

Abstract
BACKGROUND & AIMS:
G protein-coupled receptor (GPR) 120 has been implicated in regulating metabolic syndromes with anti-inflammatory function. However, the role of GPR120 in intestinal inflammation is unknown. Here, we investigated whether and how GPR120 regulates CD4+T cell function to inhibit colitis development.
METHODS:
Dextran sodium sulfate (DSS)-induced colitis model, Citrobacter rodentium infection model, and CD4+T cell adoptive transfer model were utilized to analyze the role of GPR120 in regulating colitis development. The effect of GPR120 on CD4+T cell functions was analyzed by RNA sequencing, flow cytometry, and Seahorse metabolic assays. Mice were administered GPR120 agonist for investigating the potential of GPR120 agonist in preventing and treating colitis.
RESULTS:
Deficiency of GPR120 in CD4+T cells resulted in more severe colitis in mice upon DSS insult and enteric infection. Transfer of GPR120-deficient CD4+CD45RbhiT cells induced more severe colitis in Rag−/− mice with lower intestinal IL-10+CD4+T cells. Treatment with GPR120 agonist, CpdA, promoted CD4+T cell production of IL-10 by upregulating Blimp1 and enhancing glycolysis, which was regulated by mTOR. GPR120 agonist-treated wild-type but not IL-10-deficient and Blimp1-deficient Th1 cells induced less severe colitis. Furthermore, oral administration of GPR120 agonist protected mice from intestinal inflammation in both prevention and treatment schemes. Gpr120 expression was positively correlated with Il10 expression in the human colonic mucosa, including patients with inflammatory bowel diseases (IBD).
CONCLUSIONS:
Our findings demonstrate the role of GPR120 in regulating intestinal CD4+T cell production of IL-10 to inhibit colitis development, which identifies GPR120 as a potential therapeutic target for treating IBD.
Keywords: Effector CD4+T cells, Glycolysis, Blimp1, Intestinal homeostasis, Inflammatory bowel diseases
Graphical Abstract
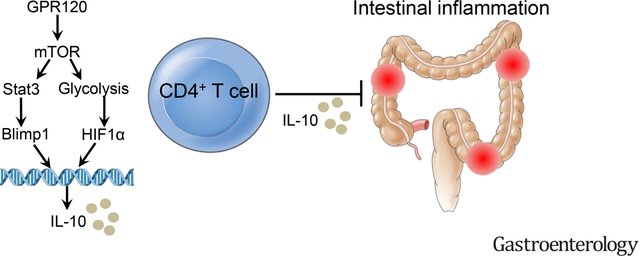
Lay Summary
Long-chain fatty acid receptor G protein-coupled receptor 120 induces interleukin-10 production in CD4+T cells to inhibit colitis development through upregulation of B lymphocyte-induced maturation protein 1 and enhancing glycolysis.
Introduction
It has been well-established that CD4+T cell responses to gut microbiota play crucial roles in the pathogenesis of inflammatory bowel disease (IBD). Among CD4+T cells, Th1 and Th17 cells are central to the pathogenesis of certain types of IBD[1], which can be inhibited by multiple mechanisms, including CD4+T cell production of IL-10, a critical immunosuppressive cytokine for regulating intestinal homeostasis. Polymorphisms in the Il10 locus confer risk for IBD[2, 3]. Deficiency in IL-10 or IL-10 receptor (IL-10R) in mice and humans causes severe intestinal inflammation[4–6]. IL-10-IL-10R signaling is essential in regulatory T cells (Tregs) for their suppressive functions in IBD, in CD4+T effector cells for inhibiting exaggerated T cell responses in mucosal compartments, and also in innate cells for regulating mucosal homeostasis[7–9]. However, how CD4+T cell production of IL-10 is regulated is still not completely understood. Accumulating evidence indicates that dietary compositions regulate health and disease, regardless of energy intake[10]. It has been shown that dietary fatty acids, the primary dietary components, affect host immune responses[11]. Long-chain fatty acids (LCFA) are the major dietary fatty acids absorbed and sensed by host cells. Omega 3 fatty acids (ω-3 FA), which belong to LCFA and are commonly consumed as fish products, dietary supplements, and pharmaceuticals, have been demonstrated to have potent anti-inflammatory effects and several health benefits, including amelioration of atherosclerosis and increased insulin sensitivity[12, 13]. The intestinal tract directly interacts with dietary components[14]. High dietary ω-3 FA has been associated with a low risk of IBD[15]. However, the underlying mechanisms are still unclear. G protein-coupled receptor (GPR) 120, which is recently identified as a receptor for ω-3 FA[16], has a critical role in various physiological homeostasis such as adipogenesis[17, 18]. Deficiency of GPR120 leads to obesity in both humans and mice, and Gpr120−/− mice are more susceptible to insulin resistance and have higher expression of the genes related to inflammation[19], whereas GPR120 agonists inhibit pro-inflammatory cytokine production by dendritic cells (DCs) and macrophages[16, 20]. However, it is still unknown how GPR120 regulates the pathogenesis of IBD.
In this study, we demonstrate that Gpr120−/− mice and CD4+T cell-specific GPR120 knockout Cd4creGpr120fl/fl mice develop more severe colitis than wild-type (WT) mice. Gpr120−/−CD4+T cells induce more severe colitis than WT CD4+T cells when transferred into Rag−/− mice. The GPR120 agonist promotes CD4+T cell production of IL-10 through upregulation of Blimp1 and enhancing glycolysis. Importantly, Gpr120 expression is positively correlated with Il10 expression in the human mucosa, including patients with IBD.
Materials and Methods
Mice
C57BL/6J WT mice, B6.129-Prdm1tm1Clme/J (Prdmfl/fl) mice, B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ (Cd4cre) mice, and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice were purchased from Jackson Laboratory. Gpr120−/− mice were provided by Bristol-Myers Squibb. Gpr120fl/fl mice were generated by inserting two loxP sites in the Gpr120 allele using CRISPER/Cas9 gene-editing technology in C57BL/6J background in Viewsolid Biotech and crossed to Cd4cre to generate Cd4creGpr120fl/fl mice. IL-10−/− mice were provided by Dr. Cohn at the University of the Texas Medical Branch (UTMB). Prdm1fl/fl mice were crossed to Cd4cre to generate Cd4crePrdmfl/fl mice. All the mice were maintained on a 12 hour-light/dark cycle with the temperature of 20–26°C and 30–70% humidity in the specific pathogen-free animal facility of UTMB. All the experimental mice were sex-matched and age-matched littermates and cohoused after weaning. All the animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of UTMB.
Human
All the patients with Crohn’s disease (CD) and ulcerative colitis (UC) were recruited at the Gastroenterology Department of the First Affiliated Hospital of Nanjing Medical University. The diagnosis of CD and UC was based on the combination of clinical signs and symptoms, endoscopic features, and histological results. Colonic tissues were collected from 14 healthy volunteers, and inflamed mucosa biopsies were obtained from 21 active CD patients and 10 active UC patients. Ethical approval was obtained from the Institutional Review Board and Ethics Committee of the First Affiliated Hospital of Nanjing Medical University, and informed consent was obtained from all the participants. The characteristics of all the human subjects were included in Supplementary Table 1.
Statistical analysis
Data were analyzed by the two-way Student’s t-test or Mann–Whitney U test or one-way ANOVA test or Spearman’s correlation analysis using GraphPad Prism 9. All data are presented as mean ± SEM, and the P-value < .05 was considered statistically significant.
All the other methods are included in the online supplemental materials.
Results
1. Gpr120−/− mice develop more severe colitis upon inflammatory insult and enteric infection.
To determine whether GPR120 regulates intestinal homeostasis, we induced colitis in WT and Gpr120−/− mice by administering DSS. First, we confirmed that Gpr120 expression was knockout in Gpr120−/− mice (Supplementary Figure 1A). Gpr120−/− mice demonstrated more weight loss, suffered from more severe colitis, and produced more TNF-α and IL-6 than WT mice (Supplementary Figure 2A–D). There were no differences in IFN-γ and IL-17A production (Supplementary Figure 2D). However, IL-10 was lower in colonic tissues of Gpr120−/− mice than WT mice (Supplementary Figure 2D). There were no differences of IFN-γ+CD4+T cells, IL-17A+CD4+T cells, and Foxp3+Tregs in the colonic lamina propria (LP) between WT and Gpr120−/− mice, but fewer intestinal IL-10+CD4+T cells were present in Gpr120−/− mice (Supplementary Figure 2E–I).
Similar results were obtained in WT and Gpr120−/− mice infected with Citrobacter rodentium (C. rodentium), an enteric bacterial strain similar to IBD-associated human enteropathogenic Escherichia coli[21]. Gpr120−/− mice demonstrated more weight loss than WT mice (Supplementary Figure 2J). While there were no differences in the bacterial burdens at day 4, colony counts of fecal C. rodentium were higher in the Gpr120−/− mice on day 7, and the difference was even more significant from day 10 (Supplementary Figure 2K). On day 14, C. rodentium was no longer detectable in WT mice, but counts were still high in Gpr120−/− mice (Supplementary Figure 2K), suggesting the importance of GPR120 in adaptive immune responses against intestinal pathogens. When sacrificed 14 days post-infection, Gpr120−/− mice developed more severe colitis and produced more TNF-α, IL-6, and IFN-γ but less IL-10 in the colon than WT mice (Supplementary Figure 2L–N). There were no differences in colonic IL-17A production (Supplementary Figure 2N). These data indicate that GPR120 inhibits intestinal inflammation.
