

Abstract
BACKGROUND AND AIMS:
A major clinical challenge for pancreatic cancer (PC) patients is metabolic adaptation. Neoplastic cells harboring molecular perturbations suffice for their increased anabolic demand and nucleotide biosynthesis to acquire chemoresistance. The mucin 5AC expressed de novo in malignant pancreas promotes cancer cell stemness and is significantly associated with poor patient survival. Identification of MUC5AC-associated drivers of chemoresistance through metabolic alterations may facilitate sculpting a new combinatorial regimen.
METHODS:
The contribution of MUC5AC on glutaminolysis and gemcitabine resistance were examined by TCGA data analysis, RNA sequencing, and immunohistochemistry analysis on pancreatic tissues of KrasG12D; Pdx1-Cre (KC) and KrasG12D; Pdx1-Cre; Muc5ac−/− (KCM) mice. These were followed by metabolite flux assays, biochemical and xenograft studies on MUC5AC-depleted human and murine PC cells. Murine and human pancreatic 3D-tumoroids were used to evaluate gemcitabine’s efficacy in combination with β-catenin and glutaminolysis inhibitors.
RESULTS:
Transcriptional analysis demonstrated that high MUC5AC-expressing human and autochthonous murine PC tumors exhibit higher resistance to gemcitabine due to enhanced glutamine utilization and nucleotide biosynthesis. Gemcitabine treatment led to MUC5AC overexpression, resulting in disruption of E-Cadherin/β-catenin junctions and the nuclear translocation of β-catenin, which increased c-Myc expression with a concomitant rise in glutamine uptake and glutamate release. MUC5AC-depletion and glutamine deprivation sensitized human PC cells to gemcitabine, which was obviated by glutamine replenishment in MUC5AC-expressing cells. Co-administration of β-catenin and glutaminolysis inhibitors with gemcitabine abrogated the MUC5AC-mediated resistance in murine and human tumoroids.
CONCLUSIONS:
MUC5AC/β-catenin/c-Myc axis increases the uptake and utilization of glutamine in PC cells and co-targeting this axis along with gemcitabine may improve the therapeutic efficacy in PC.
Keywords: glutamine, gemcitabine, c-Myc, β-catenin, pancreatic cancer
Lay Summary
Secreted mucin 5AC, overexpressed in pancreatic cancer is responsible for chemoresistance. Therefore, targeting downstream players of MUC5AC-associated drug-resistance along with the standard-of-care may improve the survival of pancreatic cancer patients.
Introduction
Despite several years of concerted efforts, pancreatic cancer (PC) continues to be a highly lethal malignancy with a dismal 5-year survival rate of 10% (pancan.org). With almost 80% of patients presenting with an unresectable disease with local or distant metastasis, the existing chemotherapeutic regimen is the only available treatment option. In spite of the 2-fold increase in the overall median survival with FOLFIRINOX as compared to gemcitabine monotherapy, the high toxicity index of FOLFIRINOX limits its utility; therefore, the gemcitabine and the nab-paclitaxel combination is the widely used first-line regimen for PC patients. Nevertheless, the intrinsic ability of cancer cells to surmount the cytotoxicity of chemotherapeutic drugs by developing resistance to gemcitabine-based therapies culminate in poor patient outcome1. Hence, identification and selective targeting of the molecular determinants of gemcitabine resistance will help in sculpting the novel combination regimen for PC management.
Constitutive activation of the oncogenic KRAS, the major initiating event of PC, is observed in 95% of PC patients. The KRAS-associated transcriptional reprogramming and aberrant signaling are assumed to play critical roles in disease pathogenesis and development of chemoresistance2, 3. Among these, the de novo expression of the secreted mucin 5AC (MUC5AC), a polymeric glycoprotein absent in the normal pancreas, has recently been implicated in PC progression via STAT3/KLF4-associated maintenance and enrichment of cancer stem cells (CSCs)4. The involvement of MUC5AC in CSC enrichment and a strong correlation with the worst clinical outcome of PC patients led us to evaluate the association between MUC5AC expression and therapy response. Our analyses from the TCGA dataset and human pancreatic tumoroids revealed that low MUC5AC-expressing patients exhibit reduced glutamine utilization and higher sensitivity to gemcitabine than the high expressing group.
Interestingly, the oncogenic KRAS upregulates the glycolytic pathway5 and reprogram glutamine metabolism6, in order to suffice for the high anabolic requirements. This KRAS-mediated metabolic rewiring includes an increase in the uptake of glutamine, anaplerotic utilization of glutamine for producing the TCA cycle intermediates, mobilization of the carbon and nitrogen of glutamine towards nucleotide biosynthesis, conversion of glutamine to glutamate, and the concurrent uptake of cysteine for glutathione synthesis7. Glutamine dependency of tumor cells increases by several folds upon administration of nucleotide analogs like gemcitabine, which impart cytotoxicity by stalling DNA synthesis and repair due to its incorporation into the replicating DNA. Tumor cells rely on de novo nucleotide biosynthesis to surpass the competitive inhibition of DNA synthesis and thereby acquire resistance to the nucleotide analogs8. Hence, limiting the intracellular glutamine availability and suppressing glutamine utilization for nucleotide synthesis by tumor cells can curb their natural nucleotide pool and efficiently augment the chemosensitivity9. However, the molecular determinants and mechanisms facilitating this metabolic adaption in the cancer cells to acquire chemoresistance are not completely understood.
Extending our findings from TCGA dataset analysis, observations from the autochthonous murine organoids and human PC cells suggest that MUC5AC depletion increases the gemcitabine sensitivity both in vitro and in vivo. The transcriptomic, biochemical, and promoter occupancy studies demonstrated that mechanistically, MUC5AC-mediated nuclear accumulation of β-catenin resulted in the transcriptional upregulation of c-Myc, which enhanced glutamine uptake and utilization, leading to enhanced deoxycytidine triphosphate (dCTP) biosynthesis and gemcitabine resistance. Further, inhibition of the MUC5AC/β-catenin/c-Myc axis using pharmacological inhibitors and genetic silencing dampened glutamine metabolism, resulting in the sensitization of MUC5AC-expressing PC cells and tumor organoids to gemcitabine treatment.
Materials and Methods
PC autochthonous mouse model:
KC (KrasG12D, Pdx1-Cre) and KCM (KrasG12D, Pdx1-Cre, Muc5ac−/−) mice were generated as mentioned before4. Pancreatic tissue sections from 50 weeks old KC and KCM mice were stained for the expression of Slc7a11, Slc1a5, and β-catenin, which were blindly scored by a pathologist. RNA seq analysis (GSE160029) was performed previously on the KrasG12D, Pdx-1-Cre (KC), and KrasG12D, Pdx-1-Cre, Muc5ac−/− (KCM) mice pancreatic tumors.
Real-time kinetics of drug efficacy on murine and human PC organoids:
KC, KCM mice, and human tumors were used to generate 3D-organoids4, 10. Murine and human organoids, in three or four independent replicates, were treated with gemcitabine alone (Gemcitabine hydrochloride, Sigma 122111-03-9) or in combination with β-catenin inhibitor (CCT 031374, Sigma 1219184-9-4) or Glutaminase 1 inhibitor (CB-839, Sigma 1439399-58-2). IncuCyte-S3 live-cell imaging system (Essen BioScience) was used to capture real-time images of the organoids, every 3–6 hours for 48 hours, and the kinetic data was analyzed using IncuCyte software (Essen BioScience). Increase in organoid darkness and decrease in organoid area normalized to the starting time-point for each well was used as the metric for cytotoxic death. For further analysis, culture supernatant and RNA were collected from the organoids at the experimental endpoint.
