

Abstract
Background
Molecular targeted therapy has been developed as an innovative treatment for metastatic cancer. Epidermal growth factor receptor (EGFR) mutation is one of the most important and frequent oncogenic drivers in non-small-cell lung cancer, and EGFR-tyrosine kinase inhibitors are indispensable drugs for mutation-positive patients. Currently, the acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) is a problem, the mechanism of which has not been elucidated. The histological transformation from original adenocarcinoma to small-cell carcinoma is rare; however, it has been detected in many cases after EGFR-TKI treatment. This study aimed to evaluate mutational status in two different histological types and further elucidate the molecular pathogenesis.
Methods
Three patients with EGFR-mutant lung cancer who underwent a histological transformation to small-cell carcinoma after growth factor receptor-TKI treatment were enrolled in this study. Two samples per patient were collected from histologically different lesions, and DNA samples were extracted from formalin-fixed, paraffin-embedded tumor tissues. The paired samples were subjected to next-generation sequencing of 160 cancer-related genes. Based on the sequencing results, the expression levels of related proteins were validated using reverse-transferase polymerase chain reaction and immunohistochemical staining.
Results
The following five variants were common among the three cases: MTOR, JAK1, NOTCH2, CSF1R, and MAP2K2. The former four variants were additive to small-cell carcinoma, and the last variant was lost. Both TP53 and Rb1 alterations were detected in adenocarcinoma. Notch2 expression was negative in small-cell carcinoma in both reverse-transcriptase polymerase chain reaction analysis and immunohistochemical staining. ASCL1 expression increased after histological transformation detected using both methods in one case, only these samples were evaluable.
Conclusions
Notch and ASCL1 signaling are the master regulators of neuroendocrine differentiation in small-cell lung carcinoma. Our results suggest that the Notch-ASCL1 axis may also play an essential role in the transformation of small-cell carcinoma under TP53 and RB1 inactivation.
Keywords: EGFR mutation, histological transformation, NOTCH, ASCL1, next-generation sequencing
Introduction
Lung cancer remains a leading cause of cancer-related deaths worldwide. Non-small-cell lung cancer (NSCLC) treatment has been one of the pioneers in solid carcinoma treatments in terms of personalized “precision medicine” using molecular targeted medicine for cancer-associated gene mutations. The discovery of epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) for the treatment of advanced NSCLC with activating EGFR mutations led to dramatic tumor shrinkage and has succeeded in prolonging survival and improving quality of life, greatly benefiting patients with EGFR mutations (1-3).
Unfortunately, tumors appear to be resistant to EGFR-TKIs vie the regrowth of new distant metastases invariably approximately one year after the initial EGFR-TKI treatment. In the past decade, genetic and histological mechanisms of acquired resistance have been investigated using resistant tumor specimens. The most frequent mechanism is an additional T790M mutation in exon 20 of EGFR. T790M was detected in approximately half of the patients with resistance to 1st and 2nd generation EGFR-TKIs, such as gefitinib, erlotinib, and afatinib (4-8). Similar to other acquired resistance mechanisms, the histological transformation from adenocarcinoma to small-cell carcinoma has been observed as one of the mechanisms in approximately 3–14% of patients with resistance to EGFR-TKIs (4,6,8). In the past few years, several genetic mechanisms underlying this transformation have been reported. Niederst et al. showed that the inactivation of RB1 and the decreased expression of EGFR are common features in transformed small-cell carcinoma compared to original adenocarcinoma (9). Lee et al. demonstrated that adenocarcinoma clones with inactivation of both TP53 and RB1 branched out to the transformed small-cell carcinoma, and proposed that both inactivations have the potential to be a predictive marker for the transformation (10). Detecting the key changes that direct the transformation may lead to a deeper understanding and utilized as therapeutic targets.
The purpose of this study was to identify key genetic changes that define the histological transformation from adenocarcinoma to small-cell carcinoma. To address this, we compared the gene alteration profile in adenocarcinoma before EGFR-TKI treatment and in transformed small-cell carcinoma. We present the following article in accordance with the STROBE reporting checklist (available at https://dx.doi.org/10.21037/tlcr-21-536).
Methods
Patients
Patients who met the following criteria were enrolled in this study: (I) patients who were diagnosed with advanced NSCLC at initial diagnosis; (II) patients who had activating mutations on EGFR at diagnosis and received EGFR-TKI treatment; (III) patients who had histologically transformed small-cell carcinoma in regrown or new tumor lesions, which are resistant to EGFR-TKIs from 2013 to 2016. All pathological diagnoses were performed by pathological experts in Kanazawa University Hospital. Immunohistochemistry (IHC) staining for neuroendocrine markers, chromogranin A, CD56, and synaptophysine was performed to support the diagnosis of small-cell carcinoma. The following patient data were obtained from patient records: age at the diagnosis of metastatic cancer or disease recurrence, sex, smoking status, tumor biopsy approaches, EGFR mutation status, response to EGFR-TKIs, and EGFR-TKI treatment period. All patients provided written informed consent, and the study was approved by the Ethics Committee of Kanazawa University Hospital (approval number: 327). The study was conducted in accordance with the Declaration of Helsinki (revised in 2013).
