

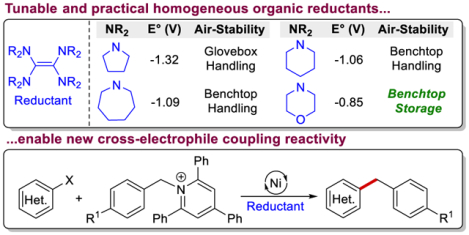
Abstract
The syntheses of four new tunable homogeneous organic reductants based on a tetraaminoethylene scaffold are reported. The new reductants have enhanced air-stability compared to current homogeneous reductants for metal mediated reductive transformations, such as cross-electrophile coupling (XEC), and are solids at room temperature. In particular, the weakest reductant is indefinitely stable in air and has a reduction potential of −0.85 V versus ferrocene, which is significantly milder than conventional reductants used in XEC. All of the new reductants are able to facilitate C(sp2)–C(sp3) Ni-catalyzed XEC reactions and are compatible with complex substrates that are relevant to medicinal chemistry. The reductants span a range of nearly 0.5 V in reduction potential, which allows for control over the rate of electron transfer events in XEC. Specifically, we report a new strategy for controlled alkyl radical generation in Ni-catalyzed C(sp2)–C(sp3) XEC. The key to our approach is to tune the rate of alkyl radical generation from Katritzky salts, which liberate alkyl radicals upon single electron reduction, by varying the redox potentials of the reductant and Katritzky salt utilized in catalysis. Using our method, we perform XEC reactions between benzylic Katritzky salts and aryl halides. The method tolerates a variety of functional groups, some of which are particularly challenging for most XEC transformations. Overall, we expect that our new reductants will both replace conventional homogeneous reductants in current reductive transformations due to their stability and relatively facile synthesis, and lead to the development of novel synthetic methods due to their tunability.
Graphical Abstract
Introduction
A variety of transition metal catalyzed processes have been developed over the last twenty years that require reductants to either generate a reactive alkyl radical from a substrate or to reduce a key transition metal intermediate.1 Ni-catalyzed cross-electrophile coupling (XEC) reactions, in which two electrophiles are coupled to form new C–C bonds, are a notable example (Figure 1a).2 XEC reactions provide a powerful method for the formation of C(sp2)–C(sp3) bonds, which are ubiquitous in active pharmaceutical ingredients but often difficult to generate using traditional synthetic methods.3 Typically, heterogeneous reductants such as Mn0 and Zn0 are used as the electron source in XEC reactions because they are inexpensive, air-tolerant, and often operationally simple to use. However, the use of these heterogeneous reductants limits the applications of XEC and related reactions because they are difficult to: (i) Use on scale due to the need for toxic amide-based solvents4 and irreproducible kinetics that arise from mass transfer limitations.5,6 (ii) Implement in flow chemistry,7 automated chemical synthesis,8 or nanomole scale reactions,9 due to engineering problems associated with heterogeneous reaction mixtures. (iii) Utilize to control the rates of reduction events in catalysis due to the large number of factors that influence electron transfer from the surface of a solid-state powder to a transition metal catalyst in solution.5,10 (iv) Tune to match the optimal redox potential of a catalyst or substrate due to the inability to readily modulate the reduction potentials of Mn0 or Zn0, respectively.
Figure 1:
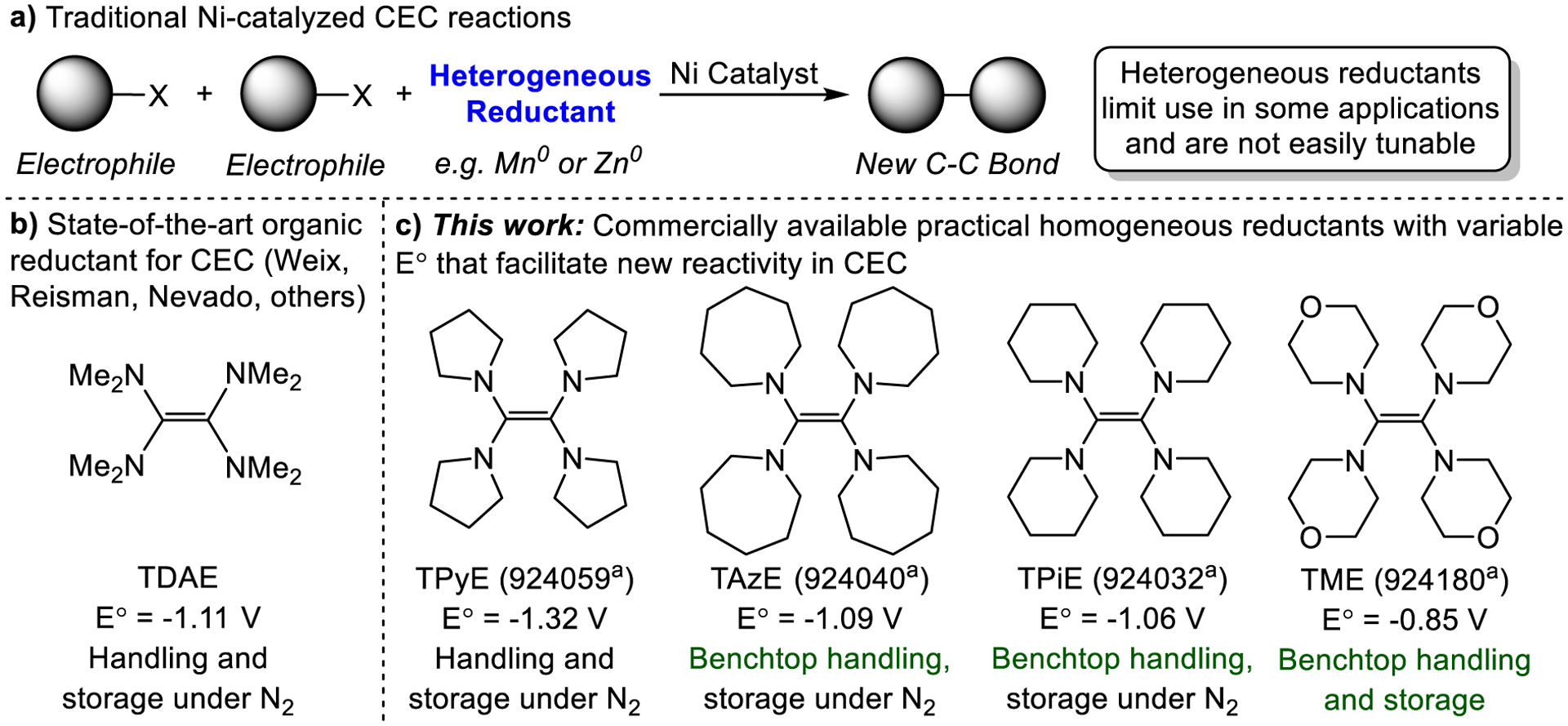
a) Traditional Ni-catalyzed XEC reactions with heterogeneous reductants. b) Current state-of-the-art homogeneous organic reductant for XEC. c) New practical homogeneous reductants that have been synthesized in this work. Potentials are reported relative to ferrocene (Fc) in DMF. aProduct number for commercially available reductants from MilliporeSigma (expected availability in early 2022).
The problems associated with heterogeneous reductants have led researchers to explore alternative electron sources in both XEC and other related transition metal mediated reactions.11 Two potentially attractive solutions are to facilitate reduction events through photocatalytic12 or electrochemical methods.13 Both of these strategies allow for tuning of the reduction potential and have led to advances in XEC, such as improved substrate scopes, extensions to new substrates, and the use of XEC in flow chemistry.12,13 Nevertheless, several challenges still persist. For example, these electron transfer methods can be difficult to implement either on multi-kilogram scale in batch,14 which is critical for the large-scale production of pharmaceuticals, fragrances, and agrochemicals, or on nanomole scale,9a,9b which is valuable for reaction optimization and first pass compound delivery in drug discovery.15 In contrast, homogeneous electron donors16 could provide a general solution to existing challenges facing XEC and related reactions. This is because, in principle, XEC reactions that use homogeneous reductants are: (i) Easier to scale-up and miniaturize, as they should result in homogeneous reaction mixtures. (ii) Compatible with many solvents,17 which is important for providing flexibility when solving synthetic problems in applied settings, achieving optimal substrate scope, and reducing toxicity concerns. (iii) Tunable like photochemical and electrochemical reductants, with electron transfer rates that can be controlled and predicted using Marcus or related theories.18 Additionally, from a fundamental perspective, homogeneous reductants could facilitate mechanistic studies of XEC reactions, which are often challenging to execute with heterogeneous, photochemical, or electrochemical electron sources.
At this stage, despite the potential advantages of homogeneous reductants, they are rarely utilized in Ni-catalyzed XEC.19,20 A major reason for this is that the small number of homogeneous reductants which have been utilized in XEC reactions are challenging to prepare and handle. For example, tetrakis(dimethylamino)ethylene (TDAE; E° = −1.11 V vs ferrocene, Fc), which is by far the most commonly utilized homogeneous organic reductant in XEC,19 is an expensive and air-sensitive liquid that is difficult to synthesize, store, and handle (Figure 1b).19d We hypothesized that if more practical homogeneous reductants were developed, they might solve some of the limitations associated with Ni-catalyzed XEC and be generally beneficial for reductive transformations. In this work, we describe the synthesis and characterization of four homogeneous organic reductants, which are based on the tetraaminoethylene scaffold found in TDAE (Figure 1c). Importantly, unlike TDAE, these reductants are straightforward to synthesize and are solids at room temperature. The new reductants span nearly 0.5 V in reduction potential, and we elucidate the factors that control their respective redox potentials through structural and computational studies, which provides design principles for future development of novel reductants. The newly synthesized reductants also display greatly enhanced air stability compared to TDAE, with the weakest reductant being indefinitely stable in air. All of the reductants facilitate metal mediated reductive transformations, such as Ni-catalyzed XEC reactions, and we demonstrate that they are compatible with substrates relevant to medicinal chemistry. Additionally, we use the tunability of our reductants to unveil a new strategy in XEC in which we achieve control of the rate of alkyl radical generation by using a reductant with the optimal redox potential. This enables the first examples of reductive coupling of aryl halides with benzylic Katritzky salts. In total, as a result of the stability and tunability of the reductants, as well as their ability to facilitate new chemistry, we expect that they will be beneficial for a variety of reductive transformations.
Results and Discussion
Development of homogeneous organic reductants
We hypothesized that by changing the substituents on the nitrogen atoms in the tetraaminoethylene scaffold of TDAE, we could develop reductants for XEC and related reactions with tunable potentials that are easy-to-handle solids under ambient conditions. Previously, derivatives of TDAE with tethered geminal amino substituents,21 such as tetraazafulvalenes,22 have been synthesized. However, these reductants are unlikely to be valuable in XEC or related reactions because: (i) They often have significantly more negative reduction potentials than TDAE, which can lead to deleterious reduction of substrates or the catalyst. (ii) They can readily form diaminocarbenes as described by the Wanzlick equilibrium,23 which can lead to poisoning of a transition metal catalyst. (iii) In many cases they have poor air stability.21 In contrast, we postulated that by modifying the tetraaminoethylene scaffold of TDAE by incorporating the nitrogen atoms into untethered heterocycles we could: (i) Limit steric interactions between the vicinal amine substituents and inhibit diaminocarbene formation, which is more likely to occur when there is a clash between the vicinal substituents. (ii) Add molecular weight by using heterocycles with relatively large ring size to give solid compounds at room temperature. (iii) Create a family of compounds with different reduction potentials by varying the identity of the heterocyclic ring. To that end, we synthesized four reductants TPyE, TAzE, TPiE, and TME (Figure 1c).24,25
The reductants were synthesized in three steps using inexpensive starting materials, and all purification steps were performed without the use of column chromatography (Figure 2). Initially, dimethylcarbamoyl chloride and anhydrous dimethylformamide were used to generate the amidinium salt 2a (which is also commercially available) in high yield according to a literature procedure.19d A subsequent transamidation-type reaction between 2a and the desired N-heterocycle 2b results in the formation of an amidinium salt 2c in good yields. The final step proceeds through a deprotonation-dimerization pathway,26 which converts 2c to the desired tetraaminoethylenes in yields ranging from 21–60%. Importantly, each reductant is a solid that can be purified primarily through filtration and washing and has good solubility in a range of common organic solvents (see SI). Additionally, for the purifications of TAzE, TPiE, and TME, filtrations can be performed under air due to their enhanced air stability. Consequently, generating high purity product on a large scale is straightforward. For example, we scaled the synthesis of TPiE to 0.04 mol, obtaining 17 g of product in a single pass (see SI). TDAE is also normally synthesized in three steps from inexpensive starting materials in generally comparable yields. However, the final step in the typical synthesis of TDAE proceeds through the thermal decomposition of tris(dimethylamino)methane at high temperatures (180 °C) over long reaction times (5 days) and requires purification through a challenging fractional distillation under reduced pressure followed by careful handling under N2.19d In contrast, the simple synthesis and purification of our reductants should lead to them being significantly easier to access than TDAE. To that end, the new reductants will be commercially available from MilliporeSigma (expected in early 2022), with product numbers 924059, 924040, 924032, 924180, for TPyE, TAzE, TPiE, and TME, respectively.
