

Abstract
Enzymatic reactions through mononuclear metal hydrides are unknown in nature, despite the prevalence of such intermediates in the reactions of synthetic transition-metal catalysts. If metalloenzymes would react through abiotic intermediates like these, then the scope of enzyme-catalyzed reactions would expand. Here we show that zinc-containing carbonic anhydrase enzymes catalyze hydride transfers from silanes to ketones with high enantioselectivity and report mechanistic data providing strong evidence that the process involves a mononuclear zinc hydride. This work shows that abiotic silanes can act as reducing equivalents in an enzyme-catalyzed process and that monomeric hydrides of electropositive metals, which are typically unstable in protic environments, can be catalytic intermediates in enzymatic processes. Overall, this work bridges a gap between the types of transformations in molecular catalysis and biocatalysis.
Graphical Abstract
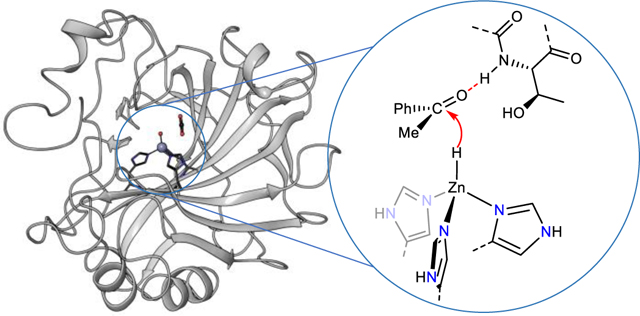
The mechanisms of reactions catalyzed by transition-metal complexes in the laboratory often differ from those in biology. If the intermediates of these abiotic reactions of synthetic systems could be transported to those of metalloenzymes, then the scope of enzymatic reactions could be broadened. Recently, interest in the application of enzymes to abiotic reactions has expanded1,2, and directed evolution of natural enzymes and of artificial metalloenzymes has led to a series of abiotic group-transfer processes3,4,5,6. However, the scope of reactions in organometallic chemistry that involve X-type ligands7 is broader than those of group transfers1,8,9. In particular, monomeric, metal-hydride complexes are important intermediates in reactions catalyzed by transition-metal complexes, but natural enzymes that react through such intermediates are unknown1,10. Instead, enzymatic reactions involving monomeric hydrides have occurred with artificial metalloenzymes in which well-established molecular iridium, rhodium or ruthenium catalysts are linked to a host protein through covalent or supramolecular, non-covalent interactions11–14. Natural enzymes that react through multimetallic metal-hydride intermediates are known, such as hydrogenases, but these enzymes contain dinuclear active sites comprising a bridging hydride ligand that are prone to undergo electron transfer instead of hydride transfer.
Zinc-enzymes are among the most prevalent families of metalloenzymes in biological systems15. Carbonic anhydrases are particularly well studied zinc enzymes16. They convert carbon dioxide to carbonic acid by a mechanism involving binding of CO2 to the active site by hydrogen bonding interactions (PDB ID 3D92) to place the electrophilic carbon center of CO2 in close proximity to the oxygen atom of the zinc hydroxide (Fig. 1a, left). Addition of the hydroxo group to CO2 generates a bicarbonate complex, and subsequent displacement of the bicarbonate with water forms carbonic acid and regenerates the zinc hydroxide. Thus, the enzyme functions by the cooperative action of a Lewis acid (the zinc ion) and a hydrogen-bonding donor (amide N-H of Thr199) to achieve the addition of a nucleophilic hydroxide to the electrophile.
Figure 1. Structure and catalytic mechanism of human carbonic anhydrase.

a, Mechanism for the conversion of CO2 to bicarbonate catalyzed by hCAII and the proposed zinc-hydride addition to ketones catalyzed by the same hCAII. b, NADPH as the reductant for natural reactions catalyzed by ketoreductases c, Zinc hydride generated from silanes was used as the reductants in this work for abiotic enzyme-catalyzed reductions of ketones.