Sample collection and DNA/RNA extraction
DNA and RNA were extracted from paired tumor samples that were collected for pathological diagnosis both at diagnosis and after transformation to small-cell carcinoma. The tumor biopsy approach is shown in Figure 1 and samples for DNA/RNA extraction were underlined. All samples were formalin-fixed paraffin-embedded (FFPE). First, FFPE tissues were micro-dissected. DNA and total RNA were extracted from tumor cells using the QIAamp® DNA FFPE Tissue Kit (QIAGEN), QIAamp® DNA Mini Kit (QIAGEN), RNeasy® Mini Kit (QIAGEN), and RNeasy® FFPE Kit (QIAGEN). DNA samples from nucleic cells in the blood were used as references for gene alteration profiles. Germline DNA samples were extracted from whole blood cells, obtained as pellets by centrifugation, using the QIAamp® DNA Mini Kit (QIAGEN). The concentration and quality of the DNA samples were measured using the Qubit® 2.0 Fluorometer (Life Technologies), dsDNA HS assay kits (Life Technologies), and QIAseq® DNA QuantiMIZE Array kit (QIAGEN).
Figure 1.

The clinical course. All three cases were histologically identified as transformed after EGFR-TKI treatment. HE, hematoxylin-eosin; DEL, deletion mutation in exon 19; EGFR, epidermal growth factor receptor; CBDCA, carboplatin; GEM, gemcitabine; PEM, pemetrexed; Bev, bevacizumab; PTX, paclitaxel; TBLB, transbronchial lung biopsy, DOC, docetaxel; Nivo, nivolumab.
Genotyping via next-generation sequencing (NGS)
Tumor genotyping via NGS were performed using the GeneRead™ DNAseq Targeted Panels V2 Human Comprehensive Cancer Panel (QIAGEN) covering 160 major oncogenes and suppressor genes, as listed in Table S1. Sequencing was performed on a Miseq™ (Illumina), and data were analyzed using QIAGEN’s GeneRead DNAseq Analysis Software (QIAGEN). Using this software, germline mutations were identified in the whole blood tissue and excluded from further analysis. The remaining somatic mutations were identified as meaningful mutations. The gene alterations detected from each sample were divided into three groups according to the QIAGEN GeneRead DNAseq Analysis Software (Qiagen) as follows: high confidence, medium confidence, and low confidence. Only gene alterations assessed with high confidence were used for further analysis.
The essential variant screening was performed for histological transformation to compare mutation profiles of adenocarcinoma with those of small-cell transformed carcinoma via an original script in “Terminal app”, a terminal emulator for the MacOS operating system, for respective three cases. We especially focused on the lost alterations in original adenocarcinoma and the additive alterations in transformed small-cell carcinoma in each case, and common alterations were extracted to combine the results of three cases. We then validated the expression of these lost and additive genes at protein and RNA levels. Analysis software also provided copy number variation (CNV) data. After comparing two paired results per case, the gene with more than two copies of difference was accepted as significant.
Gene expression analyses using real-time reverse-transcriptase polymerase chain reaction (RT-PCR)
cDNA was synthesized from RNA using PrimeScript™ RT Master Mix (Takara Bio). The primer pairs for the PCR of ASCL1, CSF1R, JAK1, MAP2K2, MTOR, NOTCH1, NOTCH2, and GAPDH were designed and purchased from Perfect Real-Time PCR Primer (Takara Bio. Shiga. Japan). The sequences of these primers are listed in Table S2. Gene expression was detected using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara Bio Inc.) using Applied Biosystems® StepOnePlus™ (Thermo Fisher). Figure S1 shows the real-time PCR curve for case 1. Expression levels were calculated according to the comparative Ct method under normalization using GAPDH as endogenous control, and relative values of tumor cells in transformed small-cell carcinoma to adenocarcinoma were calculated using ΔCt and analyzed.
Protein expressions using IHC staining
Tumor tissues were fixed in 4% formaldehyde and embedded in paraffin. Sections (5 mm) were deparaffinized and heat antigen-retrieved in citrate buffer (pH 6.4). Primary antibody staining was performed by incubating overnight at 4 °C, and endogenous peroxidase (HRP) activity was blocked by treating the sections with 3% hydrogen peroxide in methanol. Indirect IHC was performed with antispecies-specific biotinylated secondary antibodies, followed by avidin-horseradish peroxidase and developed using DAB color substrates (Nichirei Biosciences Inc., Tokyo, Japan). The sections were counterstained with haematoxylin. The following primary antibodies were used: Anti-MASH1/Achaete-scute homolog 1 (Abcam, ab211327, 1:100); anti-NOTCH1 (Abcam, ab52627, 1:100); anti-NOTCH2 (Abcam, ab8926, 1:100).
Statistical analysis
For variant calling and filtering for genotyping, the GATK Unified Genotyper program (GATKLite version 2.3-9) was used for calling variants on individual samples, and Strelka was used with default parameters to call somatic variants from matched tumor and normal samples. There was not any other statistical analysis in this study.
Results
Patient characteristics
We enrolled three patients who were histologically diagnosed with advanced EGFR-mutant NSCLC, and, after resistance to EGFR-TKIs, with small-cell carcinoma at Kanazawa University Hospital from 2013 to 2016. All patients were never-smoking females and diagnosed with adenocarcinomas harboring a deletion mutation in exon 19 of EGFR (DEL) at the initial diagnosis. The total duration of EGFR-TKI treatment until histological transformation in the three patients was 49, 42, and 32 months, respectively. The therapeutic and biopsy courses are shown in Figure 1.