Figure 2:
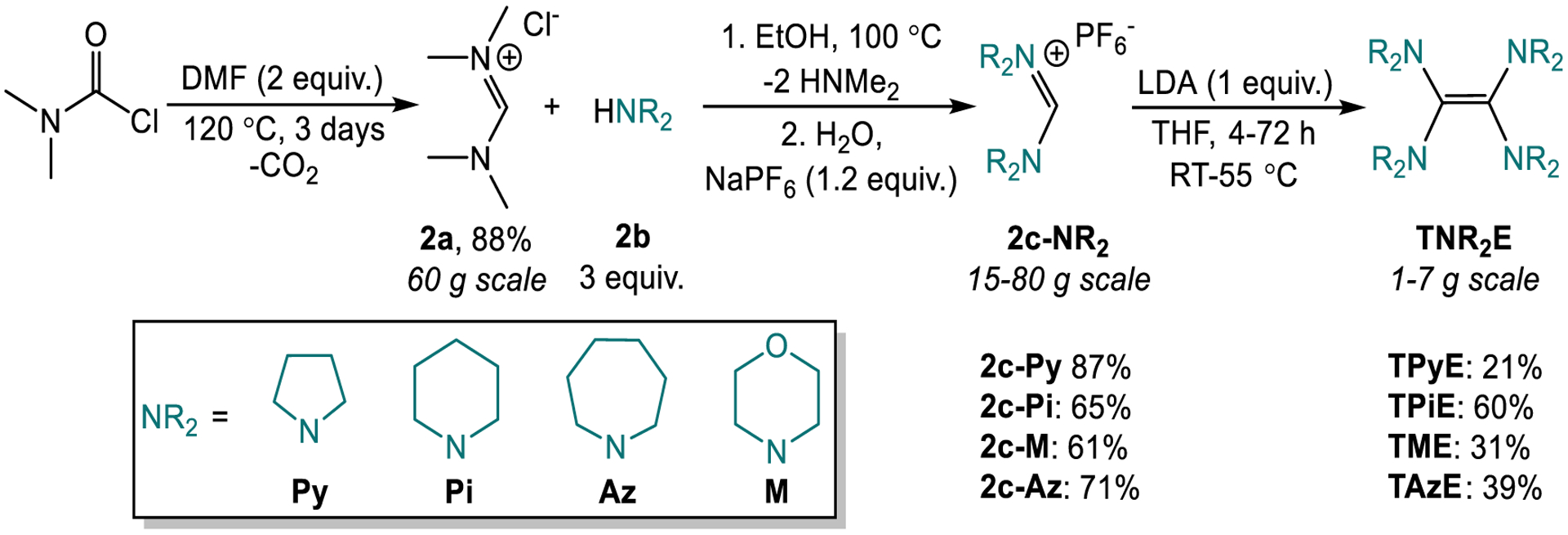
Synthesis of new homogeneous reductants. Resonance structures of 2a and 2c not depicted.
The reduction potentials of the homogeneous reductants were determined using cyclic voltammetry. In an analogous fashion to TDAE, all of the compounds display a single reversible two-electron redox event (see SI), but the reduction potentials vary by almost 500 mV. TME is the weakest reductant, with a potential of −0.85 V versus Fc in DMF. It is approximately 250 mV less reducing than TDAE (E° = −1.11 V) and provides access to a significantly weaker reductant than the traditional homogeneous or heterogeneous reductants that have been utilized in Ni-catalyzed XEC.2 TPiE and TAzE have similar reduction potentials (E° = −1.06 and −1.09 V, respectively) and are of comparable reducing power to TDAE. However, TPyE (E° = −1.32 V) is a significantly stronger reductant than TPiE, TAzE, or TDAE.
Each reductant was characterized by X-ray crystallography, which shows that all four structures contain a central C–C double bond (Figure 3). Assuming differences of greater than two standard deviations are significant, the C–C double bond in TME (1.3616(16) Å) is longer than the C–C bonds in either TPyE or TAzE (1.356(2) and 1.356(6) Å, respectively), consistent with the electron withdrawing nature of the morpholine groups. However, the error associated with the C–C bond length in TPiE (1.35(1) Å) prevents a definitive comparison about the relative C–C bond lengths in TME and TPiE. The reductants all contain a slightly non-planar core consisting of the two carbon atoms in the C–C double bond and the four nitrogen atoms incorporated in the heterocycles, but the degree of planarity in the core varies slightly. For example, in TPyE, the angle formed by the planes defined by N(1)-C(1)-C(2)-N(2) and N(3)-C(2)-C(1)-N(4) is 22.4° (Figure 3 & vide infra). In contrast, for TME, TPiE, and TAzE these angles are 29.8, 27.2, and 29.0°, respectively. This difference is likely due to steric factors because, as the size of the heterocyclic rings increases, it is more difficult for the carbon atoms in the C–C bond and the four nitrogen atoms to be in the same plane. DFT calculations indicate that a planar geometry is electronically preferred for systems with less steric bulk (vide infra and see SI).
Figure 3:
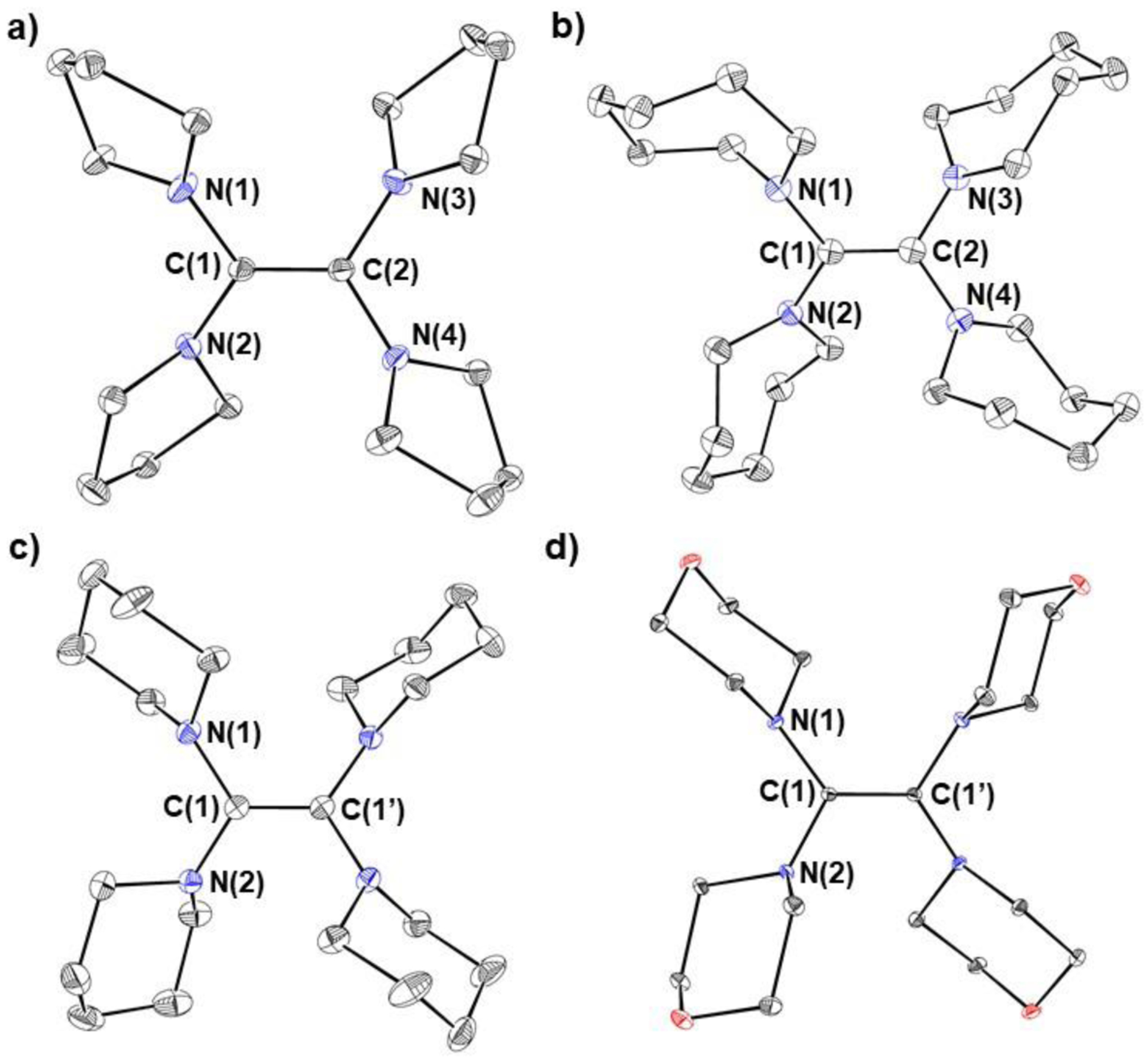
Solid-state structures with thermal ellipsoids at 30% of a) TPyE, b) TAzE, c) TPiE, and d) TME. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): TPyE: C(1)-C(2) 1.356(2), N(1)-C(1) 1.4027(15), N(2)-C(1) 1.4055(15), N(3)-C(2) 1.4044(15), N(4)-C(2) 1.3948(5), N(1)-C(1)-N(2) 115.69(10), N(3)-C(2)-N(4) 113.24(10), N(1)-C(1)-C(2) 122.00(11), N(4)-C(2)-C(1) 122.31(11). TAzE: C(1)-C(2) 1.356(6), N(1)-C(1) 1.407(5), N(2)-C(1) 1.405(5), N(3)-C(2) 1.409(5), N(4)-C(2) 1.414(5), N(1)-C(1)-N(2) 114.2(3), N(3)-C(2)-N(4) 113.2(3), N(1)-C(1)-C(2) 122.4(4), N(4)-C(2)-C(1) 123.5(4). TPiE (an inversion center is present in the molecule): C(1)-C(1’) 1.35(1), N(1)-C(1) 1.4177(11), N(2)-C(1) 1.4161(12), N(1)-C(1)-N(2) 115.79(7), N(1)-C(1)-C(1’) 122.36(10). TME (an inversion center is present in the molecule): C(1)-C(1’) 1.3616(16), N(1)-C(1) 1.4169(11), N(2)-C(1) 1.4182(10), N(1)-C(1)-N(2) 116.02(7), N(1)-C(1)-C(1’) 119.11(9).
