

Abstract
Background and Objectives
Few biochemical markers have been identified in spinocerebellar ataxia type 2 (SCA2). This study aimed to determine the levels of neurofilament light (NfL) in patients with SCA2 and to identify whether they are associated with disease severity.
Methods
Participants were recruited from 1 medical center in China, and individuals with SCA2 were genetically diagnosed. NfL levels were assessed with the single molecule array method. Disease severity was evaluated with the Scale for the Assessment and Rating of Ataxia (SARA), the International Cooperative Ataxia Rating Scale (ICARS), and the Inventory of Non-Ataxia Symptoms (INAS). Cerebellum and brainstem volumes were calculated from neuroimaging measurements. We used the Pearson correlation and partial correlation for correlation analyses.
Results
Forty-nine patients with manifest SCA2, 10 preclinical individuals with SCA2, and 92 controls were enrolled. A high consistency was identified between serum and CSF NfL (r = 0.868, p < 0.0001). In individuals with SCA2, levels of serum NfL were associated with disease severity (SARA: r = 0.425, p = 0.003; ICARS: r = 0.383, p = 0.009; INAS:, r = 0.390, p = 0.007; cerebellum volume: r = −0.393, p = 0.024) after adjustment for age. NfL levels were higher close to the expected age at onset in preclinical individuals with SCA2 (R2 = 0.43, p = 0.04).
Discussion
Levels of serum NfL were correlated with disease intensity in individuals with SCA2 and were higher close to the estimated age at onset in preclinical SCA2. Therefore, NfL is a potential serum biomarker of disease severity in SCA2.
Classification of Evidence
This study provides Class II evidence that elevated NfL levels are associated with disease severity in individuals with SCA2.
Spinocerebellar ataxia type 2 (SCA2) is one of the most common autosomal dominantly inherited degenerative ataxias worldwide, clinically characterized by progressive ataxia, slow hypometric saccades, hyporeflexia, and polyneuropathy.1 Considering the upcoming disease-modifying treatments2 and several promising therapies,3-5 accessible, reliable, and objective tools are immediately needed to stratify individuals with SCA2 with different stages, to evaluate disease severity, to track disease progression, and to further pave the way for future trials. Although extensive efforts have established neuroimaging and electrophysiology biomarkers of progression,6-11 few biochemical markers have been identified.
Neurofilament light (NfL) chain is gaining increasing attention as a candidate biomarker because the release of NfL sharply increases in response to axonal damage.12 Recent studies have validated NfL as a promising biomarker in many neurodegenerative diseases, including parkinsonian disorders, Huntington disease (HD), amyotrophic lateral sclerosis, and dementia.13-16 We have reported that NfL was higher in CSF and serum in patients with spinocerebellar ataxia type 3 and correlated with clinical severity.17 However, a previous work observed no obvious elevation of NfL level in 2 patients with SCA2.18 Thus, there is a compelling need to determine whether NfL could act as a potential biomarker of neurodegeneration in a larger cohort.
In this study, we aimed to investigate the levels of serum and CSF NfL in individuals with SCA2 with different disease stages and to identify whether NfL is associated with disease severity.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the ethics committee of the Second Affiliated Hospital, Zhejiang University School of Medicine. Written informed consent was obtained from each participant before recruitment.
Primary Research Questions/Classification of Evidence
Our primary research question is to determine the levels of NfL in individuals with SCA2 and to identify whether they are associated with disease severity. This study was designed to provide Class II evidence that elevated NfL levels were associated with clinical disease severity in individuals with SCA2.
Participants and Study Design
In the cross-sectional study, 92 controls and 59 individuals with SCA2, including 10 individuals with preclinical SCA2 (preSCA2) and 49 patients with manifest SCA2 were recruited from April 2015 to September 2020 in the Second Affiliated Hospital of Zhejiang University School of Medicine (Hangzhou). These 59 individuals with SCA2 were genetically diagnosed as previously reported.19 The unrelated neurologically healthy controls had negative genetic screening for the ATXN2 gene and no evidence of inherited, neurologic, and psychiatric diseases. Age and sex were considered in the control selection. Demographic data, clinical characteristics, rating scales, and brain MRI information were collected from individuals with SCA2. Age was defined as the age at sample collection. The preclinical stage was defined by unspecific neurologic symptoms (e.g., muscle cramps, vertigo, hyporeflexia) or mild coordination deficit with a Scale for the Assessment and Rating of Ataxia (SARA)20 score <3 according to the reported criteria.21 The patients with manifest SCA2 were classified into 2 subgroups based on median SARA scores and clinical features, including 25 patients in stage 1 (3 ≤ SARA score <11) and 24 in stage 2 (SARA score ≥11).
Blood and CSF samples were collected from participants. Forty-three individuals consisting of 23 patients with manifest SCA2 and 20 controls were classified into group A due to the availability of both CSF and serum to assess the correlation of the NfL level between them. All participants with available serum sample, including 92 controls and 59 individuals with SCA2, among whom 23 patients with manifest SCA2 and 20 controls were from group A, were classified into group B for further study.
