

Abstract
Telomeres are hot spots for mutagenic oxidative and methylation base damage due to their high guanine content. We used single-molecule fluorescence resonance energy transfer detection and biochemical assays to determine how different positions and types of guanine damage and mutations alter telomeric G-quadruplex structure and telomerase activity. We compared 15 modifications, including 8-oxoguanine (8oxoG), O-6-methylguanine (O6mG), and all three possible point mutations (G to A, T, and C) at the 3′ three terminal guanine positions of a telomeric G-quadruplex, which is the critical access point for telomerase. We found that G-quadruplex structural instability was induced in the order C < T < A ≤ 8oxoG < O6mG, with the perturbation caused by O6mG far exceeding the perturbation caused by other base alterations. For all base modifications, the central G position was the most destabilizing among the three terminal guanines. While the structural disruption by 8oxoG and O6mG led to concomitant increases in telomerase binding and extension activity, the structural perturbation by point mutations (A, T, and C) did not, due to disrupted annealing between the telomeric overhang and the telomerase RNA template. Repositioning the same mutations away from the terminal guanines caused both G-quadruplex structural instability and elevated telomerase activity. Our findings demonstrate how a single-base modification drives structural alterations and telomere lengthening in a position-dependent manner. Furthermore, our results suggest a long-term and inheritable effect of telomeric DNA damage that can lead to telomere lengthening, which potentially contributes to oncogenesis.
Graphical Abstract
DNA damage has long been linked to cytotoxicity, oncogenesis, and degenerative disorders associated with aging. Many environmental factors and byproducts of cellular metabolism can cause DNA lesions, including base loss, strand breaks, and chemical alterations (reviewed in ref 1). Chemical modification of bases is the most common DNA lesion type2 that may cause mutations, because many chemically modified bases preferentially pair with an incorrect nucleotide during DNA replication. Therefore, DNA lesions have the potential to alter the local DNA secondary structure either directly or by inducing mutations, if not removed in a timely manner by the DNA repair pathways. Many independent studies reported that 8-oxoguanine (8oxoG), the most common oxidative base lesion, destabilizes the DNA double helix and noncanonical secondary structures.3–8 8oxoG mainly induces G to T mutations by mispairing with A, although other mutations have been reported.9,10 O-6-Methylguanine (O6mG), a common base lesion caused by alkylation of the O6 atom of guanine, is highly mutagenic and carcinogenic due to mispairing with T and, thereby, induces high rates of G to A mutations.11–13 While the impact of DNA lesions on genome replication by DNA polymerases has been extensively studied, far less is known about how lesions affect telomere access and telomere elongation.
Telomeres are specialized structures that cap the ends of linear chromosomes in eukaryotes. Human telomeric DNA consists of TTAGGG tandem repeats in duplex followed by a G-rich 3′ single-stranded overhang.14 Shelterin proteins associate with telomeric DNA to regulate the structure and length of telomeres and to prevent inappropriate activation of DNA damage signaling and repair pathways.15–19 Previous work showed that telomeres are prone to oxidative DNA damage due to recurring consecutive guanines in their sequence.20,21 Other evidence indicates 8oxoG repair is less efficient in telomeres, and the 8-oxoguanine glycosylase (OGG1) repair enzyme does not act on single-stranded DNA.22 Although the repair efficiency of O6mG at telomeres is unknown, this lesion is also mutagenic and carcinogenic23,24 and can form in G runs of telomeric DNA.
In most somatic cells, telomeres are gradually shortened with rounds of cell division due to the end-replication problem. Exceptions include embryonic stem cells, pluripotent cells, and cancer cells that can replenish telomeres by a special reverse transcriptase called telomerase.25–28 Telomerase is a ribonucleoprotein complex that binds specifically to the end of the single-stranded telomere overhang and extends it. The RNA component within telomerase functions both in directing telomerase to the telomere and in templating for TTAGGG repeat addition to the 3′ overhang. When sufficiently elongated by the telomerase, a replicative DNA polymerase can then synthesize the C-rich complementary strand, known as C-strand fill in. Shelterin protein POT1 binds to the telomeric single-strand overhang to help regulate telomerase access and activity.19,29–32
Single-stranded TTAGGG repeats in the telomeric overhang can fold into G-quadruplexes (G4)33–35 in which four guanines form a quartet through Hoogsteen hydrogen bonding stabilized by a monovalent cation conjugating with the four neighboring O6 atoms (Figure 1A). The conformation of G4 depends on the loop sequence and solution conditions.36–38 The mammalian telomere sequence forms a hybrid or parallel G4 conformation at physiological ionic concentrations.33,39,40 Circular dichroism spectroscopy and ultraviolet (UV) melting experiments show that 8oxoG or O6mG can destabilize and alter telomeric G4 conformations by disrupting the Hoogsteen bonding.7,41,42 The presence of G4 structure in genomic DNA, including the telomeres, has been well-documented.43–47 More recently, immunofluorescence-based studies revealed colocalization between telomere binding proteins and the G4-specific antibody, BG4,48 strongly supporting the formation of G4 at telomeres. The folded structure of G4 can diminish telomerase loading, yet the role of G4 structures in telomere length regulation, aging, or disease development remains elusive.
Figure 1.

G-Quadruplex and base damage in the telomere terminal repeat. (A) G-Quartet formed by four guanines. (B) DNA construct design and sequence. The three guanines in the 3′ terminal repeat are denoted as G1–G3 in the 5′ to 3′ direction. The Cy5 dye is located at the phosphate group at the 5′ end. (C) 8-Oxoguanine (8oxoG), O6-methylguanine (O6mG), and the major mutations caused by these modified bases after replication.
In earlier studies, using single-molecule fluorescence resonance energy transfer (smFRET) and biochemical analyses, we reported that a single 8oxoG moiety in an oligonucleotide telomeric overhang induces G4 structural dynamics and perturbations, which significantly enhance telomerase binding and activity.49,50 We also directly compared effects of 8oxoG within the G-quartet versus thymine glycol within the loop, because these are the two most common oxidative lesions. We demonstrated that while telomerase cannot access a tightly folded G4, it binds and extends a G4 when either thymine glycol or 8oxoG is present at the second T or G in the repeat, respectively. This implies that a stable G4 in the telomeric overhang can act as a lock that prevents telomerase activity in normal cells, whereas a single lesion can disrupt the G4 and allow for telomere extension. However, how lesion location determines telomeric structural dynamics and telomerase accessibility is poorly understood.
In this study, we sought to evaluate the structural changes induced by different types and positions of base modifications or mutations. We used smFRET to investigate 15 variants of telomeric G4 that include two types of DNA lesions, 8oxoG and O6mG, and mutations of G to A, C, or T at the three different terminal guanine positions, which serve as an entry access for POT1 and telomerase. By examining the structural changes at a single-molecule level, smFRET allows us to distinguish the different structural dynamics that coexist in all of the individual molecules without having to synchronize. We developed an analytical method to categorize >100000 smFRET trajectories into different classes of dynamic conformational changes induced by various base modifications. Our data reveal that DNA lesions and mutations shift the distribution and dynamics of a series of unfolded, partially folded, or fully folded G4 conformations. Even the base modification located at the most “tolerable” position for G4 formation7 imparts a significant level of structural dynamics in our study. We correlated the distribution of structural disruption with accessibility by testing for complementary strand annealing, POT1 binding, and telomerase binding and extension activity. Our results indicate that the induced structural dynamics and disruptions, and the resulting accessibility, vary significantly depending on the type and position of the chemical base alteration. Finally, we interpret our results in the context of published G-quadruplex structural data from nuclear magnetic resonance (NMR) and circular dichroism (CD).5–7,41,42 Taken together, our results have important implications for how DNA damage and mutations at the telomeric end regulate telomerase-mediated telomere lengthening.
MATERIALS AND METHODS
DNA Sample Preparation.
The single-stranded (ss) DNA oligonucleotides containing 8oxoG or O6mG were purchased from Midland Certified Reagent Company Inc. (Midland, TX). These oligonucleotides were synthesized with a primary amine modification at the designated labeling site and then fluorescently labeled in our laboratory with Cy3-NHS-ester in 100 mM sodium bicarbonate buffer at room temperature as reported earlier.49,50 Other DNA oligonucleotides were purchased from IDT (Coralville, IA) with or without the corresponding fluorescent dye or biotin conjugation. The sequences of all of the oligonucleotides are listed in Table S1. The partial duplex DNA constructs were prepared by mixing a G-rich ss oligonucleotide with the 18-mer complementary oligonucleotide at a molar ratio of 1:1.2 in a buffer containing 20 mM Tris-HCl (pH 7.5) and 100 mM KCl. The mixtures were incubated in a Bio-Rad C1000 Touch Thermal Cycler with the following program as reported earlier:49,50 95 °C for 2 min, slowly cooled to 37 °C at a rate of 2 °C/min, and then cooled to 4 °C.
POT1 Protein and Telomerase Lysate Preparation.
Recombinant human POT1 protein was expressed in a baculovirus–insect cell system and purified as previously described.51,52 FLAG-tagged human telomerase was expressed in HEK-293T cells co-transfected with plasmids expressing hTR and FLAG-tagged hTERT in a 1:3 molar ratio as previously described.49,50,53 Briefly, the HEK-293T cells were grown in DMEM with 10% FBS and 1% penicillin-streptomycin (Gibco) to 90% confluency. The cells were then transfected using Lipofectamine 2000 Reagent (Invitrogen) and allowed to grow for 48 h. The cells were harvested by trypsin detachment, washed with phosphate-buffered saline (PBS), and lysed with CHAPS lysis buffer [10 mM Tris-HCl, 1 mM MgCl2, 1 mM EDTA, 0.5% CHAPS, 10% glycerol, 5 mM β-mercaptoethanol, 120 units of RNasin plus (Promega), 1 μg/mL pepstatin, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL chymostatin, and 1 mM AEBSF] for 30 min at 4 °C. The cell lysate was frozen in liquid nitrogen and stored at −80 °C.
Single-Molecule FRET Sample Assembly.