We hypothesized that the reactions of synthetic zinc complexes could be translated into zinc enzymes, thereby creating pathways that are distinct from those of natural enzymes. Specifically, we reasoned that reductions mediated by zinc complexes could be catalyzed by carbonic anhydrases if the zinc active site transferred hydride, instead of hydroxide, to unsaturated compounds (Fig 1a, right). These reactions could occur with simple synthetic reducing agents not found in nature, rather than the nicotinamide adenine dinucleotide phosphate (NADPH) cofactor of natural systems (Fig. 1b). Because zinc enzymes can be produced in large quantities and possess earth-abundant metals, they could be valuable replacements for precious-metal catalysts with chiral ligands17,18, and because such catalysts consist of a single component, rather than the combination of a protein and cofactor, abiotic reactions catalyzed by such enzymes could be conducted readily with whole cells. Here, we report the catalytic enantioselective transfer of the hydride of a silane to a range of ketones to form chiral alcohols enantioselectively. Mechanistic studies strongly implicate the intermediacy of a protein-bound, mononuclear zinc hydride complex that transfers this hydride to the ketone during the catalytic cycle.
Results and Discussion
Asymmetric reduction of ketone with silanes catalyzed by hCAII.
Small molecular zinc alkoxide or fluoride complexes are known to react readily with silanes to produce zinc hydrides19–21. On the basis of this reactivity of small molecules, we hypothesized that silanes can react with zinc hydroxide within carbonic anhydrase to form a zinc hydride. We tested phenylsilane, methyl phenylsilane and dimethyl phenylsilane as hydride donors for the reaction of 4-acetylpyridine in the presence of human carbonic anhydrase II (hCAII) as the catalyst (Table 1, entry 1–4). 4-Acetylpyridine was chosen as the model ketone because its solubility in water is greater than that of more typical ketones, like acetophenone.
Table 1.
Reduction of 4-Acetylpyridine with Silanes Catalyzed by Wild-Type hCAII
| |||
|---|---|---|---|
|
| |||
| Entrya | Variation from the standard conditions | Yield | e.e. |
| 1 | none | 99% | 98% |
| 2 | 3 equiv. PhMeSiH2 | 78% | 98% |
| 3 | 3 equiv. PhMe2SiH | 0% | -- |
| 4 | 10 equiv. PhMe2SiH | 0% | -- |
| 5 | 3 equiv. Et3SiH | 0% | -- |
| 6 | 3 equiv. (Me3SiO)2MeSiH | 0% | -- |
| 7 | 3 equiv. (iPr)2SiH2 | 0% | -- |
| 8 | no enzyme | 0% | -- |
| 9 | 0.2 mol% hCAVII | 67% | 97% |
| 10 | 0.2 mol% hCAXIII | 21% | 89% |
0.1 μmol of hCAII, 1 mL of buffer, 50 μmol of ketone, 150 μmol of PhSiH3 (3 equiv. w.r.t. ketone). Yields were determined by 1H NMR, e.e. values were determined by HPLC or SFC.
The identity of the silane was shown to influence the yield of alcohol. The reaction of 4-acetylpyridine with dimethyl phenylsilane, the most hindered and least hydridic silane, did not occur, but the reactions with methyl phenyl silane and phenyl silane produced the alcohol in 78% yield and 95% yield, respectively. PhSiMe2H has been computed to be less hydridic than PhSiH3 by 11 kcal·mol–1. Although several factors could cause the differences in reactivity between the silanes, the lower hydricity is consistent with the lack of reaction of PhSiMe2H22. We also tested various alkyl silanes and siloxy silanes for these reductions of ketones, but none of them gave detectable amounts of alcohol (Table 1, entry 5–7).
In addition to hCAII, we tested the activity of human carbonic anhydrase VII (hCAVII) and human carbonic anhydrase XIII (hCAXIII) as catalysts for the asymmetric reduction of ketones. Both hCAVII and hCAXIII are homologous to hCAII and contain the same zinc active site and CO2 binding site. Both hCAVII and hCAXIII catalyzed the reaction of phenylsilane with 4-acetylpyridine under the same conditions as the reactions catalyzed by hCAII, but with lower activity or enantioselectivity, or both (Table 1, entry 8–10).
In practice, biocatalytic processes on a large scale are typically performed with E. coli whole cells or cell lysates to avoid expensive and labor-intensive protein purification steps. We found that hCAII in wet whole cells catalyzed the asymmetric reduction of 4-acetylpyridine in 95% yield and 98% e.e. at the same loading of enzyme as that used for reactions with purified hCAII; this yield and enantioselectivity are almost identical to those of the purified hCAII (Fig. 2a). Although wet whole cells lost part of their catalytic activity after storing for 3 days, a dry powder formed by lyophilizing freshly cultured whole cells catalyzed the reduction of the ketone as well after three months of storage as when freshly prepared. Therefore, lyophilized whole cells were used for our studies on substrate scope.
Figure 2. Catalytic reduction of ketones with hCAII in whole cells.

