

Abstract
Single-cell RNA sequencing has revealed extensive transcriptional cell state diversity in cancer, often observed independently of genetic heterogeneity, raising the central question of how malignant cell states are encoded epigenetically. To address this, here we performed multiomics single-cell profiling–integrating DNA methylation, transcriptome and genotype within the same cells–of diffuse gliomas, tumors characterized by defined transcriptional cell state diversity. Direct comparison of the epigenetic profiles of distinct cell states revealed key switches for state transitions recapitulating neurodevelopmental trajectories and highlighted dysregulated epigenetic mechanisms underlying gliomagenesis. We further developed a quantitative framework to directly measure cell state heritability and transition dynamics based on high-resolution lineage trees in human samples. We demonstrated heritability of malignant cell states, with key differences in hierarchal and plastic cell state architectures in IDH-mutant glioma versus IDH-wild-type glioblastoma, respectively. This work provides a framework anchoring transcriptional cancer cell states in their epigenetic encoding, inheritance and transition dynamics.
Single-cell RNA sequencing (scRNA-seq) of human tumors provides a powerful means to systematically interrogate the diversity of malignant and normal cell states. Recent studies have highlighted transcriptional cell state diversity across tumor types that is often independent of genetic clonal heterogeneity1–3. Thus, tumors are composed of admixtures of cells that differ in central phenotypes1,4–7, prompting several key questions. For example, how are transcriptional cell states encoded epigenetically? How heritable are malignant cell states? Further, what are the transition dynamics between cell states? While exploration of these central aspects of cancer cell states has begun in model organisms using artificial constructs for lineage tracing8–12, these questions remain largely unexplored in primary patient samples.
Human gliomas serve as an instructive model to address these questions, as cell state diversity is an important disease hallmark of both IDH-mutant (IDH-MUT) glioma and IDH-wild-type glioblastoma (GBM), with malignant cells recapitulating trajectories of neural development13–16. Stemness-to-differentiation diversity is central to the glioma stem-cell (GSC) model, which posits that stem-like cells are uniquely capable of self-renewal, tumor propagation and preferential resistance to therapy17–19. Recent scRNA-seq profiling of gliomas provided high-resolution mapping of cell state diversity and offered additional granularity to the GSC model by revealing multiple transcriptionally defined cell states related to neurodevelopmental cell types, which are in part independent of intratumoral genetic diversity7,16,20–26. Yet, while cellular states can be precisely delineated by scRNA-seq, transcriptional information provides only a snapshot of the current state of the cell; therefore, glioma cell state heritability and transition dynamics are not readily discernable. Indeed, while malignant cell states may be propagated epigenetically27–30, the epigenetic underpinning of glioma cellular states is still largely unknown.
This question is of clinical relevance as heritable expression programs may be related to non-genetic mechanisms of therapy resistance in cancer5,6. Increased plasticity allowing for both differentiation and dedifferentiation may also offer a mechanism by which tumors could replenish their stem-cell compartment under therapeutic pressure. Attempts at addressing the dynamics of cell state transitions in glioma samples with stand-alone scRNA-seq modalities (for example, by RNA velocity31) have generated conflicting results32, suggesting that additional technological and analytical breakthroughs are required. To address these questions, we applied joint capture of transcriptional, genetic and epigenetic information at single-cell resolution33 to primary diffuse gliomas. We leveraged this approach to increase the resolution of single-cell identification of copy number alterations (CNAs), demonstrate significant DNA methylation intratumoral heterogeneity (ITH), and reveal the epigenetic encoding, heritability and plasticity of cell states in glioma.
Results
High-resolution CNA mapping by single-cell multiomics.
We profiled viable cells enriched for CD45− cells from GBM (n = 7) and IDH-MUT glioma (n = 7) primary patient samples with multimodality single-cell sequencing of DNA methylation (scDNAme; by multiplexed single-cell reduced-representation bisulfite sequencing (MscRRBS)), scRNA-seq (Smart-seq2; ref. 34) and targeted genotyping33 (Fig. 1a, Extended Data Figs. 1 and 2, and Supplementary Tables 1 and 2). After quality control, we obtained a mean of 113 cells per sample (range, 28-339 cells), with DNA methylomes with a mean ± s.e.m. of 198,345 ± 4,307 unique CpGs per cell and transcriptomes with a mean ± s.e.m. of 6,348 ± 43 genes per cell (Supplementary Table 3), comparable to results with stand-alone full-length scRNA-seq7,21,22. We then separated malignant cells from non-malignant cells on the basis of clustering of either gene expression or DNA methylation data (Fig. 1b and Extended Data Fig. 2c). Non-malignant cells expressed either typical oligodendrocytic markers (for example, PLP1) or myeloid cell markers (for example, CD14) (Extended Data Fig. 3a).
Fig. 1 |. Multiomics single-cell sequencing of primary human gliomas reveals intratumoral DNA methylation heterogeneity.
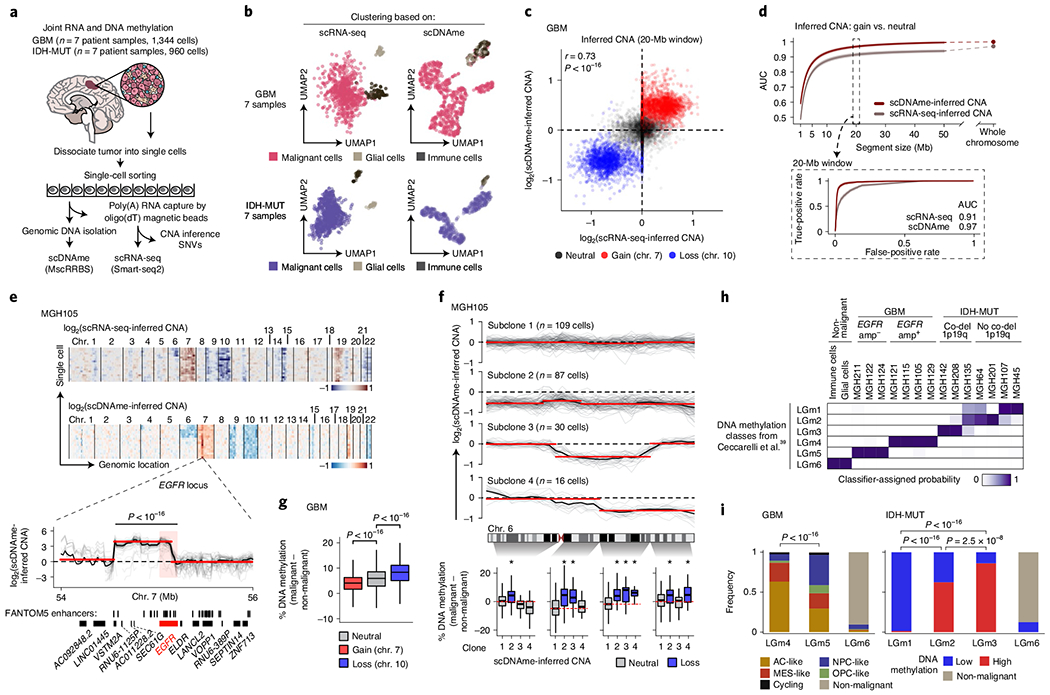
a, Joint methylomics and transcriptomics analysis applied to seven GBM and seven IDH-MUT glioma samples. b, UMAP plots of single cells that passed quality control based on data from scRNA-seq (left; GBM, n = 937; IDH-MUT, n = 809) or scDNAme (right; GBM, n = 867; IDH-MUT, n = 718). c, CNA inference by scDNAme (y axis) versus scRNA-seq (x axis) in 20-Mb windows. Pearson’s correlation coefficient is indicated. d, Performance of CNA inference by scDNAme (red line) and scRNA-seq (gray line) in correctly classifying regions of chromosome gain versus neutral regions, as assessed by the AUC of the receiver operating characteristic (ROC) curve at different genomic window resolutions. Inset, ROC curves at 20-Mb resolution. 95% confidence intervals were generated using bootstrapping. e, Top, representative example (MGH105) of CNA inference by scRNA-seq and scDNAme. Rows correspond to cells, clustered by overall CNA pattern. Bottom, CNA inference by scDNAme centered at the EGFR locus. CNA profiles for individual cells are shown in gray, with the mean per sample shown in black. Red lines represent CNA segments identified by circular binary segmentation analysis. f, Top, CNA inference by scDNAme at chromosome 6 showing distinct genetic subclones within the same tumor (MGH105). Color legend as in e. Bottom, CpG methylation changes at four regions of chromosome 6 when comparing the DNA methylation level of individual cells in each subclone to baseline. *P < 0.05. g, Percentage of CpG methylation change at regions with copy number gain (chromosome 7) or loss (chromosome 10) and neutral regions (chromosome 1) when comparing DNA methylation levels for individual GBM cells to baseline across all GBM samples. h, Heat map of probability assignment for pseudo-bulk DNA methylation profiles (based on MscRRBS) of malignant cells across all GBM and IDH-MUT glioma samples and non-malignant cells (defined in b) to previously described LGm classes39 based on a multinomial logistic regression classifier (Methods). Amp, amplification; co-del, co-deletion. i, Proportion of all single cells (defined in b) assigned to previously described DNA methylation LGm classes39. P values were determined by two-sided Mann–Whitney U test (e–g) or Fisher’s exact test (i). Boxplots represent the median and bottom and upper quartiles; whiskers correspond to 1.5 times the interquartile range.
To orthogonally validate malignant versus non-malignant classification, we identified CNAs within each cell on the basis of coverage depth imbalance in the DNA methylation data (Extended Data Figs. 1a and 2a). CNA inference by scDNAme enabled robust detection of amplifications and deletions in malignant cells, including the hallmark chromosome 7 gain and chromosome 10 loss in GBM and chromosome 1p/19q co-deletion in IDH-MUT oligodendroglioma (IDH-O) (Extended Data Figs. 1a and 2a). While CNA inference by scDNAme correlated with CNA inference by scRNA-seq7,21 (Pearsons r = 0.73; Fig. 1c), direct comparison7,21 at clonal CNAs (identified by bulk whole-exome sequencing with matched samples) (Extended Data Fig. 3b) showed that scDNAme-based CNA inference afforded greater resolution (Fig. 1d, Extended Data Fig. 3c and Supplementary Table 4) and enabled detection of focal amplifications of oncogenes (for example, EGFR, encoding epidermal growth factor receptor) and their neighboring enhancers35 (Fig. 1e and Extended Data Fig. 3d–f).
Higher-resolution scDNAme-based CNA inference further revealed the presence of genetic subclones in both GBM and IDH-MUT tumors (Extended Data Figs. 1a and 2a). For example, we identified distinct genetic subclones marked by either complete or partial chromosome 6 loss in four spatially distinct regions sampled from the same GBM tumor (MGH105) (Fig. 1f, top). Notably, copy number loss was associated with increased methylation, such that DNA methylation levels increased specifically in the chromosome 6 segments lost in each subclone ([6p25–6p11], [6q12–6q15], [6q16–6q23.2] and [6q23.3–6q27]; Fig. 1f, bottom). This pattern was observed more broadly, with increased DNA methylation with copy number loss (for example, loss of chromosome 10 in GBM or chromosomes 1p/19q in IDH-O tumors) and decreased DNA methylation with copy number gain (for example, gain of chromosome 7 in GBM or chromosomes 7/8 in IDH-MUT tumors) across patient samples (Fig. 1g and Extended Data Fig. 3g–i). While such an association between CNAs and subtle DNA methylation changes (<5% on average) has previously been observed in bulk samples36, the underlying mechanism remains unclear and may be related to recruitment of DNA methyltransferases (DNMT1 and DNMT3B) and Polycomb family members (SIRT1 and EZH2) at the chromosomal breaks that lead to CNAs37. The observed anticorrelation between copy number and DNA methylation may serve as a mechanism that amplifies gene expression changes due to CNAs38.
Single-cell DNA methylation analysis reveals significant DNA methylation ITH.
Diffuse gliomas have been classified into six distinct tumor subtypes (LGm1–LGm6) by bulk DNA methylation analysis39. LGm1–LGm3 are enriched for IDH-MUT tumors and show genome-wide hypermethylation, while LGm4–LGm6 are enriched for GBM tumors. We hypothesized that the scDNAme data might reveal ITH in these bulk DNA methylation profiles, that is, that each IDH-MUT tumor might be composed of an admixture of the LGm1–LGm3 DNA methylation subtypes, while each GBM tumor might span the LGm4–LGm6 subtypes. To test this hypothesis, we trained a classifier, robust across DNA methylation platforms, on 932 glioma samples from The Cancer Genome Atlas (TCGA)40,41 profiled with the 450K methylation array and recovered the expected bulk DNA methylation subtypes, achieving a mean accuracy of 0.94 in fivefold cross-validation. When this classifier was applied to pseudo-bulk DNA methylation profiles (based on MscRRBS) of malignant cells in our samples, it assigned each sample to its expected DNA methylation subtype, with IDH-MUT pseudo-bulk DNA methylation profiles classified as LGm1, LGm2 or LGm3 depending on their 1p/19q co-deletion status and GBM samples resolved into either LGm4 or LGm5 depending on their EGFR amplification status (Fig. 1h and Extended Data Fig. 3d–f). Notably, pseudo-bulk analysis of non-malignant glial and immune cells classified them into LGm6 (Fig. 1h), a subtype found in 77 of the 932 TCGA gliomas and associated with either GBM or pilocytic astrocytoma-like gliomas, suggesting that the tumor microenvironment may contribute to bulk subtype assignments to LGm6.
We then scored each glioma single cell to the six tumor subtypes (LGm1–LGm6; Methods) and observed that single cells within individual IDH-MUT tumors spanned the LGm1–LGm3 subtypes, while single cells within individual GBM tumors spanned the LGm4 and LGm5 subtypes (Extended Data Figs. 1c and 2d,e). Such ITH in DNA methylation subtypes is important to recognize, as bulk DNA methylation profiling is increasingly being used for clinical classification of brain tumors42. In IDH-MUT tumors, no correlation with cellular states7 was detected, but instead we found an association with genome-wide DNA methylation levels (Fisher’s exact test, P < 2.5 × 10−8; Fig. 1i and Extended Data Fig. 2d–f), as previously observed in bulk DNA methylation profiles39. By contrast, in GBM, ITH in the LGm4 and LGm5 DNA methylation subtypes correlated with recently defined GBM cellular states based on the expression of defining gene modules in matching scRNA-seq profiles21 (correlation of LGm4 with AC- and MES-like cell states and LGm5 with NPC- and OPC-like cell states; cell state definitions below; Fisher’s exact test, P < 10−16) (Fig. 1i and Extended Data Fig. 1c,d).
GBM stem-like cells exhibit PRC2 target hypomethylation.
To define the distinct DNA methylation profiles of glioma cell states, we first classified glioma cells on the basis of expression of gene modules and cell cycle programs previously defined in scRNA-seq data7,21 (Methods). GBM samples exhibited four malignant cell states, spanning stem/progenitor-like cells (neural progenitor-like (NPC-like) cells and oligodendrocyte progenitor-like (OPC-like) cells) and more differentiated states associated with astrocyte-like (AC-like) or mesenchymal-like (MES-like) programs (Fig. 2a and Supplementary Table 5), with varying representation across samples and cell cycle expression (Extended Data Fig. 1b), as previously observed21.
Fig. 2 |. PRC2 target DNA methylation is a key switch in the differentiation of malignant GBM cells.
