

Abstract
The parathyroid hormone (PTH) type 1 receptor (PTHR) is a class B G protein–coupled receptor (GPCR) that regulates mineral-ion, vitamin D, and bone homeostasis. Activation of the PTHR by PTH induces both transient cell-surface and sustained endosomal cAMP production. To address whether the spatial (location) or temporal (duration) dimension of PTHR-induced cAMP encodes distinct biological outcomes, we engineered a biased PTHR ligand (PTH7d) that elicits cAMP production at the plasma membrane but not at endosomes. PTH7d stabilized a unique active PTHR conformation that mediated sustained cAMP signaling at the plasma membrane due to impaired β-arrestin coupling to the receptor. Experiments in cells and mice revealed that sustained cAMP production by cell-surface PTHR failed to mimic the pharmacological effects of sustained endosomal cAMP production on the abundance of the rate-limiting hydroxylase catalyzing the formation of active vitamin D, as well as increases in circulating active vitamin D and Ca2+ and in bone formation in mice. Thus, similar amounts of cAMP generated by PTHR for similar lengths of time in different cellular locations – plasma membrane and endosomes– mediate distinct physiological responses. These results unveil subcellular signaling location as a means to achieve specificity in PTHR-mediated biological outcomes and raise the prospect of rational drug design based upon spatiotemporal manipulation of GPCR signaling.
INTRODUCTION
Over 800 individual genes encode G protein–coupled receptors (GPCRs) in humans, and their expression in various tissues is paramount for proper control of a wide range of physiological processes (1). These receptors reside predominantly at the cell surface, where they recognize and respond to an impressively diverse catalog of extracellular stimuli that range from photons and ions to small molecule neurotransmitters and large peptide hormones (2). To explain how these receptors achieve such exquisite specificity in their biological effects, previous reports have described receptor and functional selectivity as critical determinants. Functional selectivity originally referred to empirical observations that ligands for the same GPCR can possess differential propensities to activate downstream signaling pathways (referred to as biased agonism) (3, 4). In this classical model, GPCR activation was thought to stimulate heterotrimeric G protein signaling exclusively from the cell surface in a transient manner due to subsequent recruitment of β-arrestin, which was proposed to sterically block further G protein coupling and to mediate agonist-induced receptor internalization, preventing additional rounds of stimulation by extracellular ligand. This model has been substantially revised by the discovery of several receptors that engage in sustained G protein–dependent cyclic adenosine monophosphate (cAMP) signaling from early endosomes following endocytosis (5–7). In the case of the parathyroid hormone (PTH) type 1 receptor (PTHR), PTH and a long-acting PTH analog (hereafter referred to as LA-PTH) trigger the formation of a ternary PTHR–β-arrestin–Gβγ complex that remains active following internalization and redistribution to early endosomes. This ternary PTHR complex is thought to promote and maintain a high amount of cAMP at endosomes through a series of reactions that include its reassembly with the diffusing Gαs, the conditional activation of adenylate cyclases by Gβγ, and the inhibition of cAMP-selective phosphodiesterases by activation of extracellular signal–regulated protein kinases 1 and 2 (ERK1/2) (reviewed in (7)).
Observations that the location and duration of Gs-mediated cAMP responses can be dictated in a ligand-dependent manner for a single GPCR has led to the emergence of spatiotemporal regulation in GPCR signal transduction as a novel property of functional selectivity. Studies involving the PTHR have shown that full-length PTH (PTH1–84, a 84 amino acid peptide) or the fully functional synthetic N-terminal fragment PTH1–34 and related peptide (PTHrP1–36) stabilize distinct receptor conformations that result in cAMP responses that differ markedly in location and duration (5, 8, 9). Although all PTHR ligands stimulate transient cAMP responses from the cell surface, only PTH and PTH1–34 cause an additional sustained phase of cAMP generation derived from highly stable ligand–receptor–β-arrestin complexes that remain active in early endosomes following their internalization (5, 10–12).
