

Abstract
Here we review the motivation for creating the enhancing neuroimaging genetics through meta‐analysis (ENIGMA) Consortium and the genetic analyses undertaken by the consortium so far. We discuss the methodological challenges, findings, and future directions of the genetics working group. A major goal of the working group is tackling the reproducibility crisis affecting “candidate gene” and genome‐wide association analyses in neuroimaging. To address this, we developed harmonized analytic methods, and support their use in coordinated analyses across sites worldwide, which also makes it possible to understand heterogeneity in results across sites. These efforts have resulted in the identification of hundreds of common genomic loci robustly associated with brain structure. We have found both pleiotropic and specific genetic effects associated with brain structures, as well as genetic correlations with psychiatric and neurological diseases.
Keywords: GWAS, MRI, neuro‐imaging genetics
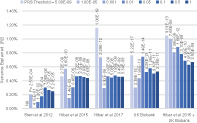
Improvement in the polygenic score prediction of hippocampal volume, as power in the discovery GWAS increases. PRS may be thought of as weighted‐sum scores that summarize the results of the GWAS to a given level of significance, these results show the increased explanatory power of the GWAS for hipocampal volume as sample size increases.
1. WHY DID WE BUILD THE ENIGMA CONSORTIUM?
The consortium was formed in 2009, largely in response to the growing evidence of a lack of reproducibility dubbed “the replication crisis” in imaging genetics. At this time, the first major works of the Psychiatric Genomics Consortium were being presented at conferences (Neale et al., 2010; The Schizophrenia Psychiatric Genome‐Wide Association Study [GWAS] Consortium, 2011a, 2011b), and we had observed the improvement in statistical power and increase in reproducibility that could be achieved through large‐scale meta‐analysis. In late 2009, we were beginning to see a series of GWAS publications using phenotypes derived from magnetic resonance imaging (MRI) attempting to answer complex and important questions in psychiatry and neurology. At that time, it was common to see GWAS papers reporting not only main effect analyses but also interactions with diagnosis or putative risk variables in sample sizes of less than 1,000 people. Insufficient power and sample size remain ongoing issues within the field, as recently highlighted by Szucs and Ioannidis (2019) who examined the sample size of clinical and experimental MRI studies published in 2017 and 2018 in four prominent neuro‐imaging journals and found that still less than 5% of these studies had sample sizes larger than 100 (Szucs & Ioannidis, 2019).
In response to these issues, Thompson and Martin sent an email to neuro‐imaging groups around the world asking for interest in being part of a collaborative meta‐analysis consortium focusing on imaging genetics. The key points in this email were that, although every group would understandably want to publish its own paper reporting their own findings, (a) the power calculations do not change just because the phenotype acquisition is expensive, (b) it was likely that the individual studies would not be large enough to find significant genetic effects, and (c) even if they did, it would still be necessary to replicate these findings in independent samples. From these beginnings, the ENIGMA consortium now involves more than 2,000 scientists from over 400 institutions in more than 40 countries (Thompson et al., 2020). While our initial focus was on GWAS and genetic analysis of imaging data, this quickly broadened, with the development of a series of clinical and methods working groups focusing on answering important questions around the impact of disorders and diseases on brain structure and function for which more power was also required to obtain more robust results.
2. WHAT WERE OUR MAJOR METHODOLOGICAL CHALLENGES?
We knew one of the first methodological challenges we would face would be addressing heterogeneity and the perceived insurmountable problems arising from heterogeneity in scanner performance and imaging protocols across groups. We addressed these issues by making a conscious decision to conduct meta—rather than mega‐analyses, which allowed the site level analyses to be run by those who knew the phenotypic data best. This also ensured that the analyses controlled for scanner and protocol effects where necessary and enabled an evaluation of heterogeneity across sites at the individual genetic variant level. By conducting analyses at the collection sites rather than bringing all imaging data to one central location for analysis, we were able not only to address site‐level heterogeneity but also ensure that contributing groups were able to quality control, process, impute, and analyse their own genotypic data, with technical and scientific support provided where necessary. This helped to disseminate these skills within the imaging community and at the same time maximized participation by avoiding challenges related to data sharing.
To reduce heterogeneity, both imaging and genetic protocols were created and made freely available from the ENIGMA webpage. The protocols specified the software versions used for data processing and analyses. For example, the imaging protocols included detailed instructions for the segmentation, extraction of volumes/areas/thicknesses and quality control procedures which included visual inspection. Following image processing, phenotypic summary statistics and histograms were shared centrally and inspected prior to running the genetic analyses. The genetic protocols included detailed instructions for genotypic quality control, estimation of ancestry components, imputation to a specific reference panel and analysis allowing for both samples of unrelated individuals and samples that included relatives. As is customary in the field, ENIGMA used fixed effect inverse SE or sample‐size weighted meta‐analyses; however, random effects meta‐analyses were also used to obtain convergent evidence and check for violations of the assumptions of fixed effects meta‐analysis. Within each meta‐analysis we further examined evidence for heterogeneity at the individual variant level using the Cochran's Q‐test (Cochran, 1954) implemented in METAL (Willer, Li, & Abecasis, 2010) and examined significant results graphically using forest plots. Recently, we have also used estimates of genetic correlations calculated using LDscore (Willer et al., 2010) to further check for evidence of heterogeneity across the genome between the ENIGMA, CHARGE and UK Biobank cohorts.
