJH, Vermeiren Y, Faassen M, et al. Age‐ and disease‐specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J Neurochem. 2019;151(5):656‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Urbańska EM, Luchowski P, Luchowska E, et al. Serum kynurenic acid positively correlates with cardiovascular disease risk factor, homocysteine: a study in stroke patients. Pharmacol Rep. 2006;58(4):507‐511. [PubMed] [Google Scholar]
- 88. Gramsbergen JBP, Schmidt W, Turski WA, Schwarcz R. Age‐related changes in kynurenic acid production in rat brain. Brain Res. 1992;588(1):1‐5. [DOI] [PubMed] [Google Scholar]
- 89. Moroni F, Russi P, Carlá V, Lombardi G. Kynurenic acid is present in the rat brain and its content increases during development and aging processes. Neurosci Lett. 1988;94(1):145‐150. [DOI] [PubMed] [Google Scholar]
- 90. Braidy N, Guillemin GJ, Mansour H, Chan‐Ling T, Grant R. Changes in kynurenine pathway metabolism in the brain, liver and kidney of aged female Wistar rats. FEBS J. 2011;278(22):4425‐4434. [DOI] [PubMed] [Google Scholar]
- 91. Wonodi I, McMahon RP, Krishna N, et al. Influence of kynurenine 3‐monooxygenase (KMO) gene polymorphism on cognitive function in schizophrenia. Schizophr Res. 2014;160(1):80‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kessler M, Terramani T, Lynch G, Baudry M. A glycine site associated with N‐Methyl‐d‐aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem. 1989;52(4):1319‐1328. [DOI] [PubMed] [Google Scholar]
- 93. Weber M, Dietrich D, Gräsel I, Reuter G, Seifert G, Steinhäuser C. 6‐Hydroxykynurenic acid and kynurenic acid differently antagonise AMPA and NMDA receptors in hippocampal neurones. J Neurochem. 2001;77(4):1108‐1115. [DOI] [PubMed] [Google Scholar]
- 94. Stone TW, Forrest CM, Darlington LG. Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection. FEBS J. 2012;279(8):1386‐1397. [DOI] [PubMed] [Google Scholar]
- 95. Urbanska EM, Chmiel‐Perzyńska I, Perzyński A, Derkacz M, Owe‐Larsson B. Endogenous Kynurenic Acid and Neurotoxicity. Handbook of Neurotoxicity. Springer; 2021:1‐31. [Google Scholar]
- 96. Urbanska E, Ikonomidou C, Sieklucka M, Turski WA. Aminooxyacetic acid produces excitotoxic lesions in the rat striatum. Synapse. 1991;9(2):129‐135. [DOI] [PubMed] [Google Scholar]
- 97. Németh H, Toldi J, Kynurenines VL. Parkinson’s Disease and Other Neurodegenerative Disorders: Preclinical and Clinical Studies. Parkinson’s Disease and Related Disorders. Springer Vienna; 2006:285‐304. [DOI] [PubMed] [Google Scholar]
- 98. Amori L, Guidetti P, Pellicciari R, Kajii Y, Schwarcz R. On the relationship between the two branches of the kynurenine pathway in the rat brain in vivo. J Neurochem. 2009;109(2):316‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Beggiato S, Tanganelli S, Fuxe K, Antonelli T, Schwarcz R, Ferraro L. Endogenous kynurenic acid regulates extracellular GABA levels in the rat prefrontal cortex. Neuropharmacology. 2014;82:11‐18. [DOI] [PubMed] [Google Scholar]
- 100. Wu H‐Q, Pereira EFR, Bruno JP, Pellicciari R, Albuquerque EX, Schwarcz R. The astrocyte‐derived alpha7 nicotinic receptor antagonist kynurenic acid controls extracellular glutamate levels in the prefrontal cortex. J Mol Neurosci. 2010;40(1–2):204‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zmarowski A, Wu H‐Q, Brooks JM, et al. Astrocyte‐derived kynurenic acid modulates basal and evoked cortical acetylcholine release. Eur J Neurosci. 2009;29(3):529‐538. [DOI] [PubMed] [Google Scholar]