These findings have subsequently been used to explain ligand-specific alterations in PTHR biological outcomes observed in vivo; however, no direct studies have been reported. For example, early studies demonstrated that injection of PTH1–34 and PTHrP1–36 into humans causes similar enhancement in the formation of new bone but discordant effects on ionized serum Ca2+ and vitamin D amounts, with lesser effects observed for PTHrP1–36 (13). Additionally, the PTHrP analog abaloparatide, which triggers transient cAMP production confined to the cell surface, has been shown to efficiently promote bone formation with a reduced hypercalcemic effect when compared to PTH1–34. Both PTH1–34 and abaloparatide are FDA-approved for the treatment of osteoporosis. Despite these advancements, whether the spatial versus the temporal dimension of cAMP signaling encodes ligand-dependent PTHR biological specificity remains unknown.
Here, we addressed this question by developing a location-biased PTHR peptide ligand through amino acid epimerization at position 7 of PTH1–34 (referred to as PTH7d). We demonstrated that this ligand stabilized a receptor conformation that failed to recruit β-arrestins and stimulated sustained cAMP exclusively from the cell surface, thus indicating that spatial bias of cAMP generation can be triggered by single amino acid epimerization of the ligand peptide. Subsequent studies in mice and polarized human epithelial kidney cells comparing the actions of PTH7d and a long-acting PTH analog (hereafter referred to as LA-PTH,) (14) that potently promotes endosomal cAMP production revealed that homeostatic control of serum Ca2+ and active vitamin D concentrations specifically requires sustained cAMP generation from endosomal compartments as opposed to sustained cAMP production from the plasma membrane. This study highlights the potential for delineating spatial and temporal signaling bias in rational drug design and the development of therapeutics with enhanced biological specificity.
RESULTS
Selection and characterization of a location-biased PTH analog
We have previously shown that translocation of PTH–PTHR signaling complexes from the cell surface to early endosomes is β-arrestin–dependent (10, 11). We reasoned that a PTHR ligand that stimulates cAMP production but fails to recruit β-arrestin would cause a shift in sustained cAMP generation from endosomes to the cell surface, thus permitting determination as to how the cellular localization of signaling responses influences PTHR biology.
To help design a PTH analog with the desired bias and to better understand the bioactive domains and key functional determinants in PTH, we analyzed an anisotropic network model (ANM) of PTH-bound receptor that we previously reported (15). In brief, the ANM probes the collective motion of the PTH–PTHR complex based on the position of Cα-atoms and thus unveils cooperative structural changes upon ligand binding (16). Here, we focused on one mode of motion that displayed opening and closing of the receptor intracellular face, reflecting a motion that occurs during receptor activation. To determine which PTH residues would elicit cooperative responses consistent with PTHR activation, we examined the cross-correlations between all PTH residues with Thr392 of the receptor, which is located near the third intracellular loop (ICL3) – a region where conformational rearrangements associated with receptor activation (fig. S1A). Notably, PTH residues 1–10 had strong positive (> 0.5) cross-correlations with the intracellular part of the receptor, which supports previous observations that this region constitutes the signaling portion of the peptide (17, 18). Our ANM analysis suggested that the opening and closing of the intracellular region is allosterically coupled to the structural changes at residues interacting with the N-terminal portion of PTH that includes residues 1–10 (fig. S1A). From our previous investigations, we discerned that PTH residues 1–4 are specifically determinant to switch ‘on’ the receptor active state (12, 14, 19). We decided to focus on PTH residues 5, 6, 7, 9, and 10, which exhibited high cross-correlation values (> 0.6) and thus we hypothesized that their substitutions in PTH1–34 (hereafter referred to as PTHWT) may bias receptor coupling to G proteins instead of β-arrestin and thus favor signaling at the plasma membrane instead of at endosomes. We aimed to introduce a minimal chemical modification of the interrogated residue; therefore, we chose the epimerization (L- to D-amino acid residue) instead of conventional alanine scanning substitution. The assumption that amino acids 5–10 of PTHWT determine signaling bias was tested by comparing time courses of cAMP production and β-arr2 recruitment to PTHR. Indeed, PTHWT and some of its diastereomers (PTH7d and PTH10d) stimulated indistinguishable increases in cAMP (fig. S1B), but only PTHWT and PTH10d stimulated the interaction of PTHR with β-arr2 (fig. S1C).