The second major methodological issue we encountered was the treatment of multiple testing and decisions regarding phenotype selection. Working in imaging genetics, we not only had to consider the multiple testing correction related to the number and the correlation structure of phenotypes being tested, but also those relating to the number of genetic variants being analyzed, while accounting for the autocorrelation patterns within the genome due to linkage disequilibrium. Moreover, we had to consider how to reconcile the two different approaches to multiple testing adopted within the fields of imaging (which typically adopts a false discovery rate approach) and genetics (which typically uses Bonferroni corrections). GWAS analyses currently analyse around 10,000,000 variants across the genome, and these analyses are often conducted for many phenotypes. As the downstream interpretation of GWAS results often uses the P values from the GWAS as input, we elected to adopt a Bonferroni approach to multiple testing correction, and used spectral decomposition analysis to determine the effective number of independent traits being analyzed (Nyholt, 2004).
3. WHAT HAVE WE DONE?
Our first major ENIGMA project was published in 2012 and focused on identifying common genetic variants associated with hippocampal and intracranial volumes. These analyses brought together data from 22 cohorts and led to a combined sample size of 21,000 individuals (Stein et al., 2012). In addition to identifying genome‐wide significant loci influencing hippocampal volume and intracranial volume, these analyses confirmed that—except for a signal relating to APOE in a cohort that included individuals with Alzheimer's disease—there were no other significant results at the individual cohort level; there were also no cohort‐level significant results when the analyses were restricted to individuals without a neurological or psychiatric diagnosis. Thus, by bringing together the data from these 22 cohorts, the 206 coauthors on this paper worked together to ensure that the resulting analysis would be well powered and robust, rather than 22 underpowered “null result” analyses. Another important finding from this paper was that the distribution of effect sizes seen on these imaging phenotypes was very similar to that which had been observed for other physiological traits in humans; with individual common genetic variants typically explaining less than 1% of the variance in the trait. As shown in Figure 1, large sample sizes are needed to robustly detect genetic variants with realistic effect sizes using conventional genome‐wide significance thresholds; however, due largely to budgetary constraints, many individual studies have been designed to detect effects in the >5% range (similar to the effect sizes seen for APOE in Alzheimer's disease). Ongoing work by multiple groups is focusing on increasing power using multivariate tests (e.g., van der Meer et al., 2020), Bayesian priors, and other conditional analyses (e.g., Smeland et al., 2018). However, as the central hypothesis of most GWAS and the bioinformatic and functional follow up of findings are essentially univariate in nature, well‐powered univariate analyses supported by replication and meta‐analyses remain crucial.
FIGURE 1.
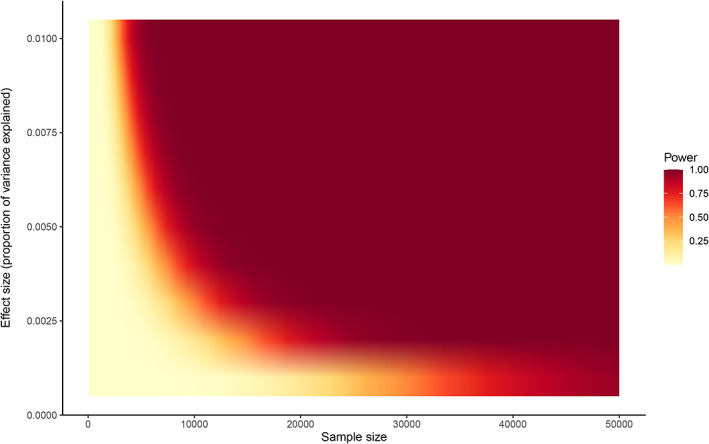
Power to detect a genome‐wide significant locus (at ∝ = 5 × 10−8) for variants explaining 0.1 to 1.0% of the variance in a quantitative trait, for sample sizes of 100 to 50,000 individuals. These analyses assume that the data come from unrelated individuals, and that additional covariates in the GWAS are not associated with the variant. See https://github.com/kaustubhad/gwas‐power for R code and a tutorial on calculating power analyses for GWAS; see also Smith and Nichols (2018), who discuss power in the context of testing multiple traits
The second major ENIGMA GWAS paper focused on a wider range of subcortical volumes as well as intracranial volume (Hibar et al., 2015). These analyses included contributions from 30,717 individuals from 50 different cohorts and identified numerous common genetic variants influencing subcortical structures. Most variants identified in these analyses influenced specific structures; however, this may to some extent be due to the way in which imaging data are analyzed and the corrections for total or intracranial volume when analyzing substructures. Despite this, at a genomic level, the results for the individual substructures clustered by developmental and functional subdivisions.