2. Gpr120−/− CD4+T cells induce more severe colitis.
While intestinal epithelial cells (IECs), DCs, and macrophages have been shown to express GPR120[16, 20, 22], its expression by CD4+T cells has not been defined. We found that activated CD4+T cells expressed GPR120 at a higher level than BMDCs and small bowel IECs, and GPR120 levels in activated CD4+T cells were higher than naïve T cells (Figure 1A–B). Besides, Th1 cells, Th17 cells, and Treg cells expressed GPR120 at similar levels but higher than DCs and IECs (Figure 1A–B).
Fig. 1. CD4+T cell-specific GPR120 KO mice develop more severe colitis upon DSS insult and C. rodentium infection.
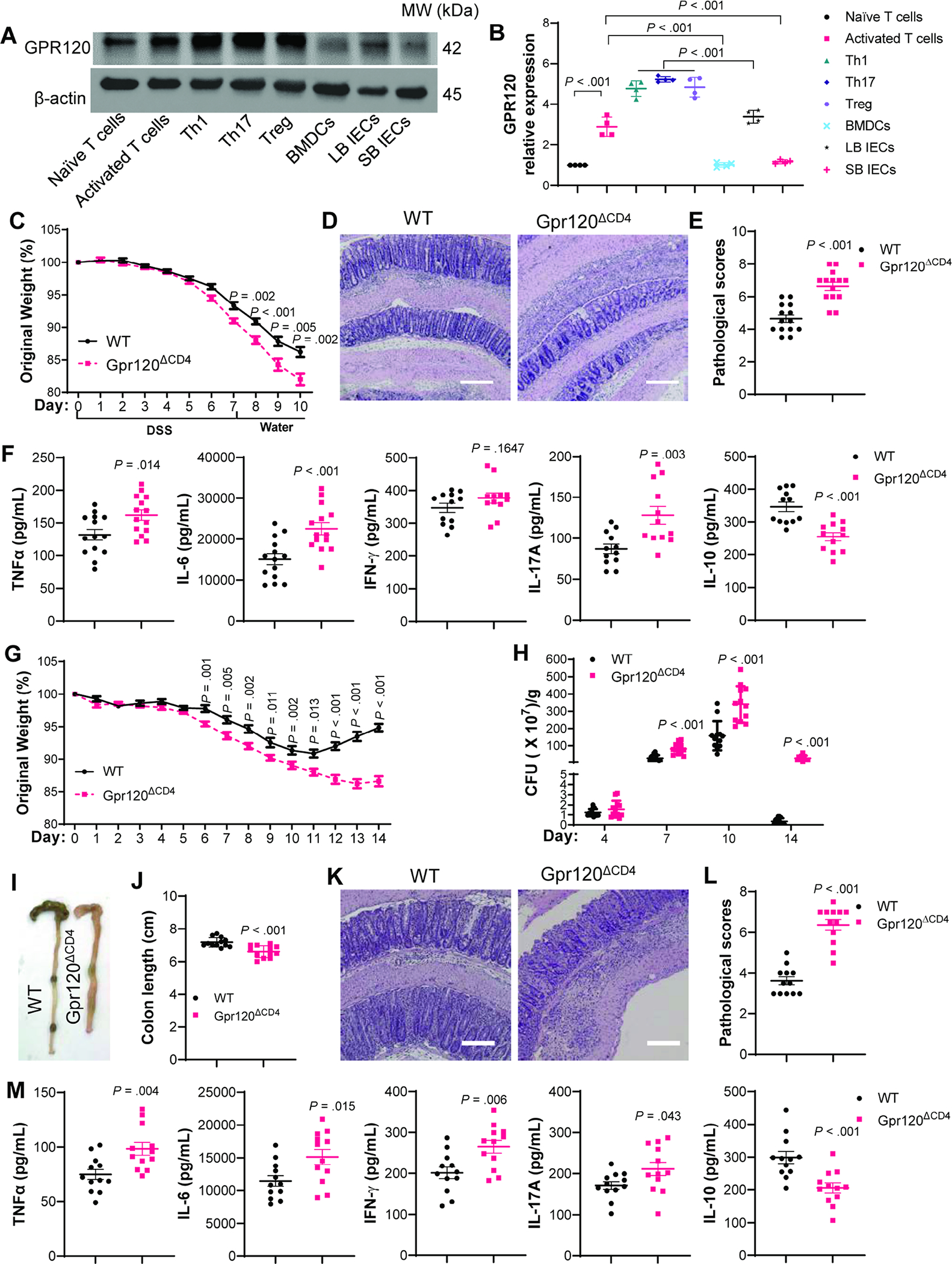
(A–B) GPR120 expression was measured in naïve CD4+T cells, activated CD4+T cells, Th1, Th17, Treg, bone-marrow dendritic cells (BMDCs), large bowl epithelial cells (LB IECs), and small bowel epithelial cells (SB IECs) (N=4/group). Western bolts (A) and GPR120 protein relative expression (B). (C–F) Cd4creGpr120fl/+ (WT) mice and Cd4creGpr120fl/fl (Gpr120ΔCD4) mice (N=14/group) were administrated with 1.65% DSS (w/v) in drinking water for 7 days followed by drinking water alone for additional 3 days. (C) Mouse weight change. (D) Representative intestinal H&E staining. (E) Pathological score. (F) Colonic secretion of cytokines. (G–M) Cd4creGpr120fl/+ (WT) mice and Cd4creGpr120fl/fl (Gpr120ΔCD4) mice (N=12/group) were orally infected with C. rodentium on day 0, and sacrificed on day 14. (G) Weight change. (H) Fecal C. rodentium counts. (I) Representative gross morphology of the colon. (J) Colon length. (K) Representative intestinal H&E staining. (L) Pathological score. (M) Colonic secretion of cytokines. All data are pooled from three independent experiments. (D and K) Scale bar, 300 μm. (A) one-way ANOVA; (C, F, G, H, J, and M) unpaired Student t-test; (E and L) Mann–Whitney U test.
We next investigated the role of CD4+T cell expression of GPR120 in intestinal homeostasis. First, we determined that Gpr120 was specifically knockout in CD4+T cells from Cd4creGpr120fl/fl mice (Supplementary Figure 1B–C). We next induced colitis in Cd4creGpr120fl/fl mice and WT Cd4creGpr120fl/+ mice using DSS. Cd4creGpr120fl/fl mice developed more severe colitis with more weight loss, higher pathological scores in the colon, and produced more TNF-α, IL-6, and IL-17A, but less IL-10 in the colon of Cd4creGpr120fl/fl mice than WT mice (Figure 1C–F). There was no difference in IFN-γ (Figure 1F). LP IL-10+CD4+T cells were decreased in Cd4creGpr120fl/fl mice, whereas there were no differences in IFN-γ+Th1 cells, IL-17+Th17 cells, and Foxp3+Tregs (Supplementary Figure 3A–E), indicating that CD4+T cell expression of GPR120 suppresses colitis, possibly through induction of IL-10.
Next, we infected Cd4creGpr120fl/fl mice and WT Cd4creGpr120fl/+ mice with C. rodentium. Cd4creGpr120fl/fl mice demonstrated more weight loss compared with WT Cd4creGpr120fl/+ mice (Figure 1G). While there were no differences in the bacterial burdens at day 4, fecal C. rodentium levels were higher in Cd4creGpr120fl/fl mice than WT mice on days 7, 10, and 14 (Figure 1H). When mice were sacrificed 14 days post-infection, Cd4creGpr120fl/fl mice developed more severe colitis and produced more TNF-α, IL-6, IFN-γ, and IL-17A, but less IL-10 in the colons (Figure 1I–M). More IFN-γ+Th1 cells and IL-17A+Th17 cells but fewer IL-10+T cells were present in LP of Cd4creGpr120fl/fl mice, while there was no difference in Foxp3+Tregs (Supplementary Figure 3F–J).
To further verify the role of GPR120 in CD4+T cells in regulating colitis, we utilized the CD4+CD45RBhiT cell adoptive transfer model[23]. WT and GPR120-deficient CD4+CD45RBhiT cells, isolated from WT Cd4creGpr120fl/+ mice and Cd4creGpr120fl/fl mice, were transferred into Rag−/− mice. The recipient mice were sacrificed 6 weeks later. GPR120-deficient CD4+CD45RBhiT cells induced more severe colitis in Rag−/− mice (Figure 2A–E), and produced more TNF-α, IL-6, IFN-γ, and IL-17A, but less IL-10 in colons than did WT CD4+CD45RBhiT cells (Figure 2F). Fewer intestinal IL-10+CD4+T cells and more IFN-γ+Th1 cells and IL-17A+Th17 cells were present in Rag−/− mice receiving GPR120-deficient T cells compared with recipient mice reconstituted with WT T cells, while there was no difference in Foxp3+Tregs between the two groups of mice (Figure 2G–K). These data indicate that GPR120 in CD4+T cells is critical in regulating intestinal homeostasis.
Fig. 2. GPR120-deficient CD4+CD45RBhiT cells induce more severe colitis in Rag−/− mice.
