

Abstract
Despite the known deleterious effects of obesity, clinical data indicate that overweight or obese patients experience higher rates of sepsis survival compared to normal and underweight patients; a phenomenon called the obesity paradox. Results from preclinical sepsis studies have not been able to replicate these findings. The objective of this study was to test the existence of the obesity paradox in a murine model of cecal slurry (CS)-induced sepsis with insulin-resistant diet-induced obese mice. Male C57BL/6 mice were provided high-fat (HFD) or low-fat (LFD) diets for 20 weeks. HFD-fed mice experienced higher rates of survival compared to LFD-fed mice after septic challenge induced by CS injection (66% vs. 25%, p=0.01, survival assessed for 14 days). Despite the survival advantage, HFD-fed mice had higher rates of positive bacterial cultures and increased markers of kidney injury. Circulating levels of IL-6, IL-1β, TNFα, and IL-23 were equivalent 24h after CS-injection; however, IL-17A was uniquely increased in HFD-fed mice. While LFD-fed mice maintained euglycemia, HFD-fed mice were hyperglycemic 6 and 12h after CS-injection. Stable isotope resolved metabolomics analysis of liver tissue showed diverging pathways of glucose utilization during sepsis, with LFD-fed mice significantly upregulating glycolytic activity and HFD-fed mice decreasing glucose entry into the TCA cycle. This murine study corroborates clinical data that obesity confers a survival benefit in sepsis, albeit at the expense of more significant organ injury. The mechanisms promoting survival in the obese remain unknown; however, this model appears to be well-poised to begin answering this question. Differences in glucose utilization are a novel target to investigate this paradox.
Keywords: glucose, inflammation, hyperglycemia, obesity, preclinical, sepsis
INTRODUCTION
Epidemiological studies have reported that falling within an overweight or obese BMI classification appears to confer a survival advantage during sepsis (1-6). This emerging phenomenon is referred to as the “obesity paradox” (5, 7, 8). While the association between higher BMI and increased survival is reflected in numerous clinical studies, including patients with sepsis as well as other critical illnesses, it is important to acknowledge that controversy exists. A few studies have found no association between obesity and survival, while some report worse outcomes in underweight and morbidly obese suggesting a U-shaped curve. Multiple methodological limitations of observational studies impose a cautious interpretation of the clinical data. Limitations of these studies include inaccuracy of using BMI as a marker of adiposity, acquisition of patient data (self-reported versus measured), timing of data acquisition (relative to initiation of resuscitation or illness-induced weight loss), differences in aggression of treatment between lean and obese patients, and inadequate adjustment for confounding variables (2, 5, 7). Nevertheless, if obesity is protective, understanding the underlying mechanisms would yield insights into sepsis pathogenesis in heterogeneous populations and allow for the development of novel therapeutics.
Preclinical animal studies have mostly been unable to confirm the obesity paradox in sepsis (9, 10). This may be due in large part to diet duration, as most diet-induced obesity (DIO) studies with sepsis models fed mice for only 3-8 weeks (9, 11-16), despite available data indicating that DIO mice, while obese and hyperglycemic, do not develop insulin resistance until at least 11-12 weeks on diet (17, 18). Additionally, studies have predominantly used single-strain bacteria or cecal ligation and puncture, the latter of which is complicated by potential differences in cecal matter consistency and microbiome resulting from different dietary regimens. These studies highlight a need for more research to ascertain the validity of the obesity paradox as well as understand the mechanisms behind it.
In this study, we sought to test the existence of the obesity paradox in a murine model of cecal slurry (CS)-induced sepsis with insulin-resistant diet-induced obese mice. In our study design, particular attention was paid to optimizing the model for clinical relevance in terms of utilizing mice having clinical features associated with obesity. Additionally, we sought to overcome the limitations of prior murine sepsis models by use of CS injection to induce sepsis. Here, we provide evidence that obese, insulin-resistant mice have improved survival in sepsis, albeit at the expense of more severe kidney injury. We further explore a link between glucose metabolism and protection in the obese.
MATERIALS AND METHODS
Animals and Husbandry
Male C57BL/6 mice were obtained from The Jackson Laboratory’s Diet-Induced Obesity colony (B6 DIO: 380050 and DIO Control: 380056, Bar Harbor, ME) at 20 weeks of age. Upon arrival, all mice were maintained in pressurized intra-ventilated cages in an environment under controlled temperature (21–23°C), humidity (30–70%), and lighting (14 hours/10 hours, light/dark) with food and water provided ad libitum. Animals were fed either a high-fat diet (HFD, 60% kcal from fat, D12492 (Research Diets, New Brunswick, NJ)) or a low-fat diet (LFD, 10% kcal from fat, D12450B (Research Diets)) continuously from 6 weeks of age (per Jax Lab protocol) until 26-27 weeks of age (diet duration 20-21 weeks). Of note, LFDs are generally not low in fat compared to standard chow. Body composition was assessed using EchoMRI Body Composition Analyzer (EchoMRI LLC, Houston, TX). All procedures were approved by the Institutional Animal Care and Use Committee at the University of Kentucky (Protocol 2019-3423) and performed in accord with the National Institutes of Health guidelines for ethical animal treatment.
Intraperitoneal Insulin and Glucose Tolerance Testing
After 20 weeks of diet, mice were fasted on wire-bottom cages for 4h or 6h prior to insulin or glucose tolerance test (ITT or GTT), respectively. For ITT, mice were injected with human insulin (Humulin R, Lilly USA, Indianapolis, IN); all mice were given the same volume of insulin based on a dose of 1 U/kg calculated for the average body weight of the LFD group. For GTT, all mice were injected with the same volume of 20% glucose solution, based on a 1g/kg dose calculated for the average body weight of the LFD group. Blood glucose levels were assessed by tail vein micropuncture using a hand-held glucometer (Accu-Check Nano, Roche, Basel, Switzerland).