In addition, we were able to demonstrate that subcortical volumes were significantly influenced by common variants with SNP based heritabilities derived from the GWAS results explaining 7 to 15% of the phenotypic variance. Since the publication of these analyses in 2015, we have worked with our collaborators in the CHARGE consortium to publish a series of updates with increased sample sizes on common genetic variants influencing hippocampal, intracranial, and other subcortical volumes; these results now account for 15 to 30% of the phenotypic variance in these structures (Adams et al., 2016; Hibar et al., 2017; Satizabal et al., 2019). Similarly, as shown in Figure 2, even when predicting into pediatric samples (aged 9–11), the performance of polygenic risk scores (PRS), derived from meta‐analyses of participants across the lifespan, has increased as the power of the GWAS have increased. However, most work in this field has focused on analysis of data from participants of European ancestry—which dramatically reduces the accuracy of polygenic prediction in non‐European samples because of differing patterns of LD and allelic effects across ancestries. Notably, as shown in Figure 3 this reduction is somewhat ameliorated through the use of unweighted polygenic scores. Focusing on increasing sample size of non‐European ancestry samples to improve the weights used in polygenic risk score calculation is likely to be the best method of increasing the predictive power in these populations.
FIGURE 2.
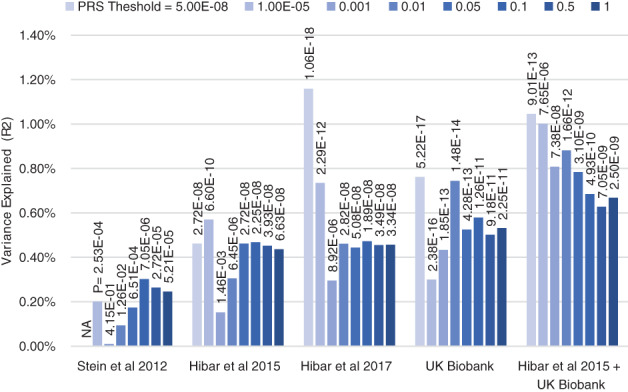
Improvement in the polygenic score prediction of hippocampal volume, as power in the discovery GWAS increases. PRS may be thought of as weighted‐sum scores that summarize the results of the GWAS to a given level of significance. PRS analyses were conducted using 4,000 randomly selected unrelated participants of European ancestry from the ABCD study, which was not included in the discovery meta‐analyses (Casey et al., 2018) (relatedness <.025; ancestry defined as <6 SD from the European reference centroid in a multidimensional scaling analysis). PRS were calculated using the traditional clumping and thresholding approach (‐‐clump‐r2 0.1 ‐‐clump‐kb 1,000). Weights for these polygenic scores were derived from GWAS for hippocampal volume controlling for ICV from Stein et al., 2012, Hibar et al., 2015 and Hibar et al., 2017, a GWAS of 20,112 UK Biobank participants of European Ancestry, and a meta‐analysis of the results from Hibar et al., 2015 and the UK Biobank GWAS of 20,111 individuals. Analyses included Age, Sex, ancestry corrections (components 1–4) and ICV as covariates
FIGURE 3.
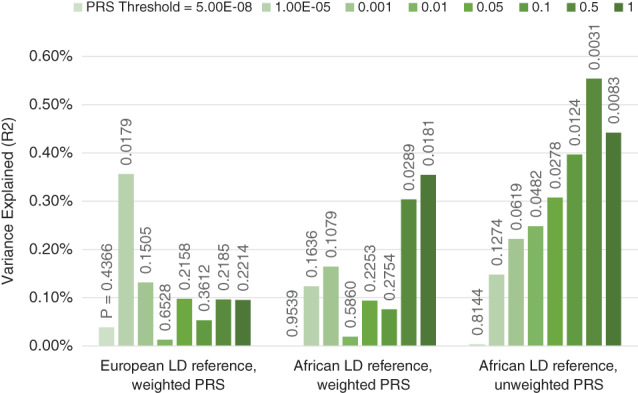
Performance of cross‐ancestry polygenic score prediction as a function of LD reference data and weighting. PRS analyses were conducted using 966 unrelated participants of African ancestry from the ABCD study (Casey et al., 2018) (relatedness <.025; ancestry defined as <6 SD from the African reference centroid in a multidimensional scaling analysis). The polygenic scores were derived from a European ancestry meta‐analysis of the hippocampal volume GWAS from Hibar et al., 2015 and a GWAS of 20,111 individuals from the UK Biobank. PRS were calculated using the traditional clumping and thresholding approach (‐‐clump‐r2 0.1 ‐‐clump‐kb 1,000) using reference sets of 1,000 participants of European or African ancestry (sampled from the ABCD study). For the weighted PRS, the SNP dosages were multiplied by the betas from the GWAS as is traditionally done. For the unweighted PRS the betas from the GWAS were recoded to 1 if β > 0, −1 if β < 0 and 0 if β = 0. Analyses included Age, Sex, ancestry corrections (components 1–4) and ICV as covariates
