


Keywords: apoptosis, cell death, infection, necroptosis, pyroptosis
Abstract
The coevolution of host-pathogen interactions underlies many human physiological traits associated with protection from or susceptibility to infections. Among the mechanisms that animals utilize to control infections are the regulated cell death pathways of pyroptosis, apoptosis, and necroptosis. Over the course of evolution these pathways have become intricate and complex, coevolving with microbes that infect animal hosts. Microbes, in turn, have evolved strategies to interfere with the pathways of regulated cell death to avoid eradication by the host. Here, we present an overview of the mechanisms of regulated cell death in Animalia and the strategies devised by pathogens to interfere with these processes. We review the molecular pathways of regulated cell death, their roles in infection, and how they are perturbed by viruses and bacteria, providing insights into the coevolution of host-pathogen interactions and cell death pathways.
CLINICAL HIGHLIGHTS
Regulated cell death is a fundamental biological process that underlies many physiological traits, including the response to infections. The regulated cell death pathways of pyroptosis, apoptosis, and necroptosis play an integral part in the immune defense against invading pathogens. We provide a comprehensive review of the molecular pathways of regulated cell death, the strategies contrived by pathogens to perturb these signaling cascades, and the evolutionary aspects of regulated cell death.
1. INTRODUCTION
Regulated cell death (RCD), cellular demise through a molecular program, is a fundamental biological process that underlies many physiological traits. RCD is involved in embryonic development, where it controls the formation of bodily structures such as the digits of the fingers, the timing of neural tube and palate closure, the selection of neuronal connections, and lymphocyte specificity. RCD mediates metamorphosis, such as the loss of the tail by developing tadpoles, and regulates homeostasis of many tissues in adult animals, including the rapid turnover of the intestinal epithelium and the cells of the blood. Moreover, RCD plays an integral part in the immune defense against invading pathogens and the control of activated immune cells (1).
Various forms of RCD exist, each with its own mode of activation, mechanistic regulation, and (patho)physiological consequences (2). Developmental and homeostatic cell death are mainly due to apoptosis, a “clean,” nonlytic, noninflammatory type of death where the cell condenses and often breaks into smaller, membrane-bound bodies, after which the corpse is rapidly cleared by phagocytic cells, mostly macrophages. Apoptosis aids in the control of infections and the immune response, mainly by mediating the removal of infected cells but also by regulating the levels of activated immune cells after these have fulfilled their function. The RCD types necroptosis and pyroptosis also help to control infections and immunological responses (3–8). Necroptosis and pyroptosis induce highly inflammatory, lytic cell death, where loss of plasma membrane integrity results in the release of the cellular contents into the surrounding milieu. Such inflammatory cellular contents are often referred to as damage-associated molecular patterns (DAMPs) and include a variety of proteins, nucleic acids, ATP, and metabolites (9), which attract innate immune cells to combat the infection.
Any organism can be infected by pathogens. Obligate, intracellular pathogens must gain access to the host cytoplasm. Viruses do this by binding to the extracellular region of transmembrane proteins present on the hosts’ cell membrane (10). Depending on the invading virus, receptor engagement induces subsequent events, such as membrane fusion or endocytosis, that eventually result in the delivery of the viral genetic material into the host cytoplasm. Intracellular bacteria, fungi, and protozoa use host cells to acquire nutrients and to shelter themselves from elimination by the host immune system (11, 12). Bacteria gain access by engaging cellular transmembrane proteins to induce their internalization or can be engulfed by phagocytes, after which they either escape to the cytosol or create a niche within the phagocytic vacuole.
For the host, the basis for the recognition and control of invading pathogens is the ability to distinguish between “self” and “nonself.” Over the course of evolution, eukaryotic cells have equipped themselves with many types of cell surface and intracellular receptors able to differentiate the wide variety of infectious organisms from “self.” Collectively, these pattern-recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) associated with many different types of microbes (13–15).
A primary function of many PRRs is to induce proinflammatory gene expression profiles that counteract an infection either directly (e.g., interferons, antibacterial peptides) or by attracting innate immune cells (e.g., cytokines, chemokines) to the site of infection to combat the invading pathogen. Moreover, this allows infected cells to “warn” neighboring cells to arm against incoming infectious agents. Another way that invading pathogens are detected is by the cellular stress associated with infection, owing to interference with cellular homeostasis. The response to such stress can be similar to that of PRR engagement, in that both can lead to inflammatory signals (9, 16, 17). Moreover, the engagement of specific PRRs and cellular stress can induce regulated cell death to destroy an infected cell (18).
PRRs are not the only receptors able to induce regulated cell death. Animal cells express death receptors (DRs), a subset of the tumor necrosis factor receptor (TNFR) superfamily, whose ligation can induce apoptosis or necroptosis, depending on the engaged receptor and the cellular context (19). Death receptors are not PRRs but recognize signals (death receptor ligands) from or on other cells. Death receptor ligation can induce cell death to control pathogenic spread and also functions in immune regulation and inflammation (20). Whether regulated cell death is activated through engagement of PRRs or DRs, by removing an infected nonessential cell the host eliminates the invading pathogens’ replicative niche, thereby halting pathogenic spread and limiting disease to improve host survival.
1.1. Scope of the Review
Over the course of evolution, many forms of regulated cell death have evolved (2). In this review, we consider only three: pyroptosis, apoptosis, and necroptosis. Although others certainly exist, our focus on these three is for several reasons. Perhaps most importantly, all three of these forms of regulated cell death have arguably evolved as forms of cellular “suicide”; that is, cells die as a consequence of engaged protein “executioners” that function to mediate the cell death. In contrast, many other forms of regulated cell death represent externally mediated subversions of cellular mechanisms to disrupt essential survival functions, such as scavenging of lipid peroxides [resulting in ferroptosis (21)] or control of vesicular transport [leading to death as a result of retrograde transport (22)]. The latter cell death modalities may be considered forms of cellular “sabotage” (23). Although it is possible that pathogens can act to cause such sabotage and thus drive evolution of survival mechanisms, we currently do not have evidence of coevolution in such interactions, and therefore they are not considered further here.
A relevant aspect of the interplay between cell death and infection is the engagement of adaptive immune responses. For example, dead and dying cells are engulfed by conventional dendritic cells, and, in a subset of the latter, proteins associated with the corpse (including microbial proteins) are degraded and their peptides presented on class I major histocompatibility molecules to CD8+ T cells (24, 25). The mode of cell death influences this “antigen cross presentation” process (25). There are excellent reviews on cell death and cross presentation (24–27), and we do not consider this further in this review.
An additional form of regulated cell death, called NETosis, appears to function in host defense, and it is quite possible that host-pathogen coevolution played important roles in its mechanisms. During NETosis, cells release DNA “nets” that trap bacteria and perhaps other microbes to control infections. We direct interested readers to other, excellent reviews of this process (28, 29).
This review describes the mechanisms of regulated cell death (here apoptosis, necroptosis, and pyroptosis) in the context of infection and host-pathogen coevolution. We begin with caspases, the regulators of the forms of RCD discussed here. The mechanisms of RCD are explored, starting with the simplest way for a cell to sense a pathogen and induce cell death (pyroptosis) and proceeding to increasingly more intricate and complex mechanisms of RCD (apoptosis and necroptosis). Next, we explore the strategies developed by pathogens to disrupt the various RCD pathways. We then investigate the evolutionary aspects of the pathways of RCD. We close by exploring the relation between coronaviruses and RCD and future directions for the field. Excellent additional reviews that consider the origins and mechanistic control of the regulated cell death pathways are available for the interested reader (30–35).
2. CASPASES CONTROL REGULATED CELL DEATH
Before we can understand how regulated cell death functions in the context of infections, we first must appreciate the proteins and their interactions that control RCD. Pyroptosis, apoptosis, and necroptosis are regulated by a family of aspartate-specific cysteine proteases called caspases (reviewed in Refs. 36, 37). Caspases function as endopeptidases that cut proteins at aspartate residues that are surrounded by other residues (defining the broad specificity of the individual caspase) (38–40).
Caspases are usually divided into three subfamilies based on their physiological functions and genomic organization: initiator, executioner (or effector), and inflammatory caspases (36, 37) (FIGURE 1). These enzymes are expressed as inactive zymogens that contain a prodomain and a caspase catalytic region, consisting of a large and a small subunit connected by a linker sequence. The short prodomains of effector caspases, in general, do not have major functions, whereas the prodomains of initiator and inflammatory caspases are required for their activation. These contain protein-protein interaction motifs that are structurally related [so-called “death folds” (41)], including a caspase-recruitment domain (CARD) (caspases-1, -2, -4, -5, -9, and -11), two tandem death effector domains (DEDs) (caspases-8 and -10), a death domain (DD) (Hydra HyDDCasp) (42), a pyrin domain (PYD) (Dania rerio caspy and caspy2) (43), or another domain that allows their activation (Hydra HyCaspA) (42) (FIGURE 1). As discussed below, these interaction domains participate in the interaction with caspase-activation platforms that assemble in response to cellular signaling events.
FIGURE 1.

Domain structure of the caspases discussed here. Schematic representation of initiator caspases-2, -9, -8, and -10 and DRONC, executioner caspases-3, -6, and -7, DRICE, and CED-3, inflammatory caspases-1, -4, -5, -11, and -12, caspase-14 (involved in cellular differentiation), a subset of caspases present in Hydra, and one of the pyrin domain (PYD)-containing caspases present in zebrafish. a.a., Amino acids; CARD, caspase-recruitment domain; Ce, Caenorhabditis elegans; DD, death domain; DED, death effector domain; Dm, Drosophila melanogaster; Dr, Dania rerio; H, Hydra spp.; Hs, Homo sapiens; Mm, Mus musculus; ?, prodomain of unknown structure.
Executioner caspases are expressed as inactive dimers that require cleavage to become active. Such cleavage is predominantly mediated by initiator caspases but can also be performed by granzyme B upon its intracellular delivery by cytotoxic lymphocytes (44, 45). Cleavage occurs at a site between the resulting large and small subunits of the procaspase. In contrast, initiator and inflammatory caspases are monomeric zymogens (procaspases) that are activated by enforced dimerization [known as induced proximity (46)]. Upstream signals induce the formation of “caspase-activation platforms,” leading to the recruitment and dimerization of procaspases via their prodomains. Dimerization endows the procaspase with enzymatic activity. The caspases then cleave their substrates, including, but not restricted to, executioner caspases (32). While initiator and inflammatory caspases undergo a series of autocleavage events that result in their release from the activation platform, the released enzyme may be active or inactive, depending on the caspase.
Caspases are solely found among the Animalia and are present throughout all animal phyla. It is therefore not surprising that caspase genes have undergone extensive diversification over the course of evolution. Caspase genes have duplicated, and some have been lost or have been rendered effectively inactive in different phyla (47–49). Caspases are members of the C14 class of cysteine proteases, which includes paracaspases and metacaspases, although the latter are not aspartic acid but arginine/lysine specific. Metacaspases and paracaspases have been found in fungi and plants (the paracaspases are also found in animals; the metacaspases are not), but their biochemistry is such that the types of pathways that activate the caspases are unlikely to function in the activation of these proteases. The functions of metacaspases and paracaspases have been reviewed elsewhere (50–54) and are not further considered here.
Humans express 12 known caspases. These include inflammatory caspases-1, -4, and -5, initiator caspases-2, -8, -9, and -10, executioner caspases-3, -6, and -7, caspase-12, and caspase-14. Caspase-10 is a gene duplication of caspase-8, although both proteins can be functionally distinguished (55), and is absent in rodents. Similarly, caspase-5 is a gene duplication of caspase-4 (56). Mice do not encode caspases-4 and -5, but instead express caspase-11, an ortholog of human caspase-4 (57). The casp12 gene has acquired multiple mutations over evolutionary time, including a single-nucleotide polymorphism that introduces a premature stop codon resulting in the expression of a truncated form of the protein, Csp12-s (58). This mutation was spread and nearly fixed in humans because of positive selection (59). However, the genomes of some individuals do not harbor the stop codon, allowing the expression of the full-length protein, apparently conferring a hypersusceptibility to sepsis (58). Nonhuman primates encode an intact casp12 gene, indicating that the nonfunctional variant evolved in humans. This is thought to coincide with prehistoric human behavioral changes that led to growth of population size and density, increasing the susceptibility for infection and sepsis (59, 60). Caspase-14 has functions in skin development (61), but no role in cell death has been identified for this caspase.
3. INFLAMMATORY CASPASES AND PYROPTOSIS
What would be the simplest way for a cell to sense a pathogen and induce cell death? This might be a protein that contains a receptor domain that upon ligation unlocks the molecule’s ability to kill the cell (a “receptor-executioner” protein). Although such a protein is not found in animals, fungi such as Podospora anserina express the HET-S/HET-s heterokaryon incompatibility system, which could be seen as one of the simplest ways to induce death upon the recognition of nonself. P. anserina expresses the HET-S protein, which contains a prion domain that upon activation folds into a β-solenoid conformation and causes cell death by forming pores in the plasma membrane (62). When P. anserina encounters another P. anserina strain that expresses the HET-S homolog HET-s, the proteins interact and form pores. The resulting death of the cell is believed to limit pathogen transmission between the strains (63).
A single-molecule receptor-executioner protein is not known to exist in animals. The shortest way to induce cell death upon pathogen recognition in animals is a two-step system in which a caspase contains a receptor domain that upon ligation activates the protease to cleave and thereby activate a substrate protein that causes death. Murine caspase-11 and human caspase-4 and caspase-5 have the ability to recognize intracellular Gram-negative bacteria through the binding of bacterial hexa-acetylated lipopolysaccharide (LPS) to a motif in their prodomains (64). LPS binding induces the dimerization and subsequent activation of the protease (65–69). The caspases then cleave the pore-forming protein gasdermin D (GSDMD), which inserts into the plasma membrane to lyse the cell (70, 71). This two-step “from recognition to death” system is known as the noncanonical inflammasome and leads to the inflammatory RCD type pyroptosis (FIGURE 2A).
FIGURE 2.
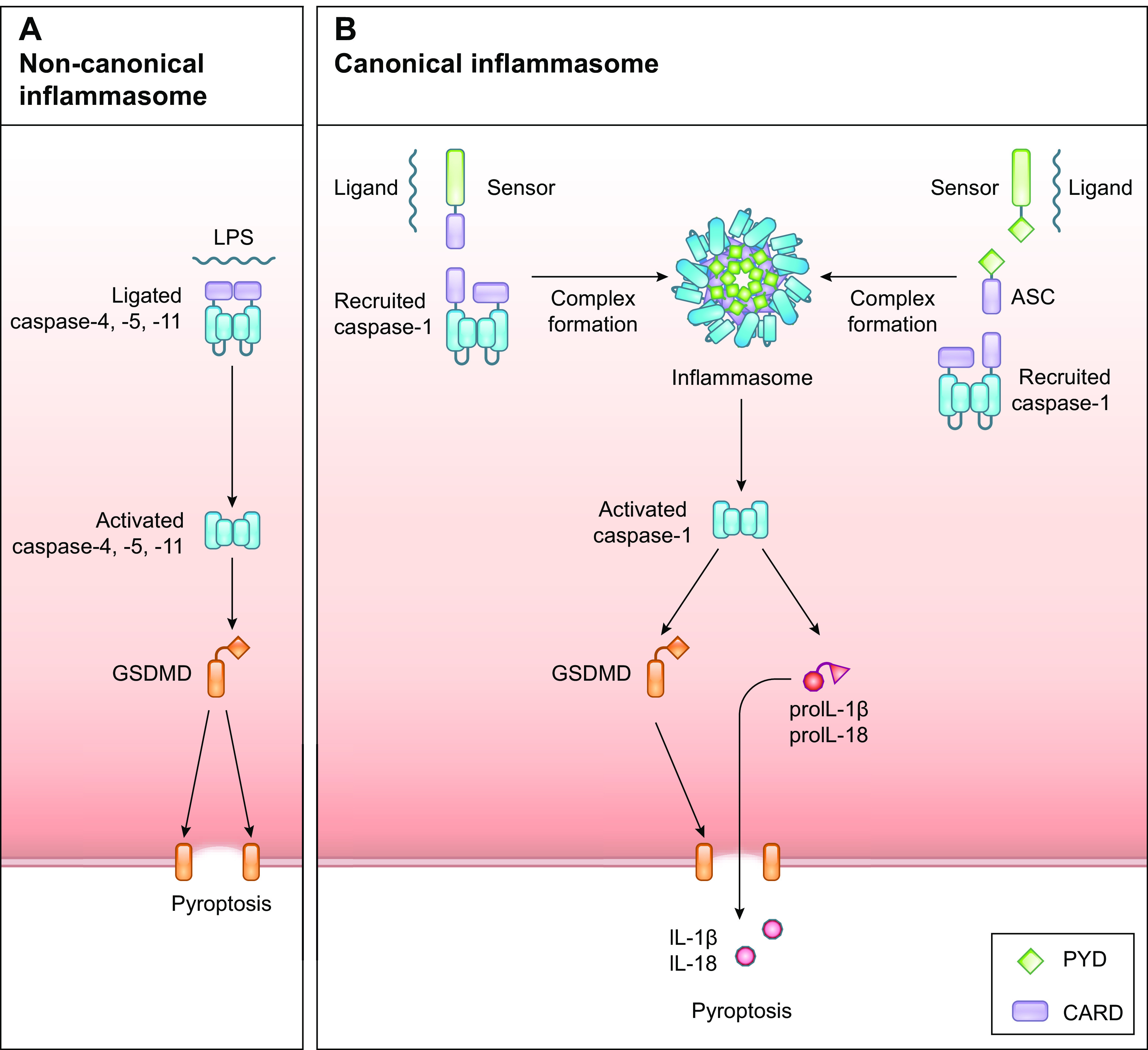
Pathways of pyroptosis. A: the noncanonical inflammasome is activated upon recognition of LPS by the inflammatory caspases-4, -5, and -11, resulting in dimerization, activation, and processing of gasdermin D (GSDMD). Activated GSDMD induces plasma membrane pore formation by oligomerization to execute pyroptosis. B: the canonical inflammasome is activated upon engagement of inflammasome-inducing receptors, which recruit the inflammatory caspase-1, either directly via caspase-recruitment domain (CARD) interactions or indirectly via pyrin domain (PYD) and CARD interactions mediated by the adaptor protein ASC, forming the inflammasome. Activated caspase-1 cleaves pro-IL-1β and pro-IL-18 into their biologically active forms and activates GSDMD to execute pyroptosis.
Most pathogens, however, do not contain LPS or have evolved ways to restrict their presence in the cytosol, e.g., by “hiding” in vacuoles, and can therefore not be controlled by the noncanonical inflammasome. In response to intracellular infections or perturbations in cellular homeostasis, some cytosolic PRRs can induce the activation of a cytosolic macromolecular platform known as the canonical inflammasome (6) (FIGURE 2B). Canonical inflammasome activation is more elaborate than noncanonical inflammasome activation as it requires specialized sensors and the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC, also known as PYCARD) to recruit and activate the inflammatory caspase-1 (72). Caspases-1, -4, -5, and -11 can all cleave GSDMD. However, unlike caspases-4, -5, and -11, caspase-1 has the additional ability to process the inflammatory cytokines pro-interleukin (IL)-1β and pro-IL-18 into their biologically active forms to further aid the immune response to control an infection. The ability of an inflammatory caspase to cleave pro-IL-1β depends on a small set of charged amino acids near the catalytic site, which are present in caspase-1 but absent in human caspase-4 (73). GSDMD cleavage does not depend on these charged residues but rather on hydrophobic interactions that are present between the COOH-terminal domain of GSDMD and all the inflammatory caspases (67, 69).
3.1. Induction of Inflammasomes
Inflammasomes can be activated by various microbial PAMPs, including flagellin, components of the bacterial type 3 secretion system (T3SS), RNA, DNA, toxins, and metabolic changes. These microbial elements are recognized by diverse inflammasome-inducing sensors, which include receptors of the Absent In Melanoma 2 (AIM2)-like receptor (ALR, aka PYHIN) gene family [the DNA sensors AIM2 and Interferon Gamma Inducible Protein 16 (IFI16)], Pyrin, and members of the nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family (74, 75). Inflammasome-inducing receptors contain a PYD or CARD through which they recruit procaspase-1 (76). PYD-containing receptors require ASC, which contains both PYD and CARD, to recruit caspase-1, whereas CARD-containing receptors can do so directly or through ASC.
Inflammasome-inducing sensors are activated through direct interaction with their respective ligands, by proteolytic processing, or by engaging and/or disrupting binding partner interactions (reviewed in Refs. 6, 76–79). AIM2, NLRP3, NLRP6, NLRP7, and NRP9b are activated upon direct interaction with their respective ligands. NLRP1 activation can be activated by proteolytic processing, although human NLRP1 can also be activated by dsRNA (80). NLRC4 is activated upon binding activated NAIP proteins that function as the actual sensors, and sequestered Pyrin is released after disruption of 14-3-3 protein interactions (all detailed below).
The best studied inflammasome-inducing sensor is NLRP3, a member of the NLR receptor family. NLRP3 is activated by perturbations in cellular homeostasis. In most cases, activation of the NLRP3 inflammasome is a two-step process. First, a priming step is required in which PAMPs, DAMPs, and certain cytokines (TNF-α, IL-1β) induce the transcriptional upregulation of inflammasome components. Priming also readies NLRP3 via posttranslational modifications to be activated (81). The second step is the activation of NLRP3. Changes in ion flux (e.g., K+ efflux or Ca2+ signaling), mitochondrial dysfunction or lysosomal rupture, and possibly other stresses activate the NLRP3 inflammasome. Cellular stress can be induced by a wide variety of stimuli, including pathogens, endogenous DAMPs during sterile inflammation, or environmental irritating factors (81–84). As such, the NLRP3 inflammasome does not appear to sense pathogens or other stressors directly but responds to their perturbing effects.
Although the exact mechanisms of NLRP3 activation by cellular stress remain to be elucidated, activated NLRP3 recruits ASC and caspase-1 to form the NLRP3 inflammasome, resulting in the processing of GSDMD and pro-IL-1β and pro-IL-18 and pyroptosis accompanied by IL-1β and IL-18 release. NLRP3 inflammasome formation occurs at the microtubule-organizing center (MTOC) (85) and further depends on the interaction with NIMA-related kinase 7 (NEK7) (86) and is promoted by DEAD-box helicase 3 X-linked (DDX3X) (87).
Cellular ion flux changes can occur as a result of disrupted plasma membrane integrity. This happens, for instance, after pore-forming proteins insert into the plasma membrane. Caspase-4 (or -11)-mediated pyroptosis following GSDMD activation causes K+ efflux, which then activates the NLRP3 inflammasome, thereby allowing the release of biologically active IL-1β and IL-18 (88). Similarly, insertion of MLKL into the plasma membrane during necroptosis (detailed below) causes K+ efflux and activates the NLRP3 inflammasome and IL-1β release independent of GSDMD (89, 90).
The NLR family member NLRP1 has been known to be a sensor for pathogen-encoded proteases, which activate NLRP1 through proteolytic cleavage. Cleavage releases the COOH-terminal CARD-containing fragment to recruit caspase-1 and induces the degradation of the formed NH2-terminal fragment (91–93). Details of the active NLRP1 structure have recently been elucidated (94, 95). The presence of ASC enhances, but is not required for, NLRP1-mediated caspase-1 activation (96). Human NLRP1 can be activated by the 3C protease protein of enteroviruses, such as human rhinovirus (HRV) (97, 98). Rodent NLRP1 can be cleaved by lethal toxin of Bacillus anthracis (99, 100), but the cleavage site has not been conserved in human NLRP1 and lethal toxin does not activate the NLRP1 inflammasome in human macrophages (101). Murine NLRP1b is also cleaved by proteases of Shigella flexneri (102), Listeria monocytogenes (102), and Toxoplasma gondii (103) and can be activated by reduction of cytosolic ATP levels (104) or possibly by autoprocessing (101, 105), although the latter remains unclear (96). No viral proteases have been identified to activate rodent NLRP1, and no identified bacterial proteases cleave human NLRP1. In addition, dsRNA has recently been observed to activate NLRP1 in human cells (80). Human NLRP1, but not murine NLRP1b, binds cytosolic dsRNA to its leucine-rich region (LRR) domain and is activated upon delivery of long dsRNA or infection with Semliki Forest virus (SMV) but not after infection with modified vaccinia virus ankara (MVA), herpes simplex virus-1 (HSV-1), or measles virus (MV) (80). Rodent NLRP1 thus seems to mainly control bacterial infections, whereas human NLRP1 controls viral infections.
The ALR family members absent in melanoma 2 (AIM2) and gamma-interferon-inducible protein Ifi-16 (IFI16) recognize pathogenic dsDNA, mainly from viruses. ALR family members contain an NH2-terminal Pyrin domain (PYD) and a DNA-sensing COOH-terminal HIN200 domain (106, 107) that under homeostatic conditions maintain an autoinhibitory conformation. Ligand binding induces the release of the autoinhibitory conformation allowing for the recruitment of ASC via PYD-PYD interactions and the subsequent ASC-mediated recruitment of caspase-1 through CARD-CARD interactions (108).