We further examined the signaling properties of PTH7d in human embryonic kidney (HEK293) cells stably expressing recombinant human PTHR. This modification did not alter binding affinity in equilibrium competition assays in intact cells (fig. S2), nor did it cause changes in the potency (EC50) or maximal efficacy (Emax) of cAMP production in luciferase-based accumulation assays (Fig. 1A, Table 1). In contrast, PTH7d displayed markedly impaired recruitment of β-arrestin compared to PTHWT or LA-PTH (a long-acting chimeric PTH/PTHrP peptide whose N- and C-terminal fragments correspond to PTH1–15 and PTHrP16–34, respectively), as measured by bioluminescence resonance energy transfer (BRET) in cells stably expressing luciferase-tagged PTHR (PTHRRluc8) and Venus-tagged βarr1 or βarr2 (Fig. 1B). This effect was confirmed by both Fӧrster resonance energy transfer (FRET)-based time courses in cells transiently coexpressing PTHR fused to cyan fluorescent protein (PTHRCFP) and βarr2 fused to yellow fluorescent protein (βarr2YFP) (Fig. 1C), in co-immunoprecipitation assays (Fig. 1D), and in photocrosslinking experiments (fig. S3).
Fig. 1. Characterization of a Gs-biased PTH analog generated by amino acid isomerization.
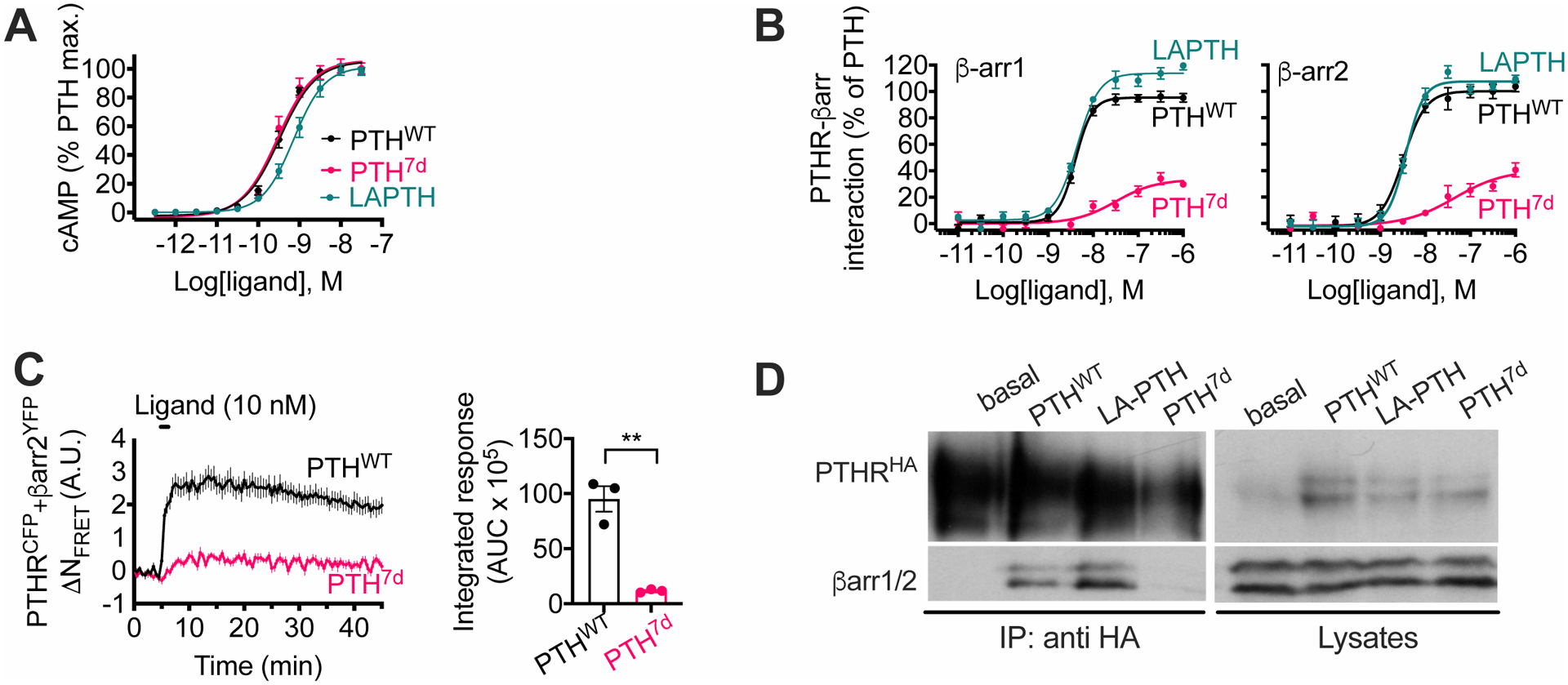
(A and B) Concentration-response curves for PTHWT, LA-PTH, and PTH7d in cAMP accumulation (A) or in β-arr1 or β-arr2 recruitment (B) assays. Data are means ± SEM from N = 5 independent experiments.