In case 1, a woman who was 50 years old at initial diagnosis by surgical resection underwent re-operation at the tumor recurrence. Both resected samples were adenocarcinoma without small cell carcinoma component. She had acquired the resistance mutation T790M in exon 20 of EGFR after the EGFR-TKI re-challenge with the combination of erlotinib and paclitaxel, and then received osimertinib as the fourth course of treatment. After resistance to osimertinib, a metastatic pulmonary tumor that regrew regardless of shrinkage after osimertinib was collected via transbronchial biopsy. It was pathologically diagnosed as small-cell carcinoma showing poorly differentiated cells with a high nuclear-to-cytoplasmic ratio and positive synaptophysin and CD56 on IHC staining. The genetic test for EGFR in small-cell carcinoma revealed the same mutation status as before osimertinib treatment (DEL and T790M). Because of a sudden exacerbation and worsening in performance status, she was unable to undergo chemotherapy for small-cell carcinoma. After her death, tumor tissues from several lesions, including adenocarcinoma from the left upper lung lobe, the liver, and the left hilar lymph node, as well as small-cell carcinoma from the left lower lung lobe, perigastric lymph node, and para-aortic lymph node were obtained, owing to her family’s consent to the autopsy.
In case 2, a woman, who was 48 years old at the initial diagnosis, presented with several progressive tumor lesions after the initial EGFR-TKI treatment with afatinib. While adenocarcinoma was cytologically diagnosed in the cancerous ascites that newly occurred after afatinib treatment, tumor tissues from abdominal lymph nodes that had already acquired afatinib resistance were collected using endoscopic ultrasound-fine needle aspiration after several treatment regimens. The samples were pathologically diagnosed as small-cell carcinoma showing poorly differentiated cells with a high nuclear-to-cytoplasmic ratio, as well as positive synaptophysin, chromogranin A, and CD56 IHC staining (Figure 1). The EGFR gene test revealed DEL only, similar to initial diagnosis, both in adenocarcinoma tissues and cancerous ascites, and small-cell carcinoma in the abdominal lymph node.
In case 3, a woman, who was 47 years old at initial diagnosis, had received gefitinib followed by erlotinib after postoperative recurrence. Tumor tissues from mediastinal lymph nodes that had already regrown after gefitinib treatment were collected via transbronchial needle biopsy after combination therapy with carboplatin, pemetrexed, and bevacizumab, and were histologically diagnosed as small-cell carcinoma showing necrotized cells with a high nuclear-to-cytoplasmic ratio and positive synaptophysin and CD56 IHC staining (Figure 1). The genetic test revealed DEL alone, which was the same as the initial diagnosis.
Comparing genetic alteration profiles in paired samples of the original adenocarcinoma and the transformed small-cell carcinoma via NGS
To identify genetic changes related to the histopathological transformation from adenocarcinoma to small-cell carcinoma after EGFR-TKI treatment, we compared gene alteration profiles in adenocarcinoma tumor tissues at initial diagnosis and small-cell carcinoma tissues after resistance to EGFR-TKIs via NGS using the GeneRead™ DNAseq Targeted Panels V2 Human Comprehensive Cancer Panel (QIAGEN) covering 160 major oncogenes and suppressor genes as listed in Table S1. At the same time, CNVs were calculated.
In all three cases, tumor tissues pathologically diagnosed as mentioned above were used in the analyses. The original EGFR mutation, DEL, was also detected in the transformed small-cell carcinoma after resistance to EGFR-TKIs in all three 3 cases. The number of genetic alterations in adenocarcinoma at initial diagnosis and in small-cell carcinoma at the time of resistance were 247 and 202 for case 1, 181 and 1,217 for case 2, and 247 and 133 for case 3, respectively. The number of additive genetic alterations found in the transformed small-cell carcinoma but not in adenocarcinoma was 105, 1,164, and 88, and that of shared alterations in both adenocarcinoma and transformed small-cell carcinoma was 97, 53, and 45 for cases 1, 2, and 3, respectively. The number of gene alterations that were absent in transformed small-cell carcinoma but present in initially diagnosed adenocarcinoma was 150, 128, and 202, respectively.
When combining all three cases with completely matching changes in the nucleotides, five genes were identified. Mammalian target of rapamycin (MTOR), Janus kinase (JAK1), NOTCH2, and colony-stimulating factor 1 receptor (CSF1R) were additive alterations common in all three cases, and mitogen-activated protein kinase kinase 2 (MAP2K2) was a lost alteration in all the cases (Table 1). These five genes were selected as candidate key genes for the transformation from adenocarcinoma to small-cell carcinoma. Duplication alterations of MTOR, JAK1, NOTCH2, and MAP2K2 were located at the 3’ untranslated region (UTR), and the CSF1R alteration was a single-nucleotide polymorphism in the intron (Table 1).
Table 1. Additive and lost alterations common in three cases.
| Added/Lost | Gene name | Chr | Position GRCh37 | COSMIC ID | Reference | Variant | Variant type | Codon change | Amino acid change | SNP effect |
|---|---|---|---|---|---|---|---|---|---|---|
| Added | MTOR | Chr1 | 11166624 | – | C | <DUP> | CNV | – | – | UTR 3 PRIME |
| JAK1 | Chr1 | 65300216 | – | C | <DUP> | CNV | – | – | UTR 3 PRIME | |
| NOTCH2 | Chr1 | 120457873 | – | T | <DUP> | CNV | – | – | UTR 3 PRIME | |
| CSF1R | Chr5 | 149435535 | – | G | T | SNP | – | – | INTRON | |
| Lost | MAP2K2 | Chr19 | 4090533 | – | G | <DUP> | CNV | – | – | UTR 3 PRIME |
Chr, chromosome; CNV, copy number variation; DUP, duplicate; SNP, single-nucleotide polymorphism; UTR, untranslated region.
Furthermore, additive or lost of genetic alterations about PI3K and AKT were investigated in each case (Table S3). There was no aberration which had in common in 3 cases in completely matching nucleotides.