Single-molecule FRET (smFRET) data were acquired using a home-built prism-type total internal reflection fluorescence (TIRF) microscope with an electron-multiplying CCD camera (EMCCD) at room temperature (23 ± 1 °C). The quartz sample slides and glass coverslips were prepared by following a polyethylene glycol (PEG)-mediated surface passivation protocol with a 40:1 mixture of m-PEG-5000-SVA and biotin-m-PEG-5000-SVA (Laysan Bio, Inc.).49 The imaging chamber formed between the quartz slide and the coverslip slide was coated with 50 μg/mL neutravidin (Thermofisher) in 10 mM Tris-HCl (pH 7.5) and 100 mM KCl for 5 min and then washed with the same buffer. Annealed biotinylated partial duplex DNA carrying both Cy3- and Cy5-labeled strands was immobilized in the PEG-coated imaging chamber via biotin–neutravidin interaction in 10 mM Tris-HCl (Ph 7.5) and 100 mM KCl. All of the smFRET experiments were performed in an imaging buffer containing 10 mM Tris-HCl (pH 7.5), 100 mM KCl, 0.5% glucose, 10 mM 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic (Trolox), and 1 mg/mL glucose oxidase and 4 μg/mL catalase to ensure stable G-quadruplex formation and to minimize photobleaching of the fluorophores.
SmFRET Binding Assay.
The real-time smFRET binding assay of the complementary strand (C strand) and POT1 was carried out in flow chambers, which have a small plastic reservoir above the hole at one end of the chamber and a syringe pump connected with silicone tubing to the hole at the other end. The DNA labeled with both Cy3 and Cy5 was immobilized in the imaging chamber and imaged alone, and then 100 μL of the C strand (10 nM) or POT1 (100 nM) in imaging buffer was loaded into the reservoir. The C strand stock was stored in 1× TE buffer [1 mM EDTA and 10 mM Tris-HCl (pH 8.0)], diluted to 1 μM in water to prevent secondary structure, and diluted to 10 nM in the imaging buffer right before the experiment. The POT1 stock was stored in storage buffer [25 mM Tris-HCl (pH 8.0) and 150 mM NaCl] and diluted in 10 mM Tris and 100 mM KCl buffer. Right before the binding experiment, POT1 was further diluted to 100 nM in the imaging buffer. The C strand or POT1 solution was passed through the imaging chamber to the silicone tubing, at a rate of 20 μL/s drawing by a Pump 11 Elite syringe pump (Harvard Apparatus, Holliston, MA) equipped with a 1 mL standard disposable plastic syringe. Real-time FRET images were collected during and after sample injection for 5 min at a frame rate of 100 or 150 ms. For the C strand and POT1 binding reactions that reached completion within 5 min of C strand or POT1 flow-in, the binding kinetics were analyzed by monitoring the time from the moment of flow to the moment of the first irreversible FRET decline event as described previously.50 For the binding reactions that did not reach completion within 5 min, short movies were collected at different time points to generate FRET histograms until the reactions were completed or 2 h had passed.
SmFRET Histograms.
A solid state 532 nm green laser (Compass 315M, Coherent) was used to generate an evanescent field of illumination for smFRET detection. Data were recorded with a time resolution of 100 ms, processed by an IDL script, and then analyzed by Matlab scripts. Each FRET histogram was generated from 15–30 independent images containing 250–350 individual molecules per image to ensure the inclusion of more than 5000 molecules. To prevent the donor-only molecules from interfering with low-FRET regions, the molecules containing both the donor and the acceptor were selected through sequential excitation of the donor (Cy3, by a 532 nm green laser) and acceptor (Cy5, by a Coherent cube 641 nm red laser). Donor leakage was corrected on the basis of the FRET value of donor-only molecules. Each corrected and normalized histogram was fitted to Gaussian distributions using Origin 2016. The histograms of DNA alone were fitted with an unrestrained peak center position. The peak center positions were then used to restrain the peak position in the C strand or POT1 bound histograms. The reported histogram fitting results were obtained from averaging two or three trials, each containing more than 6000 molecules. The uncertainty was expressed in the form of the standard error calculated from multiple trials.
SmFRET Trajectory Analysis.
We developed MatLab scripts to record and analyze the manually selected transition from smFRET trajectories because the currently available smFRET analyzing programs tend to ignore the fast (<5 s) transit state changes in some of the DNA constructs. A total of 300–600 individual molecules of each DNA construct from two or more independent experiments were initially, manually categorized to ensure the molecular dynamic behavior was consistent across different imaging areas and experiments. The detailed molecule behavior analysis was performed by incorporating more than 200 individual molecules of each DNA construct. The FRET value and time of each transition were manually assigned by the user. The recorded manually selected FRET values were then plotted against the mean FRET value calculated between two transitions. Most of the manually selected and calculated FRET values were >95% consistent. However, in the cases of fast transit states that lasted for <5 s, the manually reported FRET values better reflect the maximum FRET fluctuation of the transit states. The transition behavior analysis was designed to screen through each smFRET trajectory and identify the lowest FRET value each molecule had encountered during the 120 s recording time or before photobleaching. The fraction of molecules exhibiting different molecular behaviors was estimated by a bootstrapping procedure that randomly sampled 200 traces from each construct and repeats 200 times. Both manually selected and calculated mean FRET values of each transition were analyzed with bootstrap and generated a similar result. The data reported herein are based on the manually selected FRET values. The fraction and reported error of each population were obtained from the medium and confidential interval of 95% from the bootstrapping results.
Telomerase Binding Assay.
The single-molecule pull-down (SiMPull) assay for telomerase binding was performed on the same TIRF microscope at room temperature as smFRET. Mouse monoclonal IgM against hTERT (LS-B95) was purchased from LifeSpan Biosciences. The antibody was labeled with Alexa-647 C5 maleimide (Thermo Fisher) at a 1:35 molar ratio in PBS and 100 mM sodium bicarbonate on ice for 1 h and purified by Bio-Gel P6 columns in Tris buffer, as described previously.50 Biotinylated mouse anti-FLAG monoclonal antibody M2 at a 1:100 dilution (F9291, Sigma) was immobilized in the imaging chamber via biotin–neutravidin interaction as described above. The imaging chamber was washed with telomerase reaction buffer (TRB) containing 50 mM Tris-HCl (pH 8), 50 mM KCl, and 1 mM MgCl2. The cell lysate containing human telomerase (1:50 dilution in TRB) was added to the antibody-coated surface and incubated for 20 min at room temperature. Subsequently, 10 nM no-biotin Cy3-labeled partial duplex DNA was applied to the chamber and incubated for 20 min. Unbound DNA was washed away with TRB, which was immediately followed by adding the Alexa-647-conjugated hTERT antibody (1:5000 dilution) to the chamber. The imaging chamber was first washed with TRB and then with imaging buffer containing 0.5% glucose, 10 mM Trolox, 1 mg/mL glucose oxidase, 4 μg/mL catalase, and the same salts as TRB [50 mM Tris-HCl (pH 8), 50 mM KCl, and 1 mM MgCl2]. Thirty or more movies were recorded for each chamber with sequential excitation with a 532 nm green laser (for Cy3-labeled DNA) and a 641 nm red laser (for the Alexa 647-labeled antibody). The colocalization of the Cy3 and Cy5 signal under sequential excitation was analyzed with a MatLab script to give the binding ratio of DNA-bound telomerase to the overall telomerase signal. Each DNA construct had been tested at least three times on different dates and normalized to the mean binding ratio of the R3 DNA construct, to give the mean telomerase binding fraction and standard error between repeats. The specificity of the antibodies against telomerase was confirmed as described previously.50
Telomerase Extension Assay.
The telomerase extension assay was slightly modified from the previous protocol.49,50,54 Briefly, telomerase was eluted from the beads with 2 times the bead volume of a mix consisting of 250 μg/mL 3× FLAG peptide (Sigma-Aldrich) in 1× telomerase buffer and 150 mM KCl. The bead slurry was incubated for 30 min at 4 °C with mixing, and telomerase in the supernatant was collected using Mini Bio-Spin Chromatography columns (Bio-Rad). The same cell lysate used in our SiMPull assay was employed in the extension assay. In vitro reaction mixtures (20 μL) with oligonucleotide primers (1 μM) contained telomerase reaction buffer, 0.3 μM of 3000 Ci/mmol [α−32P]dGTP (PerkinElmer), and a cellular dNTP (Thermo Fisher) concentration mix of 37 μM dTTP, 24 μM dATP, 29 μM dCTP, and 5.2 μM dGTP. Reactions were started by adding 3 μL of an immunopurified telomerase eluent, incubated for 1 h at 37 °C, and then terminated by adding 2 μL of 0.5 M EDTA and heat inactivated for 20 min at 65 °C. An 18-mer radio end-labeled oligonucleotide was added as a loading control (LC) (8.0 fmol) to the inactivated reaction mixtures prior to purification with an Illustra MicroSpin G-25 column (GE Healthcare). Radiolabeling of LC was performed in a 20 μL reaction mixture containing 10 pmol of oligonucleotide, 1× T4 PNK buffer (NEB), 2 μL of 3000 Ci/mmol [γ−32P]ATP (PerkinElmer), and 10 units of T4 PNK (NEB), and the mixture was incubated for 60 min at 37 °C followed by heat inactivation at 65 °C for 20 min. An equal volume of loading buffer [94% formamide, 0.1× Tris-borate-EDTA (TBE), 0.1% bromophenol blue, and 0.1% xylene cyanol] was added to the reaction eluent from the G-25 spin column. The samples were heat denatured for 10 min at 100 °C, loaded onto a 10% denaturing acrylamide gel (7 M urea and 1× TBE), and electrophoresed for 70 min at a constant of 38 W. Samples were imaged using a Typhoon phosphorimager (GE Healthcare). The relative telomerase activity was quantitated using ImageQuant and normalized to the loading control as described previously.49,55,56
RESULTS
Oxidative and Alkylation Base Lesions Cause Greater Structural Disruption Than Mutations.
We designed a series of 16 DNA constructs containing four repeats of TTAGGG with five types of modifications at the three terminal guanine positions. First, we examined if the single site oxidative and alkylation base lesions, and mutations caused by these lesions, change the structure of the telomeric DNA. We substituted three guanines at the 3′ end individually with 8oxoG, O6mG, A, T, or C. All DNA constructs share the same 18 bp nontelomeric double-stranded stem. The 24-mer telomeric oligonucleotide is written as (TTAGGG)3-TTAG1G2G3, in which G1–G3 represent the three guanine positions replaced by a lesion or mutation, followed by Cy3 at the 3′ end. The annealed 18-mer oligonucleotide has a Cy5 at the 5′ end that FRET pairs with Cy3 and a biotin conjugated at the 3′ end for immobilization to a neutravidin-coated single-molecule surface (Figure 1B and Table S1). We probed the formation of G4 by FRET.19,32,38
We used a physiologically relevant potassium concentration (100 mM KCl)57 to capture FRET images and built the FRET histogram by compiling more than 4000 FRET values collected from 20 fields of view, thus representing the ensemble average FRET of all DNA molecules. The FRET histograms for all 16 DNA constructs displayed diverse patterns depending on the type of chemical modifications and their positions (Figure 2A, black). We also obtained a FRET histogram of a 25-mer polythymine (polyT) as an unstructured control, which yields a single peak centered at 0.5 FRET (Figure 2A, top orange histogram). The FRET histogram of the unmodified (TTAGGG)4 DNA (4R) has a single peak centered at 0.9 (Figure 2A, top left, black histogram), which is consistent with a tightly folded G4 structure.32
Figure 2.