- 102. Carpenedo R, Pittaluga A, Cozzi A, et al. Presynaptic kynurenate‐sensitive receptors inhibit glutamate release. Eur J Neurosci. 2001;13(11):2141‐2147. [DOI] [PubMed] [Google Scholar]
- 103. Wang J, Simonavicius N, Wu X, et al. Kynurenic acid as a ligand for orphan G protein‐coupled receptor GPR35. J Biol Chem. 2006;281(31):22021‐22028. [DOI] [PubMed] [Google Scholar]
- 104. Guo J, Williams DJ, Puhl HL 3rd, Ikeda SR. Inhibition of N‐type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J Pharmacol Exp Ther. 2008;324(1):342‐351. [DOI] [PubMed] [Google Scholar]
- 105. Berlinguer‐Palmini R, Masi A, Narducci R, et al. GPR35 activation reduces Ca2+ transients and contributes to the kynurenic acid‐dependent reduction of synaptic activity at CA3‐CA1 synapses. PLoS One. 2013;8(11):82180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cosi C, Mannaioni G, Cozzi A, et al. G‐protein coupled receptor 35 (GPR35) activation and inflammatory pain: Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology. 2011;60(7–8):1227‐1231. [DOI] [PubMed] [Google Scholar]
- 107. DiNatale BC, Murray IA, Schroeder JC, et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin‐6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115(1):89‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Moroni F, Cozzi A, Sili M, Mannaioni G. Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J Neural Transm. 2012;119(2):133‐139. [DOI] [PubMed] [Google Scholar]
- 109. Lugo‐Huitrón R, Blanco‐Ayala T, Ugalde‐Muñiz P, et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol. 2011;33(5):538‐547. [DOI] [PubMed] [Google Scholar]
- 110. Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89(1):73‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rassoulpour A, Wu H‐Q, Ferre S, Schwarcz R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J Neurochem. 2005;93(3):762‐765. [DOI] [PubMed] [Google Scholar]
- 112. Stone TW. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J Neurochem. 2020;152(6):627‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Boros FA, Vécsei L. Immunomodulatory effects of genetic alterations affecting the kynurenine pathway. Front Immunol. 2019;10:2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behav Brain Res. 2003;140(1–2):1‐47. [DOI] [PubMed] [Google Scholar]
- 115. Erhardt S, Schwieler L, Emanuelsson C, Geyer M. Endogenous kynurenic acid disrupts prepulse inhibition. Biol Psychiatry. 2004;56(4):255‐260. [DOI] [PubMed] [Google Scholar]
- 116. Chess AC, Simoni MK, Alling TE, Bucci DJ. Elevations of endogenous kynurenic acid produce spatial working memory deficits. Schizophr Bull. 2007;33(3):797‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gellért L, Varga D, Ruszka M, et al. Behavioural studies with a newly developed neuroprotective KYNA‐amide. J Neural Transm. 2012;119(2):165‐172. [DOI] [PubMed] [Google Scholar]
- 118. Kozak R, Campbell BM, Strick CA, et al. Reduction of brain kynurenic acid improves cognitive function. J Neurosci. 2014;34(32):10592‐10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ghosh R, Tabrizi SJ. Clinical features of Huntington’s disease. 2018;1‐28. [DOI] [PubMed]
- 120. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Prim. 2015;1:15005. [DOI] [PubMed] [Google Scholar]
- 121. Wilton DK, Stevens B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol Dis. 2020;143:104963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Valadão PAC, Santos KBS, Ferreira e Vieira TH, et al. Inflammation in Huntington’s disease: a few new twists on an old tale. J Neuroimmunol. 2020;348:577380. [DOI] [PubMed] [Google Scholar]