Clinical Scales and Neuroimaging Assessments
Three rating scales—SARA, the International Cooperative Ataxia Rating Scale (ICARS),22 and the Inventory of Non-Ataxia Symptoms (INAS)23—were available in the same 47 individuals to assess disease severity. SARA score ranges from 0 to 40, with 0 indicating no ataxia and 40 indicating the most severe degree of ataxia. The 100-point ICARS comprises 19 items, with 0 suggesting the absence of ataxia and 100 indicating the most severe degree of ataxia. INAS count ranges from 0 to 16 to evaluate nonataxia signs. The rating scale assessments were performed by 2 experienced neurologists who were blinded to the NfL levels of participants within 7 days after sample collection.
T1-weighted MRI scans were performed on a subgroup of 34 patients with manifest SCA2 (Signa HDx, General Electric Healthcare, Chicago, IL) within 7 days after sample collection, with the following parameters: repetition time 1750 milliseconds, echo time 14 milliseconds, field of view 24 × 24 cm, inversion time 720 milliseconds, and slice thickness 6 mm. All scans passed visual quality control check for artifacts before processing. Cerebellum, brainstem, and total intracranial regions of each scan were segmented by manual identification to ensure accuracy and calculated by ITK-SNAP software (version 3.8) to obtain volumes.24 The analyses of MRI data were carried out by investigators unaware of disease status. The cerebellum and brainstem volumes were expressed as percentages of total intracranial volumes.
Serum and CSF NfL Quantification
For each participant, peripheral blood was drawn by venipuncture from median cubital vein under fasting conditions lasting ≈10 hours and collected in BD Vacutainer tubes (Plymouth, UK). Blood samples were rested for 30 to 60 minutes at room temperature before centrifugation at 2,000g for 10 minutes. Then the supernatant was divided into aliquots and stored in Protein LoBind Tubes (Eppendorf AG, Hamburg, Germany) at −80°C immediately. These serum collection procedures were completed within 2 hours. CSF samples were collected by lumbar puncture at vertebral body L3-5 with atraumatic needles. The first 1 to 2 mL CSF was discarded, and the next 12 to 15 mL was collected and immediately placed at 4°C. CSF was centrifuged at 400g for 10 minutes to remove cells within a maximum interval of 10 minutes after collection, divided into aliquots, and immediately stored in Protein LoBind Tubes at −80°C. The serum and CSF samples were collected according to the recommended guideline25 and were sent to analysis without any thaw-freeze cycle. The NfL was quantified by ultrasensitive single-molecule array (Simoa) technique on the Simoa HD-X Analyzer (Quanterix, Billerica, MA) at GBIO (Hangzhou, China) with the Simoa NF-light Advantage Kit (product No. 103186). Serum and CSF samples were diluted at ratios of 1:4 and 1:100, respectively. One-third of the samples were detected in duplicates, and the intra-assay and interassay variabilities were <10%. All NfL values were within the linear ranges of the assay. Operators were blinded to participants' disease status.
Statistical Analysis
Demographic and clinical data were described as mean (SD) if normally distributed or median (interquartile range) if nonnormally distributed unless otherwise specified. Shapiro-Wilk test was used to test the normality. NfL levels in serum and CSF were nonnormally distributed because of biologically higher values. Natural log-transformation produced acceptable normal distribution for both serum and CSF NfL and was conducted for subsequent analyses if appropriate. Potentially confounding variables (age, sex, normal and expanded CAG repeat length, and disease duration) were analyzed preliminarily, and those with significant effect were included as covariates for subsequent analyses. To assess intergroup differences of demographics and clinical characteristics, unpaired 2-sample t test or 1-way analysis of variance was used on the premise of normality and homogeneity; otherwise, the nonparametric Mann-Whitney U test was used. Intergroup NfL level comparisons were conducted with multiple linear regressions including age and expanded CAG repeat length as covariates.
Generalized estimating equations (GEEs) were used to examine influence of familiarity on NfL levels and to repeatedly compare intergroup NfL levels while controlling for clustering of individuals with SCA2 within a family. Repeated measures on individuals within a family were included with an independent working correlation matrix. Correlation analyses were performed with the Pearson correlation and partial correlation for age adjustment. Spearman rank correlation was used for variables if appropriate. Residuals from the regression of NfL level or rating scale scores or affected brain volumes on age represented the component of those dependent variables not explained by age. Age-adjusted values were obtained by adding residuals to the mean NfL level, rating scale scores, or affected brain volumes.
To evaluate the diagnostic power of NfL, we used receiver operating characteristic curves and compared area under the curve (AUC) as suggested by a previous study.26 Analyses were performed with SPSS (IBM, Version 22; Armonk, NY). Significant level was defined as p < 0.05. After a Bonferroni correction, 0.05 is divided by the number of performed statistical tests, and a value of p < 0.017 was considered statistically significant.