To confirm the ability of the reductants to participate in chemical reduction and to develop understanding about the 260 mV difference in redox potential between TPyE and TPiE, which only differ by one atom in N-heterocycle ring size, both TPyE and TPiE were chemically oxidized. Treatment of TPyE and TPiE with iodine generates the two-electron oxidized salts [TPyE]2+2[I]− and [TPiE]2+2[I]−, respectively, in high yield (Eq 1). The solid-state structures of [TPyE]2+2[I]− and [TPiE]2+2[I]− were determined by X-ray crystallography (Figure 4). The central C–C bonds in [TPyE]2+2[I]− and [TPiE]2+2[I]− are elongated relative to the corresponding C–C bonds in TPyE and TPiE and are consistent with a C–C single bond. There is also a contraction in the C–N bond lengths between C(1) and C(2) and the nitrogen atoms of the heterocycles in [TPyE]2+2[I]− and [TPiE]2+2[I]− compared to TPyE and TPiE, respectively, which suggests a change in the C–N bond order upon oxidation from 1 to 1.5. These significant changes in bond lengths are consistent with the removal of electrons from the HOMOs of TPyE and TPiE, respectively, as DFT indicates that the HOMOs are localized on the π-bonding orbital of the C–C double bond with an anti-bonding interaction with the lone pairs of the nitrogen atoms associated with the heterocyclic rings (Figure 5).27
Figure 4:
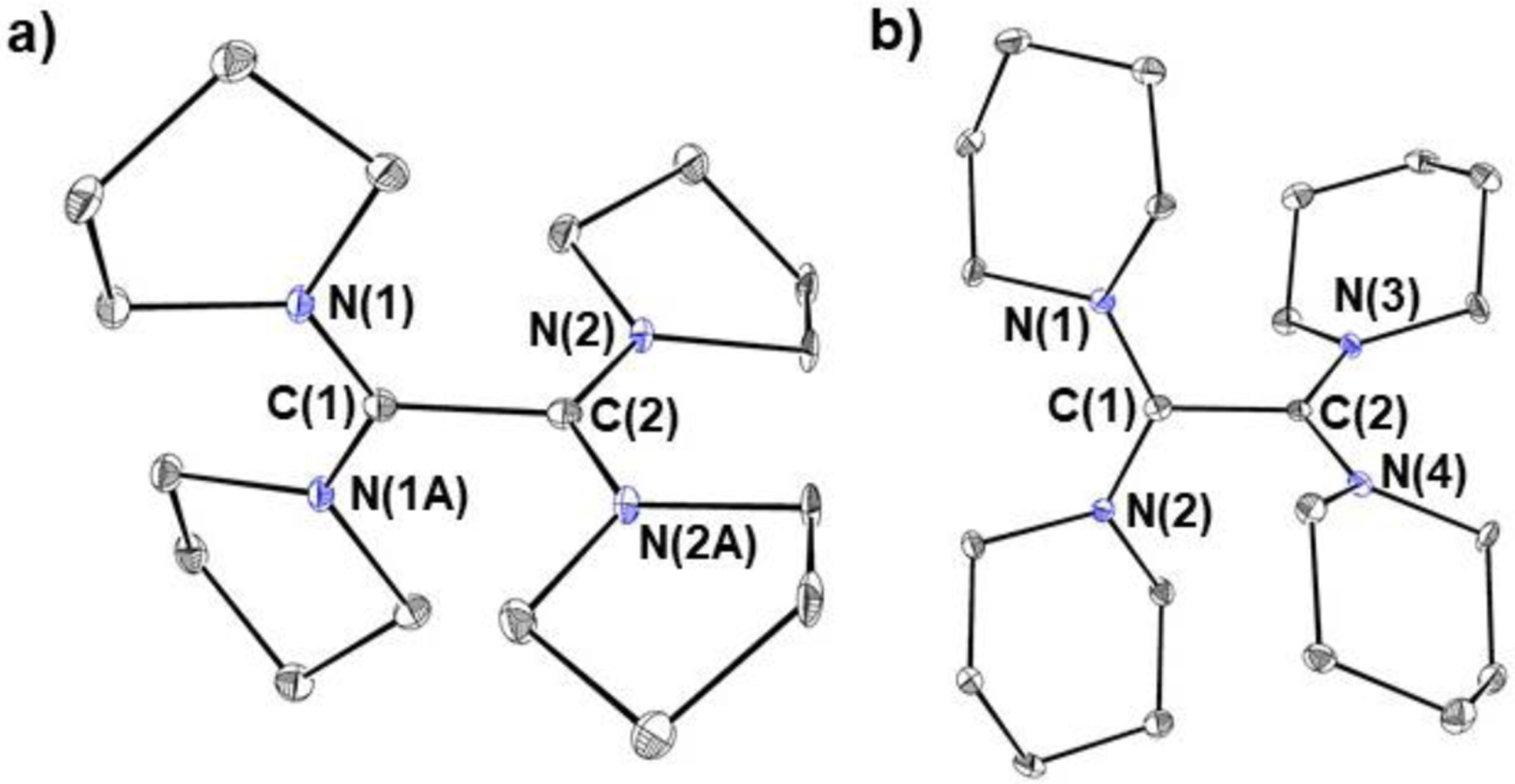
Solid-state structures with thermal ellipsoids at 30% of a) [TPyE]2+2[I]− and b) [TPiE]2+2[I]−. Hydrogen atoms and iodide anions omitted for clarity. Selected bond lengths (Å) and angles (°): [TPyE]2+2[I]− (a mirror plane is present in the molecule): C(1)-C(2) 1.524(9), N(1)-C(1) 1.4027(15), N(2)-C(2) 1.311(5), N(1)-C(1)-N(1A) 128.3(7), N(2)-C(2)-N(2A) 127.9(7), N(1)-C(1)-C(2) 115.9(3), N(2)-C(2)-C(1) 116.1(3). [TPiE]2+2[I]−: C(1)-C(2) 1.530(4), N(1)-C(1) 1.320(4), N(2)-C(1) 1.325(4), N(3)-C(2) 1.325(4), N(4)-C(2) 1.322(4), N(1)-C(1)-N(2) 125.2(3), N(3)-C(2)-N(4) 126.2(3), N(1)-C(1)-C(2) 117.4(3), N(4)-C(2)-C(1) 116.2(3).
Figure 5:
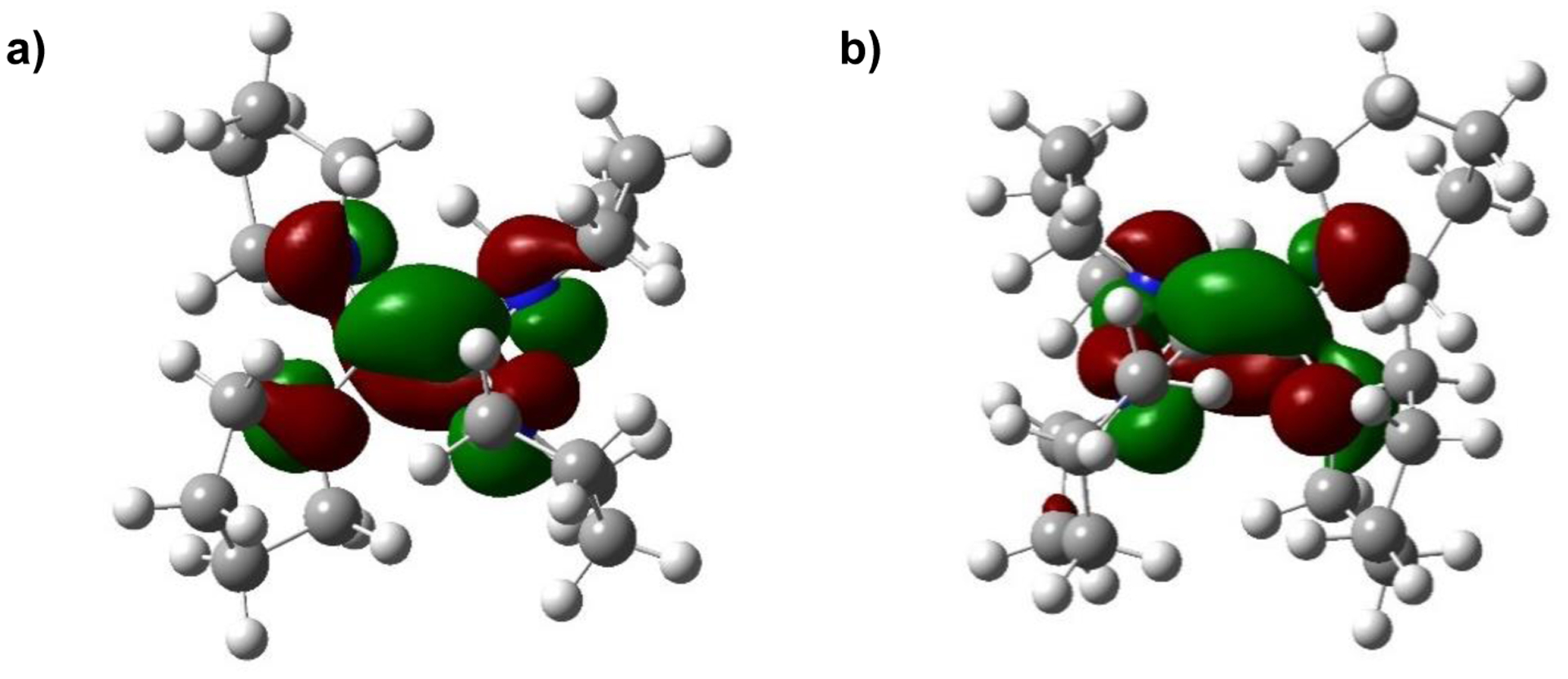
DFT calculated HOMOs of a) TPyE and b) TPiE.
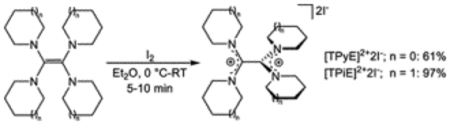 |
(Eq 1) |
The central cores of [TPyE]2+2[I]− and [TPiE]2+2[I]− are also drastically less planar than those of TPyE and TPiE. Specifically, the angles formed by the planes defined by N(1)-C(1)-C(2)-N(2) and N(3)-C(2)-C(1)-N(4) are 77.3 and 67.4° in [TPyE]2+2[I]− and [TPiE]2+2[I]−, respectively. DFT studies on a group of reductants related to TPyE and TPiE (see SI) indicate that there is a correlation between the relative difference in magnitude of this angle between the neutral and oxidized forms of the reductants and the reductant strength. TPyE and TPiE illustrate this concept, as in TPyE the angle between the planes changes by 54.9° upon oxidation (Figure 6), whereas in TPiE, which is a weaker reductant, the angle between the planes changes by 40.2° upon oxidation. This correlation is likely related to the fact that if the oxidized product is less planar, the LUMO28 is stabilized, which results in a stronger reductant. In TPiE (and TAzE), the increased steric bulk of the N-heterocyclic rings imposes constraints on the ability of the oxidized form to be as non-planar as TPyE. In contrast, although the differences in reduction potential between TPyE and TPiE are primarily determined by steric factors, the difference between TPiE and TME is likely related to electronic factors. Specifically, we propose that TME is a weaker reductant because the electron-withdrawing morpholine substituents stabilize the HOMO of TME and destabilize the LUMO of TME2+ relative to TPiE, which contains electron-neutral piperidine rings.
Figure 6:
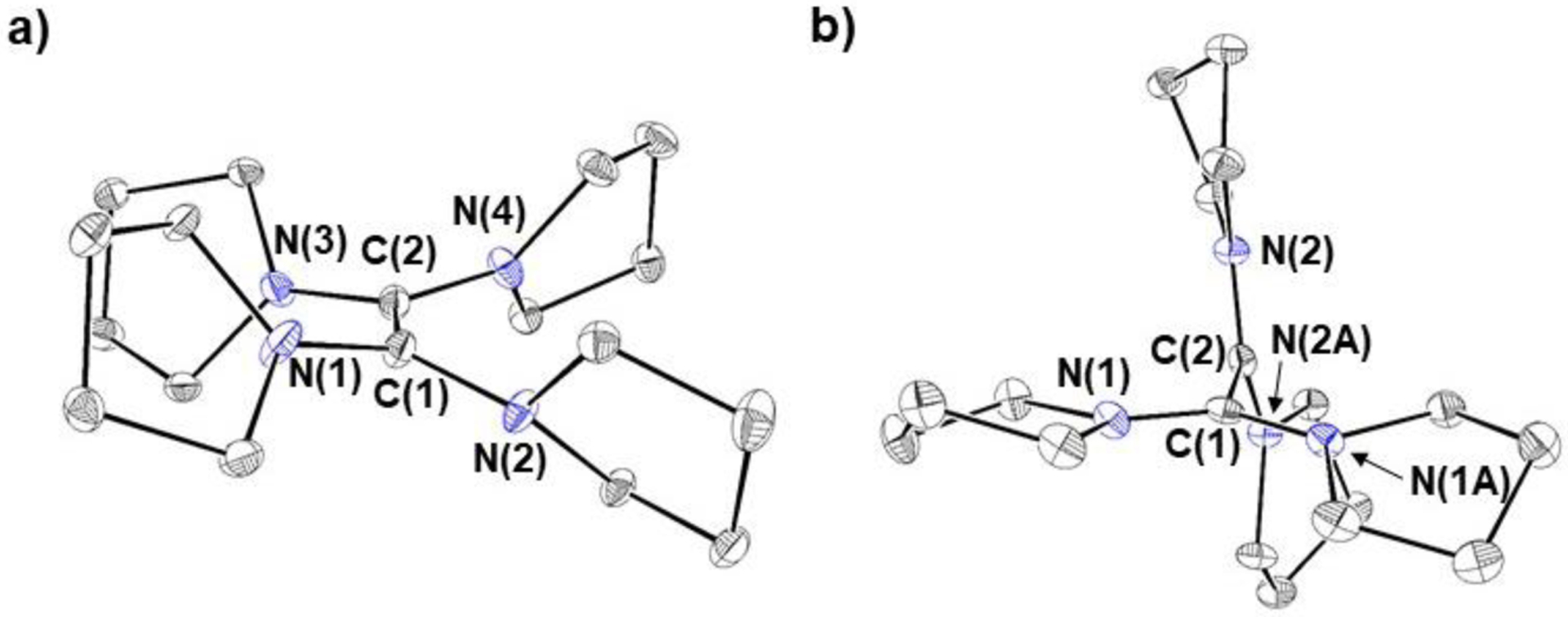
Solid-state structures of a) TPyE and b) [TPyE]2+2[I]− viewed down the C(1)-C(2) bond. This shows that in TPyE the nitrogen atoms in the heterocyclic rings are approximately eclipsed, whereas in [TPyE]2+2[I]− they are approximately staggered. This is reflected in the angles formed by the planes defined by N(1)-C(1)-C(2)-N(2) and N(3)-C(2)-C(1)-N(4). Hydrogen atoms and iodide anions omitted for clarity.
A remarkable feature of the reductants prepared in this work is their air stability (Table 1). Whereas a sample of TDAE fully decomposes in 10 minutes when exposed to air, we observed no decomposition of TME when it was stored on the benchtop for months, and TPiE only slowly decomposed over a period of two weeks (see SI). Given the similar reduction potentials of TPiE and TDAE (50 mV difference), this is likely related to kinetic factors and is presumably an additional advantage of working with solid compounds. In the case of TAzE, no decomposition was observed after 10 minutes (suggesting that it can be weighed in air), but around 23% decomposition was observed after 24 hours.29 Finally, even though TPyE is 200 mV more reducing than TDAE, it is still more stable to air. After 10 minutes of exposure, only 17% decomposition of TPyE was observed compared to the total decomposition of TDAE. This indicates that although TPyE is best handled under an inert atmosphere, exposure to small quantities of air will not result in complete loss of activity. Overall, we have synthesized a series of reductants that are easier to prepare and handle than TDAE and span nearly 500mV in reduction potential. We have also provided insights into the modulation of their redox potentials by controlling both steric and electronic factors, which may be beneficial for future reductant design. In the next sections, we explore the reactivity of these reductants in Ni-catalyzed XEC and related transformations.
Table 1.