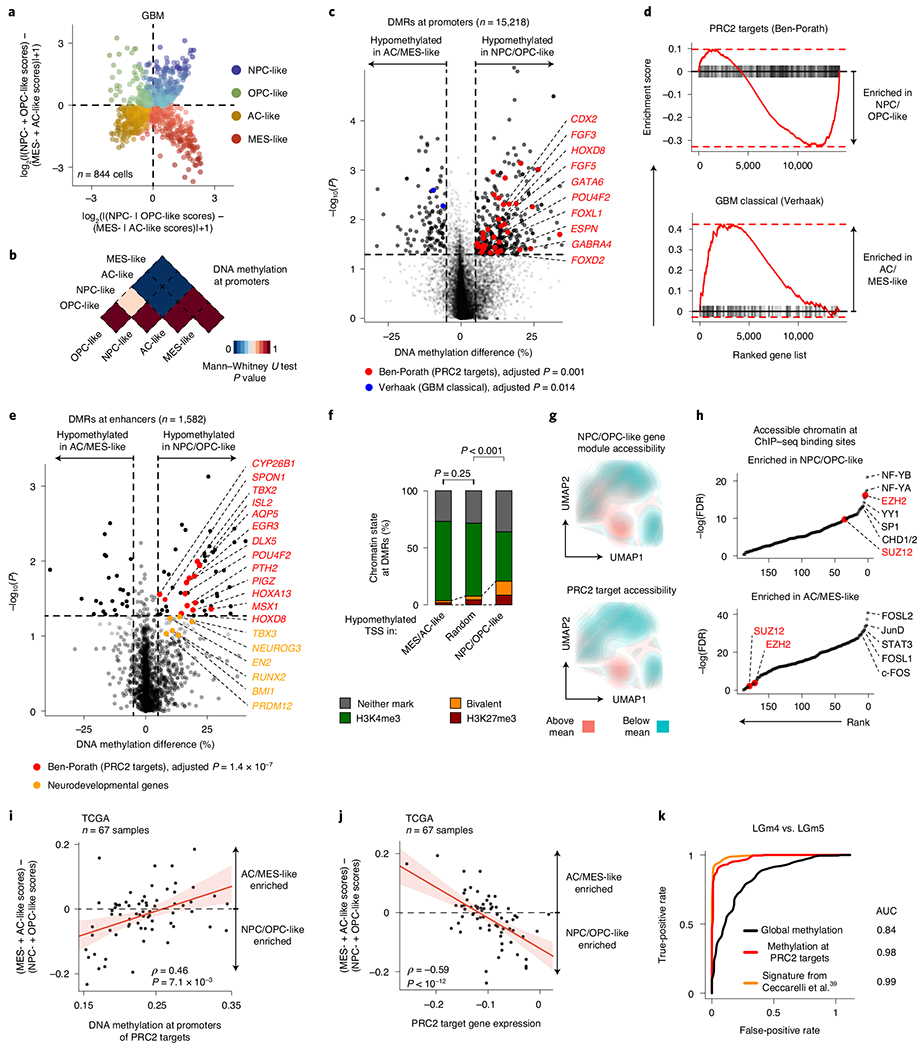
a, Two-dimensional representation of cellular states across seven GBM samples (n = 844 malignant-only cells that passed scRNA-seq quality control). b, Heat map of P values obtained when comparing mean CpG methylation at promoters (TSS ± 1 kb) between GBM cellular states (n = 706, cells in a with matched scDNAme data). c, Volcano plot of differentially methylated promoters between the NPC/OPC-like and AC/MES-like GBM cellular states. Promoters (n = 459) with an absolute mean DNA methylation difference of greater than 5% and a P value below 0.05 were defined as differentially methylated. Genes correlated with the classical TCGA GBM subtype45 (blue) and genes corresponding to PRC2 targets46 (red) are highlighted (BH FDR-adjusted permutation-based P < 0.05). d, Enrichment score plots (reflecting whether a gene set is over-represented at the top or bottom of the ranked list of genes used in c; n = 15, 218 genes) for gene sets enriched at hypomethylated promoters in NPC/OPC-like (top) or AC/MES-like (bottom) cells. e, Volcano plot of differentially methylated enhancers in comparison of NPC/OPC-like and AC/MES-like GBM cellular states. Putative gene targets51 of hypomethylated enhancers in stem-like cells that are PRC2 targets46 are labeled in red. Key neurodevelopmental genes are highlighted in orange. f, Proportion of chromatin states at hypomethylated promoters in GBM AC/MES-like cells (defined in c), randomly sampled promoters (1,000 promoters sampled) and hypomethylated promoters in GBM stem-like cells (defined in c). g, UMAP plots of scATAC-seq GBM data55 (sample SF11956) overlaid with density plots showing chromatin accessibility of genes belonging to NPC/OPC-like gene modules (top) and PRC2 targets46 (bottom). h, Comparison of rank (by P value) in the enrichment of open chromatin at transcription factor-binding sites (n = 188) between NPC/OPC-like (top) and AC/MES-like (bottom) cells. PRC2 subunits are highlighted in red. i, Spearman’s rank-order correlation between mean DNA methylation at the promoters of PRC2 targets46 and RNA differentiation score (defined as the difference in gene module scores between AC/MES-like and NPC/OPC-like cellular states) for 67 TCGA GBM samples40,41. A linear regression line (red) with its 95% confidence interval is shown. j, Same as in i, for PRC2 target46 gene expression and RNA differentiation score. k, Comparison of performance, as assessed by ROC curve, in correctly classifying bulk TCGA GBM samples40,41 to DNA methylation glioma subtypes (LGm4 or LGm5)39 using 1,300 previously defined CpG sites39, mean global DNA methylation and mean PRC2 target46 DNA methylation. P values were determined by two-sided Mann-Whitney U test (b), generalized linear model (c,e), permutation test (f), BH FDR-adjusted hypergeometric test (h) or Spearman’s rank-order correlation (i,j).
Comparison of promoter DNA methylation between transcriptional cell states revealed that, while stem-like cells were markedly different from differentiated-like cells, smaller differences were present in promoter DNA methylation levels within stem-like cells or within differentiated-like cells (Fig. 2b, Extended Data Fig. 4a and Supplementary Table 6). These data suggest that these pairs of cell states are more closely related to each other and that regulatory mechanisms other than DNA methylation, such as interaction with the tumor microenvironment, may drive certain state transitions43. In line with cross-talk between GBM cells and immune cells driving MES-like cell state transitions43,44, immune response-related genes were found to be upregulated in MES-like cells (Benjamini–Hochberg (BH) false-discovery rate (FDR)-adjusted P < 0.05; Extended Data Fig. 4b and Supplementary Table 7).
We thus focused our analysis on comparison of DNA methylation profiles between stem-like and more differentiated-like states, identifying 459 promoter differentially methylated regions (DMRs) (Fig. 2c, Extended Data Fig. 4c,d and Supplementary Table 6). Hypo-methylated promoters in AC- and MES-like cells were enriched for genes correlated with the ‘classical’ TCGA GBM subtype (TCGA-CL)45, in line with the enrichment of AC-like cells in TCGA-CL (BH FDR-adjusted permutation-based P < 0.05; Fig. 2c,d, Extended Data Fig. 4e and Supplementary Table 6).
By contrast, we identified Polycomb repressive complex 2 (PRC2) targets46 as hypomethylated in NPC- and OPC-like cells as compared to AC- and MES-like cells (BH FDR-adjusted permutation-based P < 0.05; Fig. 2d, Extended Data Fig. 4e,f and Supplementary Table 6). These hypomethylated PRC2 targets were enriched for HOX (for example, HOXD8, HOX11 and HOXA6) and homeobox (for example, CDX2 and POU4F2) genes, as well as for transcription factors (for example, GATA5, GATA6, FOXL1 and LHX2) and growth factors (for example, FGF3–FGF5) (BH FDR-adjusted Fisher’s exact test, P < 0.05; Supplementary Table 6), previously reported to have a role in the epigenetic regulation of stemness in GBM47. Notably, NPC- and OPC-like cells exhibited DNA hypomethylation of PRC2 targets as compared to AC- and MES-like cells even within GBM samples from the same patient (Extended Data Fig. 4g–i), suggesting that PRC2 target DNA hypomethylation is a key determinant of stem-like GBM cell states48,49. This was further confirmed when using chromatin immunoprecipitation and sequencing (ChIP–seq) maps50 for the PRC2 subunits EZH2 and SUZ12 (Mann–Whitney U test, P < 0.0001; Extended Data Fig. 4j). We similarly defined enhancer DMRs and found that the putative gene targets51 of hypomethylated enhancers in stem-like cells were also enriched for PRC2 targets46 (Fig. 2e and Supplementary Table 6). As direct cross-talk between PRC2 and DNA methylation has been reported52,53, these data suggest that DNA methylation marks cell states through its interaction with PRC2 and its ability to catalyze the addition of H3K27me3 marks.
To explore the link between DNA methylation and histone marks, we interrogated the differentially methylated promoters for enrichment of histone marks associated with non-overlapping regulatory functions47. While hypomethylated promoters in AC- and MES-like cells were predominantly marked by histone modifications associated with active transcription (H3K4me3, H3K27ac and H3K36me3), hypomethylated promoters in NPC- and OPC-like cells were enriched in bivalent (H3K4me3 + H3K27me3) chromatin (permutation-based P < 0.001; Fig. 2f and Extended Data Fig. 5a–e), suggesting that PRC2 complex activity may result in poised transcription at these gene promoters54. Indeed, the PRC2 subunit EZH2 and its targets46 were found to be upregulated (>2-fold increase) in NPC- and OPC-like cells in comparison to AC- and MES-like cells (Extended Data Fig. 5f,g and Supplementary Table 7).
To further validate the association between the stem-like states and PRC2 activity, we reanalyzed data from GBM single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq)55. GBM cells formed clusters associated with the four core malignant cellular states described by scRNA-seq (Extended Data Fig. 5h). Gene expression activity inferred from scATAC-seq open chromatin (Methods) revealed a positive correlation between PRC2 target accessibility and the NPC- and OPC-like cellular states in single cells (hypergeometric test, P = 0.0015; Fig. 2g and Extended Data Fig. 5i). Similarly, intersecting open chromatin with ChIP–seq maps revealed that binding sites for the PRC2 subunits EZH2 and SUZ12 were among the most enriched in NPC- and OPC-like cells as compared to AC- and MES-like cells (Fig. 2h).
To examine this association in a larger sample cohort, we leveraged 67 GBM samples from TCGA with matched bulk RNA-seq and 450K methylation profiles40,41. In line with our model, we found a positive correlation between the DNA methylation of PRC2 targets46 and glioma differentiation (Fig. 2i and Extended Data Fig. 5j), as well as an anticorrelation between the expression of PRC2 targets46 and glioma differentiation (Fig. 2j). These data confirm that PRC2 targets not only are hypomethylated but also show greater expression in stem-like cells. We note that these findings are consistent with the suppressive role of PRC2, as its targets showed lower gene expression than non-PRC2 targets across all GBM samples. However, the degree of repression was stronger in tumors enriched for differentiated-like cell states, where these gene promoters also underwent silencing through DNA methylation (Mann–Whitney U test, P < 0.0001; Extended Data Fig. 5k). As expected, PRC2 target promoter DNA methylation was lower in LGm5 cells (enriched for NPC- and OPC-like cells) than in LGm4 cells (enriched for AC- and MES-like cells) (Extended Data Fig. 6a). TCGA bulk glioma DNA methylation profiles recapitulated this finding with lower PRC2 target DNA methylation in LGm5 tumors than in LGm4 tumors (Extended Data Fig. 6b,c). In fact, using just mean PRC2 target DNA methylation as a single feature in the classifier separated bulk glioma DNA methylation subtypes (LGm4 and LGm5)39 with comparable accuracy as the multinomial logistic regression classifier (area under the curve (AUC) of 0.98 versus 0.99, respectively; Fig. 2k), suggesting that PRC2 target DNA methylation underlies the classification of GBM tumors by bulk DNA methylation.
Collectively, these data show that DNA methylation of PRC2 targets is a critical feature of GBM cell differentiation. This epigenetic encoding of glioma supports the parallels between glioma differentiation and physiological neurodevelopment where stemness is also marked by PRC2 target hypomethylation56. Maintaining PRC2 targets in a hypomethylated state in glioma stem-like states may thus preserve their stemness potential and allow their reactivation in response to stimuli.
Aberrant epigenetic and transcriptional mechanisms in IDH-MUT gliomas.
In line with previous reports, IDH-MUT malignant cells were found to be differentiated along the astrocytic (AC-like) or oligodendrocytic (OC-like) glial lineages, with a subpopulation of undifferentiated cells associated with an NPC-like expression program23 (Extended Data Fig. 2b and Supplementary Table 5). Cells with cell cycle expression signatures were enriched in this latter subpopulation, supporting a model in which stem-like cells are primarily responsible for fueling the growth of IDH-MUT tumors7 (Extended Data Fig. 2b). In contrast to GBM, differentially methylated promoters in comparisons of stem-like cells with AC- and OC-like cells in IDH-MUT samples were not enriched for PRC2 targets (Extended Data Fig. 7a–g and Supplementary Table 8). In addition, we did not observe significant enrichment of bivalent and repressive chromatin marks at hypomethylated promoters in stem-like cells as compared to AC- and OC-like cells (Extended Data Fig. 7h–l), suggesting that different epigenetic patterning is at play in the maintenance of stemness in IDH-MUT gliomas.
Mutated IDH produces 2-hydroxyglutarate (2HG), an onco-metabolite and a competitive inhibitor of the TET family of 5-methlycytosine hydroxylases57. TET enzymes oxidize 5-methylcytosines to promote demethylation, and deficiency in TET activity may lead to increased DNA methylation, primarily at regulatory elements58–61. Indeed, DNA methylation levels were highest in IDH-MUT cells as compared to GBM and non-malignant cells at gene promoters (Mann-Whitney U test, P < 10−16; Fig. 3a). Comparison of GBM and IDH-MUT samples revealed that enhancers were particularly susceptible to hypermethylation in IDH-MUT cells (Mann–Whitney U test, P < 10−16; Fig. 3b), which also affected regions enriched for H3K27ac—a histone modification marking active enhancers62 (Extended Data Fig. 8a,b). To obtain higher-coverage single-cell DNA methylomes in CpG-sparse regions, such as enhancers, we performed dual-restriction enzyme digestion (HaeIII + MspI) of cells from two IDH-MUT samples (MGH201 and MGH208). This allowed us to increase coverage to a mean of 325,492 ± 21,118 unique CpGs per cell as compared to IDH-MUT cells digested with a single restriction enzyme (Extended Data Fig. 8a), thus enabling more accurate measurement of DNA methylation in regulatory regions. Enhancer hypermethylation was observed in both subsets of cells from IDH-MUT tumors exhibiting the glioma CpG island methylator phenotype (G-CIMP-low and G-CIMP-high subsets) (Extended Data Fig. 8c), supporting the preferential involvement of TET enzymes in the regulation of DNA methylation at enhancers59–61. We observed that enhancer DNA hypermethylation increased with differentiation to AC- and OC-like cells as compared to NPC-like cells (Mann–Whitney U test, P = 0.016; Fig. 3c and Extended Data Fig. 8d).
Fig. 3 |. Increased enhancer DNA methylation, decoupling of promoter methylation-expression relationship and disruption of CTCF insulation define the IDH-MUT epigenome.