NLRC4 is activated through binding with NAIP proteins that recognize components of the T3SS of Gram-negative bacteria. Primates, including humans, express one NAIP ortholog that recognizes T3SS needle proteins. Rodents encode multiple NAIP paralogs, which recognize flagellin (NAIP5/6) or the T3SS inner rod (NAIP2) or needle (NAIP1) proteins (109). Ligand specificity of rodent NAIPs depends on the central nucleotide-binding domain (NBD)-associated domains, which have evolved under positive selection in both rodents and primates (109). Upon ligation, NAIPs undergo a conformational change that initiates the recruitment, activation, and oligomerization of the inflammasome receptor NLRC4, which contains a CARD and can recruit caspase-1 directly or through ASC (110–112).
Activation of the Pyrin inflammasome is regulated by perturbations in the phosphorylation status of Pyrin. Phosphorylation by the kinases PKN1/2 induces Pyrin’s interaction with the chaperone proteins 14-3-3ε and 14-3-3τ, resulting in restricted Pyrin activity (113, 114). Rho GTPases are required for the activity of PKN1/2. Rho GTPases can be modified or inactivated by bacterial toxins, resulting in reduced Pyrin phosphorylation and 14-3-3 protein interaction, allowing the PYD-containing Pyrin to recruit ASC and caspase-1 (115).
Other inflammasome-inducing sensors are known but have not been well characterized. NLRP6 is activated by lipoteichoic acid (LTA) derived from Gram-positive bacteria such as L. monocytogenes (116), and direct binding of LTA to NLPR6 induces dimer formation, which can oligomerize in the presence of ATP to recruit ASC (117). NLRP7 can be activated by acylated lipopeptides, which are found on microbial cell walls (118), and NLRP9b recognizes short dsRNA strands via the RNA helicase Dhx9 (119).
3.2. Execution of Pyroptosis
The best-known executioner of pyroptosis is GSDMD, but this is not the only gasdermin that can execute pyroptotic cell death (reviewed in Refs. 120, 121). The human genome encodes six gasdermin family paralogs, gsdma, gsdmb, gsdmc, gsdmd, gsdme, and pjvk. With the exception of pejvakin (PJVK), gasdermins are comprised of an NH2-terminal cytotoxic domain and a COOH-terminal inhibitory domain that are connected by a linker region. In GSDMD, this linker can be cleaved by caspase-1, -4, -5, or -11. Cleavage of the linker results in the release of the cytotoxic NH2-terminal domain, which undergoes a conformational change exposing a hydrophobic part of the protein that inserts itself into membranes and allows oligomerization with additional molecules, resulting in the formation of plasma membrane pores and pyroptotic cell death (120, 121). The NH2-terminal domains of GSDMA, GSDMB, GSDMC, GSDMD, and GSDME are all able to form such pores (122), and the structures for the GSDMA and GSDMD pores have recently been solved (122–124). Although expression of the NH2-terminal fragments of gasdermin A, B, and C induces pyroptosis, the mechanisms underlying their activation are incompletely understood. GSDMB can be cleaved by the protease granzyme A (125), released into target cells by cytotoxic lymphocytes, and GSDMB may aid pyroptosis by enhancing the ability of caspase-4 to cleave GSDMD (126), but these observations remain to be confirmed by others. Pejvakin has a short COOH-terminal domain that does not share sequence homology with the other gasdermin family members, and the exact role for PJVK and its ability to form pores remains unclear, although mutations in pjvk have been associated with hearing loss in mice and humans (127–129) due to altered functioning of peroxisomes (127).
Gasdermin D is activated by caspase-1 and caspases-4, -5, and -11 and can be cleaved by activated caspase-3, an executioner caspase in apoptosis (discussed below). The latter cleavage, however, renders GSDMD inactive, since it happens at a distinct site in the NH2-terminal domain that disrupts the ability of GSDMD to insert into membrane and thus execute pyroptosis (130). By inhibiting GSDMD, caspase-3 ensures that apoptosis remains nonlytic. However, under certain conditions, apoptotic cells can undergo secondary necrosis, which may aid in the clearance of debris at the site of infection by enhancing the attraction of innate immune cells. In GSDME-expressing apoptotic cells, activated caspase-3 can cleave the linker region of GSDME, thereby activating gasdermin E to mediate pyroptosis (131, 132). Similarly, natural killer (NK) cell-derived granzyme B, but not granzyme A, can apparently activate GSDME (133). The formation of gasdermin D or E pores not only leads to the release of intracellular contents to induce inflammation and attract immune cells, it also traps intracellular pathogens in so-called pore-induced intracellular traps (PITs), which can lead to the clearance of the infection by efferocytosis—the engulfment of dead and dying cells by phagocytes—of the cell corpses and entrapped pathogens (134).
4. APOPTOSIS AND THE ACTIVATION OF INITIATOR AND EXECUTIONER CASPASES
A common response of cells to invading pathogens is the induction of apoptotic cell death. Apoptosis as a cell death modality is unique to animals, and its features rely on the activity of caspases (2). The initiator caspases that regulate apoptosis, caspase-8 and caspase-9, cleave and thereby activate the executioner caspases-3 and -7 (FIGURES 3A and 4). The executioner caspases induce apoptosis by cleaving approximately a thousand substrates, such as the chaperone and inhibitor of Caspase-Activated DNase (iCAD, which is in a complex with the DNase CAD), gelsolin, Pac21, Xkr8, and ATP11c (32), cumulatively resulting in the characteristic phenotype of apoptotic cells: DNA fragmentation, loss of electron transport function, cell shrinkage, phosphatidylserine externalization, membrane blebbing, and other events (32).
FIGURE 3.
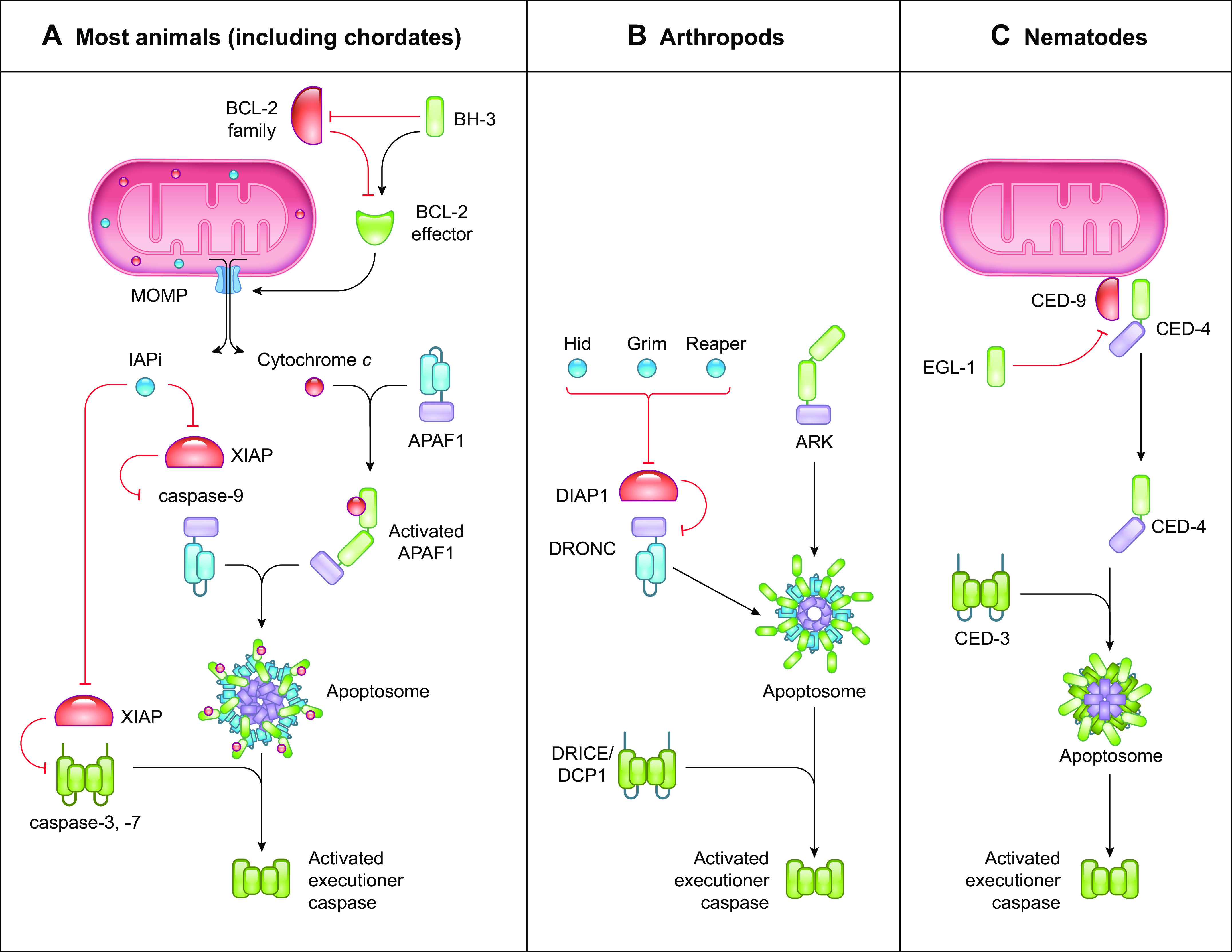
The caspase-9 pathway of apoptosis. Comparison between the caspase-9 pathway of apoptosis in most animals, arthropods, and nematodes. A: in most animals, the caspase-9 pathway of apoptosis is mediated by the BCL-2 effector proteins BAX and BAK to induce mitochondrial outer membrane permeabilization (MOMP). Activation of BCL-2 effector proteins is regulated by the balance between proapoptotic and antiapoptotic Bcl-2 proteins. MOMP releases cytochrome c and inhibitor of apoptosis (IAP) inhibitors (IAPi) into the cytosol, where cytochrome c binds and activates Apoptotic Protease Activating Factor 1 (APAF-1) to recruit the initiator caspase-9, forming the apoptosome. Activated caspase-9 cleaves and activates the executioner caspases-3 and -7 to mediate apoptosis. IAPi released by MOMP block the suppressive effects of the caspase inhibitor X-linked inhibitor of apoptosis protein (XIAP), allowing the caspases to function. B: in arthropods, the APAF-1 homolog ARK is constitutively active. The IAP inhibitors Hid, Grim, and Reaper inhibit DIAP1 to release the initiator caspase DRONC to form the apoptosome with ARK. Activated DRONC cleaves and activates the executioner caspases DRICE and DCP1 to induce apoptosis. C: in nematodes, the APAF-1 homolog CED-4 is suppressed by the anti-apoptotic protein CED-9. Binding of the BH-3-only protein EGL-1 to CED-9 releases CED-4 to form the apoptosome with the executioner caspase CED-3, resulting in CED-3 activation and apoptosis.
FIGURE 4.

The caspase-8 pathway of apoptosis. Ligation of death receptors induces the rapid association of FADD via death domain (DD) interactions with the intracellular domain of the receptor, leading to the recruitment of caspase-8 by death effector domain (DED) interactions to form the death-inducing signaling complex (DISC). Ligated TNFR1 first recruits TRADD and RIPK1 and subsequently recruits FADD through DD interactions with TRADD. Deubiquitylated RIPK1 leaves the TNFR1 complex to recruit FADD. FADD recruits caspase-8 via DED interactions leading to caspase-8 homodimerization, activation, autoproteolytic processing, and release from the complex. Receptor ligation also results in the expression of the prosurvival gene cFLIPL, which heterodimerizes with caspase-8 and inhibits the full activation of caspase-8. Fully activated caspase-8 cleaves the executioner caspases-3 and -7 to induce apoptosis. In the presence of X-linked inhibitor of apoptosis protein (XIAP) the function of activated executioner caspases is inhibited. However, fully activated caspase-8 processes BID to cleaved BID (cBID), which induces BAX, BAK-mediated mitochondrial outer membrane permeabilization (MOMP), thereby releasing inhibitor of apoptosis (IAP) inhibitors (IAPi) to block XIAP and allow the execution of apoptosis. Cyt. C, cytochrome c.
We generally think of two major pathways of apoptosis, the intrinsic or mitochondrial pathway and the extrinsic or death receptor pathway. Although useful, we know that these pathways can be intertwined, as components of the extrinsic pathway can engage the intrinsic pathway. The terms “intrinsic” and “extrinsic” are problematic, since the “intrinsic pathway” can be engaged by extrinsic signals and the “extrinsic pathway” by intrinsic signals. For this reason, we instead present the pathways in terms of how the initiator caspases specific to each are engaged. This focus on mechanism rather than characteristic description has emerged as a useful approach to terminology (135). For our purposes, we consider the activation and functions of two of the initiator caspases, caspase-9 and caspase-8 (which correspond to the “traditional” mitochondrial and death receptor pathways, respectively).
4.1. The Caspase-9 Pathway of Apoptosis
The caspase-9 pathway of apoptosis relies on the formation of an activation platform termed the apoptosome (FIGURE 3A). The adaptor protein in this complex, Apoptotic Protease Activating Factor 1 (APAF-1), is a cytosolic monomeric protein that belongs to the NLR protein family (see above) and consists of an NH2-terminal CARD, an NB-ARC ATPase domain, and a COOH-terminal domain containing WD40 repeats (WD40 domain). APAF-1 is activated by binding of holocytochrome c to the WD40 domain (136). This induces a conformational change that allows the exchange of deoxyADP for deoxyATP in the NB-ARC domain, initiating exposure of the CARD, oligomerization into septameric structures, and the recruitment of caspase-9 via CARD-CARD interactions (137, 138). Caspase-9 then cleaves and activates the executioner caspases-3 and -7 that mediate apoptosis (136).
Caspase-9 activation thus relies on the interaction between APAF-1 and cytochrome c. Under homeostatic conditions, cytochrome c resides between the inner and outer mitochondrial membranes, where it functions as an electron shuttle of the respiratory chain. During apoptosis, the mitochondrial outer membrane is permeabilized in a process called mitochondrial outer membrane permeabilization (MOMP). This results in the release of the mitochondrial intermembrane space proteins, including cytochrome c, into the cytosol. Cytochrome c then binds to APAF-1, which induces the formation of the apoptosome (136, 139, 140).
Besides cytochrome c, MOMP releases the mitochondrial intermembrane space proteins Smac/Diablo and Omi/Htra2, which are antagonists of inhibitor of apoptosis (IAP) proteins (141–144). Activated caspases-3 and -7, as well as caspase-9, are inhibited by X-linked inhibitor of apoptosis protein (XIAP). MOMP thus not only results in apoptosome activation but also induces the release of XIAP-mediated inhibition of caspases-3, -7, and -9.
The process of MOMP is regulated by the Bcl-2 family of proteins, which contain proapoptotic (MOMP promoting) and antiapoptotic (MOMP inhibiting) members (145, 146). Upon activation, the effectors of MOMP, BCL2-associated X protein (BAX) and BCL2-antagonist/killer 1 (BAK1, known as BAK), insert themselves into the outer mitochondrial membrane and oligomerize to form pores, leading to mitochondrial ultrastructure perturbation and the leakage of the intermembrane content into the cytosol. The activation of the MOMP effector proteins is tightly regulated by an interplay between the antiapoptotic Bcl-2 proteins (e.g., BCL-2, BCL-XL, and MCL-1) and the proapoptotic BH-3-only subfamily that can either neutralize the antiapoptotic proteins (e.g., BAD, BMF, and NOXA) or activate BAX and BAK (e.g., BID, BIM, and PUMA). Apoptotic stimuli, such as cellular stress, DNA damage, or pathogen recognition, can change this delicate balance in favor of the proapoptotic proteins, resulting in the initiation of MOMP (145, 146). Another Bcl-2 family protein is BCL2-like Ovarian Killer (BOK), which does not appear to be regulated by other Bcl-2 family proteins but instead is controlled by the endoplasmic reticulum (ER)-associated degradation machinery (147). Like BAX and BAK, BOK can effect MOMP to promote caspase-9 activation.
Thus, the caspase-9 pathway of apoptosis depends on the induction of the apoptosome and the balance between proapoptotic and antiapoptotic Bcl-2 proteins to harness MOMP.
4.2. The Caspase-8 Pathway of Apoptosis
The caspase-8 pathway of apoptosis depends on the interaction between caspase-8 and the adaptor protein Fas-associated Death Domain (FADD) (FIGURE 4). FADD consists of a COOH-terminal death domain (DD) and an NH2-terminal death effector domain (DED) and mediates the activation of caspase-8 by recruiting procaspase-8 via DED-DED interactions upon upstream signals that recruit FADD to DD-containing proteins (detailed below) (35, 148). Binding of procaspase-8 to FADD induces procaspase-8 oligomerization, filament formation, and proximity-induced activation. Autoproteolytic processing then yields the fully active, mature caspase-8 (35).
Caspase-8 directly cleaves and activates the executioner caspases (caspases-3 and -7). In cells that express XIAP (see above), however, the function of the activated executioner caspases is inhibited. Therefore, when caspase-8 cleaves another substrate, the proapoptotic Bcl2 family member BH3 Interacting Domain Death Agonist (BID), the cleaved BID (cBID) induces BAX-BAK-mediated MOMP (detailed above) (149, 150). The liberation of the IAP antagonists SMAC and Omi then promotes apoptosis by inhibiting the antiapoptotic function of XIAP. In such cells, antiapoptotic Bcl-2 proteins can inhibit the caspase-8 pathway of apoptosis.
FADD is engaged by DD-DD interactions. Death receptors (DRs) are a subset of the tumor necrosis factor superfamily (TNFRSF) that includes tumor necrosis factor (TNF) receptor-1 (TNFR1; TNFRSF1a), CD95 (TNFRSF6, Apo-1, Fas), TNF-related apoptosis-inducing ligand (TRAIL) receptor-1 and -2 (TRAIL-R1/2; DR4/5, TNFRSF10a/b), DR3 (TNFRSF25), and DR6 (TNFRSF21) and contains a COOH-terminal cytosolic DD (19). With the exception of TNFR1, ligation of these DRs by their cognate ligands or by agonistic antibodies results in the DD-mediated engagement of the protein FADD and the subsequent recruitment of procaspase-8 through DED interactions. DR ligation thus induces the formation of a FADD-caspase-8 activation platform at the cytosolic tail of the receptor, known as the death-inducing signaling complex (DISC) (151). TNFR1-induced apoptosis is also mediated by FADD-caspase-8, but unlike other DRs that recruit FADD directly, TNFR1 first engages the protein TNFR1-associated Death Domain (TRADD), which in turn recruits FADD (152).
FADD can also be recruited to a cytosolic protein complex called the ripoptosome that further consists of the receptor interacting serine/threonine kinase (RIPK)-1 (RIPK1) (153). RIPK1 contains an NH2-terminal kinase domain (KD), an intermediate domain (ID), a RIP homotypic interaction motif (RHIM) required for RHIM-containing protein interactions, and a COOH-terminal DD through which it interacts with FADD to promote caspase-8 activation (154).
TNFR1 ligation recruits RIPK1 to the cytoplasmic tail of the receptor, where RIPK1 is modified by phosphorylation and ubiquitylation to mediate downstream signaling, resulting in gene expression. However, when the functioning of E3 ligases that ubiquitylate RIPK1, such as cIAP1 and cIAP2, is impaired or RIPK1 deubiquitylation is enhanced, RIPK1 is released from the TNFR1 complex and then recruits the FADD-procaspase-8 complex to form the ripoptosome (155). RIPK1 may also induce the formation of the ripoptosome when engaged by retinoic acid receptor gamma (RARγ) when this protein translocates from the nucleus to the cytosol in response to DNA damage (156). Ripoptosome formation can also be induced by another protein kinase, RIPK3. RIPK3 is a homolog of RIPK1 and contains an NH2-terminal KD, an ID, and a RHIM but lacks the COOH-terminal DD found in RIPK1 (157). RIPK3 recruits RIPK1 through RHIM-RHIM interactions of the RHIMs present in both proteins, which is induced by ligation of Toll-like receptor (TLR)3, TLR4, and the cytosolic zDNA/zRNA receptor zDNA binding protein 1 [ZBP1, also known as DNA-dependent activator of IFN-regulatory factors (DAI)] (153, 158, 159). Ligation of TLR3 or TLR4 induces the recruitment of the adaptor molecule TIR-domain-containing adapter-inducing interferon-β (TRIF) to the engaged receptors. TRIF contains a RHIM through which it interacts with RIPK3 (158). Similarly, ZBP1 contains two RHIMs and can induce the ripoptosome by directly recruiting RIPK3 (159). The outcome of ripoptosome formation can be FADD-caspase-8-mediated apoptosis, although other outcomes are possible, as detailed below.
Thus, the caspase-8 pathway of apoptosis depends on FADD engagement through DD interactions with DRs, TRADD, or RIPK1 and is activated by death-inducing signals coming from within the cell or from the extracellular environment.
4.3. Regulation of the Caspase-8 Pathway of Apoptosis by cFLIP
Many death receptors, TLRs, and cytosolic receptors induce transcriptional programs upon their ligation. TNFR1 ligation induces the MAPK and NF-κB signaling pathways that induce gene expression of proinflammatory and prosurvival factors. Among the expressed genes is cflar, which encodes FLICE-like inhibitory protein (cFLIP). Depending on posttranslational mRNA splicing, two major isoforms of cFLIP are generated (160, 161). cFLIP short (cFLIPs) is a truncated form that blocks DISC-dependent procaspase-8 activation by halting DED-mediated procaspase-8 oligomer assembly and thereby blocks apoptosis. The second isoform, cFLIP long (cFLIPL), is a procaspase-8-like protein that lacks a catalytic site and thus does not possess proteolytic activity. Procaspase-8 favors heterodimerization with cFLIPL over homodimerization with other procaspase-8 zymogens (162, 163). While heterodimerization ablates the autoproteolytic processing of procaspase-8, the procaspase-8-cFLIPL complex retains proteolytic capacity, although substrate specificity is slightly changed compared to procaspase-8-procaspase-8 homodimers (164). Low cFLIPL levels can enhance apoptotic signaling by enhancing the specific activity of the heterodimer and procaspase-8 homodimer activation without disrupting the filament. High cFLIPL levels, however, disrupt procaspase-8 homodimer formation, thereby reducing the activity of procaspase-8 and blocking apoptosis (165). Thus, cFLIP isoforms and expression levels determine whether caspase-8-mediated apoptosis ensues or is inhibited.
4.4. The Caspase-2 Pathway of Apoptosis
The caspase-2 pathway of apoptosis is more elusive. Caspase-2 is an initiator caspase that has been implicated in apoptosis in response to a diverse array of triggers including genotoxic stress, heat shock, β-amyloid accumulation, and irregularities in cytoskeleton arrangement and the mitotic spindle (166). The best-known platform for the activation of caspase-2 is termed the PIDDosome, although caspase-2 can be activated outside of this complex as well (167). The PIDDosome consists of the receptor protein PIDD, which contains a COOH-terminal DD through which it interacts with the adaptor protein RAIDD. RAIDD recruits procaspase-2 via CARD-CARD interactions leading to caspase-2 activation. Activated caspase-2 likely does not directly cleave executioner caspases; rather, it induces apoptosis by cleaving substrates that mediate the activation of caspases-3 and -7. For instance, activated caspase-2 can cleave Bid, resulting in activation of the caspase-9 pathway of apoptosis (168). It is therefore not clear whether the primary function of caspase-2 is to mediate apoptosis or it has other functions (169–172). Probably the best-characterized function of the caspase-2 pathway is in mitotic quality control. The presence of supernumerary centrioles in a dividing cell activates the caspase-2 pathway (173) via the binding of PIDD to the distal appendage protein ANKRD26 (174). In this setting, activated caspase-2 functions to cleave the E3-ligase MDM2, resulting in p53 stabilization (which in turn can lead to cellular senescence or apoptosis) (173, 174). The caspase-2 pathway is also engaged in the nucleolus upon DNA damage, in this case by the binding of PIDD to nucleolar phosphoprotein nucleophosmin-1 (NPM1) (175).
5. NECROPTOSIS AND ITS REGULATION BY THE FADD-CASPASE-8 COMPLEX
Upon recognition of pathogens by certain PRRs or as a result of exacerbated interferon or TNF signaling, cells can die by necroptosis (FIGURE 5). The necroptotic pathway does not depend on the functioning of caspases but instead relies on the activity of RIPK3. Upon activation, RIPK3 autophosphorylates, oligomerizes into RHIM domain-mediated amyloid structures (176), and recruits the necroptosis executioner protein Mixed lineage kinase domain-like (MLKL), which consists of an NH2-terminal helix bundle domain (HBD), a brace region, and a COOH-terminal pseudokinase domain (psKD) (177). RIPK3 phosphorylates the psKD of MLKL, which induces a conformational change in the brace region of MLKL, allowing for oligomerization and translocation to the plasma membrane, where the HBD inserts into the lipid bilayer to induce pore formation and rupture of the cell (178–181).
FIGURE 5.