a, Catalytic activity of purified protein (green), wet whole cells (cyan), stored wet whole cell (yellow) and lyophilized whole cell (brown). b, Enantioselective reduction of 4-acetylpyridine and acetophenone by deuterated phenyl silane. c, The yield and enantioselectivity of reaction product from whole-cell reduction of 4-acetylpyridine on gram scale. The picture is a snapshot of the gram-scale reaction.
We performed deuterium labeling experiments to test whether the whole-cell reactions occurred with the silane or by NAD(P)H, potentially catalyzed by another enzyme23,24. 4-Acetylpyridine was reduced in the presence of the E. coli cells to the chiral alcohol in quantitative yield and 98% e.e. with PhSiD3 as the reductant. The same level of incorporation of deuterium was observed in the alcohol formed by reduction of acetophenone. These yields and e.e. values are similar to those achieved with PhSiH3 (Fig. 2b). Most important, the percentage of deuterium incorporation, which was analyzed by 1H NMR spectroscopy, was 99% in both cases. This experiment confirmed that the terminal reductant with whole cells, as in vitro, is the silane, not NAD(P)H.
Substrate scope of ketones and gram-scale reaction.
With optimized reaction conditions in hand, we explored the scope of whole-cell asymmetric reductions of ketones with phenylsilane catalyzed by hCAII in E. coli. This system catalyzed the reduction of a large scope of ketones. Acetophenone was reduced in 89% yield and 91% e.e. at 0.2 mol % loading of hCAII in the presence of 3 equiv. of phenylsilane. Reaction of 4’-chloroacetophenone occurred in quantitative yield and e.e. of >99%. 4-Bromoacetophenone also reacted to a high 81% conversion with undetectable amounts of the minor enantiomer (Table 2, 7-9). Acetophenones containing electron-donating groups, such as 4’-methyl, 4’-methyoxy, and 4’-thiomethoxy groups reacted to lower conversions (51–81%), but all of the ketones reacted with e.e. values above 99% (Table 2, 10-12). We presume that the electron-donating groups lead to lower yields because the lower electrophilicity of the carbonyl group reduces the rate of nucleophilic addition of the zinc hydride relative to the reaction of the hydride with water. Acetophenones containing substituents at the 2’ and 3’ positions, including 3’-bromo, 2’-methoxy, and 2’-chloro groups, were also reduced in high yields and enantioselectivities (Table 2, 13-15). Sterically hindered ketones that contain di-substituted and tri-substituted aryl groups were also converted to the corresponding chiral alcohols in high yields and enantioselectivities (Table 2, 16-18). However, methyl β-naphthyl ketone was reduced in only 51% yield, presumably by a mismatch between the size of the naphthyl group and that of the binding site within hCAII.
Table 2.
Scope of ketones that undergo enantioselective reduction with phenylsilane catalyzed by hCAII.
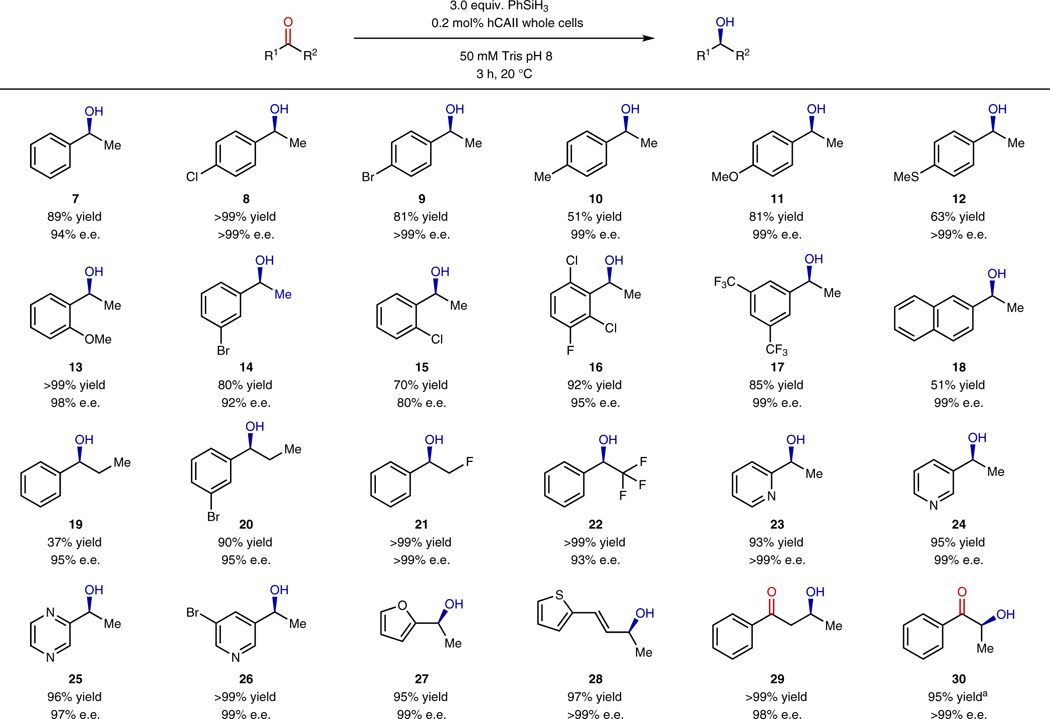
|
0.1 μmol of hCAII, 1 mL of buffer, 50 μmol of ketone, 150 μmol of PhSiH3 (3 equiv. w.r.t. ketone). Yield was determined by 1H NMR, e.e. was determined by chiral HPLC or SFC.