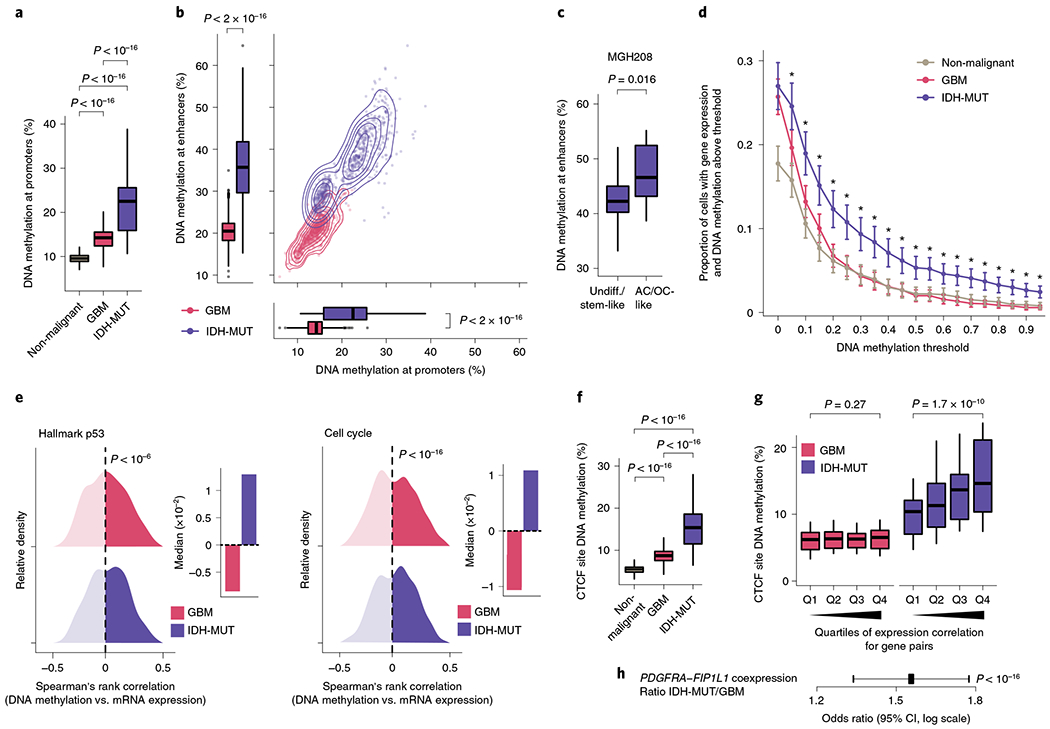
a, Mean CpG methylation at promoters (TSS±1 kb) comparing non-malignant cells (n = 148) with GBM (n = 765) and IDH-MUT (n = 670) malignant cells. b, Mean CpG methylation at promoters versus FANTOM5 enhancers for GBM (n = 765) and IDH-MUT (n = 670) malignant cells. c, Mean CpG methylation at FANTOM5 enhancers for undifferentiated/stem-like and AC/OC-like IDH-MUT cells (MGH208; n = 123 cells with matched scRNA-seq and scDNAme data). d, Proportion of cells with gene expression (RNA read count > 0) and exhibiting above-threshold DNA methylation. Data are shown as mean±s.e.m. across all genes with sufficient RNA (expression seen in >5 cells) and DNA methylation (>5 CpGs per promoter) information across non-malignant cells (n = 148) and GBM (n = 765) and IDH-MUT (n = 670) malignant cells. *P < 0.05. e, Left plot, distribution of Spearman’s rho for correlation of expression with promoter DNA methylation (n = 1,523 genes expressed in >5 cells, DNA methylation at >5 CpGs per promoter) across GBM (n = 765) and IDH-MUT (n = 670) malignant cells. The distribution of Spearman’s rho values was compared to the distribution of values obtained with randomly permuted cell labels. Right plot, median values of Spearman’s rho for correlation of expression with promoter DNA methylation. See also Extended Data Fig. 8g. f, Mean CpG methylation at CTCF-binding sites comparing non-malignant cells (n = 148) with GBM (n = 765) and IDH-MUT (n = 670) malignant cells. g, Mean CpG methylation at CTCF-binding sites per quartile of gene expression correlation (Spearman’s rho) for previously defined pairs of neighboring genes separated by CTCF-binding sites70 in GBM (n = 765) and IDH-MUT (n = 670) malignant cells. h, The log odds ratio of PDGFRA-FIP1L1 gene pair coexpression (for both genes, RNA read count > 0) in IDH-MUT and GBM malignant cells. Error bar represents the 95% confidence interval (CI). P values were determined by two-sided Mann-Whitney U test (a–d,f,g; by comparing the proportion of cells with gene expression and exhibiting above-threshold DNA methylation in IDH-MUT and GBM cells at each DNA methylation threshold in d), two-sided Kolmogorov-Smirnov test (e) and Fisher’s exact test (h). Boxplots represent the median and bottom and upper quartiles; whiskers correspond to 1.5 times the interquartile range.
Cancers are known to exhibit stochastic DNA methylation changes (epimutations), resulting in discordant DNA methylation at neighboring CpGs33,63–66. In line with this notion, single-cell epimutation at promoters was higher overall in malignant cells than in non-malignant cells (Extended Data Fig. 8e). There were more epimutations at promoters in IDH-MUT cells than in GBM cells, in line with a deficiency in TET-mediated demethylation58 (Mann–Whitney U test, P < 10−16; Extended Data Fig. 8e). This increase in promoter epimutation was associated with decoupling of the typical anticorrelation between gene expression and promoter (transcription start site (TSS) ± 1 kb) DNA methylation67 in IDH-MUT malignant cells (Mann–Whitney U test, P < 0.05; Fig. 3d and Extended Data Fig. 8f). This decoupling led to a positive correlation between DNA methylation and expression, such that expression of genes central to the oncogenic phenotype (for example, cell cycle and DNA damage response genes) persisted despite high promoter DNA methylation68,69 (Fig. 3e and Extended Data Fig. 8g).
An additional mechanism through which hypermethylation in IDH-MUT cells may cause aberrant gene activation is through stochastic hypermethylation of CTCF-binding sites (Mann–Whitney U test, P < 10−16; Fig. 3f), with loss of gene insulation between topologically associating domains (TADs) leading to aberrant enhancer–promoter interactions70. To directly assess cell-to-cell variation in CTCF-binding site methylation and insulation efficacy, we identified pairs of neighboring genes separated by TAD-boundary-associated CTCF-binding sites (<180 kb apart (the average contact domain size)70) and computed their gene expression correlation as a function of CTCF-binding site DNA methylation. Single-cell CTCF-binding site hypermethylation in IDH-MUT cells correlated with loss of gene insulation (that is, the higher the DNA methylation, the stronger the correlation in the expression of gene pairs across boundaries; Mann–Whitney U test, P = 1.7 × 10−10; Fig. 3g and Extended Data Fig. 8h,i). In line with previous work using bulk sequencing methods70, this result suggests that even small changes in DNA methylation are sufficient to disrupt CTCF binding and domain boundaries, thereby affecting gene expression in IDH-MUT gliomas. We further confirmed stronger expression correlation between PDGFRA, a prominent glioma oncogene, and FIP1L1 in IDH-MUT cells than in GBM cells (Fisher’s exact test, P < 10−16; Fig. 3h), as previously reported70. Stochastic methylation of CTCF-binding sites may thus provide the basis for higher transcriptional variation within IDH-MUT tumors by permitting malignant cells to activate alternate gene regulatory programs, eventually leading to the selection of epigenetic clones with higher fitness28.
GBM cells display higher cellular plasticity than IDH-MUT cells.
While DNA methylation changes may mark cell states, we and others have previously shown that the large majority of DNA methylation changes in cancer reflect stochastic, passenger events that do not impact gene regulation33,63–66,71,72. These heritable stochastic DNA methylation changes serve as a molecular clock33,71–73 and were therefore exploited as native barcodes to infer a high-resolution lineage history of GBM and IDH-MUT cells from primary patient samples (Fig. 4a,b and Extended Data Fig. 9a,b). Projection of information on subclonal CNAs (for example, on chromosome 6 in GBM (MGH105) and chromosome 11 in IDH-MUT glioma (MGH107)) and single-nucleotide variants (SNVs; for example, RPL5 chr1:g.93303106C>G) onto the lineage trees revealed that genetically defined subclones mapped accurately to distinct clades inferred solely on the basis of DNA methylation information (Fig. 4a,b and Extended Data Fig. 10a; note that chromosomes with CNAs were excluded from DNA methylation tree inference), providing orthogonal validation to lineage tree inference. We further validated that tree topologies were driven primarily by heritable passenger DNA methylation changes by excluding DMRs and PRC2 targets from lineage tree inference (Extended Data Fig. 10c).
Fig. 4 |. Heritability of glioma malignant cell states inferred from lineage tree architectures.
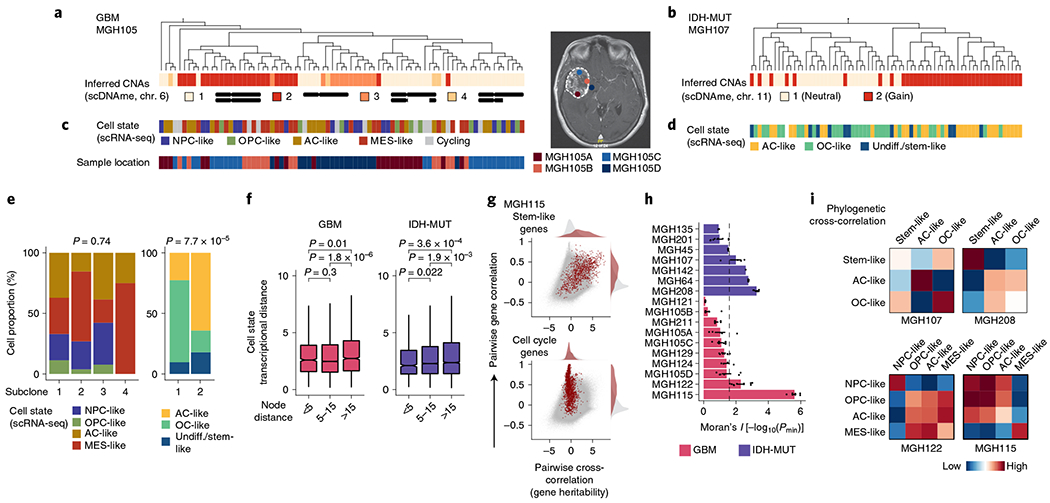
a, Representative (random cell subsampling within each of the four spatial locations sampled) DNA methylation-based lineage tree of GBM (MGH105) cells, with projection of chromosome 6 inferred CNAs (as defined in Fig. 1f). b, Representative DNA methylation-based lineage tree of IDH-MUT (MGH107) cells, with projection of chromosome 11 inferred CNAs. c, Projection onto the DNA methylation-based lineage tree of GBM MGH105 (see a) of cellular states (top) and sample collection location (bottom). Right, the magnetic resonance imaging (MRI) image from MGH105 indicates the four spatially distinct regions sampled. d, Projection onto the DNA methylation-based lineage tree of IDH-MUT MGH107 (see b) of cellular states. e, Proportion of cells belonging to GBM (MGH105; left) or IDH-MUT (MGH107; right) cellular states in distinct genetic subclones as identified by scDNAme-based CNA inference at chromosome 6 (left) or chromosome 11 (right). f, Comparison of transcriptional distances (measured as Euclidean distances between gene module scores) as a function of lineage distance (defined as <5, between 5 and 15, and >15 nodes away) for unique cell pairs from GBM (left; n = 7 patients) and IDH-MUT (right; n = 7 patients) lineage trees. g, Pairwise gene expression correlation (Pearson’s) and cross-correlation (heritability). Gray points represent all gene pair relationships, and red points represent gene pair relationships within the same selected gene module (top, stem-like; bottom, cell cycle). Correlation and cross-correlation densities are shown in the plot margins. h, Phylogenetic association of cell states on GBM (n = 7 patients; n = 10 biological replicates if considering the four spatially distinct regions sampled from MGH105) and IDH-MUT (n = 7 patients) lineage trees, as measured by cell state gene module expression autocorrelation with Moran’s I (ref. 74). Barplots represent mean±s.e.m. Moran’s I permutation-based one-sided P values (106 permutations) were calculated across lineage tree replicates. The dashed line corresponds to a P value of 0.025. i, Heat maps of pairwise cell state phylogenetic associations (gene module cross-correlation analytical z scores). Close phylogenetic associations are shown in warmer colors, and distant associations are shown in cooler colors. P values were determined by two-sided Fisher’s exact test (e) or two-sided Mann-Whitney U test (f). Boxplots represent the median and bottom and upper quartiles; whiskers correspond to 1.5 times the interquartile range.
In GBM (for example, MGH105), projection of scRNA-seq-derived cell states onto the lineage tree revealed little differential enrichment of the four core cell states in distinct clades of the tree, despite the clades also being marked by CNAs and involving spatially distinct regions of the tumor (Fig. 4c,e and Extended Data Fig. 10a). By contrast, in IDH-MUT samples (for example, MGH107), projection of cellular state onto the lineage trees revealed differential enrichment of the two main differentiated cellular states (AC- and OC-like) in separate clades of the tree, which were also marked by a distinct CNA profile on the long arm of chromosome 11 (11q; Fisher’s exact test, P = 7.7 × 10−5; Fig. 4d,e and Extended Data Fig. 10b). These observations may suggest a model of higher cellular plasticity in GBM while there is a more stable differentiation hierarchy in IDH-MUT tumors16 and raise the question of the extent to which glioma cell states are heritable.
To investigate the heritability of glioma cell states, we assessed phylogenetic association of cellular states on the lineage tree as a proxy for the heritability of gene expression programs. We observed decreased transcriptional similarity between glioma cells as a function of their lineage distance (Fig. 4f and Extended Data Fig. 10d,e). We also compared transcriptional correlation to phylogenetic cross-correlation74 for pairs of genes. As expected, genes within the same module (for example, cell cycle or stem-like genes) exhibited highly correlated transcription. However, stem-like genes (expressed in NPC- and OPC-like cells) tended to also have high phylogenetic cross-correlation, reflecting heritable expression of these lineage-specific genes over the course of cellular divisions. By contrast, cell cycle genes, despite exhibiting highly correlated expression, did not show high phylogenetic cross-correlation, reflecting their transient, non-heritable status (Fig. 4g and Extended Data Fig. 10f). To directly assess cell state heritability, we measured with Moran’s I (ref. 74) the autocorrelation between cell state gene module expression and found that the majority of IDH-MUT samples (4 of 7) and a subset of GBM samples (2 of 7) showed significant cell state heritability (Fig. 4h, Extended Data Fig. 10g,h and Supplementary Table 9).
Focusing on glioma samples with the highest degree of cell state heritability, we observed that cell state lineage proximity mirrored transcriptional similarity; in GBM, NPC- and OPC-like cells tended to cluster together on the lineage trees, and AC-like cells exhibited the closest phylogenetic proximity to MES-like cells. This pattern of phylogenetic cross-correlation may indicate that cell state heritability dynamics in GBM cohere with neurodevelopment trajectories (Fig. 4i, Extended Data Fig. 10i and Supplementary Table 10). In IDH-MUT tumors, this analysis revealed two distinct clusters of differentiated cell states in the majority of patients. This result likely reflects the branched unidirectional developmental hierarchy, with activation of neural stem-cell programs at the top of the hierarchy that branches into two distinct cellular states resembling astrocytic and oligodendrocytic lineages7 (Fig. 4i and Extended Data Fig. 10i).
These heritability findings prompted us to quantify the transition dynamics governing the distribution of glioma cell states across lineage trees. We hypothesized that plastic differentiation hierarchies (that is, those with a high degree of dedifferentiation in which differentiated cells can more easily revert to stemness) would result in lineage trees where the cell states were distributed more randomly across clades, whereas a strict unidirectional hierarchy would result in lineage trees with cell states that were more clustered, as observed in GBM and IDH-MUT tumors, respectively. In line with this hypothesis, simulated lineage trees with varying rates of dedifferentiation in comparison to stem-like cell self-renewal showed that the phylogenetic clustering of cell states (as measured by Moran’s I) decreased as the rate of dedifferentiation increased (Fig. 5a).
Fig. 5 |. GBMs exhibit higher cellular plasticity while IDH-MUT gliomas have a more stable differentiation hierarchy.
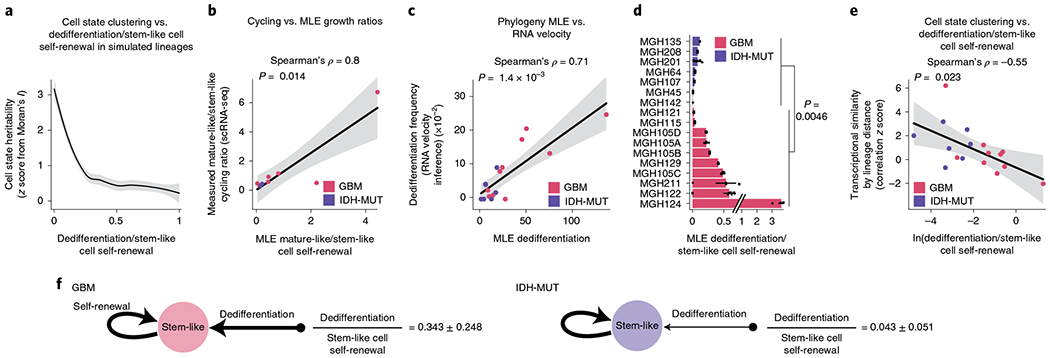
a, Simulated lineage trees (n = 1,000) with varying rates of dedifferentiation compared to stem-like cell self-renewal (x axis) as a function of phylogenetic association of cellular states (as measured by z scores from Moran’s I; y axis). The LOESS regression line (black) with its 95% confidence interval (gray) is shown. b, Comparison of the mathematical model’s estimates (MLE, maximum-likelihood estimation; weighted median across lineage tree replicates for each GBM (pink) and IDH-MUT (purple) sample) of cell state self-renewal in differentiated-like versus stem-like states with cycling rates derived from the scRNA-seq expression profiles. Spearman’s rho is indicated. Only samples with at least two cycling stem-like and two cycling differentiated-like cells were used (Methods). The linear regression line (black) with its 95% confidence interval (gray) is shown. c, Same as in b for comparison of the mathematical model’s estimates of dedifferentiation with dedifferentiation rates provided by RNA velocity estimation31 (Methods). The linear regression line (black) with its 95% confidence interval (gray) is shown. d, Rates of dedifferentiation compared to stem-like cell self-renewal (as estimated by mathematical modeling) in GBM (n = 7 patients; n = 10 if considering the four spatially distinct regions sampled from MGH105) and IDH-MUT (n = 7 patients) tumors across lineage tree replicates (Methods). Barplots represent median ± median absolute deviation (MAD) across lineage tree replicates. Medians are weighted to balance plates with a disparate number of lineage tree replicates within samples. e, Dedifferentiation/stem-like cell self-renewal ratio (weighted median across lineage tree replicates for each GBM (pink) and IDH-MUT (purple) sample; x axis) compared to cell state clustering on the lineage tree as measured by transcriptional similarity (mean across tree replicates of a gene module by lineage distance Pearson’s correlation z score; 1,000 permutations). The linear regression line (black) with its 95% confidence interval (gray) is shown. f, Data-driven model of cell state transition dynamics inferred from DNA methylation-based lineage trees. The median± MAD dedifferentiation/stem-like cell self-renewal ratios across GBM (n = 7 patients) and IDH-MUT (n = 7 patients) samples are shown. P values were determined by two-sided Mann–Whitney U test (d), comparing the weighted median dedifferentiation/stem-like cell self-renewal ratio of GBM samples with the weighted median dedifferentiation/stem-like cell self-renewal ratio of IDH-MUT samples.