Cecal Slurry (CS)-induced Polymicrobial Sepsis
CS was prepared in 10% glycerol as previously described (19, 20). Each mouse was injected with 500μL of CS (derived from 50mg cecal contents) intraperitoneally (i.p.) to induce polymicrobial sepsis. Non-sepsis control mice were injected with the same volume of 10% glycerol. For survival tests, body temperatures were monitored every 6h post-injection for the first 24 hours, and every 24h thereafter. Survival was monitored for 14 days. Blood bacterial load was assessed as previously described (19). Briefly, 10μL of blood was collected aseptically from the tail vein 12h after CS-injection, immediately diluted in 90μL of sterile saline and spread onto agar plates containing 3.7% w/v brain-heart infusion broth and 1.5 % w/v agar (Product Number 211059 and 214530, respectively, Becton Dickenson, Franklin Lakes, NJ) and incubated for 24h at 37°C. An additional small blood sample (15uL) was obtained from the tail vain 6h after CS-injection to assess plasma IL-6 levels. Two separate survival studies were done with 8-10 mice in each group and the data combined. In a separate experiment, kidneys and blood were collected 24h after CS injection; mice were anesthetized with isoflurane, exsanguinated by drawing blood from the inferior vena cava into a syringe containing 1/10 volume 0.1M sodium citrate for plasma collection, and perfused with physiological saline via the cardiac ventricles. Plasma was obtained by centrifuging blood at 2500 x g for 15 minutes at 4°C and used for subsequent creatinine and cytokine analyses. Kidneys were snap frozen in liquid N2 and used to assess renal NGAL expression.
Assessment of Kidney Injury
Creatinine concentration in plasma was assessed by isotope dilution LC-MSMS (University of Alabama O’Brien Center for Acute Kidney Injury Research Core C). Expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney was assessed by qRT-PCR using TaqMan assays (NGAL: Mm01324470_m1, HPRT: Mm03024075_m1, Applied Biosystems, ThermoFisher Scientific). Target gene expression was normalized to hypoxanthine-guanine phosphoribosyl transferase (HPRT) expression as an endogenous control, and fold change was calculated as 2−(ΔΔCT), using the mean ΔCT of the control group as a calibrator.
Plasma Cytokine Analyses
Plasma was subjected to multiplex analyses for cytokines (U-Plex Mouse Biomarker K15069L-1/298782, Mesoscale Diagnostics, Rockville, MD). Leptin concentration was assessed by ELISA (KMC2281, Invitrogen, ThermoFisher Scientific).
Glucose and Ketone Assessment during Sepsis
Blood glucose and ketone concentrations were assessed via tail vein micropuncture using a hand-held glucometer (Accu-Check Nano, Roche, Basel, Switzerland) or ketone meter (novaMax Plus, Nova Biomedical, Waltham, MA). These analyses were performed in separate sets of mice subjected to CS-induced sepsis as described above.
In Vivo 13C Glucose Labeling, Sample Preparation, and Gas Chromatography-Mass Spectrometry (GCMS) Analysis
In a separate experiment, five hours after induction of sepsis via CS injection, [U-13C]glucose (2 g/kg body weight) was injected i.p. and mice were euthanized one hour later (6h time point). Blood was collected from the inferior vena cava and tissues were collected after perfusion, snap frozen in liquid N2, and processed for GCMS analyses, as previously described (21). Briefly, liver samples were pulverized to 10 μm particles in liquid N2 and approximately 60 mg of tissue was extracted with 50% MeOH (20 μM L-norleucine) followed by BCA Protein Assay Kit (Pierce) for determination of protein concentration. The polar metabolites were dried at 10−3 mbar followed by derivatization and the dried polar metabolite pellet was derivatized by a two-step methoxyamine protocol. The electron ionization energy was set to 70 eV. Scan (m/z: 50-800) and full scan mode were used for metabolomics analysis. Mass spectra were translated to relative metabolite abundance using the Automated Mass Spectral Deconvolution and Identification System software matched to the FiehnLib metabolomics library (available through Agilent) for retention time and fragmentation pattern matching with a confidence score of > 80 (22-25). Quantitation was performed using the Data Extraction for Stable Isotope-labelled metabolites with a primary ion and at least 2 or more matching qualifying ions. Relative abundance was corrected for recovery using L-norleucine and adjusted to protein input from BCA measure.
Statistical Analyses
Survival data was analyzed using Kaplan-Meier curves and the log-rank test. Bivariate categorical data was compared using Pearson’s Chi Square test. Body temperature over time was analyzed using two-way anova with repeated measures over time. Pairwise comparisons of continuous variables were done using independent sample t-tests or paired t-tests as appropriate. Tukey’s Honest Significant or Least Significant Difference was used for post-hoc pairwise comparisons with independent samples as specified in figure legends. Blood level measurements over time were compared using Area Under the Curve (AUC). Normality and constant variance assumptions were investigated and addressed as needed. Details are provided in figure legends. All analyses were performed using JMP 15.3 statistical software (SAS, Inc., Cary, NC).