CNVs for kuppel-like factor 6 (KLF6), Harvey rat sarcoma viral oncogene homolog (HRAS), nuclear factor- kappa B1 A (NFKB1A), and G protein α subunit Gs (GNAS1) were significantly changed in transformed small-cell carcinoma compared to those in initially diagnosed adenocarcinoma (Table S4). In two cases (cases 1 and 3), CNV for all four genes was increased. In case 2, CNV for all four genes was decreased. The CNV for EGFR was approximately two in the initially diagnosed adenocarcinoma, and it did not change for the transformed small-cell carcinoma in any of the cases. Therefore, we estimated that there were no candidate genes obtained from CNV analyses in any of the three 3 cases.
TP53 alteration was detected in the paired samples of both adenocarcinoma and transformed small-cell carcinoma in all three 3 cases, and retinoblastoma 1 (RB1) alteration was detected in cases 1 and 2 (Table 2). Aberrations in TP53 were detected in all three cases as non-synonymous coding or frame-shift effects, which resulted in amino acid changes. In terms of CNVs of TP53 and RB1, there was no obvious change between the two histological tumors in any of the three cases, except that TP53 CNV was increased after transformation in case 2 (Table S4).
Table 2. Genetic aberrations in TP53 and RB1.
| Case | Gene name | Chr | Pos GRCh37 | COSMIC ID | Reference | Variant | Variant type | Transcript ID | Codon change | Amino acid change | SNP effect |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | RB1 | Chr13 | 49050852 | – | C | T | SNP | NM_000321.2 | c.2536C>T | p.Q846* | STOP_GAINED |
| TP53 | Chr17 | 7578442 | COSM3388214 | T | C | SNP | NM_000546.5 | c.488A>G | p.Y163C | NON_SYNONYMOUS_CODING | |
| 2 | RB1 | Chr13 | 48881353 | – | T | <DEL> | CNV | NM_000321.2 | – | – | SPLICE_SITE_DONOR |
| TP53 | Chr17 | 7572959 | – | TG | T | DEL | NM_000546.5 | – | p.-383 | FRAME_SHIFT | |
| 3 | TP53 | Chr17 | 7577153 | COSM216411 | C | A | SNP | NM_000546.5 | c.785G>T | p.G262V | NON_SYNONYMOUS_CODING |
Chr, chromosome; CNV, copy number variation; SNP, single-nucleotide polymorphism.
Gene expression analyses in tumor cells with transformed small-cell carcinoma compared with the original adenocarcinoma
We selected CSF1R, JAK1, MTOR, NOTCH1, MAP2K2, NOTCH2 and Achaete- scute homolog-1 (ASCL1) from the above-mentioned results using NGS and analyzed the gene expression using quantitative reverse transcriptase-polymerase chain reaction (RT-PCR). As described above, gene alterations in CSF1R, JAK1, MTOR, and NOTCH1 emerged in transformed small-cell carcinoma, and those in MAP2K2 disappeared after the transformation. In particular, we focused on NOTCH among these genes. ASCL1 and NOTCH1 were also added as candidates for transformation-related genes (11). Unfortunately, gene expression analysis using real-time RT-PCR was performed for case 1 only. A pair of tumor tissues with adenocarcinoma and small-cell carcinoma was obtained at autopsy. Since the tumor tissues obtained from the other two cases were only small tumor specimens collected for diagnosis, it was not possible to obtain a sufficient number of samples to perform additional analyses other than the genetic alteration profile detection.
The expression levels of ACSL1 and MTOR were higher in adenocarcinoma, and those of NOTCH1 and 2, CSF1R, and JAK1 were lower than those in transformed small-cell carcinoma (Figure 2).
Figure 2.

Gene expression analyzed using real-time reverse transcriptase-PCR. Y-axis represents the relative quantitation; expression levels in small-cell carcinoma compared with those in adenocarcinoma. The number in the lower column is natural logarithm of relative quantitation. ln means natural logarithm. ASCL1, Achaete-scute homolog 1; CSF1R, colony-stimulating factor 1 receptor; JAK, Janus kinase; MAP2K2, mitogen-activated protein kinase kinase 2; MTOR, mammalian target of rapamycin; ne, no examination.
NOTCH2 expression in transformed small-cell carcinoma compared to that in adenocarcinoma
We performed IHC staining to evaluate the protein expression associated with the transformation in case 1 (Figure 3). Tumor tissues representing the transformed small-cell carcinoma and adenocarcinoma in the pulmonary lesion at autopsy was used for protein expression analyses for neuroendocrine markers (chromogranin A, synaptophysin, and CD56), cytokeratin AE1/AE3, EGFR, MET, and Ki-67 (Figure 3A). Expression of synaptophysin and CD56 increased in the transformed small-cell carcinoma tissues. We confirmed that EGFR expression disappeared in the transformed small-cell carcinoma compared to that in the adenocarcinoma (9). Based on the gene mutation profile analyses and gene expression analysis as described above, Rb, NOTCH2, and ASCL1 expression was analyzed using the adenocarcinoma tissues in the pulmonary lesion and transformed small-cell carcinoma tissues in the pulmonary lesion at autopsy (Figure 3C). Rb expression was not observed in either adenocarcinoma or transformed small-cell carcinoma. NOTCH2 expression was negative in the transformed small-cell carcinoma, but positive in adenocarcinoma. ASCL1 expression was positive in the transformed small-cell carcinoma but negative in adenocarcinoma (Figure 3B).
Figure 3.