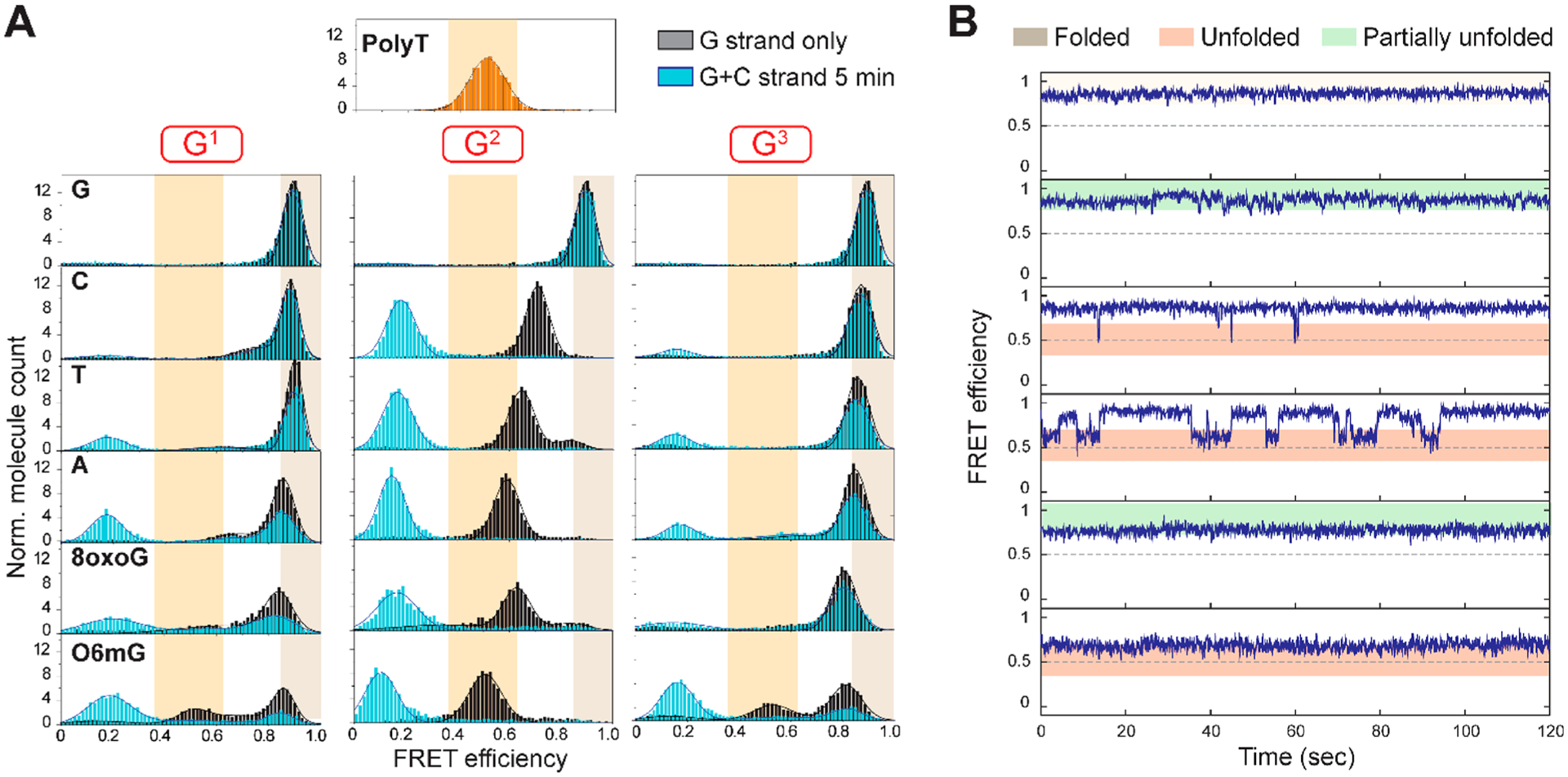
Structural disruption is dependent on both base and position. (A) FRET histograms of each DNA construct before (gray) and after (cyan) incubation for 5 min with the complementary C strand (CCCTAA)4. High FRET converts to low FRET upon annealing as an indicator of accessibility. (B) Sample single-molecule traces of a telomeric construct harboring an 8oxoG at the G1 position displaying the three major types of molecular behavior.
The constructs that harbor a point mutation (A, C, or T) at the G1 or G3 position all show a major high FRET peak similar to unmodified 4R, conferring minimal disruption in the folded G4 structure (Figure 2A). The only minor exception to this pattern is A in the G1 position that induces an additional lower FRET shoulder (23 ± 2% at 0.68 FRET calculated from the area under the Gaussian fit), suggesting a small fraction of molecules fold into a slightly altered (more open) conformation or undergo conformational dynamics. By contrast, all modifications at the G2 position induce a FRET peak shift to substantially lower values, signifying a higher degree of G4 disruption than modification at the other two positions. This is consistent with previous reports that the middle G position, denoted G2, is the most critical base for G4 thermal stability.6,42 When 8oxoG or O6mG was placed at position G1 or G3, the histograms displayed significant deviations from the G4 folded peak. 8oxoG at G1 and G3 positions induced broadening of the major high-FRET peak, which spread over to the mid-FRET range. Such shoulders become much more pronounced by O6mG substitution at both G1 and G3 positions that generate 30–40% of the peak transitioning to the mid-FRET range, reflecting a significant degree of disrupted G4 structure. The 0.5 mid-FRET peak induced by O6mG at all positions indicates that the degree of unfolding matches that of the unstructured polyT.
Taken together, our data suggest that at the G1 and G3 positions, the base lesions cause significant structural distortions while point mutations induce only minor or insignificant structural alterations. Quantification of the high-FRET and mid-FRET shifts reveals a general rank order for the extent of G4 disruption as O6mG > 8oxoG ≥ A > T > C at the G1 and G3 positions. This order likely arises from the size and chemical property of the substituted bases; the smaller pyrimidines can fit into the space of a missing guanine, while the purine and lesions cause both steric hindrance and interrupted hydrogen bonds. We observe extensive unfolding arising from modification of the central guanine, G2, in agreement with previous CD and UV melting studies.6,7,41 All FRET histograms of the G2-substituted constructs exhibit a complete disappearance of high-FRET peaks, and the concomitant appearance of mid-FRET peaks, emphasizing the maximal perturbations induced by G2 modifications (Figure 2A). Interestingly, the magnitude of the FRET peak shift of the mutant constructs follows a similar order of A (0.6) > T (0.65) > C (0.71), expressed as the main FRET peak center values. The G4 destabilization of human telomeric DNA caused by A or T substitution is consistent with previous reports.58,59 O6mG at the G2 position causes the strongest structure disruption among all of the constructs. Indeed, the FRET histogram of the O6mG substitution almost overlaps with the polyT histogram, which indicates the O6mG at the G2 construct behaves as unstructured ssDNA.
Lesion Position Determines the Accessibility of Damaged Telomeric DNA.
Having mapped the structural disruptions by the base modifications, we next tested the accessibility of all of the DNA constructs by applying a complementary strand (CCCTAA)4 that we termed the “C strand”. As shown previously, the C strand completely anneals to the G4 strand as evidenced by a single low-FRET peak when the C strand is added in a 200-fold molar excess.32,49,50 The cyan histograms in Figure 2A were collected after the addition of 10 nM C strand for 5 min (100 mM KCl). The appearance of a low-FRET peak at 0.2 indicates the formation of a long 24 bp duplex, (TTAGGG)4/(CCCTAA)4, which can form only when the C strand anneals to unfolded G4. Therefore, the FRET shift to 0.2 represents the openness or accessibility of the folded structure.32 Under our experimental condition, the unmodified 4R displayed negligible (~8%) annealing while the G2 lesion and mutant constructs all showed 100% accessibility to the C strand (Figure 2A, cyan), which is consistent with our previous report.50 Interestingly, the G1 and G3 lesion and mutant constructs exhibited varying degrees of accessibility depending on the type of base substitution, ranging from ~15% (C at G3) to ~90% (O6mG at G1), as quantified in Figure 4C (gray bars). The C strand accessibility of both G1-and G3-modified constructs roughly follows the same trend of O6mG (89% and 68%) > 8oxoG (50% and 24%) ≥ A (50% and 35%) > T (35% and 35%) > C (17% and 14%); the numbers in parentheses indicate accessibility induced by G1 and G3 modification, respectively. More interestingly, while unmodified 4R remained inaccessible to the C strand (~11%), all constructs with G1 and G3 lesions or mutations reached 84–95% accessibility after 2 h (see Table S2). This likely reflects the slow rate of G4 conformational dynamics when the chemical modification or mutation is at the first or last position of the GGG sequence. This effect correlates with the smaller degree of G4 disruption caused by G1 and G3 modifications and further emphasizes the position-dependent effect of the DNA lesions and mutations in telomeric G4.
Figure 4.

8oxoG and O6mG lesion at any of the terminal guanines that promotes telomerase binding. (A) Single-molecule pull-down experiment and a plot of normalized telomerase binding vs the percent of single-molecule traces displaying an unfolded state (FRET < 0.6). Error bars represent the SD from three or more telomerase binding assays. (B) Telomerase activity on substrates containing a base lesion. 3R and 4R represent DNA constructs with three and four (TTAGGG) repeats, which serve as unfolded and folded G-quadruplex controls, respectively. Numbers indicate the position of G modification. (C) Normalized C strand binding fraction, telomerase binding, and telomerase activity obtained from C strand annealing, single-molecule pull-down, and in vitro telomerase activity assays, respectively. The number after each mutated or modified base indicates the position.