- 123. Tobore TO. Towards a comprehensive understanding of the contributions of mitochondrial dysfunction and oxidative stress in the pathogenesis and pathophysiology of Huntington’s disease. J Neurosci Res. 2019;97(11):1455‐1468. [DOI] [PubMed] [Google Scholar]
- 124. Schwarcz R, Whetsell WO, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon‐sparing lesions in rat brain. Science. 1983;219(4582):316‐318. [DOI] [PubMed] [Google Scholar]
- 125. Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J Neurosci. 1991;11(6):1649‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Luchowski P, Luchowska E, Turski WA, Urbanska EM. 1‐Methyl‐4‐phenylpyridinium and 3‐nitropropionic acid diminish cortical synthesis of kynurenic acid via interference with kynurenine aminotransferases in rats. Neurosci Lett. 2002;330(1):49‐52. [DOI] [PubMed] [Google Scholar]
- 127. Csillik A, Knyihár E, Okuno E, Krisztin‐Péva B, Csillik B, Vécsei L. Effect of 3‐nitropropionic acid on kynurenine aminotransferase in the rat brain. Exp Neurol. 2002;177(1):233‐241. [DOI] [PubMed] [Google Scholar]
- 128. Gould DH, Gustine DL. Basal ganglia degeneration, myelin alterations, and enzyme inhibition induced in mice by the plant toxin 3‐nitropropanoic acid. Neuropathol Appl Neurobiol. 8(5):377‐393. [DOI] [PubMed] [Google Scholar]
- 129. Borlongan CV, Koutouzis TK, Sanberg PR. 3‐Nitropropionic acid animal model and Huntington’s disease. Neurosci Biobehav Rev. 1997;21(3):289‐293. [DOI] [PubMed] [Google Scholar]
- 130. Urbanska EM, Blaszczak P, Saran T, Kleinrok Z, Turski WA. Mitochondrial toxin 3‐nitropropionic acid evokes seizures in mice. Eur J Pharmacol. 1998;359(1):55‐58. [DOI] [PubMed] [Google Scholar]
- 131. Brouillet E, Jacquard C, Bizat N, Blum D. 3‐Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem. 2005;95(6):1521‐1540. [DOI] [PubMed] [Google Scholar]
- 132. Guidetti P, Reddy PH, Tagle D, Schwarcz R. Early kynurenergic impairment in Huntington’s disease and in a transgenic animal model. Neurosci Lett. 2000;283:233‐235. [DOI] [PubMed] [Google Scholar]
- 133. Sathyasaikumar KV, Stachowski EK, Amori L, Guidetti P, Muchowski PJ, Schwarcz R. Dysfunctional kynurenine pathway metabolism in the R6/2 mouse model of Huntington’s disease. J Neurochem. 2010;113(6):1416‐1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Connick JH, Carlà V, Moroni F, Stone TW. Increase in kynurenic acid in Huntington’s disease motor cortex. J Neurochem. 1989;52(3):985‐987. [DOI] [PubMed] [Google Scholar]
- 135. Connick JH, Stone TW. Quinolinic acid effects on amino acid release from the rat cerebral cortex in vitro and in vivo. Br J Pharmacol. 1988;93(4):868‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55(4):1327‐1339. [DOI] [PubMed] [Google Scholar]
- 137. Guidetti P, Luthi‐Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol Dis. 2004;17(3):455‐461. [DOI] [PubMed] [Google Scholar]
- 138. Stoy N, Mackay GM, Forrest CM, et al. Tryptophan metabolism and oxidative stress in patients with Huntington’s disease. J Neurochem. 2005;93(3):611‐623. [DOI] [PubMed] [Google Scholar]
- 139. Forrest CM, Mackay GM, Stoy N, et al. Blood levels of kynurenines, interleukin‐23 and soluble human leucocyte antigen‐G at different stages of Huntington’s disease. J Neurochem. 2010;112(1):112‐122. [DOI] [PubMed] [Google Scholar]