Pathways of necroptosis. Ligation of TNFR1 induces the recruitment of TRADD, TRAF2, cIAP1/2, and RIPK1. Within this complex, RIPK1 is ubiquitylated by cIAP1/2 to mediate the expression of proinflammatory and prosurvival genes, including cFLIPL. When RIPK1 is deubiquitylated by CYLD, it leaves the complex and associates with RIPK3 through homotypic RIP homotypic interaction motif (RHIM) domain interactions, resulting in RIPK3 activation. IFN receptor signaling induces the expression of ZBP1. ZBP1 directly recruits RIPK3 by RHIM domain interactions. Ligated TLR3 and 4 engage the RHIM domain-containing protein TRIF to recruit RIPK3. RIPK3 autoactivates and forms amyloid structures to recruit and activate MLKL, which induces plasma membrane pore formation by oligomerization to execute necroptosis. Engagement of ZBP1 and TLR3 and 4 also results in the expression of proinflammatory and prosurvival genes. The inhibitory FADD-caspase-8-cFLIPL complex is recruited to RIPK3 via RIPK1 and inhibits the activation of RIPK3 and thereby necroptosis. DD, death domain; DED, death effector domain.
Necroptosis is induced by TNFR1 ligation (182–184), engagement of TLR3 or TLR4 (both via TRIF) (185–188), interferons (185), or after RARγ translocation to the cytosol in response to DNA damage (156). Interferon signaling does not directly induce necroptosis but promotes the expression of the interferon response gene ZBP1 (189). It remains unclear how ZBP1 is engaged in uninfected cells, and endogenous ligands are yet to be defined. Activation of these receptors leads to the formation of the ripoptosome (described above) and induces FADD-caspase-8-mediated apoptosis or RIPK3-mediated necroptosis.
The engaged cell death pathways are controlled by the presence of cFLIPL and the enzymatic activity of caspase-8. Low cellular levels of cFLIPL allow for the homodimerization of caspase-8 and lead to apoptosis, whereas high cFLIPL levels induce procaspase-8-cFLIPL heterodimerization. RIPK1 and RIPK3 both contain caspase-8 cleavage sites that are processed by the catalytically active procaspase-8-cFLIPL heterodimer (164). This cleavage disrupts the ripoptosome and thereby blocks necroptosis. The procaspase-8-cFLIPL heterodimer thus inhibits both apoptosis and necroptosis upon ripoptosome formation. Inhibition of caspase-8, e.g., by pathogenic caspase inhibitors (described below) or by pharmacological inhibitors, abrogates the ability of the procaspase-8-cFLIPL heterodimer to cleave RIPK1 and RIPK3, allowing the autophosphorylation and oligomerization of RIPK3 and the induction of necroptosis.
RIPK1 has a dual role in necroptosis (154). On one hand RIPK1 mediates necroptosis induced by TNFR1 and RARγ, where FADD-mediated recruitment of the procaspase-8-cFLIPL complex blocks necroptosis, whereas on the other hand RIPK1 blocks necroptosis by recruiting the FADD-procaspase-8-cFLIPL complex to RIPK3 upon activation of TLR3/4 and ZBP1. The kinase activity of RIPK1 is required for TNFR1-induced RIPK1-mediated necroptosis but is dispensable for the inhibition of TLR- and ZBP1-induced necroptosis (154).
Thus, the ligation of TNFR1, TLR3, TLR4, or ZBP1 induces three distinct signaling pathways: gene expression, apoptosis, and necroptosis. Where apoptosis depends on the catalytic activity of initiator and executioner caspases, necroptosis relies on RHIM domain interactions and the activation of the pore-forming executioner protein MLKL. In the presence of cFLIPL, both apoptosis and necroptosis are inhibited, signifying that the apoptotic and necroptotic pathways are interconnected.
6. CROSS TALK BETWEEN RCD PATHWAYS
The mechanisms that shape the individual apoptotic, necroptotic, and pyroptotic signaling cascades are well understood, and it has been recognized that interplay between the pathways occurs (190), the best example being that necroptosis is inhibited by the apoptosis-initiating caspase-8 (FIGURE 5). However, it is now becoming apparent that the three cell death pathways are far more intertwined and intricately regulated than has previously been appreciated (FIGURE 6).
FIGURE 6.

Cross talk between the pathways of apoptosis, necroptosis, and pyroptosis. Engagement of TLR3 and 4, death receptors, or ZBP1 induces the formation of RIPK3 amyloids, which activate MLKL to form pores in the plasma membrane that may lead to the activation of the NLRP3 inflammasome. The NLRP3 inflammasome can also be activated by RIPK3 and ZBP1 independently of MLKL or after cleavage of gasdermin D (GSDMD) by caspases-4/-5/-11 or caspase-1 when caspase-1 is activated by other inflammasomes. Caspase-1 can activate BID to induce the caspase-9 pathway of apoptosis or can directly activate caspases-3/-7 to induce apoptosis. Caspase-3 can inactivate GSDMD to inhibit pyroptosis and activate gasdermin E (GSDME) to induce pyroptosis. Pores formed in the plasma membrane after GSDME or pannexin-1 activation by caspase-3 or GSDMD or pannexin-1 activation by caspase-8 can activate the NLRP3 inflammasome. Caspase-8 may form inflammasomes through mechanisms that require further elucidation. Depending on the cell type, FADD enhances or inhibits inflammasome formation. MOMP, mitochondrial outer membrane permeabilization.
The apoptotic and pyroptotic pathways can interact with each other. The apoptosis executioner caspase-3 cleaves and activates GSMDE to mediate pyroptosis or cleaves and inactivates GSDMD to block pyroptosis (130–132), and caspase-8 has been suggested to cleave and activate GSDMD (191, 192). FADD and caspase-8 are required for the optimal activation of the NLRP3 inflammasome in macrophages (193), whereas in ileal epithelial cells FADD blocks caspase-8 from its involvement in inflammasome activation and pyroptosis (194, 195). In ileal epithelial cells in which FADD is present but caspase-8 is catalytically inactive, inflammasome activation is also induced (196, 197). Moreover, apoptotic caspases can activate the NLRP3 inflammasome by activating the channel-forming protein pannexin-1 (198), and the antiapoptotic proteins BCL-2 and BCL-XL bind and inhibit NLRP1 activation, thereby reducing activation of caspase-1 and IL-1β (199). Conversely, inflammasome-activated caspase-1 can cleave caspase-3 to induce apoptosis (130, 200, 201) and Bid to release mitochondrial SMAC and drive secondary necrosis (202), and activated gasdermin pores permeabilize mitochondria to enhance caspase-3 activation (203).
The necroptotic and pyroptotic pathways also influence each other. RIPK3 can mediate NLRP3 inflammasome activation in the absence of MLKL (204) or in the presence of MLKL by inducing MLKL pores in the plasma membrane (90). In human monocytes, NLRP3 can be engaged by a TRIF-RIPK1-FADD-caspase-8 pathway, which leads to IL-1β secretion independently of inflammasome formation, K+ efflux, and pyroptosis in a process known as the alternative NLRP3 inflammasome pathway (205). ZBP1 may induce inflammasome activation, which involves a noncatalytic function of caspase-6 (206). ZBP1 activation may thus result in apoptosis, necroptosis, or pyroptosis, but how a cell mechanistically chooses its fate is not understood (207).
Recent studies indicate that proteins that mediate apoptosis, necroptosis, and/or pyroptosis may also be involved in the regulation of gene expression. Ligation of the death receptor TRAIL-R leads to the engagement of gene expression programs, which depends on the formation of a complex consisting of FADD, caspase-8, and RIPK1, which was dubbed the FADDosome (208–210). Similarly, engagement of the death receptor CD95 (also Fas-R) induces the FADDosome and gene expression (195). Thus, death receptor engagement may lead to DISC formation and apoptosis or FADDosome formation and gene expression that may lead to inflammation (194, 195).
Thus, regulated cell death pathways are not linear but interconnected, complex, and plastic signaling cascades (FIGURE 6). The plasticity between the pathways allows cells to overcome blockage posed by pathogens and as such has important implications for the control of infections and survival of the host organism.
7. PATHOGEN STRATEGIES TO INHIBIT CELL DEATH
Preventing host cells from dying secures the ability of pathogens to proliferate, spread, and survive, and pathogens have evolved a plethora of ways to dysregulate pyroptotic (FIGURE 7; TABLES 1 and 2) and apoptotic and necroptotic (FIGURE 8; TABLES 3 and 4) cell death programs in their hosts, varying from the modulation of gene expression profiles to direct interaction with cellular death-mediating proteins.
FIGURE 7.
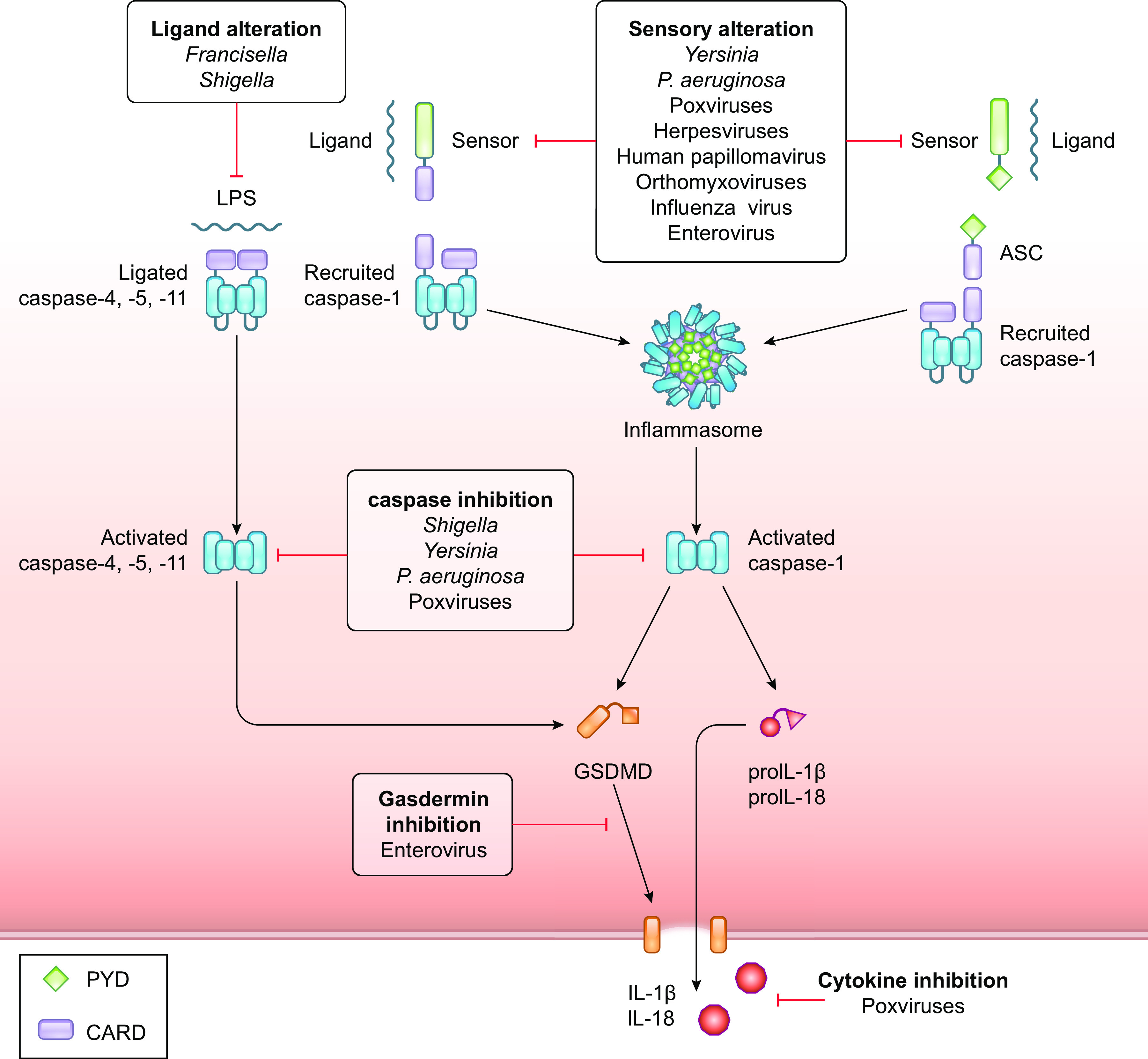
Pathogenic strategies to inhibit pyroptosis. Pathogens block inflammasomes at multiple levels. Pathogens alter their pathogen-associated molecular patterns (PAMPs) or modulate cellular ligands involved in sensor activation. Pathogens inhibit the activation of inflammasome-inducing sensors and the activity of caspases-4, -5, and -11 and caspase-1 to block pyroptosis and the release of IL-1β and IL-18. In the case that IL-1β and IL-18 are released, pathogens block the ability of these proteins to bind their receptors. Specific strategies employed by bacteria and viruses to inhibit pyroptosis are presented in TABLES 1 and 2, respectively. CARD, caspase-recruitment domain; GSDMD, gasdermin D; PYD, pyrin domain.
Table 1.
Bacterial inhibition of pyroptosis
| Bacterial Species | Protein | Mode of Action | Reference |
|---|---|---|---|
| Enteropathogenic Escherichia coli | NleF | Inhibits caspase-4 activation | (293) |
| Francisella spp. | LPS | Inhibits caspase-4 to recognize LPS | (66) |
| Pseudomonas aeruginosa | C120HSL | Inhibits NLRC4 and NLRP3 | (222) |
| ExoS | Inhibits caspase-1 activation | (223) | |
| ExoU | Inhibits caspase-1 activation | (223) | |
| Pyocyanin | Inhibits NLRC4 and NLRP3 | (222) | |
| Shigella spp. | LPS | Inhibits caspase-4 to recognize LPS | (213) |
| Osp3C | Prevents caspase-4 dimerization | (215) | |
| Yersinia spp. | YopK | Blocks NLRP3 inflammasome activation | (216) |
| YopK | Blocks NLRC4 inflammasome activation | (216) | |
| YopM | Activates PRK1/2 to keep Pyrin inactive | (219) | |
| YopM | Blocks caspase-1 | (221) |
Table 2.
Viral inhibition of pyroptosis
| Virus Family | Virus | Protein | Mode of Action | Reference |
|---|---|---|---|---|
| Herpesviridae | HHV8 (KSHV) | Orf63 | Viral NLRP1 homolog, prevents NLRP1 oligomerization | (229) |
| Hsv-1 | Unknown | Mediates degradation of the IFI16 inflammasome | (375) | |
| Unknown | Inhibits the NLRP3 inflammasome | (375) | ||
| Influenzaviridae | Influenza A virus | NS1 | Inhibits the NLRP3 inflammasome | (232) |
| Papillomaviridae | HPV | E7 | Mediates degradation of the IFI16 inflammasome | (228) |
| Paramyxovirus | Measles virus | V protein | Inhibits the NLRP3 inflammasome | (230) |
| Picornaviridae | Enterovirus 71 | 2A | Cleaves NLRP3 | (234) |
| 3C | Cleaves NLRP3 | (234) | ||
| 3C | Cleaves GSDMD | (233) | ||
| Poxviridae | Cowpox | CrmA | Blocks caspase-1 activity | (224) |
| B15R | Viral IL-1β binding protein | (376) | ||
| vIL-18BP | Viral IL-18 binding protein | (377) | ||
| MCV | MC54L | Viral IL-18 binding protein | (377) | |
| Myxoma virus | M013L | Blocks inflammasomes by binding to PYD | (227) | |
| Serp2 | Blocks caspase-1 activity | (378) | ||
| Shope fibroma virus | S013L | Blocks inflammasomes by binding to PYD | (226) | |
| VACV | B13R | Blocks caspase-1 activity | (379) | |
| B13R | Blocks caspase-4/5 activity | (282) | ||
| B15R | Viral IL-1β binding protein | (380) | ||
| vIL-18BP | Viral IL-18 binding protein | (377) |
GSDMD, gasdermin D; HHV, human herpesvirus; HPV, human papillomavirus; KSHV, Kaposi’s sarcoma-associated herpesvirus; MCV, Molluscum contagiosum virus; PYD, pyrin domain; VACV, vaccinia virus.
FIGURE 8.
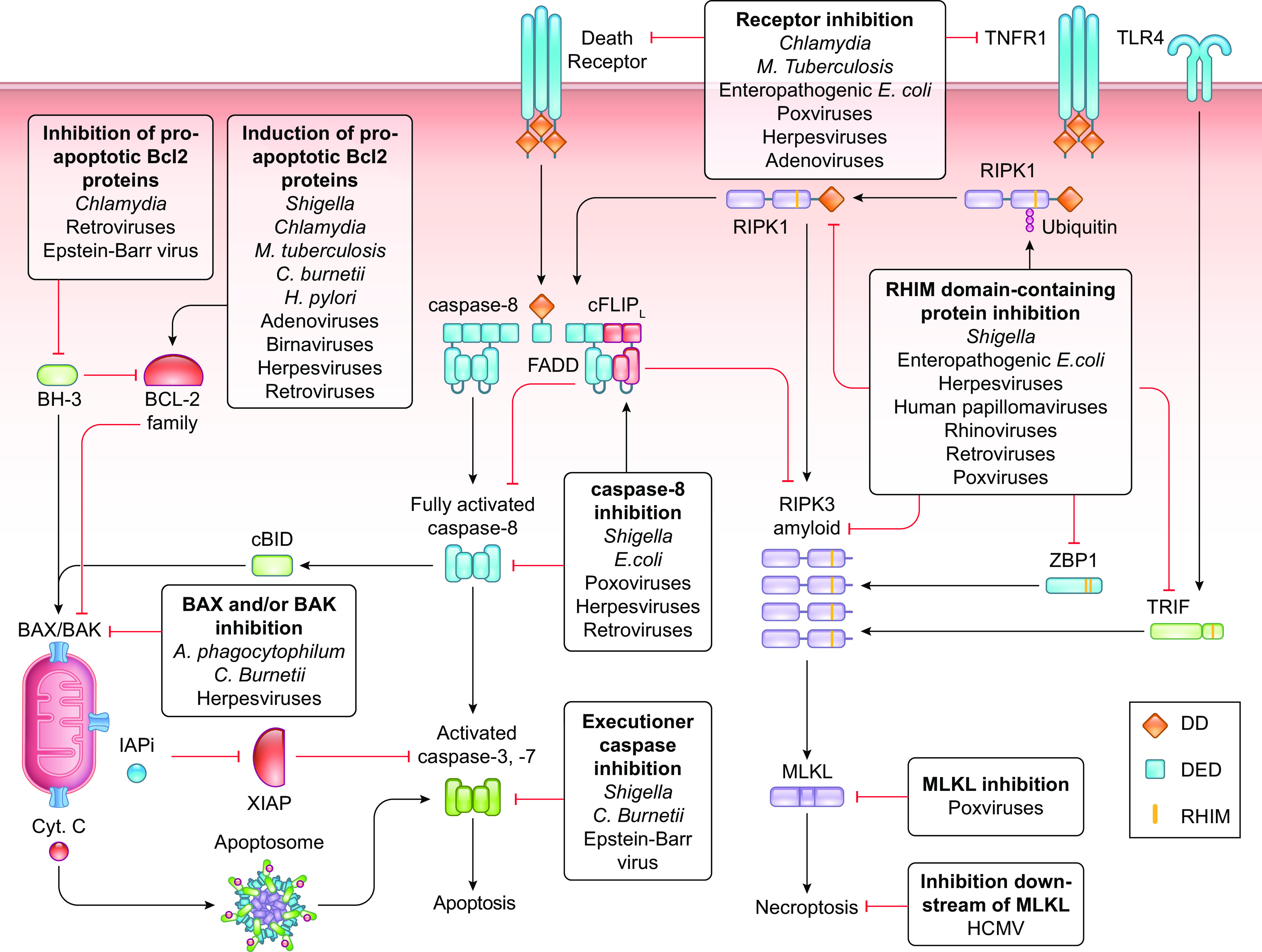
Pathogenic strategies to inhibit apoptosis and necroptosis. Pathogens block apoptosis and necroptosis at multiple levels. Pathogens block the caspase-9 pathway of apoptosis by modulating BCL-2 family proteins to restrict mitochondrial outer membrane permeabilization (MOMP) or by inhibiting the activation of executioner caspases. Pathogens disrupt the ability of receptors to sense their cognate ligands and inhibit the caspase-8 pathway of apoptosis by interfering with the functioning of caspase-8. Pathogens impair necroptosis by inhibiting the interactions between RIP homotypic interaction motif (RHIM) domain-containing proteins and the activation of MLKL. Specific strategies employed by bacteria and viruses to inhibit pyroptosis are presented in TABLES 3 and 4, respectively. Cyt. C, cytochrome c; DD, death domain; DED, death effector domain.
Table 3.
Bacterial inhibition of apoptosis and necroptosis
| Bacterial Species | Protein | Mode of Action | Reference |
|---|---|---|---|
| Anaplasma phagocytophilum | Ats-1 | Inhibits BAX activation | (250) |
| Chlamydia spp. | Cdu1 | Blocks degradation of MCL-1 | (244) |
| Unknown | Induces expression of MCL-1 | (245) | |
| Unknown | Degrades proapoptotic BH-3-only proteins | (248) | |
| Unknown | Sequesters BAD | (249) | |
| Unknown | Blocks the internalization of TNF-TNFR complexes | (268) | |
| Coxiella burnetii | Unknown | Induces expression of MCL-1 | (243) |
| AnkG | Blocks MOMP | (252) | |
| CaeA | Blocks caspase-7 activation | (253) | |
| CaeB | Blocks MOMP | (251) | |
| Enteropathogenic Escherichia coli | EspL | Cleaves RHIM domain-containing proteins | (296) |
| NleB1 | Blocks FADD recruitment | (278) | |
| NleF | Inhibits caspase-9 activation | (292) | |
| NleF | Inhibits caspase-8 activation | (292) | |
| Helicobacter pylori | Unknown | Induces expression of MCL-1 | (246) |
| Unknown | Induces expression of BCL-2 | (247) | |
| Unknown | Decreases BAX | (247) | |
| Mycobacterium tuberculosis | Unknown | Induces expression of MCL-1 | (242) |
| Unknown | Promotes secretion of soluble TNFR2 | (269) | |
| Shigella spp. | IpgD | Activates prosurvival signaling | (238) |
| IpgD | Induces expression of BCL-2 | (237) | |
| VirA | Induces expression of BCL-2 | (237) | |
| OspC1 | Inhibits caspase-8 activation | (291) | |
| OspD3 | Degrades RIPK1 and RIPK3 | (291) |
MOMP, mitochondrial outer membrane permeabilization; RHIM, RIP homotypic interaction motif.
Table 4.
Viral inhibition of apoptosis and necroptosis
| Virus Family | Virus | Protein | Mode of Action | Reference |
|---|---|---|---|---|
| Adenoviridae | Adenovirus | E1B 19K | Viral BCL-2 homolog | (381) |
| E3 | Downmodulates DRs | (275) | ||
| Birnaviridae | IPNV | VP5 | Viral BCL-2 homolog | (382) |
| Flaviviridae | HCV | NS5A | Viral BCL-2 homolog | (383) |
| Herpesviridae | EBV | BALF1 | Viral BCL-2 homolog | (384) |
| BHRF1 | Viral BCL-2 homolog | (385) | ||
| EBNA3A and EBNA3C | Block expression of the proapoptotic BCL-2 protein BIM | (386) | ||
| BART miRNAs | Block caspase-3 expression | (266) | ||
| LMP1 | Blocks RIPK1 and RIPK3 | (303) | ||
| γHV-68 | M11 | Viral BCL-2 homolog | (387) | |
| vMAP | Blocks BAX activation | (388) | ||
| HCMV | vMIA | Blocks BAX activation | (259) | |
| UL36 (aka vICA) | Blocks caspase-8 activation | (286) | ||
| UL45 | Blocks necroptosis downstream of MLKL activation | (304) | ||
| UL144 orf | Viral TNFR homolog | (274) | ||
| HHV8 (KSHV) | KSBcl-2 | Viral BCL-2 homolog | (389) | |
| K7 | Inhibits caspase-3 activation | (390) | ||
| vFLIP | Blocks caspase-8 oligomerization | (284) | ||
| HSV-1 | U(S)3 | Blocks BAD | (257) | |
| LAT | Stabilizes AKT to inactivate proapoptotic BCL-2 proteins | (391) | ||
| ICP6 | Blocks necroptosis | (302) | ||
| HSV-2 | ICP10 | Blocks caspase-8 activity | (392) | |
| ICP10 | Blocks necroptosis | (302) | ||
| HVS | Orf16 | Viral BCL-2 homolog | (393) | |
| MCMV | M38.5 | Blocks BAX activation | (260) | |
| M41.1 (aka vBIO) | Blocks BAK activation | (262) | ||
| M36 (aka vICA) | Blocks caspase-8 activation | (286) | ||
| M45 (aka vIRA) | Inhibits interaction of RHIM domain-containing proteins | (300) | ||
| MCV | MC159 (aka vFLIPs) | Blocks caspase-8 oligomerization | (394) | |
| Papillomaviridae | HPV16 and 18 | E6 | Inhibits BAK | (395) |
| E6 | Mediates degradation of caspase-8 | (288) | ||
| Unknown | Downregulates RIPK3 expression | (297) | ||
| Picornaviridae | Rhinovirus | 3C | Cleaves RIPK1 | (396) |
| Poxviridae | ASFV | A179L | Viral BCL-2 homolog | (397) |
| A224L | Viral IAP | (398) | ||
| Cowpox | CrmA | Blocks caspase-8 activity, not cFLIP activity | (282) | |
| CrmB | Viral TNFR homolog | (270) | ||
| FPV | FPV039 | Viral BCL-2 homolog | (399) | |
| Myxoma virus | M11L | Inhibits BAK | (400) | |
| M11L | Inhibits BAX | (401) | ||
| M-T2 | Viral anti-TNF protein | (272) | ||
| Serp2 | Blocks caspase-8 activity | (402) | ||
| PPVO | Orfv125 | Viral BCL-2 homolog | (403) | |
| Shope fibroma virus | T2 orf | Viral TNFR homolog | (273) | |
| Smallpox | CrmE | Viral TNFR homolog | (271) | |
| VACV | N1L | Viral BCL-2 homolog | (404) | |
| F1L | Inhibits BAK | (405) | ||
| F1L | Inhibits BAX activation | (406) | ||
| F1L | Inhibits caspase-9 | (407) | ||
| CrmE | Viral TNFR homolog | (271) | ||
| B13R | Blocks caspase-8 activity | (282) | ||
| E3 | Blocks ZBP1-RIPK3 interaction | (280) | ||
| Retroviridae | HIV | Nef | Blocks the proapoptotic BCL-2 protein BAD | (258) |
| HTLV-1 | TAX | Enhances expression of cFLIP and cIAP2 expression | (408) | |
| TAX | Enhances BCL-XL expression | (256) | ||
| TAX | Reduces BAX expression | (256) | ||
| TAX | Enhances expression of cIAP2 | (408) |
ASFV, African swine fever virus; DR, death receptor; EBV, Epstein–Barr virus; FPV, fowlpox virus; HCMV, human cytomegalovirus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HPV, human papillomavirus; HSV, herpes simplex virus; HTLV-1, human T cell leukemia virus type 1; HVS, herpesvirus saimiri; IAP, inhibitor of apoptosis; IPNV, infectious pancreatic necrosis virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; MCMV, murine cytomegalovirus; MCV, Molluscum contagiosum virus; PPVO, parapoxvirus ORF virus; RHIM, RIP homotypic interaction motif; VACV, vaccinia virus; γHV-68, gamma-herpesvirus 68.