Data Availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
Preliminary Analyses of Potentially Confounding Variables for NfL
The demographics and clinical characteristics of groups A and B are summarized in Table 1. Primary data are provided in detail in eTables 1 and 2, links.lww.com/WNL/B616. We assessed potential confounding variables for NfL levels in CSF and serum (Figure 1). In controls, NfL levels in both CSF and serum were associated with age (r = 0.607, p = 0.005; r = 0.620, p < 0.0001). No evidence for effect of sex on NfL level was observed in CSF and serum (p = 0.650, p = 0.698). In individuals with SCA2, NfL levels in serum were moderately associated with expanded CAG repeat length (r = 0.446, p = 0.0004), while those in CSF were not (p = 0.408). Age, sex, disease duration, and normal CAG repeat length were not associated with NfL levels in either CSF or serum (eTable 3). Because the 59 individuals with SCA2 were from 46 families, a GEE model was conducted to explore how much genetic and environmental backgrounds account for the variation of NfL level. It seemed that no obvious differences were found in the NfL levels between an individual with SCA2 and either affected parent or sibling (p = 0.053, p = 0.27). In group A, there was no difference in age between the controls and patients with manifest SCA2 (p = 0.13). In group B, the controls were recruited to match the mean age of patients with manifest SCA2. The preSCA2 individuals were younger than the controls and patients with manifest SCA2 (p = 0.01 and p = 0.01, respectively), a consequence of the condition that individuals with preSCA2 were too young to have developed ataxia. Thus, age adjustment was performed in all analyses to minimize the effect of age. Because expanded CAG repeat is the primary driver and the known best predictor of disease progression,1 we also included expanded CAG repeat length as a covariate in the intergroup comparison of NfL levels to explore the independent discriminatory ability of NfL.
Table 1.
Demographic, Clinical Characteristics, and NfL Levels of Participants

Figure 1. Assessments for Potential Confounding Variables for NfL.

(A and C) Correlations between age and (A) CSF neurofilament light (NfL) and (C) serum NfL. (B and D) Correlations between expanded CAG repeat length and (B) CSF NfL and (D) serum NfL. Correlations are represented by r and p values generated from Spearman rank correlation. The r and p values were generated from either controls only (gray) or individuals with spinocerebellar ataxia type 2 (SCA2) (colored) only. Only individuals with SCA2 were included in the association between expanded CAG repeat length and NfL because controls do not carry expanded CAG repeat.
High Consistency Was Identified Between Serum and CSF NfL of Patients With SCA2
To assess the association of NfL between serum and CSF, group A with 23 patients with manifest SCA2 and 20 controls was studied first. The median levels of NfL in CSF and matched serum were 6.07 and 4.05 times higher, respectively, in patients with manifest SCA2 than in controls (CSF: 2,187.86 pg/mL vs 360.50 pg/mL; serum: 27.61 pg/mL vs 6.81 pg/mL), and the difference remained significant after age adjustment (both p < 0.0001, Figure 2, A–D). In accord with CNS origin, the median NfL level was 59.85 times higher in CSF than in serum (1,113.72 pg/mL vs 18.61 pg/mL). The ratio further differed between patients with manifest SCA2 and controls (median 83.21 vs 52.29, p = 0.004). Moreover, there was an association between CSF and serum NfL levels (r = 0.868, p < 0.0001, Figure 2E), indicating a very high degree of consistency. Furthermore, we compared the discriminatory power of CSF and serum NfL using receiver operating characteristic curves and the AUC (Figure 2F). For distinguishing between patients with manifest SCA2 and controls, CSF NfL had essentially perfect accuracy (AUC, 1). Matched serum NfL also exhibited excellent accuracy (AUC, 0.987 [95% confidence interval [CI] 0.962–1).
Figure 2. NfL Levels of Manifest SCA2 and Controls in Group A and Receiver Operating Characteristic Curves for Discrimination.