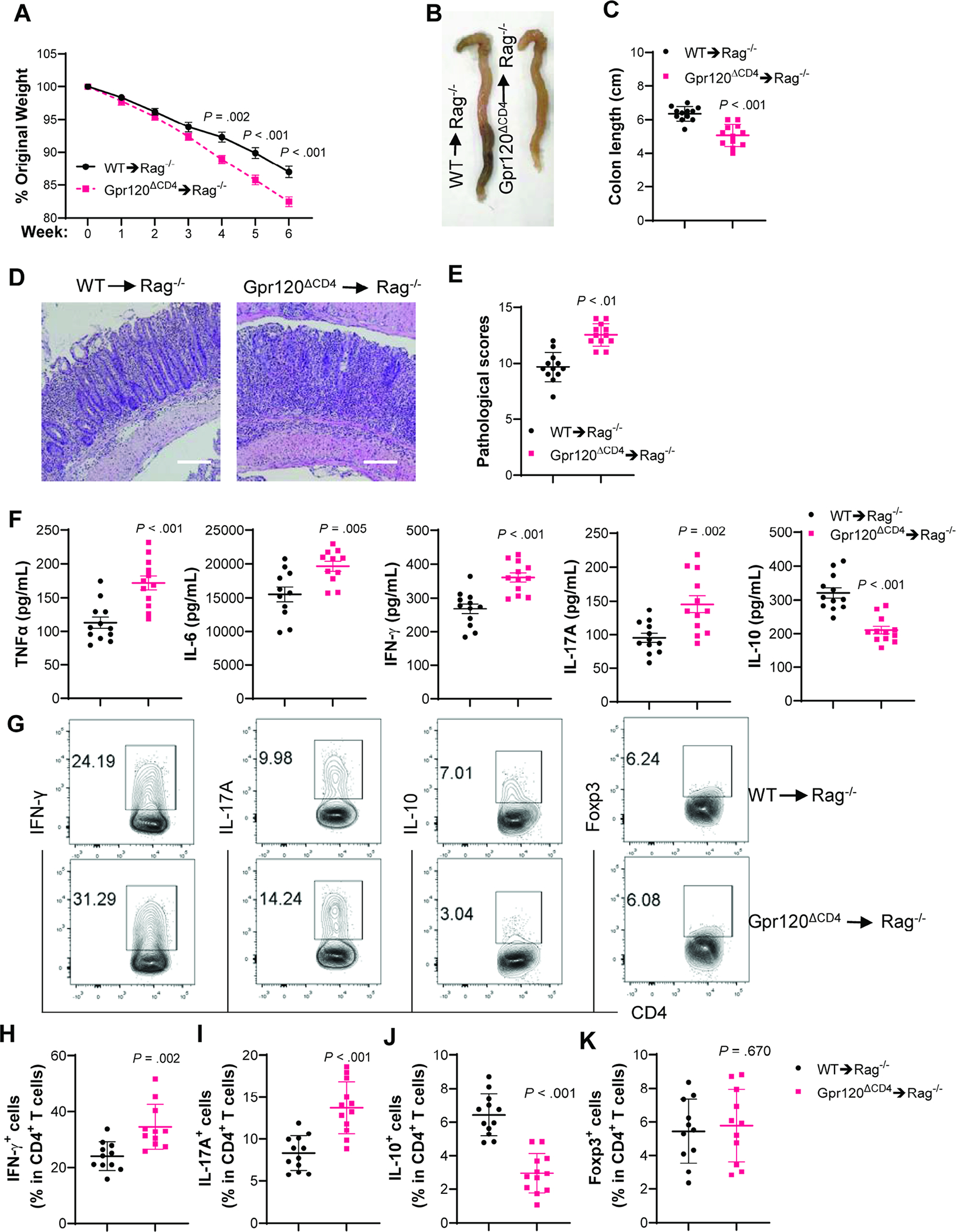
WT and GPR120-deficient CD4+CD45RbhiT cells (1×105 cells/mouse) were intravenously transferred to Rag−/− mice (N=12/group). (A) Mouse weight changes. (B) Representative gross morphology of the colon. (C) Colon length. (D) Representative intestinal H&E staining. (E) Pathological score. (F) Colonic secretion of cytokines. (G) Representative flow cytometry profile of LP CD4+T cells. (H–K) Bar charts of IFNγ+, IL-17A+, IL-10+, and Foxp3+CD4+T cells. All data are pooled from three independent experiments. (D) Scale bar, 300 μm. (A, C, F, and H–K) unpaired Student t-test; (E) Mann–Whitney U test.
3. GPR120 agonist promotes CD4+T cell production of IL-10 to suppress colitis.
To investigate whether GPR120 indeed regulates CD4+T cell functions, we cultured CD4+T cells with or without a GPR120 agonist, CpdA[20], for 2 days. Cells were collected, and the differences in the transcriptome were analyzed by RNA sequencing (RNA-Seq). GPR120 agonist altered the gene expression profile (Figure 3A and Supplementary Figure 4A), in which 1749 genes were upregulated and 1709 genes were downregulated (Supplementary Figure 4B). Gene Ontology functional enrichment analysis demonstrated that the differentially expressed genes were enriched in various functions regarding metabolism, function, and biological processes, including primary metabolic process, glycolytic process, regulation of immune system process, regulation of T cell-mediated immunity, regulation of T cell cytokine production, and cellular response to hypoxia (Supplementary Figure 4C). Furthermore, KEGG pathway enrichment analysis demonstrated that the differentially expressed genes were enriched in 46 pathways, including inflammatory bowel disease (Figure 3B). CpdA promoted Il10 but inhibited Ifng and Il17 at the mRNA levels, whereas it did not affect Foxp3 expression (Figure 3C).
Fig. 3. GPR120 promotes CD4+T cell production of IL-10 to suppress colitis.
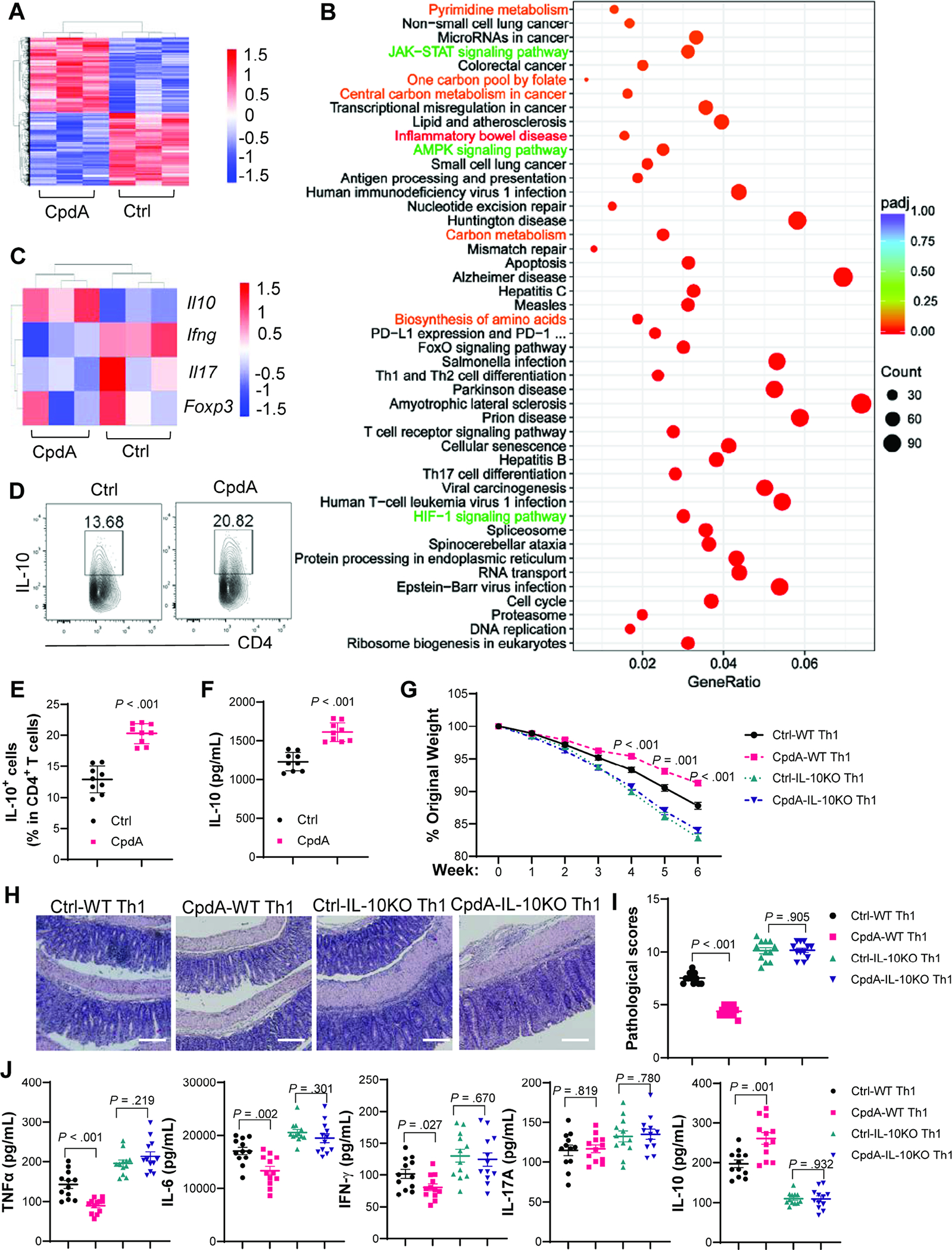
(A–C) Splenic CD4+T cells were activated with anti-CD3/anti-CD28±CpdA for 48 hours to analyze gene expression by RNA-Seq (N=3/group). (A) Differentially expressed genes between CD4+T cells treated with or without CpdA in Heatmap. Arbitrary units. (B) KEGG pathway enrichment analysis. (C) Specific gene expressions in Heatmap. Arbitrary units. (D–F) CD4+T cells were activated with anti-CD3/anti-CD28±CpdA under Th1 conditions (N=9/group). Flow cytometry profile of IL-10+CD4+T cells after 5 days (D–E). IL-10 in culture supernatants after 2 days (F). (G–J) WT and IL-10−/− CD4+T cells were activated with anti-CD3/anti-CD28±CpdA under Th1 conditions for 5 days and transferred to Rag−/− mice (N=12/group). (G) Mouse weight change. (H) Representative intestinal H&E staining. (I) Pathological score. (J) Colonic secretion of cytokines. (D–J) Data were pooled from three independent experiments. (H) Scale bar, 300 μm. (E, F, G, and J) unpaired Student’s t-test; (I) Mann–Whitney U test.