(C) Time course and integrated response of β-arr2 recruitment measured by FRET in HEK293 cells transiently expressing PTHRCFP and β-arr2YFP following brief stimulation with 10 nM PTHWT or PTH7d. Data are means ± SEM from N = 3 independent experiments with n = 28–33 cells per experiment. ***, P < 0.005 by t-test.
(D) Recruitment of endogenous β-arr1 and β-arr2 (βarr1/2) to PTHR detected by coimmunoprecipitation with HA-tagged PTHR (HAPTHR) stably expressed in HEK293 cells. Cells were challenged with 100 nM of either PTHWT, LAPTH, or PTH7d. Blot is representative of 2 independent experiments.
Table 1. Binding isotherm and signaling properties.
Binding in intact cells was performed with PTH1–34TMR and the binding affinity constant Ki calculated after the Cheng and Prusoff correction. For R0 and RG, the binding was performed in plasma membrane extracts from HEK293 cells lacking Gs by CRISPR-Cas9 deletion and transiently transfected with PTHR without (R0) or with (RG) Gs-ND. Data are means ± SEM from N = 3 (binding), N = 4 (cAMP), and N = 5 (β-arr1 and β-arr2) independent experiments.
| Ligands | Binding Intact cells |
Binding R0 |
Binding RG |
|||
|---|---|---|---|---|---|---|
| Ki (nM) | P | IC50 (nM) | P | IC50 (nM) | P | |
| PTHWT | 4.97 ± 0.92 | 1.00 | 4.70 ± 1.33 | 1.00 | 0.10 ± 0.04 | 1.00 |
| PTH7d | 4.27 ± 0.80 | 1.00 | 17.14 ± 3.28 | 0.015 | 0.10 ± 0.04 | 1.00 |
| Ligands | cAMP | βarr1 | βarr2 | |||
|---|---|---|---|---|---|---|
| pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | |
| PTHWT | 9.36 ± 0.04 | 100 | 8.32 ± 0.04 | 100 | 8.37 ± 0.05 | 100 |
| PTH7d | 9.45 ± 0.08 | 99 ± 5 | 7.76 ± 0.08 | 40 ± 2 | 7.71 ± 0.11 | 37 ± 3 |
We next sought to determine the time course of cAMP generation for PTH7d using the FRET-based sensor EpacCFP/YFP. Recordings in single cells revealed that PTH7d induced sustained cAMP responses upon ligand washout that were highly similar in magnitude and duration to those induced by PTHWT and LA-PTH (Fig. 2A). Despite similar cAMP response kinetics, we subsequently determined that prolonged cAMP generation observed for PTH7d was derived from ligand–receptor complexes retained at the cell surface, because inclusion of a cell-impermeable PTHR antagonist (D-Trp12, Tyr34-bPTH7–34) (20) in the washout buffer completely abolished the sustained phase of PTH7d-mediated cAMP generation but had moderate or no effect in analogous experiments using PTHWT or LA-PTH, respectively (Fig. 2B). Consistent with this finding, blockade of receptor internalization by expression of a dominant-negative dynamin mutant (DynK44A) significantly reduced cAMP responses induced by PTHWT, whereas those for PTH7d were unaffected (Fig. S4). Furthermore, real-time analysis of receptor trafficking in cells stably expressing PTHR N-terminally tagged with superecliptic pHluorin (SEP), a pH-sensitive green fluorescent protein (GFP) variant that exhibits fluorescence intensity reduction in acidic environments (21), showed only modest internalization upon PTH7d stimulation when compared to PTHWT and LA-PTH (Fig. 2C).