To further examine C strand accessibility, we measured the C strand annealing kinetics by collecting time lapse FRET histograms after C strand addition. All of the G2-substituted constructs completely annealed to the C strand within 3 min with a half-time (t1/2) ranging from 0.5 min (O6mG and 8oxoG) to 0.6 min (A), 0.7 min (T), and 0.8 min (C) (Figure S2 and Table S2). O6mG and 8oxoG substitution at G1 or G3 generated lower reaction rates of 2–5 min (t1/2), while T and A mutations at G1 and G3 resulted in rates of 5–9 min (t1/2). Interestingly, the C mutations showed the slowest annealing rates of 25 ± 3 min (G1) and 73 ± 4 min (G3). The fast and complete annealing observed with all of the G2 substitutions is expected on the basis of the structural destabilization caused by disrupting the central G-quartet, resulting in high accessibility (Figure 2A). However, the varying degree of accessibility obtained for G1 and G3 substitutions is more subtle and difficult to rationalize. Therefore, we sought to examine the conformational dynamics that may persist in differently folded states for each construct. We hypothesized that the accessibility of a telomeric overhang relies on (1) the reduction of global G4 compactness or (2) the dynamic conformational fluctuations (unfolding–refolding transitions) that expose DNA bases to the C strand. To test the first hypothesis, we plotted the C strand accessibility against the center of the major FRET peak or the area of the minor shoulder of each construct. This showed a poor correlation with C strand accessibility (Figure S3). This lack of correlation indicates that the reduction of global compactness is likely not the main reason for the high accessibility of damaged or mutated telomeric DNA.
The Dynamic Unfolding Population Contributes to Telomeric DNA Accessibility.
We tested the second hypothesis raised above by analyzing the real-time smFRET trajectories of all of the DNA constructs. As demonstrated previously, the 4R construct displays a steady high FRET.50 By contrast, all of the G2 mutant and lesion constructs exhibit two-step FRET fluctuations oscillating between their major peak FRET value (0.5–0.7) and high FRET of 0.8–0.9 (Figure S1). On the other hand, the G1 or G3 position variants exhibit a complicated pattern of conformational dynamics from unfolded ssDNA (FRET value of polyT), intermediate FRET states, to a fully folded G4 state (Figure S1). Interestingly, all of the lesions and mutations at G1 or G3 induced similar FRET states, albeit distributed differently (Figure S1). The main difference between various bases at the same position is the distribution of dynamic patterns. To quantify conformational dynamics, we focused on the most “unfolded” state of a molecule, which is represented by the lowest-FRET state observed in a smFRET trace, and classified the smFRET traces into three categories: folded [stable high-FRET traces without fluctuation, similar to G4 (Figure 2B, gray area)], unfolded [traces that visit the unfolded 0.5 FRET state (Figure 2B, red area)], and partially unfolded [all other traces with steady FRET interspersed with transitions above 0.6 (Figure 2B, green area)]. This classification disregards the ability of refolding in the dynamic traces and simply focuses on the maximum unfolded level of each trace.
A summary of the FRET state classification as the fractional distribution of folded (black), partially unfolded (green), and unfolded (red) populations is shown in Figure 3A. As expected, the G2 modifications led to almost entirely unfolded or partially unfolded molecules. One exception is the G2 to C mutation that displays a larger population of partially unfolded molecules, possibly due to the G:C base pair stabilizing the partially folded conformation. For the G1 or G3 substitutions, the abundance of unfolded or partially unfolded populations roughly follows the order O6mG > 8oxoG ≥ A > T > C, as seen before. To check if the molecules undergoing dynamic folding and unfolding transitions may allow for C strand annealing, we plotted the fraction of dynamic molecules against the fraction of C strand annealed molecules. This plot showed a linear correlation for both the G1 and G3 constructs (Figure 3B, black and blue, respectively), suggesting that the dynamic conformational state gives rise to C strand accessibility for constructs with G1 and G3 modifications. By contrast, the G2 modifications induced complete accessibility regardless of the conformational state of the construct (Figure 2B, red), likely due to the high degree of G-quartet disruption. The X-ray crystallography and NMR structures of telomeric G4 show that potassium ions in the central cavity interact with eight oxygen atoms from two layers of G-quartets.33,60 In our constructs, the base lesions or mutations at the G1 and G3 positions prevent formation of the top or bottom G-quartet but allow the other two consecutive layers of the G-quartet to be held together by one potassium ion. The G2 position modifications, however, interfere with the central G-quartet, leading to depletion of potassium ions and thereby disrupting all G-quartet layers. Therefore, while the 0.6–0.75 FRET states in G1- and G3-modified constructs represent partially unfolded states that are inaccessible to the C strand, the same 0.6–0.75 FRET states in G2 constructs may represent looser structures that allow the C strand to anneal.
Figure 3.

Complementary strand and POT1 binding reflect the structural instability of the telomeric G-quadruplex. (A) FRET state classification as the fractional distribution of folded (black), partially unfolded (green), and unfolded (red) populations. (B and C) Plots of the fraction of single-molecule traces displaying dynamic FRET or steady unfolded state (FRET < 0.6) vs the fraction of molecules bound to (CCCTAA)4 or POT1, respectively.
POT1 Binding Requires Structural Dynamics or Unfolding.
We next asked if all of the modified constructs differ in POT1 accessibility. POT1 is a unique shelterin component that specifically binds to the single-stranded telomeric overhang.15,16,52 In our earlier study, we reported that POT1 cannot unfold and bind to the stably folded G4 formed in 100 mM KCl, while POT1 can access a G4 with a single 8oxoG at the G2 position.50 We tested accessibility by applying POT1 (100 nM) to the constructs. POT1 binding shifts the smFRET histogram peak to a lower FRET value (0.4) (Figure S4). The FRET histograms collected 5 min after the addition of POT1 exhibited a trend highly similar to that of C strand accessibility (Figure S4). Accordingly, the plot of the dynamically unfolded fraction against the fraction of POT1 bound molecules also displays a linear correlation for the G1 and G3 constructs (Figure 3C). The O6mG constructs showed the highest bound fraction (~100%), followed by 8oxoG constructs rendering 70–90% bound. Similarly, A and T mutations showed 50–100% binding to POT1, while the C mutation displayed 10–50% binding (Table S2 and Figure S4). These data imply that the “unfolded” conformation is also responsible for binding of POT1 to constructs with damage or mutations at G1 and G3. Again, the G2 modifications all led to complete binding of POT1, consistent with a high degree of structural disruption. It is possible that binding of POT1 to the constructs with base substitutions was also affected by the mutated sequence, which reduces the availability of the minimum POT1 binding site.61,62 Nevertheless, the similarity between the POT1 and C strand correlation plots suggests that they both access their binding target passively, rather than actively invading the folded G4 structure. In both plots, the lowest accessibility point at the bottom left corner is the unmodified 4R construct (Figure 3B,C), which remains folded into a G4 without yielding accessibility to POT1 and C strand.
The binding kinetics of POT1 measured in the same way as for C strand annealing revealed that 100% of POT1 binding occurred within 3 min, with binding half-times, t1/2, ranging from 0.1 min (O6mG and 8oxoG) to 0.2 min (A and T) to 0.3 min (C) (Figure S4). The O6mG substitution at G1 and G3 yielded the shortest binding half-time (t1/2 of ~1 min) compared to those of 8oxoG (t1/2 ~ 2 min) and mutants (t1/2 ~ 2–5 min). As expected, the binding half-times obtained for the modified G1 and G3 constructs were generally longer than for modified G2 constructs (Figure S4).
Base Lesions but Not Mutations Promote Telomerase Binding and Extension Activity.
We previously showed that 8oxoG in the G2 position of a telomeric G4 enhances telomerase extension activity.49,50 To test if this stimulation depends on lesion position, we performed single-molecule pull-down (SiMPull) assays with all of the constructs tested above (Figure 4A). Briefly, the SiMPull assay entails the application of lysate from mammalian cells that overexpress FLAG-tagged human telomerase to the single-molecule slides coated with an anti-FLAG antibody.32,49,50,53 We confirmed the pull-down efficiency by adding a fluorescently (Alexa 647) labeled anti-telomerase antibody. We applied telomeric constructs (10 nM) labeled with Cy3 without biotin to the telomerase-bound surface and performed a dual excitation detection to capture both signals arising from telomerase (Alexa 647) and the bound DNA substrate (Cy3). The relative binding affinity of each construct for telomerase was calculated from the colocalization efficiency (i.e., ratio of the overlapping fluorophore signal in DNA-bound telomerase over total telomerase Alexa647 only signal). Each value was normalized against the unfolded positive control, 3R, which consists of three repeats of TTAGGG (Figure 4A, red dashed line). The tightly folded 4R (G4) showed the lowest level of binding to telomerase (Figure 4A, black dashed line) as reported previously.50,63 Surprisingly, every construct that harbors a lesion, 8oxoG or O6mG, in the G1–G3 position, uniformly displayed a very high binding affinity for telomerase, even exceeding the level of the 3R control (Figure 4A, gray area). By contrast, all of the T, A, and C mutations displayed extremely low levels of binding to telomerase, regardless of the mutation position (Figure 4A, bottom).
Plotting the telomerase binding affinity against the unfolded fraction of each telomeric construct shows that all G1–G3 constructs fall into two groups (Figure 4A). One group consists of the lesion-containing constructs (Figure 4A, gray oval) that exhibit a strikingly high telomerase binding level regardless of the status of the unfolded population. The second group consists of all of the point mutation constructs that uniformly show an unexpectedly low level of telomerase binding, matching the level seen in the highly folded 4R construct (Figure 4A, black dashed line), regardless of the degree of unfolding. Such an all-or-none pattern between the lesion and mutant constructs is in contrast to C strand and POT1 binding, which are proportional to the structural disruption of the telomeric construct. The uniformly low level of binding of telomerase to all constructs with mutations in G1–G3 clearly indicates that the mutations generate a mismatch with the telomerase RNA template, thereby lowering the base pairing capability. By contrast, 8oxoG and O6mG, although chemically modified, maintain the guanine base and may cause less disruption of the base pairing with the telomerase RNA template. On the basis of these unexpected observations, we posit that unlike the C strand and POT1, telomerase can actively disrupt partially unfolded G4 structures and exhibits high sequence selectivity.