- 140. Rodrigues FB, Byrne LM, Lowe AJ, et al. Kynurenine pathway metabolites in cerebrospinal fluid and blood as potential biomarkers in Huntington’s disease. J Neurochem. 2021.158(2):539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lang AE, Lozano AM. Parkinson’s disease. N Engl J Med. 1998;339(15):1044‐1053. [DOI] [PubMed] [Google Scholar]
- 142. Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132‐144. [DOI] [PubMed] [Google Scholar]
- 143. Klockgether T, Turski L. NMDA antagonists potentiate antiparkinsonian actions of L‐dopa in monoamine‐depleted rats. Ann Neurol. 1990;28(4):539‐546. [DOI] [PubMed] [Google Scholar]
- 144. Klockgether T, Turski L, Honoré T, et al. The AMPA receptor antagonist NBQX has antiparkinsonian effects in monoamine‐depleted rats and MPTP‐treated monkeys. Ann Neurol. 1991;30(5):717‐723. [DOI] [PubMed] [Google Scholar]
- 145. Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine‐analog synthesis. Science. 1983;219(4587):979‐980. [DOI] [PubMed] [Google Scholar]
- 146. Langston JW. The MPTP story. J Parkinsons Dis. 2017;7(s1):S11‐S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Schildknecht S, Di Monte DA, Pape R, Tieu K, Leist M. Tipping Points And Endogenous Determinants Of Nigrostriatal Degeneration by MPTP. Trends Pharmacol Sci. 2017;38(6):541‐555. [DOI] [PubMed] [Google Scholar]
- 148. Knyihár‐Csillik E, Chadaide Z, Mihály A, Krisztin‐Péva B, Fenyő R, Vécsei L. Effect of 6‐hydroxydopamine treatment on kynurenine aminotransferase‐I (KAT‐I) immunoreactivity of neurons and glial cells in the rat substantia nigra. Acta Neuropathol. 2006;112(2):127‐137. [DOI] [PubMed] [Google Scholar]
- 149. Knyihár‐Csillik E, Csillik B, Pákáski M, et al. Decreased expression of kynurenine aminotransferase‐I (KAT‐I) in the substantia nigra of mice after 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) treatment. Neuroscience. 2004;126(4):899‐914. [DOI] [PubMed] [Google Scholar]
- 150. Lee DY, Lee K‐S, Lee HJ, et al. Kynurenic acid attenuates MPP+‐induced dopaminergic neuronal cell death via a Bax‐mediated mitochondrial pathway. Eur J Cell Biol. 2008;87(6):389‐397. [DOI] [PubMed] [Google Scholar]
- 151. Ogawa T, Matson WR, Beal MF, et al. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology. 1992;42(9):1702‐1706. [DOI] [PubMed] [Google Scholar]
- 152. Caudle WM, Zhang J. Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp Neurol. 2009;220(2):230. [DOI] [PubMed] [Google Scholar]
- 153. Hartai Z, Klivenyi P, Janaky T, Penke B, Dux L, Vecsei L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J Neurol Sci. 2005;239(1):31‐35. [DOI] [PubMed] [Google Scholar]
- 154. Oxenkrug G, van der Hart M, Roeser J, Summergrad P. Peripheral tryptophan ‐ kynurenine metabolism associated with metabolic syndrome is different in Parkinson’s and Alzheimer’s diseases. Endocrinol Diabetes Metab J. 2017;1(4). [PMC free article] [PubMed] [Google Scholar]
- 155. Havelund J, Heegaard N, Færgeman N, Gramsbergen J. Biomarker research in Parkinson’s disease using metabolite profiling. Metabolites. 2017;7(3):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chang K‐H, Cheng M‐L, Tang H‐Y, Huang C‐Y, Wu Y‐R, Chen C‐M. Alternations of metabolic profile and kynurenine metabolism in the plasma of Parkinson’s disease. Mol Neurobiol. 2018;55(8):6319‐6328. [DOI] [PubMed] [Google Scholar]
- 157. Heilman PL, Wang EW, Lewis MM, et al. Tryptophan metabolites are associated with symptoms and nigral pathology in Parkinson’s disease. Mov Disord. 2020;35(11):2028‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863‐1872. [DOI] [PubMed] [Google Scholar]