Drug-efficacy on xenograft model:
FG-COLO357 (Scr and Sh5AC) cells were implanted subcutaneously in both flanks of immunocompromised mice (male and female) at 0.5×105 cells/50ul/flank (n=10/group). Tumor volume was measured every alternate day using digital calipers. The mice were randomized after 15th day of implantation to receive either saline or gemcitabine at a concentration of 12.5mg/Kg body weight, twice weekly for 2 weeks. The tumors were resected 2-days after the last injection, and tumor weight was measured. Tumor sections were immunostained for the expression of MUC5AC, cleaved PARP, SLC7A11, SLC1A5 and were blindly scored by a pathologist. Immunostaining followed by analyzing the number of β-catenin foci/5 fields/tumor section was performed for each group using ImageJ software (National Institutes of Health).
Luminescence-based metabolic flux assay:
Culture supernatant from cells and organoids, and tumor homogenates from the subcutaneous tumors (made in RIPA lysis buffer) were subjected to luminescence-based Glutamine/Glutamate Glo assay, as mentioned in the manufacturer’s protocol (J8021, Promega, USA). Briefly, the samples were diluted at 1:50 in PBS and 25 uL of diluted sample was used to measure glutamine plus glutamate concentration after incubation with glutaminase. 25 ul of diluted sample was used to measure the endogenous glutamate concentration in the sample. Glutamine concentration was obtained by subtracting the endogenous glutamate concentration from the combined glutamine plus glutamate concentration. On the basis of the sequential enzymatic process, luciferin was liberated from an inactive pro-luciferin reductase substrate leading to the generation of luminescence, which was read by BioTek Synergy H1 microplate reader (BioTek, VT, USA).
Statistical analyses:
The power analysis for the animal numbers were performed using an alpha error probability of 0.05 and a power level of 0.8. Normally distributed data were compared using a two-tailed independent sample student t-test, and the non-normally distributed data were analyzed by Wilcoxon rank-sum test. A p-value of < 0.05 was considered statistically significant. All the in vitro assays were repeated at least three times with biological and technical replicates. The detailed description of assays, reagents (source and dilutions of antibodies, the sequence of primers), and bioinformatics analyses (TCGA datasets, web tools, and cut-off criteria) used in this study are provided in the Supplementary Materials and Methods.
Results
Elevated expression of MUC5AC confers gemcitabine resistance in PC
Analysis from the TCGA and GTEx datasets demonstrated that MUC5AC is overexpressed in the pancreatic tumors as compared to the normal organ (Figure 1A) in a stage-dependent manner (Figure 1B) and is also significantly associated with poor overall survival of PC patients (Figure 1C). In order to comprehend the clinical association of MUC5AC with PC chemoresistance, patients who received gemcitabine in the TCGA cohort were segregated into high and low MUC5AC-expressors, based on the median values. While low MUC5AC-expressors were more responsive to gemcitabine, almost 85% of high MUC5AC-expressing patients exhibited a progressive disease after gemcitabine treatment (Figure 1D). To interrogate the functional contribution of MUC5AC in gemcitabine sensitivity, human PC cells FG-COLO357 and SW1990 were treated with gemcitabine after sh-RNA mediated knockdown (KD) of MUC5AC (Supplementary Figure 1A). The MUC5AC-KD cells (FG-Sh5AC and SW-Sh5AC) demonstrated a significant decline in cell viability (Supplementary Figure 1B) with a concurrent increase in an apoptotic index (Figure 1E, Supplementary Figure 1C) upon gemcitabine administration in a dose and time-dependent manner compared to scrambled control. Expression of pro-apoptotic markers like cleaved-caspase 3 and cleaved PARP were also significantly elevated in MUC5AC-depleted cells (Figure 1F, Supplementary Figure 1D). Furthermore, 48 hours of gemcitabine treatment at an IC50 concentration led to a 1.5– 2 folds increase in MUC5AC expression as well as 1.5– 4.5 folds enrichment of MUC5AC-expressing population in the murine and human PC cell lines (Figure 1G, H; Supplementary Figure 1F, G, H, I), indicating that MUC5AC indeed protects the PC cells from gemcitabine-mediated cytotoxicity.
Figure 1. MUC5AC is associated with poor prognosis and gemcitabine resistance in PC patients.
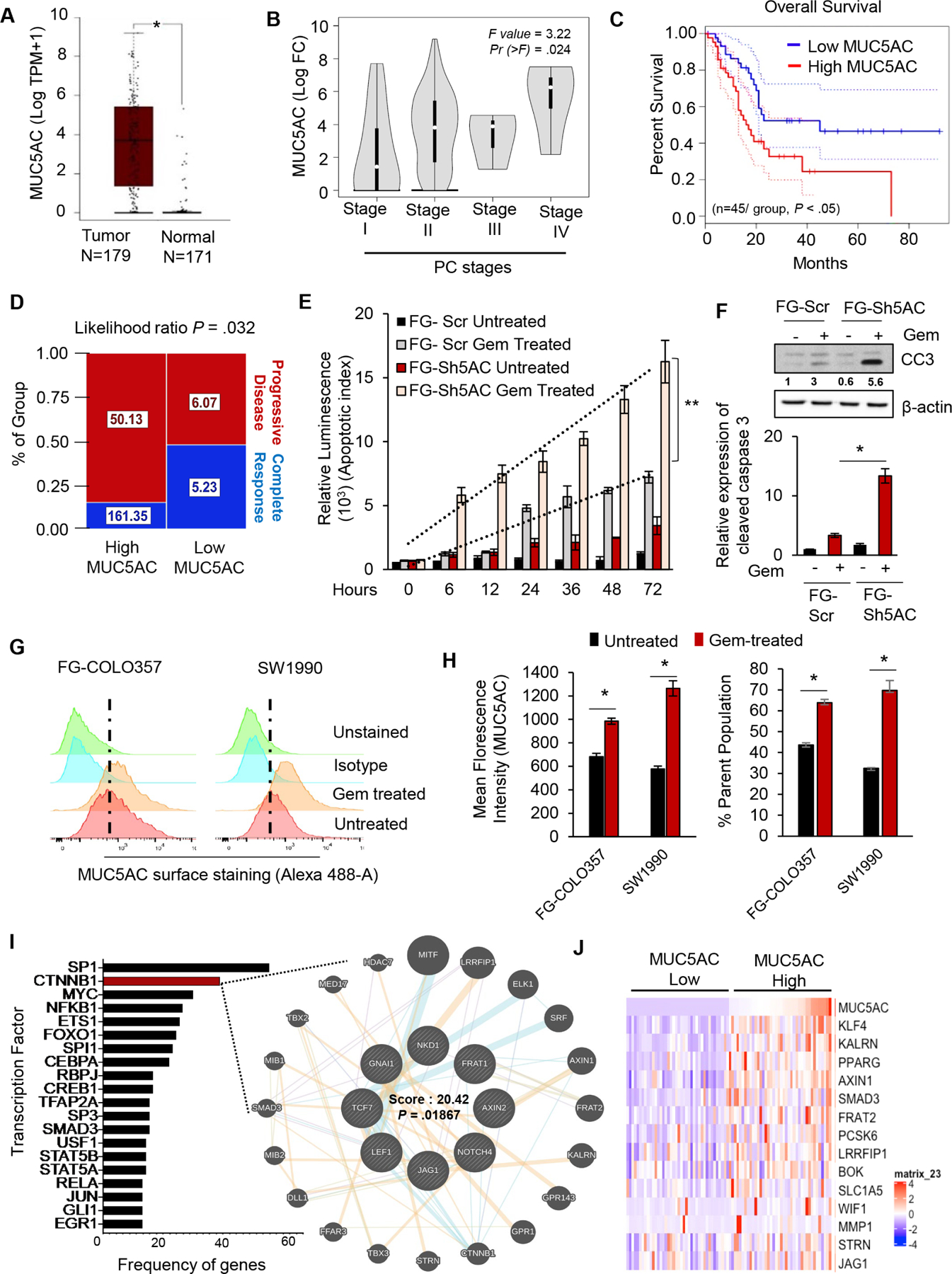
(A) Transcriptomic analysis from TCGA and GTEx datasets (gepia.org) showing high MUC5AC expression in tumor versus normal pancreas. (B) Expression of MUC5AC progressively increases with the increase of PC stages. (C) High MUC5AC expression is associated with the poor survival of PC patients. (D) Hierarchy graph demonstrating TCGA analysis on high and low MUC5AC-expressing PC patients with progressive disease and complete response after gemcitabine treatment. Median expression values (TPM) of MUC5AC for each group are mentioned in the boxes. (E) The Luminescence-based apoptotic assay demonstrated a statistically significant time-dependent increase in the percentage of apoptosis in FG-Sh5AC cells upon gemcitabine administration compared to the FG-Scr group. (F) FG-Sh5AC cells expressed significantly higher cleaved caspase-3 upon gemcitabine treatment, as compared to FG-Scr. (G) The flow-cytometry analysis revealed higher expression of MUC5AC and (H) enrichment of MUC5AC-expressing clones upon gemcitabine treatment in FG-COLO357 and SW1990 cells. (I) Bar diagram from TFacts analysis revealed a higher number of genes regulated by β-catenin among other transcription factors in KC versus KCM mice. (J) Heatmap from TCGA dataset analysis depicting differential expression of β-catenin target genes in MUC5AC-high and low expressing PC patients. * P value < .05, ** P value < .01.