RESULTS
Body Weight, Body Composition, and Insulin Resistance in Obese and Non-obese mice
After 20 weeks on respective diets, body weight, fat mass, and lean mass were significantly higher in HFD-fed mice (n=20) compared to LFD-fed mice (n=19, Fig. 1A). Body weights were 46.6 ± 4.53 g vs. 32.3 ± 2.08 g for HFD and LFD, respectively (p<0.001). HFD-fed mice had ~4-fold higher fat mass (16.0 ± 5.21 g vs. 4.1 ± 1.30 g, p<0.001) and slightly higher lean mass (30.4 ±1.85 g vs. 27.4 ± 1.96 g, p<0.001) when compared to the LFD group. In order to determine if insulin resistance, an important clinical parameter of metabolic syndrome, was present, an insulin tolerance test was performed (ITT, Fig. 1B). HFD-fed mice were insulin resistant compared to LFD-fed mice (p=0.0088 by AUC analyses). Glucose tolerance similarly showed a significant difference between the groups (Fig. 1C, p<0.001 by AUC analyses). These data clearly demonstrate that after 20 weeks on a high-fat diet, HFD-fed mice are obese and insulin resistant.
Figure 1. Body Weight, Body Composition, and Insulin Resistance in Obese and Non-obese mice.
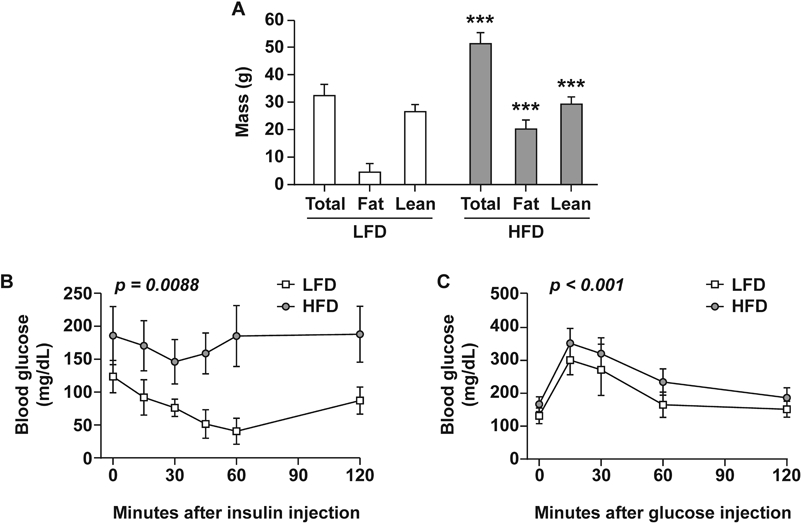
Mice were fed LFD (n=19) or HFD (n=20) from 6 weeks until 26 weeks of age (20-week duration). (A) Body weight, fat mass and lean mass after 20 weeks of diet. Statistical differences were assessed by two sample t-test comparing diet for each group; ***p < 0.001. (B) Insulin tolerance test and (C) glucose tolerance test conducted after 20 weeks of diet. Statistical differences were assessed by one-way analyses of area under the curve (AUC).
Obese mice have improved survival in sepsis despite higher bacterial burden
HFD-fed and LFD-fed mice were given CS injection to induce polymicrobial sepsis and survival was monitored for two weeks; survival was higher in HFD-fed mice compared to LFD-fed mice (66% vs. 25%, p=0.0149; Fig. 2A). Despite higher rates of survival, HFD-fed mice showed greater numbers of positive bacterial cultures (67% vs. 20%, p=0.0259; Fig. 2B). At baseline, both groups showed similar body temperatures and experienced similar sharp declines at 6h post-injection (p=0.1170 between groups, Fig. 2C) indicating equivalent initial injury. Plasma IL-6 at 6h, a predictor of survival (26), showed higher concentrations in the LFD-fed mice (167.4 ± 64.8 in LFD vs. 78.0 ± 39.4 in HFD, p=0.0080, Fig. 2D).
Figure 2. Obese mice are protected from mortality in sepsis despite higher bacterial burden.
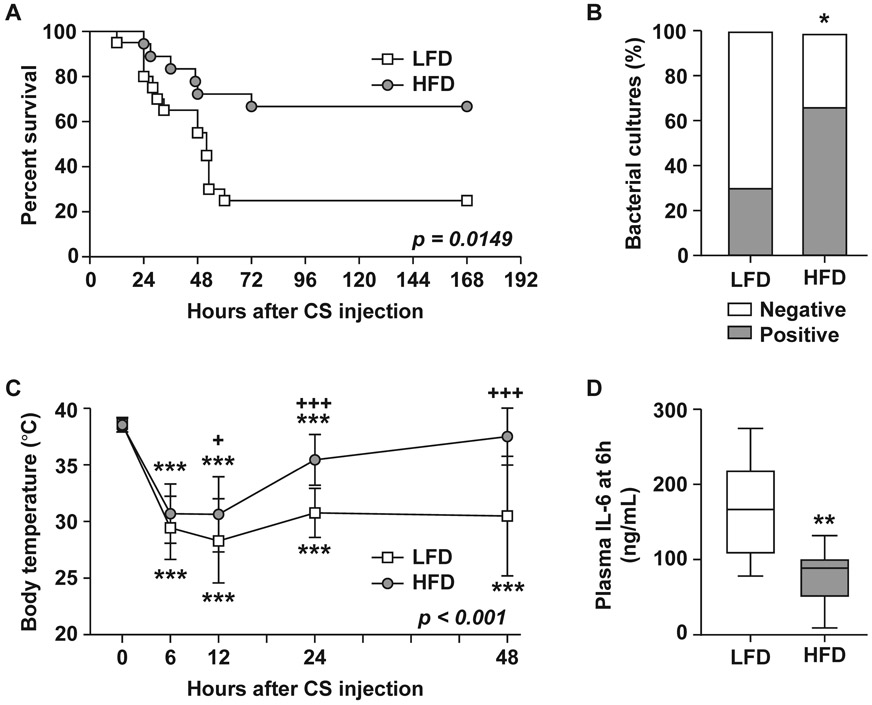
HFD-fed (n=18) and LFD-fed (n=20) mice were injected with CS (500μL, i.p.). (A) Survival was monitored for two weeks. Kaplan-Meier Log-rank test was performed. (B) Circulating bacterial load was assessed 12h after CS injection; data are represented as a proportion of positive vs. negative cultures; statistical difference was determined by chi square test. (C) Body temperature during the first 48h after CS injection; data are expressed as the mean ± SD; significant differences determined by two-way anova over diet and repeated measures over time; * compares timepoints to t=0h within a diet group, + compares diet groups at the same timepoint. (D) Plasma IL-6 concentration at 6h after CS injection. Data are expressed in box plots from minimum to maximum values with a bar representing the mean, statistical difference determined by Student’s t-test. * or + p < 0.05; ** p < 0.01; *** or +++p<0.001.