Our most recent work focused on identifying common genetic variants influencing the surface area and thickness of the cerebral cortex, across 34 commonly used regions of interest (Grasby et al., 2020). These analyses brought together a discovery sample of over 33,000 individuals from 50 cohorts with a replication sample of around 15,000 individuals. We found enrichment of loci influencing total surface area within regulatory elements active during prenatal cortical development, which supports the radial unit hypothesis (Rakic, 1988). We also found loci impacting regional surface area clustered near genes in Wnt signaling pathways known to influence progenitor cell proliferation, surface expansion and areal identity (Harrison‐Uy & Pleasure, 2012). Common variants explained 34% (SE = 3%) of the variation in total surface area and 26% (SE = 2%) of variation in average thickness across the cortex, while PRS derived from these GWAS analysis explained 2 to 3% of the variance in these traits. We also found genetic correlations (i.e., overlap in genetic effects across traits) between total cortical surface area and cognitive function, Parkinson's disease, insomnia, depression, and ADHD. The degree of genetic overlap with disease risk loci could be visualized for each cortical region, helping to map the relationships between brain structures and these disorders.
To aid in the interpretation of the meta‐analytic results we have undertaken bioinformatic follow‐up including gene and pathway analyses, fine‐mapping and Mendelian randomization analyses. These findings along with the meta‐analytic summary statistics are made freely available to provide a resource that can be used to help researchers in the field prioritize the selection of genomic targets for future follow‐up analyses which may clarify the causal variants and mechanisms underlying these effects.
Collectively, the work from the ENIGMA Genetics Working Group has identified over 460 genome‐wide significant variants influencing the structure of the human brain. As shown in Figure 4 these variants are located across the genome. In addition to the work of the ENIGMA consortium, the progress in neuroimaging genetics has also been greatly enhanced through meta‐analyses conducted by the CHARGE consortium (Adams et al., 2016; Bis et al., 2012; Hibar et al., 2017; Satizabal et al., 2019; Shin et al., 2020; van der Lee et al., 2019; Vojinovic et al., 2018). Similarly, recent analyses from the UK Biobank have allowed further advances in the field to be made (Elliott et al., 2018; Smith et al., 2020; Smith & Nichols, 2018; Zhao et al., 2020) and have facilitated a growing body of work in this area. As the strong SNP‐based heritability of MRI‐based phenotypes demonstrates, there are still substantially more common variants influencing brain structure yet to be found. As the power of neuroimaging GWAS analyses continues to grow through collaboration and meta‐analyses, we can expect to see the number of known loci and the accuracy of polygenic prediction increase markedly.
FIGURE 4.
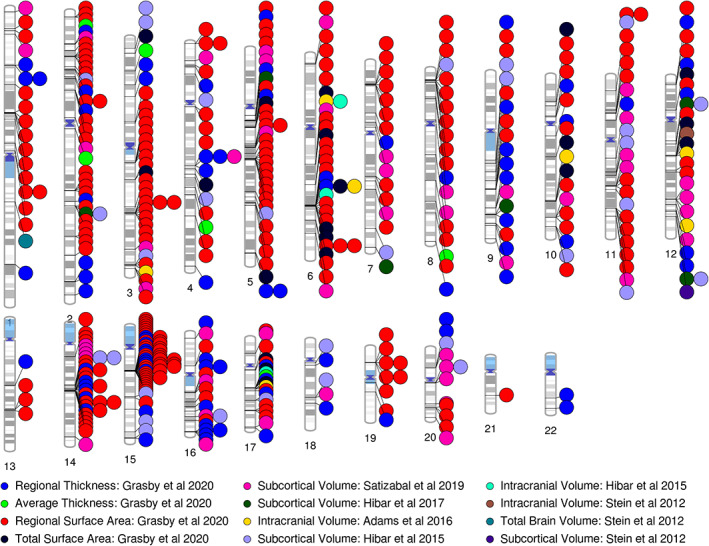
Ideogram summarizing the cortical, subcortical and ICV findings from the ENIGMA (Grasby et al., 2020; Hibar et al., 2015; Stein et al., 2012) and ENIGMA‐CHARGE (Adams et al., 2016; Hibar et al., 2017; Satizabal et al., 2019) papers
4. WHERE TO FROM HERE?
As with the vast majority of GWAS, the analytic focus has been strongly centered on single nucleotide polymorphisms located on the autosomes. However, it is important to consider other classes of genetic variants: the ENIGMA CNV working group (see Sonderby et al., 2020, this issue) is making important progress in this area. There is also a need to consider the genetic content of the allosomes or sex chromosomes. Historically, these chromosomes were excluded from GWAS meta‐analyses due to a lack of imputation reference data and complications surrounding the modeling of X‐chromosome inactivation in females, which is not necessarily complete and may be tissue and timing specific (Tukiainen et al., 2017).