To examine this hypothesis directly in patient samples, we inferred cell state growth and transition rates from glioma phylogenetic trees with leaves annotated for cell state. Specifically, we adapted a maximum-likelihood method of binary character evolution and speciation from comparative phylogenetics75,76 (Methods, Extended Data Fig. 10j,k and Supplementary Table 11). To validate the model’s parameter estimates, we used two sources of orthogonal data. First, we compared the model’s estimates of growth in differentiated-like versus stem-like states to cycling rates derived from the expression profiles and observed high correlation (Spearman’s rho = 0.8, P = 0.014; Fig. 5b). Second, we found that the model’s estimates of dedifferentiation correlated with dedifferentiation rates inferred from RNA velocity estimation31 of gene module trajectories (Spearman’s rho = 0.71, P = 0.0014; Fig. 5c). We further validated the model’s estimates by excluding DMRs and PRC2 targets from lineage tree inferences (Extended Data Fig. 10l), confirming again that DNA methylation-derived tree topology reflects stochastic passenger DNA methylation changes rather than cell state encoding.
When the binary character evolution method was applied to IDH-MUT samples, the model predicted a low rate of dedifferentiation in comparison to stem-like cell self-renewal (Fig. 5d and Extended Data Fig. 10m), in line with the highly structured lineage trees for these tumors (Fig. 5a,e). By contrast, GBM samples showed a significantly higher level of dedifferentiation (Mann–Whitney U test, P = 0.0046; Fig. 5d and Extended Data Fig. 10m), in line with the lower degree of cell state clustering on the trees and lower transcriptional similarity by lineage distance (Fig. 5a,e). Together, these data demonstrate that cell states are heritable across malignant gliomas. However, while in IDH-MUT tumors, differentiation far outpaces dedifferentiation in line with a standard hierarchical model7, GBM tumors harbor a higher degree of cell state plasticity allowing replenishment of the ranks of stem-like cells through dedifferentiation (Fig. 5f).
Discussion
Studies across cancer types have shown that heterogeneous transcriptional cell states within a single tumor contribute to tumor initiation and progression1,4–7. In glioma, cellular state diversity mirrors neurodevelopmental trajectories7,17–23. Here, through the application of multiomics single-cell sequencing to primary glioma clinical samples, we provide evidence that DNA methylation changes reflect glioma cellular states and may contribute to their propagation.
Specifically, we showed that IDH-MUT cells exhibit preferential enhancer hypermethylation with cell differentiation. Enhancers, owing to their lower transcription factor occupancy as compared to promoters77, may be less resistant to DNMTs and thus more prone to hypermethylation, which is canonically balanced by the action of TET enzymes in physiological contexts61. In IDH-MUT malignant cells, defects in TET-mediated demethylation caused by 2HG may thus lead to preferential enhancer hypermethylation60. In addition, enhancers have been shown to exhibit highly dynamic DNA methylation during differentiation78–80, in line with our data showing increased enhancer DNA methylation with glioma differentiation. While the relatively modest magnitude of DNA methylation changes observed in our study may be partly due to the sparsity of the single-cell RRBS data, our work also suggests that small increases in DNA methylation in otherwise typically unmethylated regions are sufficient to impact gene expression and can be associated with gene silencing (Fig. 3d), as previously reported across cancer types33,63,81–84, including in glioma85. Indeed, our multimodality sequencing technology that couples single-cell DNA methylomes with whole-transcriptome sequencing allowed the exploration of methylation–transcription relationships at the single-cell level, revealing that aberrant epigenetic patterning is at play in IDH-MUT gliomas. This included decoupling of promoter methylation–expression relationships, whereby expression of genes central to the oncogenic IDH-MUT phenotype persists despite high promoter DNA methylation, as well as disruption of CTCF-mediated insulation.
In GBM, direct comparison of epigenetic profiles across cell states suggests that the interaction between DNA methylation and PRC2 is an important contributor to GBM cell differentiation. The main role of PRC2 is to catalyze H3K27me3 deposition to repress lineage-specific developmental genes in both normal and neoplastic stem cells86,87. At these genes, H3K27me3 is largely enriched at promoters along with H3K4me3, an activating histone mark88. These bivalent poised promoters in stem cells largely resolve to either an active (H3K4me3-only) or repressed (H3K27me3-only) state during differentiation. While PRC2 target hypermethylation has previously been extensively reported in cancer52, we observed that stem-like GBM cells are protected from this phenomenon, likely owing to PRC2 binding protecting these sites from DNA methylation, in line with data from neurodevelopment89,90. This may also underlie the enhanced chromatin accessibility signal that we observed at hypomethylated PRC2 targets in GBM stem-like cells91. By contrast, differentiated-like GBM cells may reinforce gene silencing by increasing the length of H3K27me3 domains or through complementary silencing mechanisms involving DNA methylation87,92. In line with this model, we observed more than twofold-higher expression of PRC2 targets in stem-like cell states in comparison to more robust silencing involving DNA methylation in more differentiated cell states. Thus, our multimodal single-cell analyses support a critical role for PRC2 in maintaining GBM cellular states, suggesting a model in which PRC2 targets are maintained in a hypomethylated state in glioma stem-like cells, allowing their reactivation in response to stimuli, thereby ultimately providing a key mechanism for stemness maintenance90 and tumor progression93.
The observed parallels between glioma differentiation and neural development invoke the question of whether gliomas follow unidirectional differentiation hierarchies or more reversible bidirectional cell state transitions21,55. As we seek to therapeutically target defined glioma cell states, such as stem-like cells94, it is critical to dissect the relative rates at which other cells revert to assume the role of stem cells. To address this question, we integrated lineage histories derived from heritable stochastic DNA methylation changes with scRNA-seq-derived cell states in single-cell multiomics data. We demonstrated that in IDH-MUT glioma differentiation far outpaces dedifferentiation, in line with a model in which stem-like cells are self-renewing and reside at the apex of the cellular hierarchy7. By contrast, in GBM, cells demonstrated the capacity to dedifferentiate into stem-like states, providing evidence for plastic bidirectional cell state transitions, as also observed in other cancer types95,96. Such plastic differentiation topologies may result from relaxation of epigenetic identity barriers28,63,80 and in turn may empower positive selection97 to enhance the evolutionary capacity of gliomas.
Our work has several limitations. The MscRRBS platform only captures approximately 10% of the targeted methylome for a single cell owing to the sparsity of single-cell data33. We have thus implemented several analytical approaches to mitigate the sparsity of the single-cell methylomes, including averaging DNA methylation levels across defined genomic windows and regions or aggregating DNA methylation signal over multiple single cells within a sample. We further note that, while DNA methylation is one of the central mechanisms for propagating stable epigenetic information across cell division54 and accumulating data suggest that malignant cell states are propagated epigenetically4,27–29, the nature of the causal relationship between DNA methylation and the establishment of stable cellular identity is still under debate98. Nonetheless, we envision that future advances in both experimental technologies and data analysis methods99 will enable more accurate measurement of DNA methylation across the genome in single cells, as well as a better understanding of the causal relationship between DNA methylation and transcriptional cell states.
In conclusion, cell state diversity and tumor evolution are often studied independently. The data presented herein show that single-cell multiomics analysis of clinical samples can help draw together these disparate frameworks, through the unique lens of a high-resolution phylogenetic tree coupled with leaf annotation for current phenotypic states. This new perspective allows transcriptional cell state diversity to be connected with fundamental evolutionary properties such as heritability and cell state transition dynamics, opening up new horizons for the study of human somatic evolution in both malignant and healthy tissues.
Methods
Study participants.
Adult patients included in this work provided preoperative informed consent to take part in the study according to institutional review board protocol Dana-Farber/Harvard Cancer Center 10-417. Patients were male and female. Clinical characteristics are summarized in Supplementary Table 2.
Tumor acquisition and single-cell sorting.
Fresh tumor specimens were collected on PBS (Gibco) and mechanically dissociated into small pieces of 0.5–1 mm with a disposable sterile scalpel. They were further dissociated into single-cell suspensions using the enzymatic brain tumor dissociation kit (P) from Miltenyi Biotec, following the manufacturer’s protocol. Viable single cells were sorted into individual wells of a 96-well twin.tec PCR plate (Eppendorf) that contained 10 μl per well of TCL buffer (Qiagen) with 1% β-mercaptoethanol (see the Supplementary Note and Supplementary Fig. 1 for details). Plates were frozen on dry ice immediately after sorting and stored at −80 °C before joint MscRRBS and whole-transcriptome library preparation and sequencing.
Joint MscRRBS and scRNA-seq library construction.
MscRRBS and whole-transcriptome library preparation and sequencing were performed as previously described33 (see the Supplementary Note for details). To obtain higher-coverage single-cell DNA methylomes, dual-restriction enzyme digestion of cells from two IDH-MUT samples (MGH201 and MGH208) was performed. This allowed us to increase coverage to 325,492 ± 21,118 unique CpGs per cell (~2-fold increase) as compared to IDH-MUT cells digested with a single restriction enzyme, thus enabling more accurate measurement of DNA methylation in regions that are captured less efficiently with standard RRBS, such as enhancers and CTCF-binding sites (Extended Data Fig. 8a).
MscRRBS read alignment.
Each pool of 96 cells was first demultiplexed by Illumina i7 barcodes (Supplementary Table 1), resulting in four pools of 24 cells. Each pool of 24 cells was further demultiplexed by unique cell barcodes (Supplementary Table 1). Quality control, trimming and alignment of MscRRBS data were then performed33 (see the Supplementary Note for details). Cells with coverage of at least 50,000 unique CpGs and a bisulfite conversion rate of at least 99% were retained for downstream analyses (Supplementary Tables 2 and 3).
scRNA-seq and differential gene expression analysis.
Sequenced read fragments were mapped against the GRCh38 (hg38 Ensembl version 94) genome assembly using the 2pass default mode of STAR100 (v2.5.2a). The number of read counts overlapping annotated genes was determined using RSEM101 v1.3.1 (rsem-calculate-expression). Cells with mitochondrial and ribosomal read counts of less than 20% and a minimum of 2,000 detected genes were retained for downstream analyses (Supplementary Tables 2 and 3). Differential gene expression analyses were performed using a negative binomial model with observational weights to account for zero inflation102. Specifically, we used ZINB-WaVE103 (v1.6) to estimate a set of observational weights and edgeR (v3.26.8) to test for differential expression using a weighted F-statistic approach104. We defined differentially expressed genes by adjusting nominal P values using a BH FDR procedure (cutoff of adjusted P value < 0.05), with an additional criterion of an absolute log2(fold change) value of >1 (Extended Data Figs. 4b and 5f).
Identification of non-malignant cell types.
To classify all cells passing scRNA-seq quality control (Supplementary Table 3) into malignant or non-malignant cells (Fig. 1b), we normalized gene count matrices, performed dimensionality reduction and corrected for patient batch effects using the ZINB-WaVE method103 (v1.6; parameters: K = 30, X = “~ patient sample”). To classify all cells passing scDNAme quality control (Supplementary Table 3) into malignant or non-malignant cells (Fig. 1b), we focused on 1,300 CpG sites that were identified as glioma related by a previous TCGA bulk DNA methylation study39. We generated a window of 1,000 bp around each CpG (resulting in 996 windows) and averaged the DNA methylation within each window. We then imputed the missing values in the windows using KNN with N = 5. We used the scanpy package105 (v1.4.4) to cluster cells. For visualization, we generated a UMAP cell embedding using the umap function (v0.2.3.1) with default settings.
Single-cell differential methylation analysis.
For each cell, Bismark methylation extractor output files (containing information on the methylation state of each individual CpG) were intersected with different genomic regions investigated (for example, promoters and enhancers) using BEDTools106 (v2.27.1). A generalized linear model was then built to predict the DNA methylation for a given genomic region between groups of cells on the basis of transcriptionally defined malignant cellular states (see the Supplementary Note for details). We defined regions with a Student’s t-test P value < 0.05 and an absolute DNA methylation difference of ≥5% as differentially methylated to nominate candidate genes for subsequent gene set enrichment analysis.
CNA inference from single-cell DNA methylation data.
To estimate CNAs using scDNAme data, we first split the genome of each cell into windows of equal length (20 Mb) and obtained the number of CpGs per window with a sliding window of 5 Mb. We subsequently normalized the number of CpGs per window by the total number of CpGs for each cell. Cells classified to each of the non-malignant cell types (see “Identification of non-malignant cell types”) were used to define a baseline normal karyotype. We then divided the number of normalized CpGs per window in each malignant cell by the median normalized number of CpGs in the set of non-malignant cells. The resulting copy number estimates were log2 transformed. Missing values were replaced by the value zero (Extended Data Figs. 1a and 2a). For CNA analysis of the EGFR locus, we applied the above-described approach using a 0.1-Mb window (with a sliding window of 0.02 Mb) centered on the EGFR locus on chromosome 7 (Fig. 1e and Extended Data Fig. 3d,f). We further localized the start and end points of aberrant copy number regions of the pseudo-bulk averages (mean of CNAs across individual malignant cells) using the circular binary segmentation algorithm implemented in the R package DNAcopy107 (v1.60.0). See the Supplementary Note for further details.
Glioma DNA methylation subtype (LGm1-LGm6) single-cell projection.
To bridge the 450K methylation array and MscRRBS technologies, we created a window of 1,000 bp around each 450K probe obtained for 932 glioma samples from TCGA40,41, averaging the DNA methylation within each window (450K probes for the TCGA samples and single CpGs for MscRRBS), resulting in 996 windows. We further filtered the data by retaining (1) 450K probes that were detected in at least 20 bulk TCGA samples; (2) single cells with at least 50,000 detected CpG sites; (3) windows containing more than 5 CpGs per cell; and (4) windows for which more than 10 single cells had at least 1 CpG in them. After filtering, we retained 979 windows and imputed missing values in the windows using KNN with N = 5. We then trained a logistic regression multiclass classifier on the 932 TCGA glioma samples, achieving 0.94 accuracy, and applied it to pseudo-bulk DNA methylation profiles for malignant cells in our samples (Fig. 1h) to assign each glioma single cell to one of the six bulk DNA methylation subtypes (Fig. 1i).
Definition of single-cell gene signature scores.
Single-cell gene signature scores were defined as previously described1,7,21 (see the Supplementary Note for details).
Assignment of glioma cells to expression cell states.
We classified glioma cells by expression cell state on the basis of gene modules and cell cycle programs as previously described7,21 (see the Supplementary Note for details).
Analysis of TCGA patient samples.