Acute kidney injury markers are significantly higher in obese mice with sepsis
Markers of kidney injury were assessed 24h after CS or vehicle injection. Plasma creatinine concentration (Fig. 3A) and renal NGAL expression (Fig. 3B) were similar between control groups and increased in septic groups, but showed significantly greater concentrations in the HFD-fed sepsis group compared to LFD sepsis (p=0.0135 and 0.0011 LFD vs. HFD for creatinine and NGAL, respectively).
Figure 3. Markers of acute kidney injury are significantly higher in obese mice with sepsis.
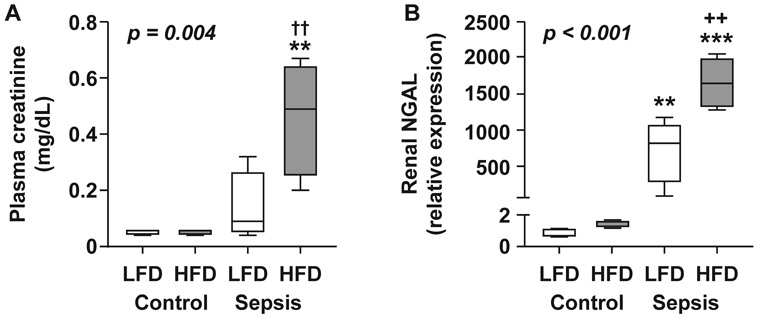
Markers of kidney injury were assessed 24h after injection with CS (LFD n=4, HFD n=5) or 10% glycerol as a control (LFD n=3, HFD n=3) (A) Plasma creatinine concentration. (B) NGAL gene expression in kidney tissue. Data are expressed in box plots from minimum to maximum values with a bar representing the mean; statistical differences were determined by two-way anova of logged response with Tukey’s Honest Significant Difference for multiple comparisons. * compares sepsis vs. control within a diet group, + compares diet groups with the same treatment ** or ++p < 0.01 ***p<0.001.
Inflammatory cytokine profile among obese and non-obese mice with sepsis
To determine if reduced systemic inflammation was a factor contributing to improved survival in the obese, plasma concentrations of pro-inflammatory cytokines were measured 24h after CS or vehicle injection (Fig. 4 A-E). TNFα, IL-1β, IL-6, and IL-23 levels were significantly increased by sepsis in both groups with no significant difference between groups. Interestingly, plasma concentration of IL-17A (Fig. 4D) was significantly higher in the HFD sepsis vs. LFD sepsis (33.7 ± 20.0 pg/mL vs. 7.4 ± 6.9 pg/mL, p=0.0042). Adipose-derived hormone leptin (Fig. 4F) was measured in parallel and showed the typical well-characterized increase during sepsis in the LFD group and blunted increase in the HFD group.
Figure 4. Inflammatory cytokine profile among obese and non-obese mice with sepsis.

Plasma concentrations of (A) TNFα, (B) IL-1β, (C) IL-6 (D) IL-17A, (E) IL-23, and (F) leptin were measured 24h after CS or 10% glycerol (control) injection. Data are expressed in box plots from minimum to maximum values with a bar representing the mean, statistical differences were determined by two-way anova over treatment and diet with the Least Significand Difference method for multiple comparisons. * compares sepsis vs. control within a diet group, + compares diet groups with the same treatment * or +p < 0.05; ** or ++ p < 0.01; *** or +++p<0.001.
Obese mice experience acute hyperglycemia and ketogenesis during sepsis
Because of the impact of metabolic syndrome on glucose regulation, and the known association between deregulated glucose metabolism and metabolic and energetic failure in sepsis, we measured the kinetics of glucose and ketone bodies in the blood following sepsis induction. While LFD-fed mice maintained euglycemia during the acute phase of illness, HFD-fed mice exhibited acute hyperglycemia (p=0.0082 HFD vs. LFD by AUC analyses, Fig. 5A). Ketone body concentrations were significantly increased in both groups at 24-48h; however, HFD mice showed significant elevations as early as 6h; AUC analyses demonstrated a modest significant difference between LFD and HFD groups (p=0.0259, Fig. 5B).
Figure 5. Obese mice experience acute hyperglycemia during sepsis.
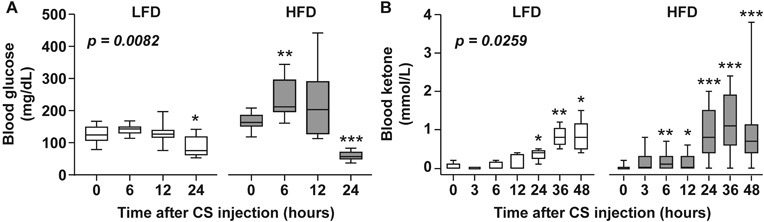
Longitudinal kinetics of blood glucose (A) and ketone (B) levels after CS injection in LFD and HFD-fed mice. For glucose measurements, n=10 for each of LFD and HFD groups. For ketone measurements, n=5 for LFD and 15 for HFD groups, respectively. Data are expressed in box plots from minimum to maximum values with a bar representing the mean, statistical differences between diet groups were determined by AUC analyses, paired t-tests were used to compare baseline (0h) vs. each timepoint within each diet group. * p < 0.05; ** p < 0.01; ***p<0.001.