0.1 μmol of hCAII, 1 mL of buffer, 50 μmol of ketone, 20 μmol of PhSiH3 (0.4 equiv. w.r.t. ketone).
In addition to methyl aryl ketones, ethyl aryl ketones and fluoromethyl aryl ketones were readily reduced by phenylsilane in high yields and enantioselectivities when catalyzed by hCAII in whole cells (Table 2, 19-22). Various heterocyclic ketones, including 2- and 3-acetylpyridine, 3-acetylpyrazine, and 1-acetylfuran were readily reduced in high yields and exceptionally high enantioselectivity (2, 23-27). It is striking that the electron pair of the pyridyl, pyrazyl, and furanyl groups did not disrupt the hydrogen-bonding of the carbonyl group in the binding site of hCAII (vide infra). hCAII also catalyzed the 1,2-reduction of an α,β-unsaturated ketone to form the corresponding allylic alcohol (28) in high yield and enantioselectivity.
Encouraged by the high enantioselectivity of hCAII toward a broad scope of substrates, we tested the regioselectivity of the enzyme for the reduction of diketones. The selective reduction of one ketone within a diketone is a challenge for molecular catalysts25,26. The reaction of 1-phenylbutane-1,3-dione with phenylsilane catalyzed by hCAII occurred at the terminal acetyl group in >99% yield and 98% e.e., leading to the S-enantiomer (29). 1-Phenylpropane-1,2-dione is a more challenging substrate for semi-reduction than 1-phenylbutane-1,3-dione because the internal ketone is sterically more hindered but electronically more activated. Again, hCAII selectively reduced the terminal ketone. The terminal carbonyl group was selectively reduced in this case with 0.4 equiv. of silane, showing that multiple silane hydrogens can be transferred in the reduction process. The corresponding α-hydroxyketone was obtained in 95% yield and >99% e.e. (30).
To assess the ability of hCAII in E. coli to catalyze reactions on a large scale, we conducted the asymmetric reduction of 4-acetylpyridine on a gram scale with lyophilized cell powder. The reaction was run in Tris buffer at ambient conditions for 3 h, leading to complete conversion of the starting ketone to the corresponding chiral alcohol. The alcohol was isolated simply by extraction and evaporation of the solvent in 93% yield (1.2 g) and 97% e.e. (Fig. 2c).
Mechanistic experiments indicating the intermediacy of zinc hydride.
Control experiments with mutants of hCAII were conducted to confirm that the catalyst for the whole-cell reactions was hCAII, rather than another cell component, and to determine if the reaction occurred in the same binding site as the natural one for addition of hydroxide to CO2. We prepared ten mutants in which four different residues in the enzyme active site were modified to various non-polar or polar residues (Supplementary Table 1). Based on our computational models (vide infra), all four nonpolar residues of wild-type hCAII (V121, V143, L198, and W209) interact with the aryl group of the bound ketones. Changing the Val to Ile, a larger non-polar residue, significantly reduced the activity of the enzyme for the reaction of phenylsilane with acetophenone, while changing Val to much larger non-polar residues, including Phe and Trp, completely eliminated the activity of the enzyme. Mutants in which any of these non-polar residues had been changed to polar ones eliminated the enzyme activity of hCAII for the reaction of silane with acetophenone. These results show clearly that the reactions in whole cells containing hCAII are catalyzed by this enzyme and not by an NAD(P)H-dependent alcohol dehydrogenase, other class of zinc enzyme, or other component of these cells, and the effect of mutations in the native active site are consistent with reductions by the silane occurring in the same site of hCAII.