Many viral species, especially large DNA viruses, dedicate sizable parts of their genomes to the inhibition of cell death pathways. Six bacterial species, Burkholderia spp., enteroinvasive Escherichia coli, Francisella spp., Listeria spp., Rickettsia spp., and Shigella spp., have been identified to proliferate within the cytosol (211), and other bacteria invade host cells but reside and multiply within vacuoles or other structures (212) and deliver their effector proteins that regulate host cellular mechanisms delivered via a T3SS or other secretion systems into the host’s cytosol. Regardless of their cellular localization, the intricate cross talk between RCD pathways means that pathogens must simultaneously block multiple RCD pathways to effectively inhibit cell death.
7.1. Pathogen Blockade of Pyroptosis
One of the first defenses against intracellular bacteria is the activation of the noncanonical inflammasome upon the detection of LPS. To avoid detection by caspase-4, -5, or -11, Francisella and Shigella modulate their LPS composition by decreasing the degree of acetylation of the lipid A moiety (66, 213). Interestingly, hypoacetylation of Francisella LPS abrogates detection by caspase-11, whereas caspase-4 remains able to detect hypoacetylated LPS, albeit less efficiently (214). This indicates that human cells are better equipped to control cytosolic bacteria than murine cells and suggests that intracellular bacteria that modulate their LPS composition either must do so in such a way that can no longer lead to recognition by caspase-4 or must have evolved additional mechanisms to block the noncanonical inflammasome in human cells. Shigella does so by preventing the LPS-induced dimerization of caspase-4, but not caspase-1 or caspase-11, by its T3SS-delivered Osp3C effector protein (215).
Microbes modulate various aspects of inflammasome signaling (FIGURE 7; TABLE 1). The virulence factors of pathogenic Yersinia species, named Yersinia outer protein (Yop) proteins, are delivered via the T3SS, which can be recognized by NAIPs to activate the NLRC4 inflammasome. YopK of Yersinia pseudotuberculosis prevents the recognition of the T3SS by NAIPs and also blocks the activation of the NLRP3 inflammasome (216). Yersinia enterocolitica YopE and YopT proteins inactivate Rho GTPases and thereby induce the Pyrin inflammasome (217, 218). YopM, however, recruits and activates PRK1/2 (219, 220), effectively taking over one of the functions of Rho GTPases to keep Pyrin inactive. Furthermore, YopM interacts with caspase-1 to restrict IL-1β maturation and pyroptosis (221).
Proteases secreted by Pseudomonas aeruginosa into the extracellular space degrade host defense proteins, including complement components and proinflammatory cytokines, and degrade extracellular inflammasome components and flagellin, which would trigger the NLRC4 inflammasome upon cellular uptake. The P. aeruginosa virulence factors pyocyanin and C120HSL prevent inflammasome activation by directly inhibiting NLRC4 and NLRP3 (222). The effector proteins ExoU and ExoS inhibit caspase-1 activation and thereby prevent the processing of pro-IL-1β and pro-IL-18 (223), although precise mechanisms are unclear.
Viruses also disrupt the functioning of multiple inflammasome components (FIGURE 7; TABLE 2). Poxviral serpins block the catalytic site of caspase-1, thereby blocking pyroptosis and the release of IL-1β and IL-18 (224). Poxviruses also neutralize bioactive IL-1β and IL-18 by expressing scavenger receptors and soluble proteins that bind these cytokines, preventing them from activating their cognate receptors (225). Poxviruses Myxoma virus (M013L) and Shope fibroma virus (S013L) express pyrin-only proteins (POPs) that resemble human POPs and inhibit inflammasome activation by binding the PYDs of ASC and inflammasome-inducing receptors, thereby disrupting the ability of these proteins to effectively form an inflammasome and activate caspase-1 (226, 227). Human papillomaviruses dampen pyroptotic cell death responses by enhancing TRIM21-mediated proteasomal degradation of the IFI16 inflammasome (228). Kaposi’s sarcoma-associated herpesvirus (KSHV) inhibits the formation of NLRP1 and NLRP3 inflammasomes by expressing a viral NLRP1 homolog, Orf63, that lacks the NH2-terminal PYD and COOH-terminal CARD of human NLRP1 but can bind to NLRP1 and NLRP3 (229). The Measles virus V protein (230) and NS1 protein of Influenza virus (231, 232) also interact with NLRP3, whereas the viral proteases 2A and 3C of Enterovirus 71 inactivate NLRP3 and GSDMD by cleavage (233–235).
7.2. Pathogen Blockade of the Caspase-9 Pathway of Apoptosis
The presence of pathogens in a cell disturbs cellular homeostasis, and the associated stress can trigger the caspase-9 pathway of apoptosis. Pathogens therefore developed several strategies to block the caspase-9 pathway of apoptosis (FIGURE 8; TABLES 3 and 4). Shigella infection, for instance, induces necrotic death and provokes cellular genotoxic stress, which induces p53-mediated apoptosis. Shigella counteracts this by activating a prosurvival program that induces expression of BCL-2 (236). Moreover, Shigella mediates the degradation of p53 by its T3SS-delivered virulence factor VirA, which induces calpain proteases to cleave p53, and its virulence factor IpgD that induces MDM2 to ubiquitylate p53 for proteasomal degradation (237). IpgD also generates phosphatidylinositol 5-phosphate (PI5P) to activate additional prosurvival signaling (238). VirA-activated calpain proteases cleave and activate BID, leading to mitochondrial permeabilization and the release of proapoptotic factors including SMAC to block the inflammatory effects of XIAP (236, 239). However, mitochondrial permeabilization does not lead to apoptosis, since the O-antigen moiety of LPS from Shigella inhibits the activity of caspases-3 and -7 by directly binding to these executioner caspases (240, 241).
Many bacterial species regulate the expression and degradation of cellular pro- and antiapoptotic Bcl-2 proteins to inhibit the activation of MOMP. Chlamydia, Mycobacterium tuberculosis, and Coxiella burnetii induce the transcription and block the degradation of the antiapoptotic Bcl-2 protein MCL-1 (242–245), whereas Helicobacter pylori upregulates MCL-1 (246) and BCL-2 and decreases BAX (247). Chlamydia degrades proapoptotic BH-3-only proteins, including Bad, Bim, and PUMA (248), and recruits Bad to the inclusion vacuole, thereby sequestering Bad from mitochondria (249). The Anaplasma phagocytophilum T4SS effector Ats-1 enters mitochondria, where it inhibits redistribution of BAX, thereby inhibiting MOMP and cytochrome c release (250). Coxiella burnetii employs three T4SS effector proteins, AnkG, CaeA, and CaeB, to inhibit apoptosis. AnkG and CaeB act at the mitochondrial level (251, 252), whereas CaeA prevents caspase-7 activation (253).
A similar approach to block MOMP is employed by viruses belonging to the families of Adenoviridae, Birnaviridae, Herpesviridae, and Poxviridae. These viruses express viral BCL-2 (vBcl2) proteins that show structural similarities with antiapoptotic BCL-2 proteins and block MOMP by inhibiting the activation of BAK and BAX (254). vBcl2 proteins from Herpesviridae and Poxviridae contain transmembrane domains through which they integrate into the mitochondrial membrane, where they interact with cellular pro- and antiapoptotic Bcl2 proteins to modulate apoptosis (255). Among the retroviruses, human T cell leukemia virus type 1 (HTLV-1) promotes BCL-XL expression and represses BAX expression via its TAX protein (256), whereas human immunodeficiency virus (HIV) Nef and HSV-1 U(S)3 proteins inhibit the proapoptotic BH-3-only protein Bad by mediating its phosphorylation (257, 258). Human cytomegalovirus (HCMV) vMIA and murine cytomegalovirus (MCMV) m38.5 inhibit the proapoptotic activity of BAX, but not BAK, by sequestering BAX at mitochondria (259–261). MCMV also inhibits BAK-mediated MOMP by its vIBO protein (262). Epstein–Barr virus (EBV) blocks apoptosis by sequestering BH-3-only proteins by its viral BCL-2 homolog protein BHRF-1 (263–265) and expresses viral miRNAs that repress the transcriptional levels of caspase-3 (266).
Apoptosis in insect cell does not rely on MOMP but rather on expression of IAP antagonist proteins (detailed below). Therefore, viruses that infect insect cells would not benefit from expressing vBcl2 proteins. The insect poxvirus Amsacta moorei Entomopoxvirus (AmEPV) does not contain the apoptosis suppressor genes expressed by vertebrate poxviruses but encodes a viral IAP (vIAP), AMV-IAP, that binds and impairs two IAP antagonist proteins in D. melanogaster cells (267). AmEPV AMV-IAP is not present in vertebrate poxviruses, even though AMV-IAPs can inhibit the enzymatic activities of human caspases-3 and -9 in vitro (267). In fact, with the exception of African swine fever virus, vIAPs have not been identified in mammalian viruses, indicating that viral mimicry of IAPs is not an evolutionarily selected strategy to block apoptosis in mammalian cells.
7.3. Pathogen Blockade of Receptor-Induced Apoptosis and Necroptosis
Pathogens employ many strategies to inhibit apoptosis and necroptosis (FIGURE 8; TABLES 3 and 4). One such strategy is to hamper the activation of death-inducing receptors. Chlamydia inhibits DR-mediated cell death by blocking the internalization of TNF-TNFR complexes (268), and M. tuberculosis promotes secretion of soluble TNFR2 to prevent TNF from binding to cellular receptors (269). The cowpox (270), smallpox (271), myxoma (272), Shope fibroma (273), and VACV (271) poxviruses and the herpesvirus HCMV (274) express viral TNFR orthologs that neutralize TNF-α, whereas adenoviruses promote the internalization and lysosomal degradation of Fas, TNFR, and TRAIL-R1 (275) and the herpesviruses HCMV and MCMV suppress the expression and function of Fas (276). EPEC NleB1 blocks DR-induced apoptosis by adding an N-acetylglucosamine (GlcNac) to death domain-containing proteins, preferentially to Arg117 in the death domain of FADD, to prevent DISC formation (277–279). The E3 protein of Vaccinia virus (VACV) inhibits ZBP1-RIPK3 to prevent cell death (280).
A frequently used approach to block the caspase-8 pathway of apoptosis is through inhibition of the proteolytic activity or the activation of caspase-8 at the DISC or in the ripoptosome. Serpins expressed by poxviruses, e.g., VACV B13R and Cowpox virus crmA, bind and block the catalytic sites of caspase-8 and caspase-1 (224, 281, 282), whereas the poxvirus Molluscum contagiosum virus (MCV) (283, 284) and several gamma-herpesviruses (284, 285) encode a viral ortholog of cFLIPs (vFLIP) that blocks caspase-8 oligomerization and apoptosis. HCMV and MCMV express the viral inhibitor of caspase-8-induced apoptosis (vICA) (MCMV M36, HCMV UL36) (286), and HSV-1 and HSV-2 contain ICP-6 and ICP-10 (287), respectively, to bind and inhibit caspase-8. Human papillomavirus type 16 (HPV16) E6 binds to DEDs of caspase-8 and inhibits or activates caspase-8, depending on the E6 splice variant (288, 289). The TAX protein of HTLV-1 induces the upregulation of cFLIP expression by enhancing NF-κB signaling (290). Bacteria also block caspase activation. The Shigella effector protein OspC1 inhibits caspase-8 activation (291), whereas EPEC NleF inhibits the activity of caspases-4, -8, and -9 (278, 292, 293).
Although blocking the activity of caspase-8 is an efficient strategy to block the caspase-8 pathway of apoptosis, this also disrupts the ability of caspase-8 to inhibit RIPK1 and RIPK3, and consequently may result in the induction of necroptosis. The necroptotic pathway was therefore originally believed to have evolved as a “back-up” cell death mechanism for when the extrinsic apoptosis pathway is blocked by pathogenic caspase-8 inhibitors or vFLIPs. It is therefore interesting that the poxvirus crmA is more effective in inhibiting caspase-8 homodimer activity than caspase-8-cFLIPL heterodimer activity (294), allowing it to block caspase-8-mediated apoptosis without sensitizing for necroptosis (295).
Several pathogens express proteins that specifically target necroptosis by inhibiting RIPK1, RIPK3, MLKL, or effects downstream of MLKL (FIGURE 8; TABLES 3 and 4). The EPEC cysteine protease EspL degrades all cellular and viral RHIM domain-containing proteins (296), and Shigella employs its OspD3 protease to degrade RIPK1 and RIPK3 to block necroptosis (291). Human papillomaviruses downregulate the expression of ripk3 (297), whereas Rhinovirus 3C cleaves RIPK1 (298) and HTLV-1 TAX induces the expression of cIAP2 by enhancing NF-κB signaling (299) to dampen necroptotic cell death responses. Herpesviruses express functional RHIM domain-containing proteins to inhibit necroptosis. Alpha- and gamma-herpesviruses encode an enzymatically active large subunit (R1) of the ribonucleotide reductase (RNR) complex that functions in viral DNA synthesis, whereas beta-herpesviruses encode an enzymatically inactive R1 homolog. These R1 proteins contain RHIM domains through which they suppress signal transduction to RIPK3. Indeed, viral inhibitor of RIP activation (vIRA) proteins of MCMV (M45) can interact with TRIF, ZBP1, RIPK1, and RIPK3 (300, 301) to impair cell death signaling. Where MCMV expresses two proteins to inhibit caspase-8 and necroptosis, HSV-1 ICP-6 and HSV-2 ICP-10 can simultaneously inhibit both caspase-8 and the RIP kinases through direct interaction (302). The latent membrane protein 1 (LMP1) of Epstein–Barr virus (EBV) inhibits necroptosis by interacting with RIPK1 and RIPK3 through RHIM domain-independent interactions (303). Unlike its MCMV M45 counterpart, HCMV UL45 does not contain a RHIM domain and is not able to block RIPK3-mediated activation of MLKL. However, HCMV UL45 blocks necroptosis by acting downstream of MLKL activation, although it is currently unknown how this is mechanistically achieved (304).
Poxviruses have several ways to inhibit necroptosis, depending on the viral genus. Many orthopoxviruses, including Cowpox virus, express viral inducer of RIPK3 degradation (vIRD) proteins that bind to and promote K48-linked ubiquitylation of RIPK3, resulting in proteasomal degradation of RIPK3 and repression of necroptosis (305). Other poxviruses express viral MLKL-like proteins (vMLKL), which lack the NH2-terminal 4HB domain and the brace region but maintain the pseudokinase domain (306). vMLKLs of BeAN 58058 poxvirus (BAV), originally isolated from an Oryzomys rodent (307), and mouse-infecting Cotia poxvirus (COTV) block necroptosis, but not apoptosis, in human and mouse cells by sequestering RIPK3. Swine poxvirus (SPV) vMLKL, however, does not block necroptosis in human and mouse cells, indicating that vMLKLs have evolved to target necroptosis in specific hosts (306). Interestingly, avipoxviruses, which infect birds that do not harbor ripk3, express vMLKLs, whereas poxviruses that infect necroptosis-deficient mammals lacking RIPK3 or MLKL have lost the virulence factors that inhibit necroptosis (305, 306, 308), exemplifying how well poxviruses are adapted to their specific host. Such findings underscore the dynamic interactions between viral infection and the necroptosis machinery.
7.4. Is Necroptosis a Back-Up or Stand-Alone Cell Death Modality?
Studies using influenza A virus (IAV) suggest that necroptosis can be viewed as a stand-alone cell death mechanism to limit infections (207). IAV activates ZBP1-dependent RIPK3-mediated cell death, which can be necroptosis, apoptosis (207, 309–311), or pyroptosis (79, 312) and is dependent on the presence but not the kinase activity of RIPK3 (207, 309, 311). Interestingly, infected individual cells each seem to undergo only one type of cell death—that is, cells ultimately die by apoptosis or necroptosis. It is unknown how a cell mechanistically decides which type of cell death it will engage (207). Deletion of zbp1 or ripk3 renders cells resistant to IAV-induced death, and, as a result, zbp1- and ripk3-deficient mice cannot control viral replication and succumb to the infection (309–311), although this may depend on the viral dose (313). Abrogation of necroptosis by pharmacological inhibition of RIPK3 or mlkl deficiency does not block IAV-induced cell death, and mlkl-deficient mice control and survive IAV infection, since ZBP1 now engages apoptosis. Pharmacological or genetic inhibition of both caspase-8-mediated apoptosis and necroptosis renders cells unable to die upon IAV infection, resulting in apoptosis-necroptosis double-deficient mice that succumb to the infection (207, 309, 311). These data suggest that the apoptotic and necroptotic pathways can independently control IAV infection, indicating that necroptosis is a stand-alone cell death modality and should not be seen as “just” a back-up mechanism, engaged only when apoptosis fails.
8. EVOLUTIONARY ASPECTS OF REGULATED CELL DEATH
Host-pathogen interactions are among the major driving forces for organismal evolution. Hosts must adapt to survive infection with an invading pathogen, and, in turn, the pathogen must adapt to avoid destruction by the host’s immune defenses. The resulting arms race selects for increasingly complex defense mechanisms in the host, while the pathogen is selected to evolve equally complex evasion strategies. In evolutionary biology, this is known as the Red Queen effect (314), which states that an organism must continuously adapt and evolve to maintain its relative fitness among the systems with which it is coevolving.
In accordance with the Red Queen effect, pathogens that coevolved with vertebrates have gained a multitude of strategies to avoid detection or interfere with signaling leading to a cell’s demise, effectively rendering pathways of RCD incapable to control the infection. It therefore may seem curious that the components of regulated cell death remain present in the genomes of evolving vertebrates. It has been speculated that inflammasomes are not maintained for defense against vertebrate-adapted pathogens but to defend against ubiquitous opportunistic pathogens that reside in the environment (315, 316). Such microbes are not dependent on vertebrate hosts and therefore have not evolved strategies to inhibit pyroptotic cell death. Infection with these environmental pathogens could be harmful to a vertebrate host, but by retaining inflammasomes hosts can cope with ubiquitous opportunistic pathogens (315, 316). This hypothesis can be seen as a corollary to the Red Queen effect and was dubbed the Red Pawn effect (315). It should be noted that not all host-microbe interactions result in a Red Queen effect; some scenarios invoke a cooperative defense strategy that can result in commensalism (317, 318). Although much of our thinking, herein, relies on antagonistic coevolution as a driving force, it is likely that cooperative defense strategies and Red Pawn effects also played a major role in the evolution and maintenance of cell death mechanisms.
In light of the Red Queen effect, it is not surprising that the activation and execution mechanisms of RCD have evolved to be elaborate and tightly controlled. At first sight RCD may only seem advantageous to multicellular organisms. However, unicellular organisms also benefit from regulated cell death to control infection within a colony. Invading intracellular pathogens use the host’s resources to replicate and spread, often killing the host cell in the process. The individual cell is thus fated to die. By undergoing a more rapid death, the altruistic individual cell can limit the spread of the pathogen within its identical clone mates, effectively “saving” its genome. In a process called kin selection (319), colonies in which kin evolved to undergo rapid, regulated cell death upon pathogen recognition would thus be less susceptible to eradication by that pathogen. As such, RCD forms a first line of immune defense that protects the host organism and its kin from extinction. From this perspective, regulated cell death may have evolved as early immune defense mechanisms to allow organismal survival at the single cell level (33, 320).
Many studies have investigated the evolutionary origins of the components involved in regulated cell death signaling as present in humans today. The components of the regulated cell death signaling machinery have highly diversified over evolutionary time and continue to evolve, arguably as a consequence of the Red Queen effect as pathogens similarly evolve strategies to subvert the cell death pathways. Gene ontology studies have identified homologs of mammalian genes involved in regulated cell death in many animal phyla. However, it has to be noted that sequence similarity does not predict conserved functionality and cellular and biochemical approaches are required to understand whether conserved genes also have conserved functions. From this perspective, apoptosis and pyroptosis are functional in basal metazoans and therefore are ancient forms of regulated cell death, whereas necroptosis appears to be evolutionarily “younger,” arising with the vertebrates. Together, these studies have led to surprising insights into the origins, regulation, and conservation of the apoptotic, necroptotic, and pyroptotic cell death pathways throughout the animal kingdom.
8.1. Pyroptosis in Animal Evolution
Pyroptosis in humans, as mediated by the inflammatory caspases-1, -4, and -5 and mainly executed by GSDMD, is a relatively recent evolutionary innovation, since gsdmd, like gsdmb and gsdmc, is only found in mammals (321). GSDMA homologs are present in many vertebrates, but no studies have been performed to confirm the functional conservation of gsdma in nonmammalian model systems. Pjvk and gsdme are evolutionarily ancient, and homologs have been identified in cnidaria, arthropods, and placozoans in addition to vertebrates (322). Phylogenetically, pjvk and gsdme from invertebrate species form distinct clusters. Moreover, both clusters are distinctly separated from clusters of vertebrate gsdme, gsdmb, and gsdmd. It is thus unclear whether all gasdermins are derived from gsdme or if the vertebrate GSDM family and invertebrate gsdme arose from a common (unidentified) ancestor. The possibility that the other gasdermins evolved from pejvakin seems unlikely because of a lack of sequence homology. Although the structure of GSDMA3 resembles structures formed by aerolysins (123), cytolytic pore-forming toxins secreted by the Gram-negative bacterium Aeromonas hydrophila, gasdermins and aerolysins are likely not related, since the proteins do not share any sequence homology (123).
Although the evolutionary relation between gsdme and the other gasdermins is unclear, GSDME-mediated cell death, and thus pyroptosis, is functional in Cnidaria. Cnidarian GSDME is able to mediate pyroptosis in coral species, including Orbicella faveolata (322). Similar to human caspase-3, coral caspase-3 is involved in apoptosis, and apoptotic and necrotic death have been observed to occur in parallel in corals (323). Coral GSDME can be cleaved and activated by both human and coral caspase-3 (322), indicating that the mechanisms of GSDME activation are evolutionarily conserved. In the coral P. damicornis, GSDME-mediated pyroptosis appears to control infection with the coral pathogen V. coralliilyticus. Infection with this pathogen induces caspase-dependent GSDME cleavage and tissue necrosis in coral polyps (322). Although further studies are required to elucidate the exact mechanisms of coral caspase-3-mediated coral GSDME activation, cnidarian and mammalian GSDME-mediated pyroptotic death are remarkably similar, suggesting that GSDME-mediated pyroptosis is an evolutionarily ancient form of regulated cell death.
Zebrafish (Dania rerio) contain all components of the pyroptotic machinery, including many NLRs, four inflammatory caspases, ASC, and three gasdermin genes (pjvk, gsdmea, and gsdmeb), and both the noncanonical and canonical inflammasomes are functional in these vertebrates (324). The inflammatory caspases caspy and caspy2 consist of an NH2-terminal PYD followed by the catalytic domain, which is unlike mammalian inflammatory caspases that contain NH2-terminal CARDs (FIGURE 1). The PYD of caspy2 is a receptor for cytosolic LPS and activates the noncanonical inflammasome upon ligation, similar to human caspases-4 and -5 (325). The PYD of caspy, but not of caspy2, directly interacts with the zebrafish ortholog of ASC (43, 326). Similar to mammalian NLRP1 and NLRP3, zebrafish DrNLRP1 and DrNLRP3 induce the formation of a canonical, ASC-dependent inflammasome leading to caspy and caspy2 recruitment and activation, zebrafish IL-1β processing, and pyroptosis (327, 328). DrNLRP3 can also recruit caspy2 without ASC and caspy, leading to caspy2 activity, but not autoproteolytic processing, which suffices to cleave GSDMEa/b and execute pyroptosis (328). Thus, zebrafish caspy and caspy2 are functional orthologs of human caspase-1 and caspase-5, respectively. Moreover, these studies in teleost fish signify that NLRP3 inflammasomes are conserved in vertebrates.