Percent decomposition of tetraaminoethylenes under air at room temperature over time.
| Time | TME E° = −0.85 Va |
TPiE E° = −1.06 Va |
TAzE E° = −1.09 Va |
TDAE E° = −1.11 Va |
TPyE E° = −1.32 Va |
|---|---|---|---|---|---|
| 10 minutes | <1% | <1% | <1% | 100% | 17% |
| 24 hours | <1% | <1% | 23% | -- | 100% |
| 5 days | <1% | <1% | 70% | -- | -- |
| 2 weeks | <1% | 10% | -- | -- | -- |
| 4 weeks | <1% | 68% | -- | -- | -- |
| 6 weeks | <1% | -- | -- | -- | -- |
Reduction potential versus Fc.
Application of reductants in existing C(sp2)–C(sp3) XEC reactions
We recently reported a Ni/Co dual catalytic method for C(sp2)–C(sp3) XEC using TDAE.19g This method has advantages over conventional systems for C(sp2)–C(sp3) XEC reactions that use heterogeneous reductants3b,30 because it is compatible with a range of complex medicinal substrates and can be rationally optimized by changing the relative catalyst loading of Ni and Co based on the selectivity of the reaction. The use of TDAE, however, decreases the practicality of the method. To evaluate the compatibility of the new family of reductants in XEC reactions, we performed a Ni/Co co-catalyzed reaction using the simple substrates methyl 4-bromobenzoate and 1-bromo-3-phenylpropane with all of the new reductants (Table 2).31 To our surprise, despite the 0.5 V range in E°, all of the reductants give high yields of product, under analogous reaction conditions to those optimized for TDAE. This shows that the reaction is compatible with a relatively wide range of reduction potentials. In general, the tolerance of XEC reactions to the reduction potential of the reductant is challenging to evaluate because heterogeneous reductants are not tunable; however, our family of homogenous reductants facilitates this type of experiment. Further, from a synthetic perspective, the ability to tune the reduction potential of the reductant without sacrificing yield may be particularly valuable for coupling substrates that contain functional groups that are easy to reduce.
Table 2:
XEC of methyl 4-bromobenzoate with 1-bromo-3-phenylpropane using different homogeneous reductants. Reaction conditions: methyl 4-bromobenzoate (0.0625 mmol), 1-bromo-3-phenylpropane (0.075 mmol), reductant (0.075 mmol), (dtbbpy)NiIIBr2 (2.5 mol%), CoII(Pc) (0.5 mol%) in DMAc (0.25 mL) at 80 °C for 24 h. Yields were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
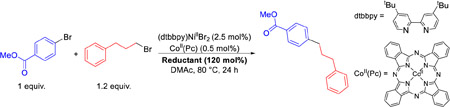
| ||
|---|---|---|
| Reductant | E° (V vs Fc) | Yield (%) |
| TME | −0.85 | 80 |
| TPiE | −1.06 | 97 |
| TAzE | −1.09 | 85 |
| TDAE | −1.11 | 92 |
| TPyE | −1.32 | 81 |
A feature of the Ni/Co dual catalytic method with TDAE is its compatibility with substrates relevant to medicinal chemistry and its amenability to optimization using high-throughput experimentation (HTE).19g We performed a series of C(sp2)–C(sp3) XEC reactions with complex aryl halides that were once intermediates in drug discovery programs from the MSD Aryl Halide Informer Library using our most practical reductants, TPiE and TME (Figure 7).32 High-throughput experimentation (HTE) was used to identify the optimal Ni and Co loadings for each substrate (see SI). Despite the presence of a diverse range of functional groups, we observed moderate to high yields (58%–93% by 1H NMR spectroscopy) for the coupling of 7a-7e with 1-bromo-3-phenylpropane using both TPiE and TME (Figure 7). Lower, but still medicinally useful, yields were observed in the coupling of aryl halides 7f-7h, which are difficult substrates for XEC due to the presence of alcohol, thiophene, and amine functional groups, respectively. We also performed the reactions described in Figure 7 using TDAE as the reductant (see SI). Comparable yields are observed between TPiE, TME, and TDAE, but due to their higher stability there are practical advantages to using TPiE and TME. Additionally, these results suggest that TPiE and TME can in some cases be used as direct replacements for TDAE, without the need for any modification of the reaction conditions. This is also demonstrated by the ability of TPiE and TPyE to facilitate the intermolecular three-component dicarbofunctionalization of alkenes,19c,19e,19f which is another notable example of a Ni-catalyzed reductive transformation that uses TDAE as the electron source (see SI).
Figure 7.
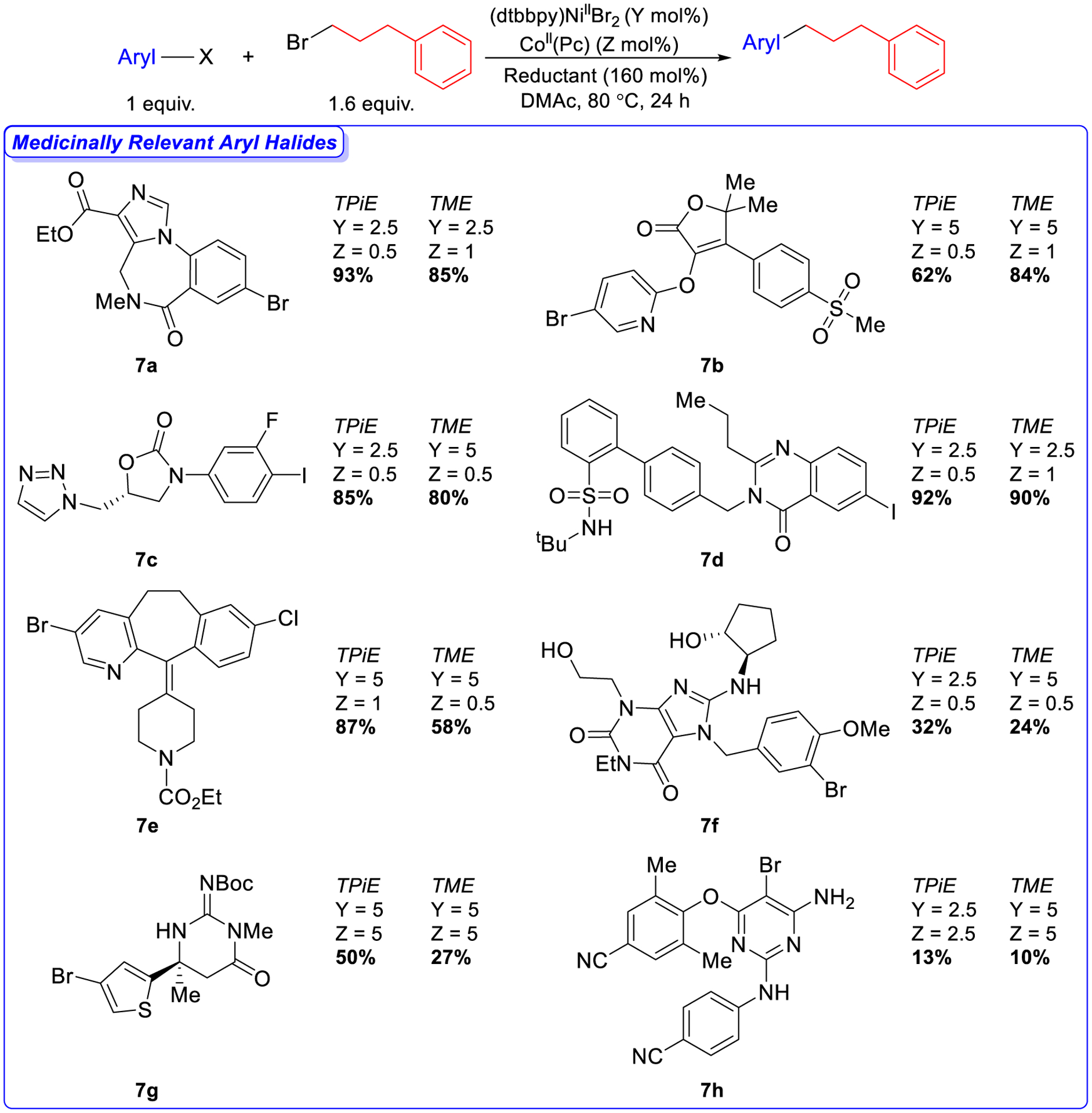
Dual-catalyzed XEC of medicinally relevant aryl halides with 1-bromo-3-phenylpropane using TPiE and TME as the reductants. Reaction conditions: aryl halides (0.0300 mmol), 1-bromo-3phenylpropane (0.0480 mmol), reductant (0.0480 mmol), (dtbbpy)NiIIBr2 (Y mol%), CoII(Pc) (Z mol%) in DMAc (0.3 mL) at 80 °C for 24 h. The yields were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
A new strategy for controlled XEC reactions based on reductant tunability
Alkyl Katritzky salts, which can be generated from reactions between primary amines and pyrylium cations,33 are desirable alkyl electrophiles in XEC and related reactions because of the ubiquity of primary amines as building blocks in synthetic chemistry. In XEC, they are proposed to produce alkyl radicals upon single electron reduction with Mn0 (or Zn0).34 However, it is often difficult to control the rate of alkyl radical generation in these reactions because of our inability to control the rates of electron transfer between a heterogeneous reductant and a substrate in solution (vide supra).5,10 This is problematic because in C(sp2)–C(sp3) XEC reactions with Katritzky salts, it is proposed that a highly reactive alkyl radical is trapped by an intermediate of the type LnNiII(Ar)X (X = halide) that is unstable under the reaction conditions, which can complicate reaction development and optimization (Figure 8a).2d,19g,35 For example, if the radical is generated faster than the LnNiII(Ar)X intermediate, the radical will undergo undesired side reactions that deplete the alkyl electrophile, whereas if the radical is generated slower than the LnNiII(Ar)X intermediate, the NiII intermediate may decompose, deleteriously consuming aryl halide. Our inability to control the rate of electron transfer reactions in catalysis with Mn0 or Zn0 can prevent some substrates from being utilized in XEC. For instance, it is typically not possible to couple benzylic Katritzky salts with aryl halides because the benzyl radical is formed too quickly (vide infra) relative to the LnNiII(Ar)X intermediate.36 We hypothesized that we could use the new reductants we developed, with different redox potentials, to control the relative rate of radical generation from Katritzky salts in catalysis (Figure 8b). This would enable matching of the rate of alkyl radical generation with the rate of formation of the LnNiII(Ar)X intermediate (Figure 8), which should facilitate the development of novel reactions. From a fundamental perspective, modulation of the rate of alkyl radical production in catalysis by varying the reductant strength represents a new strategy for controlling XEC reactions.
Figure 8:
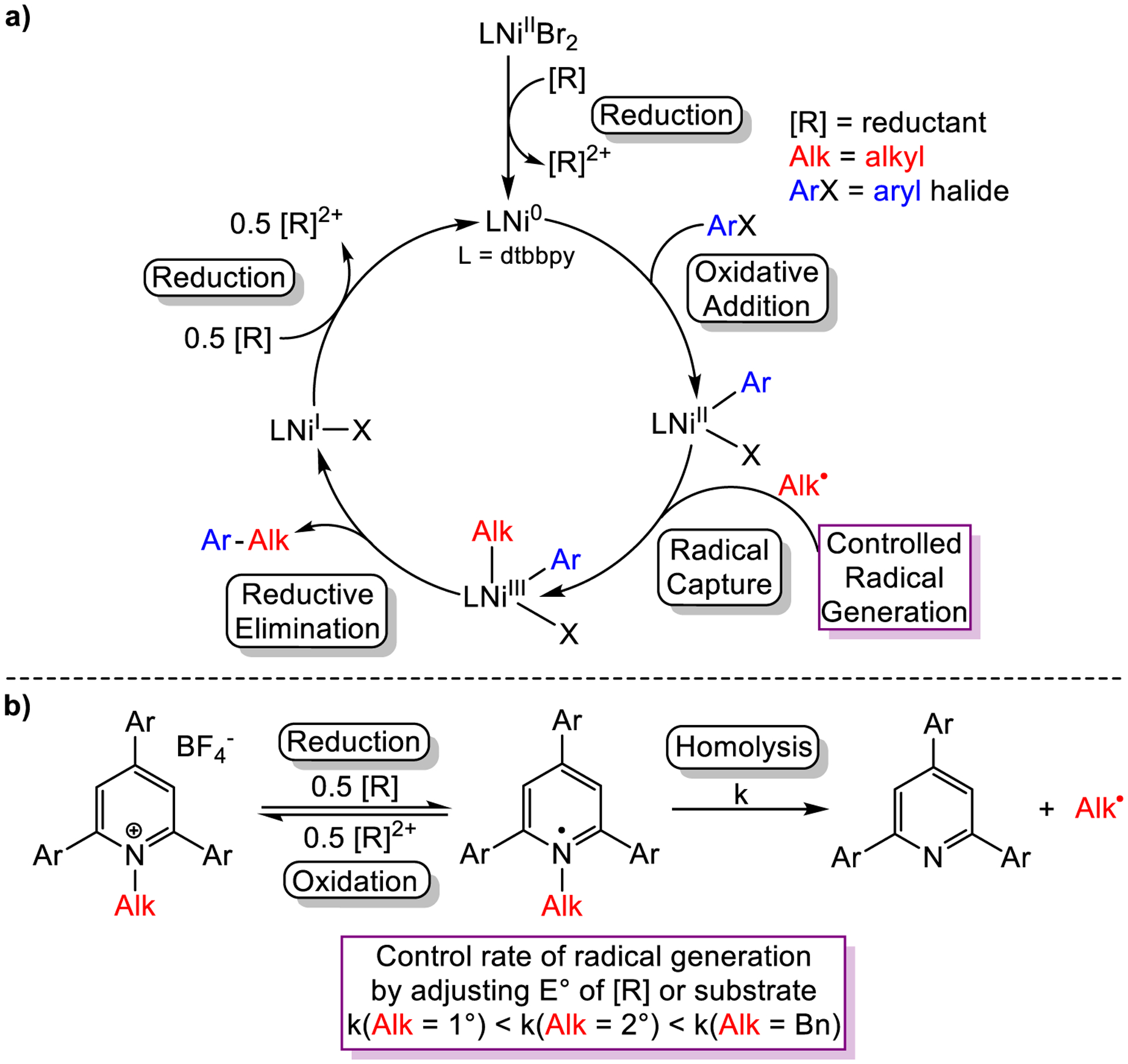
a) Proposed mechanism of Ni-catalyzed XEC using dtbbpy as the ligand and a homogeneous reductant. b) Proposed mechanism of radical generation from Katritzky salts under reductive conditions.