We next performed in vitro primer extension assays to measure telomerase activity on all of the DNA constructs tested above. The telomerase extension products appeared as well-distinguished bands in the gel, demonstrating the addition of hexameric repeats. As shown in Figure 4B, the telomeric substrates harboring 8oxoG or O6mG at the G1 and G2 positions were elongated to lengths similar to those of 3R, suggesting that telomerase can extend a damaged telomeric substrate with similar processivity. We quantified the extension products and plotted with the telomerase binding and C strand accessibility values (Figure 4C). Overall, the telomerase binding and extension activity show a high level of correlation. Exceptions are the G3-8oxoG and G3-O6mG constructs that show low extension activity despite a high telomerase binding level (Figure 4B), clearly indicating the importance of the terminal guanine for extension. This agrees well with our previous finding and work from others that the incorporation of 8oxoG from the cellular dNTP pool terminates telomerase extension.49,64 We also noticed a mild discrepancy between telomerase binding and activity for the constructs with A mutations at G1 and G2, which show a low level of telomerase binding, but intermediate levels of telomerase activity (55% and 38%) (Figure 4C and Figure S5). This may be explained by telomerase tolerating a purine-pyrimidine mismatch, which partially compensates for the lower level of telomerase binding. On the other hand, an A mutation at G3 has ~45% telomerase binding but <10% telomerase activity, which resembles the pattern for 8oxoG and O6mG at G3. Taken together, these results suggest that either an incorrect or damaged base can inhibit further extension when incorporated at the end of a telomere.
8oxoG at G1 or G3 Causes Partial Unfolding of the Telomeric DNA That Allows Telomerase to Engage.
Our previous and current findings corroborate a model that accessibility of telomeric DNA to external factors such as a C strand and POT1 depends on the structural status (i.e., any modification that unlocks the folded G4 structure can increase the accessibility depending on the extent of the disruption). Nevertheless, telomerase binding does not follow this order as demonstrated by the constructs with 8oxoG at G1 and G3, which are largely inaccessible to the C strand but attain 100% telomerase binding (Figure 4A). To further examine a possible rearrangement of the DNA conformation that may not be visible in our current design, we prepared alternatively labeled DNA FRET constructs that provide a better resolution of G4 conformations.38,50 The Cy5 dye was relocated away from the end of the duplex to a phosphate between the fourth and fifth base from the 5′ end (Top4.5) (Figure 5A). This new configuration enables us to distinguish between different G4 conformations as demonstrated by the smFRET histogram of undamaged 4R splitting into one dominant peak at 0.7 FRET (80%) and a minor peak at 0.85, while polyT has a single peak at 0.35 (Figure 5B).
Figure 5.
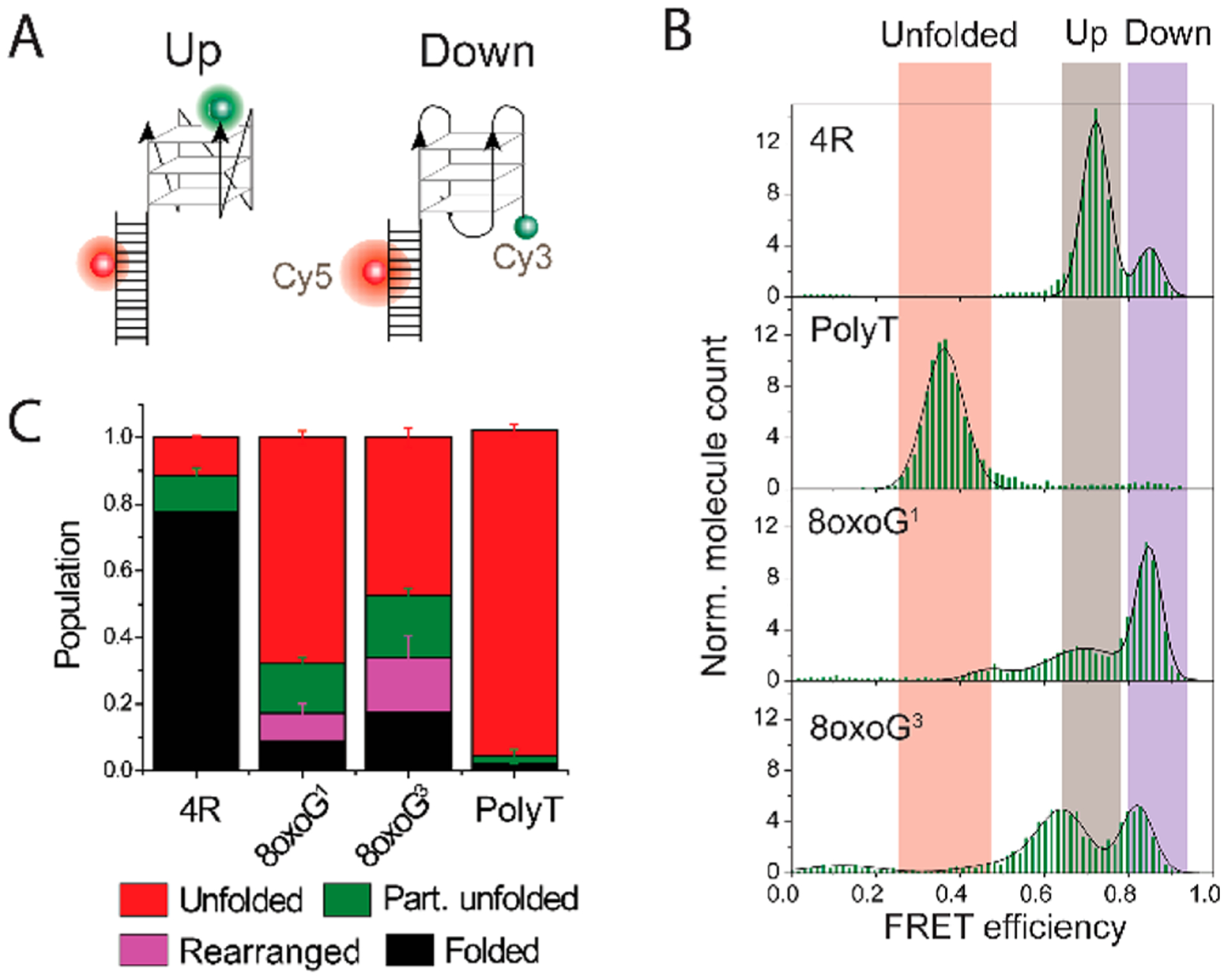
8oxoG at G1 and G3 induces subtle conformational changes that allow telomerase to engage. (A) DNA construct design with an alternative dye position. The Cy5 dye is at the phosphate group between the fourth and fifth bases from the 5′ end (Top4.5). (B) FRET histogram of undamaged DNA and constructs with 8oxoG at G1 or G3 displaying up (gray), down (purple), and unfolded populations (orange). (C) Distribution of single-molecule traces observed with the alternative (Top4.5) Cy5 dye position. The molecules with subtle conformational changes (magenta), which were not detectable with the original Cy5 position, may allow telomerase binding to transiently unfolded molecules. Error bars represent the standard error obtained from randomized sampling process of trace distributions.
In the Top4.5 FRET constructs, the G1-8oxoG and G3-8oxoG substitutions display a different pattern of FRET histograms. While the G1-8oxoG construct shows a dominant FRET peak at the lower value, 0.7 with a small shoulder, the G3-8oxoG construct produces slightly shifted FRET peaks at 0.65 and 0.8 with similar peak sizes (Figure 5B). It is difficult, and not our intention, to distinguish which FRET peak corresponds to which particular G4 conformer, because our FRET construct can report only on the distance between the 5′ and 3′ ends of the (TTAGGG)4 sequence. In Top4.5, all of the G4 conformations that have the same relative orientation between the first and fourth TTAGGG repeat may have a similar FRET value. Therefore, parallel and hybrid-1 G4 [both first and fourth repeats have the same 3′ to 5′ orientation (“up” in Figure 5A)] as well as antiparallel and hybrid-2 [fourth strand has the opposite 3′ to 5′ orientation (“down” in Figure 5A)] will be in one FRET peak. This bias might be explained by an 8oxoG preference for the syn conformation as reported in a recent NMR study.7 While the smFRET assay cannot define the precise G4 structure, it can reveal structural dynamics caused by the lesion. We performed analysis of single-molecule trajectories with the same algorithm used in the previous analysis with the adjusted FRET cutoff values. In addition to the previous categories of folded, partially unfolded, and unfolded, we assigned the additional fluctuating FRET transitions between the two FRET states as “rearranged” (Figure 5C and Figure S6). The fraction of the rearranged population (purple in Figure 5C) is calculated from the difference between the partial unfolded population obtained from the Top4.5 construct (brown in Figure S6) and the construct with the original Cy5 position (green in Figure S1). This “rearranged” population was not identified in the original dye position because the FRET fluctuation range was mixed with the rest of the partially unfolded species. Interestingly, the comparison between this FRET construct (Figure 5A) with the previous FRET construct (Figure 1B) reveals that the constructs with 8oxoG in both G1 and G3 positions undergo high levels of dynamic conformational exchange (Figure 5B). This was masked in the original FRET construct due to the dye locations, which were insensitive to this range of distance change (Figure 3A). Taken together, the increased population of molecules undergoing rearrangement (purple), partial unfolding (green), and unfolding (red) observed in both Top4.5 G1-8oxoG and G3-8oxoG constructs reveals a high degree of dynamics induced by both single-site lesions, which may contribute to the unusually high accessibility of telomerase.
Telomerase Binding Depends on an Open Telomeric Structure and Base Pairing with the RNA Template.
Having shown that telomerase can bind to damaged telomeric DNA with the conformational rearrangement, we asked if the low level of telomerase binding to mutant telomeric DNA (Figure 4A) is due primarily to mismatches between the telomeric primer and the telomerase RNA template. To test this, we moved the mutation to an internal, second repeat (R2) location such that the structural disruption is induced while allowing for correct base pairing with the telomerase RNA template (Figure 6A). We observed that the constructs with a G to T mutation at G1 and G2 positions in the R2 repeat produce FRET histograms and dynamic trace distributions similar to the terminal repeat T mutations (Figure 6A and Figure S7). Consistently, the accessibilities of the C strand and POT1 to constructs in both the second (R2) and terminal (R4) repeats were also comparable, confirming the same degree of structural disruption induced by both mutant positions (Figure S8A,B). Strikingly, the level of binding of telomerase to the R2 mutants reached 100%, dramatically higher than that of the corresponding mutation in the terminal repeat of R4 (Figure S8C). Likewise, the telomerase extension activity of all C, T, and A mutations at G1 and G2 positions in R2 was substantially enhanced (Figure 6B,C), revealing recovered extension activity by all six R2 mutants. This result strongly indicates that telomerase extension activity is greatly enhanced when the G4 structure is disrupted, yet recovery of telomerase binding and extension activity requires correct base pairing between the telomeric overhang and RNA template. In other words, mutations can have a significant impact up- or downregulating telomerase activity in a manner that is highly dependent on position.