Although it remains unclear why the gasdermin family diversified only later in evolution, the functionality of GSDME-mediated pyroptosis in zebrafish and cnidaria indicates that pyroptosis can be seen as an ancient form of regulated cell death in response to infection. Furthermore, murine caspase-11 can be involved in canonical inflammasome-mediated pyroptosis, since, similar to the ability of caspy2 to be recruited to zebrafish NLRP3 inflammasomes, caspase-11 can be recruited to NLRP6-ASC inflammasomes (116). Thus, inflammatory caspases that are involved in noncanonical inflammasome activation may have more elaborate functions that are yet to be fully understood.
Inflammasome-inducing sensors seem to continuously diversify. For instance, the ALR gene family, present in mammals, shows wide species diversity, and functional redundancy between family members can act as a compensatory mechanism when specific receptors are absent in a species (329). Similarly, NLRP1 molecules differ between mammalian species. The linker regions of primate NLRP1 proteins show high levels of sequence diversification, indicating that this region continues to evolve under positive selection (101, 330), likely increasing the chances for species-specific NLRP1 to detect proteases expressed by host-specific pathogens. Moreover, NLRP1 of primates and various other mammals contain an NH2-terminal PYD with currently unknown function, which has been lost in rodent NLRP1 (101). Pyrin is evolving under episodic positive selection in humans and primates, and the function of Pyrin seems to be modulating over evolutionary time (331). Interestingly, humans suffering from familial Mediterranean fever (FMF) contain an amino acid sequence similar to a domain sequence in Pyrins of nonhuman primates. These mutations lower the threshold for human Pyrin activation, although how this mechanistically works is not understood (332, 333). Since FMF has high carrier frequencies in people with origins associated with Mediterranean countries, the mutations are speculated to protect against endemic pathogens and as such became prevalent by selection in this population. It thus seems that the receptors that induce inflammasomes continue to evolve with harmful pathogens.
Not all mammalian species seem to benefit from all aspects of the pyroptotic pathway. Members of the order Carnivora have several alterations in inflammasome-related proteins. Felidae (cats and relatives) species do not encode NLRP1, whereas Canidae (dogs and relatives) and Mustelidae (weasels and relatives) lost NLRC4 and NAIPs, and mustelids are additionally deficient for the dsRNA sensor NLRP9 (334). Cats, tigers, and dogs are also deficient for AIM2 and lack the GSDMB activation site, with cats and tigers additionally lacking the activation site of GSDME (335). All carnivores lost the individual genes for caspases-1, -4, and -5. Instead, they encode a casp1/casp4 fusion gene that is made up of the CARD of the ancestral casp1 and the CARD and enzymatic domain of the ancestral casp4 gene. Alternative splicing provides two isoforms: casp1/4/a contains the casp1 CARD and the enzymatic domain, and casp1/4/b harbors both CARDs and the enzymatic domain (56). Although the enzymatic domains are homologs of human caspase-4, which is able to cleave GSDMD but not pro-IL-1β, both dog (Canis lupus familiaris) casp1/4a and -b isoforms can process GSDMD and pro-IL-1β upon multiple inflammasome stimuli (73). However, the casp1/4/b isoform is not able to sense cytosolic LPS and induce pyroptosis, indicating that canine casp1/4a and -b function like human caspase-1, although being homologs to human caspase-4 (73). Feline casp1/4/b is able to sense cytosolic LPS and induce subsequent pyroptosis and, unlike human caspase-4, can additionally process pro-IL1β. Thus, pyroptosis functions in carnivores, although several inflammasome receptors are lacking. Carnivore species are known to carry many zoonotic pathogens (334, 336), which suggests that the lack of certain inflammasome-inducing receptors may contribute to the presence of these pathogens.
The best-known mammalian species to carry zoonotic pathogens are bats (336). Bats can tolerate high viral loads and live full life spans, asymptomatically hosting viruses, including SARS-CoV-2, SARS-CoV-1, and Ebolavirus, that can be lethal to humans or other mammals. Bats have naturally dampened stress-related and virus-induced inflammatory responses, and it is therefore not surprising that bats evolved multiple ways to diminish inflammasome activation or the outcome thereof.
Bats lack the entire locus that harbors the AIM2-like receptor gene family, which includes the DNA sensors AIM2 and IFI16 (337), rendering bat cells incapable of cytosolic dsDNA-induced ASC-speck formation (338). Bats express four NLRP3 splice variants, including a splice variant that skips exon 7. This exon 7-deficient isoform is the most commonly expressed variant in Pteropus alecto tissues and contains an alteration in the LRR domain that results in a reduced ability of NLRP3 to induce ASC speck formation and IL-1β processing in response to ATP or the RNA viruses PRV3M, IAV, and MERS-CoV, known inducers of NLRP3 inflammasome activation. The LRR domain alterations probably are conserved features in bats (339). Bats control the level of IL1β-associated inflammation. Caspase-1 of the bat species P. alecto and P. vampyrus contains two inactivating mutations that prevent substrate cleavage, presumably by weakening homodimerization. Two other bat species, E. spelaea and M. davidii, do not contain these mutations and, accordingly, retain caspase-1 activity. However, M. davidii harbors three mutations in IL-1β, resulting in defective IL-1β cleavage. Interestingly, IL-1β cleavage potential inversely correlates with caspase-1 activity in bats; bat species with high caspase-1 activity have low IL-1β cleavage potential and vice versa (338). Thus, bats harbor significant alterations in inflammasome-related proteins, thereby dampening the ability of inflammasomes to induce inflammation. Bats harbor a functional caspase-4 homolog, indicating that pyroptosis in response to bacteria may be beneficial. It remains unclear, however, whether the loss of canonical inflammasomes evolved because of the selective pressure posed by the various residing viruses or other mechanisms that are unique to bats, such as their metabolically costly ability to fly or their relative longevity (338).
8.2. Apoptosis in Animal Evolution
The presence of the apoptotic machinery has been observed throughout the animal kingdom, and an increasing number of mechanistic studies, mainly by means of expression of apoptotic protein orthologs into mammalian cell culture systems, suggest that the apoptotic pathways are functionally present in the earliest metazoans. Bcl-2 proteins from the cnidarian species Acropora (coral) are able to inhibit cell death (340), and caspases and Bcl-2 family members from Hydra function similarly to their mammalian orthologs (42, 341). Platyhelminth (Planaria) Bak localizes to mitochondria and induces MOMP and cell death, and addition of cytochrome c to cytosolic extracts from Planaria and echinoderms (sea urchin and sand dollar) induces caspase activation (342). Functional TNF-induced apoptosis has been observed in the cniderian Acropora (coral) (343) and seems to signal through FADD and caspase-8, since Acropora FADD and caspase-8 functionally interact (344). Moreover, all components of the caspase-8 and caspase-9 pathways of apoptosis are present and functional in zebrafish (345). Collectively, these studies suggest that the complex apoptotic machinery present in mammals is functionally conserved across many animal phyla.
However, not all animals execute apoptosis similarly. The apoptotic mechanisms in D. melanogaster (arthropod) and C. elegans (nematode) function differently than in other animals (346). The D. melanogaster APAF-1 homolog ARK is constitutively active (FIGURE 3B). Recruitment and activation of the initiator caspase DRONC (FIGURE 1) are controlled by DIAP1, a homolog of the mammalian IAP proteins (e.g., XIAP). Apoptotic signals regulate the expression of the DIAP1 inhibitors HID, Grim, and Reaper, which antagonize DIAP1, resulting in the release, recruitment to ARK, and activation of DRONC. DRONC then cleaves the executioner caspases DRICE (FIGURE 1) and DCP-1, resulting in cellular demise (347).
Apoptosis in C. elegans is mediated by cell death defective (ced) genes ced-3, ced-4, and egg laying defective-1 (egl-1), whereas ced-9 inhibits death (348) (FIGURE 3C). CED-3 functions as the caspase (FIGURE 1) and is activated by the APAF-1 homolog CED-4. CED-9 is homologous to antiapoptotic Bcl-2 proteins and is anchored in the mitochondrial outer membrane, where it sequesters CED-4. Developmental signals induce the expression of the BH-3-only protein EGL-1, which disrupts the interaction between CED-4 and CED-9, permitting CED-4 to recruit CED-3 via CARD-CARD interactions, resulting in the direct demise of the cell. Thus, despite the similarities to the caspase-9 pathway in other phyla, including those basal to the arthropods and nematodes, the molecular mechanisms of caspase activation have diverged. MOMP and cytochrome c-induced activation of the APAF-1 homolog do not occur in these animals (342), and the ability of the BCL-2 homolog CED-9 to directly bind and suppress the APAF-1 homolog CED-4 is not observed in Bcl-2 family proteins from other phyla (including arthropods).
8.3. Necroptosis in Animal Evolution
Although components of the TNF signaling pathway are present in the most basal animals, the TNF-induced necroptotic pathway seems to have arisen only later in animal evolution. With the exception of RIPK1-like molecules, RHIM domain-containing proteins (RIPK3, TRIF, and ZBP1) are not found in nonvertebrates (349), and although RIPK1-like proteins are present throughout the Deuterostomia they are likely involved in mediating gene expression, not cell death. The necroptotic pathway, as defined by RIPK3-mediated activation of MLKL, therefore likely arose with the vertebrates (349). MLKL homologs are also found throughout the Deuterostomia, but since RIPK3 is the only known activator of MLKL, it seems unlikely that necroptosis is a cell death modality in invertebrate species (349). This view has been debated, however, based on homology between an MLKL domain and a domain of the fungal protein HELLP that acts similarly to HET-s to mediate cell death (350, 351). This speculatively suggests that MLKL activation may occur in the absence of RIPK3 and implies that necroptosis should be defined as death induced upon domain-dependent activation of MLKL.
The possibility that MLKL may be activated independently of RIPK3 is supported by the finding that all sequenced avipoxviruses contain a viral homolog of MLKL (vMLKL), suggesting that inhibition of MLKL is a necessary step for these viruses to establish an infection. The fact that avipoxviruses infect birds, which lack the gene for ripk3 but encode mlkl, indicates that avian MLKL may be activated by one or more unidentified kinases (306). This suggests that MLKL of other vertebrate species, including human MLKL, may also be activated by such kinases. Although human and mouse MLKL can be directly phosphorylated by TAM kinases, this requires prior MLKL phosphorylation by RIPK3 (352), and as such TAM kinases are not RIPK3-independent MLKL activators. Thus, MLKL may be activated by RIPK3-independent mechanisms in some species, but whether human MLKL can be activated independently of RIPK3 remains to be determined. Identifying such a mechanism may have implications for the treatment of cancers, since many cancer types downregulate RIPK3 expression and protein levels and as such may overcome control by necroptosis (353, 354).
Although necroptosis likely arose with the vertebrates, several species have lost components of the necroptotic machinery. All species of the class Aves (birds) lack ripk3 but encode mlkl, whereas Marsupialia lack both genes. Although the evolutionary reasons for this are not understood, this suggested that MLKL functions independently of RIPK3, perhaps to promote necroptosis (349). ZBP1 is also lacking in birds and marsupials, suggesting that ZBP1- and RIPK3-induced necroptosis are evolutionarily linked (349). TRIF is present in these species, indicating that TLR-induced, TRIF-mediated activation of gene expression programs is required for organismal survival, whereas ZBP1-induced gene expression may be controlled by other nucleic acid sensors and can therefore be seen as redundant (349). Several other mammalian species lack mlkl or harbor disrupting mutations in ripk3 and/or mlkl. Members of the order Carnivora, which also carry alterations in inflammasome-related genes (see above), lack the gene for mlkl. It has been speculated that their raw meat diet, comprising an abundant presence of microbes and viruses, would cause exuberant inflammation and excessive damage in the digestive tract of the foraging carnivore, which could have driven evolution to counterselect for the highly inflammatory necroptotic (and pyroptotic) pathways (349). A similar reasoning may explain why cetaceans (whales, orcas, dolphins, and narwhals), who are also strictly carnivorous, harbor mutations in mlkl and ripk3 that introduce frameshifts or premature stop codons in these genes (308). However, several herbivorous species also lost necroptosis. Lagomorpha (such as hares, rabbits, and pikas) lack mlkl and harbor a mutation in ripk3 that introduces a premature stop codon (308). An exception is Ochotona curzoniae (the plateau pika), which does harbor mlkl and functional ripk3 genes but lacks ripk1 (308). A small number of species from afrotheria (Trichechus manatus) and rodentia (Heterocephalus glaber, Fukomys damarensis, and Octodon degus) contain premature stop codons in ripk3 and mlkl, and Loxodonta africana (the African bush elephant) is deficient in mlkl (308). Why these herbivorous species lack a necroptotic pathway is currently unclear, but the convergent loss of necroptosis in different species demonstrates that necroptosis is not necessary for organismal development and survival. This raises the question of why necroptosis evolved and is retained in other species. It may be that Red Pawn effects are responsible for the evolution and retainment of necroptosis, although to date no opportunistic pathogens have been identified in animals in which necroptosis has been experimentally ablated. The reasons for the evolution and retainment of necroptosis thus remain enigmatic.
9. REGULATED CELL DEATH AND CORONAVIRUSES
The causative agent for the current coronavirus disease-2019 (COVID-19) pandemic is the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). Like SARS-CoV-1 (here SARS-1) and Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV-2 is a member of the Betacoronavirus genus within the Coronaviridae family. Infection with betacoronaviruses may remain asymptomatic in early stages but may progress to severe disease that encompasses inflammation, organ damage, and sometimes death of the infected individuals. Disease severity of COVID-19 has been associated with inflammasome activation (355), which occurs in various tissues including lung epithelium and circulating monocytes (355–357), and the ability of the lung epithelium to undergo apoptosis (358).
Studies investigating SARS-CoV-2-mediated inflammasome activation are currently ongoing, and our current knowledge of how betacoronaviruses activate inflammasomes mainly comes from studies on SARS-1. SARS-1 seems to actively engage inflammasomes and has several strategies to do so. The envelope (E) protein of SARS-1 forms Ca2+-permeable ion channels in the endoplasmic reticulum (ER) Golgi apparatus intermediate compartment (ERGIC) and Golgi membranes, which activates the NLRP3 inflammasome (359). SARS-1 Orf8b activates the NLRP3 inflammasome by directly interacting with the LRR domain of NLRP3 (360). Moreover, Orf8b proteins can form insoluble intracellular aggregates that induce ER stress and lysosomal damage that causes cellular demise (360). The viral Orf3a protein of SARS-1 is a Viroporin that forms potassium-sensitive ion channels upon oligomerization (361). Orf3a oligomerization and subsequent cell death depend on its interaction with RIPK3, which it binds through the NH2-terminal kinase domain of RIPK3, although the detailed mechanism of RIPK3-Orf3a pore formation remains unclear (362). The kinase activity of RIPK3 is not required for Orf3a oligomerization and subsequent cell death, and the latter is independent of MLKL or the activity of caspase-8, indicating that cell death is not apoptotic or necroptotic (362). Instead, the Orf3a ion channel seems to activate the NLRP3-inflammasome by facilitating K+ efflux (362, 363). Orf3a-mediated NLRP3 inflammasome activation also depends on the production of mitochondrial reactive oxygen species (ROS) (363), and, independent of its ion channel activity, Orf3a directly binds to TRAFs and ASC, which mediates the ubiquitylation of ASC by TRAF3 (364).
Most of these studies focus on loss of plasma membrane integrity and IL-1β release and do not analyze cleavage of caspase-1 and/or gasdermin D. Although most studies conclude that inflammasomes mediate IL-1β release through pyroptosis, SARS-CoV-2 was suggested to activate caspase-8 to mediate IL-1β cleavage and necroptosis to facilitate IL-1β release (365). However, inflammasome activation does not necessarily lead to pyroptosis (defined by gasdermin-mediated membrane disruption), and other, currently unknown, mechanisms may lead to the release of IL-1β. For instance, ssRNA sequences of SARS-CoV-2 and SARS-1 can activate the NLRP3 inflammasome in macrophages, leading to IL-1β maturation and release while GSDMD remains uncleaved (366). IL-1β is apparently released through a noncanonical pathway that involves caspase-8, caspase-1, the NLRP3 inflammasome, potassium efflux, and autophagy (366). Although the complete sequence of events remains to be determined, the process is suggested to be activated by binding of coronaviral GU-rich ssRNA to TLR8 (366). However, since TLR8-MyD88-mediated activation of a FADD-caspase-8-RIPK1-RIPK3 complex has not been demonstrated to date, we speculate that the GU-rich ssRNA from SARS-CoV-2 (and SARS-1 and possibly MERS-CoV) may activate ZBP1 to induce the NLRP3 inflammasome upon gaining access to the cytosol.
Betacoronaviruses not only induce inflammasome activation but also actively induce apoptosis. MERS-CoV M protein induces PERK-mediated apoptosis to augment viral titers, which contributes to disease severity and lethality (367). SARS-1 proteins Orf3b, Orf6, and Orf7a induce caspase-3-mediated apoptosis (368), whereas SARS-CoV-2 induces apoptosis by an ER stress response (358). Orf3a of SARS-CoV-2 and SARS-1 mediate activation of caspase-8, leading to apoptosis via tB499, caspase-9, and caspase-3 (369), although the exact mechanisms underlying apoptosis induction remain elusive, since, given the effects of Orf3a on inflammasome activation, it is unclear whether both the apoptotic and inflammasome pathways are activated by Orf3a or if apoptosis is an effect of activated inflammasomes.
Our current knowledge of coronaviral strategies to block the pathways of regulated cell death is limited. It seems that SARS-CoV-2 NSP1 and NSP13 are able to block the activation of caspase-1 and the subsequent release of cleaved IL-1β (370), but how this mechanistically occurs is not understood. SARS-1 Orf3a reduces phosphorylation of RIPK3 and MLKL, indicating that it may suppress necroptosis (362), but whether SARS-CoV-2 suppresses necroptosis remains to be determined. The extent of coronaviral blockage of regulated cell death currently is unclear, but we postulate that these potential mechanisms may have implications for disease severity.
The active engagement of inflammasomes and apoptosis by highly pathogenic betacoronaviruses suggests that betacoronavirus infections may be therapeutically controlled by targeting apoptosis and/or inflammasome activation (371). The disease severity and lethality of MERS-CoV infection can be controlled by inhibiting intrinsic apoptosis by PERK and caspase-3 and -7 inhibitors in murine infection models (367). The infection outcome of SARS-CoV-2 and SARS-1 was also improved by an inhibitor of caspases-3 and -7 in these murine models (367), but more studies are needed to confirm that apoptosis inhibition is a potent strategy for the treatment of COVID-19. Interestingly, lung cells in young individuals are more susceptible to apoptosis than cells of older individuals. Older individuals are at higher risk for developing severe COVID-19, possibly suggesting that the propensity for cells to undergo apoptosis helps to control the infection (358). These observations also indicate that the age of a patient should be considered when potential therapeutic strategies to combat COVID-19 and other diseases are developed.
The pronounced activation of cell death pathways during betacoronavirus infections seems paradoxical since these viruses risk losing their replicative niche by inducing rapid cellular demise. However, humans are not the natural reservoir for coronaviruses, and the current COVID-19 as well as the SARS-1 and MERS pandemics stem from zoonotic infections that likely originated from bats (372, 373). Bats are natural reservoirs for SARS-like coronaviruses (373) and are speculated to be the ancestral hosts for coronaviruses (372). The reduction of inflammasome functioning in bats (see above and Ref. 374) leads to an overall reduction in inflammation activation and an increase in tolerance of resident microbes, allowing bats to live healthy lives while hosting pathogens that are lethal to other species. How bats do this is unclear but of great interest (374). Bats have an intact necroptosis pathway, and that may be sufficient to control pathogenic infections, although these for now are speculations. Fully suppressing inflammatory responses in humans may lead to significantly increased pathogenic titers that may be detrimental for the patient. Learning from bats (374) as to how to fine-tune immune tolerance to keep infections in check will help us find therapeutic strategies to overcome the current COVID-19 pandemic and may be instrumental in defining approaches for the treatment of inflammatory diseases, autoimmune diseases, neuropathies, cancers, and other zoonotic diseases that are likely to arise in the future.
10. FUTURE PERSPECTIVES
Over evolutionary time, the human pathways of regulated cell death have become intricate and complex. But how effective are cell death strategies in combatting infections, and how well do pathogens evade these mechanisms? The coevolution of pathogens and their hosts provides clues, as does the existence of “Red Pawn” microbes that only manifest pathogenicity in the absence of cell death pathways (see above and Ref. 315). However, understanding the clinical significance of cell death and its evasion by pathogens will require new probes and markers for the activation of cell death pathways and the interrogation of clinical samples in the setting of infection. At present, such analyses are in their infancy. Unlike many biological events, cell death pathways cannot be elucidated from gene expression data, as our discussion of these pathways, their engagement, and their mechanisms of action makes clear. Caution should be exercised in evaluating any gene expression studies that purport to elucidate cell death mechanisms on this basis.
There is much to be learned about the extent to which the pathways of RCD interact and how these interactions affect organismal health in contexts of homeostasis and infection. This interplay is not only important for the control of infectious diseases but also affects immune homeostasis (190, 194, 195) and organismal development (196, 197). These studies show that deregulation of the genes involved in RCD may lead to severe inflammation that may prove lethal to an organism. Determining whether inflammation happens under sterile conditions or is induced by a resident microbiome (195) is of interest, and molecular and genetic studies aiming to understand how the RCD pathways interact are ongoing. Whether the effects of pathogenic virulence factors that interfere with the death-mediating functions of RCD proteins may also directly contribute to inflammation that is associated with a given pathogenic infection is an important area of ongoing study.
The recent realization that the pathways of regulated cell death extensively interact indicates that we are only at the beginning of fully understanding of how infections are controlled by regulated cell death. We now know that the interplay between the RCD pathways may have important implications for the outcome of infections, exemplified by the recent observation that plasticity between the caspase-8 pathway of apoptosis and necroptosis is required for the control of influenza A virus infections (207). This is particularly relevant in settings of coinfections, where an established microbe may block certain pathways of RCD, potentially adding to the pathogenicity of a coinfecting pathogen. This illustrates the need for a deeper understanding of the interplay between pathogens and cell death, the specific signaling pathways that trigger one type of cell death over another, and whether they manifest as a benefit to the host or to the pathogen. This understanding will be vital for the development of therapeutics aimed to control infections and/or immunological diseases that are affected by RCD.
GRANTS
D.R.G. is supported by grants AI40646, AI44828, AI12322, CA231620, and CA20635 from NIH and the National Cancer Institute (NCI) and by American Lebanese Syrian Associated Charities (ALSAC).
DISCLOSURES
D.R.G. consults for Ventus Therapeutics and Inzen Therapeutics. B.T. has no conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
B.T. and D.R.G. prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Patrick Fitzgerald for insightful discussions and wish him a happy retirement.