To verify the effect of CpdA on CD4+T cell cytokine production at the protein level, we cultured CD4+T cells with or without CpdA for 5 days to measure cytokine production using flow cytometry. CpdA promoted CD4+T cell IL-10 production, but it did not affect IFN-γ and IL-17A under neutral conditions (Supplementary Figure 5A–F), which might be attributed to low protein expressions of IFN-γ and IL-17A in CD4+T cells under neutral conditions. GPR120 agonist promoted IL-10 in CD4+T cells under Th1 conditions (Figure 3D–F), but not under Th17 and Treg conditions (Supplementary Figure 5A–B). Furthermore, CD4+T cells expressed higher levels of IL-10 under Th1 conditions than under other polarization conditions (Figure 3D–E and Supplementary Figure 5A–B). Interestingly, CpdA promoted IFN-γ+IL-10+T cells but not IFN-γ+IL-10−T cells under Th1 conditions (Supplementary Figure 5G–I), indicating that CpdA renders self-regulatory function to Th1 effector cells. CpdA suppressed IL-17A production under Th17 conditions (Supplementary Figure 5J–K), but it did not affect Treg differentiation (Supplementary Figure 5L–M). Besides, CpdA at the indicated dose did not affect CD4+T cell viability and proliferation (Supplementary Figure 6A–C). To determine whether the natural ligand for GPR120 also promotes IL-10 production in CD4+T cells, we treated CD4+T cells with DHA, a ω-3 FA, under Th1 conditions. As shown in Supplementary Figure 7A–B, DHA increased T cell IL-10 production in a dose-dependent manner.
As GPR120 promoted CD4+T cell production of IL-10, especially under Th1 conditions, and Th1 cells expressed higher levels of IL-10, we next investigated whether GPR120 agonist-treated Th1 cells induced less severe colitis through induction of IL-10. We treated WT and IL-10-deficient CD4+T cells with or without the GPR120 agonist CpdA for 5 days, then transferred them into Rag−/− mice. The mice were sacrificed 6 weeks later. The Rag−/− recipient mice of CpdA-treated WT Th1 cells developed less severe colitis and produced less TNF-α, IL-6, and IFN-γ, but more IL-10 in colons compared with Rag−/− mice reconstituted with untreated WT Th1 cells (Figure 3G–J). However, the Rag−/− mice receiving control IL-10-deficient Th1 cells or CpdA-treated IL-10-deficient Th1 cells developed severe colitis at similar levels with no differences in colonic cytokines (Figure 3G–J). We also used the anti-IL-10R antibody to inhibit the IL-10-IL-10R pathway in Rag−/− mice receiving CpdA-treated Th1 cells or control Th1 cells. Anti-IgG antibody was used as a control. CpdA-treated Th1 cells induced less severe colitis than control Th1 cells in Rag−/− mice treated with control anti-IgG antibody (Supplementary Figure 8A–F). However, there were no differences in disease severity and cytokine production when the mice were treated with anti-IL-10R antibody (Supplementary Figure 8A–F).
As GPR120 did not affect Treg differentiation (Supplementary Figure 5L–M), we then investigated whether GPR120 affects Treg suppressive functions. CD45.1 WT CD4+CD45RBhiT cells were transferred into Rag−/− mice together with or without CD45.2 WT or GPR120-deficient Tregs, differentiated from naϊve CD4+T cells of WT Cd4creGpr120fl/+ mice or Cd4creGpr120fl/fl mice. Both WT and GPR120-deficient Tregs suppressed colitis induced by CD4+CD45RBhi T cells at similar levels (Supplementary Figure 9A–F). These data indicate that GPR120 does not affect Treg function in suppressing colitis.
4. Blimp1 mediates GPR120 induction of IL-10 through the mTOR-Stat3 pathway.
Next, we investigated the potential mechanisms underlying the GPR120 induction of IL-10 in CD4+T cells. RNA-Seq showed an increased tendency of transcription factors Irf4, Prdm1 (encodes Blimp1), and Maf, but a decreased expression of Ahr (Supplementary Figure 10A), all of which have been implicated in regulating IL-10 production in CD4+T cells[24, 25]. To evaluate the transcriptome data, we treated CD4+T cells with or without CpdA with increased sample size. We found that CpdA significantly increased Prdm1 expression (Figure 4A), but not Irf4 and Maf (Supplementary Figure 10B–C). To determine if Blimp1 mediates GPR120 induction of IL-10, we utilized Cd4crePrdm1fl/fl mice, in which GPR120 was specifically knockout in CD4+T cells (Supplementary Figure 1D–E). We found that CpdA induction of IL-10 was compromised in Blimp1-deficient CD4+T cells (Figure 4B–D), indicating that Blimp1, at least partially, mediates GPR120 induction of IL-10.
Fig. 4. Blimp1 mediates GPR120 induction of IL-10 through the mTOR-Stat3 pathway.
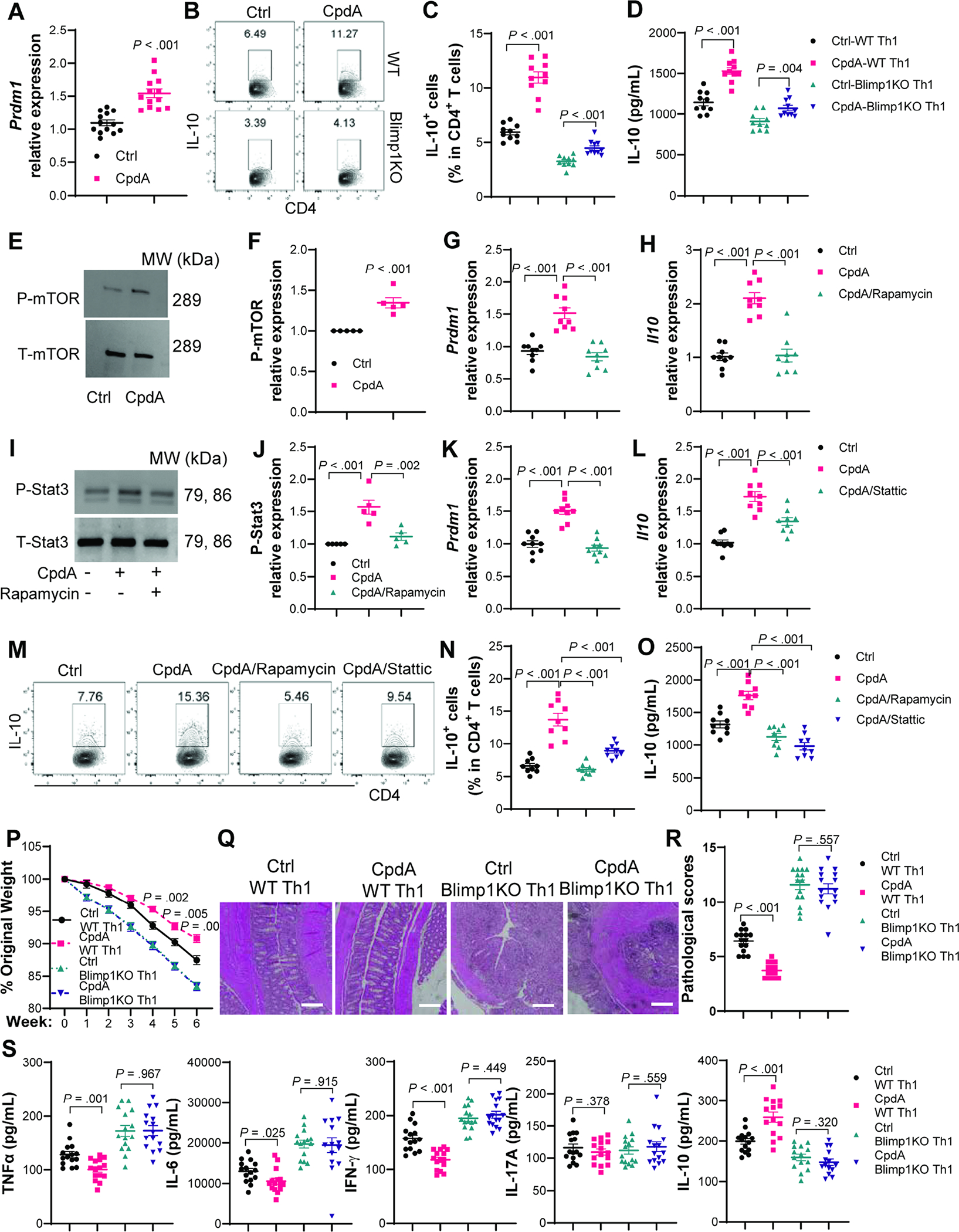
Splenic CD4+T cells were activated with anti-CD3/anti-CD28±CpdA and various inhibitors under Th1 conditions. (A) Prdm1 mRNA expression on day 2 (N=13/group). (B–C) Flow cytometry profile of IL-10+CD4+T cells in WT and Blimp−/− CD4+T cells on day 5. (D) IL-10 in culture supernatants on day 2 (N=10/group). (E–F) Phosphorylated and total mTOR expressions at 30 minutes (N=5/group). (G) Prdm1 and (H) Il10 mRNA expression in CD4+T cells treated with or without CpdA±rapamycin on day 2 (N=9/group). (I–J) Phosphorylated and total Stat3 expressions at 2 hours in CD4+T cells treated with or without CpdA±rapamycin (N=5/group). (K) Prdm1 and (L) Il10 mRNA expression in CD4+T cells treated with or without CpdA±Static (N=9/group). (M–N) Flow cytometry profile of IL-10+CD4+T cells on day 5, and (O) IL-10 in culture supernatants on day 2 (N=9/group). (P–S) WT and Blimp1−/− CD4+T cells from Cd4crePrdm1fl/+ mice and Cd4crePrdm1fl/fl mice were activated with anti-CD3/anti-CD28±CpdA under Th1 conditions for 5 days and transferred to Rag−/− mice (N=15/group). (P) Mouse weight change. (Q) Representative intestinal H&E staining. (R) Pathological score. (S) Colonic secretion of cytokines. All data are pooled from three independent experiments. (A, C, D, F, J, P, and S) unpaired Student’s t-test; (G, H, N, O) one-way ANOVA; (R) Mann–Whitney U test.