Abrogation of MUC5AC-mediated nuclear accumulation of β-catenin sensitizes PC cells to gemcitabine
To characterize the mechanistic involvement of MUC5AC during PC development, RNA-seq. analysis was performed on the pancreatic tumors from KC and KCM mice4. Using TFactS, a prediction algorithm for transcription-factor network from the RNA-seq., a significant downregulation of well-characterized β-catenin targets was observed in KCM pancreatic tumors11 (Figure 1I), which was corroborated by microarray-based transcriptomic analysis on the human PC cells (Supplementary Figure 2A). Concurrently, pathway analysis from the TCGA data, using Enrichr12, revealed Wnt-β catenin signaling as the second-most predominant pathway in the high MUC5AC-expressors (Supplementary Figure 2B). Furthermore, high MUC5AC-expressing patients demonstrated increased expression of the β-catenin target genes, compared to low-expressors (Figure 1J); with a strong-to-moderate positive correlation between the MUC5AC expression and β-catenin target genes, like KLF4, KALRN, AXIN1, SLC1A5 (Supplementary 1J, Supplementary Figure 2C). While the majority of the β-catenin remains associated with E-Cadherin in adherens junctions at the plasma membrane, several oncogenic stimuli can release and localize β-catenin to the nucleus where it can modulate gene transcription. Indeed, while almost 40% of the β-catenin was localized in the nucleus of the ductal epithelial cells of the KC mice pancreatic tissues, only 10% of β-catenin was present in the nucleus in the KCM group (Figure 2A). Next, upon gemcitabine administration, MUC5AC-expressing PC cells and those ectopically treated with MUC5AC (FG-Sh5AC+FG-Scr CM) demonstrated a significant increase in the nuclear accumulation of β-catenin, as compared to their corresponding Sh5AC groups (Figure 2B, Supplementary Figure 2D). Utilizing TOPFLASH assay, we observed that upon gemcitabine treatment, the FG-Scr cells and FG-Sh5ac treated with the conditioned media (CM) from FG-Scr cells demonstrate the significantly higher transcriptional activity of β-catenin compared to the FG-Sh5AC cells (Figure 2C). Furthermore, impeding the nuclear localization of β-catenin using pharmacological inhibitor, CCT 031374 enhanced the susceptibility of these cells to gemcitabine-mediated apoptosis (Figure 2D, Supplementary Figure 2E), thereby suggesting the role of the MUC5AC/β-catenin axis in gemcitabine resistance. Next, we investigated how MUC5AC, being a secreted mucin, can mediate nuclear localization of β-catenin. A previous study demonstrated that MUC5AC at the intercellular junctions interferes with the membrane localization of E-cadherin, which correlated with β-catenin’s nuclear localization13. Utilizing co-immunoprecipitation experiments, we observed a strong physical interaction of MUC5AC and E-Cadherin in PC cells (Figure 2E, Supplementary Figure 2F), and MUC5AC-depletion (FG-Sh5AC cells) increased the association of β-catenin with E-Cadherin (Figure 2F), while reducing the interaction between β-catenin and TCF/LEF (the nuclear co-activator required for β-catenin activity) (Figure 2G), as compared to the FG-Scr cells or FG-Sh5AC cells treated with FG-Scr-CM, indicating that MUC5AC disrupts the E-cadherin/β-catenin junctions. The interaction of MUC5AC and E-cadherin on the surface of cancer cells was also evident from the murine organoids derived from KC mice pancreas (Figure 2H). In concordance to the findings from human PC cells, Muc5ac-depleted (KCM) murine organoids were significantly more sensitive to gemcitabine, as compared to the KC organoids, which turned susceptible to gemcitabine-mediated cytotoxicity upon co-administration of β-catenin inhibitor (Figure 2I, J).
Figure 2. MUC5AC-mediated disruption of E-Cadherin/β-catenin junctions leads to nuclear localization of β-catenin.