To examine the association between GBM stem-like states and PRC2 target activity in a larger sample cohort, we leveraged 67 GBM samples from the TCGA collection with matched bulk RNA-seq and 450K methylation profiles (Fig. 2i,j and Extended Data Fig. 5j). We computed differentiation scores (defined as the difference in gene module scores between AC/MES-like and NPC/OPC-like cellular states) where the gene signatures for each of the four states were taken from the previously described gene module signatures21. We calculated the mean DNA methylation at PRC2 target promoters by averaging the DNA methylation for the 450K probes mapping within PRC2 target genes46.
Chromatin state analysis.
To explore the link between differentially methylated promoters (see “Single-cell differential methylation analysis”) and histone marks, we interrogated differentially methylated promoters for enrichment of histone marks with non-overlapping regulatory functions (H3K4me3, H3K27ac, H3K4me1, H3K36me3 and H3K27me3) using previously published ChIP–seq maps47 of GBM cancer stem cells (n = 4 lines derived from different human gliomas (MGG23CSC, MGGG4CSC, MGG6CSC and MGG8CSC)). In Extended Data Figs. 5c–e and 7k,l, chromatin states across the genome were defined using ChromHMM108 (v1.20), which is based on a multivariate hidden Markov model (HMM), using H3K4me3, H3K27ac, H3K27me3, H3K36me3 and H3K4me1 from the above-described previously published datasets47 as input (the MGG8CSC sample was used as it was the only one where all five main histone marks were profiled). See the Supplementary Note for further details.
Single-cell DNA methylation–gene expression correlation analysis.
Single-cell DNA methylation–gene expression correlation analysis was performed as previously described33 (see the Supplementary Note for details).
Lineage tree inference.
We generated DNA methylation-based lineage trees by applying a tree searching maximum-likelihood algorithm based on binary DNA methylation values as previously described33 (see the Supplementary Note for details). Projection of information on subclonal CNAs (for example, on chromosome 6 in GBM (MGH105) and chromosome 11 in IDH-MUT glioma (MGH107)) and SNVs (for example, in RPL5) onto the lineage trees revealed that genetically defined subclones mapped accurately to distinct clades inferred solely on the basis of DNA methylation information, providing orthogonal validation to lineage tree inferences (Fig. 4a,b and Extended Data Fig. 10a). We further validated that lineage tree topologies were driven by heritable stochastic passenger DNA methylation changes by excluding CpGs belonging to DMRs and PRC2 targets from lineage tree inference (Extended Data Fig. 10c). To compare inferred lineage trees, we computed the pairwise Robinson–Foulds (RF) distance—a measure of tree structure similarity between two given trees109. RF distances were normalized by the total number of internal edges in respective pairs of trees (normalized RF distance).
Phylogenetic association.
To quantify the association of different cell states and transcriptional patterns on the DNA methylation-based lineage trees, we used Moran’s I (ref. 74), a classic measure of spatial association (that is, autocorrelation) used to detect phylogenetic signal110, as well as its multivariate generalization, a measure of spatial cross-correlation111,112. Conceptually, Moran’s I is a weighted correlation metric, as its calculation is similar to that of Pearson’s correlation coefficient but with measurements weighted by proximity. To compute Moran’s I for an n-cell lineage tree, we first organize single-cell measurements into a column-standardized matrix X (centered with mean 0 and population standard deviation of 1), consisting of n rows corresponding to cells and m columns corresponding to single-cell measurements. Then, the data matrix X and its transpose (notated with superscript T) is right and left multiplied with the proximity matrix W,
Each element of the n×n proximity matrix Wij records the inverse node distance between cells i and j, with diagonal elements set to 0, and normalized such that . Measurements contained in matrix X could correspond to gene expression (as in Fig. 4g), gene module scores (for example, in Fig. 4h) or cell states (for example, in Fig. 5a). When m = 1, this metric becomes the classic univariate Moran’s I. When m = 1, each element of the m×m matrix Iyx measures the phylogenetic cross-correlation between measurements y and x. High values within I indicate phylogenetic co-clustering, whereas low values indicate phylogenetic dispersion.
To assess the heritability of glioma cell states, we measured the phylogenetic autocorrelation of each cell state gene module (using univariate Moran’s I) and assessed significance with a one-sided permutation test (with 106 leaf permutations) for each tree replicate. To improve resolution, we first recomputed GBM module scores, pooling the NPC1-like and NPC2-like gene sets and the MES1-like and MES2-like gene sets, and removed cells without matching scRNA-seq information. As both GBM and IDH-MUT samples contained multiple cell states at different frequencies, to summarize a sample’s transcriptional heritability, we used the most heritable gene module for each tree, as represented by its permutation test −log10(P value). As we had multiple lineage tree replicates per sample plate, we arrived at a plate heritability score by averaging the tree replicate −log10(P values). For patient samples with multiple plates, scores for only the least variable plate (measured by RF distance; see “Lineage tree inference”) are shown (Fig. 4h). Heritability scores for all plates are shown in Extended Data Fig. 10g and included in Supplementary Table 9.
To further understand how cell states were co-distributed/dispersed across lineage trees, we also measured gene module cross-correlation (multivariate Moran’s I). Cross-correlations for each tree replicate were transformed into z scores using moments of the statistic derived by Czaplewski and Reich112 and were then averaged for each sample. Analytical z scores were used to increase computational efficiency and closely matched leaf-permutation-based z score estimates. Moran’s I z score heat maps for representative lineages are shown in Fig. 4i. These heat maps illustrate which cell states form clusters and how pairs of different cell states cluster together on lineage trees. Close and distant phylogenetic associations are shown in warmer and cooler colors, respectively.
Finally, to study the phylogenetic distribution of transcription at the single-gene level, we compared cross-correlations and correlations for all available (2,000 most variable genes selected with Seurat), stem-like (that is, NPC-like and OPC-like) and cell cycle genes in glioma samples with high gene module transcriptional heritability (MGH115 and MGH122) (Fig. 4g and Extended Data Fig. 10f). For each available gene, all pairwise Pearson’s correlations and cross-correlations (mean tree replicate analytical z scores) were plotted, with self-correlations and autocorrelations omitted. Densities are shown for all gene pairs (gray) and for genes from the selected module (red) in plot margins.
Mathematical model of glioma evolutionary dynamics.
To model glioma evolutionary dynamics, we adapted a mathematical model of binary state speciation and extinction (BiSSE) from comparative phylogenetics75. The BiSSE approach models speciation, extinction and character transition rates as a dynamical system, where species in character state k (either 0 or 1) speciate at rate λk. Species transition from state 0 (1) to state 1 (0) at rate q01 (q10). This mathematical framework can be translated to tumor dynamics, where λ and q measure cell-state-specific growth (self-renewal) and transition (that is, differentiation and dedifferentiation) rates. In this application, we set the binary character trait to be the tumor cell state, either stem-like (k = 0) or mature-like (k = 1). As we are interested in net cell state growth rates, we use a Yule (pure birth) version of the model. The change in the number of cells nk(t) in state k at time t is described by the following dynamical system (Extended Data Fig. 10j,k):
To apply this method to samples from patients with GBM, we binned cells with a maximum gene module score of NPC- or OPC-like as ‘stem-like’ and those with a score of AC- or MES-like as ‘mature-like’. For IDH-MUT samples, cells with a maximum gene module score of AC- or OC-like were binned to the mature-like cell state and cells with a maximum module score of stem-like remained classified as stem-like. Before assigning cell states, cells without scRNA-seq data were removed and gene module scores for GBM samples were recomputed by first pooling NPC1/NPC2-like and MES1/MES2-like genes into one module each.
Maximum-likelihood estimation of evolutionary dynamics.
To infer the tumor growth and transition rates that generated the observed phylogenies, we used maximum-likelihood estimation. We generated a likelihood function using make. bisse() from diversitree v0.9.15 (ref.113), using a Yule version of the model and a sampling fraction of 10−6, as our lineages represent a tumor sampling. We assumed that the root of each tree was in the stem-like state and otherwise used default settings. As the BiSSE method requires ultrametric trees, we converted our trees using force.ultrametric() with the ‘extend’ method in the R package phytools (v0.7.70)114.
To minimize the chances of reaching a local, instead of a global, maximum estimate, we initiated the maximum-likelihood searches from 100 randomly generated starting points (initial BiSSE parameter values) using simulated annealing with the R package GenSA (v1.1.7)115, searching parameter values bounded by 10−4 and 500, and allowed for a maximum of 1,000 iterations with a stop threshold of 10−8. After these initial searches, we used mle2() from the bbmle R package (v1.0.23.1)116, initializing each maximum-likelihood search with a simulated annealing estimate, using the L-BFGS-B optimization method, lower and upper bounds of 10−4 and 500, and a maximum of 1,000 iterations.
After 100 maximum-likelihood searches per tree replicate, the BiSSE parameter scheme with the highest likelihood among the individual runs that converged without error was selected. To arrive at a final parameter set estimate for each biological sample, we used the weighted median of the maximum-likelihood estimates (Supplementary Table 11), weighting each plate for a sample equally Outlier tree replicates estimated from the same cells with an estimated dedifferentiation/stem-like cell self-renewal (q10/λ0) ratio greater than 5 MAD above the median were removed. Maximum-likelihood estimates for GBM and IDH-MUT dynamics (Extended Data Fig. 10j) represent the median of patient-sample-weighted median estimates. The ratio of median replicate estimates of q10/λ0 was significantly larger in GBM than in IDH-MUT samples (Fig. 5f). For patient samples with multiple plates, q10/λ0 for only the least variable plate (measured by RF distance; see “Lineage tree inference”) is shown (Fig. 5d), and the weighted median of q10/λ0 for all plates is shown in Extended Data Fig. 10m and included in Supplementary Table 11. The P value in Fig. 5d was calculated by Mann–Whitney U test by comparing the weighted median of q10/λ0 between GBM and IDH-MUT samples using all plates. Lastly, we validated the maximum-likelihood estimates by excluding DMRs and PRC2 targets from lineage tree inference (Extended Data Fig. 10l), confirming that DNA methylation-derived tree topology reflects stochastic passenger DNA methylation changes rather than marking cell states.
Statistical methods.
Statistical analysis was performed with Python 3.0 and R version 3.6.1. Categorical variables were compared using the hypergeometric test or Fisher’s exact test. Continuous variables were compared using the Mann–Whitney U test, Student’s t test, nonparametric permutation test or Kolmogorov–Smirnov test, as appropriate. P values were adjusted for multiple comparisons using the BH FDR adjustment procedure. All P values are two sided and were considered significant at the 0.05 level unless otherwise noted.
Extended Data
Extended Data Fig. 1 |. Multi-omics single-cell sequencing of GBM reveals intra-tumoral DNAme heterogeneity.
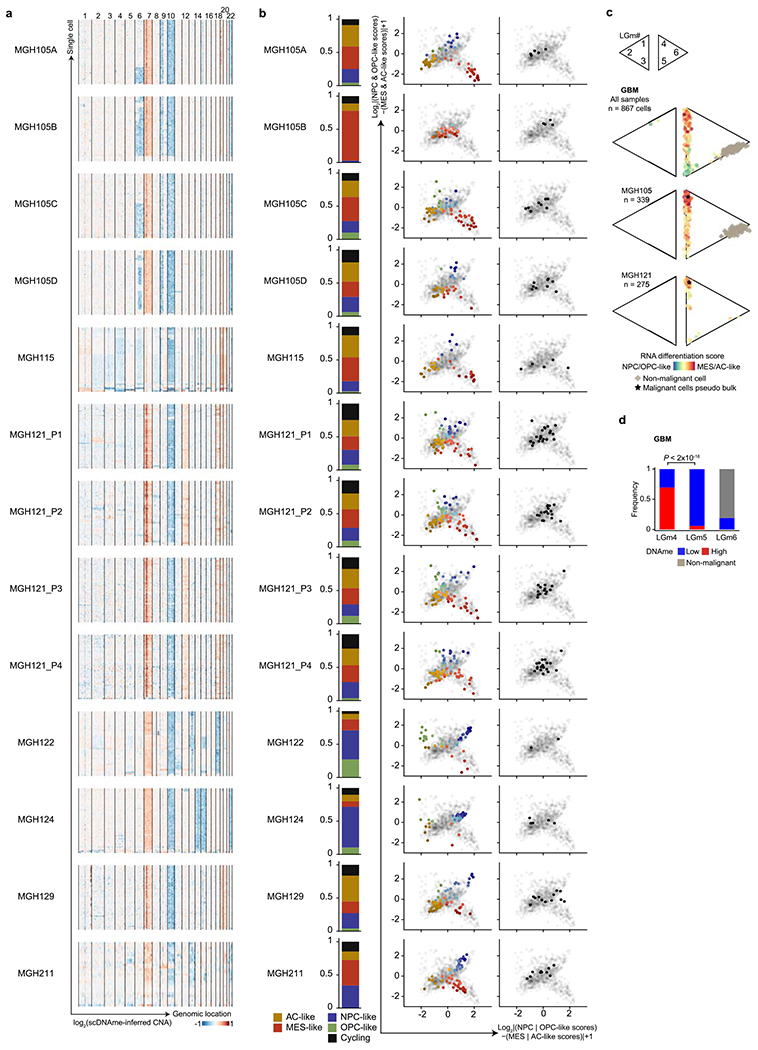
a, CNA inference based on coverage depth imbalance in the scDNAme data in windows of 20 Mb (sliding window of 5 Mb). Rows correspond to cells, clustered by overall CNA pattern. b, Proportion of single cells belonging to GBM cellular states (left) and two-dimensional representation of GBM cellular states (middle) or cycling cells based on the relative expression of gene-sets associated with G1.S and G2.M (right) for each GBM patient sample (including MGH105 biological replicates and MGH121 technical replicates). Each quadrant corresponds to one cellular state and the exact position of malignant cells (dots) reflect their relative scores for pairs of gene modules previously defined in scRNAseq data21. Light grey dots in the background represent all GBM samples (n = 844 malignant-only cells that passed quality control based on scRNAseq). c, Two-dimensional representation of single cells assigned to previously described LGm classes39, visualized as triangle plots (where each vertex corresponds to one LGm class) across all 7 GBM samples (n = 867 cells [malignant and non-malignant] that passed quality control based on scDNAme, top), and the two samples harboring the highest number of cells: MGH105 (n = 339, middle) and MGH121 (n = 275, bottom). RNA differentiation score (defined as the difference in gene module scores between AC-/MES-like and NPC-/OPC-like cells) is overlaid. d, Proportion of GBM cells (n = 867 cells [malignant and non-malignant]) with high or low DNAme (defined as above or below the median of mean DNAme across windows of 1,000 bp around 450K array probes from TCGA glioma samples used in the analysis, respectively; Methods) assigned to previously described LGm classes39. P value was determined by two-sided Fisher’s exact test (d).
Extended Data Fig. 2 |. Multi-omics single-cell sequencing of IDH-MUT reveals intra-tumoral DNAme heterogeneity.