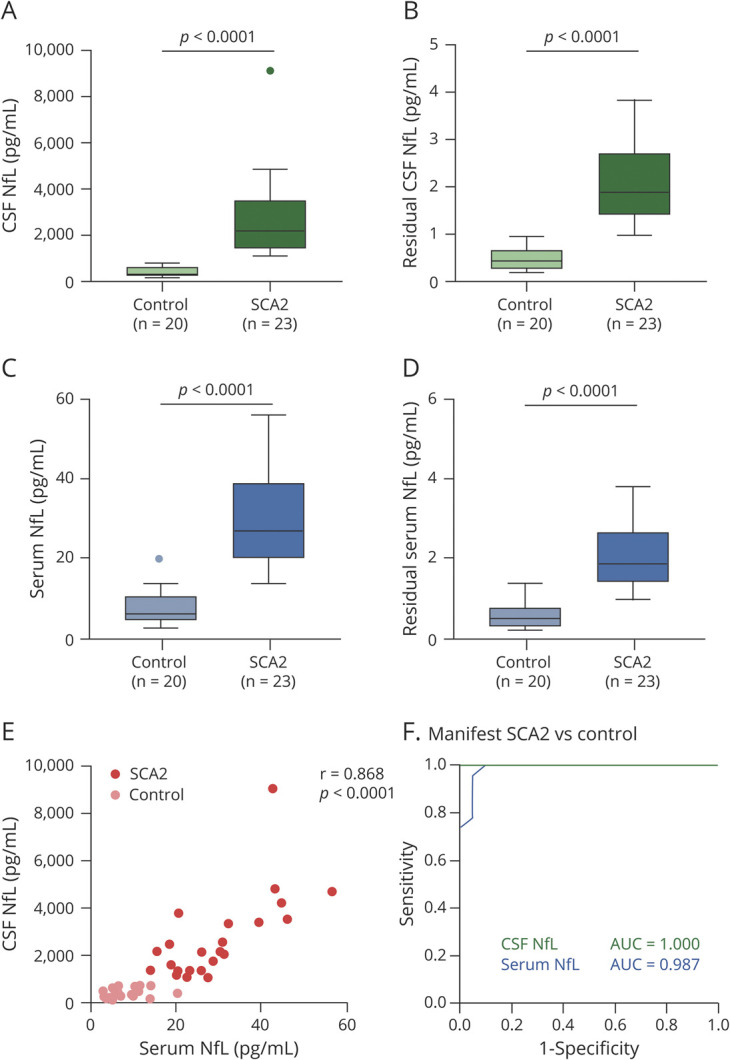
(A and C) Neurofilament light (NfL) levels in CSF and matched serum were significantly higher in individuals with manifest spinocerebellar ataxia type 2 (SCA2) than in controls. NfL levels are shown by boxplots with Tukey whiskers. (B and D) Residual NfL levels in CSF and matched serum were significantly higher in those with manifest SCA2 than in controls. Residuals were generated from the regression of CSF NfL or serum NfL on age representing the component of CSF NfL or serum NfL not explained by age. Residual NfL levels are shown by boxplots with Tukey whiskers. (E) Very high degree of consistency was identified between CSF and serum NfL levels. (F) Area under a receiver operating characteristic curve (AUC) gives a measure of the discriminatory ability of a test; 0.8 indicates an 80% probability of the test giving correct answer, and 1 indicates a 100% probability. CSF NfL had essentially perfect accuracy to distinguish between those with manifest SCA2 and controls. In addition, serum NfL exhibited excellent accuracy. NfL values were natural log-transformed for statistical analyses and are displayed geometrically.
Serum NfL Was Elevated in Individuals With SCA2
In a larger serum group (group B) consisting of 59 individuals with SCA2 and 92 controls, the level of serum NfL was higher in individuals with SCA2 compared to controls after age adjustment using a multiple linear regression model (p < 0.0001; median 23.36 pg/mL vs 6.79 pg/mL). As shown in Figure 3A, level of serum NfL was remarkably elevated in patients with manifest SCA2 compared with those with preSCA2 (p < 0.0001, Bonferroni-corrected threshold 0.017, median 26.07 pg/mL vs 15.56 pg/mL). The significance existed after Bonferroni correction. The level of serum NfL was also higher in those with preSCA2 than in controls (p < 0.0001, Bonferroni-corrected threshold 0.017, median 15.56 pg/mL vs 6.79 pg/mL), surviving rigorous Bonferroni correction. The difference remained significant after age adjustment (Figure 3B). After adjustment for age and expanded CAG repeat length, the manifest SCA2 subgroup had a higher level of serum NfL compared to the preSCA2 subgroup (p = 0.007). Likewise, a GEE model adjusted for familiarity and age revealed that NfL levels between patients with manifest SCA2 and those with preSCA2, between patients with manifest SCA2 and controls, and between those with preSCA2 and controls were different (p < 0.0001, p < 0.0001, p < 0.0001).
Figure 3. Serum NfL Levels of Controls and Individuals With SCA2 in Group B and Receiver Operating Characteristic Curves for Discrimination.

(A and B) Serum neurofilament light (NfL) and residual serum NfL were higher in those with manifest spinocerebellar ataxia type 2 (SCA2) than in controls and individuals with preclinical SCA2 (preSCA2) and higher in those with preSCA2 than in controls. Residuals were generated from the regression of serum NfL on age representing the component of serum NfL not explained by age. NfL levels and residual NfL levels are shown by boxplots with Tukey whiskers. NfL values were natural log-transformed for statistical analyses and are displayed geometrically. The p values were generated from multiple linear regressions, surviving Bonferroni correction. (C) Serum NfL had high accuracy to distinguish among patients with manifest SCA2, those with preSCA2, and controls and between individuals with SCA2 and controls.
For distinguishing between controls and individuals with SCA2, serum NfL had high accuracy (AUC, 0.945 [95% CI, 0.906–0.984], Figure 3C). It also exhibited great accuracy to discriminate between patients with manifest SCA2 and controls [AUC, 0.973 (95% CI, 0.947–1]. Serum NfL showed moderate accuracy to distinguish patients with manifest SCA2 from those with preSCA2 with an AUC of 0.847 (95% CI, 0.724–0.970) and those with preSCA2 from controls with an AUC of 0.811 (95% CI, 0.660–0.962). In addition, a level of 14.5 pg/mL as the cutoff value displayed good sensitivity and specificity for SCA2 detection (88% and 95%, respectively).