REFERENCES
- 1.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17: 97–111, 2017. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25: 486–541, 2018. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kearney CJ, Martin SJ. An inflammatory perspective on necroptosis. Mol Cell 65: 965–973, 2017. doi: 10.1016/j.molcel.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 5.Linkermann A, Green DR. Necroptosis. N Engl J Med 370: 455–465, 2014. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277: 61–75, 2017. doi: 10.1111/imr.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 517: 311–320, 2015. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 8.Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352: aaf2154, 2016. doi: 10.1126/science.aaf2154. [DOI] [PubMed] [Google Scholar]
- 9.Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20: 95–112, 2020. doi: 10.1038/s41577-019-0215-7. [DOI] [PubMed] [Google Scholar]
- 10.Poranen MM, Daugelavicius R, Bamford DH. Common principles in viral entry. Annu Rev Microbiol 56: 521–538, 2002. doi: 10.1146/annurev.micro.56.012302.160643. [DOI] [PubMed] [Google Scholar]
- 11.Bliska JB, Casadevall A. Intracellular pathogenic bacteria and fungi—a case of convergent evolution? Nat Rev Microbiol 7: 165–171, 2009. doi: 10.1038/nrmicro2049. [DOI] [PubMed] [Google Scholar]
- 12.Casadevall A. Evolution of intracellular pathogens. Annu Rev Microbiol 62: 19–33, 2008. doi: 10.1146/annurev.micro.61.080706.093305. [DOI] [PubMed] [Google Scholar]
- 13.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33: 257–290, 2015. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meunier E, Broz P. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol 38: 744–757, 2017. doi: 10.1016/j.it.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 140: 805–820, 2010. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 16.Martin SJ. Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J 283: 2599–2615, 2016. doi: 10.1111/febs.13775. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan GP, O’Connor H, Henry CM, Davidovich P, Clancy DM, Albert ML, Cullen SP, Martin SJ. TRAIL receptors serve as stress-associated molecular patterns to promote ER-stress-induced inflammation. Dev Cell 52: 714–730.e5, 2020. doi: 10.1016/j.devcel.2020.01.031. [DOI] [PubMed] [Google Scholar]
- 18.Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern recognition receptors and the host cell death molecular machinery. Front Immunol 9: 2379, 2018. doi: 10.3389/fimmu.2018.02379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Annibaldi A, Walczak H. Death receptors and their ligands in inflammatory disease and cancer. Cold Spring Harb Perspect Biol 12: a036384, 2020. doi: 10.1101/cshperspect.a036384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol 5: a008698, 2013. doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Annu Rev Cancer Biol 3: 35–54, 2019. doi: 10.1146/annurev-cancerbio-030518-055844. [DOI] [Google Scholar]
- 22.Green DR. Death by retrograde transport: avoiding the apoptosis default. Cell Chem Biol 26: 1636–1638, 2019. doi: 10.1016/j.chembiol.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Green DR, Victor B. The pantheon of the fallen: why are there so many forms of cell death? Trends Cell Biol 22: 555–556, 2012. doi: 10.1016/j.tcb.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsson M, Fonteneau JF, Bhardwaj N. Dendritic cells resurrect antigens from dead cells. Trends Immunol 22: 141–148, 2001. doi: 10.1016/S1471-4906(01)01860-9. [DOI] [PubMed] [Google Scholar]
- 25.Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol 17: 262–275, 2017. doi: 10.1038/nri.2017.9. [DOI] [PubMed] [Google Scholar]
- 26.Fonteneau JF, Larsson M, Bhardwaj N. Interactions between dead cells and dendritic cells in the induction of antiviral CTL responses. Curr Opin Immunol 14: 471–477, 2002. doi: 10.1016/S0952-7915(02)00358-8. [DOI] [PubMed] [Google Scholar]
- 27.Giodini A, Albert ML. A whodunit: an appointment with death. Curr Opin Immunol 22: 94–108, 2010. doi: 10.1016/j.coi.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 28.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18: 134–147, 2018. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 29.Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol 36: 191–218, 2020. doi: 10.1146/annurev-cellbio-020520-111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ameisen JC. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ 9: 367–393, 2002. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 31.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 9: 378–390, 2008. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 32.Green DR. Cell Death: Apoptosis and Other Means to an End (2nd ed.). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2018, p. 278. [Google Scholar]
- 33.Green DR, Fitzgerald P. Just so stories about the evolution of apoptosis. Curr Biol 26: R620–R627, 2016. doi: 10.1016/j.cub.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Legrand AJ, Konstantinou M, Goode EF, Meier P. The diversification of cell death and immunity: memento mori. Mol Cell 76: 232–242, 2019. doi: 10.1016/j.molcel.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev 277: 76–89, 2017. doi: 10.1111/imr.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kesavardhana S, Malireddi RK, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol 38: 567–595, 2020. doi: 10.1146/annurev-immunol-073119-095439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity 50: 1352–1364, 2019. doi: 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Julien O, Wells JA. Caspases and their substrates. Cell Death Differ 24: 1380–1389, 2017. doi: 10.1038/cdd.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poreba M, Strózyk A, Salvesen GS, Drag M. Caspase substrates and inhibitors. Cold Spring Harb Perspect Biol 5: a008680, 2013. doi: 10.1101/cshperspect.a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salvesen GS. Programmed cell death and the caspases. APMIS 107: 73–79, 1999. doi: 10.1111/j.1699-0463.1999.tb01528.x. [DOI] [PubMed] [Google Scholar]
- 41.Lahm A, Paradisi A, Green DR, Melino G. Death fold domain interaction in apoptosis. Cell Death Differ 10: 10–12, 2003. doi: 10.1038/sj.cdd.4401203. [DOI] [PubMed] [Google Scholar]
- 42.Lasi M, Pauly B, Schmidt N, Cikala M, Stiening B, Käsbauer T, Zenner G, Popp T, Wagner A, Knapp RT, Huber AH, Grunert M, Söding J, David CN, Böttger A. The molecular cell death machinery in the simple cnidarian Hydra includes an expanded caspase family and pro- and anti-apoptotic Bcl-2 proteins. Cell Res 20: 812–825, 2010. doi: 10.1038/cr.2010.66. [DOI] [PubMed] [Google Scholar]
- 43.Masumoto J, Zhou W, Chen FF, Su F, Kuwada JY, Hidaka E, Katsuyama T, Sagara J, Taniguchi S, Ngo-Hazelett P, Postlethwait JH, Nuñez G, Inohara N. Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J Biol Chem 278: 4268–4276, 2003. doi: 10.1074/jbc.M203944200. [DOI] [PubMed] [Google Scholar]
- 44.Goping IS, Barry M, Liston P, Sawchuk T, Constantinescu G, Michalak KM, Shostak I, Roberts DL, Hunter AM, Korneluk R, Bleackley RC. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity 18: 355–365, 2003. doi: 10.1016/S1074-7613(03)00032-3. [DOI] [PubMed] [Google Scholar]
- 45.Metkar SS, Wang B, Ebbs ML, Kim JH, Lee YJ, Raja SM, Froelich CJ. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol 160: 875–885, 2003. doi: 10.1083/jcb.200210158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA 96: 10964–10967, 1999. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grinshpon RD, Williford A, Titus-McQuillan J, Clark AC. The CaspBase: a curated database for evolutionary biochemical studies of caspase functional divergence and ancestral sequence inference. Protein Sci 27: 1857–1870, 2018. doi: 10.1002/pro.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lamkanfi M, Declercq W, Kalai M, Saelens X, Vandenabeele P. Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ 9: 358–361, 2002. doi: 10.1038/sj.cdd.4400989. [DOI] [PubMed] [Google Scholar]
- 49.Sakamaki K, Satou Y. Caspases: evolutionary aspects of their functions in vertebrates. J Fish Biol 74: 727–753, 2009. doi: 10.1111/j.1095-8649.2009.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hachmann J, Salvesen GS. The paracaspase MALT1. Biochimie 122: 324–338, 2016. doi: 10.1016/j.biochi.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaworski M, Thome M. The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell Mol Life Sci 73: 459–473, 2016. doi: 10.1007/s00018-015-2059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minina EA, Coll NS, Tuominen H, Bozhkov PV. Metacaspases versus caspases in development and cell fate regulation. Cell Death Differ 24: 1314–1325, 2017. doi: 10.1038/cdd.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsiatsiani L, Van Breusegem F, Gallois P, Zavialov A, Lam E, Bozhkov PV. Metacaspases. Cell Death Differ 18: 1279–1288, 2011. doi: 10.1038/cdd.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 6: 961–967, 2000. doi: 10.1016/S1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 55.Wachmann K, Pop C, van Raam BJ, Drag M, Mace PD, Snipas SJ, Zmasek C, Schwarzenbacher R, Salvesen GS, Riedl SJ. Activation and specificity of human caspase-10. Biochemistry 49: 8307–8315, 2010. doi: 10.1021/bi100968m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eckhart L, Ballaun C, Hermann M, VandeBerg JL, Sipos W, Uthman A, Fischer H, Tschachler E. Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol Biol Evol 25: 831–841, 2008. doi: 10.1093/molbev/msn012. [DOI] [PubMed] [Google Scholar]
- 57.Ng TM, Monack DM. Revisiting caspase-11 function in host defense. Cell Host Microbe 14: 9–14, 2013. doi: 10.1016/j.chom.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT, Hotchkiss RS, Buchman TG, Zehnbauer BA, Hayden MR, Farrer LA, Roy S, Nicholson DW. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429: 75–79, 2004. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 59.Xue Y, Daly A, Yngvadottir B, Liu M, Coop G, Kim Y, Sabeti P, Chen Y, Stalker J, Huckle E, Burton J, Leonard S, Rogers J, Tyler-Smith C. Spread of an inactive form of caspase-12 in humans is due to recent positive selection. Am J Hum Genet 78: 659–670, 2006. doi: 10.1086/503116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Grus WE, Zhang J. Gene losses during human origins. PLoS Biol 4: e52, 2006. doi: 10.1371/journal.pbio.0040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirchmeier P, Zimmer A, Bouadjar B, Rösler B, Fischer J. Whole-exome-sequencing reveals small deletions in CASP14 in patients with autosomal recessive inherited ichthyosis. Acta Derm Venereol 97: 102–104, 2017. doi: 10.2340/00015555-2510. [DOI] [PubMed] [Google Scholar]
- 62.Seuring C, Greenwald J, Wasmer C, Wepf R, Saupe SJ, Meier BH, Riek R. The mechanism of toxicity in HET-S/HET-s prion incompatibility. PLoS Biol 10: e1001451, 2012. doi: 10.1371/journal.pbio.1001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riek R, Saupe SJ. The HET-S/s Prion Motif in the Control of Programmed Cell Death. Cold Spring Harb Perspect Biol 8: a023515, 2016. doi: 10.1101/cshperspect.a023515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rathinam VA, Zhao Y, Shao F. Innate immunity to intracellular LPS. Nat Immunol 20: 527–533, 2019. doi: 10.1038/s41590-019-0368-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, Miao EA. Caspase-11 protects against bacteria that escape the vacuole. Science 339: 975–978, 2013. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341: 1250–1253, 2013. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu M, Zhou K, Xu Z, Ma H, Cao X, Yin X, Zeng W, Zahid A, Fu S, Ni K, Ye X, Zhou Y, Bai L, Zhou R, Jin T. Crystal structure of caspase-11 CARD provides insights into caspase-11 activation. Cell Discov 6: 70, 2020. doi: 10.1038/s41421-020-00201-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192, 2014. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 69.Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, Li Z, Wang Y, Zhao Q, Shao F, Ding J. Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell 180: 941–955.e20, 2020. doi: 10.1016/j.cell.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 70.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526: 666–671, 2015. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 71.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665, 2015. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 72.Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, Kanneganti TD. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep 7: 45126, 2017. doi: 10.1038/srep45126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Devant P, Cao A, Kagan JC. Evolution-inspired dissection of caspase activities enables the redesign of caspase-4 into an LPS sensing interleukin-1 converting enzyme (Preprint). bioRxiv 2020.12.07.413732, 2020. doi: 10.1101/2020.12.07.413732. [DOI]
- 74.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687, 2015. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022, 2014. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 76.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411, 2013. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jing W, Lo Pilato J, Kay C, Man SM. Activation mechanisms of inflammasomes by bacterial toxins. Cell Microbiol 23: e13309, 2021. doi: 10.1111/cmi.13309. [DOI] [PubMed] [Google Scholar]
- 78.Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol 29: 201–210, 2017. doi: 10.1093/intimm/dxx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev 297: 26–38, 2020. doi: 10.1111/imr.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bauernfried S, Scherr MJ, Pichlmair A, Duderstadt KE, Hornung V. Human NLRP1 is a sensor for double-stranded RNA. Science, 371: eabd0811, 2021. doi: 10.1126/science.abd0811. [DOI] [PubMed] [Google Scholar]
- 81.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19: 477–489, 2019. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He Y, Hara H, Nuñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41: 1012–1021, 2016. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20: 3328, 2019. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moretti J, Blander JM. Increasing complexity of NLRP3 inflammasome regulation. J Leukoc Biol 109: 561–571, 2021. doi: 10.1002/JLB.3MR0520-104RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, Sharif H, Hu JJ, Evavold CL, Kagan JC, Schmidt FI, Fitzgerald KA, Kirchhausen T, Li Y, Wu H. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369: eaas8995, 2020. doi: 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357, 2016. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RK, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, Sharma BR, Nourse A, King SV, Pitre A, Burton AR, Pelletier S, Gilbertson RJ, Kanneganti TD. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573: 590–594, 2019. doi: 10.1038/s41586-019-1551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121, 2011. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 89.Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, Masters SL, Murphy JM, Schroder K, Vaux DL, Lawlor KE, Lindqvist LM, Vince JE. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA 114: E961–E969, 2017. doi: 10.1073/pnas.1613305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SW, Gale M Jr, Oberst A. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of Gasdermin-D. J Immunol 198: 2156–2164, 2017. doi: 10.4049/jimmunol.1601757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA, Bachovchin DA. N-terminal degradation activates the NLRP1B inflammasome. Science 364: 82–85, 2019. doi: 10.1126/science.aau1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364: eaau1330, 2019. doi: 10.1126/science.aau1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, Dong N. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J 38: e101996, 2019. doi: 10.15252/embj.2019101996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gong Q, Robinson K, Xu C, Huynh PT, Chong KH, Tan EY, Zhang J, Boo ZZ, Teo DE, Lay K, Zhang Y, Lim JS, Goh WI, Wright G, Zhong FL, Reversade B, Wu B. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat Commun 12: 188, 2021. doi: 10.1038/s41467-020-20319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang M, Zhang X, Toh GA, Gong Q, Wang J, Han Z, Wu B, Zhong F, Chai J. Structural and biochemical mechanisms of NLRP1 inhibition by DPP9. Nature 592: 773–777, 2021. doi: 10.1038/s41586-021-03320-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25: 713–724, 2007. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 97.Robinson KS, Teo DE, Tan KS, Toh GA, Ong HH, Lim CK, Lay K, Au BV, Lew TS, Chu JJ, Chow VT, Wang Y, Zhong FL, Reversade B. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 370: eaay2002, 2020. doi: 10.1126/science.aay2002. [DOI] [PubMed] [Google Scholar]
- 98.Tsu BV, Beierschmitt C, Ryan AP, Agarwal R, Mitchell PS, Daugherty MD. Diverse viral proteases activate the NLRP1 inflammasome. Elife 10: e60609, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38: 240–244, 2006. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 100.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8: e1002638, 2012. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chavarría-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9: e1003452, 2013. doi: 10.1371/journal.ppat.1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Neiman-Zenevich J, Stuart S, Abdel-Nour M, Girardin SE, Mogridge J. Listeria monocytogenes and Shigella flexneri activate the NLRP1B inflammasome. Infect Immun 85: e00338-17, 2017. doi: 10.1128/IAI.00338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ewald SE, Chavarria-Smith J, Boothroyd JC. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun 82: 460–468, 2014. doi: 10.1128/IAI.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liao KC, Mogridge J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun 81: 570–579, 2013. doi: 10.1128/IAI.01003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin J, Bertin JJ, Gough PJ. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem 287: 25030–25037, 2012. doi: 10.1074/jbc.M112.378323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458: 514–518, 2009. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323: 1057–1060, 2009. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 108.Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, Hornung V, Latz E, Bowie AG, Fitzgerald KA, Xiao TS. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36: 561–571, 2012. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell 54: 17–29, 2014. doi: 10.1016/j.molcel.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, Shao F, Wang HW, Sui SF, Chai J. Structural and biochemical basis for induced self-propagation of NLRC4. Science 350: 399–404, 2015. doi: 10.1126/science.aac5489. [DOI] [PubMed] [Google Scholar]
- 111.Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, Li Y, David L, Lu A, Wang WL, Marks C, Ouyang Q, Zhang X, Mao Y, Wu H. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350: 404–409, 2015. doi: 10.1126/science.aac5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600, 2011. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 113.Gao W, Yang J, Liu W, Wang Y, Shao F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci USA 113: E4857–E4866, 2016. doi: 10.1073/pnas.1601700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17: 914–921, 2016. doi: 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, Wang F, Shao F. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513: 237–241, 2014. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 116.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY, Núñez G. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates Gram-positive pathogen infection. Cell 175: 1651–1664.e14, 2018. doi: 10.1016/j.cell.2018.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leng F, Yin H, Qin S, Zhang K, Guan Y, Fang R, Wang H, Li G, Jiang Z, Sun F, Wang DC, Xie C. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci Rep 10: 198, 2020. doi: 10.1038/s41598-019-57043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, Rojanasakul Y, Stehlik C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36: 464–476, 2012. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, Yang Y, Yu H, Li HB, Wang G, Lei X, de Zoete MR, Zhao J, Zheng Y, Chen H, Zhao Y, Jurado KA, Feng N, Shan L, Kluger Y, Lu J, Abraham C, Fikrig E, Greenberg HB, Flavell RA. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546: 667–670, 2017. doi: 10.1038/nature22967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol 20: 143–157, 2020. doi: 10.1038/s41577-019-0228-2. [DOI] [PubMed] [Google Scholar]
- 121.Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol 430: 3068–3080, 2018. doi: 10.1016/j.jmb.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 122.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116, 2016. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 123.Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557: 62–67, 2018. doi: 10.1038/s41586-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593: 607–611, 2021. doi: 10.1038/s41586-021-03478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, Shen L, Han W, Shen L, Ding J, Shao F. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368: eaaz7548, 2020. doi: 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- 126.Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, Hu Q, Zou Y, Huang Z, Ren J, Lin Z, Gao X. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol 11: 496–508, 2019. doi: 10.1093/jmcb/mjy056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Delmaghani S, Defourny J, Aghaie A, Beurg M, Dulon D, Thelen N, Perfettini I, Zelles T, Aller M, Meyer A, Emptoz A, Giraudet F, Leibovici M, Dartevelle S, Soubigou G, Thiry M, Vizi ES, Safieddine S, Hardelin JP, Avan P, Petit C. Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell 163: 894–906, 2015. doi: 10.1016/j.cell.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 128.Harris SL, Kazmierczak M, Pangršič T, Shah P, Chuchvara N, Barrantes-Freer A, Moser T, Schwander M. Conditional deletion of pejvakin in adult outer hair cells causes progressive hearing loss in mice. Neuroscience 344: 380–393, 2017. doi: 10.1016/j.neuroscience.2016.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, Van de Heyning P, McGuirt WT, Smith RJ, Willems PJ, Legan PK, Richardson GP, Van Camp G. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet 20: 194–197, 1998. doi: 10.1038/2503. [DOI] [PubMed] [Google Scholar]
- 130.Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol 24: 507–514.e4, 2017. doi: 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8: 14128, 2017. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547: 99–103, 2017. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 133.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TM, Ansara J, Sengupta S, Yao Y, Wu H, Lieberman J. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579: 415–420, 2020. doi: 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med 213: 2113–2128, 2016. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Núñez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 19: 107–120, 2012. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489, 1997. doi: 10.1016/S0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 137.Hu Y, Benedict MA, Ding L, Núñez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J 18: 3586–3595, 1999. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274: 11549–11556, 1999. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 139.Kulikov AV, Shilov ES, Mufazalov IA, Gogvadze V, Nedospasov SA, Zhivotovsky B. Cytochrome c: the Achilles' heel in apoptosis. Cell Mol Life Sci 69: 1787–1797, 2012. doi: 10.1007/s00018-011-0895-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol 9: 532–542, 2008. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 141.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42, 2000. doi: 10.1016/S0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 142.Muñoz-Pinedo C, Guio-Carrión A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci USA 103: 11573–11578, 2006. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 8: 613–621, 2001. doi: 10.1016/S1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 144.van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, Gevaert K, Vandekerckhove J, Declercq W, Vandenabeele P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ 9: 20–26, 2002. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- 145.Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25: 46–55, 2018. doi: 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem Sci 39: 101–111, 2014. doi: 10.1016/j.tibs.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH, Brown SA, Pelletier S, Moldoveanu T, Chen T, Green DR. BOK Is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165: 421–433, 2016. doi: 10.1016/j.cell.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Seyrek K, Ivanisenko NV, Richter M, Hillert LK, König C, Lavrik IN. Controlling cell death through post-translational modifications of DED proteins. Trends Cell Biol 30: 354–369, 2020. doi: 10.1016/j.tcb.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 149.Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, Strasser A, Kaufmann T. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460: 1035–1039, 2009. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ 19: 42–50, 2012. doi: 10.1038/cdd.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14: 5579–5588, 1995. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190, 2003. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 153.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Häcker G, Leverkus M. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43: 449–463, 2011. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell 56: 469–480, 2014. doi: 10.1016/j.molcel.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43: 432–448, 2011. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 156.Xu Q, Jitkaew S, Choksi S, Kadigamuwa C, Qu J, Choe M, Jang J, Liu C, Liu ZG. The cytoplasmic nuclear receptor RARgamma controls RIP1 initiated cell death when cIAP activity is inhibited. Nat Commun 8: 425, 2017. doi: 10.1038/s41467-017-00496-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ 22: 225–236, 2015. doi: 10.1038/cdd.2014.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 174: 4942–4952, 2005. doi: 10.4049/jimmunol.174.8.4942. [DOI] [PubMed] [Google Scholar]
- 159.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 10: 916–922, 2009. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature 388: 190–195, 1997. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 161.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 274: 1541–1548, 1999. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 162.Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by FLIPL. Biochem J 382: 651–657, 2004. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yu JW, Jeffrey PD, Shi Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci USA 106: 8169–8174, 2009. doi: 10.1073/pnas.0812453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, Salvesen GS. FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J 433: 447–457, 2011. doi: 10.1042/BJ20101738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, MacFarlane M. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell 61: 834–849, 2016. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fava LL, Bock FJ, Geley S, Villunger A. Caspase-2 at a glance. J Cell Sci 125: 5911–5915, 2012. doi: 10.1242/jcs.115105. [DOI] [PubMed] [Google Scholar]