- 159. Ting KK, Brew B, Guillemin G. The involvement of astrocytes and kynurenine pathway in Alzheimer’s disease. Neurotox Res. 2007;12(4):247‐262. [DOI] [PubMed] [Google Scholar]
- 160. Baran H, Jellinger K, Deecke L. Kynurenine metabolism in Alzheimer’s disease. J Neural Transm. 1999;106(2):165‐181. [DOI] [PubMed] [Google Scholar]
- 161. Zwilling D, Huang S‐Y, Sathyasaikumar KV, et al. Kynurenine 3‐monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145(6):863‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Widner B, Leblhuber F, Walli J, Tilz GP, Demel U, Fuchs D. Tryptophan degradation and immune activation in Alzheimer’s disease. J Neural Transm. 2000;107(3):343‐353. [DOI] [PubMed] [Google Scholar]
- 163. Linderholm KR, Skogh E, Olsson SK, et al. Increased levels of kynurenine and kynurenic acid in the CSF of patients with schizophrenia. Schizophr Bull. 2012;38(3):426‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wennström M, Nielsen HM, Orhan F, Londos E, Minthon L, Erhardt S. Kynurenic acid levels in cerebrospinal fluid from patients with Alzheimer’s disease or dementia with Lewy bodies. Int J Tryptophan Res. 2014;7:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. González‐Sánchez M, Jiménez J, Narváez A, et al. Kynurenic acid levels are increased in the CSF of Alzheimer’s disease patients. Biomolecules. 2020;10(4):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Jacobs KR, Lim CK, Blennow K, et al. Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer’s disease and relationship to amyloid‐β and tau. Neurobiol Aging. 2019;80:11‐20. [DOI] [PubMed] [Google Scholar]
- 167. Van Der Velpen V, Teav T, Gallart‐Ayala H, et al. Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimer’s Res Ther. 2019;11(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Heyes MP, Saito K, Crowley JS, et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non‐inflammatory neurological disease. Brain A J Neurol. 1992;115 (Pt 5):1249‐1273. [DOI] [PubMed] [Google Scholar]
- 169. Gulaj E, Pawlak K, Bien B, Pawlak D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv Med Sci. 2010;55(2):204‐211. [DOI] [PubMed] [Google Scholar]
- 170. Whiley L, Chappell KE, D’Hondt E, et al. Metabolic phenotyping reveals a reduction in the bioavailability of serotonin and kynurenine pathway metabolites in both the urine and serum of individuals living with Alzheimer’s disease. Alzheimers Res Ther. 2021;13(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Muench G, Schinzel R, Loske C, et al. Alzheimer’s disease ‐ Synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm. 1998;105:439‐461. [DOI] [PubMed] [Google Scholar]
- 172. Hirsch EC, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in parkinson’s disease: a role in neurodegeneration? Ann Neurol. 1998;44(S1):S115‐S120. [DOI] [PubMed] [Google Scholar]
- 173. Pitter KL, Tamagno I, Feng X, et al. The SHH/Gli pathway is reactivated in reactive glia and drives proliferation in response to neurodegeneration‐induced lesions. Glia. 2014;62(10):1595‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Robinson KSL, Stewart AM, Cachat J, Landsman S, Gebhardt M, Kalueff AV. Psychopharmacological effects of acute exposure to kynurenic acid (KYNA) in zebrafish. Pharmacol Biochem Behav. 2013;108:54‐60. [DOI] [PubMed] [Google Scholar]