A series of experiments provided strong evidence that this abiotic reaction occurred through a monomeric zinc hydride in this active site. Reductions of ketones with silanes catalyzed by synthetic, molecular zinc catalysts occur by one of two classes of mechanisms27. In the first, the carbonyl group is activated by zinc acting as a Lewis acid. In the second, the hydride of the silane is activated. This activation of the hydride can occur by the association of the silane with a basic group or by transfer of the hydride to zinc.
To begin to distinguish between these pathways, we conducted the reactions of silane with hCAII in the absence of substrate and in D2O. If the reaction occurred by a zinc hydride formed from the reaction of a zinc hydroxide or alkoxide with silane, then hydrogen would form in the absence of substrate and, if the reaction is conducted in D2O, then HD, rather than H2 or D2, would form. Indeed, the reaction of silane alone without ketone evolved gas, which was determined to be H2 by dissolving the headspace gases in benzene-d6 and analysis by 1H NMR spectroscopy. A resonance at 4.47 ppm corresponding to H2 was observed (Fig. 3a, top). A similar evolution of gas was observed in the catalytic reaction, indicating that generation of H2 competes with reduction of the ketone. The same reaction in the absence of ketone in D2O formed HD (87% vs H2), as characterized by the distinct 1H NMR signal of HD at 4.43 ppm and H-D coupling constant of 43 Hz (Fig. 3a, bottom).
Figure 3. Mechanistic study for the reduction of ketone catalyzed by carbonic anhydrase.

a, 1H NMR spectra of H2 and HD generated by reaction of hCAII with phenylsilane in H2O and D2O. The NMR signal at 4.48 ppm is residual PhSiH3. b, Two potential mechanisms for reduction of the ketone with PhSiH3, including a ternary mechanism (left) and a stepwise mechanism (right). c, hCAII-catalyzed reduction of 4-acetylpyridine (green and cyan) and acetophenone (yellow and brown) by different silanes. Data are presented as average values and error bars are calculated as the standard deviation. n=3 independent experiments, individual data points are shown as gray circles. d, Formation of zinc hydride from the reaction of zinc alkoxide (top) and zinc fluoride (bottom) with silanes.
The two pathways that involve activation of a hydride can be distinguished by the effect of the silane on enantioselectivity. The first pathway occurs by the formation of a ternary complex, in which a zinc hydroxide is associated with the silane (Fig. 3b, left). The silane in this complex would be more hydridic, thereby enabling transfer of the hydride to the ketone. The second pathway is a stepwise mechanism involving the formation of a zinc hydride from the reaction of zinc hydroxide with phenyl silane and transfer of the zinc-bound hydride to the ketone (Fig. 3b, right).
If the mechanism involves the ternary complex, then the identity of the silane should influence the enantioselectivity of the reduction of the ketone; however, if the reaction occurs by formation of a zinc hydride, then the silane is absent in the step that involves reaction with the ketone and the enantioselectivity should be independent of the identity of the silane. To test the effect of the silane, the reactions of 4-acetylpyridine with phenylsilane and methyl phenylsilane were performed under identical reaction conditions in three replicates (Fig. 3c). The reaction of phenylsilane as the reductant led to 99% yield and 98±0.7% e.e., while the reaction of methylphenylsilane led to 78% yield of the alcohol with an identical 98±0.4% e.e.. Likewise, the reaction of acetophenone with phenylsilane occurred with the same enantioselectivity as the reaction of methylphenylsilane (94±0.2% and 94±0.2% e.e.). These results are inconsistent with the intermediacy of the ternary complex. Instead, they are consistent with formation of a zinc hydride in the first step, followed by transfer of the hydride to the ketone in the absence of a silicon-containing unit in a second step. The slightly lower reactivity of methyl phenylsilane than that of phenylsilane is consistent with slower formation of the zinc hydride in the rate-determining step.
The formation of zinc hydrides from silanes is well established in molecular systems. In many molecular complexes, zinc ions are coordinated to tridentate nitrogen-donor ligands, which were initially designed as model compounds to study the mechanisms of the reactions of zinc enzymes. Metathesis reactions of zinc alkoxides with silanes were shown to form zinc hydrides and silyl ethers19,28. Similarly, silane rapidly converts zinc fluorides to zinc hydrides while forming a silyl fluoride as the side product (Fig. 3d)20,29. The formation of zinc hydride from the metathesis of zinc hydroxide with boron hydride has been observed even in protic solvents.30,31 We propose that a similar reaction occurs with the zinc hydroxide inside the active site of carbonic anhydrase. In our proposed mechanism, silanes abstract the hydroxide from ZnOH through an intermediate or transition state containing a pentacoordinate silicon, and the resulting zinc hydride reacts with the ketone to form a zinc alkoxide that hydrolyzes to the alcohol and the starting zinc hydroxide.