Autopsy findings in case 1. (A) Immunohistochemical staining at the border of coexistent metastasis in the left upper lung lobe, indicated as arrow in (C). Adenocarcinoma in the upper region is in contact with the small-cell carcinoma in the lower region. (B) Immunohistochemical staining of isolated histological lesions indicated as arrowheads in (C). (C) Schema of tumor distribution at case 1 autopsy. HE, hematoxylin-eosin; CK, cytokeratin; EGFR, epidermal growth factor receptor; MET, mesenchymal-to-epithelial transition; ASCL1, Achaete-scute homolog 1; Rb, retinoblastoma.
The differences in NOTCH/ASCL pathway between the original adenocarcinoma and transformed small-cell carcinoma
The results obtained from the three cases showed that first, in the genetic alteration profile analysis via NGS, NOTCH2 mutation was commonly detected in the transformed small-cell carcinoma, but not in the original adenocarcinoma. In the additional study on case 1 only, a decrease in NOTCH2 and an increase in ASCL expression were observed in transformed small-cell carcinoma using gene expression analysis via RT-PCR. In addition, IHC of transformed small-cell carcinoma tissues confirmed a decrease in NOTCH2 expression and an increase in ASCL expression. TP53 mutations were found in both the original adenocarcinoma and the transformed small-cell carcinoma in all cases. Rb1 expression was only examined in case 1, but was similarly reduced in both adenocarcinoma and transformed small-cell carcinoma (Figure 3B).
Discussion
Here, we analyzed additional gene alterations in transformed small-cell carcinoma after resistance to EGFR-TKI treatment, compared with the original adenocarcinoma. To begin with, it is fundamental problem whether the histological transformation is real or the actual initial diagnosis is just combined small cell carcinoma and adenocarcinoma. Because 2 out of 3 our cases were initially diagnosed by surgical resection, it must be suitable there were no small cell carcinoma component. Furthermore, FFPE samples were micro-dissected. Thus, purity of tumor samples must be extremely 100%. Among the additional gene alterations that were common in all cases, we focused on NOTCH mutations as a possible alteration representing the transformation to small-cell carcinoma from adenocarcinoma with EGFR mutations. Additionally, we found that the expression of ASCL, which is downstream of Notch signaling, was increased in tissues of transformed small-cell carcinoma compared to that in the original adenocarcinoma. Notably, this study showed for the first time that the NOTCH- and ASCL-dependent pathway was one of the key processes in the transformation using the actual tumor tissues from patients with the transformed small-cell carcinoma after resistance to EGFR-TKI treatment.
The transformation from adenocarcinoma to small-cell carcinoma has attracted attention as one of the outcomes of EGFR-TKI resistance. Meanwhile, the histological transformation from adenocarcinoma to small-cell carcinoma is rare in patients with NSCLC without EGFR mutations. In a previous study to search for molecular mechanisms of the transformation from adenocarcinoma to small-cell carcinoma in patients with NSCLC with EGFR mutations, Niederst et al. reported that the inactivation of RB1 increases the expression of neuroendocrine markers, and decreases EGFR expression, which is commonly detected in the transformed tissues compared with the original adenocarcinoma (9). Furthermore, a study by Lee et al., describing 21 patients with the transformation, indicated that inactivation of both RB1 and TP53 is required for the transformation from adenocarcinoma to small-cell carcinoma after resistance to EGFR-TKI treatment, and the molecular features already existed just before branching, suggesting that both inactivations in patients treated with EGFR-TKIs may serve as predictive markers for the transformation (10). The inactivation of both TP53 and RB1 is well known in almost all SCLCs (12,13). In integrative genome analyses using NGS in tumor cells from SCLC, inactivating gene alterations (mutations, loss, or loss of heterozygosity) of both TP53 and RB1 were detected in all SCLC tissues, and the mutations were described in five groups: receptor-tyrosine kinase signaling (e.g., FGFR1 amplification in 6%), PI3K and p53 pathway including TP53 mutation and loss, cell cycle control including RB1 mutation or loss, histone modification (e.g., CREBBP mutation in 18%), and regulation of the actin cytoskeleton (e.g., SLIT2 mutation in 10% and EPHA7 mutation in 10%) (13). In a recent review article, SCLC subtypes were defined by differential expression of four key transcription regulators; ASCL1, neurogenic differentiation factor 1 (NeuroD1), yes-associated protein 1 (YAP1) and POU class 2 homeobox 3 (POU2F3) (14). As YAP1 or POU2F3 expression was appeared in non-neuroendocrine SCLC (14), transformed small cell tumors in our study were supposed to be lack of the expression of both gene. In a study on the pathogenesis of SCLC using a mouse model, Meuwissen et al. reported that bi-allelic inactivation of both Rb1 and Tp53 in lung epithelial cells leads to the formation of small-cell NE tumors (15). They concluded that both inactivations are prerequisite for the pathogenesis of SCLC (15). Additionally, Sutherland et al. demonstrated the loss of TP53 and Rb1 using adenoviral vectors to transform neuroendocrine cells to SCLC cells and indicated that neuroendocrine cells play a role as the predominant cell of origin of SCLC, along with a subset of alveolar type 2 cells (16). Based on these previous findings, the inactivation of TP53 and RB1 seem necessary events in the development of SCLC from neuroendocrine cells. Our results also coincide with the previous ones. We detected TP53 alterations that induced amino acid changes in all cases (Table 2) and decreased RB1 expression in both adenocarcinoma and transformed small-cell carcinoma in autopsy samples from case 1 (Figure 3). Our findings were in line with a previous report by Lee et al. (10), and both inactivations were identified in both small-cell carcinoma and adenocarcinoma.