Glucose tracing shows upregulation of glycolysis in lean, but not obese, mice
Using stable isotope tracing we measured glucose utilization in downstream metabolic pathways in liver tissue isolated from septic or non-septic mice fed either LFD or HFD. Mice received an i.p. injection of [U-13C]glucose and 13C was tracked through glycolysis, highlighting glycolytic intermediates, pyruvate conversion to lactate, and into the TCA cycle (Fig. 6A). A striking increase in glycolytic flux was observed in LFD fed mice upon induction of sepsis; notably this change was not apparent in the HFD-fed mice. Specifically, septic LFD-fed mice displayed significantly higher 13C enrichment from glucose in the glycolytic intermediate phosphoenolpyruvate (PEP) as well as in the end-product lactate (Fig. 6D and F). Similar trends were observed in glycolytic intermediates glycerol-3-phosphate (G3P), 3-phosphoglycerate (3PG), and pyruvate, although these did not achieve statistical significance (Fig. 6B, C, and E). Interestingly, no differences were observed between HFD control and HFD sepsis mice across all glycolytic metabolites measured, notably the HFD mice had increased baseline levels of these intermediates compared to LFD control mice (Fig. 6B-F).
Figure 6. Effects of sepsis on liver glucose metabolism in obese and non-obese mice.

Glucose metabolism measured 6h after sepsis induction. A, Tracing of 13C carbon atoms (grey circles) from [U-13C]glucose through metabolites in glycolysis and into the TCA cycle. (B-F) Fractional enrichment of 13C in glycerol-3-phosphate (G3P), 3-phosphoglycerate (3PG), phosphoenolpyruvate (PEP), pyruvate, and lactate. Fractional 13C labeling denotes the fraction of fully labeled isotopologue relative to the total respective metabolite pool size (13C / 13C + 12C). (G-J) Percentage of 13C-labeling in metabolites associated with the TCA cycle; citrate, fumarate, malate, and aspartate (shown as sum of all labeled isotopologues). Values shown are mean ± SEM (n=3 per control, n=6 per sepsis group). Statistical differences were analyzed by two-way anova over treatment and diet followed by Tukey’s Honest Significant Difference for pairwise comparisons. * p < 0.05; ** p < 0.01.
Conversely, 13C enrichment in TCA cycle metabolites was decreased by sepsis in HFD-fed mice, but not LFD-fed mice. Specifically, septic HFD fed mice showed significantly lower 13C labeling in TCA intermediates fumarate and aspartate, with similar trends observed in citrate and malate (Fig. 6G-J). Together, these results suggest that sepsis increases glycolytic activity among LFD-fed mice, but has no effect in HFD fed mice. Additionally, induction of sepsis leads to a decrease in glucose entry into the TCA cycle in HFD mice, but no change in the LFD groups. In sum, these data suggest that sepsis differentially alters glucose utilization in the background of obesity.
DISCUSSION
In this study we show that our murine model of CS-induced sepsis with diet-induced and insulin resistant obese mice reflects the obesity paradox observed in the clinical literature derived from sepsis patients. Specifically, obese mice exhibited improved survival during sepsis, despite a higher bacterial burden and increased kidney injury. These results are consistent with a large body of clinical literature showing lower rates of mortality in obese and overweight patients, despite longer hospital stays and more significant organ dysfunction. Here, we describe aspects of our study design which allowed us to replicate the obesity paradox preclinically. We also provide avenues related to glucose metabolism for further exploration as to potential mechanisms promoting survival in the obese.
In testing the existence of the obesity paradox in mice, we prioritized parameters that best replicate measures of metabolic syndrome. Previous studies have shown that despite being obese HFD-fed mice do not develop insulin resistance until at least 11-12 weeks on diet (17, 18). Additionally, alterations in adipose tissue physiology, such as cell death and macrophage infiltration, do not peak until 16 weeks on diet (18). Therefore, we extended the duration of our HFD to 20 weeks; this is a far longer diet duration than typical for most studies (only 3-8 weeks (11-14, 16), potentially leading to the different results obtained by those groups. Indeed, one study carrying out the diet for 12 weeks reported a survival advantage in HFD-fed mice after CLP-induced sepsis (27). Moreover, we uniquely induced sepsis via CS injection; this reduced confounding factors that CLP may produce when comparing heterogeneous groups of mice with different intestinal microbiome, cecum size and shape, or stool consistency which may be altered by diet (20, 28).
A puzzling facet of the obesity paradox, as it pertains to sepsis, is the systemic inflammatory process. Many would expect chronic, low-grade inflammation exhibited by obese individuals to compound the acute inflammation produced by sepsis. However, the results of our study do not reflect this dogma as the three major cytokines often blamed for exaggerated inflammation (TNFα, IL-1β, IL-6) did not show significant differences between the groups (24h analyses). Others have shown similar findings, with unchanged or even suppressed levels of IL-6 and TNFα in HFD-fed mice with sepsis depending on the time point of analyses (11-13, 27). Indeed, despite equivalent levels at 24h, our analyses of plasma IL-6 at 6h showed lower levels in HFD mice, which reflects survival based on the six at six principle (26). One clinical study also showed lower IL-6 levels in obese and overweight septic patients compared to non-obese (6). It has been postulated that the obese have a blunted inflammatory response during acute insults due to inflammatory preconditioning. That is, the heightened state of inflammation prior to the onset of sepsis provides a primed system where anti-inflammatory modulators are already upregulated to combat the chronic pro-inflammatory effects of obesity, thus suppressing the typical exaggerated pro-inflammatory response (5, 7, 8). In addition to prompting secretion of anti-inflammatory mediators, the abundance of pro-inflammatory mediators produced by adipose tissue could also confer protection in the obese due to priming of the innate immune system. However, our results showing similar levels of inflammatory mediators, despite improved survival in the obese, does not provide evidence that the inflammatory response is responsible for improved survival.