- 167.Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F, Logette E, Tschopp J, Villunger A. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol 185: 291–303, 2009. doi: 10.1083/jcb.200811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem 277: 13430–13437, 2002. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 169.Bouchier-Hayes L, Green DR. Caspase-2: the orphan caspase. Cell Death Differ 19: 51–57, 2012. doi: 10.1038/cdd.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sladky V, Schuler F, Fava LL, Villunger A. The resurrection of the PIDDosome—emerging roles in the DNA-damage response and centrosome surveillance. J Cell Sci 130: 3779–3787, 2017. doi: 10.1242/jcs.203448. [DOI] [PubMed] [Google Scholar]
- 171.Sladky VC, Villunger A. Uncovering the PIDDosome and caspase-2 as regulators of organogenesis and cellular differentiation. Cell Death Differ 27: 2037–2047, 2020. doi: 10.1038/s41418-020-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vigneswara V, Ahmed Z. The role of caspase-2 in regulating cell fate. Cells 9: 1259, 2020. doi: 10.3390/cells9051259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, Villunger A. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31: 34–45, 2017. doi: 10.1101/gad.289728.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A, Maffini S, Fava LL. Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. EMBO J 40: e104844, 2021. doi: 10.15252/embj.2020104844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ando K, Parsons MJ, Shah RB, Charendoff CI, Paris SL, Liu PH, Fassio SR, Rohrman BA, Thompson R, Oberst A, Sidi S, Bouchier-Hayes L. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J Cell Biol 216: 1795–1810, 2017. doi: 10.1083/jcb.201608095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wu XL, Hu H, Dong XQ, Zhang J, Wang J, Schwieters CD, Liu J, Wu GX, Li B, Lin JY, Wang HY, Lu JX. The amyloid structure of mouse RIPK3 (receptor interacting protein kinase 3) in cell necroptosis. Nat Commun 12: 1627, 2021. doi: 10.1038/s41467-021-21881-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453, 2013. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 178.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM, Silke J. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA 111: 15072–15077, 2014. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell 61: 589–601, 2016. doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ 23: 76–88, 2016. doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133–146, 2014. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 182.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123, 2009. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137: 1100–1111, 2009. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 184.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332–336, 2009. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 185.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157: 1189–1202, 2014. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA 108: 20054–20059, 2011. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288: 31268–31279, 2013. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, Finger JN, Polykratis A, Gough PJ, Bertin J, Whalen M, Pasparakis M, Balachandran S, Kelliher M, Poltorak A, Degterev A. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by toll-like receptor 4. Immunity 45: 46–59, 2016. doi: 10.1016/j.immuni.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ingram JP, Thapa RJ, Fisher A, Tummers B, Zhang T, Yin C, Rodriguez DA, Guo H, Lane R, Williams R, Slifker MJ, Basagoudanavar SH, Rall GF, Dillon CP, Green DR, Kaiser WJ, Balachandran S. ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J Immunol 203: 1348–1355, 2019. doi: 10.4049/jimmunol.1900216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schwarzer R, Laurien L, Pasparakis M. New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8. Curr Opin Cell Biol 63: 186–193, 2020. doi: 10.1016/j.ceb.2020.02.004. [DOI] [PubMed] [Google Scholar]
- 191.Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher CA, Radaelli E, Fettrelet T, Mack V, Linkermann A, Roger T, Brodsky IE, Chen KW, Broz P. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv 6: eabc3465, 2020. doi: 10.1126/sciadv.abc3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA 115: E10888–E10897, 2018. doi: 10.1073/pnas.1809548115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192: 1835–1846, 2014. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and caspase-8 regulate gut homeostasis and inflammation by controlling MLKL- and GSDMD-mediated death of intestinal epithelial cells. Immunity 52: 978–993.e6, 2020. doi: 10.1016/j.immuni.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 195.Tummers B, Mari L, Guy CS, Heckmann BL, Rodriguez DA, Rühl S, Moretti J, Crawford JC, Fitzgerald P, Kanneganti TD, Janke LJ, Pelletier S, Blander JM, Green DR. Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 52: 994–1006.e8, 2020. doi: 10.1016/j.immuni.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fritsch M, Günther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM, Lamkanfi M, Krönke M, Pasparakis M, Kashkar H. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575: 683–687, 2019. doi: 10.1038/s41586-019-1770-6. [DOI] [PubMed] [Google Scholar]
- 197.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, Roose-Girma M, Modrusan Z, Sagolla MS, Webster JD, Dixit VM. Activity of caspase-8 determines plasticity between cell death pathways. Nature 575: 679–682, 2019. doi: 10.1038/s41586-019-1752-8. [DOI] [PubMed] [Google Scholar]
- 198.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 38: e101638, 2019. doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129: 45–56, 2007. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 200.Sagulenko V, Vitak N, Vajjhala PR, Vince JE, Stacey KJ. Caspase-1 is an apical caspase leading to caspase-3 cleavage in the AIM2 inflammasome response, independent of caspase-8. J Mol Biol 430: 238–247, 2018. doi: 10.1016/j.jmb.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 201.Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, Kinoshita T, Kushiyama H, Sakurai M, Shiroishi T, Suda T. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun 10: 2091, 2019. doi: 10.1038/s41467-019-09753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Heilig R, Dilucca M, Boucher D, Chen KW, Hancz D, Demarco B, Shkarina K, Broz P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci Alliance 3: e202000735, 2020. doi: 10.26508/lsa.202000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10: 1689, 2019. doi: 10.1038/s41467-019-09397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, Rashidi M, Wicks IP, Alexander WS, Mitsuuchi Y, Benetatos CA, Condon SM, Wong WW, Silke J, Vaux DL, Vince JE. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6: 6282, 2015. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, Hornung V. Human monocytes engage an alternative inflammasome pathway. Immunity 44: 833–846, 2016. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 206.Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell 181: 674–687.e13, 2020. doi: 10.1016/j.cell.2020.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shubina M, Tummers B, Boyd DF, Zhang T, Yin C, Gautam A, Guo XJ, Rodriguez DA, Kaiser WJ, Vogel P, Green DR, Thomas PG, Balachandran S. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J Exp Med 217: e20191259, 2020. doi: 10.1084/jem.20191259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Grunert M, Gottschalk K, Kapahnke J, Gündisch S, Kieser A, Jeremias I. The adaptor protein FADD and the initiator caspase-8 mediate activation of NF-kappaB by TRAIL. Cell Death Dis 3: e414, 2012. doi: 10.1038/cddis.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, Taraborrelli L, Draber P, Lafont E, Arce Vargas F, El-Bahrawy MA, Quezada SA, Walczak H. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol Cell 65: 730–742.e5, 2017. doi: 10.1016/j.molcel.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Henry CM, Martin SJ. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol Cell 65: 715–729.e5, 2017. doi: 10.1016/j.molcel.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 211.Ray K, Marteyn B, Sansonetti PJ, Tang CM. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7: 333–340, 2009. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- 212.Kumar Y, Valdivia RH. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5: 593–601, 2009. doi: 10.1016/j.chom.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Paciello I, Silipo A, Lembo-Fazio L, Curcurù L, Zumsteg A, Noël G, Ciancarella V, Sturiale L, Molinaro A, Bernardini ML. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc Natl Acad Sci USA 110: E4345–E4354, 2013. doi: 10.1073/pnas.1303641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, Martin A, Di Lorenzo F, Py BF, Molinaro A, Henry T. Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat Commun 9: 242, 2018. doi: 10.1038/s41467-017-02682-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, Akakura R, Yoshida M, Kawalec M, Reichhart JM, Mizushima T, Sasakawa C. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13: 570–583, 2013. doi: 10.1016/j.chom.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 216.Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7: 376–387, 2010. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Aepfelbacher M, Trasak C, Wilharm G, Wiedemann A, Trulzsch K, Krauss K, Gierschik P, Heesemann J. Characterization of YopT effects on Rho GTPases in Yersinia enterocolitica-infected cells. J Biol Chem 278: 33217–33223, 2003. doi: 10.1074/jbc.M303349200. [DOI] [PubMed] [Google Scholar]
- 218.Medici NP, Rashid M, Bliska JB. Characterization of pyrin dephosphorylation and inflammasome activation in macrophages as triggered by the Yersinia effectors YopE and YopT. Infect Immun 87: e00822-18, 2019. doi: 10.1128/IAI.00822-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chung LK, Park YH, Zheng Y, Brodsky IE, Hearing P, Kastner DL, Chae JJ, Bliska JB. The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 20: 296–306, 2016. doi: 10.1016/j.chom.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ratner D, Orning MP, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, Kayagaki N, Goguen JD, Lien E. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog 12: e1006035, 2016. doi: 10.1371/journal.ppat.1006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12: 799–805, 2012. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Yang J, Lee KM, Park S, Cho Y, Lee E, Park JH, Shin OS, Son J, Yoon SS, Yu JW. Bacterial secretant from Pseudomonas aeruginosa dampens inflammasome activation in a quorum sensing-dependent manner. Front Immunol 8: 333, 2017. doi: 10.3389/fimmu.2017.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204: 3235–3245, 2007. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell 69: 597–604, 1992. doi: 10.1016/0092-8674(92)90223-Y. [DOI] [PubMed] [Google Scholar]
- 225.Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3: 36–50, 2003. doi: 10.1038/nri980. [DOI] [PubMed] [Google Scholar]
- 226.Dorfleutner A, Talbott SJ, Bryan NB, Funya KN, Rellick SL, Reed JC, Shi X, Rojanasakul Y, Flynn DC, Stehlik C. A Shope Fibroma virus PYRIN-only protein modulates the host immune response. Virus Genes 35: 685–694, 2007. doi: 10.1007/s11262-007-0141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Johnston JB, Barrett JW, Nazarian SH, Goodwin M, Ricciuto D, Wang G, McFadden G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 23: 587–598, 2005. doi: 10.1016/j.immuni.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 228.Song Y, Wu X, Xu Y, Zhu J, Li J, Zou Z, Chen L, Zhang B, Hua C, Rui H, Zheng Q, Zhou Q, Wang Q, Cheng H. HPV E7 inhibits cell pyroptosis by promoting TRIM21-mediated degradation and ubiquitination of the IFI16 inflammasome. Int J Biol Sci 16: 2924–2937, 2020. doi: 10.7150/ijbs.50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331: 330–334, 2011. doi: 10.1126/science.1199478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Komune N, Ichinohe T, Ito M, Yanagi Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol 85: 13019–13026, 2011. doi: 10.1128/JVI.05942-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chung WC, Kang HR, Yoon H, Kang SJ, Ting JP, Song MJ. Influenza A virus NS1 protein inhibits the NLRP3 inflammasome. PLoS One 10: e0126456, 2015. doi: 10.1371/journal.pone.0126456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Moriyama M, Chen IY, Kawaguchi A, Koshiba T, Nagata K, Takeyama H, Hasegawa H, Ichinohe T. The RNA- and TRIM25-binding domains of influenza virus ns1 protein are essential for suppression of NLRP3 inflammasome-mediated interleukin-1beta secretion. J Virol 90: 4105–4114, 2016. doi: 10.1128/JVI.00120-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lei X, Zhang Z, Xiao X, Qi J, He B, Wang J. Enterovirus 71 inhibits pyroptosis through cleavage of gasdermin D. J Virol 91: e01069-17, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wang H, Lei X, Xiao X, Yang C, Lu W, Huang Z, Leng Q, Jin Q, He B, Meng G, Wang J. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep 12: 42–48, 2015. doi: 10.1016/j.celrep.2015.05.047. [DOI] [PubMed] [Google Scholar]
- 235.Xiao X, Qi J, Lei X, Wang J. Interactions between enteroviruses and the inflammasome: new insights into viral pathogenesis. Front Microbiol 10: 321, 2019. doi: 10.3389/fmicb.2019.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Carneiro LA, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT, Plotkowski MC, Sansonetti PJ, Molkentin JD, Philpott DJ, Girardin SE. Shigella induces mitochondrial dysfunction and cell death in nonmyeloid cells. Cell Host Microbe 5: 123–136, 2009. doi: 10.1016/j.chom.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 237.Bergounioux J, Elisee R, Prunier AL, Donnadieu F, Sperandio B, Sansonetti P, Arbibe L. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium's epithelial niche. Cell Host Microbe 11: 240–252, 2012. doi: 10.1016/j.chom.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 238.Pendaries C, Tronchère H, Arbibe L, Mounier J, Gozani O, Cantley L, Fry MJ, Gaits-Iacovoni F, Sansonetti PJ, Payrastre B. PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J 25: 1024–1034, 2006. doi: 10.1038/sj.emboj.7601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Andree M, Seeger JM, Schüll S, Coutelle O, Wagner-Stippich D, Wiegmann K, Wunderlich CM, Brinkmann K, Broxtermann P, Witt A, Fritsch M, Martinelli P, Bielig H, Lamkemeyer T, Rugarli EI, Kaufmann T, Sterner-Kock A, Wunderlich FT, Villunger A, Martins LM, Krönke M, Kufer TA, Utermöhlen O, Kashkar H. BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. EMBO J 33: 2171–2187, 2014. doi: 10.15252/embj.201387244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Clark CS, Maurelli AT. Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells. Infect Immun 75: 2531–2539, 2007. doi: 10.1128/IAI.01866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gunther SD, Fritsch M, Seeger JM, Schiffmann LM, Snipas SJ, Coutelle M, Kufer TA, Higgins PG, Hornung V, Bernardini ML, Höning S, Krönke M, Salvesen GS, Kashkar H. Cytosolic Gram-negative bacteria prevent apoptosis by inhibition of effector caspases through lipopolysaccharide. Nat Microbiol 5: 354–367, 2020. doi: 10.1038/s41564-019-0620-5. [DOI] [PubMed] [Google Scholar]
- 242.Arnett E, Weaver AM, Woodyard KC, Montoya MJ, Li M, Hoang KV, Hayhurst A, Azad AK, Schlesinger LS. PPARgamma is critical for Mycobacterium tuberculosis induction of Mcl-1 and limitation of human macrophage apoptosis. PLoS Pathog 14: e1007100, 2018. doi: 10.1371/journal.ppat.1007100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Cherla R, Zhang Y, Ledbetter L, Zhang G. Coxiella burnetii inhibits neutrophil apoptosis by exploiting survival pathways and antiapoptotic protein Mcl-1. Infect Immun 86: e00504-17, 2018. doi: 10.1128/IAI.00504-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Fischer A, Harrison KS, Ramirez Y, Auer D, Chowdhury SR, Prusty BK, Sauer F, Dimond Z, Kisker C, Hefty PS, Rudel T. Chlamydia trachomatis-containing vacuole serves as deubiquitination platform to stabilize Mcl-1 and to interfere with host defense. Elife 6: e21465, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Sharma M, Machuy N, Böhme L, Karunakaran K, Mäurer AP, Meyer TF, Rudel T. HIF-1alpha is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell Microbiol 13: 1573–1585, 2011. doi: 10.1111/j.1462-5822.2011.01642.x. [DOI] [PubMed] [Google Scholar]
- 246.Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, Haas R, Sasakawa C. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2: 250–263, 2007. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 247.Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ, Peek RM Jr, Cover TL, Washington MK, Wilson KT, Polk DB. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology 136: 1297–1307, 2009. doi: 10.1053/j.gastro.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ying S, Seiffert BM, Häcker G, Fischer SF. Broad degradation of proapoptotic proteins with the conserved Bcl-2 homology domain 3 during infection with Chlamydia trachomatis. Infect Immun 73: 1399–1403, 2005. doi: 10.1128/IAI.73.3.1399-1403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Verbeke P, Welter-Stahl L, Ying S, Hansen J, Häcker G, Darville T, Ojcius DM. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog 2: e45, 2006. doi: 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Niu H, Kozjak-Pavlovic V, Rudel T, Rikihisa Y. Anaplasma phagocytophilum Ats-1 is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog 6: e1000774, 2010. doi: 10.1371/journal.ppat.1000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Klingenbeck L, Eckart RA, Berens C, Lührmann A. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol 15: 675–687, 2013. doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- 252.Lührmann A, Nogueira CV, Carey KL, Roy CR. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci USA 107: 18997–19001, 2010. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bisle S, Klingenbeck L, Borges V, Sobotta K, Schulze-Luehrmann J, Menge C, Heydel C, Gomes JP, Lührmann A. The inhibition of the apoptosis pathway by the Coxiella burnetii effector protein CaeA requires the EK repetition motif, but is independent of survivin. Virulence 7: 400–412, 2016. doi: 10.1080/21505594.2016.1139280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Cuconati A, White E. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev 16: 2465–2478, 2002. doi: 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- 255.García-Murria MJ, Duart G, Grau B, Diaz-Beneitez E, Rodríguez D, Mingarro I, Martínez-Gil L. Viral Bcl2s’ transmembrane domain interact with host Bcl2 proteins to control cellular apoptosis. Nat Commun 11: 6056, 2020. doi: 10.1038/s41467-020-19881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Tsukahara T, Kannagi M, Ohashi T, Kato H, Arai M, Nunez G, Iwanaga Y, Yamamoto N, Ohtani K, Nakamura M, Fujii M. Induction of Bcl-x(L) expression by human T-cell leukemia virus type 1 Tax through NF-kappaB in apoptosis-resistant T-cell transfectants with Tax. J Virol 73: 7981–7987, 1999. doi: 10.1128/JVI.73.10.7981-7987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Munger J, Roizman B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc Natl Acad Sci USA 98: 10410–10415, 2001. doi: 10.1073/pnas.181344498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wolf D, Witte V, Laffert B, Blume K, Stromer E, Trapp S, d’Aloja P, Schürmann A, Baur AS. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat Med 7: 1217–1224, 2001. doi: 10.1038/nm1101-1217. [DOI] [PubMed] [Google Scholar]
- 259.Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, Sharpe J, Youle RJ, Goldmacher VS. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci USA 101: 7988–7993, 2004. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Arnoult D, Skaletskaya A, Estaquier J, Dufour C, Goldmacher VS. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis 13: 1100–1110, 2008. doi: 10.1007/s10495-008-0245-2. [DOI] [PubMed] [Google Scholar]
- 261.Poncet D, Larochette N, Pauleau AL, Boya P, Jalil AA, Cartron PF, Vallette F, Schnebelen C, Bartle LM, Skaletskaya A, Boutolleau D, Martinou JC, Goldmacher VS, Kroemer G, Zamzami N. An anti-apoptotic viral protein that recruits Bax to mitochondria. J Biol Chem 279: 22605–22614, 2004. doi: 10.1074/jbc.M308408200. [DOI] [PubMed] [Google Scholar]
- 262.Cam M, Handke W, Picard-Maureau M, Brune W. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ 17: 655–665, 2010. doi: 10.1038/cdd.2009.147. [DOI] [PubMed] [Google Scholar]
- 263.Desbien AL, Kappler JW, Marrack P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc Natl Acad Sci USA 106: 5663–5668, 2009. doi: 10.1073/pnas.0901036106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Flanagan AM, Letai A. BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ 15: 580–588, 2008. doi: 10.1038/sj.cdd.4402292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kvansakul M, Wei AH, Fletcher JI, Willis SN, Chen L, Roberts AW, Huang DC, Colman PM. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog 6: e1001236, 2010. doi: 10.1371/journal.ppat.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Harold C, Cox D, Riley KJ. Epstein-Barr viral microRNAs target caspase 3. Virol J 13: 145, 2016. doi: 10.1186/s12985-016-0602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li Q, Liston P, Moyer RW. Functional analysis of the inhibitor of apoptosis (iap) gene carried by the entomopoxvirus of Amsacta moorei. J Virol 79: 2335–2345, 2005. doi: 10.1128/JVI.79.4.2335-2345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Waguia Kontchou C, Tzivelekidis T, Gentle IE, Häcker G. Infection of epithelial cells with Chlamydia trachomatis inhibits TNF-induced apoptosis at the level of receptor internalization while leaving non-apoptotic TNF-signalling intact. Cell Microbiol 18: 1583–1595, 2016. doi: 10.1111/cmi.12598. [DOI] [PubMed] [Google Scholar]
- 269.Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol 161: 2636–2641, 1998. [PubMed] [Google Scholar]
- 270.Hu FQ, Smith CA, Pickup DJ. Cowpox virus contains two copies of an early gene encoding a soluble secreted form of the type II TNF receptor. Virology 204: 343–356, 1994. doi: 10.1006/viro.1994.1539. [DOI] [PubMed] [Google Scholar]
- 271.Reading PC, Khanna A, Smith GL. Vaccinia virus CrmE encodes a soluble and cell surface tumor necrosis factor receptor that contributes to virus virulence. Virology 292: 285–298, 2002. doi: 10.1006/viro.2001.1236. [DOI] [PubMed] [Google Scholar]
- 272.Upton C, Macen JL, Schreiber M, McFadden G. Myxoma virus expresses a secreted protein with homology to the tumor necrosis factor receptor gene family that contributes to viral virulence. Virology 184: 370–382, 1991. doi: 10.1016/0042-6822(91)90853-4. [DOI] [PubMed] [Google Scholar]
- 273.Smith CA, Davis T, Anderson D, Solam L, Beckmann MP, Jerzy R, Dower SK, Cosman D, Goodwin RG. A receptor for tumor necrosis factor defines an unusual family of cellular and viral proteins. Science 248: 1019–1023, 1990. doi: 10.1126/science.2160731. [DOI] [PubMed] [Google Scholar]
- 274.Benedict CA, Butrovich KD, Lurain NS, Corbeil J, Rooney I, Schneider P, Tschopp J, Ware CF. Cutting edge: a novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J Immunol 162: 6967–6970, 1999. [PubMed] [Google Scholar]
- 275.Benedict CA, Norris PS, Prigozy TI, Bodmer JL, Mahr JA, Garnett CT, Martinon F, Tschopp J, Gooding LR, Ware CF. Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2. J Biol Chem 276: 3270–3278, 2001. doi: 10.1074/jbc.M008218200. [DOI] [PubMed] [Google Scholar]
- 276.Seirafian S, Prod’homme V, Sugrue D, Davies J, Fielding C, Tomasec P, Wilkinson GW. Human cytomegalovirus suppresses Fas expression and function. J Gen Virol 95: 933–939, 2014. doi: 10.1099/vir.0.058313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, Lung TW, Mansell A, Riedmaier P, Oates CV, Zaid A, Mühlen S, Crepin VF, Marches O, Ang CS, Williamson NA, O'Reilly LA, Bankovacki A, Nachbur U, Infusini G, Webb AI, Silke J, Strasser A, Frankel G, Hartland EL. Type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501: 247–251, 2013. doi: 10.1038/nature12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Pollock GL, Oates CV, Giogha C, Wong Fok Lung T, Ong SY, Pearson JS, Hartland EL. Distinct roles of the antiapoptotic effectors NleB and NleF from enteropathogenic Escherichia coli. Infect Immun 85: e01071-16, 2017. doi: 10.1128/IAI.01071-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Scott NE, Giogha C, Pollock GL, Kennedy CL, Webb AI, Williamson NA, Pearson JS, Hartland EL. The bacterial arginine glycosyltransferase effector NleB preferentially modifies Fas-associated death domain protein (FADD). J Biol Chem 292: 17337–17350, 2017. doi: 10.1074/jbc.M117.805036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, Mocarski ES, Jacobs BL. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci USA 114: 11506–11511, 2017. doi: 10.1073/pnas.1700999114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Komiyama T, Ray CA, Pickup DJ, Howard AD, Thornberry NA, Peterson EP, Salvesen G. Inhibition of interleukin-1 beta converting enzyme by the cowpox virus serpin CrmA. An example of cross-class inhibition. J Biol Chem 269: 19331–19337, 1994. doi: 10.1016/S0021-9258(17)32171-3. [DOI] [PubMed] [Google Scholar]
- 282.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem 272: 7797–7800, 1997. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 283.Shisler JL, Moss B. Molluscum contagiosum virus inhibitors of apoptosis: The MC159 v-FLIP protein blocks Fas-induced activation of procaspases and degradation of the related MC160 protein. Virology 282: 14–25, 2001. doi: 10.1006/viro.2001.0834. [DOI] [PubMed] [Google Scholar]
- 284.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schröter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386: 517–521, 1997. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 285.Bélanger C, Gravel A, Tomoiu A, Janelle ME, Gosselin J, Tremblay MJ, Flamand L. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J Hum Virol 4: 62–73, 2001. [PubMed] [Google Scholar]
- 286.Skaletskaya A, Bartle LM, Chittenden T, McCormick AL, Mocarski ES, Goldmacher VS. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA 98: 7829–7834, 2001. doi: 10.1073/pnas.141108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Chabaud S, Sasseville AM, Elahi SM, Caron A, Dufour F, Massie B, Langelier Y. The ribonucleotide reductase domain of the R1 subunit of herpes simplex virus type 2 ribonucleotide reductase is essential for R1 antiapoptotic function. J Gen Virol 88: 384–394, 2007. doi: 10.1099/vir.0.82383-0. [DOI] [PubMed] [Google Scholar]
- 288.Filippova M, Johnson MM, Bautista M, Filippov V, Fodor N, Tungteakkhun SS, Williams K, Duerksen-Hughes PJ. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J Virol 81: 4116–4129, 2007. doi: 10.1128/JVI.01924-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Tungteakkhun SS, Filippova M, Fodor N, Duerksen-Hughes PJ. The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J Virol 84: 1453–1463, 2010. doi: 10.1128/JVI.01331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Krueger A, Fas SC, Giaisi M, Bleumink M, Merling A, Stumpf C, Baumann S, Holtkotte D, Bosch V, Krammer PH, Li-Weber M. HTLV-1 Tax protects against CD95-mediated apoptosis by induction of the cellular FLICE-inhibitory protein (c-FLIP). Blood 107: 3933–3939, 2006. doi: 10.1182/blood-2005-06-2567. [DOI] [PubMed] [Google Scholar]