Fig. 2. Location bias of PTH7d signaling.
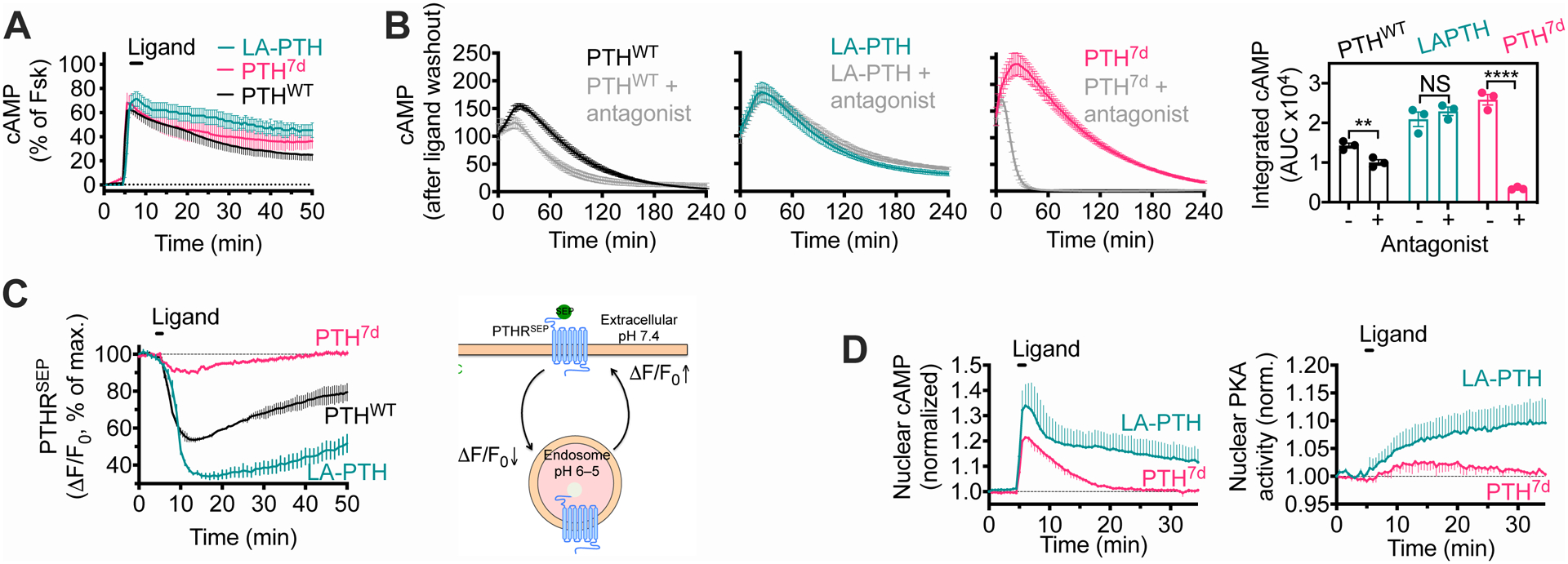
(A) Averaged cAMP time-course responses following brief stimulation with 10 nM PTHWT, LAPTH, or PTH7d. Percentage of cAMP responses is relative to the response in the presence of forskolin (Fsk). Data are means ± SEM from N = 3 independent experiments with n = 20–26 cells per experiment.
(B) Time courses and integrated responses after washout of 1 nM PTHWT, PTH7d, or LA-PTH in the presence or absence of cell-impermeable antagonist. Data are means ± SEM from N = 3 experiments. P = 0.01 (PTHWT), 0.39 (LA-PTH), and < 0.0001 (PTH7d) by t-test.