Computational study of the formation and transfer of zinc hydride.
To assess the feasibility of a mechanism involving a zinc hydride and the potential origins of enantioselectivity, we conducted hybrid quantum mechanics/molecular mechanics (QM/MM) calculations of the proposed reaction pathway. QM/MM is an established computational method for studying the mechanisms of enzymatic reactions32–35. We first investigated the docking of phenylsilane to hCAII. The zinc hydroxide, three histidine side chains, and phenylsilane were chosen as the core structure for quantum calculations by DFT with the B3LYP functional, while the structure of the remaining protein was calculated by molecular mechanics with the OPLS force field. Geometry optimization initiated with the most populated structures from molecular mechanics converged to a structure in which the phenyl silane docks into the substrate-binding site. Driven by the oxophilicity of the silane, the silicon atom associates with the zinc hydroxide in the computed structure with an O–Si bond distance of 1.77 Å. The silicon hydride was also close to the zinc center, with a H–Zn distance of 3.58 Å. This structure is well poised for the metathesis reaction between the zinc hydroxide and the silicon hydride (Fig. 4a).
Figure 4. Computational study of hydride generation and transfer.

a, QMMM optimized structure of phenylsilane docked into zinc hydroxide hCAII. b, QMMM optimized structure of acetophenone docked into zinc hydride hCAII. c, Hydrogen bonding interaction of Thr199 and Thr200 with the carbonyl group of acetophenone. d, Hydrogen bonding interaction of Thr199 with the carbonyl group of CO2. e, Proposed overall mechanism for hCAII-catalyzed reduction of ketone with phenylsilane. f, hCAII-catalyzed reduction of 2’-bromoacetophenone with phenylsilane. g, The docking modes of 2’-bromoacetophenone in hCAII through carbonyl hydrogen bonding h, The docking modes of 2’-bromoacetophenone in hCAII through halogen bonding with the bromine. All distances between atoms were shown in Ångström.
We also calculated the docking of acetophenone to hCAII containing a zinc hydride by applying the QM/MM method to the most populated structures from molecular mechanics (Fig. 4b). In the resulting structure, acetophenone docks in the substrate-binding site, and the carbonyl group of acetophenone associates with the amide N–H of Thr199 by a hydrogen-bonding interaction in which the CO–HN distance is 2.28 Å (Fig. 4c). Such docking of acetophenone positions the carbonyl carbon close to the zinc hydride, with an H–C distance of 3.02 Å. This proximity facilitates the subsequent hydride transfer. This docking mode resembles the docking of the natural substrate, CO2, in the same active site of hCAII (Fig. 4d, PDB ID 3D92). Moreover, the computational model predicts that the hydride transfers to the prochiral re face of the ketone, matching the experimental formation of the S configuration of the alcohol. Thus, both experimental and computational studies are consistent with a catalytic cycle that involves the formation of a zinc hydride intermediate and reaction of the hydride with the ketone that interacts with the amide N–H bond of Thr199, leading to the highly enantioselective reductions (Fig. 4e).
Among the 26 ketones we investigated for reduction with PhSiH3, only 2’-bromoacetophenone reacted with low e.e. of 10% (Fig. 4f). To determine if the low selectivity for reaction of this ketone could be explained by our proposed mechanism, we computed the docking of this substrate to hCAII. These calculations revealed two major docking modes of 2’-bromoacetophenone, and hydride transfer would occur to opposite faces of the ketone in these two modes (Fig. 4g). In the first mode, 2’-bromoacetophenone docks with hydrogen bonding of the carbonyl with the amide hydrogen of Thr199 in an analogous fashion to the docking of acetophenone and leading to the (S)-enantiomer. However, in the second mode, the 2’-bromo group participates in a halogen bonding interaction with the hydroxy group of Thr200, with a linear arrangement of the C–O–Br atoms and an O–Br distance of 3.35 Å to form a structure in which the hydride would transfer to the si face of the ketone to form the R enantiomer (Fig. 4h). Such halogen bonding interactions are frequently observed in the structures of enzyme-inhibitor complexes36. While this halogen bonding interaction is not captured by the molecular dynamics calculations that do not include such orbital interactions, competitive binding of the ketone in these two orientations and addition of the hydride to them can tentatively explain the distinct enantioselectivity of this ketone containing a bromine atom in an appropriate position for a halogen bond to override the hydrogen bond.