As is the case for many other domains, neuroimaging genetics as a field has typically focused on genotyping cohorts in which MRI data had already been collected. This has led to the majority of analyses focusing on participants of European ancestry. There is a pressing need to address the issue of diversity. Large studies, such as the UK Biobank (Littlejohns et al., 2020) and ABCD (Casey et al., 2018), which have been designed to be more representative of the communities in which the research is being conducted are an important advance in this area. However, more work is needed, and disparities in prediction accuracy cannot be addressed until large‐scale genetic studies are undertaken in other ancestral populations.
To provide results with the widest generalizability, our work to date has focused on commonly studied imaging phenotypes derived from cortical and subcortical structures using the most commonly used atlases and parcellation tools. Moving forward, there are obvious avenues for further work comparing results from other atlases and image processing methodologies. Work has also begun on image‐wide GWAS (Medland, Jahanshad, Neale, & Thompson, 2014), also known as voxelwise GWAS or vGWAS (Hibar et al., 2011; Stein et al., 2010). This approach combines the spatial resolution of brain‐wide statistical parametric mapping with the genome‐wide search afforded by GWAS. Until recently power was insufficient for simultaneous genome‐wide screening of very high dimensional phenotypes, leading to novel strategies for phenotype prioritization and sorting (Jahanshad et al., 2019; Thompson, Ge, Glahn, Jahanshad, & Nichols, 2013) Similarly, there is a need to study other important structural phenotypes such as fractional anisotropy measures derived from diffusion tensor imaging; there is work underway on this within the ENIGMA DTI Working Group (Jahanshad et al., 2013). Beyond brain structure, the genetics of brain activity remains a subject of interest within ENIGMA. This involves ongoing work on (resting state) functional MRI as well as use of other imaging modalities, such as electroencephalography (EEG), where the ENIGMA EEG Working Group has already provided first results (Smit et al., 2018). At the same time, there is also interest in new methods for the analysis of imaging genomics data. Some of these attempt to bring in other sources of bioinformatics data to constrain and guide the search for mechanisms (Shen & Thompson, 2020). A key lesson learned in all these efforts, which will no doubt apply to future machine learning and deep learning efforts in imaging genomics, has been the need for multi‐cohort replication in diverse data sets. Multi‐site replication can also help address the need to safeguard against false positives arising from too many “researcher degrees of freedom” when many methods and parameters are tried on the same data sets.
Finally, in addition to raising awareness of the importance of conducting well‐powered analysis in imaging genetics, ENIGMA has greatly increased the awareness of the need for collaborations among researchers in the field. As shown in Figure 5a, from 2000 to 2010, the coauthorship network for published research being conducted in imaging genetics was perhaps best characterized as a sparse network with relatively few connections. However, as shown in Figure 5b, since this time, the overall characteristics of the network have largely evolved, and there is now much tighter collaboration among researchers. We look forward to seeing this trend continue as the field of imaging genetics continues to advance.
FIGURE 5.
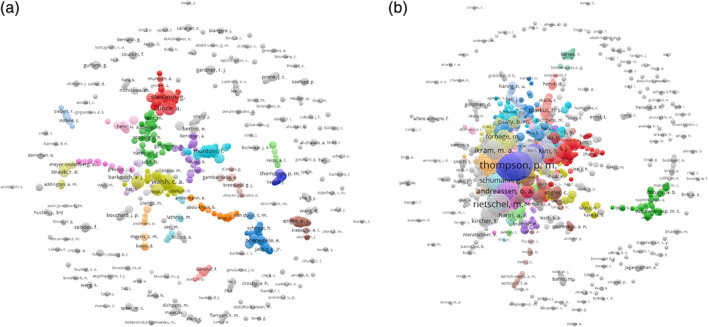
Coauthorship network diagrams for imaging genetics publications indexed in PubMed: (a) from 2000 to 2010, and (b) from 2010 to 2020. Network analyses were conducted using VOS viewer (van Eck & Waltman, 2010) using the default clustering methods, normalized by association strength, using a repulsion strength of 0 in the layout. Data extracted August 14, 2020, using the following search syntax (a) (((("2000/01/01"[Date‐ Publication]: "2010/01/01" [Date ‐ Publication])) AND (((linkage) OR GWAS) AND imaging genetics)) AND MRI) AND English[Language] (b) (((("2010/01/01"[Date ‐ Publication]: "2021/01/01" [Date ‐Publication])) AND ((linkage) OR GWAS) AND imaging genetics)) AND MRI) AND English[Language]
ACKNOWLEDGMENTS
SEM was supported by NHMRC grants APP1103623, APP1158127, and APP1172917. N. J., S. I. T., B. F., and P. T. are supported in part by U54 EB020403 from the NIH Big Data to Knowledge (BD2K) program, a multi‐institute NIH initiative. L. C. ‐C. is supported by a QIMR Berghofer fellowship.