Other investigators have explored the possibility of the omentum playing a role in the immune response against sepsis. The omentum is a layer of adipose tissue covering the peritoneum. Much like other visceral fat depots, it contains various immune cells including macrophages and mast cells that eliminate bacteria upon phagocytosis (29). Historically, this tissue has been called the “watchdog” or “policeman” of the abdomen due to its ability to promote wound healing and seal visceral perforations thus preventing bacterial spread (30-32). While study of the omentum in mice is often difficult due to its small size in this species, some studies using large CD-1 mice or rats found that removal of the omentum negatively impacted bacterial growth, cytokine expression, and mortality in murine models (33, 34). Further, clinical observations suggest that patients undergoing omentectomy have increased risk of peritonitis (30). In essence, the omentum appears to contain and mitigate the effects of peritoneal infections, possibly playing a role in manifestation of sepsis from abdominal infections. While the omentum of C57BL/6 mice (used in our study) is small, the function of other larger fat depots, such as epididymal fat (which also covers the abdomen, especially in obese mice), may be similar. Nevertheless, in our study, the HFD group had a higher proportion of positive bacterial cultures indicating enhanced bacterial dissemination as opposed to diminished spread supported by the “watchdog” theory.
Many other mechanisms by which obesity could provide a survival advantage during sepsis have been theorized in the literature (5, 7). Among them is the notion that obese and overweight patients utilize excess metabolic reserves which combat the hypercatabolic nature of acute illnesses. The current paradigm regarding glucose metabolism is that there is a shift from oxidative phosphorylation to aerobic glycolysis which, while energetically less favorable, allows for faster ATP production and generation of intermediates necessary for nucleotide, amino acid, and lipid synthesis to support downstream proliferation and inflammatory mediator production (35). Consistent with this model, we found that septic LFD-fed mice appear to increase glycolysis with a shift towards lactate production. However, this shift towards increased aerobic glycolysis (Warburg effect) was not evident in the HFD-fed group. Instead, sepsis in HFD-fed mice was associated with decreased glucose entry into the TCA cycle, a change not observed in the LFD mice. Together, these findings suggest that sepsis disrupts metabolic homeostasis in the obese in a distinct manner. Whether or not these obesity-associated differences in glucose utilization could confer protection remains to be determined.
We found that obese mice experience acute hyperglycemia, perhaps similar to stress hyperglycemia observed clinically. Historically, due to an association between stress hyperglycemia and poor outcomes, tight glycemic control was made standard of care for critically ill patients. However, the association was never proven to be causal, and more recent studies have indicated that hyperglycemia does not always predict mortality and that intensive glycemic control does not improve outcomes (36, 37). One of the largest clinical trials to examine this relationship, NICE-SUGAR, demonstrated that intensive glucose control actually increased mortality when compared to conventional glucose control (38). Other clinical studies have demonstrated that among patients with septic shock, those with stress hyperglycemia had significantly lower mortality than those with normal blood glucose levels (39). This is still an area of intense debate and whether or not acute hyperglycemia in our mouse model is related to improved survival in the obese requires further study. In the setting of sepsis, hyperglycemia is thought to arise primarily from increased hepatic glucose production (i.e. glycogen metabolism or gluconeogenesis), rather than decreased glucose utilization (36, 40-42). Following in vivo administration of fully labeled 13C-glucose (m+6) (as performed in the current study) gluconeogenesis can be estimated from plasma levels of m+1-m+5 labeled glucose (43). However, we did not detect measurable quantities of any m+1-m+5 isotopologues of glucose in the plasma in any group. While one interpretation of this lack of signal is that hyperglycemia in the HFD-fed mice during sepsis is not mediated by increased hepatic glucose production, the possibility cannot be ruled out because: i) the time point and 13C-glucose dose injected may not be optimal to detect gluconeogenesis, and ii) we are unable to distinguish endogenous circulating glucose (12C; m+0) from glucose produced de novo via hepatic glycogenolysis (also m+0).
Despite obesity being protective in terms of survival, HFD-fed mice experienced greater incidence of kidney injury, as evidenced by increased expression of renal NGAL and elevated plasma creatinine. These results are in line with murine and clinical data (9, 44). IL-17A, which is the only inflammatory cytokine having significantly higher levels in the septic HFD-fed mice in our study, has been shown to contribute to kidney injury in CLP-induced sepsis as knockout of IL-17A improved functional and morphological measures of AKI, and exogenous administration of IL-17A aggravated AKI (45). Data derived from septic-AKI patients and human cells stimulated ex vivo support the hypothesis that IL-17A drives AKI in sepsis (46). While we did not directly test the effects of IL-17A, prior work in this area could explain why obese mice have greater incidence of kidney injury despite having improved overall survival.
Although we strove to bolster the clinical relevance of our study by making modifications to the animal modeling which are not apparent in other related studies, there were limitations worth noting. First, while we used a longer diet duration than most studies, the duration of high-fat feeding is still not matching that which occurs in obese humans who tend to have poor diets spanning years to decades. Use of adult mice resembling middle to old age humans with high-fat feeding over the entire life course would be more representative; however, the length of time necessary to complete such a study precluded its use as a first attempt in modeling the obesity paradox. Moreover, despite our use of a purified HFD with a nutrient- and ingredient-matched purified LFD, which are well-regarded in the obesity field but often left out of sepsis studies, the diets themselves are homogeneous and not accurately reflecting the diets of obese or lean humans. With regard to the sepsis model, we chose the CS model as it avoided limitations of other studies; however, it is still an imperfect animal model only replicating in part an abdominal peritonitis induced by frozen inocula and not capturing sepsis of other etiologies. Further, in this study we did not provide antibiotics or fluid resuscitation which would have provided more clinical relevance. Our rationale for eliminating these additions was a desire to evaluate differences in bacterial burden which would have been masked if antibiotics were applied. As resuscitation influences the outcome of sepsis, further studies using this model should incorporate a resuscitation protocol prior to proposing therapeutic interventions based on new findings.