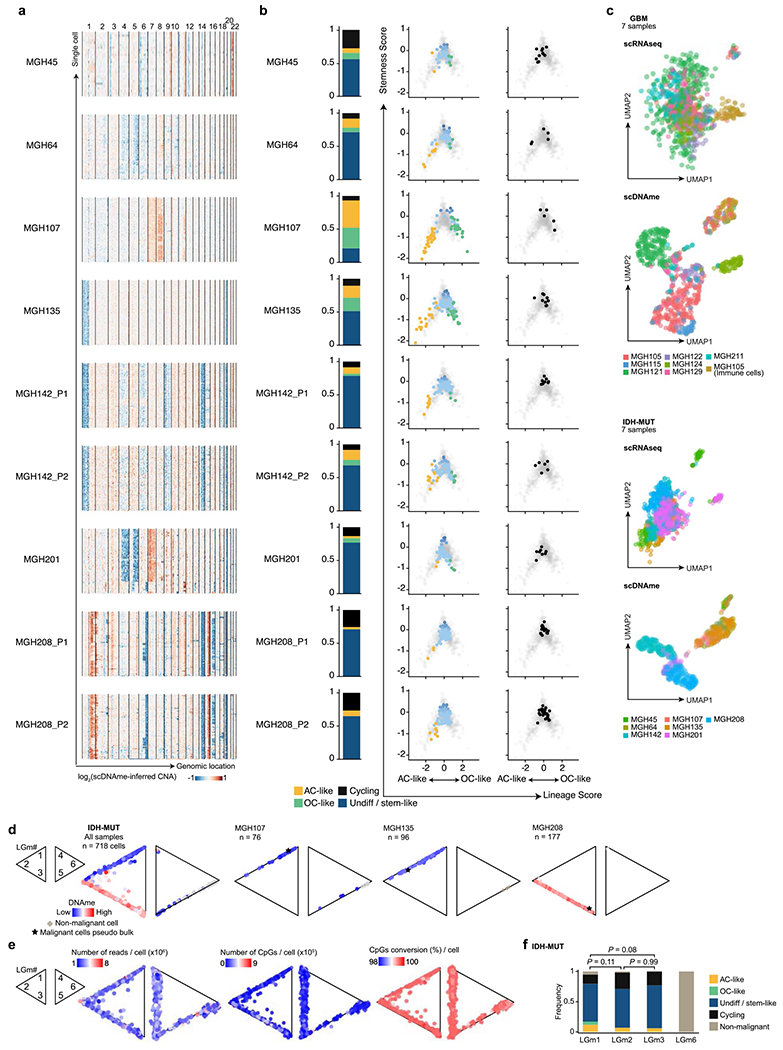
a, CNA inference based on coverage depth imbalance in the scDNAme data in windows of 20 Mb. Rows correspond to cells, clustered by overall CNA pattern. b, Proportion of single cells belonging to IDH-MUT cellular states (left) and developmental hierarchy representation of IDH-MUT cellular states (middle) or cycling cells based on the relative expression of gene-sets associated with G1.S and G2.M (right) for each IDH-MUT patient sample. Lineage and stemness scores define the exact position of malignant cells (dots) as computed from scRNAseq data. Light grey dots in the background represent all IDH-MUT samples (n = 739 malignant-only cells that passed quality control based on scRNAseq). c, UMAP of all single cells that passed quality control based on scRNAseq (GBM n = 937, IDH-MUT n = 809) or scDNAme (GBM n = 867, IDH-MUT n = 718). Each patient sample is indicated. See also Fig. 1b. d, Two-dimensional representation of single cells assigned to previously described LGm classes39, visualized as triangle plots (where each vertex corresponds to one LGm class) across all 7 IDH-MUT samples (n = 718 cells [malignant and non-malignant] that passed quality control based on scDNAme, left), and three representative samples: MGH107 (n = 76), MGH135 (n = 96), and MGH208 (n = 177). DNAme value is overlaid. e, Same as (d) for the 7 GBM and 7 IDH-MUT samples (n = 867 cells [malignant and non-malignant]; IDH-MUT, n = 718 cells [malignant and non-malignant] that passed quality control based on scDNAme). Number of reads per cell (left), number of CpGs per cell (middle), and CpG conversion rate per cell (right) are overlaid. f, Proportion of IDH-MUT cellular states or cycling cells (n = 718 cells [malignant and non-malignant]) assigned to previously described LGm classes39. P values were determined by two-sided Fisher’s exact test (f).
Extended Data Fig. 3 |. High-resolution copy number alteration mapping enabled by single-cell multi-omics.
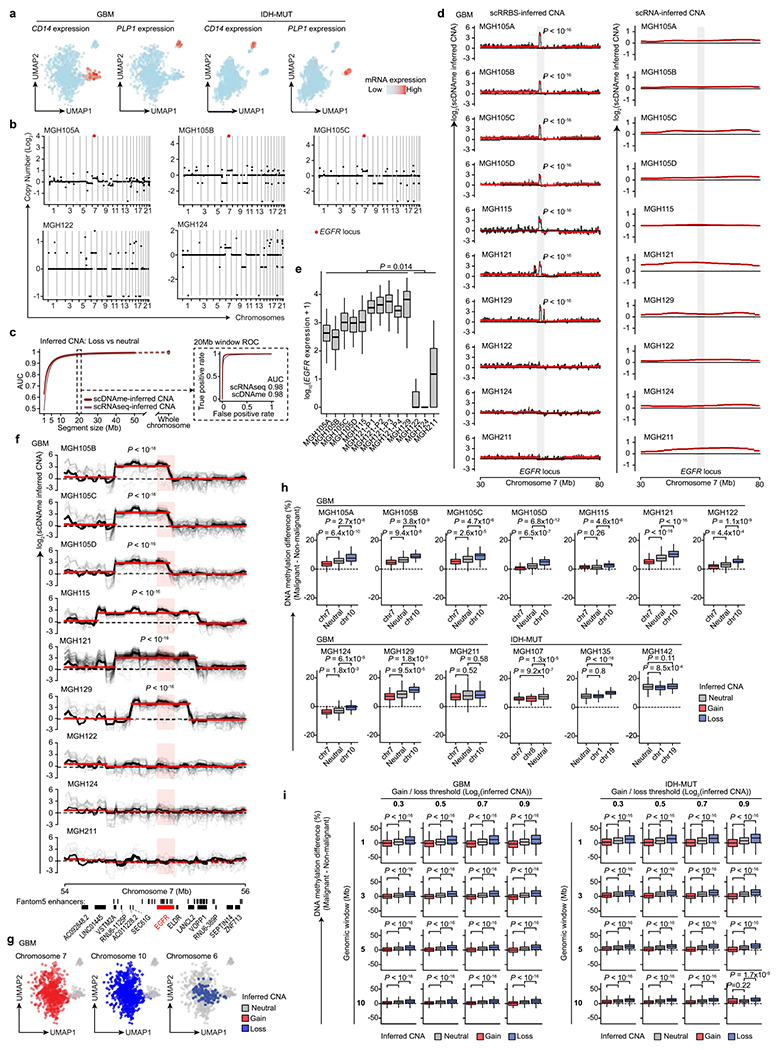
a, UMAP of single cells that passed quality control based on scRNAseq (GBM n = 937, IDH-MUT n = 809). b, CNA inference based on bulk WES for GBM samples MGH105A/B/C, MGH122, and MGH124. EGFR locus is highlighted. c, CNA inference by scDNAme (red line) and scRNAseq (grey line) performance in correctly classifying chr. loss vs. neutral, as assessed by the AUC of ROC curve at different genomic window resolutions. ROC curve at 20 Mb resolution is shown (inset). 95% confidence intervals were generated using bootstrapping. d, CNA inferred by scDNAme (left) and scRNAseq (right) at a 50 Mb region centered at EGFR locus. Mean CNA profile per sample is shown in black. Red lines represent CNA segments identified by circular binary segmentation (CBS) analysis. e, EGFR expression as assessed by scRNAseq for each GBM patient sample (n = 844 malignant-only cells that passed quality control based on scRNAseq). f, Same as (d) for CNA inference by scDNAme at a 2 Mb region centered at EGFR locus. Individual cell CNA profiles are shown in grey. g, UMAP of single cells as defined in (a). Clonal chr. 7 gain (left) and chr. 10 loss (middle), as inferred by scDNAme, along with sub-clonal loss of chr. 6 (right), are indicated. h, Percentage of CpG methylation change at copy number gain, loss, and neutral chromosomal regions when comparing DNAme level of individual malignant cells to baseline for GBM (n = 7) and IDH-MUT (n = 3) samples. i, Same as (h) across all GBM and IDH-MUT samples for different thresholds adopted to define copy number gain vs. loss genomic window resolutions. P values were determined by two-sided Mann-Whitney U-test (d-f, h-i), comparing the EGFR expression median values across samples (e). Boxplots represent the median, bottom and upper quartiles, whiskers correspond to 1.5 times the interquartile range.
Extended Data Fig. 4 |. GBM stem-like states exhibit PRC2 target hypomethylation compared with more differentiated-like cell states.
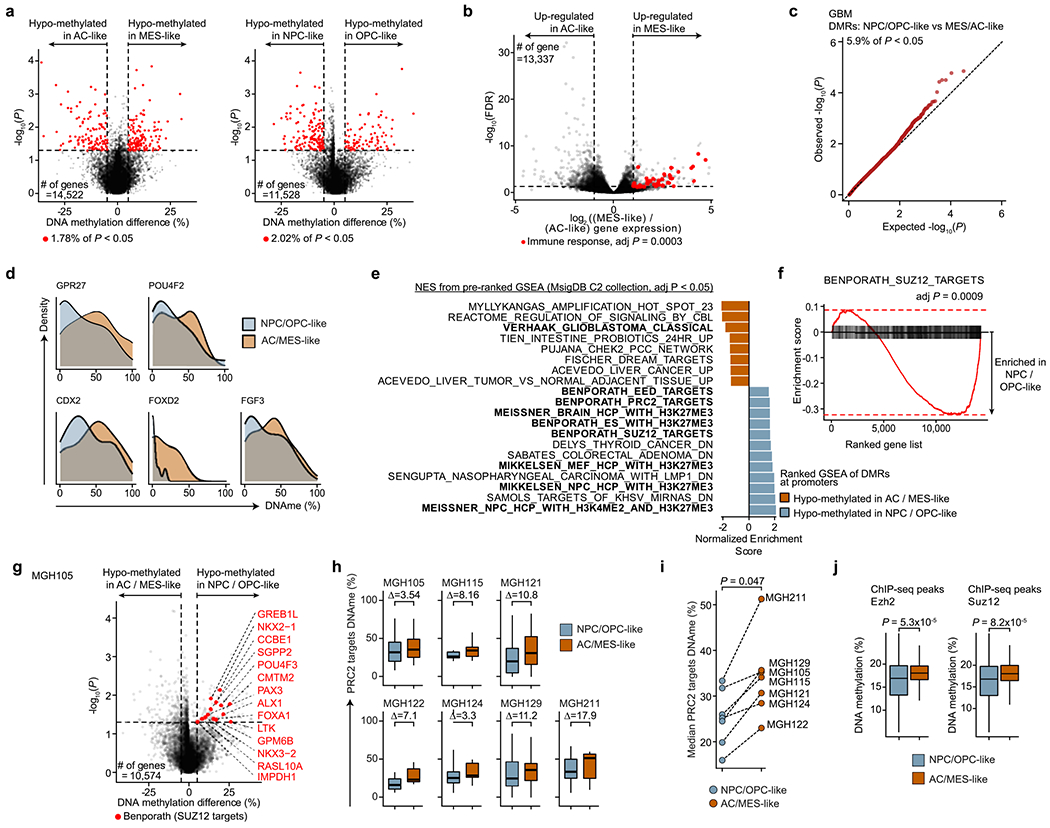
a, Differentially methylated TSS (±1Kb) between stem-like (NPC-like, n = 175 vs. OPC-like, n = 51; left) and differentiated GBM cellular states (MES-like, n = 201; AC-like, n = 168; right). b, Differential gene expression between AC-like (n = 205) and MES-like cells (n = 232). Genes with an absolute log2(fold-change) > 1 and BH-FDR < 0.05 were defined as differentially expressed (DE). DE genes belonging to immune response pathways are highlighted. c, Q-Q plot comparing the observed −log10P values of all genes used in the differential methylation analysis between GBM cellular states (Fig. 2c) to expected −log10P values. d, Distribution of mean promoter DNAme values in stem-like and differentiated cells for representative differentially methylated PRC2 target genes (Fig. 2c). e, Normalized enrichment scores for gene sets (MSigDB C2) enriched at hypomethylated promoters in NPC/OPC-like (turquoise) or MES-/AC-like (orange) cells (Fig. 2c; n = 15,218 genes). f, Enrichment score plot for SUZ12 targets46 gene set enriched at hypomethylated promoters in NPC-/OPC-like cells (Fig. 2c; n = 15,218 genes). g, Same as (a) for a representative GBM sample (MGH105; NPC-/OPC-like, n = 50 cells; MES-/AC-like, n = 138 cells). Genes belonging to PRC2 targets46 are labelled. h, Mean CpG methylation at promoters of PRC2 targets46 between cell states for each of the 7 GBM samples. Difference in median promoter DNAme at PRC2 targets46 between cell states is indicated. i, Median promoter DNAme at PRC2 targets46 of MES-/AC-like and NPC-/OPC-like cells for each of the 7 GBM samples. j, Mean CpG methylation at ChIP-seq maps50 of EZH2 and SUZ12 between GBM cell states (n = 706 cells). P values were determined by generalized linear model (a, c, g), weighted F-test (b), permutation test (f), two-sided Mann-Whitney U test (i, j). Boxplots represent the median, bottom and upper quartiles, whiskers correspond to 1.5 times the interquartile range.
Extended Data Fig. 5 |. Validation of PRC2 hypomethylation in GBM stem-like states using histone marks, single-cell ATACseq and TCGA bulk data.
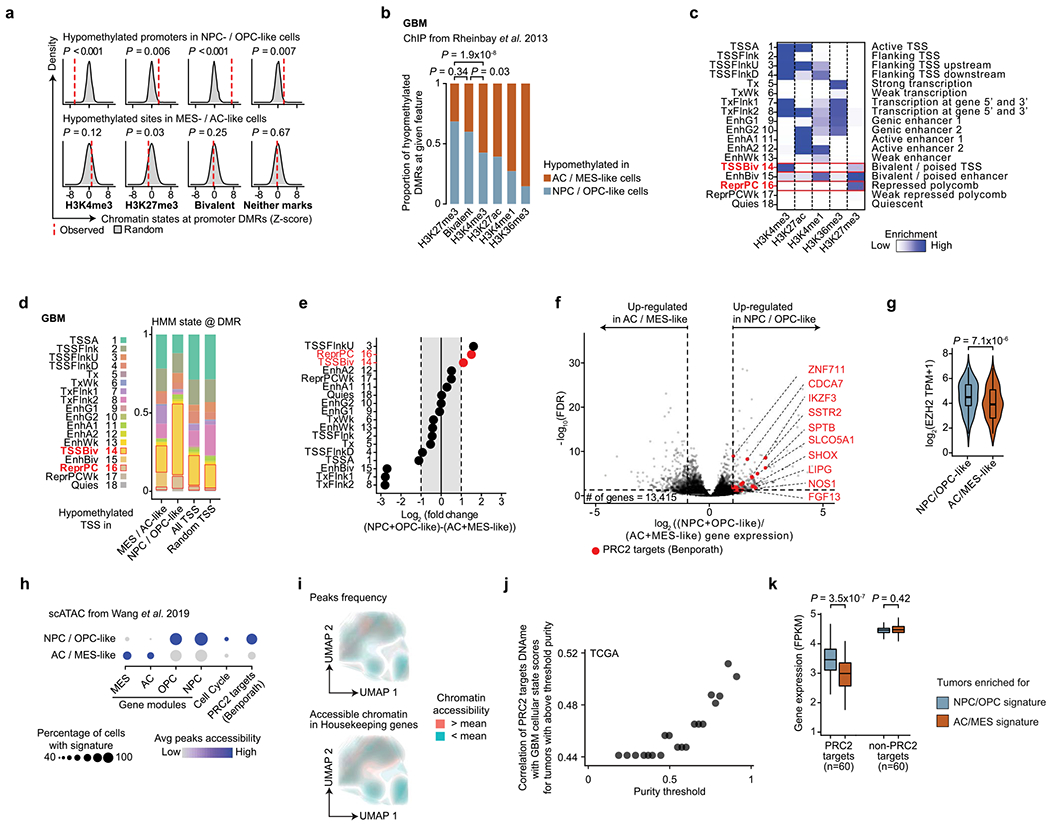
a, Proportion of chromatin states at randomly sampled promoters (1,000 random samplings) and hypomethylated promoters in GBM stem-like (top) vs. AC/MES-like (bottom) cells. b, Proportion of ChIP-seq peaks47 at hypomethylated promoters in GBM stem-like vs. AC/MES-like cells. c, Heatmap of emission parameters for a HMM 18-state model derived from GBM ChIP-seq maps47. Chromatin states of interest are highlighted in red. d, Proportion of chromatin states (see (c)) at hypomethylated promoters in GBM stem-like and AC/MES-like cells (Fig. 2c), all genes used in differential methylation promoter analysis (n = 15,218 genes), and randomly sampled promoters. e, Fold-change (log2) of chromatin states (see (c)) between hypomethylated promoters in GBM stem-like vs. AC/MES-like cells. Chromatin states of interest are highlighted in red. f, Differential gene expression between NPC/OPC-like (n = 270) and AC-/MES-like cells (n = 437). PRC2 target46 genes are highlighted. g, EZH2 expression (scRNAseq) between NPC-/OPC-like and MES-/AC-like cells across GBM samples. h, Gene expression activity derived from scATAC-seq open chromatin for GBM cellular states, cell cycle-related genes, and PRC2 targets46 at distinct NPC-/OPC-like and AC-/MES-like clusters identified based on scATACseq GBM data55. i, UMAP of scATACseq GBM data55 (sample SF11956) overlaid with density plot of peaks frequency (top) and chromatin accessibility of housekeeping genes1 (bottom). j, Spearman’s rank-order correlation between mean DNAme at promoters of PRC2 targets46 and RNA differentiation score and bulk sample purity for 67 TCGA GBM samples40,41. k, Mean gene expression of hypomethylated PRC2 targets in stem-like cells (n = 60; Fig. 2c) and randomly selected non-PRC2 targets (n = 60) in TCGA GBM samples40,41 enriched for NPC-/OPC-like vs. AC-/MES-like signature. P values were determined by permutation test (a), two-sided Fisher’s exact test (b), weighted F-test (f), two-sided Mann-Whitney U test (g, k). Boxplots represent the median, bottom and upper quartiles, whiskers correspond to 1.5 times the interquartile range.