Serum NfL Was Associated With Disease Severity
In the 47 individuals with SCA2 with rating scales (8 individuals at preclinical stage, 19 individuals at stage 1, and 20 individuals at stage 2), levels of serum NfL were associated with SARA score (r = 0.412, p = 0.004), ICARS score (r = 0.368, p = 0.011), and INAS score (r = 0.360, p = 0.013). The correlations remained significant after adjustment for age (SARA score: r = 0.425, p = 0.003, Figure 4A; ICARS score: r = 0.383, p = 0.009, Figure 4B; INAS score: r = 0.390, p = 0.007, Figure 4C). In the subgroup of 34 patients with manifest SCA2 with a brain MRI scan (16 individuals at stage 1, 18 individuals at stage 2), we observed negative correlation between level of serum NfL and cerebellum volume (r = −0.555, p = 0.001), expressed as percentage of intracranial volume. The correlation survived adjustment for age (r = −0.393, p = 0.024, Figure 4D). No significant correlation was observed between level of serum NfL and brainstem volume. Therefore, higher level of serum NfL was associated with more severe disease condition and smaller cerebellar volume. A post hoc analysis showed that a sample size of 34 would achieve 86% power to detect a correlation (r = 0.45) between levels of NfL in serum and cerebellum volumes using a 1-sided hypothesis with a significance level of 5%.
Figure 4. Association Between NfL Levels and Disease Severity in Individuals With SCA2.

Elevated serum neurofilament light (NfL) levels were significantly associated with worse Scale for the Assessment and Rating of Ataxia (SARA) scores (A), worse International Cooperative Ataxia Rating Scale (ICARS) scores (B), worse Inventory of Non-Ataxia Signs (INAS) scores (C), and smaller cerebellum volumes (D). The patients with manifest spinocerebellar ataxia type 2 (SCA2) were classified into 2 subgroups based on median SARA scores and clinical features: stage 1 (3 ≤ SARA score <11) and stage 2 (SARA score ≥11). All volumetric measures were calculated as percentages of total intracranial volumes. Partial p and r values were obtained after adjustment for age.
Higher Serum NfL Levels Indicated Proximity to the Estimated Age at Onset in Preclinical SCA2
For 10 individuals at the preclinical stage, nystagmus was detected in 4 individuals, reduced reflexes were seen in 2, abnormal finger-to-nose test was noted in 1, abnormal heel-to-knee test was observed in 1, and abnormal tandem gait was noted in 1, indicating the presence of subclinical dysfunction. Therefore, we further explored the NfL levels in preSCA2. We calculated the time to the estimated age at ataxia onset according to expanded CAG repeat length and age at the time of evaluation, as previously established.27 Levels of serum NfL in preSCA2 were higher close to the predicted age at ataxia onset, as revealed by a linear regression between level of NfL and time from expected age at onset (R2 = 0.59, p = 0.01, Figure 5A). To compare preSCA2 with controls at the same age, measured NfL levels were expressed as NfL z scores, which were calculated as the difference between the measured NfL level and the NfL level estimated for controls at the same age, and then were standardized relative to the NfL distribution in controls (Figure 5B). The NfL z score were higher close to expected age at onset in preSCA2 (R2 = 0.43, p = 0.04, Figure 5C), not overlapping the 95% CI of controls (z score > 1.96) already 4.8 years before the estimated age at onset. This meant that the NfL levels in preSCA2 were elevated significantly 4.8 years before ataxia onset compared with controls. Residual plots for the linear regression of serum NfL and its z score exhibited a random scatter (Figure 5D).
Figure 5. Association Between Serum NfL and Time From Expected Onset in preSCA2.

(A) Serum neurofilament light (NfL) levels were significantly higher close to the predicted age at ataxia onset in preclinical spinocerebellar ataxia type 2 (preSCA2), as revealed by a linear regression between NfL levels and time from expected onset. (B) To compare individuals with preSCA2 and controls at the same age, measured NfL levels were expressed as NfL z scores. Difference between the measured NfL level (green dot) and the NfL level estimated for controls at the same age (solid gray line) is visualized by the length of vertical line. Standardization of this difference relative to the NfL distribution in controls yields the individual NfL z score. (C) The NfL z score was significantly higher close to the expected age at onset in preSCA2, not overlapping the 95% confidence interval of controls (i.e., z score > 1.96, dashed black line) already 4.8 years before the estimated age at onset. (D) Residual plot for the linear regression of serum NfL z score exhibits a random scatter.
Discussion
This study identified that serum NfL could be a great alternative to CSF NfL in SCA2, although the origin of NfL is the CNS. Serum NfL was significantly higher not only at the manifest but also at the preclinical stage of SCA2 compared to the controls. Moreover, NfL level was higher in preclinical individuals approaching to the onset of ataxia. The levels of serum NfL were correlated with disease severity in terms of both ataxia symptoms and nonataxia signs. These findings suggest that serum NfL is a potential biomarker of disease onset and severity in SCA2.