We then investigated how GPR120 regulates Blimp1 expression in CD4+T cells. It has been reported that GPR120 activates mTOR in bone marrow-derived mesenchymal stem cells[26], which has been shown to regulate IL-10 production in T cells[27]. KEGG pathway enrichment analysis demonstrated that GPR120 agonist altered mTOR upstream AMPK signaling pathway in T cells (Figure 3B). Treatment with GPR120 agonist CpdA promoted mTOR activation in CD4+T cells (Figure 4E–F). Treatment with rapamycin, the mTOR inhibitor, decreased T cell expression of Prdm1 (Figure 4G) and IL-10 production induced by CpdA (Figure 4H and M–O). Given that Stat3 promotes Blimp1 expression and mTOR positively regulates Stat3[28–30], and RNA-seq data suggested an altered JAK-STAT signaling pathway in CpdA-treated T cells (Figure 3B), we hypothesized that GPR120 activation of mTOR induces IL-10 production through the Stat3-Blimp1 pathway. Indeed, CpdA activated Stat3, which was inhibited by the mTOR inhibitor rapamycin (Figure 4I–J). Stattic, a selective Stat3 inhibitor, suppressed CpdA-induced Prdm1 expression and IL-10 production (Figure 4K–O). Furthermore, both mTOR and Stat3 inhibitors suppressed IL-10 production induced by DHA (Supplementary Figure 7C–D). These data suggest that Blimp1 mediates GPR120 induction of IL-10 through the mTOR-Stat3 pathway.
5. Blimp1 is essential for GPR120 inhibition of T effector cell induction of colitis.
We then investigated whether Blimp1 is essential for GPR120 inhibition of T effector cell induction of colitis. We treated Blimp1-deficient and WT CD4+T cells from Cd4crePrdm1fl/fl mice and Cd4crePrdm1fl/+ mice with or without the CpdA for 5 days, then transferred them into Rag−/− mice. Rag−/− recipient mice of CpdA-treated WT Th1 cells developed less severe colitis and produced less TNF-α, IL-6, and IFN-γ, but more IL-10 in the colon than those with WT Th1 cells (Figure 4P–S). However, both CpdA-treated Blimp1-deficient Th1 cells and control Blimp1-deficient Th1 cells induced severe colitis and led to similar levels of pro-inflammatory cytokines in the colon of Rag−/− recipient mice (Figure 4P–S). While intestinal IFN-γ+CD4+T cells were decreased, and IL-10+CD4+T cells were increased in Rag−/− mice reconstituted with CpdA-treated WT Th1 cells compared with the mice receiving control WT Th1 cells, similar levels of intestinal IL-10+CD4+T cells and IFN-γ+CD4+T cells were present in recipient mice receiving control Blimp1-deficient Th1 cells or CpdA-treated Blimp1-deficient Th1 cells (Supplementary Figure 11A–B, and E). All the recipient mice showed similar levels of intestinal IL-17+CD4+T cells and Foxp3+Tregs (Supplementary Figure 11A, and C–D).
6. Glycolysis is involved in GPR120 induction of IL-10 in CD4+T cells.
Because transcriptome data indicated that GPR120 affects the metabolism of CD4+T cells (Figure 3B and Supplementary Figure 4C), we next investigated whether GPR120 modulates mitochondrial oxidation and glycolysis, the major events of metabolism, which differentially regulate T cell functions[31]. First, we measured the oxygen consumption rate (OCR), primarily attributed to mitochondrial oxidation, and the extracellular acidification rate (ECAR), which represents glycolysis, in CD4+T cells using an extracellular flux Seahorse analyzer. GPR120 agonist, CpdA, enhanced OCR and ECAR levels in both naïve T cells (Supplementary Figure 12A–D) and anti-CD3/anti-CD28-activated T cells (Supplementary Figure 12E–H). We also checked the key parameters of mitochondrial oxidation using the Mito stress test kit. CpdA-treated CD4+T cells exhibited enhanced OCR with higher basal respiration, ATP-related respiration, maximal respiration, and spare respiratory capacity than control T cells (Figure 5A–B). To further assess the key parameters of glycolytic flux and exclude the non-glycolytic acidification, we measured ECAR levels in activated CD4+T cells using the Glycolysis stress test kit. CpdA promoted glycolysis and glycolytic capacity but not glycolytic reserve, and slightly increased the ECAR levels related to the non-glycolytic activity (Figure 5C–D). In line with elevated glycolysis, CpdA promoted CD4+T cell glucose uptake (Supplementary Figure 12I–J).
Fig. 5. GPR120 agonist promotes IL-10 production through the glycolysis-HIF1α pathway.
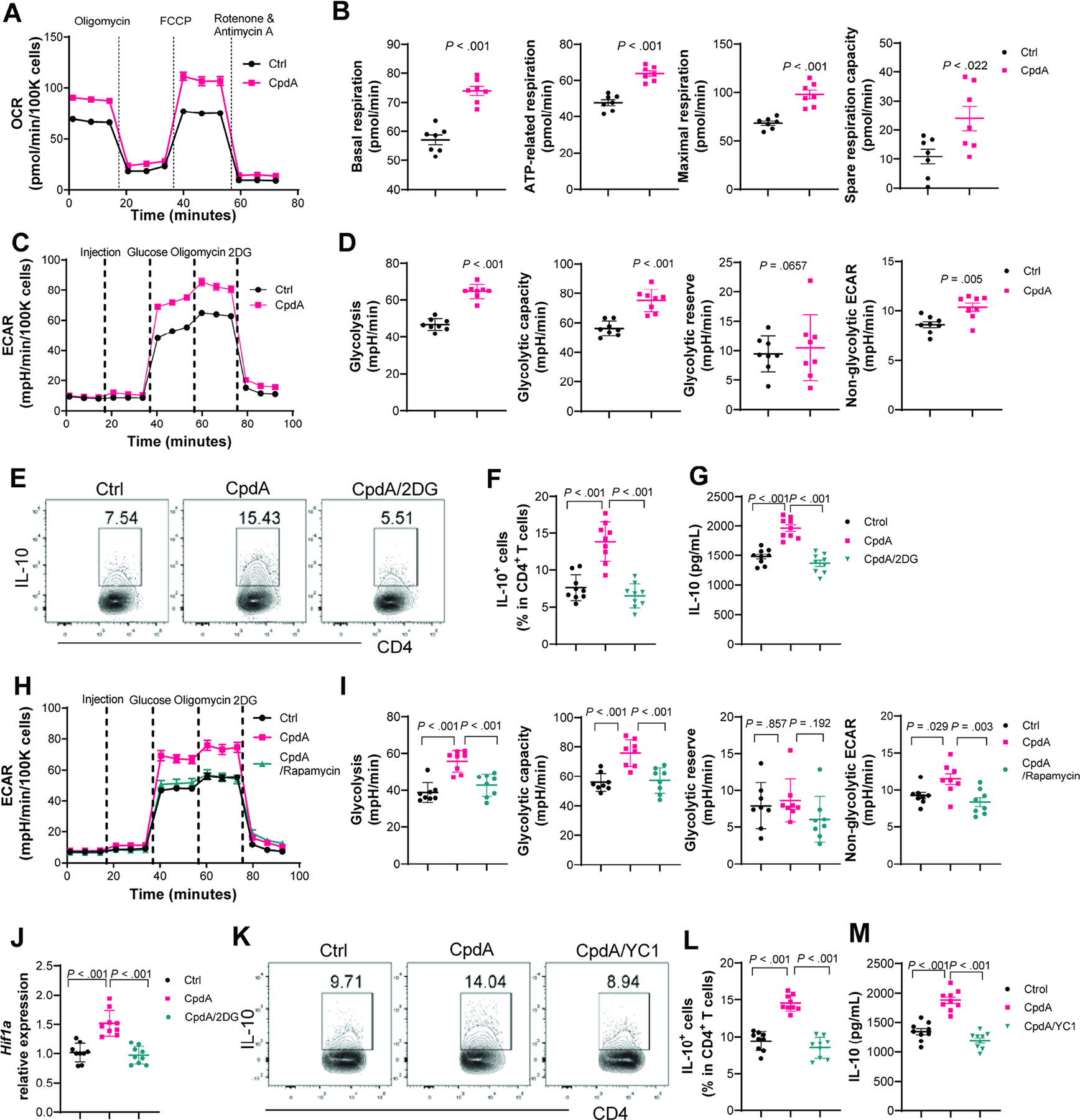
(A–B) Splenic CD4+T cells were activated with anti-CD3/anti-CD28±CpdA under Th1 conditions for 48 hours, and the Mito stress test kit was used to measure the parameters of mitochondrial respiration (N=7/group). The OCR profile, basal respiration, ATP-related respiration, maximum respiration, and spare respiration capacity. (C–D, and H–I) Splenic CD4+T cells were activated with anti-CD3/anti-CD28 under Th1 conditions for 48 hours, and a Glycolytic stress test kit was used to measure the key parameters of glycolysis (N=8/group). The ECAR profile, glycolysis, glycolytic capacity, glycolytic reserve, and non-glycolytic ECAR. (E–G) CD4+T cells were treated with or without CpdA±2-DG (N=9/group). IL-10+CD4+T cells on day 5 (E–F), and IL-10 in culture supernatants on day 2 (G). (J) Hif1a mRNA expression in CD4+T cells treated with or without CpdA±2-DG at 48 hours (N=9/group). (K–M) CD4+T cells were treated with or without CpdA±YC-1 (N=9/group). Flow cytometry profile of IL-10+CD4+T cells at day 5 (K–L) and IL-10 production in culture supernatants on day 2 (M). (E–G, and J-M) Data are pooled from three independent experiments. (B and D) Unpaired Student’s t-test (F, G, I, L, and M) one-way ANOVA.