In our gene profiling analysis comparing the pre- and post-transformation status, alterations in MTOR, JAK1, NOTCH2, CSF1R, and MAP2K2 were detected in all three tissues with transformed small-cell carcinoma. The former four genes were detected as additive alterations, and the last one was lost (Table 1). Gene expression analysis confirmed that NOTCH2 and JAK1 expression decreased after the transformation of small-cell carcinoma compared with the original adenocarcinoma. Among these genes, we focused on NOTCH as a marker for the transformation, inferring that Notch signaling includes ASCL1. ASCL1 is downstream of Notch signaling, regulated through hairy and enhancer of split 1 (HES-1) and HES related with a YRPW motif 1 (HEY-1), which are targets of Notch signaling and have a function as transcriptional repressors of ASCL1 (12).
In the present study, as IHC analyses of autopsy samples from case 1 showed, the expression of NOTCH2 decreased in the transformed small-cell carcinoma in the pulmonary lesion compared with that in the pulmonary lesion. NOTCH2 expression was not analyzed in the other two cases because we could not obtain both transformed small-cell carcinoma and adenocarcinoma tissues for IHC staining. Additionally, the expression of ASCL1 was elevated in the transformed small-cell carcinoma of case 1.
Depending on the above results, we focused on the abnormality in NOTCH expression, which seemed to be the key mechanism for the transformation. According to a comprehensive genomic analysis of classic SCLC, 25% of human SCLCs are affected by genomic alterations of the NOTCH family, and the majority (77%) of SCLCs with high expression of neuroendocrine markers show a gene expression pattern suggestive of low Notch signaling activity, including a high level of ASCL1 (12). In the analysis for detection of NOTCH mutations in 65 patients with late-stage SCLC, mutations were found in 19 of 65 cases (29%). NOTCH1 mutations were the most frequent (n=12), followed by NOTCH2 (n=6), and NOTCH3 (n=5) (17). Notch signaling is a key regulator of neuroendocrine differentiation in SCLC and plays a critical role in early SCLC development (12). In the Trp53, Rb1, and Rbl2 triple-knockout SCLC mouse model, an activated form of Notch2 intracellular domain (N2ICD) led to a reduction in the number of tumors and better survival, and overexpression of N2ICD inhibited the expression of neuroendocrine markers ASCL1 and synaptophysin (12). ASCL1 is an essential factor for neuroendocrine differentiation of normal pulmonary neuroendocrine cells and it promotes neuroendocrine lung carcinogenesis as a precursor of SCLC tumor-initiating cells (18,19). In an in vitro study, a lung cancer cell line transfected with an ASCL1-coding plasmid led to the small-cell carcinoma morphology and overexpression of CD56, detected via flow cytometry (20,21).
Based on several previous studies, inactivation of both p53 and RB1 is essential for the tumorigenesis of small-cell carcinoma, and inactivation of Notch signaling leads to neuroendocrine differentiation. The genetic changes causing lung epithelial cells to transform into SCLC can be explained in several steps (17,20). First, inactivation of the Notch signaling/upregulated ASCL1 is involved in the cell differentiation of neuroendocrine precursor early lung stem cells. The loss of TP53 and RB1 causes neuroendocrine precursor cells to transform into small-cell carcinoma cells. The case of secondary SCLC transformed from NSCLC at initial diagnosis even develops by adding NOTCH inactivating mutations and loss of p53/RB during the clinical course. Notch signaling is also involved in the intratumoral heterogeneity of small-cell carcinomas and non-small-cell carcinomas (22). In a study using a mouse model, activation of the Notch pathway has been linked to the neuroendocrine to non-neuroendocrine switch in SCLC cells (22). In previous analyses of actual tumor specimens from combined SCLC, Notch1 expression was negative in the SCLC component, whereas it was positive in the NSCLC component (23). Inactivation of RB1 and TP53 occur before branching from adenocarcinoma, as previously reported by Lee et al. (10). Intriguingly, RB1 alterations, including point mutation and rearrangement of the protein coding regions, were identified more frequently in EGFR-mutant adenocarcinoma than in wild-type adenocarcinoma (24). It was also reported that inactivation of RB1 preceded the acquisition of EGFR-TKI, and they assumed that the alteration did not confer resistance to EGFR-TKI (24). EGFR mutation-positive adenocarcinoma cells that harbor inactivation of both RB1 and TP53 at baseline have a chance of future transformation. Our autopsy analysis confirmed the inactivation of p53 and RB1 in both adenocarcinoma and transformed small-cell carcinoma. NOTCH mutation was detected only after the transformation, and its expression at the protein level was also reduced after the transformation. Combining our results with previous reports, we speculated that inactivation of p53 and RB1 emerged during cancer progression, and tissues that acquired Notch inactivation subsequently transformed (Figure 4). In the tissues without Notch inactivation, neuroendocrine differentiation did not occur. For example, in case 1, the metastatic lesions in the left upper lung lobe and the liver remained as adenocarcinoma (Figure 3C).
Figure 4.