Initially, we performed a XEC reaction between a 1° alkyl Katritzky salt, 1, and 2-iodoanisole using (dtbbpy)NiIIBr2 (dtbbpy = 4,4’-ditertbutyl-2,2’-bipyridine) as a catalyst with all four of our reductants, as well as TDAE (Table 3). A high yield of product, 77%, is only obtained using TPyE, the strongest of our reductants. When weaker reductants are used, radical generation is likely too slow, and the 1° alkyl Katritzky salt is recovered after all the aryl iodide is consumed, resulting in a negligible yield of product and predominantly biaryl formation through homocoupling of the aryl iodide (see SI). In contrast, when the same reaction is performed with a 2° alkyl Katritzky salt, 2, under the same conditions, TPyE gives only a 34% yield of product. This is likely because it is easier to generate radicals from a Katritzky salt with a 2° alkyl substituent compared to a 1° alkyl substituent,37 so the strongest reductant TPyE generates 2° alkyl radicals too quickly, and the radicals decompose before they are trapped by LnNiII(Ar)X. Conversely, higher yields are obtained when TDAE (77%) and TPiE (83%) are used as the reductant because the rate of radical generation is slower and better matched to the rate of LnNiII(Ar)X formation. Nevertheless, the weakest reductant TME still gives a low yield (43%) with the 2° alkyl containing Katritzky salt, likely because the rate of radical generation is too slow, so the aryl iodide is consumed before the Katritzky salt leading to the formation of biaryl. To further explore this trend, we performed a XEC reaction between 2-iodoanisole and a benzylic Katritzky salt, 3, under the same conditions. Even though XEC coupling reactions involving benzylic Katritzky salts and aryl halides are unknown in the literature, we obtained near quantitative yields of product using TDAE, TAzE and TPiE as the reductant, respectively. Since benzylic Katritzky salts generate radicals faster than 2° Katritzky salts, it is not surprising that the weakest reductant TME gives an improved yield of 75% compared to the corresponding reactions with 1° or 2° Katritzky salts. However, in this case, biaryl is still produced as a by-product because LnNiII(Ar)X is consumed too fast relative to radical generation. In principle, the rate of LnNiII(Ar)X formation can be decreased by using a lower catalyst loading, and when the reaction with 3 is performed using 5 mol% catalyst loading with TME, the yield of coupled product increases to 93%. This demonstrates that catalyst loading and reductant strength can both be used to optimize reaction conditions by controlling the relative concentrations of LnNiII(Ar)X and the alkyl radical, respectively.
Table 3:
Yields of XEC reactions between 1°, 2°, and benzylic Katritzky salts and alkyl iodides with different homogeneous reductants. Reaction conditions: 2-iodoanisole (0.03125 mmol), Katritzky salt (0.0375 mmol), reductant (0.0375 mmol), (dtbbpy)NiIIBr2 (10 mol%), in DMAc (0.25 mL) at 80 °C for 24 h. The yields were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
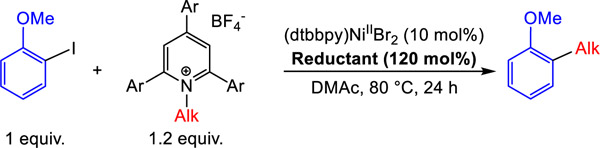
| |||||
|---|---|---|---|---|---|
| Reductant | E° (V vs Fc) | Yield (%) with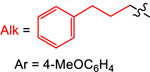
|
Yield (%) with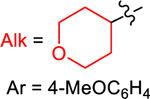
|
Yield (%) with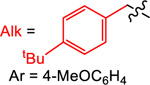
|
Yield (%) with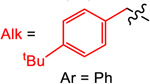
|
| 1 | 2 | 3 | 4 | ||
| TPyE | −1.32 | 77 | 34 | 57 | 25 |
| TDAE | −1.11 | 10 | 77 | 99 | 29 |
| TAzE | −1.09 | <1 | 66 | 93 | 29 |
| TPiE | −1.06 | <1 | 83 | 99 | 30 |
| TME | −0.85 | <1 | 43 | 75 (93a) | 90 |
5 mol% (dtbbpy)NiIIBr2 used.
We hypothesized that changing the ancillary aromatic substituents on the Katritzky salt would provide an alternative handle to control the rate of radical generation. Specifically, instead of using the benzylic Katritzky salt 3, which contains electron donating methoxy substituents, we used an unsubstituted Katritzky salt, 4. It is known that the reduction of the unsubstituted Katritzky salt 4 is easier than 3,33 and therefore it was expected that a weaker reductant would be needed to generate the radical at an appropriate rate to match the production of LnNiII(Ar)X. In agreement with this proposal, only TME gives a high yield (93%) of product in a XEC reaction between 2-iodoanisole and 4. Synthetically, the unsubstituted Katritzky salt, 4, is more accessible because the starting pyrylium cation is commercially available. Overall, the series of reactions described in Table 3 demonstrate that we can use the differences in the reducing power of reductants and the substrates themselves, as well as the catalyst loading, to control the outcome of XEC reactions. We anticipate that this strategy may facilitate the discovery of new XEC reactions, given the growing number of substrates,3i,19d,38 such as N-hydroxyphthalimide esters, that can generate carbon-centered radicals upon single electron reduction. Specifically, using this approach, we were able to perform the first XEC reactions between aryl halides and Katritzky salts using homogeneous reductants, which have practical advantages in comparison to other reduction methods (vide supra). Further, the XEC reaction between a benzylic Katritzky salt and an aryl halide is a rare example of a reaction of this type with either a homogeneous or heterogeneous reductant, and therefore we selected this transformation for further experiments.
The coupling of benzylic Katritzky salts with aryl halides represents a novel method to prepare diarylmethanes, which are present in antibacterial, anti-HIV, and antitumor agents.39 Further, benzylic Katritzky salts can be readily prepared from benzyl amines, which are generally stable, inexpensive, and widely available in comparison to more commonly utilized electrophiles, such as benzyl halides. Therefore, we optimized the XEC reaction between aryl iodides and benzylic Katritzky salts shown in Table 3, focusing on derivatives of the unsubstituted Katritzky salt 4 because they can be accessed in one step from commercially available materials (see SI). The substrate scope for this transformation is shown in Figure 9. The reaction is tolerant of aryl iodides with electron neutral (9a), electron-rich (9b), and electron-deficient (9c) substituents in the para-position, with yields of above 60% being observed in each case. A major advantage of systems for XEC that feature homogeneous reductants is their compatibility with sterically bulky aryl halides.19g Our system is tolerant of aryl iodides with a methoxy (9d) or iso-propyl (9e) substituent in the ortho position but is unable to couple di-ortho substituted aryl iodides, which is a common problem in XEC.30 The reaction is also tolerant of a range of different functional groups. For example, it can couple aryl iodides containing ester (9f), phenol (9g), bromo (9h), or BPin (9i) functional groups in relatively high yields. The compatibility of the reaction with acidic functional groups, such as phenol, is noteworthy as many transition metal catalyzed routes to prepare diarylmethanes are not tolerant of alcoholic functional groups.40 Additionally, the ability of the reaction to couple substrates with both bromo and BPin functional groups means that the products can be used as either the electrophile or the nucleophile in subsequent cross-coupling reactions. In particular, the selectivity of the reaction for the aryl iodide over the aryl bromide in 9h is crucial for facilitating orthogonal reactivity. In general, our method is not compatible with aryl bromides because the rate of oxidative addition of an aryl bromide to generate the LnNiII(Ar)X intermediate is too slow compared to the rate of radical generation from the benzylic Katritzky salt even when TME is used as the reductant (see SI). However, when 4-bromophenyl methyl sulfone, which contains a strongly electron withdrawing sulfone group, is used as the substrate, the rate of oxidative addition is sufficiently fast for the reaction to be compatible with an aryl bromide. Specifically, a reaction between 4-bromophenyl methyl sulfone and the unsubstituted Katritzky salt 4 provides the cross-electrophile coupled product in 51% yield by NMR spectroscopy, but it was not possible to separate the product from the unreacted aryl bromide (see SI). To slow down the rate of radical generation and reduce the amount of aryl bromide left at the end of the reaction, we coupled 4-bromophenyl methyl sulfone with the methoxy substituted Katritzky salt 3. This led to a 96% percent yield of the sulfone-containing product 9j by NMR spectroscopy and also demonstrates how we can use our knowledge of the reaction mechanism to improve the reaction yield beyond our optimized conditions. Heteroaryl halides are important substrates in medicinal chemistry,41 but it has been traditionally difficult to couple heteroaryl halides in XEC reactions.3g,3r We are able to couple several heteroaryl iodides (9k, 9l, 9m) in yields of 50–71% yield. Particularly notable are the successful coupling of 2-iodo-pyridine, as pyridines are problematic substrates in most XEC methods,3m,30 and 5-iodo-1H-indole, which contains both an acidic functionality and a heterocycle that can potentially coordinate to the Ni center. Finally, we were able to successfully reproduce a reaction between methyl 4-iodobenzoate and Katritzky salt 4 on 1 mmol scale to generate 250 mg of 9f demonstrating that the reaction is scalable.
Figure 9:
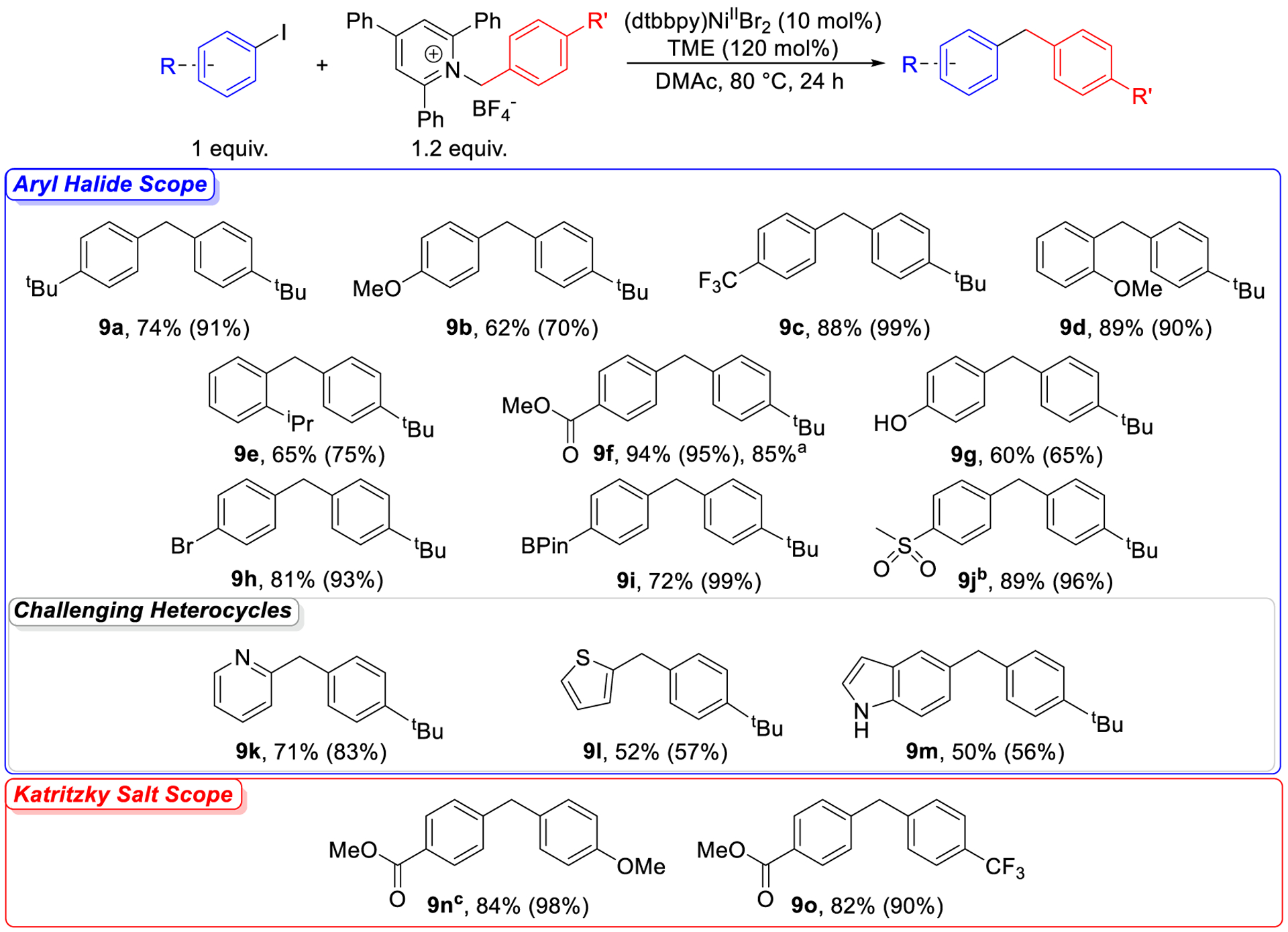
Substrate scope for XEC reactions between benzylic Katritzky salts and aryl iodides. Values outside of parentheses are isolated yields, and values inside of parentheses are NMR yields, which were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard. aIsolated yield from a reaction performed on a 1 mmol scale. bBenzylic Katritzky salt 3 coupled with 4-bromophenyl methyl sulfone. c20 mol% (dtbbpy)NiIIBr2 used.