- 175. Akagbosu CO, Evans GC, Gulick D, Suckow RF, Bucci DJ. Exposure to kynurenic acid during adolescence produces memory deficits in adulthood. Schizophr Bull. 2012;38(4):769‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Alexander KS, Pocivavsek A, Wu H‐Q, Pershing ML, Schwarcz R, Bruno JP. Early developmental elevations of brain kynurenic acid impair cognitive flexibility in adults: reversal with galantamine. Neuroscience. 2013;238:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Pocivavsek A, Thomas MAR, Elmer GI, Bruno JP, Schwarcz R. Continuous kynurenine administration during the prenatal period, but not during adolescence, causes learning and memory deficits in adult rats. Psychopharmacology. 2014;231(14):2799‐2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Bahn A, Ljubojević M, Lorenz H, et al. Murine renal organic anion transporters mOAT1 and mOAT3 facilitate the transport of neuroactive tryptophan metabolites. Am J Physiol ‐ Cell Physiol. 2005;289(5):1075‐1084. [DOI] [PubMed] [Google Scholar]
- 179. Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM. Indoleamine 2,3 dioxygenase and quinolinic acid Immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol Appl Neurobiol. 2005;31(4):395‐404. [DOI] [PubMed] [Google Scholar]
- 180. Huang Y‐S, Ogbechi J, Clanchy FI, Williams RO, Stone TW. IDO and kynurenine metabolites in peripheral and CNS disorders. Front Immunol. 2020;11:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Zhang S, Collier MEW, Heyes DJ, Giorgini F, Scrutton NS. Advantages of brain penetrating inhibitors of kynurenine‐3‐monooxygenase for treatment of neurodegenerative diseases. Arch Biochem Biophys. 2021;697:108702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Phillips RS, Iradukunda EC, Hughes T, Bowen JP. Modulation of enzyme activity in the kynurenine pathway by kynurenine monooxygenase inhibition. Front Mol Biosci. 2019;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Jacobs KR, Castellano‐Gonzalez G, Guillemin GJ, Lovejoy DB. Major developments in the design of inhibitors along the kynurenine pathway. Curr Med Chem. 2017;24(23):2471‐2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Connick JH, Heywood GC, Sills GJ, Thompson GG, Brodie MJ, Stone TW. Nicotinylalanine increases cerebral kynurenic acid content and has anticonvulsant activity. Gen Pharmacol. 1992;23(2):235‐239. [DOI] [PubMed] [Google Scholar]
- 185. Miranda AF, Boegman RJ, Beninger RJ, Jhamandas K. Protection against quinolinic acid‐mediated excitotoxicity in nigrostriatal dopaminergic neurons by endogenous kynurenic acid. Neuroscience. 1997;78(4):967‐975. [DOI] [PubMed] [Google Scholar]
- 186. Pellicciari R, Natalini B, Costantino G, et al. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m‐Nitrobenzoyl)alanine, a potent inhibitor of kynurenine‐3‐hydroxylase. J Med Chem. 1994;37(5):647‐655. [DOI] [PubMed] [Google Scholar]
- 187. Speciale C, Wu HQ, Cini M, Marconi M, Varasi M, Schwarcz R. (R, S)‐3,4‐dichlorobenzoylalanine (FCE 28833A) causes a large and persistent increase in brain kynurenic acid levels in rats. Eur J Pharmacol. 1996;315(3):263‐267. [DOI] [PubMed] [Google Scholar]
- 188. Phillips RS, Anderson AD, Gentry HG, Güner OF, Bowen JP. Substrate and inhibitor specificity of kynurenine monooxygenase from Cytophaga hutchinsonii . Bioorg Med Chem Lett. 2017;27(8):1705‐1708. [DOI] [PubMed] [Google Scholar]
- 189. Pellicciari R, Amori L, Costantino G, et al. Modulation of the kynurine pathway of tryptophan metabolism in search for neuroprotective agents. Focus on kynurenine‐3‐hydroxylase. Adv Exp Med Biol. 2003;527:621‐628. [DOI] [PubMed] [Google Scholar]