Figure 6.
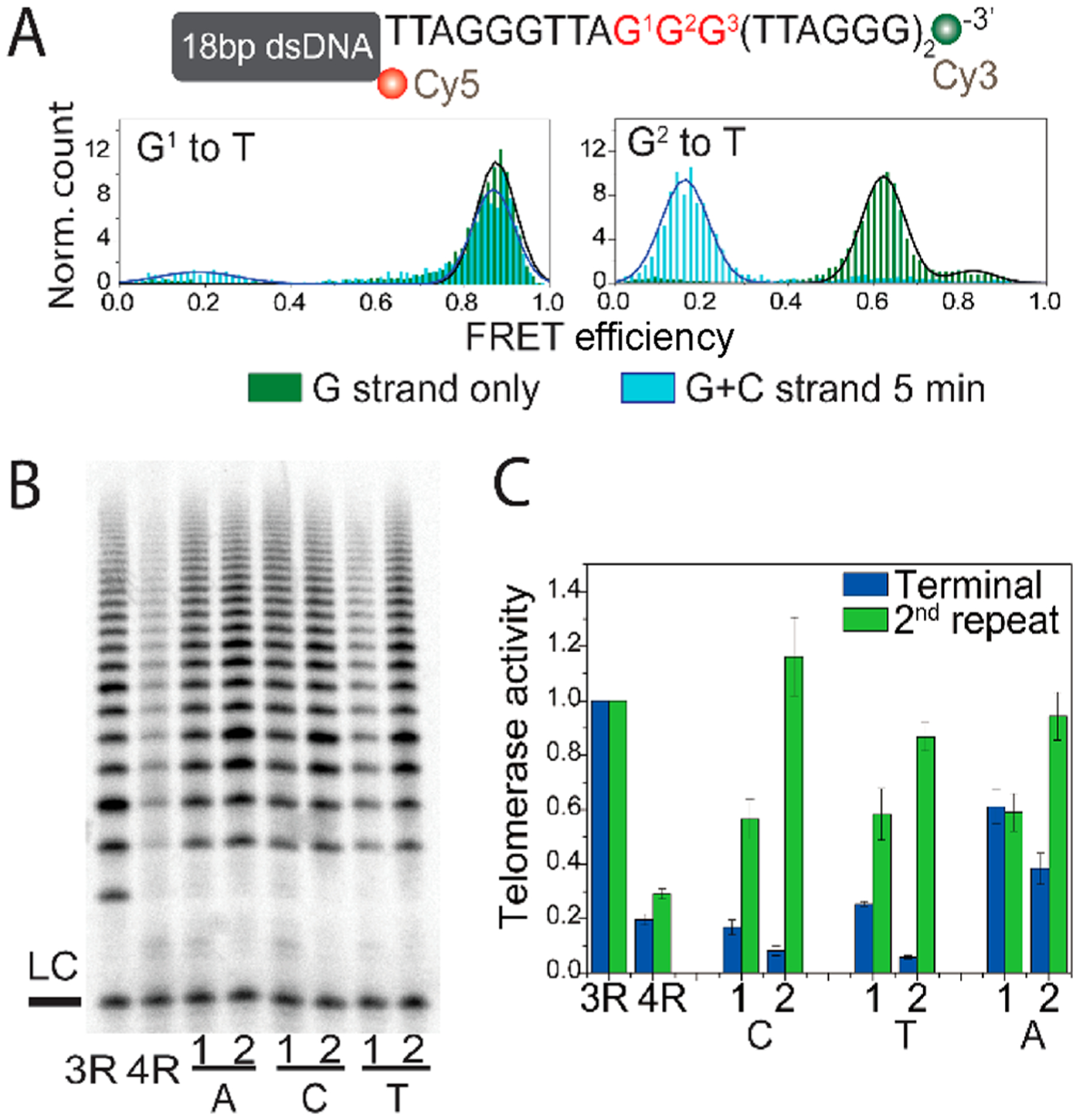
Mutations at the upstream TTAGGG induce conformational changes that allow telomerase to engage. (A) FRET histogram of T mutations at G1 and G2 of the second TTAGGG repeat alone (green) or upon C strand addition (light blue). (B) Gel image of the telomerase activity assay for constructs with the indicated base mutation at G1 and G2 of the second TTAGGG repeat. (C) Quantified telomerase activity from constructs with mutations at G1 and G2 of the second and terminal repeats. The telomerase activity of terminal repeat mutant constructs (blue) was extracted from Figure 4C and is shown here for comparison.
DISCUSSION
The G-quadruplex (G4) is a common noncanonical secondary structure that forms in telomeric single-stranded overhangs, yet the physiological role of this structure in telomeres remains ambiguous. Studies of G4-stabilizing ligands and G4-destabilizing base modifications suggest G4 might play a regulatory role in telomere integrity.49,65,66 Many structural studies reported that base modifications disrupt the hydrogen bonding between guanines and, therefore, destabilize the G4 formed in telomeric overhangs.5–7,41,42 In this study, we systematically tested at the single-molecule level the effects on the structural dynamics of two common DNA lesions, 8oxoG and O6mG, and all possible point mutations, C, T, and A, at the three terminal guanine positions of telomeric DNA overhangs. We took advantage of single-molecule resolution to confirm the formation of unimolecular structure in all of the constructs, because ensemble assays cannot distinguish bimolecular G4 from unimolecular G4.41,42 We further discerned whether multiple conformations and structural dynamics coexist in molecules sharing the exact same base sequence and chemical configuration. Using our newly developed smFRET analytical tool, we categorized >100000 smFRET trajectories into different classes of dynamic conformational changes based on the most unfolded state (lowest FRET value) each single molecule experienced. The FRET values of the unstructured polyT and stably folded G4 were used as markers for unfolded and fully folded G4, respectively. Our data reveal that DNA lesions and mutations shift the distribution of the unfolded, partially folded, or fully folded G4 conformations, to varying extents. Our single-molecule approach reveals how base modifications that affect Hoogsteen hydrogen bonding alter the dynamics of G4 structure, in addition to causing the conformational changes shown by NMR and CD approaches.5–7,41,42
Our results strongly support the conclusion from earlier studies that modification of guanines in telomeric overhang disrupts and destabilizes the G4 structure,6,42 which is most pronounced when the central guanine (G2) is modified or mutated.6 We also showed that lesions and mutations at G1 and G3 cause a G4 structural distortion that differs from the undamaged 4R. This result is consistent with a recent NMR study that shows that G4 with 8oxoG at the central guanine (G2) prefers a different strand orientation from 8oxoG at the other two guanines (G1 and G3).7 Interestingly, our new data reveal that this position-dependent effect extends to all of the modified guanines and mutations we tested, although the extent of G4 disruption and the corresponding accessibility vary with the type of modification. Our results suggest that the structural disruption caused by lesions and mutations might be a general phenomenon arising from any base changes in the telomeric overhang GGG runs.
Using our new method of classifying structural dynamics, we discovered two general rankings that affect the strength of G4 structure disruption independently: (1) G2 > G3 ≥ G1, and (2) O6mG > 8oxoG ≥ A > T > C. This trend of different guanine positions can be explained by the shared potassium ion shown in the X-ray crystal structure.33,67 Losing or misaligning one guanine reduced the number of oxygens that interact with the K+ between adjacent G-quartets. Guanine lesions or mutations at either end, G1 or G3, allow the other two G-quartets to form and be stabilized by one shared K+, while losing a guanine in the central G-quartet makes it unable to hold the K+ between G-quartets. The differences observed among various base modifications can be explained by steric hindrance and electrostatic repulsion that interfere with Hoogsteen base pairing. Such effects will be more prominent with larger purines than pyrimidines. In addition, the smallest distortion caused by cytosine substitution may arise from the Watson–Crick base pairing with the guanine that may stabilize G4. In contrast, substitution with adenine or modified guanines not only eliminates the proper electron donor and acceptor needed for Hoogsteen base pairing but also repels the other three guanines from the G-quartet, perturbing sugar puckering and further disrupting G4 structure. It is worth noting that reports68,69 show telomeric G4 in buffer containing K+ is kinetically more stable and is less accessible than the G4 in Na+. Throughout our study, we used 100 mM K+, which is similar to the intracellular environment and physiologically relevant. At this K+ concentration, G4 is stable and undergoes very few conformational changes. However, we found the base lesions and mutations can still unfold this stable G4, which is reported to be a poor telomerase substrate.61 We reason this destabilization effect and the consequential increase in telomerase accessibility would not be as pronounced in a low-K+ or no K+ environment.
Remarkably, the correlation between structural disruption and telomeric DNA accessibility did not apply for telomerase binding and extension activity. First, all lesions led to a high level of telomerase binding regardless of the damage positions. In addition, our results suggest that telomerase can gain access to weakly folded structures that POT1 or the C strand cannot bind, suggesting an active unfolding mechanism of telomerase. Despite the chemical base changes, telomerase may engage with the damaged guanine that can base pair with the RNA template in telomerase. The binding and extension of telomerase were correlated in all cases except for 8oxoG and O6mG at the G3 position, which showed reduced activity due to the lesions acting as terminating bases.49,64 In contrast, all mutations (except A in the terminal repeat) showed greatly diminished telomerase binding and extension activity. We tested two possible reasons: structural distortion versus imperfect base pairing. Our data support the second scenario. First, all of the G2 mutant constructs have high accessibility to the C strand, arguing against the structural disruption explanation. Second, the detrimental effects of mismatches were confirmed by relocating the mutation to an upstream repeat (R2). A construct with structural disruption lacking a mismatch with the RNA template was sufficient to fully recover telomerase binding and activity, implicating the imperfect base pairing as the primary cause.