Neurofilaments are particularly abundant in axons with constant low-level release from axons in an age-dependent manner and are essential for structural stability and radial growth of axons, thereby achieving an optimal nerve conduction velocity. NfL is the most abundant and soluble neurofilament subunit and hence the most reliably measurable one in biofluids. The abnormal elevated levels in either CSF or serum are acknowledged as markers of axonal injury, axonal loss, and neuronal death.12 With regard to the neuropathology of SCA2, the abnormally elongated polyglutamine tract encoded by a CAG expansion in the ATXN2 gene is believed to drive pathogenesis.1 The role of accumulation and aggregation of polyglutamine proteins is yet to be fully understood. Pathologic changes usually occur in cerebellum, brainstem, basal ganglia, and peripheral nerves, eventually leading to neuronal dysfunction, loss, and death throughout the affected brain regions in patients with SCA2.2 Therefore, the loss of cerebellum volumes underlying the neuropathologic changes in SCA2 was associated with the elevation of NfL levels resulting from axonal loss. The released NfL level in CSF was 60-fold higher than that in serum in this study, roughly in accordance with the previous report.28 As a consequence, NfL changes in CSF might be more responsive and sensitive in identifying a neurodegenerative progression. Blood NfL measurement, however, would be more feasible and ideal due to its easier accessibility and lower invasiveness. Furthermore, our results demonstrated that the discriminatory power of NfL measurement in serum was slightly inferior to that in CSF but sufficiently excellent, laying a theoretical foundation for the application of serum.
In this study, the elevation of NfL level was found in the preclinical stage of the disease and preceded the projected conversion to manifest stage by 4.8 years in SCA2. These findings might be consistent with the presence of subclinical dysfunction and degeneration of the cerebellum and other neural structures before the onset of ataxia, of which brainstem, basal ganglia, and peripheral nerves are notable,29-31 indicating NfL measurement as a biomarker to predict the time to onset in preclinical individuals. In addition, in a longitudinal study on individuals with SCA2 at risk,31 the occurrence of cramps and sensory symptoms was most frequent in 5 and 4 years, respectively, before ataxia onset. Mean onset time between cramps and ataxia was 6.5 years, and mean time from sensory symptoms onset to ataxia onset was 4.27 years, which were close to 4.8 years in this study. In other neurodegenerative diseases such as HD and spinocerebellar ataxia type 3, NfL was also found to be elevated in presymptomatic individuals.32,33 In longitudinal studies on familial Alzheimer disease (AD), NfL could discriminate individuals with preSCA2 from controls as early as 1 or 2 decades before estimated age at onset.34,35 Those studies suggested that NfL measurement had the potential ability to capture the onset of axonal degeneration and to monitor the subclinical progression in the preclinical stage, aiding in stratifying individuals with preSCA2 and determining the optimal opportunity for therapeutic intervention. Longitudinal investigations on NfL measurement involving preSCA2 with larger samples and multiple populations are needed to confirm our results.
A recently prospective study enrolling 13 patients with SCA2 found that NfL levels were higher in patients and could predict disease progression.36 Those findings were consistent with our results in terms of diagnostic value of NfL, and further highlighted its importance as a prognostic marker. That work had definite advantages for its 2-year prospective analysis and relatively broad enrollment of several SCA genotypes. Our study had unique advantages as follows: (1) Because 59 individuals with SCA2 and 92 controls were enrolled, our study provided a more powerful evidence. (2) We included a broader range of individuals with SCA2, including individuals with preSCA2, and found that NfL was higher close to the estimated age at onset in preSCA2. (3) Because the origin of NfL is the CNS, we analyzed NfL levels in CSF and identified that serum NfL could be a great alternative to CSF NfL in SCA2. Consequently, our findings extend the knowledge of NfL in SCA2 and provide independent insights into the role of NfL as a potential biomarker of capturing onset of ataxia and assessing disease severity.
Because the diagnostic value of elevated NfL level has been found in a variety of neurodegenerative diseases (summarized in eTable 4, links.lww.com/WNL/B616), NfL is a sensitive but unspecific biomarker of axonal damage. However, NfL seems to function in the differential diagnosis between Parkinson disease (PD) and atypical parkinsonian disorders.37 In addition, the elevation of NfL level was reported to be a discriminative biomarker between frontotemporal dementia (FTD) and primary psychiatric disorders.38 As a result, the potential diagnostic value of NfL does not consist of the ability to distinguish between neurologic diseases with similar degree of axonal loss but rather with a different degree of axon loss and disease severity. In addition to diagnostic value, several longitudinal studies have revealed that NfL might have important prognostic implications. In HD and PD, baseline level of NfL could predict clinical progression.16,39 Furthermore, the value of NfL as a dynamic marker of clinical progression was highlighted in a series of neurodegenerative diseases, including FTD, AD, and PD.40-42 That was not supported, however, in amyotrophic lateral sclerosis and SCAs because NfL maintained a relatively constant expression during follow-up.36,43 The controversy might be relevant to different affected tissues releasing NfL underlying different neuropathology and the involvement of clearing mechanisms.44 Moreover, the dynamics of NfL might be more sensitive to early stage of neurodegeneration and more suitable to capture disease conversion, as indicated by longitudinal studies of AD and genetic FTD.34,40 Despite the limitation of nonspecificity, NfL may still be used in many potential contexts such as a tool as an outcome measure and for tracking disease progression and treatment response. Therefore, studies on the association between longitudinal changes in NfL and clinical assessments, especially radiologic measures, are encouraged in SCA2.