Glycolysis has been shown to regulate IL-10 production in several types of immune cells[32, 33]. We then investigated whether GPR120-enhanced glycolysis contributes to upregulated IL-10 production in CD4+T cells. We found that blocking glycolysis suppressed IL-10 production induced by CpdA and DHA (Figure 5E–G, and Supplementary Figure 7C–D). Additionally, inhibition of the mTOR using rapamycin inhibited CpdA-induced glycolysis and glycolytic capacity (Figure 5H–I) and glucose uptake (Supplementary Figure 12K–L), suggesting that GPR120 activation of mTOR regulates glycolysis. Glycolysis has been reported to promote HIF1α expression[34], which increases IL-10 production in B cells [33]. KEGG pathway enrichment analysis suggested that GPR120 regulates the HIF1 signaling pathway in T cells (Figure 3B), and GPR120 agonist CpdA treatment promoted HIF1α expression in CD4+T cells (Supplementary Figure 10), which was suppressed by the glycolysis inhibitor, 2-DG (Figure 5J). Furthermore, treatment with YC-1, a HIF1α inhibitor, decreased CpdA-induced IL-10 production (Figure 5K–M). Collectively, these data indicate that GPR120-elevated HIF1α expression also contributes to GPR120 induction of IL-10.
7. Oral feeding of GPR120 agonist inhibits colitis.
As the GPR120 agonist promoted CD4+T cell production of IL-10 and GPR120 regulates intestinal inflammation, we then investigated whether administering the GPR120 agonist could prevent and treat colitis. We first transferred WT CD4+CD45RBhiT cell to Rag−/− mice and gave the mice orally with or without CpdA daily from the day of cell transfer until the mice were sacrificed. Administering CpdA suppressed colitis, characterized by less weight loss, increased colon length, decreased pathology scores, and inhibited TNF-α, IL-6, IFN-γ, and IL-17A, but promoted IL-10 production in the colon (Figure 6A–F). However, CpdA did not alleviate colitis severity in Rag−/− recipients of GPR120-deficient CD4+CD45RBhi T cells (Supplementary Figure 13A–F), suggesting that CD4+T cells are indispensable in GPR120 regulation of colitis. Similar results were obtained in mice infected with C. rodentium, which were treated orally with or without CpdA daily for 10 days. The CpdA-treated mice developed less severe colitis with less weight loss, lower intestinal bacteria load, decreased pathological scores, and lower pro-inflammatory cytokines in the intestine than those treated with carrier alone (Supplementary Figure 14A–E). Administering CpdA promoted IL-10 and IL-22 production in the colon and increased intestinal expression of antimicrobial peptide regenerating islet-derived 3 gamma (Reg3γ) (Supplementary Figure 14E–F). Furthermore, CpdA treatment decreased intestinal IFN-γ+CD4+T cells and IL-17A+CD4+T cells but increased intestinal IL-10+CD4+T cells and IL-22+CD4+T cells (Supplementary Figure 14 G–K). However, CpdA treatment did not affect the levels of intestinal Foxp3+Treg (Supplementary Figure 14 G and L).
Fig. 6. Oral feeding of GPR120 agonist prevents and treats colitis.
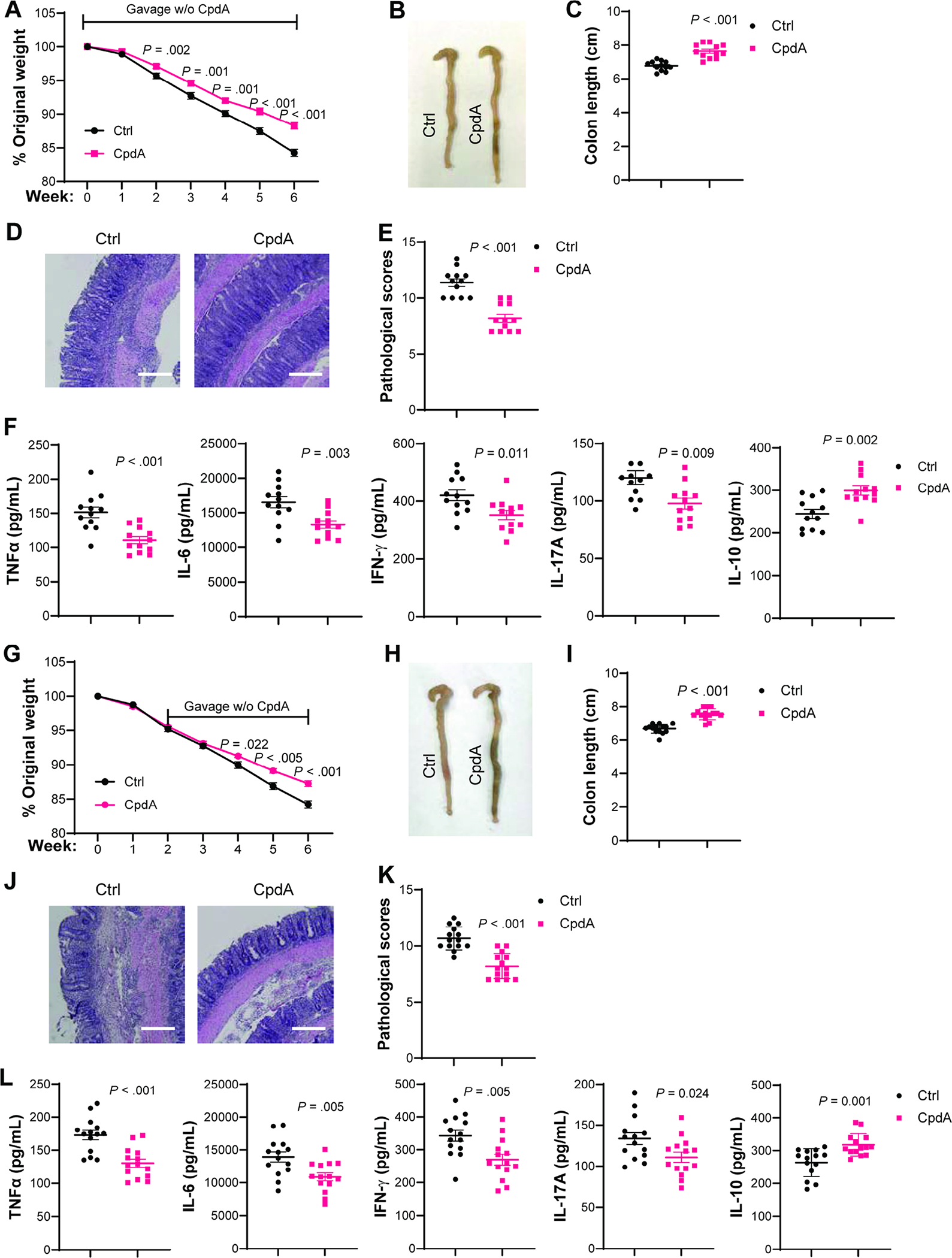
(A–F) WT CD4+CD45RbhiT cells (1×105 cells/mouse) were intravenously transferred to Rag−/− mice and orally administered with or without CpdA (N=12/group) daily from the day of cell transfer until the mice were sacrificed 6 weeks after cell transfer. (A) Mouse weight change. (B) Gross morphology of colon. (C) Colon length. (D) Representative intestinal H&E staining. (E) Pathological score. (F) Colonic secretion of cytokines. (G–L) WT CD4+CD45RbhiT cells (1×105 cells/mouse) were intravenously transferred to Rag−/− mice and orally administered with or without CpdA (N=12/group) daily from 2 weeks post cell transfer for additional 4 weeks. (G) Mouse weight change. (H) Gross morphology of colon. (I) Colon length. (J) Representative intestinal H&E staining. (K) Pathological score. (L) Colonic secretion of cytokines. All data are pooled from two independent experiments. (D and J) Scale bar, 300 μm. (A, C, F, G, I, and L) unpaired Student t-test; (E and K) Mann–Whitney U test.
Next, we investigated whether GPR120 agonist treats colitis. We transferred WT CD4+CD45RBhiT cells into Rag−/− mice. Two weeks after cell transfer, when mice have developed mild colitis (Supplementary Figure 15), the mice were treated orally with or without CpdA daily for additional 4 weeks. Administering CpdA inhibited colitis progression with decreased weight loss, increased colon length, lower pathological scores, and lower pro-inflammatory cytokine production but increased IL-10 production in the intestine than carrier alone-treated mice (Figure 6 G–L). Collectively, these data suggest that GPR120 agonist prevents and treats colitis.