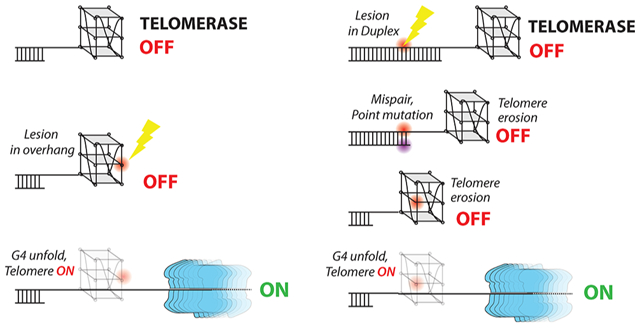
Importantly, our findings provide evidence for a longer-lasting and inheritable consequence of telomeric base damage. First, when a lesion occurs at the telomeric overhang, it can disrupt G4 structure, allowing telomerase to access the 3′ end and triggering telomerase extension activity, which can lead to oncogenic consequences in premalignant cells (Scheme 1A). The damaged base positioned upstream after several cycles of TTAGGG addition can destabilize the G4 structure forming in the DNA product between contacts with the telomerase catalytic site and the anchor site that resides in the TEN domain,70 which prevents DNA dissociation and enhances telomerase processivity. This hypothesis is supported by a report that proposed nascent telomeric DNA folding in the product can trigger product dissociation.71 Moreover, unless repaired, 8oxoG and O6mG lesions arising in the extended overhang, or in the duplex telomeric region, can cause mutations upon C strand fill-in or replication of the complementary C-rich strand, respectively. For example, O6meG will mispair with T during replication and cause a G to A mutation in the G-rich strand. In other words, base lesions in the duplex can be converted to point mutations within telomere DNA. The mutation may reach the overhang through telomere erosion if telomerase is absent or inhibited, and thereby disrupt G4 formation and enable telomerase extension. Therefore, DNA lesions in the telomeres that are converted to mutations may upregulate telomerase activity after rounds of cell replication (Scheme 1B). In summary, our biophysical studies reveal that DNA lesions and consequent mutations can promote or inhibit telomerase activity depending on where the base modification arises.
Scheme 1.
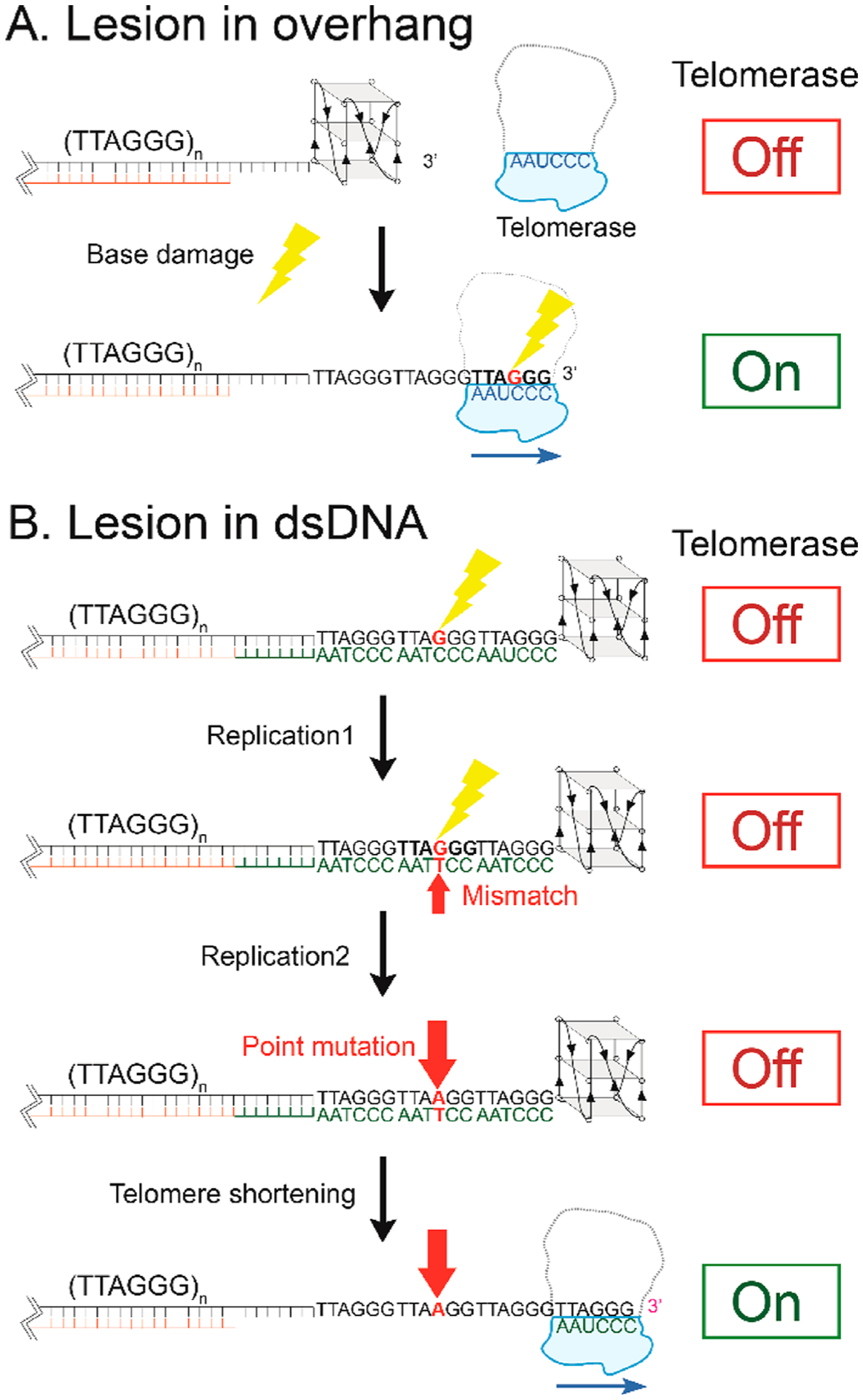
(A) Lesions in an Overhang May Induce Telomerase Activation, and (B) a Base Modification Lesion Might Induce a Long-Term “Unlocking” Effect in the Telomere
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Taekjip Ha for helpful discussion about telomere G-quadruplexes. The authors especially appreciate Ms. Olivia Yang’s help in developing the MatLab script used for semiautomatic analysis of smFRET trajectories. The authors thank Xander Orenstein for assistance with POT1 purification.
Funding
This work was supported by Grant R01 CA 207342-01A1 (all members of the Myong and Opresko laboratory) and Grant R35 ES030396 to P.L.O.
ABBREVIATIONS
- G4
G-quadruplex
- smFRET
single-molecule Förster resonance energy transfer
- SiMPull
single-molecule pull-down
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00434.
List of oligonucleotides, table of binding kinetics, and extra figures of data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.0c00434
The authors declare no competing financial interest.
Contributor Information
Hui-Ting Lee, Thomas C. Jenkins Department of Biophysics, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Samantha Sanford, Department of Environmental and Occupational Health, University of Pittsburgh Graduate School of Public Health and University of Pittsburgh Medical Center Hillman Cancer Center, Pittsburgh, Pennsylvania 15261, United States.
Tapas Paul, Thomas C. Jenkins Department of Biophysics, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Joshua Choe, Thomas C. Jenkins Department of Biophysics, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Arindam Bose, Department of Environmental and Occupational Health, University of Pittsburgh Graduate School of Public Health and University of Pittsburgh Medical Center Hillman Cancer Center, Pittsburgh, Pennsylvania 15261, United States.
Patricia L. Opresko, Department of Environmental and Occupational Health, University of Pittsburgh Graduate School of Public Health and University of Pittsburgh Medical Center Hillman Cancer Center, Pittsburgh, Pennsylvania 15261, United States
Sua Myong, Thomas C. Jenkins Department of Biophysics, Johns Hopkins University, Baltimore, Maryland 21218, United States; Physics Frontier Center (Center for Physics of Living Cells), University of Illinois, Urbana, Illinois 61801, United States.
REFERENCES
- (1).Cadet J, and Wagner JR (2013) DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harbor Perspect. Biol 5, a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- (3).Lipscomb LA, Peek ME, Morningstar ML, Verghis SM, Miller EM, Rich A, Essigmann JM, and Williams LD (1995) X-ray structure of a DNA decamer containing 7,8-dihydro-8-oxoguanine. Proc. Natl. Acad. Sci. U. S. A 92, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Singh SK, Szulik MW, Ganguly M, Khutsishvili I, Stone MP, Marky LA, and Gold B (2011) Characterization of DNA with an 8-oxoguanine modification. Nucleic Acids Res. 39, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cheong VV, Heddi B, Lech CJ, and Phan AT (2015) Xanthine and 8-oxoguanine in G-quadruplexes: formation of a G.G.X.O tetrad. Nucleic Acids Res. 43, 10506–10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vorlickova M, Tomasko M, Sagi AJ, Bednarova K, and Sagi J (2012) 8-oxoguanine in a quadruplex of the human telomere DNA sequence. FEBS J. 279, 29–39. [DOI] [PubMed] [Google Scholar]
- (7).Bielskute S, Plavec J, and Podbevsek P (2019) Impact of Oxidative Lesions on the Human Telomeric G-Quadruplex. J. Am. Chem. Soc 141, 2594–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Takahashi S, Kim KT, Podbevsek P, Plavec J, Kim BH, and Sugimoto N (2018) Recovery of the Formation and Function of Oxidized G-Quadruplexes by a Pyrene-Modified Guanine Tract. J. Am. Chem. Soc 140, 5774–5783. [DOI] [PubMed] [Google Scholar]
- (9).Kamiya H, Miura K, Ishikawa H, Inoue H, Nishimura S, and Ohtsuka E (1992) c-Ha-ras containing 8-hydroxyguanine at codon 12 induces point mutations at the modified and adjacent positions. Cancer Res. 52, 3483–3485. [PubMed] [Google Scholar]
- (10).Kamiya H, Murata-Kamiya N, Koizume S, Inoue H, Nishimura S, and Ohtsuka E (1995) 8-Hydroxyguanine (7,8-dihydro-8-oxoguanine) in hot spots of the c-Ha-ras gene: effects of sequence contexts on mutation spectra. Carcinogenesis 16, 883–889. [DOI] [PubMed] [Google Scholar]
- (11).Cooke MS, Evans MD, Dizdaroglu M, and Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- (12).Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, and Krokan HE (2004) Alkylation damage in DNA and RNA–repair mechanisms and medical significance. DNA Repair 3, 1389–1407. [DOI] [PubMed] [Google Scholar]
- (13).Bauer NC, Corbett AH, and Doetsch PW (2015) The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 43, 10083–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cimino-Reale G, Pascale E, Battiloro E, Starace G, Verna R, and D’Ambrosio E (2001) The length of telomeric G-rich strand 3′-overhang measured by oligonucleotide ligation assay. Nucleic Acids Res. 29, 35e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Palm W, and de Lange T (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet 42, 301–334. [DOI] [PubMed] [Google Scholar]
- (16).Baumann P, and Cech TR (2001) Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292, 1171–1175. [DOI] [PubMed] [Google Scholar]
- (17).Rodriguez R, Muller S, Yeoman JA, Trentesaux C, Riou JF, and Balasubramanian S (2008) A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres. J. Am. Chem. Soc 130, 15758–15759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Abreu E, Aritonovska E, Reichenbach P, Cristofari G, Culp B, Terns RM, Lingner J, and Terns MP (2010) TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol. Cell. Biol 30, 2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hwang H, Buncher N, Opresko PL, and Myong S (2012) POT1-TPP1 regulates telomeric overhang structural dynamics. Structure 20, 1872–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kawanishi S, and Oikawa S (2004) Mechanism of telomere shortening by oxidative stress. Ann. N. Y. Acad. Sci 1019, 278–284. [DOI] [PubMed] [Google Scholar]
- (21).Oikawa S, Tada-Oikawa S, and Kawanishi S (2001) Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 40, 4763–4768. [DOI] [PubMed] [Google Scholar]
- (22).Rhee DB, Ghosh A, Lu J, Bohr VA, and Liu Y (2011) Factors that influence telomeric oxidative base damage and repair by DNA glycosylase OGG1. DNA Repair 10, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yarosh DB (1985) The role of O6-methylguanine-DNA methyltransferase in cell survival, mutagenesis and carcinogenesis. Mutat. Res., DNA Repair Rep 145, 1–16. [DOI] [PubMed] [Google Scholar]
- (24).Rasouli-Nia A, Sigbhat-Ullah Mirzayans, R., Paterson MC, and Day RS 3rd. (1994) On the quantitative relationship between O6-methylguanine residues in genomic DNA and production of sister-chromatid exchanges, mutations and lethal events in a Mer-human tumor cell line. Mutat. Res., DNA Repair 314, 99–113. [DOI] [PubMed] [Google Scholar]
- (25).Morin GB (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521–529. [DOI] [PubMed] [Google Scholar]