Given the successful coupling of a range of aryl iodides, we explored the impact of changing the electronic properties of the benzylic Katritzky salt by introducing different substituents to the para-position of the benzyl amine. Specifically, a Katritzky salt bearing an electron-poor substituent (9o) is successfully coupled with methyl 4-iodobenzoate in high yield under our optimized conditions. However, although a Katritzky salt bearing an electron-rich substituent (9n) is also compatible with our system, the reaction requires higher catalyst loading presumably because the methoxy substituent stabilizes the radical through a resonance effect and leads to relatively faster alkyl radical generation. As a result, a higher concentration of LnNiII(Ar)X, which can be achieved by increasing the catalyst loading, is needed in this case. This again demonstrates how understanding of the reaction mechanism enables the optimization of reactions with challenging substrates.
On the whole, our substrate scope demonstrates that our method is both versatile and compatible with a number of functional groups that have traditionally been challenging for XEC. Unlike TDAE and common heterogeneous reductants such as Mn0 or Zn0, TME has a significantly lower reduction potential, which allows it to generate radicals from an easily accessible benzylic Katritzky salt at a rate that is compatible with the elementary steps at Ni. In fact, given the paucity of easy-to-use reductants of comparable strength to TME that are compatible with Ni-catalyzed XEC, the development of this reductant may allow other substrates that are rapidly reduced with conventional heterogeneous reductants to be used in XEC. More broadly, our results represent a proof-of-concept for the potential utility of our rational optimization strategy using tunable homogeneous reductants.
Conclusions
In this work, we have synthesized four new commercially available homogeneous reductants based on a tetraaminoethylene scaffold. In comparison to TDAE, which is currently the most utilized homogeneous reductant for Ni-catalyzed XEC, the new reductants are more practical because they are easier to synthesize, are solids at room temperature, and display increased air stability. In fact, the weakest reductant, TME, is indefinitely stable when stored in air for an extended period of time. The new reductants span 0.5 V in reduction potential and through structural characterization and DFT calculations, we have developed a model that explains the differences in reduction potentials and provides guidance for the design of future reductants with a greater range of potentials. All of our new reductants are able to facilitate Ni-catalyzed XEC reactions that are currently performed with TDAE, and we have demonstrated that TPiE and TME are compatible with complex, medicinally relevant substrates. Additionally, one of the main benefits of our new reductants is that their different reduction potentials allow us to rationally modulate the rate of electron transfer processes in XEC. We demonstrate this by tuning the rate of alkyl radical generation in C(sp2)–C(sp3) XEC with Katritzky salts to match the rate of elementary reactions that occur at Ni by changing the reductant. We have utilized this new strategy to develop a novel reaction between benzylic Katritzky salts and aryl iodides, which exhibits high functional group compatibility. Overall, we expect that our new reductants will provide more practical alternatives to current homogeneous electron sources such as TDAE in XEC and related reductive reactions due to their stability and also enable the development of new synthetic methodology due to their tunability. This is the subject of ongoing research in our laboratory.
Supplementary Material
Acknowledgements
NH acknowledges support from the NIHGMS under Award Number R01GM120162. All computational work was supported by the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center. We thank Dr. Fabian Menges for help with mass spectrometry and Dr. Xiaofan Jia for valuable advice on substrate purification.
Footnotes
Supporting Information
Experimental procedures, additional experimental results, NMR spectra, cyclic voltammetry data, X-ray crystallographic information, and computational data.
X-ray data for TME, TPiE, TAzE, TPyE, [TPyE]2+2[I]− and [TPiE]2+2[I]−. (CIF)
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.For selected examples of recently developed transition metal mediated processes that use Zn or Mn as reductants, see:; (a) Gui J; Pan C-M; Jin Y; Qin T; Lo JC; Lee BJ; Spergel SH; Mertzman ME; Pitts WJ; La Cruz TE; Schmidt MA; Darvatkar N; Natarajan SR; Baran PS Practical Olefin Hydroamination with Nitroarenes. Science 2015, 348, 886–891; [DOI] [PubMed] [Google Scholar]; (b) Pal S; Zhou Y-Y; Uyeda C Catalytic Reductive Vinylidene Transfer Reactions. J. Am. Chem. Soc 2017, 139, 11686–11689; [DOI] [PubMed] [Google Scholar]; (c) Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJE; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chem. Int. Ed 2017, 56, 260–265; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fang Y; Rogge T; Ackermann L; Wang S-Y; Ji S-J Nickel-Catalyzed Reductive Thiolation and Selenylation of Unactivated Alkyl Bromides. Nat. Commun 2018, 9, 2240; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Green SA; Vásquez-Céspedes S; Shenvi RA Iron–Nickel Dual-Catalysis: A New Engine for Olefin Functionalization and the Formation of Quaternary Centers. J. Am. Chem. Soc 2018, 140, 11317–11324; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ni S; Garrido-Castro AF; Merchant RR; de Gruyter JN; Schmitt DC; Mousseau JJ; Gallego GM; Yang S; Collins MR; Qiao JX; Yeung K-S; Langley DR; Poss MA; Scola PM; Qin T; Baran PS A General Amino Acid Synthesis Enabled by Innate Radical Cross-Coupling. Angew. Chem. Int. Ed 2018, 57, 14560–14565; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wu X; Hao W; Ye K-Y; Jiang B; Pombar G; Song Z; Lin S Ti-Catalyzed Radical Alkylation of Secondary and Tertiary Alkyl Chlorides Using Michael Acceptors. J. Am. Chem. Soc 2018, 140, 14836–14843; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhou Y-Y; Uyeda C Catalytic Reductive [4 + 1]-Cycloadditions of Vinylidenes and Dienes. Science 2019, 363, 857–862; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ni S; Padial NM; Kingston C; Vantourout JC; Schmitt DC; Edwards JT; Kruszyk MM; Merchant RR; Mykhailiuk PK; Sanchez BB; Yang S; Perry MA; Gallego GM; Mousseau JJ; Collins MR; Cherney RJ; Lebed PS; Chen JS; Qin T; Baran PS A Radical Approach to Anionic Chemistry: Synthesis of Ketones, Alcohols, and Amines. J. Am. Chem. Soc 2019, 141, 6726–6739; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Lu X; Wang X-X; Gong T-J; Pi J-J; He S-J; Fu Y Nickel-Catalyzed Allylic Defluorinative Alkylation of Trifluoromethyl Alkenes with Reductive Decarboxylation of Redox-Active Esters. Chem. Sci 2019, 10, 809–814; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Pang H; Wang Y; Gallou F; Lipshutz BH Fe-Catalyzed Reductive Couplings of Terminal (Hetero)Aryl Alkenes and Alkyl Halides under Aqueous Micellar Conditions. J. Am. Chem. Soc 2019, 141, 17117–17124; [DOI] [PubMed] [Google Scholar]; (l) Anthony D; Lin Q; Baudet J; Diao T Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes. Angew. Chem. Int. Ed 2019, 58, 3198–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moragas T; Correa A; Martin R Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds. Chem. Eur. J 2014, 20, 8242–8258; [DOI] [PubMed] [Google Scholar]; (c) Knappke CEI; Grupe S; Gärtner D; Corpet M; Gosmini C; Jacobi von Wangelin A Reductive Cross-Coupling Reactions Between Two Electrophiles. Chem. Eur. J 2014, 20, 6828–6842; [DOI] [PubMed] [Google Scholar]; (d) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gu J; Wang X; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Alkyl Halides with Other Electrophiles: Concept and Mechanistic Considerations. Org. Chem. Front 2015, 2, 1411–1421; [Google Scholar]; (f) Richmond E; Moran J Recent Advances in Nickel Catalysis Enabled by Stoichiometric Metallic Reducing Agents. Synthesis 2018, 50, 499–513; [Google Scholar]; (g) Tortajada A; Juliá-Hernández F; Börjesson M; Moragas T; Martin R Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem. Int. Ed 2018, 57, 15948–15982; [DOI] [PubMed] [Google Scholar]; (h) Poremba KE; Dibrell SE; Reisman SE Nickel-Catalyzed Enantioselective Reductive Cross-Coupling Reactions. ACS Catal. 2020, 10, 8237–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For selected leading examples, see:; (a) Everson DA; Shrestha R; Weix DJ Nickel-Catalyzed Reductive Cross-Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2010, 132, 920–921; [DOI] [PubMed] [Google Scholar]; (b) Everson DA; Jones BA; Weix DJ Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cherney AH; Reisman SE Nickel-Catalyzed Asymmetric Reductive Cross-Coupling Between Vinyl and Benzyl Electrophiles. J. Am. Chem. Soc 2014, 136, 14365–14368; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Molander GA; Wisniewski SR; Traister KM Reductive Cross-Coupling of 3-Bromo-2,1-borazaronaphthalenes with Alkyl Iodides. Org. Lett 2014, 16, 3692–3695; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhao C; Jia X; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Alkyl Acids with Unactivated Tertiary Alkyl and Glycosyl Halides. J. Am. Chem. Soc 2014, 136, 17645–17651; [DOI] [PubMed] [Google Scholar]; (f) Arendt KM; Doyle AG Dialkyl Ether Formation by Nickel-Catalyzed Cross-Coupling of Acetals and Aryl Iodides. Angew. Chem. Int. Ed 2015, 54, 9876–9880; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kadunce NT; Reisman SE Nickel-Catalyzed Asymmetric Reductive Cross-Coupling between Heteroaryl Iodides and α-Chloronitriles. J. Am. Chem. Soc 2015, 137, 10480–10483; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang X; Wang S; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Aryl Bromides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2015, 137, 11562–11565; [DOI] [PubMed] [Google Scholar]; (i) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016–5019; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Konev MO; Hanna LE; Jarvo ER Intra- and Intermolecular Nickel-Catalyzed Reductive Cross-Electrophile Coupling Reactions of Benzylic Esters with Aryl Halides. Angew. Chem. Int. Ed 2016, 55, 6730–6733; [DOI] [PubMed] [Google Scholar]; (k) Liu J; Ren Q; Zhang X; Gong H Preparation of Vinyl Arenes by Nickel-Catalyzed Reductive Coupling of Aryl Halides with Vinyl Bromides. Angew. Chem. Int. Ed 2016, 55, 15544–15548; [DOI] [PubMed] [Google Scholar]; (l) Woods BP; Orlandi M; Huang C-Y; Sigman MS; Doyle AG Nickel-Catalyzed Enantioselective Reductive Cross-Coupling of Styrenyl Aziridines. J. Am. Chem. Soc 2017, 139, 5688–5691; [DOI] [PubMed] [Google Scholar]; (m) Hansen EC; Li C; Yang S; Pedro D; Weix DJ Coupling of Challenging Heteroaryl Halides with Alkyl Halides via Nickel-Catalyzed Cross-Electrophile Coupling. J. Org. Chem 2017, 82, 7085–7092; [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Peng L; Li Y; Li Y; Wang W; Pang H; Yin G Ligand-Controlled Nickel-Catalyzed Reductive Relay Cross-Coupling of Alkyl Bromides and Aryl Bromides. ACS Catal. 