In summary, using a modified model of cecal slurry-induced sepsis with HFD-fed, obese, insulin-resistant mice, we replicated features of the obesity paradox which are often observed in patients. Our model includes further paradoxical aspects such as increased bacterial dissemination and increased kidney injury despite reduced mortality. Further, our data points to stress hyperglycemia and differential glucose utilization as potential mechanisms mediating the protective effects of obesity in sepsis.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the Markey Cancer Center Research Communications Office for illustrative expertise.
Conflicts of Interest and Sources of Funding
All authors declare no relevant conflicts of interest. This study was supported by NIH grants R01 GM129532 (awarded to MES), R01 GM126181 (awarded to HS), T32 AG057461 (HCW), and R01 AG060056 and R01 AG062550 (awarded to LAJ). Support for core facilities utilized in this study were provided by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (NIH) under grant number P30 GM127211, the National Center for Research Resources and the National Center for Advancing Translational Sciences (NIH) through Grant UL1TR001998, and by the National Institute of Diabetes and Digestive and Kidney Diseases P30 DK 079337 awarded to the O’Brien Center for Acute Kidney Injury Research.
REFERENCES
- 1.Pepper DJ, Sun J, Welsh J, Cui X, Suffredini AF, Eichacker PQ. Increased body mass index and adjusted mortality in ICU patients with sepsis or septic shock: a systematic review and meta-analysis. Crit Care 20(1):181, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pepper DJ, Demirkale CY, Sun J, Rhee C, Fram D, Eichacker P, Klompas M, Suffredini AF, Kadri SS. Does Obesity Protect Against Death in Sepsis? A Retrospective Cohort Study of 55,038 Adult Patients. Crit Care Med 47(5):643–50, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li S, Hu X, Xu J, Huang F, Guo Z, Tong L, Lui KY, Cao L, Zhu Y, Yao J, et al. Increased body mass index linked to greater short- and long-term survival in sepsis patients: A retrospective analysis of a large clinical database. Int J Infect Dis 87:109–16, 2019. [DOI] [PubMed] [Google Scholar]
- 4.Wang S, Liu X, Chen Q, Liu C, Huang C, Fang X. The role of increased body mass index in outcomes of sepsis: a systematic review and meta-analysis. BMC Anesthesiol 17(1):118, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karampela I, Chrysanthopoulou E, Christodoulatos GS, Dalamaga M. Is There an Obesity Paradox in Critical Illness? Epidemiologic and Metabolic Considerations. Curr Obes Rep 9(3):231–44, 2020. [DOI] [PubMed] [Google Scholar]
- 6.Wacharasint P, Boyd JH, Russell JA, Walley KR. One size does not fit all in severe infection: obesity alters outcome, susceptibility, treatment, and inflammatory response. Crit Care 17(3):R122, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng PY, Eikermann M. The obesity conundrum in sepsis. BMC Anesthesiol 17(1):147, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson J, Swift-Scanlan T, Salyer J, Jones T. The Obesity Paradox in Sepsis: A Theoretical Framework. Biol Res Nurs 22(2):287–94, 2020. [DOI] [PubMed] [Google Scholar]
- 9.Xu W, Pepper D, Sun J, Welsh J, Cui X, Eichacker PQ. The Effects of Obesity on Outcome in Preclinical Animal Models of Infection and Sepsis: A Systematic Review and Meta-Analysis. J Obes 2020:1508764, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittwede PN, Clemmer JS, Bergin PF, Xiang L. Obesity and critical illness: insights from animal models. Shock 45(4):349–58, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strandberg L, Verdrengh M, Enge M, Andersson N, Amu S, Onnheim K, Benrick A, Brisslert M, Bylund J, Bokarewa M, et al. Mice chronically fed high-fat diet have increased mortality and disturbed immune response in sepsis. PLoS One 4(10):e7605, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan JM, Nowell M, Lahni P, O'Connor MP, Hake PW, Zingarelli B. Short-term high fat feeding increases organ injury and mortality after polymicrobial sepsis. Obesity 20(10):1995–2002, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan JM, Nowell M, Lahni P, Shen H, Shanmukhappa SK, Zingarelli B. Obesity enhances sepsis-induced liver inflammation and injury in mice. Obesity 24(7):1480–8, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svahn SL, Grahnemo L, Pálsdóttir V, Nookaew I, Wendt K, Gabrielsson B, Schéle E, Benrick A, Andersson N, Nilsson S, et al. Dietary polyunsaturated fatty acids increase survival and decrease bacterial load during septic Staphylococcus aureus infection and improve neutrophil function in mice. Infect Immun 83(2):514–21, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svahn SL, Ulleryd MA, Grahnemo L, Ståhlman M, Borén J, Nilsson S, Jansson JO, Johansson ME. Dietary Omega-3 Fatty Acids Increase Survival and Decrease Bacterial Load in Mice Subjected to Staphylococcus aureus-Induced Sepsis. Infect Immun 84(4):1205–13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan T, Yuan G, Ren Y, Zuo Z, Wang Z, Jia Y, Cui H, Peng X, Fang J, Deng J, et al. Diet-induced obese mice exhibit altered immune responses to acute lung injury induced by Escherichia coli. Obesity 24(10):2101–10, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta D, Jetton TL, LaRock K, Monga N, Satish B, Lausier J, Peshavaria M, Leahy JL. Temporal characterization of β cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. J Biol Chem 292(30):12449–59, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heydemann A An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J Diabetes Res 