- 190. Röver S, Cesura AM, Huguenin P, Kettler R, Szente A. Synthesis and biochemical evaluation of N‐(4‐phenylthiazol‐2‐yl)benzenesulfonamides as high‐affinity inhibitors of kynurenine 3‐hydroxylase. J Med Chem. 1997;40(26):4378‐4385. [DOI] [PubMed] [Google Scholar]
- 191. Carpenedo R, Chiarugi A, Russi P, et al. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anticonvulsant activities. Neuroscience. 1994;61(2):237‐243. [DOI] [PubMed] [Google Scholar]
- 192. Cozzi A, Carpenedo R, Moroni F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: studies with (m‐nitrobenzoyl)‐alanine (mNBA) and 3,4‐dimethoxy‐[‐N‐4‐(nitrophenyl)thiazol‐2yl]‐benzenesulfonamide (Ro 61–8048) in models of focal or global brain ischemia. J Cereb Blood Flow Metab off J Int Soc Cereb Blood Flow Metab. 1999;19(7):771‐777. [DOI] [PubMed] [Google Scholar]
- 193. Amaral M, Levy C, Heyes DJ, et al. Structural basis of kynurenine 3‐monooxygenase inhibition. Nature. 2013;496(7445):382‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Kim HT, Na BK, Chung J, et al. Structural Basis for Inhibitor‐Induced Hydrogen Peroxide Production by Kynurenine 3‐Monooxygenase. Cell Chem Biol. 2018;25(4):426‐438.e4. [DOI] [PubMed] [Google Scholar]
- 195. Campesan S, Green EW, Breda C, et al. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr Biol. 2011;21(11):961‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Beaumont V, Mrzljak L, Dijkman U, et al. The novel KMO inhibitor CHDI‐340246 leads to a restoration of electrophysiological alterations in mouse models of Huntington’s disease. Exp Neurol. 2016;282:99‐118. [DOI] [PubMed] [Google Scholar]
- 197. Breda C, Sathyasaikumar KV, Idrissi SS, et al. Tryptophan‐2,3‐dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc Natl Acad Sci USA. 2016;113(19):5435‐5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Turski L, Bressler K, Rettig KJ, Löschmann PA, Wachtel H. Protection of substantia nigra from MPP+ neurotoxicity by N‐methyl‐D‐aspartate antagonists. Nature. 1991;349(6308):414‐418. [DOI] [PubMed] [Google Scholar]
- 199. Brotchie JM, Mitchell IJ, Sambrook MA, Crossman AR. Alleviation of parkinsonism by antagonism of excitatory amino acid transmission in the medial segment of the globus pallidus in rat and primate. Mov Disord. 1991;6(2):133‐138. [DOI] [PubMed] [Google Scholar]
- 200. Butler EG, Bourke DW, Finkelstein DI, Horne MK. The effects of reversible inactivation of the subthalamo‐pallidal pathway on the behaviour of naive and hemiparkinsonian monkeys. J Clin Neurosci. 1997;4(2):218‐227. [DOI] [PubMed] [Google Scholar]
- 201. Moroni F, Cozzi A, Carpendo R, Cipriani G, Veneroni O, Izzo E. Kynurenine 3‐mono‐oxygenase inhibitors reduce glutamate concentration in the extracellular spaces of the basal ganglia but not in those of the cortex or hippocampus. Neuropharmacology. 2005;48(6):788‐795. [DOI] [PubMed] [Google Scholar]
- 202. Ouattara B, Belkhir S, Morissette M, et al. Implication of NMDA receptors in the antidyskinetic activity of cabergoline, CI‐1041, and Ro 61–8048 in MPTP monkeys with levodopa‐induced dyskinesias. J Mol Neurosci. 2009;38(2):128‐142. [DOI] [PubMed] [Google Scholar]
- 203. Szabó N, Kincses ZT, Toldi J, Vécsei L. Altered tryptophan metabolism in Parkinson’s disease: a possible novel therapeutic approach. J Neurol Sci. 2011;310(1–2):256‐260. [DOI] [PubMed] [Google Scholar]