2018, 8, 310–313; [Google Scholar]; (o) Xu C; Guo W-H; He X; Guo Y-L; Zhang X-Y; Zhang X Difluoromethylation of (hetero)aryl Chlorides with Chlorodifluoromethane Catalyzed by Nickel. Nat. Commun 2018, 9, 1170; [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Yue H; Zhu C; Shen L; Geng Q; Hock KJ; Yuan T; Cavallo L; Rueping M Nickel-Catalyzed C–N Bond Activation: Activated Primary Amines as Alkylating Reagents in Reductive Cross-Coupling. Chem. Sci 2019, 10, 4430–4435; [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Komeyama K; Michiyuki T; Osaka I Nickel/Cobalt-Catalyzed C(sp3)–C(sp3) Cross-Coupling of Alkyl Halides with Alkyl Tosylates. ACS Catal. 2019, 9, 9285–9291; [Google Scholar]; (r) Yi J; Badir SO; Kammer LM; Ribagorda M; Molander GA Deaminative Reductive Arylation Enabled by Nickel/Photoredox Dual Catalysis. Org. Lett 2019, 21, 3346–3351; [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Kim S; Goldfogel MJ; Gilbert MM; Weix DJ Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Chlorides with Primary Alkyl Chlorides. J. Am. Chem. Soc 2020, 142, 9902–9907; [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Wang J; Hoerrner ME; Watson MP; Weix DJ Nickel-Catalyzed Synthesis of Dialkyl Ketones from the Coupling of N-Alkyl Pyridinium Salts with Activated Carboxylic Acids. Angew. Chem. Int. Ed 2020, 59, 13484–13489; [DOI] [PMC free article] [PubMed] [Google Scholar]; (u) Sanford AB; Thane TA; McGinnis TM; Chen P-P; Hong X; Jarvo ER Nickel-Catalyzed Alkyl–Alkyl Cross-Electrophile Coupling Reaction of 1,3-Dimesylates for the Synthesis of Alkylcyclopropanes. J. Am. Chem. Soc 2020, 142, 5017–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koenig SG; Leahy DK; Wells AS Evaluating the Impact of a Decade of Funding from the Green Chemistry Institute Pharmaceutical Roundtable. Org. Proc. Res. Dev 2018, 22, 1344–1359. [Google Scholar]
- 5.Nimmagadda SK; Korapati S; Dasgupta D; Malik NA; Vinodini A; Gangu AS; Kalidindi S; Maity P; Bondigela SS; Venu A; Gallagher WP; Aytar S; González-Bobes F; Vaidyanathan R Development and Execution of an Ni(II)-Catalyzed Reductive Cross-Coupling of Substituted 2-Chloropyridine and Ethyl 3-Chloropropanoate. Org. Proc. Res. Dev 2020, 24, 1141–1148. [Google Scholar]
- 6.For an extended discussion of the problems associated with heterogeneous reduction conditions in the Buchwald-Hartwig reaction, many of which are also relevant to CEC, see:; Beutner GL; Coombs JR; Green RA; Inankur B; Lin D; Qiu J; Roberts F; Simmons EM; Wisniewski SR Palladium-Catalyzed Amidation and Amination of (Hetero)aryl Chlorides under Homogeneous Conditions Enabled by a Soluble DBU/NaTFA Dual-Base System. Org. Proc. Res. Dev 2019, 23, 1529–1537. [Google Scholar]
- 7.(a) Hartman RL Managing Solids in Microreactors for the Upstream Continuous Processing of Fine Chemicals. Org. Proc. Res. Dev 2012, 16, 870–887; [Google Scholar]; (b) Plutschack MB; Pieber B; Gilmore K; Seeberger PH The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev 2017, 117, 11796–11893; [DOI] [PubMed] [Google Scholar]; (c) Jensen KF Flow Chemistry—Microreaction Technology Comes of Age. AlChE J. 2017, 63, 858–869. [Google Scholar]
- 8.(a) Godfrey AG; Masquelin T; Hemmerle H A Remote-Controlled Adaptive Medchem Lab: An Innovative Approach to Enable Drug Discovery in the 21st Century. Drug Discov. Today 2013, 18, 795–802; [DOI] [PubMed] [Google Scholar]; (b) Li J; Ballmer SG; Gillis EP; Fujii S; Schmidt MJ; Palazzolo AM; Lehmann JW; Morehouse GF; Burke MD Synthesis of Many Different Types of Organic Small Molecules Using One Automated Process. Science 2015, 347, 1221–1226; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Perera D; Tucker JW; Brahmbhatt S; Helal CJ; Chong A; Farrell W; Richardson P; Sach NW A Platform for Automated Nanomole-Scale Reaction Screening and Micromole-Scale Synthesis in Flow. Science 2018, 359, 429–434. [DOI] [PubMed] [Google Scholar]
- 9.(a) Santanilla AB; Regalado EL; Pereira T; Shevlin M; Bateman K; Campeau L-C; Schneeweis J; Berritt S; Shi Z-C; Nantermet P Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules. Science 2015, 347, 49–53; [DOI] [PubMed] [Google Scholar]; (b) Uehling MR; King RP; Krska SW; Cernak T; Buchwald SL Pharmaceutical Diversification via Palladium Oxidative Addition Complexes. Science 2019, 363, 405–408; [DOI] [PubMed] [Google Scholar]; (c) Aguirre AL; Loud NL; Johnson KA; Weix DJ; Wang Y ChemBead Enabled High-Throughput Cross-Electrophile Coupling Reveals a New Complementary Ligand. Chem. Eur. J 2021, 27, 12981–12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Q; Diao T Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2019, 141, 17937–17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beutner GL; Simmons EM; Ayers S; Bemis CY; Goldfogel MJ; Joe CL; Marshall J; Wisniewski SR A Process Chemistry Benchmark for sp2–sp3 Cross Couplings. J. Org. Chem 2021, 86, 10380–10396. [DOI] [PubMed] [Google Scholar]
- 12.For selected leading examples, see:; (a) Zhang P; Le CC; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Duan Z; Li W; Lei A Nickel-Catalyzed Reductive Cross-Coupling of Aryl Bromides with Alkyl Bromides: Et3N as the Terminal Reductant. Org. Lett 2016, 18, 4012–4015; [DOI] [PubMed] [Google Scholar]; (c) Paul A; Smith MD; Vannucci AK Photoredox-Assisted Reductive Cross-Coupling: Mechanistic Insight into Catalytic Aryl–Alkyl Cross-Couplings. J. Org. Chem 2017, 82, 1996–2003; [DOI] [PubMed] [Google Scholar]; (d) Lévêque C; Corcé V; Chenneberg L; Ollivier C; Fensterbank L Photoredox/Nickel Dual Catalysis for the C(sp3)-C(sp3) Cross-Coupling of Alkylsilicates with Alkyl Halides. Eur. J. Org. Chem 2017, 2017, 2118–2121; [Google Scholar]; (e) Smith RT; Zhang X; Rincón JA; Agejas J; Mateos C; Barberis M; García-Cerrada S; de Frutos O; MacMillan DWC Metallaphotoredox-Catalyzed Cross-Electrophile Csp3–Csp3 Coupling of Aliphatic Bromides. J. Am. Chem. Soc 2018, 140, 17433–17438; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Peng L; Li Z; Yin G Photochemical Nickel-Catalyzed Reductive Migratory Cross-Coupling of Alkyl Bromides with Aryl Bromides. Org. Lett 2018, 20, 1880–1883; [DOI] [PubMed] [Google Scholar]; (g) Zhang R; Li G; Wismer M; Vachal P; Colletti SL; Shi Z-C Profiling and Application of Photoredox C(sp3)–C(sp2) Cross-Coupling in Medicinal Chemistry. ACS Med. Chem. Lett 2018, 9, 773–777; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Milligan JA; Phelan JP; Badir SO; Molander GA Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem. Int. Ed 2019, 58, 6152–6163; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yu W; Chen L; Tao J; Wang T; Fu J Dual Nickel-and Photoredox-Catalyzed Reductive Cross-Coupling of Aryl Vinyl Halides and Unactivated Tertiary Alkyl Bromides. Chem. Commun 2019, 55, 5918–5921; [DOI] [PubMed] [Google Scholar]; (j) Brill ZG; Ritts CB; Mansoor UF; Sciammetta N Continuous Flow Enables Metallaphotoredox Catalysis in a Medicinal Chemistry Setting: Accelerated Optimization and Library Execution of a Reductive Coupling between Benzylic Chlorides and Aryl Bromides. Org. Lett 2019, 22, 410–416; [DOI] [PubMed] [Google Scholar]; (k) Sakai HA; Liu W; Le CC; MacMillan DWC Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 2020, 142, 11691–11697; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Parasram M; Shields BJ; Ahmad O; Knauber T; Doyle AG Regioselective Cross-Electrophile Coupling of Epoxides and (Hetero)aryl Iodides via Ni/Ti/Photoredox Catalysis. ACS Catal. 2020, 10, 5821–5827; [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Kariofillis SK; Shields BJ; Tekle-Smith MA; Zacuto MJ; Doyle AG Nickel/Photoredox-Catalyzed Methylation of (Hetero)aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc 2020, 142, 7683–7689; [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Kerackian T; Reina A; Bouyssi D; Monteiro N; Amgoune A Silyl Radical Mediated Cross-Electrophile Coupling of N-Acylimides with Alkyl Bromides under Photoredox/Nickel Dual Catalysis. Org. Lett 2020, 22, 2240–2245. [DOI] [PubMed] [Google Scholar]
- 13.For selected leading examples, see:; (a) Perkins RJ; Pedro DJ; Hansen EC Electrochemical Nickel Catalysis for sp2-sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides. Org. Lett 2017, 19, 3755–3758; [DOI] [PubMed] [Google Scholar]; (b) Li H; Breen CP; Seo H; Jamison TF; Fang Y-Q; Bio MM Ni-Catalyzed Electrochemical Decarboxylative C–C Couplings in Batch and Continuous Flow. Org. Lett 2018, 20, 1338–1341; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Perkins RJ; Hughes AJ; Weix DJ; Hansen EC Metal-Reductant-Free Electrochemical Nickel-Catalyzed Couplings of Aryl and Alkyl Bromides in Acetonitrile. Org. Proc. Res. Dev 2019, 23, 1746–1751; [Google Scholar]; (d) DeLano TJ; Reisman SE Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis. ACS Catal. 2019, 9, 6751–6754; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Koyanagi T; Herath A; Chong A; Ratnikov M; Valiere A; Chang J; Molteni V; Loren J One-Pot Electrochemical Nickel-Catalyzed Decarboxylative sp2–sp3 Cross-Coupling. Org. Lett 2019, 21, 816–820; [DOI] [PubMed] [Google Scholar]; (f) Kumar GS; Peshkov A; Brzozowska A; Nikolaienko P; Zhu C; Rueping M Nickel-Catalyzed Chain-Walking Cross-Electrophile Coupling of Alkyl and Aryl Halides and Olefin Hydroarylation Enabled by Electrochemical Reduction. Angew. Chem. Int. Ed 2020, 59, 6513–6519; [DOI] [PubMed] [Google Scholar]; (g) Truesdell BL; Hamby TB; Sevov CS General C(sp2)–C(sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc 2020, 142, 5884–5893; [DOI] [PubMed] [Google Scholar]; (h) Jiao K-J; Liu D; Ma H-X; Qiu H; Fang P; Mei T-S Nickel-Catalyzed Electrochemical Reductive Relay Cross-Coupling of Alkyl Halides to Aryl Halides. Angew. Chem. Int. Ed 2020, 59, 6520–6524; [DOI] [PubMed] [Google Scholar]; (i) Li Z; Sun W; Wang X; Li L; Zhang Y; Li C Electrochemically Enabled, Nickel-Catalyzed Dehydroxylative Cross-Coupling of Alcohols with Aryl Halides. J. Am. Chem. Soc 2021, 143, 3536–3543; [DOI] [PubMed] [Google Scholar]; (j) Kammer LM; Badir SO; Hu R-M; Molander GA Photoactive Electron Donor–Acceptor Complex Platform for Ni-Mediated C(sp3)–C(sp2) Bond Formation. Chem. Sci 2021, 12, 5450–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Maljuric S; Jud W; Kappe CO; Cantillo D Translating Batch Electrochemistry to Single-Pass Continuous Flow Conditions: An Organic Chemist’s Guide. J. Flow Chem 2020, 10, 181–190; [Google Scholar]; (b) Corcoran EB; McMullen JP; Lévesque F; Wismer MK; Naber JR Photon Equivalents as a Parameter for Scaling Photoredox Reactions in Flow: Translation of Photocatalytic C–N Cross-Coupling from Lab Scale to Multikilogram Scale. Angew. Chem. Int. Ed 2020, 59, 11964–11968. [DOI] [PubMed] [Google Scholar]
- 15.Gesmundo NJ; Sauvagnat B; Curran PJ; Richards MP; Andrews CL; Dandliker PJ; Cernak T Nanoscale Synthesis and Affinity Ranking. Nature 2018, 557, 228–232. [DOI] [PubMed] [Google Scholar]
- 16.In this work, we define a homogeneous electron donor as a molecule that transfers electrons to a catalyst or substrate in the same phase (for example liquid to liquid) and does not require activation by light.