- (26).Cristofari G, and Lingner J (2006) Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 25, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Blasco MA (2005) Telomeres and human disease: ageing, cancer and beyond. Nat. Rev. Genet 6, 611–622. [DOI] [PubMed] [Google Scholar]
- (28).Zhu X, Han W, Xue W, Zou Y, Xie C, Du J, and Jin G (2016) The association between telomere length and cancer risk in population studies. Sci. Rep 6, 22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lei M, Zaug AJ, Podell ER, and Cech TR (2005) Switching human telomerase on and off with hPOT1 protein in vitro. J. Biol. Chem 280, 20449–20456. [DOI] [PubMed] [Google Scholar]
- (30).de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110. [DOI] [PubMed] [Google Scholar]
- (31).Ray S, Bandaria JN, Qureshi MH, Yildiz A, and Balci H (2014) G-quadruplex formation in telomeres enhances POT1/TPP1 protection against RPA binding. Proc. Natl. Acad. Sci. U. S. A 111, 2990–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hwang H, Kreig A, Calvert J, Lormand J, Kwon Y, Daley JM, Sung P, Opresko PL, and Myong S (2014) Telomeric overhang length determines structural dynamics and accessibility to telomerase and ALT-associated proteins. Structure 22, 842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Parkinson GN, Lee MP, and Neidle S (2002) Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 417, 876–880. [DOI] [PubMed] [Google Scholar]
- (34).Dai J, Punchihewa C, Ambrus A, Chen D, Jones RA, and Yang D (2007) Structure of the intramolecular human telomeric G-quadruplex in potassium solution: a novel adenine triple formation. Nucleic Acids Res. 35, 2440–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Phan AT, Kuryavyi V, Luu KN, and Patel DJ (2007) Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucleic Acids Res. 35, 6517–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schultze P, Hud NV, Smith FW, and Feigon J (1999) The effect of sodium, potassium and ammonium ions on the conformation of the dimeric quadruplex formed by the Oxytricha nova telomere repeat oligonucleotide d(G(4)T(4)G(4)). Nucleic Acids Res. 27, 3018–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Burge S, Parkinson GN, Hazel P, Todd AK, and Neidle S (2006) Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 34, 5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tippana R, Xiao W, and Myong S (2014) G-quadruplex conformation and dynamics are determined by loop length and sequence. Nucleic Acids Res. 42, 8106–8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lee JY, Okumus B, Kim DS, and Ha T (2005) Extreme conformational diversity in human telomeric DNA. Proc. Natl. Acad. Sci. U. S. A 102, 18938–18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ambrus A, Chen D, Dai J, Bialis T, Jones RA, and Yang D (2006) Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 34, 2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhao A, Zhao C, Tateishi-Karimata H, Ren J, Sugimoto N, and Qu X (2016) Incorporation of O(6)-methylguanine restricts the conformational conversion of the human telomere G-quadruplex under molecular crowding conditions. Chem. Commun. (Cambridge, U. K.) 52, 1903–1906. [DOI] [PubMed] [Google Scholar]
- (42).Mekmaysy CS, Petraccone L, Garbett NC, Ragazzon PA, Gray R, Trent JO, and Chaires JB (2008) Effect of O6-Methylguanine on the Stability of G-Quadruplex DNA. J. Am. Chem. Soc 130, 6710–6711. [DOI] [PubMed] [Google Scholar]
- (43).Johnson JE, Smith JS, Kozak ML, and Johnson FB (2008) In vivo veritas: using yeast to probe the biological functions of G-quadruplexes. Biochimie 90, 1250–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Maizels N (2006) Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat. Struct. Mol. Biol 13, 1055–1059. [DOI] [PubMed] [Google Scholar]
- (45).Macrae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, and Doudna JA (2006) Structural basis for double-stranded RNA processing by Dicer. Science 311, 195–198. [DOI] [PubMed] [Google Scholar]
- (46).Huppert JL (2008) Hunting G-quadruplexes. Biochimie 90, 1140–1148. [DOI] [PubMed] [Google Scholar]
- (47).Lipps HJ, and Rhodes D (2009) G-quadruplex structures: in vivo evidence and function. Trends Cell Biol. 19, 414–422. [DOI] [PubMed] [Google Scholar]
- (48).Biffi G, Tannahill D, McCafferty J, and Balasubramanian S (2013) Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem 5, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Fouquerel E, Lormand J, Bose A, Lee HT, Kim GS, Li J, Sobol RW, Freudenthal BD, Myong S, and Opresko PL (2016) Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol 23, 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lee HT, Bose A, Lee CY, Opresko PL, and Myong S (2017) Molecular mechanisms by which oxidative DNA damage promotes telomerase activity. Nucleic Acids Res. 45, 11752–11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Sowd G, Lei M, and Opresko PL (2008) Mechanism and substrate specificity of telomeric protein POT1 stimulation of the Werner syndrome helicase. Nucleic Acids Res. 36, 4242–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lei M, Podell ER, and Cech TR (2004) Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol 11, 1223–1229. [DOI] [PubMed] [Google Scholar]
- (53).Hwang H, Opresko P, and Myong S (2015) Single-molecule real-time detection of telomerase extension activity. Sci. Rep 4, 6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hwang H, and Myong S (2014) Protein induced fluorescence enhancement (PIFE) for probing protein-nucleic acid interactions. Chem. Soc. Rev 43, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Latrick CM, and Cech TR (2010) POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 29, 924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zaug AJ, Podell ER, Nandakumar J, and Cech TR (2010) Functional interaction between telomere protein TPP1 and telomerase. Genes Dev. 24, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Andersen OS (2013) Cellular electrolyte metabolism, Springer, New York. [Google Scholar]
- (58).Pedroso IM, Duarte LF, Yanez G, Burkewitz K, and Fletcher TM (2007) Sequence specificity of inter- and intramolecular G-quadruplex formation by human telomeric DNA. Biopolymers 87, 74–84. [DOI] [PubMed] [Google Scholar]
- (59).Tomaško M, Vorlíčková M, and Sagi J (2009) Substitution of adenine for guanine in the quadruplex-forming human telomere DNA sequence G3(T2AG3)3. Biochimie 91, 171–179. [DOI] [PubMed] [Google Scholar]
- (60).Bao HL, Liu HS, and Xu Y (2019) Hybrid-type and twotetrad antiparallel telomere DNA G-quadruplex structures in living human cells. Nucleic Acids Res. 47, 4940–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zaug AJ, Podell ER, and Cech TR (2005) Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc. Natl. Acad. Sci. U. S. A 102, 10864–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Loayza D, Parsons H, Donigian J, Hoke K, and de Lange T (2004) DNA binding features of human POT1: a nonamer 5′-TAGGGTTAG-3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J. Biol. Chem 279, 13241–13248. [DOI] [PubMed] [Google Scholar]
- (63).Zahler AM, Williamson JR, Cech TR, and Prescott DM (1991) Inhibition of telomerase by G-quartet DNA structures. Nature 350, 718–720. [DOI] [PubMed] [Google Scholar]
- (64).Aeby E, Ahmed W, Redon S, Simanis V, and Lingner J (2016) Peroxiredoxin 1 Protects Telomeres from Oxidative Damage and Preserves Telomeric DNA for Extension by Telomerase. Cell Rep. 17, 3107–3114. [DOI] [PubMed] [Google Scholar]
- (65).Moye AL, Porter KC, Cohen SB, Phan T, Zyner KG, Sasaki N, Lovrecz GO, Beck JL, and Bryan TM (2015) Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun 6, 7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Yaku H, Murashima T, Miyoshi D, and Sugimoto N (2012) Specific binding of anionic porphyrin and phthalocyanine to the G-quadruplex with a variety of in vitro and in vivo applications. Molecules 17, 10586–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Luu KN, Phan AT, Kuryavyi V, Lacroix L, and Patel DJ (2006) Structure of the human telomere in K+ solution: an intramolecular (3 + 1) G-quadruplex scaffold. J. Am. Chem. Soc 128, 9963–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wang ZF, Li MH, Hsu ST, and Chang TC (2014) Structural basis of sodium-potassium exchange of a human telomeric DNA quadruplex without topological conversion. Nucleic Acids Res. 42, 4723–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Siebenmorgen T, and Zacharias M (2017) Origin of Ion Specificity of Telomeric DNA G-Quadruplexes Investigated by Free-Energy Simulations. Biophys. J 112, 2280–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, and Collins K (2018) Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557, 190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Jansson LI, Hentschel J, Parks JW, Chang TR, Lu C, Baral R, Bagshaw CR, and Stone MD (2019) Telomere DNA G-quadruplex folding within actively extending human telomerase. Proc. Natl. Acad. Sci. U. S. A 116, 9350–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.