In summary, we show that carbonic anhydrases catalyze the enantioselective reduction of ketones with silanes at an active site in which the metal is directly bound to the protein without an exogenous cofactor by the intermediacy of a monomeric hydride of an electropositive metal, which is a class of intermediate unknown in nature. These reactions are the first enzymatic reactions with silanes as reducing agents. The process of hydride transfer from silanes to zinc and subsequent transfer of the hydride to the ketone also lacks precedent in biological systems. This biocompatible approach to form metal hydrides can be envisioned to enable the generation of additional types of metal hydrides as reactive intermediates inside enzymes to create new pathways for additional abiotic enzymatic reactions. Future studies will seek to generate analogous metal hydrides that are more stable in the protein than the zinc hydride and that could be characterized directly.
Supplementary Material
Acknowledgement
This work on enantioselective reductions was supported by the NIH (grant no. R37 GM130387) and the studies pertaining to the intermediacy of the metal hydride were supported by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DEAC02-05CH11231. We thank the Berkeley DNA Sequencing Facility for plasmid sequencing. We thank the College of Chemistry’s Molecular Graphics and Computing Facility for resources provided and Dr. Kathleen Durkin for her assistance. The COC-MGCF is supported in part by the NIH (grant no. S10OD023532). P. Ji acknowledges support from the Miller Institute for Basic Research in Science at the University of California Berkeley for postdoctoral fellowship.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
The data supporting the findings of this study are available within the article and its Supplementary Information.
References
- 1.Schwizer F. et al. Artificial metalloenzymes: Reaction scope and optimization strategies. Chem. Rev. 118, 142–231 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Rosati F. & Roelfes G. Artificial metalloenzymes. ChemCatChem 2, 916–927 (2010). [Google Scholar]
- 3.Coelho PS, Brustad EM, Kannan A. & Arnold FH Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Key HM et al. Beyond iron: Iridium-containing P450 enzymes for selective cyclopropanations of structurally diverse alkenes. ACS Cent. Sci. 3, 302–308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Key HM, Dydio P, Clark DS & Hartwig JF Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 534, 534–537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kan SBJ, Lewis RD, Chen K. & Arnold FH Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science 354, 1048–1051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green MLH A new approach to the formal classification of covalent compounds of the elements. J. Organomet. Chem. 500, 127–148 (1995). [Google Scholar]
- 8.Jing Q. & Kazlauskas RJ Regioselective hydroformylation of styrene using rhodium-substituted carbonic anhydrase. ChemCatChem 2, 953–957 (2010). [Google Scholar]
- 9.Horitani M. et al. Radical SAM catalysis via an organometallic intermediate with an Fe-[5’-c]-deoxyadenosyl bond. Science 352, 822–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gloaguen F. & Rauchfuss TB Small molecule mimics of hydrogenases: Hydrides and redox. Chem. Soc. Rev. 38, 100–108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skander M. et al. Artificial metalloenzymes: (strept)avidin as host for enantioselective hydrogenation by achiral biotinylated rhodium−diphosphine complexes. J. Am. Chem. Soc. 126, 14411–14418 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Lin C-C, Lin C-W & Chan ASC Catalytic hydrogenation of itaconic acid in a biotinylated Pyrphos–rhodium(I) system in a protein cavity. Tetrahedron Asymmetry 10, 1887–1893 (1999). [Google Scholar]
- 13.Rebelein JG, Cotelle Y, Garabedian B. & Ward TR Chemical optimization of whole-cell transfer hydrogenation using carbonic anhydrase as host protein. ACS Catal. 9, 4173–4178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chevalley A, Cherrier MV, Fontecilla-Camps JC, Ghasemi M. & Salmain M. Artificial metalloenzymes derived from bovine β-lactoglobulin for the asymmetric transfer hydrogenation of an aryl ketone – synthesis, characterization and catalytic activity. Dalton Trans. 43, 5482–5489 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Merkler DJ & Schramm VL Catalytic mechanism of yeast adenosine 5’-monophosphate deaminase. Zinc content, substrate specificity, ph studies, and solvent isotope effects. Biochemistry 32, 5792–5799 (1993). [DOI] [PubMed] [Google Scholar]