- 17.(a) Jessop PG Searching for Green Solvents. Green Chem. 2011, 13, 1391–1398; [Google Scholar]; (b) Diorazio LJ; Hose DRJ; Adlington NK Toward a More Holistic Framework for Solvent Selection. Org. Proc. Res. Dev 2016, 20, 760–773; [Google Scholar]; (c) Prat D; Wells A; Hayler J; Sneddon H; McElroy CR; Abou-Shehada S; Dunn PJ CHEM21 Selection Guide of Classical- and Less Classical-Solvents. Green Chem. 2016, 18, 288–296; [Google Scholar]; (d) Yin J; Maguire CK; Yasuda N; Brunskill APJ; Klapars A Impact of Lead Impurities in Zinc Dust on the Selective Reduction of a Dibromoimidazole Derivative. Org. Proc. Res. Dev 2017, 21, 94–97. [Google Scholar]
- 18.Marcus RA Chemical and Electrochemical Electron-Transfer Theory. Ann. Rev. Phys. Chem 1964, 15, 155–196. [Google Scholar]
- 19.For examples that use TDAE as a homogeneous reductant in CEC, see:; (a) Kuroboshi M; Tanaka M; Kishimoto S; Goto K; Mochizuki M; Tanaka H Tetrakis(dimethylamino)ethylene (TDAE) as a Potent Organic Electron Source: Alkenylation of Aldehydes using an Ni/Cr/TDAE Redox System. Tetrahedron Lett. 2000, 41, 81–84; [Google Scholar]; (b) Anka-Lufford LL; Huihui KMM; Gower NJ; Ackerman LKG; Weix DJ Nickel-Catalyzed Cross-Electrophile Coupling with Organic Reductants in Non-Amide Solvents. Chem. Eur. J 2016, 22, 11564–11567; [DOI] [PubMed] [Google Scholar]; (c) García-Domínguez A; Li Z; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2017, 139, 6835–6838; [DOI] [PubMed] [Google Scholar]; (d) Suzuki N; Hofstra JL; Poremba KE; Reisman SE Nickel-Catalyzed Enantioselective Cross-Coupling of N-Hydroxyphthalimide Esters with Vinyl Bromides. Org. Lett 2017, 19, 2150–2153; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shu W; García-Domínguez A; Quirós MT; Mondal R; Cárdenas DJ; Nevado C Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights. J. Am. Chem. Soc 2019, 141, 13812–13821; [DOI] [PubMed] [Google Scholar]; (f) Wei X; Shu W; García-Domínguez A; Merino E; Nevado C Asymmetric Ni-Catalyzed Radical Relayed Reductive Coupling. J. Am. Chem. Soc 2020, 142, 13515–13522; [DOI] [PubMed] [Google Scholar]; (g) Charboneau DJ; Barth EL; Hazari N; Uehling MR; Zultanski SL A Widely Applicable Dual Catalytic System for Cross-Electrophile Coupling Enabled by Mechanistic Studies. ACS Catal. 2020, 10, 12642–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For examples that use homogeneous reductants other than TDAE in CEC, see:; (a) Yurino T; Ueda Y; Shimizu Y; Tanaka S; Nishiyama H; Tsurugi H; Sato K; Mashima K Salt-Free Reduction of Nonprecious Transition-Metal Compounds: Generation of Amorphous Ni Nanoparticles for Catalytic C–C Bond Formation. Angew. Chem. Int. Ed 2015, 54, 14437–14441; [DOI] [PubMed] [Google Scholar]; (b) Lv L; Qiu Z; Li J; Liu M; Li C-J N2H4 as Traceless Mediator for Homo- and Cross-Aryl Coupling. Nature Commun. 2018, 9, 4739; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ueda Y; Tsujimoto N; Yurino T; Tsurugi H; Mashima K Nickel-Catalyzed Cyanation of Aryl Halides and Triflates using Acetonitrile via C–CN Bond Cleavage Assisted by 1, 4-bis (trimethylsilyl)-2,3,5,6-tetramethyl-1,4-dihydropyrazine. Chem. Sci 2019, 10, 994–999; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ishida N; Masuda Y; Sun F; Kamae Y; Murakami M A Strained Vicinal Diol as a Reductant for Coupling of Organyl Halides. Chem. Lett 2019, 48, 1042–1045; [Google Scholar]; (e) Charboneau DJ; Brudvig GW; Hazari N; Lant HMC; Saydjari AK Development of an Improved System for the Carboxylation of Aryl Halides through Mechanistic Studies. ACS Catal. 2019, 9, 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broggi J; Terme T; Vanelle P Organic Electron Donors as Powerful Single-Electron Reducing Agents in Organic Synthesis. Angew. Chem. Int. Ed 2014, 53, 384–413. [DOI] [PubMed] [Google Scholar]
- 22.For selected leading examples, see:; (a) Wanzlick HW; Schikora E Ein Nucleophiles Carben. Chem. Ber 1961, 94, 2389–2393; [Google Scholar]; (b) Wanzlick HW Aspects of Nucleophilic Carbene Chemistry. Angew. Chem. Int. Ed 1962, 1, 75–80; [Google Scholar]; (c) Wanzlick HW; Esser F; Kleiner HJ Nucleophile Carben-Chemie, III. Neue Verbindungen vom Typ des Bis-[1,3-diphenyl-imidazolidinylidens-(2)]. Chem. Ber 1963, 96, 1208–1212; [Google Scholar]; (d) Thummel RP; Goulle V; Chen B Bridged Derivatives of 2, 2’-Biimidazole. J. Org. Chem 1989, 54, 3057–3061; [Google Scholar]; (e) Shi Z; Thummel RPN, N’-Bridged Derivatives of 2, 2’-Bibenzimidazole. J. Org. Chem 1995, 60, 5935–5945; [Google Scholar]; (f) Taton TA; Chen P A Stable Tetraazafulvalene. Angew. Chem. Int. Ed 1996, 35, 1011–1013; [Google Scholar]; (g) Shi Z; Thummel RP Bridged Dibenzimidazolinylidenes as New Derivatives of Tetraaminoethylene. Tetrahedron Lett. 1995, 36, 2741–2744; [Google Scholar]; (h) Shi Z; Goulle V; Thummel RP An Aza-Analogue of TTF1: 1,1’; 3,3’-Bistrimethylene-2,2’-Diimidazolinylidine. Tetrahedron Lett. 1996, 37, 2357–2360. [Google Scholar]
- 23.(a) Wanzlick HW; Schikora E Ein Neuer Zugang zur Carben-Chemie. Angew. Chem 1960, 72, 494–494; [Google Scholar]; (b) Böhm VP; Herrmann WA The “Wanzlick Equilibrium”. Angew. Chem. Int. Ed 2000, 39, 4036–4038; [DOI] [PubMed] [Google Scholar]; (c) Alder RW; Blake ME; Chaker L; Harvey JN; Paolini F; Schütz J When and How do Diaminocarbenes Dimerize? Angew. Chem. Int. Ed 2004, 43, 5896–5911; [DOI] [PubMed] [Google Scholar]; (d) Hahn FE; Jahnke MC Heterocyclic Carbenes: Synthesis and Coordination Chemistry. Angew. Chem. Int. Ed 2008, 47, 3122–3172. [DOI] [PubMed] [Google Scholar]
- 24.Prior reports have described the structures of TPyE, TPiE, and TME, however in these articles there is limited characterization, which is not consistent with our characterization of these compounds using modern analytical methods. Therefore, it is not clear that the correct compounds were actually prepared in the earlier reports. See:; Winberg HE; Carnahan J; Coffman D; Brown M Tetraaminoethylenes. J. Am. Chem. Soc 1965, 87, 2055–2056; [Google Scholar]; Scheeren JW; Nivard RJF Synthesis and Stability of Tri-sec-aminomethanes. Recl. Trav. Chim. Pays-Bas 1969, 88, 289–300; [Google Scholar]; Otohiko T; Kiyoshi Y; Masako H Tetraaminoethylenes. I. The Preparation and Some Reactions of Tetrapiperidino- and Tetramorpholinoethylene. Bull. Chem. Soc. Jpn 1971, 44, 2171–2176. [Google Scholar]
- 25.The naming of reductants is based on the following convention: The first letter T (for tetra) is associated with the fact that there are four N-heterocycles in the molecules. The second (and in some cases third letter) designates the type of heterocycle that is present, so for example Py for pyrrolidine and M for morpholine. The final letter E stands for ethylene as all of the reductants use ethylene as a building block.
- 26.W. Alder R; E. Blake M Bis(N-piperidyl)carbene and its Slow Dimerisation to Tetrakis(N-piperidyl)ethene. Chem. Commun 1997, 1513–1514. [Google Scholar]
- 27. The calculated HOMOs of TME and TAzE are similar to those of TPyE and TPiE (see SI).
- 28. The LUMOs of the oxidized compounds are similar to the HOMOs of the reductants (see SI).
- 29.The significant difference in the air sensitivity of TPiE and TAzE is surprising given there is only a 30 mV difference in their reduction potential. The difference is clearly related to kinetic factors but we do not have an explanation for why the solid-state packing of TPiE results in less sensitivity to air comapred to the packing of TAzE.
- 30.Hansen EC; Pedro DJ; Wotal AC; Gower NJ; Nelson JD; Caron S; Weix DJ New Ligands for Nickel Catalysis From Diverse Pharmaceutical Heterocycle Libraries. Nature Chem. 2016, 8, 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The only major change to the reaction conditions reported in reference 19g was the use of DMAc as the solvent instead of 1,4-dioxane. This change was made because it is easier to perform HTE using DMAc (due to its higher boiling point) and the new reductants display greater solubility in DMAc.
- 32.Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau L-C; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry Informer Libraries: A Chemoinformatics Enabled Approach to Evaluate and Advance Synthetic Methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Pang Y; Moser D; Cornella J Pyrylium Salts: Selective Reagents for the Activation of Primary Amino Groups in Organic Synthesis. Synthesis 2020, 52, 489–503; [Google Scholar]; (b) M. Correia JT; A. Fernandes V; Matsuo BT; Delgado JAC; de Souza WC; Paixão MW Photoinduced Deaminative Strategies: Katritzky Salts as Alkyl Radical Precursors. Chem. Commun 2020, 56, 503–514. [DOI] [PubMed] [Google Scholar]
- 34.(a) Martin-Montero R; Yatham VR; Yin H; Davies J; Martin R Ni-Catalyzed Reductive Deaminative Arylation at sp3 Carbon Centers. Org. Lett 2019, 21, 2947–2951; [DOI] [PubMed] [Google Scholar]; (b) Tcyrulnikov S; Cai Q; Twitty JC; Xu J; Atifi A; Bercher OP; Yap GPA; Rosenthal J; Watson MP; Kozlowski MC Dissection of Alkylpyridinium Structures to Understand Deamination Reactions. ACS Catal. 2021, 11, 8456–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.For examples of coupling reactions between benzylic Katritzky salts and acyl chlorides see:; Pulikottil FT; Pilli R; Suku RV; Rasappan R Nickel-Catalyzed Cross-Coupling of Alkyl Carboxylic Acid Derivatives with Pyridinium Salts via C–N Bond Cleavage. Org. Lett 2020, 22, 2902–2907. [DOI] [PubMed] [Google Scholar]
- 37.(a) Ociepa M; Turkowska J; Gryko D Redox-Activated Amines in C(sp3)–C(sp) and C(sp3)–C(sp2) Bond Formation Enabled by Metal-Free Photoredox Catalysis. ACS Catal. 2018, 8, 11362–11367; [Google Scholar]; (b) Kong D; Moon PJ; Lundgren RJ Radical Coupling From Alkyl Amines. Nat. Catal 2019, 2, 473–476. [Google Scholar]
- 38.(a) Liao J; Basch CH; Hoerrner ME; Talley MR; Boscoe BP; Tucker JW; Garnsey MR; Watson MP Deaminative Reductive Cross-Electrophile Couplings of Alkylpyridinium Salts and Aryl Bromides. Org. Lett 2019, 21, 2941–2946; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cong F; Lv X-Y; Day CS; Martin R Dual Catalytic Strategy for Forging sp2–sp3 and sp3–sp3 Architectures via β-Scission of Aliphatic Alcohol Derivatives. J. Am. Chem. Soc 2020, 142, 20594–20599; [DOI] [PubMed] [Google Scholar]; (c) Li J; Chen J; Sang R; Ham W-S; Plutschack MB; Berger F; Chabbra S; Schnegg A; Genicot C; Ritter T Photoredox Catalysis with Aryl Sulfonium Salts Enables Site-Selective Late-Stage Fluorination. Nature Chem. 2020, 12, 56–62. [DOI] [PubMed] [Google Scholar]
- 39.Liao J; Guan W; Boscoe BP; Tucker JW; Tomlin JW; Garnsey MR; Watson MP Transforming Benzylic Amines into Diarylmethanes: Cross-Couplings of Benzylic Pyridinium Salts via C–N Bond Activation. Org. Lett 2018, 20, 3030–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.For selected leading examples, see:; (a) Maity P; Shacklady-McAtee DM; Yap GPA; Sirianni ER; Watson MP Nickel-Catalyzed Cross Couplings of Benzylic Ammonium Salts and Boronic Acids: Stereospecific Formation of Diarylethanes via C–N Bond Activation. J. Am. Chem. Soc 2013, 135, 280–285; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao F; Tan Q; Xiao F; Zhang S; Deng G-J Palladium-Catalyzed Desulfitative Cross-Coupling Reaction of Sodium Sulfinates with Benzyl Chlorides. Org. Lett 2013, 15, 1520–1523; [DOI] [PubMed] [Google Scholar]; (c) Ackerman LK; Anka-Lufford LL; Naodovic M; Weix DJ Cobalt Co-Catalysis for Cross-Electrophile Coupling: Diarylmethanes from Benzyl Mesylates and Aryl Halides. Chem. Sci 2015, 6, 1115–1119; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang J; Lu G; Xu J; Sun H; Shen Q Nickel-Catalyzed Reductive Cross-Coupling of Benzyl Chlorides with Aryl Chlorides/Fluorides: A One-Pot Synthesis of Diarylmethanes. Org. Lett 2016, 18, 2860–2863. [DOI] [PubMed] [Google Scholar]
- 41.Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.