Medland SE, Grasby KL, Jahanshad N, et al. Ten years of enhancing neuro‐imaging genetics through meta‐analysis: An overview from the ENIGMA Genetics Working Group . Hum Brain Mapp. 2022;43:292–299. 10.1002/hbm.25311
Funding information Foundation for the National Institutes of Health, Grant/Award Number: EB020403; National Health and Medical Research Council, Grant/Award Numbers: APP1103623, APP1158127, APP1172917; QIMR Berghofer Medical Research Institute, Grant/Award Number: Fellowship
DATA AVAILABILITY STATEMENT
Summary statistics from the ENIGMA and ENIGMA‐CHARGE meta‐analyses are available from http://enigma.ini.usc.edu/research/download‐enigma‐gwas‐results/ UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number ‘11559’. Data is available from https://www.ukbiobank.ac.uk/ ABCD: Data used in the preparation of this article were obtained from the Adolescent Brain Cognitive Development (ABCD) Study (https://abcdstudy.org), held in the NIMH Data Archive (NDA). This is a multisite, longitudinal study designed to recruit more than 10,000 children age 9–10 and follow them over 10 years into early adulthood. The ABCD Study is supported by the National Institutes of Health and additional federal partners under award numbers U01DA041048, U01DA050989, U01DA051016, U01DA041022, U01DA051018, U01DA051037, U01DA050987, U01DA041174, U01DA041106, U01DA041117, U01DA041028, U01DA041134, U01DA050988, U01DA051039, U01DA041156, U01DA041025, U01DA041120, U01DA051038, U01DA041148, U01DA041093, U01DA041089, U24DA041123, U24DA041147. A full list of supporters is available at https://abcdstudy.org/federal‐partners.html. A listing of participating sites and a complete listing of the study investigators can be found at https://abcdstudy.org/consortium_members/. ABCD consortium investigators designed and implemented the study and/or provided data but did not necessarily participate in analysis or writing of this report. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or ABCD consortium investigators. Data are available from https://nda.nih.gov/abcd
REFERENCES
- Adams, H. H. , Hibar, D. P. , Chouraki, V. , Stein, J. L. , Nyquist, P. A. , Rentería, M. E. , … Thompson, P. M. (2016). Novel genetic loci underlying human intracranial volume identified through genome‐wide association. Nature Neuroscience, 19(12), 1569–1582. 10.1038/nn.4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bis, J. C. , DeCarli, C. , Smith, A. V. , van der Lijn, F. , Crivello, F. , Fornage, M. , … Seshadri, S. (2012). Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nature Genetics, 44(5), 545–551. 10.1038/ng.2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey, B. J. , Cannonier, T. , Conley, M. I. , Cohen, A. O. , Barch, D. M. , Heitzeg, M. M. , … Dale, A. M. (2018). The adolescent brain cognitive development (ABCD) study: Imaging acquisition across 21 sites. Developmental Cognitive Neuroscience, 32, 43–54. 10.1016/j.dcn.2018.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran, W. G. (1954). The combination of estimates from different experiments. Biometrics, 10(1), 101–129. 10.2307/3001666 [DOI] [Google Scholar]
- Elliott, L. T. , Sharp, K. , Alfaro‐Almagro, F. , Shi, S. , Miller, K. L. , Douaud, G. , … Smith, S. M. (2018). Genome‐wide association studies of brain imaging phenotypes in UKbiobank. Nature, 562(7726), 210–216. 10.1038/s41586-018-0571-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasby, K. L. , Jahanshad, N. , Painter, J. N. , Colodro‐Conde, L. , Bralten, J. , Hibar, D. P. , … Medland, S. E. (2020). The genetic architecture of the human cerebral cortex. Science, 367(6484), eaay6690. 10.1126/science.aay6690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison‐Uy, S. J. , & Pleasure, S. J. (2012). Wnt signaling and forebrain development. Cold Spring Harbor Perspectives in Biology, 4(7), a008094. 10.1101/cshperspect.a008094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar, D. P. , Adams, H. H. H. , Jahanshad, N. , Chauhan, G. , Stein, J. L. , Hofer, E. , … Ikram, M. A. (2017). Novel genetic loci associated with hippocampal volume. Nature Communications, 8, 13624. 10.1038/ncomms13624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar, D. P. , Stein, J. L. , Kohannim, O. , Jahanshad, N. , Saykin, A. J. , Shen, L. , … Thompson, P. M. (2011). Voxelwise gene‐wide association study (vGeneWAS): Multivariate gene‐based association testing in 731 elderly subjects. NeuroImage, 56(4), 1875–1891. 10.1016/j.neuroimage.2011.03.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar, D. P. , Stein, J. L. , Renteria, M. E. , Arias‐Vasquez, A. , Desrivières, S. , Jahanshad, N. , … Medland, S. E. (2015). Common genetic variants influence human subcortical brain structures. Nature, 520(7546), 224–229. 