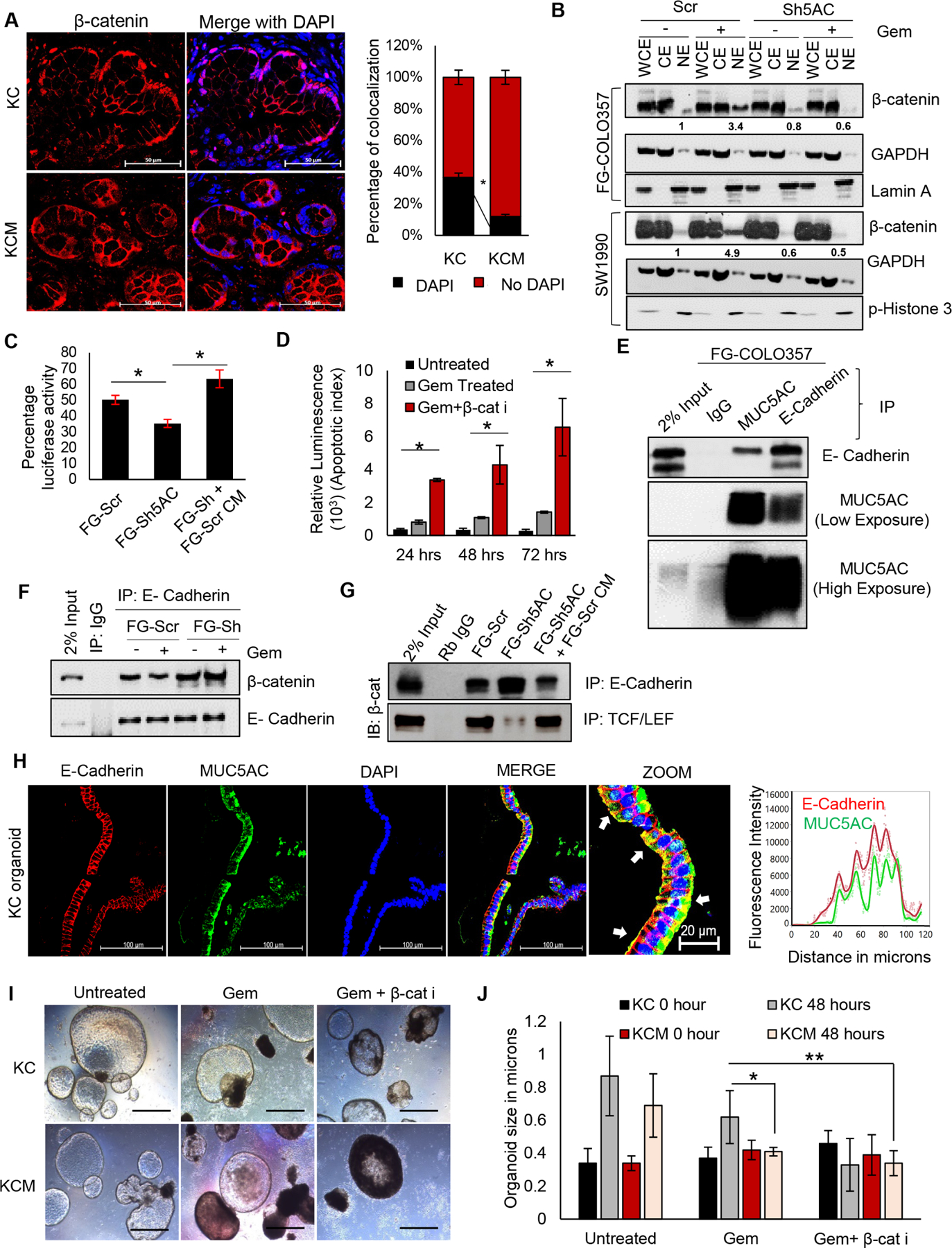
(A) Immunofluorescence analysis for β-catenin demonstrating nuclear versus membranous distribution, with corresponding quantifications in KC and KCM pancreatic tumors. The percentage of colocalization of β-catenin with DAPI was analyzed using ImageJ software. (B) Western blot analysis showing β-catenin enrichment in cytoplasmic versus nuclear fractions in FG-Scr, FG-Sh5AC, SW-Scr, and SW-Sh5AC cells with or without gemcitabine treatment. GAPDH and Lamin A or Phospho-Histone-3 represent the purity of cytoplasmic and nuclear fractions, respectively. (C) Percentage luciferase activity from TOPFLASH-assay demonstrating the transcriptional activity of β-catenin in gemcitabine treated-FG-Scr and FG-Sh5AC cells (with or without treatment with MUC5AC-enriched conditioned medium from FG-Scr cells). (D) Luminescence assay demonstrating a time-dependent increase in apoptosis of FG-Scr cells treated with a combination of gemcitabine and β-catenin inhibitor. (E) Co-immunoprecipitation of MUC5AC with E-cadherin from FG-COLO357 cells, 2% whole cell lysate was taken as input. (F) Co-immunoprecipitation of E-cadherin with β-catenin from FG-Scr and FG-Sh5AC cells, with or without gemcitabine treatment, 2% whole cell lysate was taken as input. (G) Co-immunoprecipitation of β-catenin with E-cadherin and TCF/LEF from gemcitabine treated-FG-Scr and FG-Sh5AC cells (with or without treatment with MUC5AC-enriched conditioned medium from FG-Scr cells), 2% whole cell lysate was taken as input. (H) Immunofluorescence analysis and quantitation demonstrating colocalization of Muc5ac and E-Cadherin on the cell surface of KC murine organoids, white arrows indicate yellow zones of colocalization. (I) Light-microscopic and (J) quantitative representation of KC and KCM organoids’ viability upon treatment with gemcitabine alone or in a combination with β-catenin inhibitor. * P value < .05, ** P value < .01, scale bars (unless otherwise mentioned): 400 microns, magnified images: 200 microns.
MUC5AC-depletion decreases β-catenin-mediated c-Myc expression and curbs glutamine utilization by PC cells upon gemcitabine administration
Next, we analyzed the mechanistic outcome of MUC5AC-mediated β-catenin’s translocation to the nucleus. Based on our RNA-seq. analysis, we took the candidate-based approach and evaluated the expression of the β-catenin target gene, c-Myc. Chromatin immunoprecipitation (ChIP) analysis showed a 10 folds higher recruitment of β-catenin on c-Myc promoter in FG-Scr compared to the FG-Sh5AC cells (Figure 3A) as well as a significant increase in the expression of c-Myc in FG-Scr cells as compared to FG-Sh5AC cells upon gemcitabine treatment. C-Myc expression was drastically declined upon β-catenin inhibition in the FG-Scr cells (Figure 3A). Furthermore, there was a significant increase in the expression of c-Myc in the neoplastic ducts of KC pancreas, compared to KCM (Figure 3B). The chemoresistance of pancreatic tumors is majorly contributed by oncogene-associated de novo nucleotide biosynthesis, which support the cancer cells in ameliorating DNA damage caused by nucleotide analogues14. Studies have shown that c-Myc transcriptionally upregulates the genes involved in the uptake and utilization of glutamine15, which serves as the critical nitrogen donor during nucleotide biosynthesis16. Indeed, transient silencing of β-catenin and c-myc individually in the FG-Scr cells significantly enhanced their gem-mediated apoptosis, which was significantly reverted by the ectopic supplementation of a cell-penetrable recombinant c-Myc protein (Figure 3C). Transcriptomic analysis from the TCGA dataset also revealed a significant downregulation of genes involved in glutamine metabolism and nucleotide biosynthesis in the low MUC5AC-expressors (Figure 3D). Further, RNA seq. data demonstrated significant enrichments of the pathways associated with glutamine, glutamate, and glutathione metabolic processes in the KC mice pancreas, compared to KCM (Figure 3E, Supplementary Figure 2I, J). The TCGA data analysis also revealed a significant, strong-to-moderate positive correlation between MUC5AC and the amino acid transporters (SLC1A5, SLC6A14, SLC7A11, and SLC7A5), that are associated with glutamine/glutamate/cysteine transport and are direct transcriptional targets of oncogenic c-Myc (Supplementary Figure 2H). The expressions of Slc7a11 and Slc1a5 were also significantly low in the KCM mice, as demonstrated by a 2-folds decrease in their histoscore (Figure 3F), suggesting the contribution of MUC5AC in regulation of glutamine/glutamate metabolism in PC cells. The mechanistic involvement of MUC5AC/β-catenin axis in c-Myc-associated glutamine uptake and glutamate release in the context of gemcitabine resistance was further validated when the FG-Scr cells demonstrated a significant increase in the expression of SLC1A5, SLC6A14, SLC7A11, and SLC7A5 transporters, compared to FG-Sh5AC cells, upon gemcitabine treatment. In contrast, treatment with the combination of gemcitabine and β-catenin inhibitor in the FG-Scr cells demonstrated a significant, 4–10 folds decrease in the expression of the glutamine transporters, as compared to gemcitabine treatment alone (Figure 3G).
Figure 3. MUC5AC/β -catenin axis upregulates the expression of c-Myc and glutamine transporters upon gemcitabine treatment.