As a quantitative measure of ongoing axonal injury, NfL level is unable to help locate specific site of damage. On one hand, the lack of anatomic characterization of NfL suggests that its measurement in CSF and blood cannot replace MRI in the disease evaluation. On the other hand, NfL measurement indeed may provide more indications on the degree of ongoing axonal damage in normal-appearing MRI, especially in individuals with preclinical disease. Therefore, NfL measurement cannot be used alone as a substitute for MRI assessment but could be used as a complementary tool for detecting and monitoring axonal damage.
Our results revealed that NfL levels were elevated in individuals with SCA2 in combined biofluids, including CSF and serum. In individuals with preclinical disease, NfL levels were higher close to the estimated age at onset. In individuals with SCA2, NfL levels were correlated with disease intensity, reflected by rating scales and neuroimaging changes. Therefore, we identified NfL measurement as a potential biomarker in SCA2. The value of NfL levels might lie in their potential to capture the onset of ataxia, to monitor disease severity and progression, and to further aid in stratification and therapeutic response for upcoming treatment trials. Nevertheless, our study had several limitations. First, the sample size was relatively small because of this rare disease. Second, longitudinal assessment of NfL levels, especially involving more preclinical disease, is warranted to confirm our cross-sectional findings. Third, MRI examinations of individuals with preclinical disease were lacking due to the participants' personal willingness. In addition, considering that NfL levels are influenced by age, it is crucial to establish an age-dependent reference range in a large and multicenter cohort of healthy individuals before the application of NfL.
Acknowledgment
The authors gratefully acknowledge all participants for their help and willingness to participate this study.
Glossary
- AD
Alzheimer disease
- AUC
area under the curve
- CI
confidence interval
- FTD
frontotemporal dementia
- GEE
generalized estimating equation
- HD
Huntington disease
- ICARS
International Cooperative Ataxia Rating Scale
- INAS
Inventory of Non-Ataxia Symptoms
- NfL
neurofilament light
- PD
Parkinson disease
- preSCA2
preclinical SCA2
- SARA
Scale for the Assessment and Rating of Ataxia
- SCA2
spinocerebellar ataxia type 2
- Simoa
single-molecule array
Appendix. Authors

Footnotes
Class of Evidence: NPub.org/coe
Study Funding
This work was supported by the National Natural Science Foundation of China to Zhi-Ying Wu (82071260) and the research foundation for distinguished scholar of Zhejiang University to Zhi-Ying Wu (188020–193810101/089).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Sanpei K, Takano H, Igarashi S, et al. . Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14(3):277-284. [DOI] [PubMed] [Google Scholar]
- 2.Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5(1):24. [DOI] [PubMed] [Google Scholar]
- 3.Scoles DR, Meera P, Schneider MD, et al. . Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544(7650):362-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai YA, Liu RS, Lirng JF, et al. . Treatment of spinocerebellar ataxia with mesenchymal stem cells: a phase I/IIa clinical study. Cell Transpl. 2017;26(3):503-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romano S, Coarelli G, Marcotulli C, et al. . Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(10):985-991. [DOI] [PubMed] [Google Scholar]
- 6.Klockgether T, Skalej M, Wedekind D, et al. . Autosomal dominant cerebellar ataxia type I. MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain. 1998;121(pt 9):1687-1693. [DOI] [PubMed] [Google Scholar]
- 7.Mandelli ML, De Simone T, Minati L, et al. . Diffusion tensor imaging of spinocerebellar ataxias types 1 and 2. AJNR Am J Neuroradiol. 2007;28(10):1996-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adanyeguh IM, Henry PG, Nguyen TM, et al. . In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3, and 7. Mov Disord. 2015;30(5):662-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boesch SM, Donnemiller E, Muller J, et al. . Abnormalities of dopaminergic neurotransmission in SCA2: a combined 123I-betaCIT and 123I-IBZM SPECT study. Mov Disord. 2004;19(11):1320-1325. [DOI] [PubMed] [Google Scholar]
- 10.van de Warrenburg BP, Notermans NC, Schelhaas HJ, et al. . Peripheral nerve involvement in spinocerebellar ataxias. Arch Neurol. 2004;61(2):257-261. [DOI] [PubMed] [Google Scholar]
- 11.Mascalchi M, Toschi N, Giannelli M, et al. . Progression of microstructural damage in spinocerebellar ataxia type 2: a longitudinal DTI study. AJNR Am J Neuroradiol. 2015;36(6):1096-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870-881. [DOI] [PubMed] [Google Scholar]
- 13.Hansson O, Janelidze S, Hall S, et al. . Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88(10):930-937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrne LM, Rodrigues FB, Blennow K, et al. . Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol. 2017;16(8):601-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weydt P, Oeckl P, Huss A, et al. . Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol. 2016;79(1):152-158. [DOI] [PubMed] [Google Scholar]