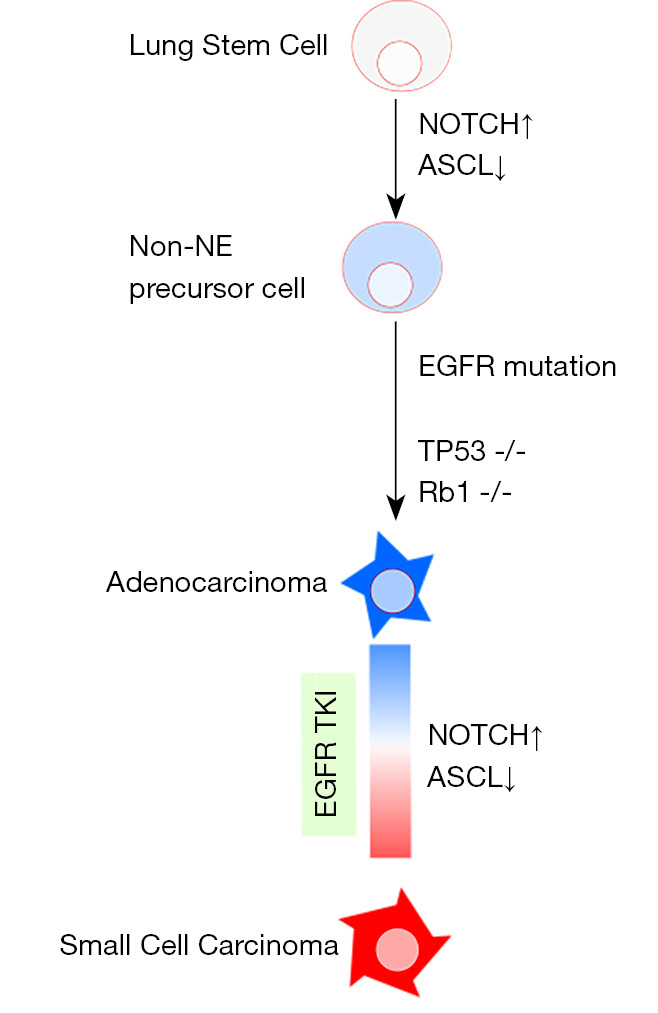
Conceptual scheme of histological transformation. ASCL1, Achaete-scute homolog 1; NE, neuroendocrine; EGFR, epidermal growth factor receptor; Rb, retinoblastoma; TKI, tyrosine kinase inhibitor.
As described above, the NOTCH-ASCL1 axis might have a potential therapeutic target in transformed small cell carcinoma from adenocarcinoma with oncogenic driver mutation, which has few treatment options except for the regimen to the classical SCLC. Further investigation is needed to confirm whether pharmacological inhibitor of these pathways is effective to the neuroendocrine transformed tumor of patient derived xenograft model. Additionally, Quintanal-Villalonga et al. pointed out the involvement of genes expression, such as PRC2 complex, PI3K/AKT and NOTCH pathways (25). As PI3K and AKT were included in gene list of cancer-panel in this time, both genetic alterations were detected in all three cases (Table S4). These results also lead that AKT inhibitor is expected to delay transformed neuroendocrine lung carcinoma (25).
This study has several limitations. The sample size in this study was too small, and it remains unclear whether the findings were consistent in all transformations. As reported in a few reports so far, NOTCH alterations are rarely detected in transformations (26). Even in classic SCLC, NOTCH alteration is not detected in all cases, so the behavior shown in Figure 4 may apply to some SCLC cases. However, since the evaluation of Notch inactivation at the protein level has not been examined, Notch inactivation may be involved in transformations more than expected. Lee et al. showed that APOBEC-induced hypermutation is frequent in various genetic abnormalities found in the tissues that transformed after branching (10). The next question is why NOTCH and ASCL1 expression levels are altered during treatment. Further studies are warranted to determine whether EGFR-TKI exposure of cancer cells leads to variations in NOTCH and ASCL1 expression levels. However, we believe that Notch signaling-dependent small-cell carcinoma transformation occurs, as shown in Figure 4. According to the autopsy investigation in case 1, there were adenocarcinoma, combined- small cell and adenocarcinoma and transformed small cell carcinoma under the clinical course in one patient. Thus, tumor progression has heterogeneity among the multi-metastatic lesions. In clinical setting, comprehensive biopsy to every metastatic site is so hard, due to the invasiveness. Further investigation is needed to grasp whole status of malignancy in the patient more easily, such as using cell-free DNA technique. We recently observed that cfDNA harbors essential genetic alterations that are representative of the whole cancer; the most detectable tumor-derived genetic alterations in cfDNA were truncation mutations with a high-variant allele frequency (27). Detecting of the sign of histological transformation using cfDNA is expected. The other deciding limitation is that our study was mainly deal with genomic alterations. The number of genetic alterations were quite low and did not increase after histological transformation in 2 among 3 cases. Transcriptional reprogramming, epigenetic mechanisms or DNA methylation may have another important involvement to drive transformation. Recent report presented neuroendocrine transformation is primarily driven by transcriptional reprogramming, including loss of the 3p chromosome arm (25).
In summary, we have provided gene alteration profiles in transformed small-cell carcinoma after resistance to EGFR-TKI compared to the original adenocarcinoma. NOTCH mutations were detected as additional alterations in all three cases. It is suggested that Notch inactivation is one of the key conditions leading to small-cell transformation under RB1 and p53 inactivation.
Supplementary
The article’s supplementary files as
Acknowledgments
We would like to thank M Kashiwano and Y Iwauchi for their technical assistance.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (revised in 2013). The study was approved by the Ethics Committee of Kanazawa University Hospital (approval number: 327), and informed consent was obtained from all participants.