(A) Schematic and quantitative representation of chromatin immunoprecipitation demonstrating the recruitment of β-catenin on the c-Myc promoter. Fold enrichment is represented after normalization with IgG control. Western blot analysis showing c-Myc expression in FG-Scr and FG-Sh5AC cells upon treatment with gemcitabine alone or in a combination of β-catenin inhibitor. (B) Representative images and quantitation from immunohistochemistry showing c-myc expression in the nuclei of neoplastic ducts in KC and KCM pancreas. (C) Real-time apoptosis assay demonstrating the relative caspase 3/7 incorporation in gem-treated FG-Scr cells upon transient knockdowns of β-catenin and c-Myc, in presence or absence of recombinant Myc protein. (D) Heatmap from TCGA dataset analysis depicting differential expression of genes involved in glutamine metabolism, nucleotide metabolism, and pyrimidine biosynthesis. (E) GSEA analysis showing a negative association of glutamine metabolic pathway in KCM mice. (F) Immunohistochemistry analysis demonstrating the expression of Muc5ac, Slc7a11, and Slc1a5 in KC and KCM mice pancreatic tumors. Black arrowheads represent neoplastic ducts that were considered during histologic scoring. (G) The qRT-PCR analysis for expression of SLC1A5, SLC6A14, SLC&A11, SLC7A5 in FG-Scr and FG-Sh5AC cells upon treatment with gemcitabine alone or in combination of β-catenin inhibitor. * P value < .05, ** P value < .01, scale bars: Muc5ac (100 microns, magnified images: 400 microns), Slc7a11 and Slc1a5 (400 microns, magnified images: 200 microns)
After entry into the cells via SLC1A5 and SLC6A14, glutamine is deaminated to glutamate by the enzyme glutaminase (GLS)., which can be expelled out in the extracellular space via SLC7A11 in exchange for cysteine, which aids in relieving the cancer cells from oxidative stress via glutathione synthesis. Glutamate can further feed into the nucleotide biosynthetic pathway via Aspartate. The c-Myc has been demonstrated to transcriptionally regulate and functionally couple the transporters and enzymes involved in glutamine/glutamate metabolism in the cancer cells17,18, as shown in Figure 4A. In luminescence-based metabolite flux assay, MUC5AC-expressing cells demonstrated a significantly higher uptake of glutamine and efflux of glutamate in the culture media upon gemcitabine treatment (Figure 4B), compared to knockdown cells. Similar observations were obtained from murine PC cells (Supplementary Figure 3C, D). To investigate the dependence of gemcitabine resistance of PC cells on the β-catenin/c-myc/glutaminolysis axis, we exclusively ablated the important nodes of proposed signaling cascade, including β-catenin, c-Myc and SLC1A5 in MUC5AC-expressing (Scr control) cells, by transient gene silencing. While silencing β-catenin significantly reduced the expressions of c-myc and SLC1A5, c-Myc silencing caused a marginal decrease in β-catenin but a significant decline in SLC1A5 levels, and SLC1A5 ablation did not affect the expressions of β-catenin and c-Myc (Supplementary Figure 3E). Importantly, the silencing of all three molecular nodes individually led to a significant decline in glutamine utilization (Supplementary Figure 4A, B, C, D) and gemcitabine resistance (Figure 4C), comparable to the levels observed in MUC5AC KD cells. Further, treatment with cell-penetrable recombinant c-Myc protein was able to significantly rescue the expression of SLC1A5 (Supplementary Figure 3G), levels of glutamine uptake and glutamate efflux (Supplementary Figure 4A, B, C, D), and gemcitabine resistance (Figure 4C) in the β-catenin and c-Myc-silenced cells. To interrogate whether MUC5AC-mediated gemcitabine resistance is predominantly contingent on glutamine utilization, PC cell lines were deprived of glutamine prior to and during gemcitabine treatment. The glutamine-deprived FG-Scr cells demonstrated a drastic decline in viability upon gemcitabine treatment, which was significantly rescued upon glutamine replenishment. In contrast, FG-Sh5AC cells could not salvage their viability upon glutamine replenishment (Figure 4D, E), suggesting that in the absence of MUC5AC, PC cells demonstrate significantly less glutamine uptake and utilization. Glutamine serves as the nitrogen donor in the cascade of nucleotide biosynthesis; hence upon gemcitabine treatment, glutaminolysis and nucleotide metabolism enhanced several folds as a survival mechanism of the cancer cells to combat the cytotoxic effect of gemcitabine. Hence, in our metabolic flux experiment, MUC5AC KD cells demonstrated a significant decline in the incorporation of N15-label into deoxycytidine triphosphate (dCTP) from glutamine upon gemcitabine treatment, compared to the FG-Scr group (Supplementary Figure 4E). Since dCTP competitively inhibits gemcitabine’s incorporation in the DNA, the lower concentration of N15 -labelled dCTP in the FG-Sh5AC cells suggest their increased susceptibility towards the gemcitabine-mediated cytotoxicity. Therefore, we sought to examine the efficacy of targeting MUC5AC-mediated glutaminolysis in murine PC organoids using specific GLS1 inhibitor CB-839 (6 uM), which is in clinical trials for colorectal cancer and hematological malignancies (clinicaltrials.gov). As indicated by the real-time kinetics of organoid viability, KC organoids were significantly more resistant to gemcitabine than the KCM group, which was obviated upon co-administration with CB-839. However, the combinatorial effect of gemcitabine and CB-839 was not evident in the KCM group (Figure 4F, G, Supplementary video file 1), suggesting that MUC5AC-expression through c-Myc-associated glutaminolysis provide resistance to the standard nucleotide analog-based chemotherapies.
Figure 4. MUC5AC enhances glutamine uptake and utilization in human PC cells and murine organoids.
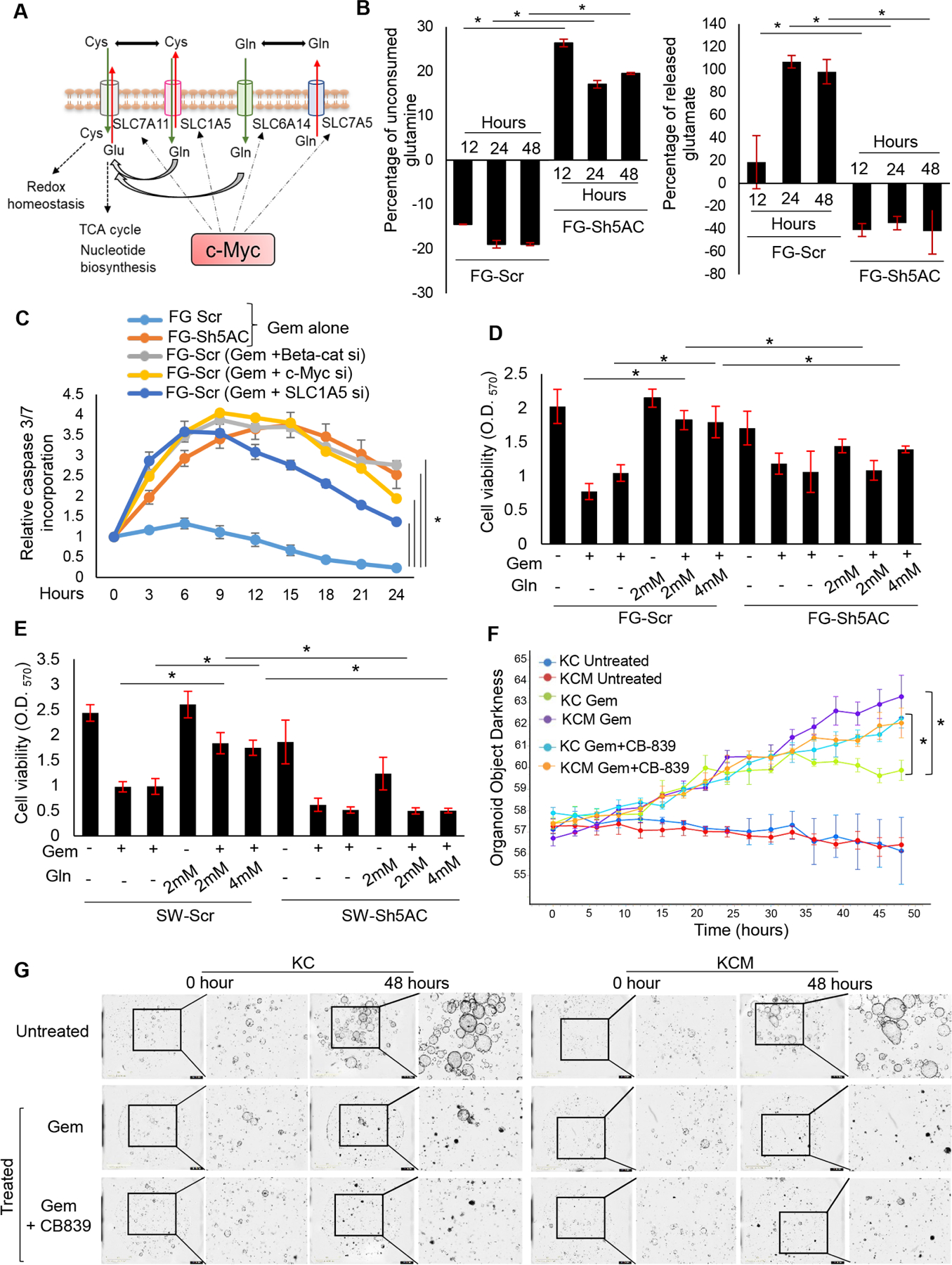
(A) Schematic representation of c-Myc-mediated coupling of glutamine transporters for glutamine/glutamate flux. (B) Luminescence-based metabolic flux assay to demonstrate unconsumed glutamine and effluxed glutamate in the culture supernatant of FG-Scr and FG-Sh5AC cells with gemcitabine treatment (the data at each time-point is normalized by the respective untreated FG-Scr controls). (C) Real-time apoptosis assay demonstrating the relative caspase 3/7 incorporation in gem-treated FG-Scr cells upon transient knockdowns of β-catenin, c-Myc, and SLC1A5, compared to gem-treated FG-Sh5AC group (all datapoints normalized to untreated FG-Scr controls). (D-E) Cell viability assay demonstrating gemcitabine-mediated cytotoxicity of FG-Scr, FG-Sh5AC, SW-Scr, and SW-Sh5AC cells upon glutamine deprivation, followed by the rescue of viability with glutamine replenishment. (F-G) Quantitation and representative organoid pictures demonstrating the real-time kinetics the loss of viability of KC and KCM organoids after treatment with gemcitabine and CB-839. * P value < .05, ** P value < .01, scale bars 1000 microns for organoid images.