10.1038/nature14101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanshad, N. , Faskowitz, J. , Roshchupkin, G. , Hibar, D. P. , Gutman, B. A. , Tustison, N. J. , … Thompson, P. M. (2019). Multi‐site meta‐analysis of morphometry. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 16(5), 1508–1514. 10.1109/tcbb.2019.2914905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahanshad, N. , Kochunov, P. V. , Sprooten, E. , Mandl, R. C. , Nichols, T. E. , Almasy, L. , … Glahn, D. C. (2013). Multi‐site genetic analysis of diffusion images and voxelwise heritability analysis: A pilot project of the ENIGMA‐DTI working group. NeuroImage, 81, 455–469. 10.1016/j.neuroimage.2013.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlejohns, T. J. , Holliday, J. , Gibson, L. M. , Garratt, S. , Oesingmann, N. , Alfaro‐Almagro, F. , … Allen, N. E. (2020). The UKbiobank imaging enhancement of 100,000 participants: Rationale, data collection, management and future directions. Nature Communications, 11(1), 2624. 10.1038/s41467-020-15948-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medland, S. E. , Jahanshad, N. , Neale, B. M. , & Thompson, P. M. (2014). Whole‐genome analyses of whole‐brain data: Working within an expanded search space. Nature Neuroscience, 17(6), 791–800. 10.1038/nn.3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, B. M. , Medland, S. E. , Ripke, S. , Asherson, P. , Franke, B. , Lesch, K. P. , … Nelson, S. (2010). Meta‐analysis of genome‐wide association studies of attention‐deficit/hyperactivity disorder. Journal of the American Academy of Child and Adolescent Psychiatry, 49(9), 884–897. 10.1016/j.jaac.2010.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyholt, D. R. (2004). A simple correction for multiple testing for single‐nucleotide polymorphisms in linkage disequilibrium with each other. American Journal of Human Genetics, 74(4), 765–769. 10.1086/383251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic, P. (1988). Specification of cerebral cortical areas. Science, 241(4862), 170–176. 10.1126/science.3291116 [DOI] [PubMed] [Google Scholar]
- Satizabal, C. L. , Adams, H. H. H. , Hibar, D. P. , White, C. C. , Knol, M. J. , Stein, J. L. , … Ikram, M. A. (2019). Genetic architecture of subcortical brain structures in 38,851 individuals. Nature Genetics, 51(11), 1624–1636. 10.1038/s41588-019-0511-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, L. , & Thompson, P. M. (2020). Brain imaging genomics: Integrated analysis and machine learning. Proceedings of the IEEE, 108(1), 125–162. 10.1109/JPROC.2019.2947272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J. , Ma, S. , Hofer, E. , Patel, Y. , Vosberg, D. E. , Tilley, S. , … Paus, T. (2020). Global and regional development of the human cerebral cortex: Molecular architecture and occupational aptitudes. Cerebral Cortex, 30(7), 4121–4139. 10.1093/cercor/bhaa035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeland, O. B. , Wang, Y. , Frei, O. , Li, W. , Hibar, D. P. , Franke, B. , … Andreassen, O. A. (2018). Genetic overlap between schizophrenia and volumes of hippocampus, putamen, and intracranial volume indicates shared molecular genetic mechanisms. Schizophrenia Bulletin, 44(4), 854–864. 10.1093/schbul/sbx148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, D. J. A. , Wright, M. J. , Meyers, J. L. , Martin, N. G. , Ho, Y. Y. W. , Malone, S. M. , … Boomsma, D. I. (2018). Genome‐wide association analysis links multiple psychiatric liability genes to oscillatory brain activity. Human Brain Mapping, 39(11), 4183–4195. 10.1002/hbm.24238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. M. , Elliott, L. T. , Alfaro‐Almagro, F. , McCarthy, P. , Nichols, T. E. , Douaud, G. , & Miller, K. L. (2020). Brain aging comprises many modes of structural and functional change with distinct genetic and biophysical associations. eLife, 9, e52677. 10.7554/eLife.52677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. M. , & Nichols, T. E. (2018). Statistical challenges in “big data” human neuroimaging. Neuron, 97(2), 263–268. 10.1016/j.neuron.2017.12.018 [DOI] [PubMed] [Google Scholar]
- Stein, J. L. , Hua, X. , Lee, S. , Ho, A. J. , Leow, A. D. , Toga, A. W. , … Thompson, P. (2010). Voxelwise genome‐wide association study (vGWAS). NeuroImage, 53(3), 1160–1174. 10.1016/j.neuroimage.2010.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, J. L. , Medland, S. E. , Vasquez, A. A. , Hibar, D. P. , Senstad, R. E. , Winkler, A. M. , … Thompson, P. M. (2012). Identification of common variants associated with human hippocampal and intracranial volumes. Nature Genetics, 44(5), 552–561. 10.1038/ng.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs, D. , & Ioannidis, J. P. (2019). Sample size evolution in neuroimaging research: an evaluation of highly‐cited studies (1990–2012) and of latest practices (2017–2018) in high‐impact journals. bioRxiv , 809715. doi: 10.1101/809715 [DOI] [PubMed]