Extended Data Fig. 6 |. PRC2 target DNAme underlies the classification of GBM tumors by bulk DNAme.
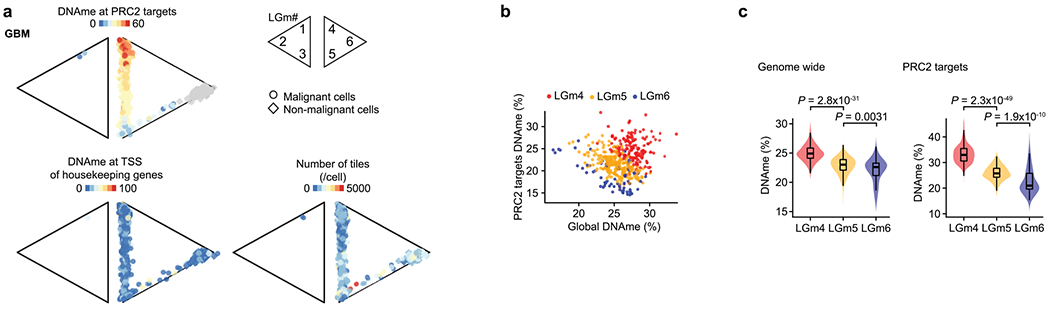
a, Two-dimensional representation of single cells assigned to previously described LGm classes39, visualized as triangle plots (where each vertex corresponds to one LGm class) across 7 GBM samples (n = 867 cells [malignant and non-malignant] that passed quality control based on scDNAme). Mean DNAme at promoters of PRC2 targets46 (top), mean DNAme at promoters of housekeeping genes1, and number of tiles per cell (bottom) are overlaid for each triangle plot. b, Comparison between mean genome wide DNAme (defined as the mean DNAme across windows of 1,000 bp around 450K array probes, Methods) and mean DNAme at promoters (TSS ± 1Kb) of PRC2 targets46 for the 478 TCGA GBM samples that were classified as LGm4-6 by Ceccarelli et al.39. LGm classes assignment for each sample is shown. c, Left: mean genome wide DNAme for TCGA GBM samples (n = 478) previously classified as either LGm4, LGm5, or LGm6 by Ceccarelli et al.39 Right: mean DNAme at promoters (TSS ± 1Kb) of PRC2 targets46 for TCGA GBM samples (n = 478) previously classified as either LGm4, LGm5, or LGm6 by Ceccarelli et al.39. P values were determined by two-sided Mann-Whitney U test (c). Boxplots represent the median, bottom and upper quartiles, whiskers correspond to 1.5 times the interquartile range.
Extended Data Fig. 7 |. Comparison of DNA methylation and chromatin state patterns between transcriptional cell states in IDH-MUT.
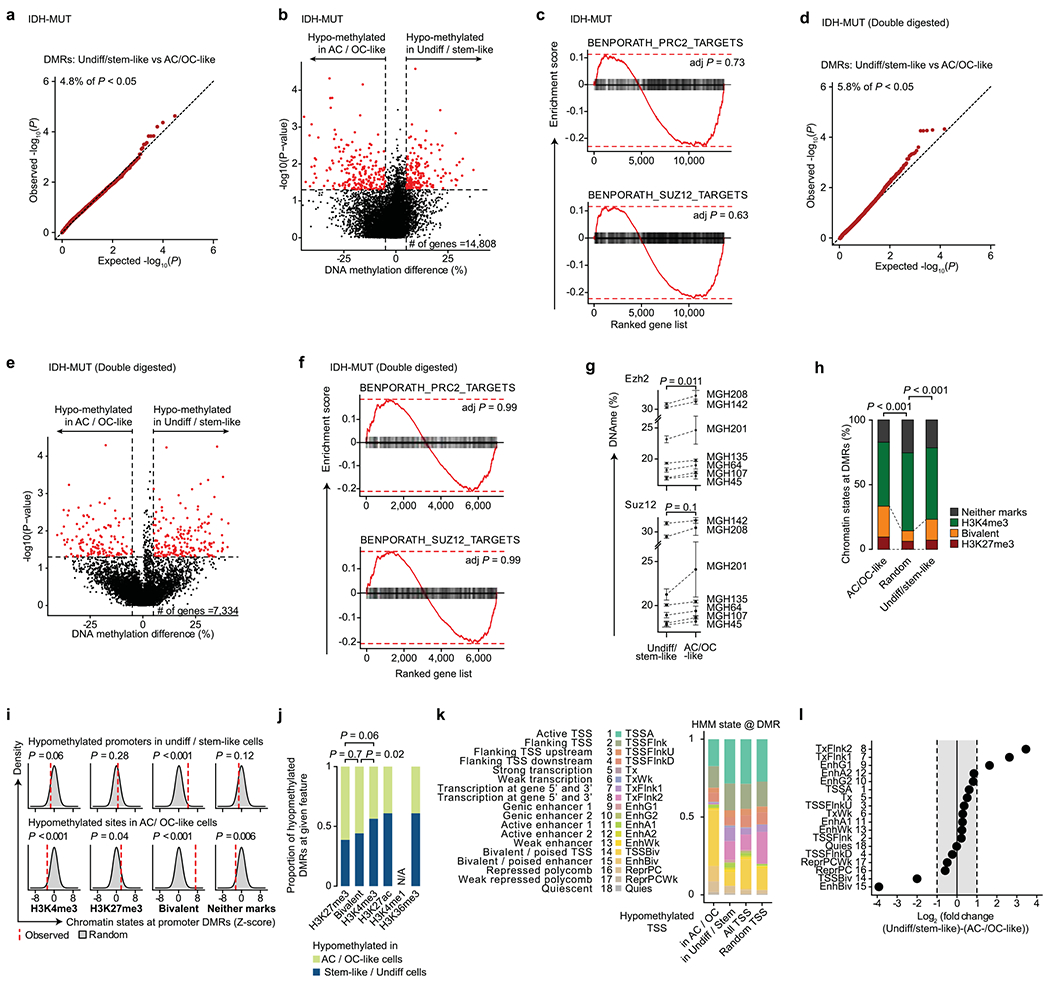
a, Q-Q plot comparing the observed −log10P values of genes used in the differential methylation analysis of promoters (n = 14,808 genes) between undiff/stem-like and AC-/OC-like IDH-MUT cellular states (defined in (b)) to expected −log10P values. b, Differentially methylated promoters between undiff/stem-like (n = 251) and AC-/OC-like (n = 133) cells with matched scRNAseq and scDNAme data across IDH-MUT samples. Promoters with absolute mean DNAme difference > 5% and P values < 0.05 were defined as differentially methylated (red). c, Enrichment score plots (n = 14,808 genes, as in (b)) for PRC2 and SUZ12 targets46 between stem-like/undifferentiated cells and AC-/OC-like cells in IDH-MUT samples. d-f, Same as (a-c), for single-cell DNA methylomes obtained performing double digestion with HaeIII+MspI on cells from two IDH-MUT samples (MGH201 and MGH208). g, Mean (±s.e.m.) CpG methylation at ChIP-seq maps50 of EZH2 and SUZ12 between undiff/stem-like and AC-/OC-like cells in each IDH-MUT sample. h, Proportion of chromatin states at hypomethylated promoters in IDH-MUT AC-/OC-like cells (defined in (b)), randomly sampled promoters (1,000 random samplings), and hypomethylated promoters in IDH-MUT undiff/stem-like (defined in (b)). i, Proportion of chromatin states at randomly sampled promoters (1,000 random samplings) and hypomethylated promoters in IDH-MUT undiff/stem-like (top) vs. AC-/OC-like cells (bottom.) j, Proportion of ChIP-seq peaks47 at hypomethylated promoters in IDH-MUT undiff/stem-like vs. AC-/OC-like cells. k, Proportion of each of the chromatin states (defined in Extended Data Fig. 5c) at hypomethylated promoters in IDH-MUT undiff/stem-like (defined in (b)), hypomethylated promoters in IDH-MUT AC-/OC-like cells (defined in (b)), all genes used in differential methylation promoter analysis (n = 14,808 genes), and randomly sampled promoters, respectively. l, Fold-change (log2) of chromatin states between hypomethylated promoters in IDH-MUT undiff/stem-like vs. AC-/OC-like cells. P values were determined by generalized linear model (a-b, d-e), Fisher’s combined probability test (g), permutation test (c, f, h-i), two-sided Fisher’s exact test (j).
Extended Data Fig. 8 |. IDH-MUT cells exhibit preferential enhancer hypermethylation, decoupling of the promoter methylation-expression relationship and disruption of CTCF insulation.
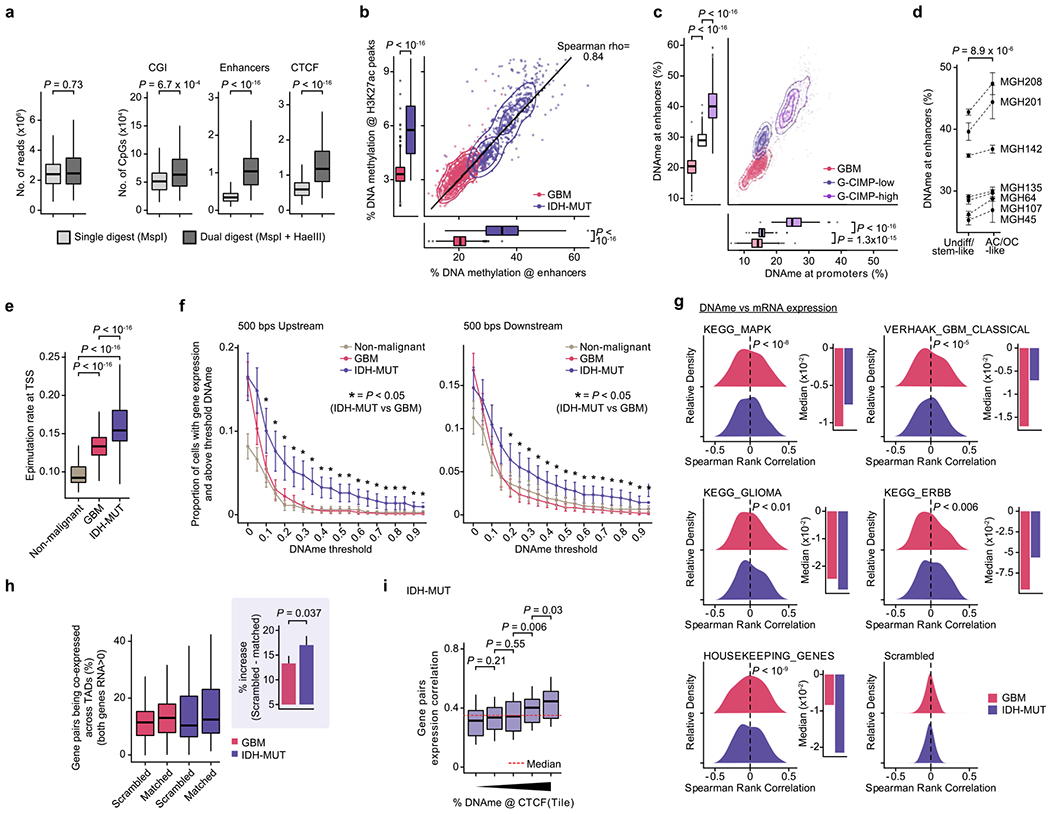
a, Number of aligned reads and unique CpGs for MspI (n=476) and HaeIII+MspI digested IDH-MUT cells (n=242; MGH201 and MGH208). b, Mean CpG methylation at FANTOM5 enhancers vs. H3K27ac ChIP-seq peaks47,70 between GBM (n=765) and IDH-MUT (n=670) cells. c, Mean CpG methylation at TSS (±1Kb) vs. FANTOM5 enhancers between GBM (n=765) and IDH-MUT (n=670) cells (G-CIMP-low [MGH107, MGH135, MGH45, MGH64]; G-CIMP-high [MGH142, MGH201, MGH208]). d, Mean (±SEM) CpG methylation at FANTOM5 enhancers for stem-like/undifferentiated and AC-/OC-like IDH-MUT cells. e, Epimutation rate across non-malignant (n=148), GBM (n=765) and IDH-MUT (n=670) cells. f, Proportion of cells with gene expression (read count >0) and above-threshold DNAme at 500 base-pairs regions upstream (left) or downstream (right) of TSS. Data are mean (±s.e.m.) across all genes (expression seen in > 5 cells, DNAme >5 CpGs per region) for non-malignant cells (n=148), GBM (n=765) and IDH-MUT (n=670) cells. ’*’ P-value < 0.05. g, Left: Distribution of Spearman’s rho of expression and promoter DNAme correlation (n=1,523 genes expressed >5 cells, DNAme >5 CpGs per promoter); GBM (n=765) and IDH-MUT (n=670) cells. Right: Median values of Spearman’s rho of expression and promoter DNAme correlation. h, Percentage of genes pairs across CTCF sites70 being co-expressed (both RNA read count >0); GBM (n=765) and IDH-MUT (n=670) cells. Scrambled represents randomly permuted cell labels for the expression values. Inset: Increase in percentage of genes pairs across CTCF sites70 being co-expressed when comparing matched vs. scrambled groups. Error bars represent 95% CIs. i, Gene expression correlation (Spearman’s rho) of genes pairs across CTCF sites70 per tile of mean CpG methylation at CTCF binding sites (low-to-high); IDH-MUT (n=670) cells. P values are two-sided Mann-Whitney U test (a-c, e-f, h-i), Fisher’s combined probability test (d), two-sided Kolmogorov-Smirnov test (g). Boxplots represent the median, bottom and upper quartiles, whiskers correspond to 1.5 times the interquartile range.
Extended Data Fig. 9 |. High-resolution DNAme-based lineage trees coupled with leaf annotation of cellular states.
a, Representative (random cell subsampling) DNAme-based lineage tree for each GBM patient sample (including MGH105 biological replicates and MGH121 technical replicates), with projection of GBM cellular states. b, Representative (random cell subsampling) DNAme-based lineage tree for each IDH-MUT patient sample (including MGH142 and MGH208 technical replicates), with projection of IDH-MUT cellular states. Throughout the figure, scale represents DNAme changes per site.
Extended Data Fig. 10 |. Cell state transition dynamics inference from lineage tree architectures revealed higher cellular plasticity in GBM compared to a more stable differentiation hierarchy in IDH-MUT.