MUC5AC/β-catenin/c-Myc axis exacerbates the aggressiveness and chemoresistance of PC cells in vivo
To investigate the contribution of glutaminolysis via MUC5AC/β-catenin/c-Myc axis in gemcitabine resistance of PC cells in vivo, the FG-Scr and FG-Sh5AC cells were subcutaneously implanted in athymic mice to generate tumors followed by intraperitoneal administration of gemcitabine after 15 days post-implantation. Our previous study demonstrated that abrogation of MUC5AC reduces the growth and tumorigenic potential of PC cells4. Hence, gemcitabine response in the FG-Scr and FG-Sh5AC groups have been represented as the percentage change in tumor volume in each group. There was a significant 2 and 3-folds decline in the mean percentage change in tumor volume in the FG-Sh5AC tumors, compared to the FG-Scr group, after the second and third doses of gemcitabine, respectively (Figure 5A). Concurrently, at euthanasia, a 50% shrinkage in the mean tumor size was observed in the FG-Sh5AC group with gemcitabine treatment, while the reduction in the FG-Scr group was only 30% (Figure 5B). Immunohistochemical analysis demonstrated a significant 2-folds increase in MUC5AC expression in the gemcitabine-treated FG-Scr group, thereby corroborating the observation from our in vitro experiments. Consecutively, compared to the FG-Scr group, there was a significant 2.2-folds increase in the expression of pro-apoptotic marker cleaved PARP in FG-Sh5AC tumors (Figure 5B), which was further corroborated by the autochthonous murine tumors (Supplementary Figure 5A). The gemcitabine-treated FG-Scr tumors also demonstrated a significant 3–4 folds higher expression of SLC1A5 and SLC7A11 than the FG-Sh5AC tumors (Figure 5C, D; Supplementary Figure 5B). Further, immunofluorescence studies revealed a 3.5 folds higher number of nuclear β-catenin foci in the gemcitabine-treated FG-Scr tumors than the FG-Sh5AC group (Figure 5E, F). Furthermore, there was a significant 1.5 folds higher accumulation of glutamate in the gemcitabine-treated FG-Scr tumors than FG-Sh5AC tumors (Figure 5G). These findings cumulatively substantiate our hypothesis and validate our in vitro observations that the MUC5AC/β-catenin axis promotes upregulation of glutamine uptake leading to resistance against nucleotide analogues like gemcitabine, and can also lead to glutamate synthesis, which in turn favors the redox-homeostasis of PC cells.
Figure 5. Depletion of MUC5AC sensitizes PC cells to gemcitabine in vivo.

(A) Experimental schema and treatment strategy for xenograft from FG-Scr and FG-Sh5AC cells. Line graph demonstrating temporal mean percentage change in tumor volume in gemcitabine treated groups compared to the respective control groups. (B) Pictorial and quantitative representation of xenograft tumor volumes at euthanasia. (C-D) Immunohistochemistry analysis and quantitative histoscores demonstrating the expression of MUC5AC, SLC1A5, SLC7A11, and cleaved PARP in FG-Scr and FG-Sh5AC tumors treated with gemcitabine. (E) Representative and (F) quantitative analysis of β-catenin nuclear foci in gemcitabine-treated FG-Scr and FG-Sh5AC tumors. For visualization of the β-catenin foci, Gemcitabine-treated FG-Scr tumor images are represented at higher magnification as depicted by the scale bars. (G) Luminescence-based metabolic flux assay to demonstrate glutamate enrichment in the vehicle and gemcitabine-treated FG-Scr and FG-Sh5AC tumors. * P value < .05, ** P value < .01, scale bars (unless otherwise mentioned): 400 microns, magnified images: 200 microns
Disruption of MUC5AC/β-catenin/c-Myc axis using pharmacological inhibitors restricts glutaminolysis and augments chemotherapeutic efficacy in human PC organoids
In order to clinically validate the MUC5AC-mediated resistance to nucleotide analogs, organoids derived from human pancreatic tumors were segregated based on MUC5AC expression (Figure 6A) and were subjected to gemcitabine treatment. As the growth kinetics and complexity of the organoid structures in high and low MUC5AC-expressing groups were diverse, the observation is represented as a percentage increase in organoid darkness (or decrease in organoid viability) with time, which demonstrates that the low MUC5AC group was significantly more susceptible to gemcitabine treatment (Figure 6B; Supplementary video file 2). Furthermore, the high-MUC5AC group exhibited significant 4–5 folds upregulation in the expression of c-Myc, SLC1A5, and SLC7A11 (Supplementary Figure 5C), with a concurrent enhanced glutaminolysis, as exemplified by an increase in glutamine uptake and glutamate release (Figure 6C, D), upon gemcitabine administration.
Figure 6. Abrogation of MUC5AC/β-catenin/c-Myc axis sensitizes human organoids to gemcitabine.