- The Schizophrenia Psychiatric Genome‐Wide Association Study (GWAS) Consortium . (2011a). Genome‐wide association study identifies five new schizophrenia loci. Nature Genetics, 43(10), 969–976. 10.1038/ng.940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Schizophrenia Psychiatric Genome‐Wide Association Study (GWAS) Consortium . (2011b). Large‐scale genome‐wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature Genetics, 43(10), 977–983. 10.1038/ng.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, P. M. , Ge, T. , Glahn, D. C. , Jahanshad, N. , & Nichols, T. E. (2013). Genetics of the connectome. NeuroImage, 80, 475–488. 10.1016/j.neuroimage.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, P. M. , Jahanshad, N. , Ching, C. R. K. , Salminen, L. E. , Thomopoulos, S. I. , Bright, J. , … for the, E. C . (2020). ENIGMA and global neuroscience: A decade of large‐scale studies of the brain in health and disease across more than 40 countries. Translational Psychiatry, 10(1), 100. 10.1038/s41398-020-0705-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukiainen, T. , Villani, A.‐C. , Yen, A. , Rivas, M. A. , Marshall, J. L. , Satija, R. , … Visualization—Ucsc Genomics Institute, U. o. C. S. C . (2017). Landscape of X chromosome inactivation across human tissues. Nature, 550(7675), 244–248. 10.1038/nature24265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee, S. J. , Knol, M. J. , Chauhan, G. , Satizabal, C. L. , Smith, A. V. , Hofer, E. , … DeCarli, C. (2019). A genome‐wide association study identifies genetic loci associated with specific lobar brain volumes. Communications Biology, 2, 285. 10.1038/s42003-019-0537-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer, D. , Frei, O. , Kaufmann, T. , Shadrin, A. A. , Devor, A. , Smeland, O. B. , … Dale, A. M. (2020). Understanding the genetic determinants of the brain with MOSTest. Nature Communications, 11(1), 3512. 10.1038/s41467-020-17368-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eck, N. J. , & Waltman, L. (2010). Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics, 84(2), 523–538. 10.1007/s11192-009-0146-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojinovic, D. , Adams, H. H. , Jian, X. , Yang, Q. , Smith, A. V. , Bis, J. C. , … Fornage, M. (2018). Genome‐wide association study of 23,500 individuals identifies 7 loci associated with brain ventricular volume. Nature Communications, 9(1), 3945. 10.1038/s41467-018-06234-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer, C. J. , Li, Y. , & Abecasis, G. R. (2010). METAL: Fast and efficient meta‐analysis of genomewide association scans. Bioinformatics (Oxford, England), 26(17), 2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, B. , Li, T. , Smith, S. M. , Xiong, D. , Wang, X. , Yang, Y. , … Zhu, H. (2020). Common variants contribute to intrinsic functional architecture of human brain. bioRxiv , 2020.2007.2030.229914. doi: 10.1101/2020.07.30.229914 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Summary statistics from the ENIGMA and ENIGMA‐CHARGE meta‐analyses are available from http://enigma.ini.usc.edu/research/download‐enigma‐gwas‐results/ UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number ‘11559’. Data is available from https://www.ukbiobank.ac.uk/ ABCD: Data used in the preparation of this article were obtained from the Adolescent Brain Cognitive Development (ABCD) Study (https://abcdstudy.org), held in the NIMH Data Archive (NDA). This is a multisite, longitudinal study designed to recruit more than 10,000 children age 9–10 and follow them over 10 years into early adulthood. The ABCD Study is supported by the National Institutes of Health and additional federal partners under award numbers U01DA041048, U01DA050989, U01DA051016, U01DA041022, U01DA051018, U01DA051037, U01DA050987, U01DA041174, U01DA041106, U01DA041117, U01DA041028, U01DA041134, U01DA050988, U01DA051039, U01DA041156, U01DA041025, U01DA041120, U01DA051038, U01DA041148, U01DA041093, U01DA041089, U24DA041123, U24DA041147. A full list of supporters is available at https://abcdstudy.org/federal‐partners.html. A listing of participating sites and a complete listing of the study investigators can be found at https://abcdstudy.org/consortium_members/. ABCD consortium investigators designed and implemented the study and/or provided data but did not necessarily participate in analysis or writing of this report. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or ABCD consortium investigators. Data are available from https://nda.nih.gov/abcd