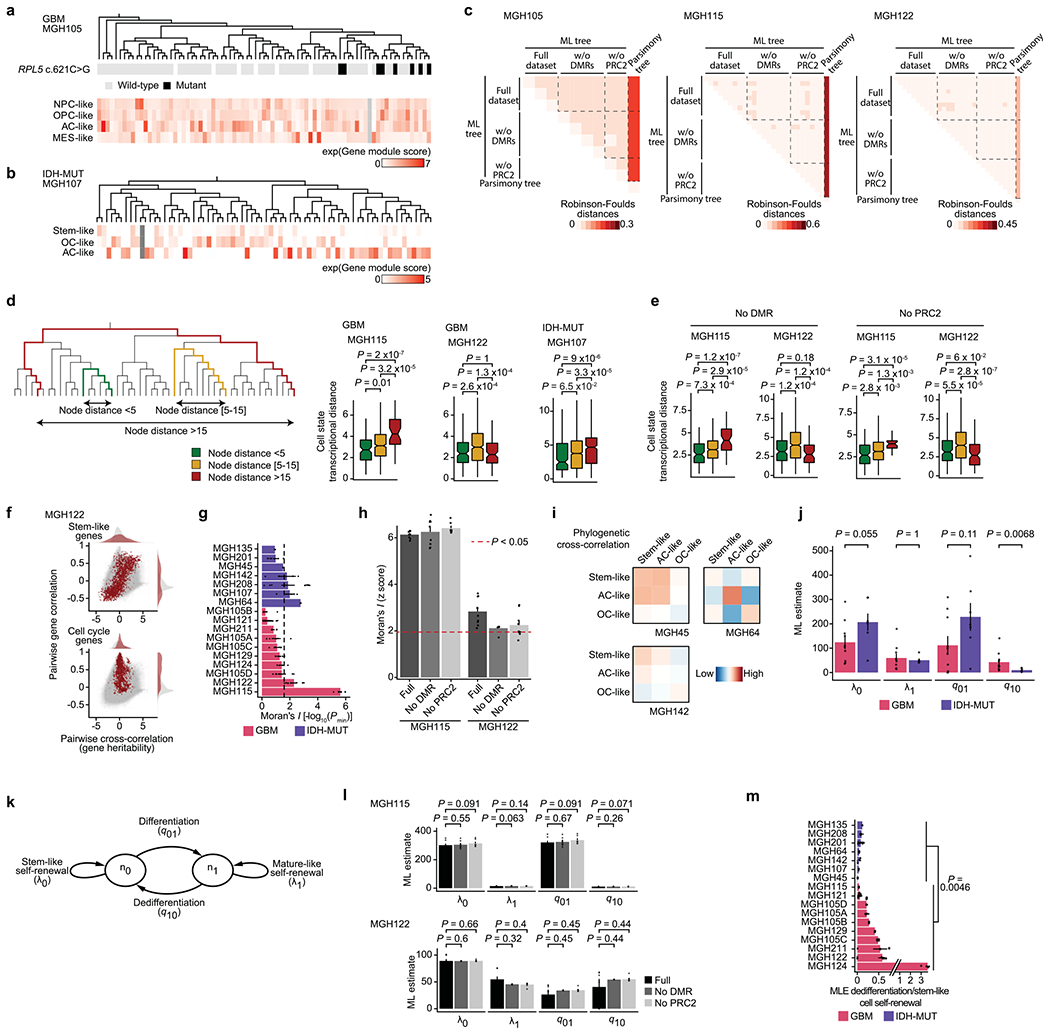
a, Top: GBM DNAme-based lineage tree (MGH105) with RPL5 c.621 C>G genotyping. Bottom: GBM gene module scores. b, IDH-MUT DNAme-based lineage tree (MGH107) with IDH-MUT gene module scores. c, Normalized Robinson-Foulds between GBM tree replicates (from same sample; full dataset or removing CpGs from DMRs (Fig. 2c) or PRC2 targets46) reconstructed by maximum-likelihood (ML) vs. maximum parsimony. d, Transcriptional distances as function of lineage distance between unique cell pairs for MGH115, MGH122 and MGH107. e, As (d), for DNAme-based lineage tree of MGH115 and MGH122 (n=47 and 46 cells, respectively) reconstructed removing CpGs from DMRs (Fig. 2c) or PRC2 targets46. f, Pairwise gene expression correlation (Pearson’s) and cross-correlation (heritability). Grey points=all gene pair relationships; red points=gene pair relationships within selected gene module (top: stem-like; bottom: cell cycle). g, Phylogenetic association of cell states on GBM (n=7 patients; n=10 samples with MGH105A-D) and IDH-MUT (n=7 patients). Barplots=weighted mean±s.e.m. Moran’s I permutation-based one-sided P values (106 permutations) across replicates. Dashed line: P=0.025. h, As (g), comparing DNAme-based lineage tree reconstruction of MGH115 and MGH122, using replicates from same sample with full dataset or removing CpGs from DMRs (Fig. 2c) or PRC2 targets46. Barplots=mean±s.e.m. i, Heat maps of pairwise cell state phylogenetic associations. Close phylogenetic associations are shown in warmer colors. j, ML estimate (median±MAD across tree replicates; samples as in (g)) rates of cell state growth and transition. k, Mathematical model of glioma evolutionary dynamics. l, ML estimate (mean±s.e.m. across tree replicates of MGH115 and MGH122) rates of cell state self-renewal and transition, using replicates from same sample (full dataset or removing CpGs from DMRs analysis (Fig. 2c) or PRC2 targets46). m, Weighted median±weighted MAD rates of dedifferentiation compared to stem-like cell self-renewal across lineage tree replicates (sample as in (g)). P values: two-sided Mann-Whitney U test (d-e, j, l-m). Boxplots: median, bottom and upper quartiles, whiskers: 1.5 times the interquartile range.
Supplementary Material
Acknowledgements
We thank members of the Landau and Suva laboratories for constructive discussions, the Epigenomics Core Facility at Weill Cornell Medical College for technical help and E. Rheinbay at the Massachusetts General Hospital Cancer Center for whole-exome sequencing data processing. This project and R.C. have received funding from the European Union’s Horizon 2020 research and innovation program under Marie Skłodowska-Curie grant agreement no. 750345. F.G. was supported by NIH K99/R00 Pathway to Independence Award (NCI K99CA248955). D.S. was supported by EMBO long-term fellowship ALTF (570-2017) and by the Schmidt Family Foundation. J.K. was supported by an HFSP long-term fellowship (LT000452/2019-L). A.R. was supported by funds from the Howard Hughes Medical Institute, the Klarman Cell Observatory, the STARR Cancer Consortium, NCI grant 1U24CA180922, NCI grant R33CA202820, Koch Institute support (core) grant P30CA14051 from the NCI, the Ludwig Center and the Broad Institute. L.N.G.C. was supported by NIH award K12CA090354. This work was supported by grants to M.L.S. from the Mark Foundation (Emerging Leader Award), the Sontag Foundation (Distinguished Scientist Award), the MGH Research Scholars, and NCI R37CA245523 and NCI R01CA258763 (to M.L.S. and D.A.L.). D.A.L. was supported by the Burroughs Wellcome Fund Career Award for Medical Scientists, the Pershing Square Sohn Prize for Young Investigators in Cancer Research, the NIH Director’s New Innovator Award (DP2-CA239065), the Sontag Foundation (Distinguished Scientist Award, SFI 203261-01), the William Rhodes and Louise Tilzer-Rhodes Center for Glioblastoma at NewYork-Presbyterian Hospital (NYPH 203205-01) and NHGRI RM1HG011014-01. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41588-021-00927-7.
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The analytic code used for this work is provided for noncommercial use at https://doi.org/10.5281/zenodo.4776456 (ref.117).
Competing interests
M.L.S. is an equity holder, scientific cofounder and advisory board member of Immunitas Therapeutics. A.R. is a founder and equity holder of Celsius Therapeutics, is an equity holder in Immunitas Therapeutics and, until 31 July 2020, was a scientific advisory board member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. Since 1 August 2020, A.R. has been an employee of Genentech. Since 19 October 2020, O.R.-R. has been an employee of Genentech. D.A.L. is an equity holder, scientific cofounder and advisory board member of C2i Genomics and a scientific advisory board member for Mission Bio. The authors declare that these activities are not related to the research reported in this publication and have not influenced the conclusions in this manuscript. The remaining authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41588-021-00927-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41588-021-00927-7.
Peer review information Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
Processed data generated for this study are available through the NCBI Gene Expression Omnibus (GEO) under accession number GSE151506. Raw data access can be requested through the Data Use Oversight System (DUOS) Dataset Catalog with dataset ID DUOS-000133 as well as the European Genome–phenome Archive (EGA) with dataset ID EGAS00001005472. The data can be visualized and interrogated through the Broad Institute’s Single-Cell Portal at https://singlecell.broadinstitute.org/single_cell/study/SCP936. scATAC-seq data are available at the EGA repository under EGAS00001002185, EGAS00001001900 and EGAS00001003845 and at NCBI GEO under accession number GSE138794. TCGA data (DNA methylation, gene expression and clinical profiles) are available from the TCGA database (https://cancergenome.nih.gov/). ChIP–seq data are available at NCBI GEO under accession number GSE46016.
References
- 1.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nam AS et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature 571, 355–360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puram SV et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hata AN et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med 22, 262–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaffer SM et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaffer SM et al. Memory sequencing reveals heritable single-cell gene expression programs associated with distinct cellular behaviors. Cell 182, 947–959 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frieda KL et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spanjaard B et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol 36, 469–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raj B et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol 36, 442–450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alemany A, Florescu M, Baron CS, Peterson-Maduro J & van Oudenaarden A Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL & Rich JN Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gimple RC, Bhargava S, Dixit D & Rich JN Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 33, 591–609 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suvà ML et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suvà ML & Tirosh I The glioma stem cell model in the era of single-cell genomics. Cancer Cell 37, 630–636 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Bao S et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Liau BB et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell 20, 233–246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neftel C et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garofano L et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2, 141–156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards LM et al. Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2, 157–173 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Castellan M et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in glioblastoma. Nat. Cancer 2, 174–188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latil M et al. Cell-type-specific chromatin states differentially prime squamous cell carcinoma tumor-initiating cells for epithelial to mesenchymal transition. Cell Stem Cell 20, 191–204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flavahan WA, Gaskell E & Bernstein BE Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meir Z, Mukamel Z, Chomsky E, Lifshitz A & Tanay A Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat. Genet 52, 709–718 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guilhamon P et al. Single-cell chromatin accessibility profiling of glioblastoma identifies an invasive cancer stem cell population associated with lower survival. eLife 10, e64090 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fine HA Malignant gliomas: simplifying the complexity. Cancer Discov. 9, 1650–1652 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Gaiti F et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569, 576–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Morton AR et al. Functional enhancers shape extrachromosomal oncogene amplifications. Cell 179, 1330–1341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun W et al. The association between copy number aberration, DNA methylation and gene expression in tumor samples. Nucleic Acids Res. 46, 3009–3018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Hagan HM, Mohammad HP & Baylin SB Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 4, e1000155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davoli T et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ceccarelli M et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLendon R et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brennan CW et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capper D et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pine AR et al. Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov. 10.1158/2159-8290.CD-20-0057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verhaak RGW et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ben-Porath I et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet 40, 499–507 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rheinbay E et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 3, 1567–1579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suvà M-L et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 69, 9211–9218 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Natsume A et al. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 73, 4559–4570 (2013). [DOI] [PubMed] [Google Scholar]
- 50.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Connor T, Grant CE, Bodén M & Bailey TL T-Gene: improved target gene prediction. Bioinformatics 10.1093/bioinformatics/btaa227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reddington JP, Sproul D & Meehan RR DNA methylation reprogramming in cancer: Does it act by re-configuring the binding landscape of Polycomb repressive complexes? Bioessays 36, 134–140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Douillet D et al. Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat. Genet 10.1038/s41588-020-0618-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bintu L et al. Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 9, 1708–1719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffmann A, Sportelli V, Ziller M & Spengler D Switch-like roles for Polycomb proteins from neurodevelopment to neurodegeneration. Epigenomes 1, 21 (2017). [Google Scholar]
- 57.Xu W et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turcan S et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu F, Liu Y, Jiang L, Yamaguchi S & Zhang Y Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 10.1101/gad.248005.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hon GC et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 56, 286–297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ginno PA et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat. Commun 11, 2680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Creyghton MP et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21931–21936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Landau DA et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 26, 813–825 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shipony Z et al. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 513, 115–119 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Landan G et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet 44, 1207–1214 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Pan H et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat. Commun 6, 6921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jones PA Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Turcan S et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet 50, 62–72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Núñez FJ et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med 11, eaaq1427 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brocks D et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep. 8, 798–806 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Roerink SF et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Shibata D Mutation and epigenetic molecular clocks in cancer. Carcinogenesis 32, 123–128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moran PAP Notes on continuous stochastic phenomena. Biometrika 37, 17–23 (1950). [PubMed] [Google Scholar]
- 75.Maddison WP, Midford PE & Otto SP Estimating a binary character’s effect on speciation and extinction. Syst. Biol 56, 701–710 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Stadler T & Bonhoeffer S Uncovering epidemiological dynamics in heterogeneous host populations using phylogenetic methods. Philos. Trans. R. Soc. B Biol. Sci 368, 20120198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boyle AP et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bell RE et al. Enhancer methylation dynamics contribute to cancer plasticity and patient mortality. Genome Res. 26, 601–611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ziller MJ et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pastore A et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun 10, 1874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Irizarry RA et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet 41, 178–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polak P et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet 49, 1476–1486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Izzo F et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet 52, 378–387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Challen GA et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet 44, 23–31 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klughammer J et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med 24, 1611–1624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boyer LA et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Margueron R & Reinberg D The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bernstein BE et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Boulard M, Edwards JR & Bestor TH FBXL10 protects Polycomb-bound genes from hypermethylation. Nat. Genet 47, 479–485 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Meissner A et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Domcke S et al. A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mohn F et al. Lineage-specific Polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30, 755–766 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Suvà ML, Riggi N & Bernstein BE Epigenetic reprogramming in cancer. Science 339, 1567–1570 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alcantara Llaguno SR & Parada LF Cell of origin of glioma: biological and clinical implications. Br. J. Cancer 115, 1445–1450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaffer CL et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl Acad. Sci. USA 108, 7950–7955 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morris V et al. Single-cell analysis reveals mechanisms of plasticity of leukemia initiating cells. Preprint at bioRxiv 10.1101/2020.04.29.066332 (2020). [DOI] [Google Scholar]
- 97.Lieberman E, Hauert C & Nowak MA Evolutionary dynamics on graphs. Nature 433, 312–316 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Lappalainen T & Greally JM Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet 18, 441–451 (2017). [DOI] [PubMed] [Google Scholar]
- 99.Angermueller C, Lee HJ, Reik W & Stegle O DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 18, 67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Van den Berge K et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 19, 24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Risso D, Perraudeau F, Gribkova S, Dudoit S & Vert J-P A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun 9, 284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van den Berge K, Soneson C, Robinson MD & Clement L stageR: a general stage-wise method for controlling the gene-level false discovery rate in differential expression and differential transcript usage. Genome Biol. 18, 151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seshan VE & Olshen AB DNAcopy: a package for analyzing DNA copy data (v1.60.0). R package. (2021). [Google Scholar]
- 108.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Robinson DF & Foulds LR Comparison of phylogenetic trees. Math. Biosci 53, 131–147 (1981). [Google Scholar]
- 110.Gittleman JL & Kot M Adaptation: statistics and a null model for estimating phylogenetic effects. Syst. Biol 39, 227–241 (1990). [Google Scholar]
- 111.Wartenberg D Multivariate spatial correlation: a method for exploratory geographical analysis. Geographical Anal. 17, 263–283 (1985). [Google Scholar]
- 112.Czaplewski RL Expected Value and Variance of Moran’s Bivariate Spatial Autocorrelation Statistic for a Permutation Test (US Department of Agriculture, Forest Service, Rocky Mountain Forest and Range Experiment Station, 1993). [Google Scholar]
- 113.FitzJohn RG Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol 3, 1084–1092 (2012). [Google Scholar]
- 114.Revell LJ phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol 3, 217–223 (2012). [Google Scholar]
- 115.Xiang Y, Gubian S, Suomela B & Hoeng J Generalized simulated annealing for global optimization: the GenSA package. R Journal 5, 13 (2013). [Google Scholar]
- 116.Bolker B Maximum likelihood estimation and analysis with the bbmle package (v1.0.23.1). R package. (2021). [Google Scholar]
- 117.Gaiti F, Silverbush D, Schiffman J & Kluegel L Single-cell multi-omics profiling of human gliomas. Zenodo 10.5281/zenodo.4776456 (2021). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data generated for this study are available through the NCBI Gene Expression Omnibus (GEO) under accession number GSE151506. Raw data access can be requested through the Data Use Oversight System (DUOS) Dataset Catalog with dataset ID DUOS-000133 as well as the European Genome–phenome Archive (EGA) with dataset ID EGAS00001005472. The data can be visualized and interrogated through the Broad Institute’s Single-Cell Portal at https://singlecell.broadinstitute.org/single_cell/study/SCP936. scATAC-seq data are available at the EGA repository under EGAS00001002185, EGAS00001001900 and EGAS00001003845 and at NCBI GEO under accession number GSE138794. TCGA data (DNA methylation, gene expression and clinical profiles) are available from the TCGA database (https://cancergenome.nih.gov/). ChIP–seq data are available at NCBI GEO under accession number GSE46016.