(A) Representative images and (B) quantitation of real-time kinetics of viability of high and low MUC5AC-expressing human PC tumoroids after treatment with gemcitabine. (C-D) Glutamine/glutamate flux assay demonstrated a significant decline in glutamine uptake and glutamate release in the low-MUC5AC group compared to the high-MUC5AC group. (E) Representative images and (F) quantitation of immunohistochemistry-based correlation of MUC5AC expression with that of SLC7A11 in 30 PC patients demonstrating strong significant correlations in untreated and gemcitabine-treated patients. (G) Pictorial and (H) graphical representation of viability kinetics for MUC5AC-high organoids demonstrating a significant increase in organoid mortality upon treatment with a combination of gemcitabine and GLS1 inhibitor CB-839 compared to gemcitabine alone. (I-J) The luminescence-based glutamine/glutamate flux assay demonstrates a significant decrease in glutamine uptake upon β-catenin inhibition in gemcitabine treated MUC5AC-high organoids, a significant decrease in glutamate release upon β-catenin and GLS1 inhibition in gemcitabine treated MUC5AC-high organoids. (K) Schematic representation depicting that de novo expressed, abundantly accumulated MUC5AC breached E-Cadherin/β catenin junctions on PC cell surfaces, leading to nuclear accumulation of β-catenin, upregulation of c-Myc and glutamine/glutamate transporters, and, in turn, leading to enhanced glutamine uptake and utilization. Enhanced glutaminolysis contributes towards chemoresistance via de novo nucleotide biosynthesis and surpassing the cytotoxic effect of nucleotide analogs like gemcitabine. * P value < .05, ** P value < .01, scale bars (unless otherwise mentioned): 400 microns, magnified images: 200 microns, organoid images: 1000 microns.
The transcriptomic correlation between MUC5AC and glutamate/cysteine transporter SLC7A11, as observed in TCGA patient data, was further corroborated at the protein level by immunohistochemical analysis on tissue arrays of 30 PC patients, which demonstrated significant strong correlations of R=0.77 and R=0.56 between MUC5AC and SLC7A11 in the untreated and the treated groups, respectively (Figure 6E, F). Next, we sought to explore the relevance of targeting the MUC5AC/β-catenin axis-associated glutaminolysis in order to potentiate chemotherapeutic efficacy. Co-administration of β-catenin inhibitor and GLS inhibitor significantly sensitized the high-MUC5AC expressing human PC organoids to gemcitabine-mediated cytotoxicity (Figure 6G, H; Supplementary Figure 5D, E, F; Supplementary video file 3). The identical kinetics of the loss of viability of the organoids imparted by the two inhibitors in the presence of gemcitabine further advocate for MUC5AC-mediated mechanism through β-Catenin and c-Myc to enhance glutaminolysis and drug-resistance in PC. Furthermore, a significant reversal in glutamine uptake upon β-catenin inhibition with a concurrent decrease in glutamine uptake and glutamate efflux upon GLS inhibition (Figure 6I, J).
Discussion
Nucleotide analogs like gemcitabine and 5-FU have been integral parts of the chemotherapeutic regimen for cancer patients; however, most of the patients eventually succumb to the disease owing to resistance to the therapeutic drugs. Years of concerted efforts have led to identifying several resistance mechanisms in the tumor, for example, upregulation of drug-efflux proteins, enrichment of CSCs, increase in DNA repair, and downregulation of apoptotic pathways19. Recently, metabolic adaptations have been identified as a critical event in acquiring resistance by the cancer cells. Most resistant cells are dependent on glycolysis and glutaminolysis to overcome pharmacological stress, oxidative stress, and DNA replication arrest by nucleotide analogs9, 20. To maintain the intracellular nucleotide pool and thereby compete with the nucleotide analogs for DNA incorporation, tumor cells heavily rely on de novo nucleotide biosynthesis. While glucose serves as the primary carbon donor in the metabolic pathways leading to nucleotide generation, glutamine is utilized as the nitrogen donor, alongside generating glutamate to feed into the TCA cycle, enhance glutathione synthesis for redox homeostasis and mobilize carbon and nitrogen backbones for nucleotide biosynthesis.
Mucins, the aberrantly expressed, high molecular-weight glycoproteins, serve as the hallmarks of PC progression. These highly glycosylated, metabolically expensive molecules21 not only serve as rheological barriers to the chemotherapeutic drugs but mediate molecular perturbations to promote intrinsic drug resistance in several solid tumors22. For example, MUC4 has been demonstrated to cause gemcitabine resistance via downregulation of concentrative nucleoside transporter (hCNT1) that is responsible for intracellular uptake of gemcitabine23. Recently, transmembrane mucin, MUC1 was implicated in gemcitabine resistance by HIF1α-mediated upregulation of glycolytic flux and deoxycytidine synthesis24. Recently identified role of MUC5AC in the enrichment of CSCs during PC led us to delineate its contribution and identify the associated molecular partners in PC chemoresistance.
The TCGA analysis revealed that high MUC5AC-expressing tumors are less responsive to gemcitabine and rely significantly on β-catenin-regulated genes for their oncogenic signaling. From the autochthonous murine model and human PC cells, we observed that MUC5AC associates with E-Cadherin and disrupts the E-Cadherin/β-catenin complex, which releases β-catenin from the junctional complex leading to its nuclear localization. The RNA-seq. analysis from the murine model demonstrated a concurrent downregulation of genes transcribed by β-catenin and c-Myc in the Muc5ac-knockout group, which drove us to investigate the mutual dependence of these transcription factors in mediating MUC5AC-associated chemoresistance. Most KRAS-mutated tumors exhibit c-Myc addiction for reprogramming metabolic pathways associated with glutamine utilization25. Oncogenic c-Myc transcriptionally upregulates glutamine/glutamate transporters and the enzymes responsible for the utilization of glutamine for de novo nucleotide biosynthesis18. MUC5AC-high patients demonstrated a significant upregulation of genes involved in glutamine/glutamate metabolism, pyrimidine biosynthesis, and nucleotide metabolism. Mechanistically, gemcitabine administration enriches β-catenin on the c-Myc promoter leading to its increased expression and metabolic adaptation in MUC5AC-expressing PC cells. Glutamine deprivation significantly impaired the viability of gemcitabine-treated MUC5AC-expressing cells without much impact on the MUC5AC-depleted cells, reinstating the involvement of the MUC5AC/β-catenin/c-Myc axis in glutaminolysis and chemoresistance. A recent study demonstrated that CD44, the receptor for extracellular matrix protein hyaluronan, bolsters the membrane stability of SLC7A1126. Interestingly, MUC5AC has been shown to physically interact with CD44 and enhance the stemness and chemoresistance of colon cancer cells27. A secreted yet glycocalyx-associated bulky molecule, MUC5AC can be envisioned as a molecular scaffold on the PC cells involved in dynamic interactions with multiple cell membrane-tethered molecules, including the glutamine transporters prolonging their surface availability and priming their oncogenic signaling. The C-terminus of MUC1 transcriptionally activates various genes involved in redox homeostasis, utilization of glucose, and amino acids henceforth contributing to gemcitabine resistance of PC cells via glycolytic induction and upregulation nucleotide biosynthesis24, 28. While MUC1 is present in the normal pancreas, MUC5AC is abundantly expressed de novo during oncogenesis29. Being an integral part of the glycocalyx, the sudden emergence of MUC5AC can augment the MUC1/HIF1α axis via rheological or chemical associations during gemcitabine resistance. Several studies have indicated that oncogenic c-Myc collaborates with HIF1α for hypoxic adaptation and metabolic advantages of cancer cells30. Intriguingly, a recent study demonstrated that overexpression of c-Myc stabilized and enhanced the cellular accumulation of HIF1α, leading to the induction of HIF1α target genes31. Hence, it is tempting to speculate that the MUC5AC/β-catenin axis-mediated upregulation of c-Myc can promote glutamine utilization and set the stage for MUC1/HIF1α axis and the associated glycolytic shift for gemcitabine resistance. However, further work is required to evaluate this crosstalk.