- 204. Silva‐Adaya D, Pérez‐De La Cruz V, Villeda‐Hernández J, et al. Protective effect of L‐kynurenine and probenecid on 6‐hydroxydopamine‐induced striatal toxicity in rats: implications of modulating kynurenate as a protective strategy. Neurotoxicol Teratol. 2011;33(2):303‐312. [DOI] [PubMed] [Google Scholar]
- 205. Samadi P, Grégoire L, Rassoulpour A, et al. Effect of kynurenine 3‐hydroxylase inhibition on the dyskinetic and antiparkinsonian responses to levodopa in parkinsonian monkeys. Mov Disord. 2005;20(7):792‐802. [DOI] [PubMed] [Google Scholar]
- 206. Grégoire L, Rassoulpour A, Guidetti P, et al. Prolonged kynurenine 3‐hydroxylase inhibition reduces development of levodopa‐induced dyskinesias in parkinsonian monkeys. Behav Brain Res. 2008;186(2):161‐167. [DOI] [PubMed] [Google Scholar]
- 207. Luchowska E, Luchowski P, Wielosz M, Turski WA, Urbanska EM. FK506 attenuates 1‐methyl‐4‐phenylpyridinium‐ and 3‐nitropropionic acid‐evoked inhibition of kynurenic acid synthesis in rat cortical slices. Acta Neurobiol Exp. 2003;63(2):101‐108. [DOI] [PubMed] [Google Scholar]
- 208. Guo X, Dawson VL, Dawson TM. Neuroimmunophilin ligands exert neuroregeneration and neuroprotection in midbrain dopaminergic neurons. Eur J Neurosci. 2001;13(9):1683‐1693. [DOI] [PubMed] [Google Scholar]
- 209. Crozier‐Reabe KR, Phillips RS, Moran GR. Kynurenine 3‐monooxygenase from pseudomonas fluorescens : substrate‐like inhibitors both stimulate flavin reduction and stabilize the flavin−peroxo intermediate yet result in the production of hydrogen peroxide. Biochemistry. 2008;47(47):12420‐12433. [DOI] [PubMed] [Google Scholar]
- 210. Hokari M, Wu HQ, Schwarcz R, Smith QR. Facilitated brain uptake of 4‐chlorokynurenine and conversion to 7‐chlorokynurenic acid. NeuroReport. 1996;8(1):15‐18. [DOI] [PubMed] [Google Scholar]
- 211. Battaglia G, La Russa M, Bruno V, et al. Systemically administered d‐glucose conjugates of 7‐chlorokynurenic acid are centrally available and exert anticonvulsant activity in rodents. Brain Res. 2000;860(1):149‐156. [DOI] [PubMed] [Google Scholar]
- 212. Zádori D, Nyiri G, Szonyi A, et al. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J Neural Transm. 2011;118(6):865‐875. [DOI] [PubMed] [Google Scholar]
- 213. Demeter I, Nagy K, Farkas T, et al. Paradox effects of kynurenines on LTP induction in the Wistar rat. An in vivo study. Neurosci Lett. 2013;553:138‐141. [DOI] [PubMed] [Google Scholar]
- 214. Deora GS, Kantham S, Chan S, et al. Multifunctional analogs of kynurenic acid for the treatment of Alzheimer’s disease: synthesis, pharmacology, and molecular modeling studies. ACS Chem Neurosci. 2017;8(12):2667‐2675. [DOI] [PubMed] [Google Scholar]
- 215. Li C, Xue L, Liu Y, Yang Z, Chi S, Xie A. Zonisamide for the treatment of parkinson disease: a current update. Front Neurosci. 2020;14:574652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Fukuyama K, Tanahashi S, Hoshikawa M, Shinagawa R, Okada M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology. 2014;76(Pt A):137‐145. [DOI] [PubMed] [Google Scholar]
- 217. Van der Perren A, Macchi F, Toelen J, et al. FK506 reduces neuroinflammation and dopaminergic neurodegeneration in an α‐synuclein‐based rat model for Parkinson’s disease. Neurobiol Aging. 2015;36(3):1559‐1568. [DOI] [PubMed] [Google Scholar]
- 218. Skorobogatov K, De Picker L, Verkerk R, et al. Brain versus blood: a systematic review on the concordance between peripheral and central kynurenine pathway measures in psychiatric disorders. Front Immunol. 2021;12: 716980 [DOI] [PMC free article] [PubMed] [Google Scholar]