2016:2902351, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steele AM, Starr ME, Saito H. Late Therapeutic Intervention with Antibiotics and Fluid Resuscitation Allows for a Prolonged Disease Course with High Survival in a Severe Murine Model of Sepsis. Shock 47(6):726–34, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starr ME, Steele AM, Saito M, Hacker BJ, Evers BM, Saito H. A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS One 9(12):e115705, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams HC, Piron MA, Nation GK, Walsh AE, Young LEA, Sun RC, Johnson LA. Oral Gavage Delivery of Stable Isotope Tracer for In Vivo Metabolomics. Metabolites 10(12), 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiehn O, Kopka J, Dörmann P, Altmann T, Trethewey RN, Willmitzer L. Metabolite profiling for plant functional genomics. Nat Biotechnol 18(11):1157–61, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Fiehn O Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr Protoc Mol Biol 114:30.4.1–4.2, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kind T, Tolstikov V, Fiehn O, Weiss RH. A comprehensive urinary metabolomic approach for identifying kidney cancer. Anal Biochem 363(2):185–95, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Kind T, Wohlgemuth G, Lee DY, Lu Y, Palazoglu M, Shahbaz S, Fiehn O. FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal Chem 81(24):10038–48, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17(6):463–7, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Siegl D, Annecke T, Johnson BL, Schlag C, Martignoni A, Huber N, Conzen P, Caldwell CC, Tschöp J. Obesity-induced hyperleptinemia improves survival and immune response in a murine model of sepsis. Anesthesiology 121(1):98–114, 2014. [DOI] [PubMed] [Google Scholar]
- 28.Nagpal R, Mishra SP, Yadav H. Unique Gut Microbiome Signatures Depict Diet-Versus Genetically Induced Obesity in Mice. Int J Mol Sci 21(10), 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang AW, Prieto JM, Cauvi DM, Bickler SW, De Maio A. The Greater Omentum-A Vibrant and Enigmatic Immunologic Organ Involved in Injury and Infection Resolution. Shock 53(4):384–90, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ambroze WL, Wolff BG, Kelly KA, Beart RW, Dozois RR, Ilstrup DM. Let sleeping dogs lie: role of the omentum in the ileal pouch-anal anastomosis procedure. Dis Colon Rectum 34(7):563–5, 1991. [DOI] [PubMed] [Google Scholar]
- 31.Liebermann-Meffert D The greater omentum. Anatomy, embryology, and surgical applications. Surg Clin North Am 80(1):275–93, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Collins D, Hogan AM, O'Shea D, Winter DC. The omentum: anatomical, metabolic, and surgical aspects. J Gastrointest Surg 13(6):1138–46, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Wang AW, Cauvi DM, Hawisher D, Reyes T, Coimbra R, Bickler S, De Maio A. The Contribution of the Omentum to the Outcome From Sepsis: An Experimental Animal Study. Shock 52(6):604–11, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uzunköy A, Ozbilge H, Horoz M. The influence of omentectomy on bacterial clearance: an experimental study. Ulus Travma Acil Cerrahi Derg 15(6):541–5, 2009. [PubMed] [Google Scholar]
- 35.Van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med 10(8), 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marik PE, Bellomo R. Stress hyperglycemia: an essential survival response! Crit Care 17(2):305, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Green JP, Berger T, Garg N, Horeczko T, Suarez A, Radeos MS, Hagar Y, Panacek EA. Hyperlactatemia affects the association of hyperglycemia with mortality in nondiabetic adults with sepsis. Acad Emerg Med 19(11):1268–75, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, Henderson WR, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 360(13):1283–97, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Tiruvoipati R, Chiezey B, Lewis D, Ong K, Villanueva E, Haji K, Botha J. Stress hyperglycemia may not be harmful in critically ill patients with sepsis. J Crit Care 27(2):153–8, 2012. [DOI] [PubMed] [Google Scholar]
- 40.Dungan KM, Braithwaite SS, Preiser JC. Stress hyperglycaemia. Lancet 373(9677):1798–807, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bar-Or D, Rael LT, Madayag RM, Banton KL, Tanner A, Acuna DL, Lieser MJ, Marshall GT, Mains CW, Brody E. Stress Hyperglycemia in Critically Ill Patients: Insight Into Possible Molecular Pathways. Front Med 6:54, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Revelly JP, Tappy L, Martinez A, Bollmann M, Cayeux MC, Berger MM, Chioléro RL. Lactate and glucose metabolism in severe sepsis and cardiogenic shock. Crit Care Med 33(10):2235–40, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Chung ST, Chacko SK, Sunehag AL, Haymond MW. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes 64(12):3996–4010, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiffl H. Obesity and the Survival of Critically Ill Patients with Acute Kidney Injury: A Paradox within the Paradox? Kidney Dis 6(1):13–21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naito Y, Tsuji T, Nagata S, Tsuji N, Fujikura T, Ohashi N, Kato A, Miyajima H, Yasuda H. IL-17A activated by Toll-like receptor 9 contributes to the development of septic acute kidney injury. Am J Physiol Renal Physiol 318(1):F238–F47, 2020. [DOI] [PubMed] [Google Scholar]
- 46.Maravitsa P, Adamopoulou M, Pistiki A, Netea MG, Louis K, Giamarellos-Bourboulis EJ. Systemic over-release of interleukin-17 in acute kidney injury after septic shock: Clinical and experimental evidence. Immunol Lett 178:68–76, 2016. [DOI] [PubMed] [Google Scholar]