Footnotes
Reporting Checklist: The authors have completed the STROBE reporting checklist. Available at https://dx.doi.org/10.21037/tlcr-21-536
Data Sharing Statement: Available at https://dx.doi.org/10.21037/tlcr-21-536
Peer Review File: Available at https://dx.doi.org/10.21037/tlcr-21-536
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/tlcr-21-536). HKo reports the relevant financial activities outside the submitted work; personal fees from AstraZeneca K.K., Chugai Pharmaceutical Co. Ltd., Taiho Pharmaceutical Co., Ltd. and Boehringer Ingelheim GmbH. HKi reports that this work was financially supported by the JSPS KAKENHI Grant-in-Aid for Scientific Research (C) Grant Number: 19K07727 by HKi. KK reports the JSPS KAKENHI Grant-in-Aid for Scientific Research (C) Grant Number: 17K09606, outside the submitted work. The other authors have no conflicts of interest to declare.
References
- 1.Enewold L, Thomas A. Real-World Patterns of EGFR Testing and Treatment with Erlotinib for Non-Small Cell Lung Cancer in the United States. PLoS One 2016;11:e0156728. 10.1371/journal.pone.0156728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998-2006. 10.1001/jama.2014.3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takano T, Fukui T, Ohe Y, et al. EGFR mutations predict survival benefit from gefitinib in patients with advanced lung adenocarcinoma: a historical comparison of patients treated before and after gefitinib approval in Japan. J Clin Oncol 2008;26:5589-95. 10.1200/JCO.2008.16.7254 [DOI] [PubMed] [Google Scholar]
- 4.Westover D, Zugazagoitia J, Cho BC, et al. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol 2018;29:i10-9. 10.1093/annonc/mdx703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu SG, Liu YN, Tsai MF, et al. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 2016;7:12404-13. 10.18632/oncotarget.7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240-7. 10.1158/1078-0432.CCR-12-2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campo M, Gerber D, Gainor JF, et al. Acquired Resistance to First-Line Afatinib and the Challenges of Prearranged Progression Biopsies. J Thorac Oncol 2016;11:2022-6. 10.1016/j.jtho.2016.06.032 [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26. 10.1126/scitranslmed.3002003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niederst MJ, Sequist LV, Poirier JT, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 2015;6:6377. 10.1038/ncomms7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JK, Lee J, Kim S, et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J Clin Oncol 2017;35:3065-74. 10.1200/JCO.2016.71.9096 [DOI] [PubMed] [Google Scholar]
- 11.Dorantes-Heredia R, Ruiz-Morales JM, Cano-García F. Histopathological transformation to small-cell lung carcinoma in non-small cell lung carcinoma tumors. Transl Lung Cancer Res 2016;5:401-12. 10.21037/tlcr.2016.07.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47-53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peifer M, Fernández-Cuesta L, Sos ML, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 2012;44:1104-10. 10.1038/ng.2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudin CM, Poirier JT, Byers LA, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19:289-97. 10.1038/s41568-019-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meuwissen R, Linn SC, Linnoila RI, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003;4:181-9. 10.1016/S1535-6108(03)00220-4 [DOI] [PubMed] [Google Scholar]
- 16.Sutherland KD, Proost N, Brouns I, et al. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011;19:754-64. 10.1016/j.ccr.2011.04.019 [DOI] [PubMed] [Google Scholar]
- 17.Leonetti A, Facchinetti F, Minari R, et al. Notch pathway in small-cell lung cancer: from preclinical evidence to therapeutic challenges. Cell Oncol (Dordr) 2019;42:261-73. 10.1007/s13402-019-00441-3 [DOI] [PubMed] [Google Scholar]
- 18.Borges M, Linnoila RI, van de Velde HJ, et al. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 1997;386:852-5. 10.1038/386852a0 [DOI] [PubMed] [Google Scholar]
- 19.Linnoila RI, Zhao B, DeMayo JL, et al. Constitutive achaete-scute homologue-1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res 2000;60:4005-9. [PubMed] [Google Scholar]
- 20.Meder L, König K, Ozretić L, et al. NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int J Cancer 2016;138:927-38. 10.1002/ijc.29835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osada H, Tomida S, Yatabe Y, et al. Roles of achaete-scute homologue 1 in DKK1 and E-cadherin repression and neuroendocrine differentiation in lung cancer. Cancer Res 2008;68:1647-55. 10.1158/0008-5472.CAN-07-5039 [DOI] [PubMed] [Google Scholar]
- 22.Lim JS, Ibaseta A, Fischer MM, et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017;545:360-4. 10.1038/nature22323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hassan WA, Takebayashi SI, Abdalla MOA, et al. Correlation between histone acetylation and expression of Notch1 in human lung carcinoma and its possible role in combined small-cell lung carcinoma. Lab Invest 2017;97:913-21. 10.1038/labinvest.2017.36 [DOI] [PubMed] [Google Scholar]
- 24.Pros E, Saigi M, Alameda D, et al. Genome-wide profiling of non-smoking-related lung cancer cells reveals common RB1 rearrangements associated with histopathologic transformation in EGFR-mutant tumors. Ann Oncol 2020;31:274-82. 10.1016/j.annonc.2019.09.001 [DOI] [PubMed] [Google Scholar]
- 25.Quintanal-Villalonga A, Taniguchi H, Zhan YA, et al. Multi-omic analysis of lung tumors defines pathways activated in neuroendocrine transformation. Cancer Discov 2021. [Epub ahead of print]. 10.1158/2159-8290.CD-20-1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcoux N, Gettinger SN, O'Kane G, et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J Clin Oncol 2019;37:278-85. 10.1200/JCO.18.01585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koba H, Kimura H, Yoneda T, et al. Molecular features of tumor-derived genetic alterations in circulating cell-free DNA in virtue of autopsy analysis. Sci Rep 2021;11:8398. 10.1038/s41598-021-87094-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as