8. GPR120 is positively correlated with IL-10 in IBD patients.
To investigate whether GPR120 is differentially expressed in the intestinal mucosa of IBD patients, we retrieved the data from GSE11223 in the GEO database. Gpr120 expression values were increased in the colonic mucosa of UC patients compared with healthy controls (Figure 7A). The higher expression of intestinal GPR120 in UC patients suggests the potential for oral GPR120 agonists as therapeutics in IBD patients. Additionally, colonic Gpr120 expression values were positively correlated with Il10 in healthy controls, and there was a tendency correlation in UC patients (Figure 7B–C). To further verify the correlation between colonic GPR120 and IL-10 expression, we collected colonic biopsies from 14 healthy controls, 10 active UC patients, and 21 active CD patients, and measured GPR120 and IL-10 expression. Consistent with the result retrieved from the database, Gpr120 expression was positively correlated with Il10 expression in colonic biopsies of healthy controls and UC patients (Figure 7D–E). CD patients also showed the correlation between Gpr120 and Il10 in the mucosa (Figure 7F).
Figure 7. Gpr120 is positively correlated with Il10 in human colonic mucosa.
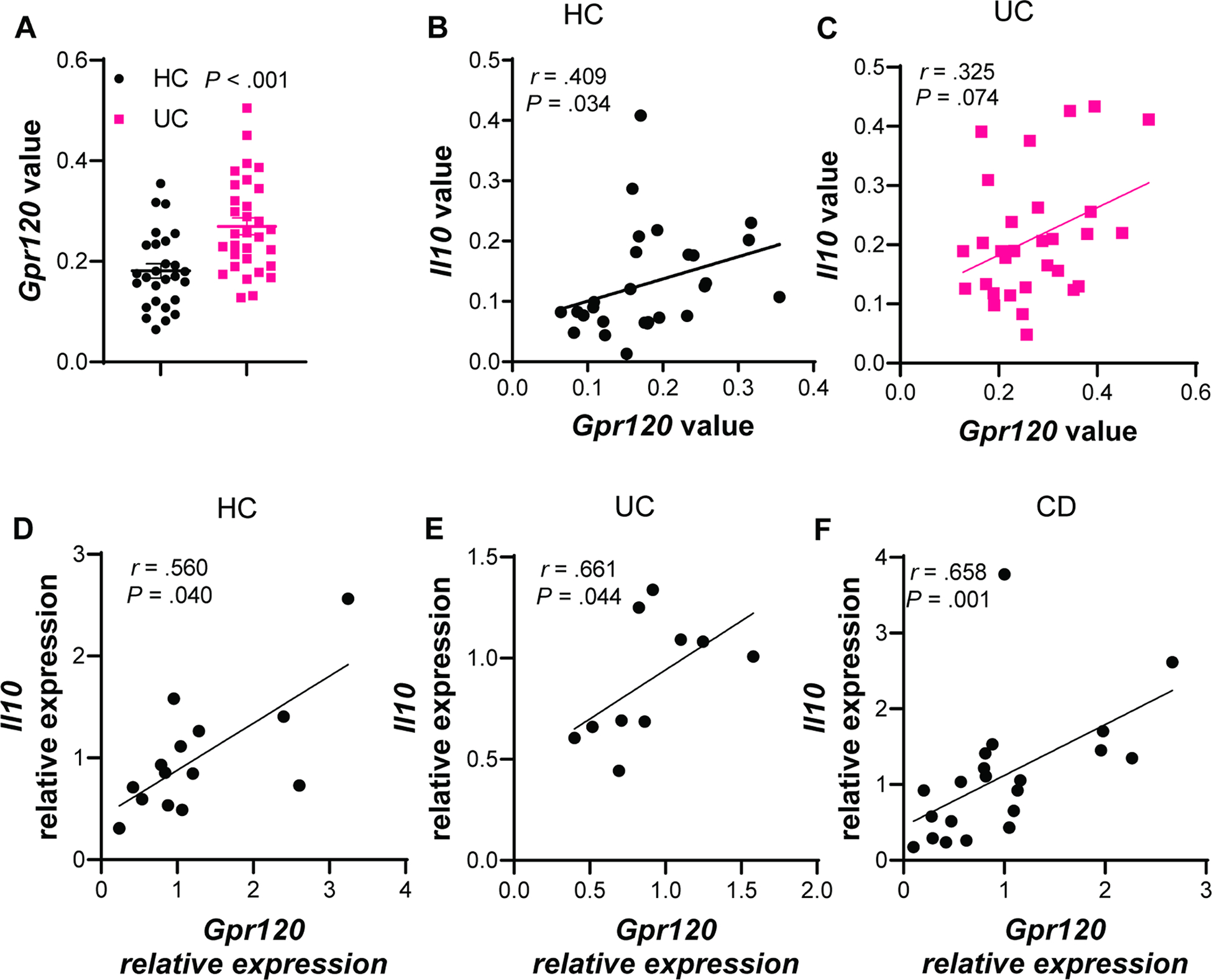
(A–C) The data from GSE11223 in the GEO database were retrieved. (A) Gpr120 expression in the colonic mucosa of healthy controls (HC, N=27) and patients with ulcerative colitis (UC, N=31). (B–C) The correlation between colonic Gpr120 and Il10 in HC (B) and UC patients (C). (D–F) Intestinal biopsies were collected from HC (N=14), UC patients (N=10), and patients with Crohn’s disease (CD, N=21). The correlation between intestinal Gpr120 and Il10 in HC (D), UC patients (E), and CD patients (F). (A) unpaired Student t-test; (B–F) Spearman correlation.
Discussion
Dietary regulation has been shown to play essential roles in health and diseases. GPR120, the receptor for ω-3 FA, has been implicated in regulating metabolic diseases and several types of immune cells[16, 19]. However, the role of GPR120 in the regulation of intestinal inflammation is unknown. In this study, we demonstrated that GPR120 promotes CD4+T cell production of IL-10 by upregulating Blimp1 and enhancing glycolysis to inhibit colitis, thus, providing novel insights into a critical role for GPR120 in maintaining intestinal homeostasis through the modulation of CD4+T cell functions.
Dietary ω-3 FA are beneficial for preventing metabolic syndrome and inflammation[12]. As a receptor of ω-3 FA, GPR120 regulates insulin signaling and inflammation in adipose tissue and controls the energy balance[17, 19]. GPR120 agonist increases insulin sensitivity, indicating that GPR120 might be a promising target for diabetes[16, 20, 35]. The intestinal tract directly interacts with dietary components, and several intestinal disorders have been linked to diet[14]. Nevertheless, the exact role of diet in IBD pathogenesis remains poorly understood. ω-3 FA consumption is negatively associated with IBD incidence[15]. We found that DHA, a ω-3 FA, promoted T cell production of IL-10. It has been reported that dietary intake of ω-3 FA increased the blood levels of ω-3 FA[36], suggesting that dietary ω-3 FA could be absorbed in the intestine and transferred into circulation to regulate circulating CD4+T cell production of IL-10. IL-10+CD4+T cells could be recruited to the inflamed intestines to suppress colitis. It is also possible that dietary ω-3 FA might directly affect CD4+T cell functions in the gut.
GPRs are actively involved in most physiological responses in humans and have become one of the successful targets of pharmaceuticals[37]. Previous studies have demonstrated the potential treatment of the GPR120 agonist for treating diabetes and metabolic syndrome[16, 20, 35, 37]. We showed that Gpr120−/− mice developed more severe colitis upon inflammatory insult and enteric infection, and oral administration of the GPR120 agonist prevented and treated colitis, indicating that GPR120 agonists might be potential therapeutic targets for IBD.
Excessive CD4+T cell responses have been considered critical in driving intestinal inflammation, whereas IL-10 restricts pro-inflammatory responses to maintain intestinal homeostasis. IL-10 produced by CD4+ effector T cells exerts a self-limiting mechanism to prevent an exaggerated T cell response in the intestines, which otherwise would be detrimental[38]. Although several types of cells, including IECs, macrophages, and adipocytes, have been shown to express GPR120, it was unknown whether CD4+T cells express GPR120. We demonstrated that activated CD4+T cells express GPR120 at high levels, and CD4+T cell-specific GPR120 knockout mice developed more severe colitis. Furthermore, transfer of GPR120-deficient CD4+CD45RbhiT cells induced more severe colitis than WT CD4+CD45RbhiT cells with lower levels of IL-10 in the intestine. GPR120 agonist treatment inhibited WT but not IL-10-deficient T cell induction of colitis, indicating that GPR120 promotes T cell IL-10 production to inhibit colitis development. It has been shown that the lack of IL-10 in CD4+T cells worsens colitis but does not affect or slightly increases the clearance of C. rodentium[39, 40]. In our study, GPR120 agonist also increases IL-22 production and Reg3γ expression in the intestine, which could promote the intestinal clearance of C. rodentium[41]. Meanwhile, higher levels of IL-10 suppress excessive inflammation. The combination of IL-10 and IL-22 might contribute to the decreased severity of colitis and enhanced C. rodentium clearance. Variants in Prdm1, which encodes Blimp1, have been associated with the susceptibility of developing IBD[42]. Blimp1 plays an essential role in promoting IL-10 production in CD4+T cells[43]. We showed that GPR120 agonist inhibited WT but not Blimp−/− Th1 cell induction of colitis with higher levels of IL-10 production, indicating that Blimp1 mediates GPR120 regulation of colitis through IL-10 production.