- 291.Ashida H, Sasakawa C, Suzuki T. A unique bacterial tactic to circumvent the cell death crosstalk induced by blockade of caspase-8. EMBO J 39: e104469, 2020. doi: 10.15252/embj.2020104469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Blasche S, Mörtl M, Steuber H, Siszler G, Nisa S, Schwarz F, Lavrik I, Gronewold TM, Maskos K, Donnenberg MS, Ullmann D, Uetz P, Kögl M. The E. coli effector protein NleF is a caspase inhibitor. PLoS One 8: e58937, 2013. doi: 10.1371/journal.pone.0058937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Pallett MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, Di Santo JP, Shenoy AR, Berger CN, Frankel G. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol 10: 602–612, 2017. doi: 10.1038/mi.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367, 2011. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol 12: 757–763, 2011. doi: 10.1038/nrm3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Pearson JS, Giogha C, Mühlen S, Nachbur U, Pham CL, Zhang Y, Hildebrand JM, Oates CV, Lung TW, Ingle D, Dagley LF, Bankovacki A, Petrie EJ, Schroeder GN, Crepin VF, Frankel G, Masters SL, Vince J, Murphy JM, Sunde M, Webb AI, Silke J, Hartland EL. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol 2: 16258, 2017. doi: 10.1038/nmicrobiol.2016.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ma W, Tummers B, van Esch EM, Goedemans R, Melief CJ, Meyers C, Boer JM, van der Burg SH. Human papillomavirus downregulates the expression of IFITM1 and RIPK3 to escape from IFNgamma- and TNFalpha-mediated antiproliferative effects and necroptosis. Front Immunol 7: 496, 2016. doi: 10.3389/fimmu.2016.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Croft SN, Walker EJ, Ghildyal R. Human Rhinovirus 3C protease cleaves RIPK1, concurrent with caspase 8 activation. Sci Rep 8: 1569, 2018. doi: 10.1038/s41598-018-19839-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zane L, Sibon D, Jeannin L, Zandecki M, Delfau-Larue MH, Gessain A, Gout O, Pinatel C, Lançon A, Mortreux F, Wattel E. Tax gene expression and cell cycling but not cell death are selected during HTLV-1 infection in vivo. Retrovirology 7: 17, 2010. doi: 10.1186/1742-4690-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem 283: 16966–16970, 2008. doi: 10.1074/jbc.C800051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11: 290–297, 2012. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17: 243–251, 2015. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Liu X, Li Y, Peng S, Yu X, Li W, Shi F, Luo X, Tang M, Tan Z, Bode AM, Cao Y. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis 9: 53, 2018. doi: 10.1038/s41419-017-0081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem 290: 11635–11648, 2015. doi: 10.1074/jbc.M115.646042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Liu Z, Nailwal H, Rector J, Rahman MM, Sam R, McFadden G, Chan FK. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54: 247–258.e7, 2021. doi: 10.1016/j.immuni.2020.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Petrie EJ, Sandow JJ, Lehmann WIL, Liang LY, Coursier D, Young SN, Kersten WJ, Fitzgibbon C, Samson AL, Jacobsen AV, Lowes KN, Au AE, Jousset Sabroux H, Lalaoui N, Webb AI, Lessene G, Manning G, Lucet IS, Murphy JM. Viral MLKL homologs subvert necroptotic cell death by sequestering cellular RIPK3. Cell Rep 28: 3309–3319.e5, 2019. doi: 10.1016/j.celrep.2019.08.055. [DOI] [PubMed] [Google Scholar]
- 307.Fonseca FG, Lanna MC, Campos MA, Kitajima EW, Peres JN, Golgher RR, Ferreira PC, Kroon EG. Morphological and molecular characterization of the poxvirus BeAn 58058. Arch Virol 143: 1171–1186, 1998. doi: 10.1007/s007050050365. [DOI] [PubMed] [Google Scholar]
- 308.Águeda-Pinto A, Alves LQ, Neves F, McFadden G, Jacobs BL, Castro LF, Rahman M, Esteves PJ. Convergent loss of the necroptosis pathway in disparate mammalian lineages shapes virus countermeasures (Preprint). bioRxiv 2021.06.08.447400, 2021. doi: 10.1101/2021.06.08.447400. [DOI] [PMC free article] [PubMed]
- 309.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, Verbist K, Gough PJ, Bertin J, Hartmann BM, Sealfon SC, Kaiser WJ, Mocarski ES, López CB, Thomas PG, Oberst A, Green DR, Balachandran S. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20: 13–24, 2016. doi: 10.1016/j.chom.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, Andrake M, Vogel P, Sigal LJ, tenOever BR, Thomas PG, Upton JW, Balachandran S. DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20: 674–681, 2016. doi: 10.1016/j.chom.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, Ragan KB, Ishizuka T, Crawford JC, Tummers B, Rodriguez DA, Xue J, Peri S, Kaiser WJ, López CB, Xu Y, Upton JW, Thomas PG, Green DR, Balachandran S. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180: 1115–1129.e3, 2020. doi: 10.1016/j.cell.2020.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Kuriakose T, Zheng M, Neale G, Kanneganti TD. IRF1 is a transcriptional regulator of ZBP1 promoting NLRP3 inflammasome activation and cell death during influenza virus infection. J Immunol 200: 1489–1495, 2018. doi: 10.4049/jimmunol.1701538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Oltean T, Van San E, Divert T, Vanden Berghe T, Saelens X, Maelfait J, Takahashi N, Vandenabeele P. Viral dosing of influenza A infection reveals involvement of RIPK3 and FADD, but not MLKL. Cell Death Dis 12: 471, 2021. doi: 10.1038/s41419-021-03746-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Van Valen L. A new evolutionary law. Evol Theory 1: 1–30, 1973. [Google Scholar]
- 315.Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol 17: 151–164, 2017. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Maltez VI, Miao EA. Reassessing the evolutionary importance of inflammasomes. J Immunol 196: 956–962, 2016. doi: 10.4049/jimmunol.1502060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Ayres JS. Cooperative microbial tolerance behaviors in host-microbiota mutualism. Cell 165: 1323–1331, 2016. doi: 10.1016/j.cell.2016.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Sanchez KK, Chen GY, Schieber AMP, Redford SE, Shokhirev MN, Leblanc M, Lee YM, Ayres JS. Cooperative metabolic adaptations in the host can favor asymptomatic infection and select for attenuated virulence in an enteric pathogen. Cell 175: 146–158.e15, 2018. doi: 10.1016/j.cell.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Vostinar AE, Goldsby HJ, Ofria C. Suicidal selection: programmed cell death can evolve in unicellular organisms due solely to kin selection. Ecol Evol 9: 9129–9136, 2019. doi: 10.1002/ece3.5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.James ER, Green DR. Infection and the origins of apoptosis. Cell Death Differ 9: 355–357, 2002. doi: 10.1038/sj.cdd.4400986. [DOI] [PubMed] [Google Scholar]
- 321.De Schutter E, Roelandt R, Riquet FB, Van Camp G, Wullaert A, Vandenabeele P. Punching holes in cellular membranes: biology and evolution of gasdermins. Trends Cell Biol 31: 500–513, 2021. doi: 10.1016/j.tcb.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 322.Jiang S, Zhou Z, Sun Y, Zhang T, Sun L. Coral gasdermin triggers pyroptosis. Sci Immunol 5: eabd2591, 2020. doi: 10.1126/sciimmunol.abd2591. [DOI] [PubMed] [Google Scholar]
- 323.Kvitt H, Kramarsky-Winter E, Maor-Landaw K, Zandbank K, Kushmaro A, Rosenfeld H, Fine M, Tchernov D. Breakdown of coral colonial form under reduced pH conditions is initiated in polyps and mediated through apoptosis. Proc Natl Acad Sci U S A 112: 2082–2086, 2015. doi: 10.1073/pnas.1419621112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Forn-Cuní G, Meijer AH, Varela M. Zebrafish in inflammasome research. Cells 8: 901, 2019. doi: 10.3390/cells8080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Yang D, Zheng X, Chen S, Wang Z, Xu W, Tan J, Hu T, Hou M, Wang W, Gu Z, Wang Q, Zhang R, Zhang Y, Liu Q. Sensing of cytosolic LPS through caspy2 pyrin domain mediates noncanonical inflammasome activation in zebrafish. Nat Commun 9: 3052, 2018. doi: 10.1038/s41467-018-04984-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Li Y, Huang Y, Cao X, Yin X, Jin X, Liu S, Jiang J, Jiang W, Xiao TS, Zhou R, Cai G, Hu B, Jin T. Functional and structural characterization of zebrafish ASC. FEBS J 285: 2691–2707, 2018. doi: 10.1111/febs.14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Li JY, Gao K, Shao T, Fan DD, Hu CB, Sun CC, Dong WR, Lin AF, Xiang LX, Shao JZ. Characterization of an NLRP1 Inflammasome from zebrafish reveals a unique sequential activation mechanism underlying inflammatory caspases in ancient vertebrates. J Immunol 201: 1946–1966, 2018. doi: 10.4049/jimmunol.1800498. [DOI] [PubMed] [Google Scholar]
- 328.Li JY, Wang YY, Shao T, Fan DD, Lin AF, Xiang LX, Shao JZ. The zebrafish NLRP3 inflammasome has functional roles in ASC-dependent interleukin-1beta maturation and gasdermin E-mediated pyroptosis. J Biol Chem 295: 1120–1141, 2020. doi: 10.1016/S0021-9258(17)49920-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J Exp Med 209: 1969–1983, 2012. doi: 10.1084/jem.20121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.George RD, McVicker G, Diederich R, Ng SB, MacKenzie AP, Swanson WJ, Shendure J, Thomas JH. Trans genomic capture and sequencing of primate exomes reveals new targets of positive selection. Genome Res 21: 1686–1694, 2011. doi: 10.1101/gr.121327.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Schaner P, Richards N, Wadhwa A, Aksentijevich I, Kastner D, Tucker P, Gumucio D. Episodic evolution of pyrin in primates: human mutations recapitulate ancestral amino acid states. Nat Genet 27: 318–321, 2001. doi: 10.1038/85893. [DOI] [PubMed] [Google Scholar]
- 332.Jamilloux Y, Lefeuvre L, Magnotti F, Martin A, Benezech S, Allatif O, Penel-Page M, Hentgen V, Seve P, Gerfaud-Valentin M, Duquesne A, Desjonqueres M, Laurent A, Remy-Piccolo V, Cimaz R, Cantarini L, Bourdonnay E, Walzer T, Py BF, Belot A, Henry T. Familial Mediterranean fever mutations are hypermorphic mutations that specifically decrease the activation threshold of the Pyrin inflammasome. Rheumatology (Oxford) 57: 100–111, 2018. doi: 10.1093/rheumatology/kex373. [DOI] [PubMed] [Google Scholar]
- 333.Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The pyrin inflammasome in health and disease. Front Immunol 10: 1745, 2019. doi: 10.3389/fimmu.2019.01745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Digby Z, Rooney J, Boyle JP, Bibo-Verdugo B, Pickering R, Webster SJ, Monie TP, Hopkins LJ, Kayagaki N, Salvesen GS, Warming S, Weinert L, Bryant CE. Evolutionary loss of inflammasomes in carnivores to facilitate carriage of zoonotic infections (Preprint). bioRxiv 2020.12.07.398529, 2020. doi: 10.1101/2020.12.07.398529. [DOI] [PMC free article] [PubMed]
- 335.Cui H, Zhang L. key components of inflammasome and pyroptosis pathways are deficient in canines and felines, possibly affecting their response to SARS-CoV-2 infection. Front Immunol 11: 592622, 2020. doi: 10.3389/fimmu.2020.592622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Han BA, Kramer AM, Drake JM. Global patterns of zoonotic disease in mammals. Trends Parasitol 32: 565–577, 2016. doi: 10.1016/j.pt.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, Wynne JW, Xiong Z, Baker ML, Zhao W, Tachedjian M, Zhu Y, Zhou P, Jiang X, Ng J, Yang L, Wu L, Xiao J, Feng Y, Chen Y, Sun X, Zhang Y, Marsh GA, Crameri G, Broder CC, Frey KG, Wang LF, Wang J. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science 339: 456–460, 2013. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Goh G, Ahn M, Zhu F, Lee LB, Luo D, Irving AT, Wang LF. Complementary regulation of caspase-1 and IL-1beta reveals additional mechanisms of dampened inflammation in bats. Proc Natl Acad Sci USA 117: 28939–28949, 2020. doi: 10.1073/pnas.2003352117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Ahn M, Anderson DE, Zhang Q, Tan CW, Lim BL, Luko K, Wen M, Chia WN, Mani S, Wang LC, Ng JH, Sobota RM, Dutertre CA, Ginhoux F, Shi ZL, Irving AT, Wang LF. Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat Microbiol 4: 789–799, 2019. doi: 10.1038/s41564-019-0371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Moya A, Sakamaki K, Mason BM, Huisman L, Forêt S, Weiss Y, Bull TE, Tomii K, Imai K, Hayward DC, Ball EE, Miller DJ. Functional conservation of the apoptotic machinery from coral to man: the diverse and complex Bcl-2 and caspase repertoires of Acropora millepora. BMC Genomics 17: 62, 2016. doi: 10.1186/s12864-015-2355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Lasi M, David CN, Böttger A. Apoptosis in pre-Bilaterians: hydra as a model. Apoptosis 15: 269–278, 2010. doi: 10.1007/s10495-009-0442-7. [DOI] [PubMed] [Google Scholar]
- 342.Bender CE, Fitzgerald P, Tait SW, Llambi F, McStay GP, Tupper DO, Pellettieri J, Sánchez Alvarado A, Salvesen GS, Green DR. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci USA 109: 4904–4909, 2012. doi: 10.1073/pnas.1120680109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Quistad SD, Stotland A, Barott KL, Smurthwaite CA, Hilton BJ, Grasis JA, Wolkowicz R, Rohwer FL. Evolution of TNF-induced apoptosis reveals 550 My of functional conservation. Proc Natl Acad Sci USA 111: 9567–9572, 2014. doi: 10.1073/pnas.1405912111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Sakamaki K, Shimizu K, Iwata H, Imai K, Satou Y, Funayama N, Nozaki M, Yajima M, Nishimura O, Higuchi M, Chiba K, Yoshimoto M, Kimura H, Gracey AY, Shimizu T, Tomii K, Gotoh O, Akasaka K, Sawasaki T, Miller DJ. The apoptotic initiator caspase-8: its functional ubiquity and genetic diversity during animal evolution. Mol Biol Evol 31: 3282–3301, 2014. doi: 10.1093/molbev/msu260. [DOI] [PubMed] [Google Scholar]
- 345.Eimon PM, Ashkenazi A. The zebrafish as a model organism for the study of apoptosis. Apoptosis 15: 331–349, 2010. doi: 10.1007/s10495-009-0432-9. [DOI] [PubMed] [Google Scholar]
- 346.Yan N, Shi Y. Mechanisms of apoptosis through structural biology. Annu Rev Cell Dev Biol 21: 35–56, 2005. doi: 10.1146/annurev.cellbio.21.012704.131040. [DOI] [PubMed] [Google Scholar]
- 347.Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ 15: 1132–1138, 2008. doi: 10.1038/cdd.2008.50. [DOI] [PubMed] [Google Scholar]
- 348.Yan N, Chai J, Lee ES, Gu L, Liu Q, He J, Wu JW, Kokel D, Li H, Hao Q, Xue D, Shi Y. Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature 437: 831–837, 2005. doi: 10.1038/nature04002. [DOI] [PubMed] [Google Scholar]
- 349.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJ, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol 26: 721–732, 2016. doi: 10.1016/j.tcb.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 350.Daskalov A, Habenstein B, Sabaté R, Berbon M, Martinez D, Chaignepain S, Coulary-Salin B, Hofmann K, Loquet A, Saupe SJ. Identification of a novel cell death-inducing domain reveals that fungal amyloid-controlled programmed cell death is related to necroptosis. Proc Natl Acad Sci USA 113: 2720–2725, 2016. doi: 10.1073/pnas.1522361113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Hofmann K. The evolutionary origins of programmed cell death signaling. Cold Spring Harb Perspect Biol 12: a036442, 2020. doi: 10.1101/cshperspect.a036442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, Li Y, Lu Q, Yuan J. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell 75: 457–468.e4, 2019. doi: 10.1016/j.molcel.2019.05.022. [DOI] [PubMed] [Google Scholar]
- 353.Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, Gough PJ, Feoktistova M, Leverkus M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis 6: e1884, 2015. doi: 10.1038/cddis.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, Levy JM, Pollyea DA, Jordan CT, Yan P, Frankhouser D, Nicolet D, Maharry K, Marcucci G, Choi KS, Cho H, Thorburn A, Kim YS. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25: 707–725, 2015. doi: 10.1038/cr.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Rodrigues TS, de Sá KS, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med 218: e20201707, 2021. doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S, Fintelman-Rodrigues N, Sacramento CQ, Mattos M, de Freitas CS, Temerozo JR, Teixeira L, Damaceno Hottz E, Barreto EA, Pao CR, Palhinha L, Miranda M, Bou-Habib DC, Bozza FA, Bozza PT, Souza TM. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov 7: 43, 2021. doi: 10.1038/s41420-021-00428-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Junqueira C, Crespo A, Ranjbar S, Ingber J, Parry B, Ravid S, de Lacerda LB, Lewandrowski M, Clark S, Ho F, Vora SM, Leger V, Beakes C, Margolin J, Russell N, Gehrke L, Adhikari UD, Henderson L, Janssen E, Kwon D, Sander C, Abraham J, Filbin M, Goldberg MB, Wu H, Mehta G, Bell S, Goldfeld AE, Lieberman J. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release (Preprint). medRxiv 2021.03.06.2125279, 2021. doi: 10.1101/2021.03.06.21252796. [DOI]
- 358.Inde Z, Yapp C, Joshi GN, Spetz J, Fraser C, Deskin B, Ghelfi E, Sodhi C, Hackam D, Kobzik L, Croker B, Brownfield D, Jia H, Sarosiek KA. Age-dependent regulation of SARS-CoV-2 cell entry genes and cell death programs correlates with COVID-19 disease severity (Preprint). bioRxiv 2020.09.13.27692, 2020. doi: 10.1101/2020.09.13.276923. [DOI]
- 359.Nieto-Torres JL, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, Torres J, Aguilella VM, Enjuanes L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485: 330–339, 2015. doi: 10.1016/j.virol.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov 5: 101, 2019. doi: 10.1038/s41420-019-0181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Lu W, Zheng BJ, Xu K, Schwarz W, Du L, Wong CK, Chen J, Duan S, Deubel V, Sun B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc Natl Acad Sci USA 103: 12540–12545, 2006. doi: 10.1073/pnas.0605402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Yue Y, Nabar NR, Shi CS, Kamenyeva O, Xiao X, Hwang IY, Wang M, Kehrl JH. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis 9: 904, 2018. doi: 10.1038/s41419-018-0917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a activates the NLRP3 inflammasome. Front Microbiol 10: 50, 2019. doi: 10.3389/fmicb.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, Yuan S, Chan CP, Yuen KY, Enjuanes L, Jin DY. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J 33: 8865–8877, 2019. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X, Shen J, Zhou Y, Shi ZL, Zhou P, Peng K. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther 5: 235, 2020. doi: 10.1038/s41392-020-00334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Campbell GR, To RK, Hanna J, Spector SA. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience 24: 102295, 2021. doi: 10.1016/j.isci.2021.102295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Chu H, Shuai H, Hou Y, Zhang X, Wen L, Huang X, Hu B, Yang D, Wang Y, Yoon C, Wong BH, Li C, Zhao X, Poon VK, Cai JP, Wong KK, Yeung ML, Zhou J, Au-Yeung RK, Yuan S, Jin DY, Kok KH, Perlman S, Chan JF, Yuen KY. Targeting highly pathogenic coronavirus-induced apoptosis reduces viral pathogenesis and disease severity. Sci Adv 7: eabf8577, 2021. doi: 10.1126/sciadv.abf8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Ye Z, Wong CK, Li P, Xie Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim Biophys Acta 1780: 1383–1387, 2008. doi: 10.1016/j.bbagen.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, Han Y, Zhang XY, Zhou W, Qiu Y, Zhou X. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol 17: 881–883, 2020. doi: 10.1038/s41423-020-0485-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Kim NE, Kim DK, Song YJ. SARS-CoV-2 nonstructural proteins 1 and 13 suppress caspase-1 and the NLRP3 inflammasome activation. Microorganisms 9: 494, 2021. doi: 10.3390/microorganisms9030494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Freeman TL, Swartz TH. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol 11: 1518, 2020. doi: 10.3389/fimmu.2020.01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Anthony SJ, Gilardi K, Menachery VD, Goldstein T, Ssebide B, Mbabazi R, Navarrete-Macias I, Liang E, Wells H, Hicks A, Petrosov A, Byarugaba DK, Debbink K, Dinnon KH, Scobey T, Randell SH, Yount BL, Cranfield M, Johnson CK, Baric RS, Lipkin WI, Mazet JA. Further evidence for bats as the evolutionary source of Middle East Respiratory Syndrome Coronavirus. mBio 8: e00373-17, 2017. doi: 10.1128/mBio.00373-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. Bats are natural reservoirs of SARS-like coronaviruses. Science 310: 676–679, 2005. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 374.Irving AT, Ahn M, Goh G, Anderson DE, Wang LF. Lessons from the host defences of bats, a unique viral reservoir. Nature 589: 363–370, 2021. doi: 10.1038/s41586-020-03128-0. [DOI] [PubMed] [Google Scholar]
- 375.Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 87: 5005–5018, 2013. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Spriggs MK, Hruby DE, Maliszewski CR, Pickup DJ, Sims JE, Buller RM, VanSlyke J. Vaccinia and cowpox viruses encode a novel secreted interleukin-1-binding protein. Cell 71: 145–152, 1992. doi: 10.1016/0092-8674(92)90273-F. [DOI] [PubMed] [Google Scholar]
- 377.Smith VP, Bryant NA, Alcamí A. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J Gen Virol 81: 1223–1230, 2000. doi: 10.1099/0022-1317-81-5-1223. [DOI] [PubMed] [Google Scholar]
- 378.Petit F, Bertagnoli S, Gelfi J, Fassy F, Boucraut-Baralon C, Milon A. Characterization of a myxoma virus-encoded serpin-like protein with activity against interleukin-1 beta-converting enzyme. J Virol 70: 5860–5866, 1996. doi: 10.1128/jvi.70.9.5860-5866.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Kettle S, Alcamí A, Khanna A, Ehret R, Jassoy C, Smith GL. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J Gen Virol 78: 677–685, 1997. doi: 10.1099/0022-1317-78-3-677. [DOI] [PubMed] [Google Scholar]
- 380.Alcamí A, Smith GL. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell 71: 153–167, 1992. doi: 10.1016/0092-8674(92)90274-G. [DOI] [PubMed] [Google Scholar]
- 381.Chiou SK, Tseng CC, Rao L, White E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J Virol 68: 6553–6566, 1994. doi: 10.1128/jvi.68.10.6553-6566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Hong JR, Gong HY, Wu JL. IPNV VP5, a novel anti-apoptosis gene of the Bcl-2 family, regulates Mcl-1 and viral protein expression. Virology 295: 217–229, 2002. doi: 10.1006/viro.2001.1336. [DOI] [PubMed] [Google Scholar]
- 383.Chung YL, Sheu ML, Yen SH. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int J Cancer 107: 65–73, 2003. doi: 10.1002/ijc.11303. [DOI] [PubMed] [Google Scholar]
- 384.Marshall WL, Yim C, Gustafson E, Graf T, Sage DR, Hanify K, Williams L, Fingeroth J, Finberg RW. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J Virol 73: 5181–5185, 1999. doi: 10.1128/JVI.73.6.5181-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci USA 90: 8479–8483, 1993. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 27: 421–433, 2008. doi: 10.1038/sj.onc.1210668. [DOI] [PubMed] [Google Scholar]
- 387.Loh J, Huang Q, Petros AM, Nettesheim D, van Dyk LF, Labrada L, Speck SH, Levine B, Olejniczak ET, Virgin HW 4th.. A surface groove essential for viral Bcl-2 function during chronic infection in vivo. PLoS Pathog 1: e10, 2005. doi: 10.1371/journal.ppat.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Feng P, Liang C, Shin YC, Xiaofei E, Zhang W, Gravel R, Wu TT, Sun R, Usherwood E, Jung JU. A novel inhibitory mechanism of mitochondrion-dependent apoptosis by a herpesviral protein. PLoS Pathog 3: e174, 2007. doi: 10.1371/journal.ppat.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS, Hardwick JM. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci USA 94: 690–694, 1997. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Wang HW, Sharp TV, Koumi A, Koentges G, Boshoff C. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J 21: 2602–2615, 2002. doi: 10.1093/emboj/21.11.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Carpenter D, Hsiang C, Jiang X, Osorio N, BenMohamed L, Jones C, Wechsler SL. The herpes simplex virus type 1 (HSV-1) latency-associated transcript (LAT) protects cells against cold-shock-induced apoptosis by maintaining phosphorylation of protein kinase B (AKT). J Neurovirol 21: 568–575, 2015. doi: 10.1007/s13365-015-0361-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 16: 256–271, 2011. doi: 10.1007/s10495-010-0560-2. [DOI] [PubMed] [Google Scholar]
- 393.Nava VE, Cheng EH, Veliuona M, Zou S, Clem RJ, Mayer ML, Hardwick JM. Herpesvirus saimiri encodes a functional homolog of the human bcl-2 oncogene. J Virol 71: 4118–4122, 1997. doi: 10.1128/jvi.71.5.4118-4122.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci USA 94: 1172–1176, 1997. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Liu HC, Chen GG, Vlantis AC, Tse GM, Chan AT, van Hasselt CA. Inhibition of apoptosis in human laryngeal cancer cells by E6 and E7 oncoproteins of human papillomavirus 16. J Cell Biochem 103: 1125–1143, 2008. doi: 10.1002/jcb.21490. [DOI] [PubMed] [Google Scholar]
- 396.Lötzerich M, Roulin PS, Boucke K, Witte R, Georgiev O, Greber UF. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis 9: 272, 2018. doi: 10.1038/s41419-018-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Brun A, Rivas C, Esteban M, Escribano JM, Alonso C. African swine fever virus gene A179L, a viral homologue of bcl-2, protects cells from programmed cell death. Virology 225: 227–230, 1996. doi: 10.1006/viro.1996.0592. [DOI] [PubMed] [Google Scholar]
- 398.Nogal ML, González de Buitrago G, Rodríguez C, Cubelos B, Carrascosa AL, Salas ML, Revilla Y. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J Virol 75: 2535–2543, 2001. doi: 10.1128/JVI.75.6.2535-2543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Banadyga L, Gerig J, Stewart T, Barry M. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J Virol 81: 11032–11045, 2007. doi: 10.1128/JVI.00734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Wang G, Barrett JW, Nazarian SH, Everett H, Gao X, Bleackley C, Colwill K, Moran MF, McFadden G. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J Virol 78: 7097–7111, 2004. doi: 10.1128/JVI.78.13.7097-7111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Su J, Wang G, Barrett JW, Irvine TS, Gao X, McFadden G. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J Virol 80: 1140–1151, 2006. doi: 10.1128/JVI.80.3.1140-1151.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Turner PC, Sancho MC, Thoennes SR, Caputo A, Bleackley RC, Moyer RW. Myxoma virus Serp2 is a weak inhibitor of granzyme B and interleukin-1beta-converting enzyme in vitro and unlike CrmA cannot block apoptosis in cowpox virus-infected cells. J Virol 73: 6394–6404, 1999. doi: 10.1128/JVI.73.8.6394-6404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Westphal D, Ledgerwood EC, Hibma MH, Fleming SB, Whelan EM, Mercer AA. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J Virol 81: 7178–7188, 2007. doi: 10.1128/JVI.00404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Cooray S, Bahar MW, Abrescia NG, McVey CE, Bartlett NW, Chen RA, Stuart DI, Grimes JM, Smith GL. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gen Virol 88: 1656–1666, 2007. doi: 10.1099/vir.0.82772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Postigo A, Cross JR, Downward J, Way M. Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell Death Differ 13: 1651–1662, 2006. doi: 10.1038/sj.cdd.4401853. [DOI] [PubMed] [Google Scholar]
- 406.Taylor JM, Quilty D, Banadyga L, Barry M. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J Biol Chem 281: 39728–39739, 2006. doi: 10.1074/jbc.M607465200. [DOI] [PubMed] [Google Scholar]
- 407.Zhai D, Yu E, Jin C, Welsh K, Shiau CW, Chen L, Salvesen GS, Liddington R, Reed JC. Vaccinia virus protein F1L is a caspase-9 inhibitor. J Biol Chem 285: 5569–5580, 2010. doi: 10.1074/jbc.M109.078113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Okamoto K, Fujisawa J, Reth M, Yonehara S. Human T-cell leukemia virus type-I oncoprotein Tax inhibits Fas-mediated apoptosis by inducing cellular FLIP through activation of NF-kappaB. Genes Cells 11: 177–191, 2006. doi: 10.1111/j.1365-2443.2006.00927.x. [DOI] [PubMed] [Google Scholar]