The pancreatic cancer tumor microenvironment (PC-TME) harbors a plethora of cell types and an intense desmoplasia. Apart from the physical impediment in drug availability inside the bulk tumor32, several studies have identified the cancer-associated fibroblasts (CAFs) as an accomplice to the malignant cells during the tumor growth and acquiring chemoresistance33. Activated CAFs have been shown to promote chemoresistance by inducing ERK, AKT, or STAT3 pathways and secreting insulin-growth factor (IGF) and insoluble ECM proteins like hyaluronic acid34. Moreover, CAFs undergo autophagy and release glutamine-loaded exosomes, which are received by PC cells via micropinocytosis35. Tumor-associated macrophages (TAMs) in the PC-TME have been recently shown to contribute to gemcitabine resistance via the release of pyrimidine nucleosides that are taken up by the cancer cells36. Understanding the molecular contributions and mechanistic involvement of MUC5AC in the genesis of PC-TME will be interesting and is currently underway in the lab.
The clinical significance of MUC5AC in imparting chemoresistance of PC patients was corroborated when MUC5AC-high patient tumoroids demonstrated significantly lesser mortality compared to the MUC5AC-low group upon gemcitabine treatment. Moreover, gemcitabine co-administration with the pharmacologic inhibitor (CB-839) of GLS 1, the enzyme responsible for the conversion of glutamine to glutamate, significantly sensitized the high-MUC5AC expressing human and murine tumoroids to gemcitabine. In contrast, the effect was less evident in the Muc5ac-knockout organoids. Cumulatively, the current study establishes the pathological role of the MUC5AC/β-catenin/c-Myc axis in PC gemcitabine resistance (Figure 6K). Our findings also usher a prospective avenue of improved targeting of MUC5AC-expressing pancreatic tumors with the standard nucleotide analogs in combination with CB-839. The CB-839 is in the clinical trials for hematological malignancies and colorectal cancer and hence can be expected to show a faster transition to the clinics. Recently, a retrospective gene sequencing-based analysis on 1856 patients revealed that patients harboring an actionable mutation had a significantly better clinical outcome when treated with a matched therapy as compared to the ones treated conventionally37, thereby holding promise for precision medicine. Several groups, including ours, have recognized MUC5AC as a potential serum biomarker in PC38. Hence, identifying the molecular perturbations involved in MUC5AC-associated PC chemoresistance may be beneficial in selective stratification and targeting of PC patients based on their actionable alterations, thereby improving their chemotherapeutic response.
Supplementary Material
What You Need to Know.
BACKGROUND AND CONTEXT
A major challenge for pancreatic cancer (PC) patients is the resistance to conventional chemotherapeutics. The de novo expression of secreted mucin 5AC (MUC5AC) is significantly associated with poor patient survival.
NEW FINDINGS
High MUC5AC-expressing PC exhibit resistance to nucleotide analogues like gemcitabine due to enhanced glutamine utilization and nucleotide biosynthesis. The co-administration of β-catenin and glutaminolysis inhibitors with gemcitabine abrogate MUC5AC-mediated chemoresistance.
LIMITATIONS
Future studies should investigate the combined contribution of mucin family members to drug resistance in PC and role of MUC5AC in metabolic inter-dependence of cancer cells and associated stroma.
IMPACT
Identifying molecular perturbations involved in MUC5AC-associated PC chemoresistance will help in patient stratification for targeting with specific inhibitors of β-catenin and glutamine utilization to improve the chemotherapeutic response in PC.
Acknowledgements:
C57BL/6 Muc5ac−/− mice were obtained from Dr. Christopher Evans at the University of Colorado Denver, Colorado, USA. We thank the UNMC Mass Spectrometry Core Facility for their help in the metabolomic experiment.
Funding:
The authors/work on this manuscript were supported, in parts, by grants from the NIH (R01 CA247471, RO1 CA210637, RO1CA206444, RO1 CA183459, UO1 CA200466, PO1 CA217798, R44 CA235991, NIAID RO1AI125588, PO1 AI83211).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: SKB is one of the co-founders of Sanguine Diagnostics and Therapeutics, Inc. The other authors declare no competing interests.
References
- 1.Neoptolemos JP, Kleeff J, Michl P, et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nature reviews Gastroenterology & hepatology 2018;15:333–348. [DOI] [PubMed] [Google Scholar]
- 2.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nature Reviews Gastroenterology & Hepatology 2020:1–16. [DOI] [PubMed] [Google Scholar]
- 3.Tao S, Wang S, Moghaddam SJ, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer research 2014;74:7430–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganguly K, Krishn SR, Rachagani S, et al. Secretory mucin 5AC promotes neoplastic progression by augmenting KLF4-mediated pancreatic cancer cell stemness. Cancer Research 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013;496:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cluntun AA, Lukey MJ, Cerione RA, et al. Glutamine metabolism in cancer: understanding the heterogeneity. Trends in cancer 2017;3:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santana-Codina N, Roeth AA, Zhang Y, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nature communications 2018;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X, Chen S, Yu D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites 2020;10:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaushik G, Seshacharyulu P, Rauth S, et al. Selective inhibition of stemness through EGFR/FOXA2/SOX9 axis reduces pancreatic cancer metastasis. Oncogene 2020:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. The EMBO journal 2012;31:2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research 2016;44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inaguma S, Kasai K, Ikeda H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011;30:714–723. [DOI] [PubMed] [Google Scholar]
- 14.Galmarini C, Mackey J, Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia 2001;15:875–890. [DOI] [PubMed] [Google Scholar]
- 15.Bott AJ, Peng I-C, Fan Y, et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell metabolism 2015;22:1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends in biochemical sciences 2010;35:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhutia YD, Ganapathy V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2016;1863:2531–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences 2008;105:18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng S, Pöttler M, Lan B, et al. Chemoresistance in pancreatic cancer. International Journal of Molecular Sciences 2019;20:4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grasso C, Jansen G, Giovannetti E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Critical reviews in oncology/hematology 2017;114:139–152. [DOI] [PubMed] [Google Scholar]
- 21.Ganguly K, Rauth S, Marimuthu S, et al. Unraveling mucin domains in cancer and metastasis: when protectors become predators. Cancer Metastasis Reviews 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao CV, Janakiram NB, Mohammed A. Molecular pathways: mucins and drug delivery in cancer. Clinical Cancer Research 2017;23:1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skrypek N, Duchêne B, Hebbar M, et al. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene 2013;32:1714–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer cell 2017;32:71–87. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay S, Goswami D, Adiseshaiah PP, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer research 2020;80:1630–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc– and thereby promotes tumor growth. Cancer cell 2011;19:387–400. [DOI] [PubMed] [Google Scholar]
- 27.Pothuraju R, Rachagani S, Krishn SR, et al. Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Molecular cancer 2020;19:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaika NV, Gebregiworgis T, Lewallen ME, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proceedings of the National Academy of Sciences 2012;109:13787–13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur S, Kumar S, Momi N, et al. Mucins in pancreatic cancer and its microenvironment. Nature reviews Gastroenterology & hepatology 2013;10:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang CV, Kim J-w, Gao P, et al. The interplay between MYC and HIF in cancer. Nature Reviews Cancer 2008;8:51–56. [DOI] [PubMed] [Google Scholar]
- 31.Doe MR, Ascano JM, Kaur M, et al. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer research 2012;72:949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schober M, Jesenofsky R, Faissner R, et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014;6:2137–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCarroll JA, Naim S, Sharbeen G, et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Frontiers in physiology 2014;5:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang C, Shi S, Meng Q, et al. Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: where we are and where we are going. Experimental & molecular medicine 2017;49:e406–e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Yang L, Baddour J, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. elife 2016;5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halbrook CJ, Pontious C, Kovalenko I, et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell metabolism 2019;29:1390–1399. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. The Lancet Oncology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaur S, Smith LM, Patel A, et al. A combination of MUC5AC and CA19–9 improves the diagnosis of pancreatic cancer: a multicenter study. The American journal of gastroenterology 2017;112:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.