Different CD4+T cells require distinct metabolic programs compatible with their functional demands. While quiescent CD4+T cells are characterized by mixed fuel oxidative phosphorylation, activated T cells become more glycolytic with increased oxidative phosphorylation for fulfilling the requirement of rapid cell growth and proliferation[31]. Metabolic programs have been shown to involve and regulate CD4+T cell functions. However, how the metabolic pathways affect T cell production of IL-10 has not been fully defined. Our study found that the GPR120 agonist increased CD4+T cell oxygen consumption and glycolysis, leading to higher ATP production and energy levels. Blocking glycolysis compromised GPR120-induced IL-10 production, indicating that enhanced glycolysis mediates GPR120-promotion of IL-10 production in CD4+T cells. Additionally, the GPR120 agonist increased T cell expression of HIF1α expression. Although HIF1α has been reported to suppress mitochondrial respiration[44], the increased oxidation in CD4+T cells might be attributed to other pathways affected by GPR120. A recent report showed that distinct mitochondrial metabolism determines CD4+T cell differentiation and function[45]; thus, it will be interesting to investigate whether the mode of mitochondrial metabolism is also involved in GPR120 induction of IL-10 in T cells.
In summary, our study demonstrated that GPR120 suppresses CD4+T cell induction of colitis through promoting IL-10 production. Oral administration of the GPR120 agonist inhibits colitis, which presents GPR120 as a potential therapeutic target for IBD treatment.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT:
G protein-coupled receptor (GPR) 120 has been implicated in regulating metabolic syndromes with anti-inflammatory function. However, the role of GPR120 in the regulation of intestinal inflammation is unknown.
NEW FINDINGS:
We discover that GPR120 induces IL-10 production in CD4+T cells through upregulation of Blimp1 and enhancing glycolysis to inhibit colitis. Administration of GPR120 agonist protected mice against intestinal inflammation.
LIMITATIONS:
A limitation of the current study is that it was mostly performed in animal models. The effect of GPR120 in human CD4+T cells and patients with inflammatory bowel disease (IBD) needs to be further validated in the clinical setting.
IMPACT:
This study identifies GPR120 as a potential therapeutic target for treating IBD.
Acknowledgment:
We appreciate Dr. Sherry Haller of The University of Texas Medical Branch for proofreading the manuscript.
Grant Support:
This work was supported in part by the National Institutes of Health grants DK125011, AI150210, and DK124132, the University of Texas System STARs award (Y.C.), and the James W. McLaughlin Fellowship Fund from The University of Texas Medical Branch at Galveston (W.Y.).
Abbreviations:
- BMDCs
Bone-marrow dendritic cells
- C. rodentium
Citrobacter rodentium
- DCs
Dendritic cells
- DSS
Dextran sulfate sodium
- DHA
Docosahexaenoic acid
- ECAR
Extracellular acidification rate
- GPR
G protein-coupled receptor
- IL-10R
IL-10 receptor
- IBD
Inflammatory bowel disease
- IECs
Intestinal epithelial cells
- LP
Lamina propria
- LCFA
Long-chain fatty acids
- ω-3 FA
Omega 3 fatty acids
- OCR
Oxygen consumption rate
- Tregs
Regulatory T cells
- RNA-Seq
RNA sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: No authors have conflicting financial, professional, or personal interests.
RNA sequencing data:
RNA sequencing data in this study is available in the GEO database under the accession number GSE158782.
References
- [1].Globig AM, Hennecke N, Martin B, et al. Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN-gamma+IL-17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm Bowel Dis 2014;20(12): 2321–9. [DOI] [PubMed] [Google Scholar]
- [2].Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet 2008;40(11): 1319–23. [DOI] [PubMed] [Google Scholar]
- [3].Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 2010;42(12): 1118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Begue B, Verdier J, Rieux-Laucat F, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol 2011;106(8): 1544–55. [DOI] [PubMed] [Google Scholar]
- [5].Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 2009;361(21): 2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993;75(2): 263–74. [DOI] [PubMed] [Google Scholar]
- [7].Kamanaka M, Huber S, Zenewicz LA, et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J Exp Med 2011;208(5): 1027–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Murai M, Turovskaya O, Kim G, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 2009;10(11): 1178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shouval DS, Biswas A, Goettel JA, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014;40(5): 706–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nature Reviews Gastroenterology & Hepatology 2019;16(1): 35–56. [DOI] [PubMed] [Google Scholar]
- [11].Kumar NG, Contaifer D, Madurantakam P, et al. Dietary Bioactive Fatty Acids as Modulators of Immune Function: Implications on Human Health. 2019;11(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sijben JW, Calder PC. Differential immunomodulation with long-chain n-3 PUFA in health and chronic disease. Proc Nutr Soc 2007;66(2): 237–59. [DOI] [PubMed] [Google Scholar]
- [13].Neschen S, Morino K, Dong J, et al. n-3 Fatty acids preserve insulin sensitivity in vivo in a peroxisome proliferator-activated receptor-alpha-dependent manner. Diabetes 2007;56(4): 1034–41. [DOI] [PubMed] [Google Scholar]
- [14].Moreira AP, Texeira TF, Ferreira AB, et al. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 2012;108(5): 801–9. [DOI] [PubMed] [Google Scholar]
- [15].Ananthakrishnan AN, Khalili H, Konijeti GG, et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2014;63(5): 776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010;142(5): 687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gotoh C, Hong YH, Iga T, et al. The regulation of adipogenesis through GPR120. Biochem Biophys Res Commun 2007;354(2): 591–7. [DOI] [PubMed] [Google Scholar]
- [18].Hilgendorf KI, Johnson CT, Mezger A, et al. Omega-3 Fatty Acids Activate Ciliary FFAR4 to Control Adipogenesis. Cell 2019;179(6): 1289–1305. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ichimura A, Hirasawa A, Poulain-Godefroy O, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012;483(7389): 350–354. [DOI] [PubMed] [Google Scholar]
- [20].Oh DY, Walenta E, Akiyama TE, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med 2014;20(8): 942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006;441(7090): 231–4. [DOI] [PubMed] [Google Scholar]
- [22].Kim JM, Lee KP, Park SJ, et al. Omega-3 fatty acids induce Ca(2+) mobilization responses in human colon epithelial cell lines endogenously expressing FFA4. Acta Pharmacol Sin 2015;36(7): 813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Powrie F, Correa-Oliveira R, Mauze S, et al. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med 1994;179(2): 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kubo M, Motomura Y. Transcriptional regulation of the anti-inflammatory cytokine IL-10 in acquired immune cells. Front Immunol 2012;3(275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ng TH, Britton GJ, Hill EV, et al. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol 2013;4(129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gao B, Han YH, Wang L, et al. Eicosapentaenoic acid attenuates dexamethasome-induced apoptosis by inducing adaptive autophagy via GPR120 in murine bone marrow-derived mesenchymal stem cells. Cell Death & Disease 2016;7(5): e2235–e2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sun M, Wu W, Chen L, et al. Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nature Communications 2018;9(1): 3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Diehl SA, Schmidlin H, Nagasawa M, et al. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol 2008;180(7): 4805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou J, Wulfkuhle J, Zhang H, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proceedings of the National Academy of Sciences 2007;104(41): 16158–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Poholek AC, Jankovic D, Villarino AV, et al. IL-10 induces a STAT3-dependent autoregulatory loop in T(H)2 cells that promotes Blimp-1 restriction of cell expansion via antagonism of STAT5 target genes. Sci Immunol 2016;1(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology 2020;20(1): 55–70. [DOI] [PubMed] [Google Scholar]
- [32].Suzuki H, Hisamatsu T, Naganuma M, et al. Glycolysis regulates macrophage differentiation into IL-10 producing phenotype (INM6P.342). The Journal of Immunology 2015;194(1 Supplement): 193.16–193.16. [Google Scholar]
- [33].Meng X, Grötsch B, Luo Y, et al. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nature Communications 2018;9(1): 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xu Y, Chaudhury A, Zhang M, et al. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J Clin Invest 2016;126(7): 2678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Paschoal VA, Walenta E, Talukdar S, et al. Positive Reinforcing Mechanisms between GPR120 and PPARγ Modulate Insulin Sensitivity. Cell Metab 2020;31(6): 1173–1188. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Superko HR, Superko SM, Nasir K, et al. Omega-3 fatty acid blood levels: clinical significance and controversy. Circulation 2013;128(19): 2154–61. [DOI] [PubMed] [Google Scholar]
- [37].Sriram K, Insel PA. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol 2018;93(4): 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jankovic D, Kugler DG, Sher A. IL-10 production by CD4+ effector T cells: a mechanism for self-regulation. Mucosal Immunology 2010;3(3): 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Krause P, Morris V, Greenbaum JA, et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nature Communications 2015;6(1): 7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Seiffart V, Zoeller J, Klopfleisch R, et al. IL10-Deficiency in CD4+ T Cells Exacerbates the IFNγ and IL17 Response During Bacteria Induced Colitis. Cell Physiol Biochem 2015;36(4): 1259–73. [DOI] [PubMed] [Google Scholar]
- [41].Yang W, Yu T, Huang X, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nature Communications 2020;11(1): 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ellinghaus D, Zhang H, Zeissig S, et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 2013;145(2): 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Neumann C, Heinrich F, Neumann K, et al. Role of Blimp-1 in programing Th effector cells into IL-10 producers. J Exp Med 2014;211(9): 1807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Okamoto A, Sumi C, Tanaka H, et al. HIF-1-mediated suppression of mitochondria electron transport chain function confers resistance to lidocaine-induced cell death. Scientific Reports 2017;7(1): 3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bailis W, Shyer JA, Zhao J, et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019;571(7765): 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.