- 16.Håkansson K, Carlsson M, Svensson LA & Liljas A. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol. 227, 1192–1204 (1992). [DOI] [PubMed] [Google Scholar]
- 17.Friedfeld MR, Zhong H, Ruck RT, Shevlin M. & Chirik PJ Cobalt-catalyzed asymmetric hydrogenation of enamides enabled by single-electron reduction. Science 360, 888–893 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Knowles WS Asymmetric hydrogenations (Nobel lecture). Angew. Chem. Int. Ed. 41, 1998–2007 (2002). [PubMed] [Google Scholar]
- 19.Mukherjee D, Ellern A. & Sadow AD Conversion of a zinc disilazide to a zinc hydride mediated by licl. J. Am. Chem. Soc. 132, 7582–7583 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Kläui W, Schilde U. & Schmidt M. Fluoro[η3-hydrotris(3-R-5-methylpyrazol-1-yl)borato]zinc(II): The first TpZnF complexes, convenient precursors to zinc hydride complexes. Inorg. Chem. 36, 1598–1601 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Sattler W. & Parkin G. Zinc catalysts for on-demand hydrogen generation and carbon dioxide functionalization. J. Am. Chem. Soc. 134, 17462–17465 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Ilic S, Alherz A, Musgrave CB & Glusac KD Thermodynamic and kinetic hydricities of metal-free hydrides. Chem. Soc. Rev. 47, 2809–2836 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Sun Z. et al. Catalytic asymmetric reduction of difficult-to-reduce ketones: Triple-code saturation mutagenesis of an alcohol dehydrogenase. ACS Catal. 6, 1598–1605 (2016). [Google Scholar]
- 24.Paul CE, Lavandera I, Gotor-Fernández V, Kroutil W. & Gotor V. Escherichia coli/ADH-A: An all-inclusive catalyst for the selective biooxidation and deracemisation of secondary alcohols. ChemCatChem 5, 3875–3881 (2013). [Google Scholar]
- 25.Kitamura M. et al. Homogeneous asymmetric hydrogenation of functionalized ketones. J. Am. Chem. Soc. 110, 629–631 (1988). [Google Scholar]
- 26.Li W, Lu B, Xie X. & Zhang Z. Ru-catalyzed chemo- and enantioselective hydrogenation of β-diketones assisted by the neighboring heteroatoms. Org. Lett. 21, 5509–5513 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Gajewy J, Gawronski J. & Kwit M. Mechanism and enantioselectivity of [zinc(diamine)(diol)]-catalyzed asymmetric hydrosilylation of ketones: DFT, NMR and ECD studies. Eur. J. Org. Chem. 2013, 307–318 (2013). [Google Scholar]
- 28.Mukherjee D, Thompson RR, Ellern A. & Sadow AD Coordinatively saturated tris(oxazolinyl)borato zinc hydride-catalyzed cross dehydrocoupling of silanes and alcohols. ACS Catal. 1, 698–702 (2011). [Google Scholar]
- 29.Rauch M, Rong Y, Sattler W. & Parkin G. Synthesis of a terminal zinc hydride compound, [TpBut,Me]ZnH, from a hydroxide derivative, [TpBut,Me]ZnOH: Interconversions with the fluoride complex, [TpBut,Me]ZnF. Polyhedron 103, 135–140 (2016). [Google Scholar]
- 30.Bergquist C, Koutcher L, Vaught AL & Parkin G. Reactivity of the B−H bond in tris(pyrazolyl)hydroborato zinc complexes: Unexpected example of zinc hydride formation in a protic solvent and its relevance towards hydrogen transfer to NAD+ mimics by tris(pyrazolyl)hydroborato zinc complexes in alcoholic media. Inorg. Chem. 41, 625–627 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Bergquist C. & Parkin G. Modeling the catalytic cycle of liver alcohol dehydrogenase: Synthesis and structural characterization of a four-coordinate zinc ethoxide complex and determination of relative Zn−OR versus Zn−OH bond energies. Inorg. Chem. 38, 422–423 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Senn HM & Thiel W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 48, 1198–1229 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Field MJ, Bash PA & Karplus M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 11, 700–733 (1990). [Google Scholar]
- 34.Lewis RD et al. Catalytic iron-carbene intermediate revealed in a cytochromec carbene transferase. Proc. Natl. Acad. Sci. 115, 7308–7313 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker Dockrey SA, Lukowski AL, Becker MR & Narayan ARH Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem. 10, 119–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howard EI et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase–inhibitor complex at 0.66 Å. Proteins 55, 792–804 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.