- 16.Olsson B, Portelius E, Cullen NC, et al. . Association of cerebrospinal fluid neurofilament light protein levels with cognition in patients with dementia, motor neuron disease, and movement disorders. JAMA Neurol. 2019;76(3):318-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li QF, Dong Y, Yang L, et al. . Neurofilament light chain is a promising serum biomarker in spinocerebellar ataxia type 3. Mol Neurodegener. 2019;14(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilke C, Bender F, Hayer SN, et al. . Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat-expansion spinocerebellar ataxias: a pilot study. J Neurol. 2018;265(7):1618-1624. [DOI] [PubMed] [Google Scholar]
- 19.Yang L, Dong Y, Ma Y, Ni W, Wu ZY. Genetic profile and clinical characteristics of Chinese patients with spinocerebellar ataxia type 2: a multicenter experience over ten years. Eur J Neurol. 2021;28(3):955-964. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. . Scale for the Assessment and Rating of Ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717-1720. [DOI] [PubMed] [Google Scholar]
- 21.Maas RP, van Gaalen J, Klockgether T, van de Warrenburg BP. The preclinical stage of spinocerebellar ataxias. Neurology. 2015;85(1):96-103. [DOI] [PubMed] [Google Scholar]
- 22.Trouillas P, Takayanagi T, Hallett M, et al. . International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. J Neurol Sci. 1997;145(2):205-211. [DOI] [PubMed] [Google Scholar]
- 23.Jacobi H, Rakowicz M, Rola R, et al. . Inventory of Non-Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum. 2013;12(3):418-428. [DOI] [PubMed] [Google Scholar]
- 24.Yushkevich PA, Piven J, Hazlett HC, et al. . User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31(3):1116-1128. [DOI] [PubMed] [Google Scholar]
- 25.Teunissen CE, Petzold A, Bennett JL, et al. . A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology. 2009;73(22):1914-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837-845. [PubMed] [Google Scholar]
- 27.Tezenas du Montcel S, Durr A, Rakowicz M, et al. . Prediction of the age at onset in spinocerebellar ataxia type 1, 2, 3 and 6. J Med Genet. 2014;51(7):479-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Disanto G, Barro C, Benkert P, et al. . Serum neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol. 2017;81(6):857-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobi H, Reetz K, du Montcel ST. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol. 2013;12(7):650-658. [DOI] [PubMed] [Google Scholar]
- 30.Kim JM, Hong S, Kim GP, et al. . Importance of low-range CAG expansion and CAA interruption in SCA2 parkinsonism. Arch Neurol-Chicago. 2007;64(10):1510-1518. [DOI] [PubMed] [Google Scholar]
- 31.Velázquez-Pérez L, Rodríguez-Labrada R, Canales-Ochoa N, et al. . Progression of early features of spinocerebellar ataxia type 2 in individuals at risk: a longitudinal study. Lancet Neurol. 2014;13(5):482-489. [DOI] [PubMed] [Google Scholar]
- 32.Scahill RI, Zeun P, Osborne-Crowley K, et al. . Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington's Disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19(6):502-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilke C, Haas E, Reetz K, et al. . Neurofilaments in spinocerebellar ataxia type 3: blood biomarkers at the preataxic and ataxic stage in humans and mice. EMBO Mol Med. 2020;12(7):e11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Preische O, Schultz SA, Apel A, et al. . Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25(2):277-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quiroz YT, Zetterberg H, Reiman EM, et al. . Plasma neurofilament light chain in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross-sectional and longitudinal cohort study. Lancet Neurol. 2020;19(6):513-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coarelli G, Darios F, Petit E, et al. . Plasma neurofilament light chain predicts cerebellar atrophy and clinical progression in spinocerebellar ataxia. Neurobiol Dis. 2021;153:105311. [DOI] [PubMed] [Google Scholar]
- 37.Marques TM, van Rumund A, Oeckl P, et al. . Serum NFL discriminates Parkinson disease from atypical parkinsonisms. Neurology. 2019;92(13):e1479-e1486. [DOI] [PubMed] [Google Scholar]
- 38.Katisko K, Cajanus A, Jääskeläinen O, et al. . Serum neurofilament light chain is a discriminative biomarker between frontotemporal lobar degeneration and primary psychiatric disorders. J Neurol. 2020;267(1):162-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodrigues FB, Byrne LM, Tortelli R, et al. . Mutant huntingtin and neurofilament light have distinct longitudinal dynamics in Huntington's disease. Sci Transl Med. 2020;12(574):eabc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Ende EL, Meeter LH, Poos JM, et al. . Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 2019;18(12):1103-1111. [DOI] [PubMed] [Google Scholar]
- 41.Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76(7):791-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mollenhauer B, Dakna M, Kruse N, et al. . Validation of serum neurofilament light chain as a biomarker of Parkinson's disease progression. Mov Disord. 2020;35(11):1999-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu CH, Macdonald-Wallis C, Gray E, et al. . Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84(22):2247-2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puentes F, Topping J, Kuhle J, et al. . Immune reactivity to neurofilament proteins in the clinical staging of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2014;85(3):274-278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.