(C) Time courses of internalization and recycling of PTHR tagged with super-ecliptic pHluorin (PTHRSEP) in response to 100 nM ligand measured by time-lapse confocal microscopy. A schematic depicting the measured values is also shown. Data are means ± SEM from N = 3 experiments with n = 11–14 cells per experiment.
(D) Averaged nuclear cAMP and PKA activity time-course experiments following brief stimulation with 30 nM PTH7d or LA-PTH. Data are means ± SEM from N = 3 independent experiments with n = 9–14 cells per experiment.
Finally, we previously reported that the endosomal cAMP pool diffuses more effectively into the nucleus than does the plasma membrane–adjacent pool and thus represents a rate-limiting step for nuclear cAMP-mediated protein kinase A (PKA) activation (22, 23). We verified that here by comparing nuclear cAMP accumulation and PKA activity over time in response to LA-PTH compared with PTH7d (Fig. 2D). Collectively, our signaling characterization data indicate that amino acid epimerization at position 7 of PTH results in a location and Gs-biased peptide ligand that differs from PTHWT and LA-PTH solely by the cellular localization of active signaling complexes involved in sustained cAMP responses. Specifically, PTH7d stimulates G protein–dependent production of cAMP at the plasma membrane but does not stimulate β-arrestin–dependent internalization of PTHR and subsequent generation of cAMP at endosomes. We subsequently examined the molecular basis for these signaling discrepancies, as well as their consequences in vivo.
Molecular basis for impaired PTH7d function
Given the well-established importance of β-arrestin coupling to the PTHR in permitting sustained cAMP generation at endosomes in response to PTHWT, we sought to investigate the molecular mechanisms that underlie the relative inability of PTH7d to promote this interaction, as well as its consequences for both cell biological and pharmacological processes. Recruitment of β-arrestin to activated GPCRs is thought to occur following phosphorylation of serine and threonine residues within intracellular receptor domains, specifically the third intracellular loop (ICL3) and C-terminal tail, by GPCR kinases (GRKs). Furthermore, it has been proposed that receptor phosphorylation status serves as the foremost determinant for GPCR–β-arrestin interactions, a paradigm commonly referred to as the phosphorylation barcode hypothesis (24–26). Accordingly, we examined whether PTHWT and PTH7d triggered distinct phosphorylation patterns in the PTHR that differentially permitted distinct interactions with β-arrestins by performing stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative proteomics (fig. S5A). By liquid chromatography with tandem mass spectrometry (LC-MS/MS), we identified 10 phosphorylation sites located in the C-terminal tail and ICL3 in response to 5 min of PTHWT or PTH7d stimulation at a concentration of 30 nM (fig. S5B, and fig. S6A and B). Although β-arrestin recruitment is usually considered to be driven by the phosphorylation status of a GPCR (27), we found no significant differences in the sites or extent of phosphorylation between PTHWT and PTH7d treatments. In an effort to confirm our hypothesis that β-arrestin recruitment to hormone-activated PTHR may occur independently of receptor phosphorylation, we assessed the extent of PTHWT-triggered β-arrestin recruitment to phosphorylation-deficient PTHR mutants, where identified phosphorylated serine and threonine residues in either ICL3 (PTHRPD-ICL3) or the C-terminal tail (PTHRPD-Ct) were mutated to alanine. Our immunoprecipitation analysis revealed that whereas the absence of phosphorylation sites in the receptor’s C-terminus had no effect on β-arrestin recruitment, the elimination of phosphorylation sites within ICL3 severely impaired β-arrestin recruitment to PTHR (fig. S7A). Taking into account the lack of quantitative LC/MS-MS data on ICL3 phosphorylation status, this observation suggests that ICL3 might be a determinant of initial β-arrestin recruitment to PTHR, whereas the C-terminus itself contributes to the stabilization of β-arrestin–receptor complexes as previously proposed (28). The latter hypothesis was supported by FRET time-course experiments using the PTHRPD-Ct mutant, which showed that receptor phosphorylation did not contribute to the initial coupling of the PTHR to β-arr2 (fig. S7B), but rather to the stability of the complex over time (fig. S7C), a result consistent with earlier work (28). Overall, this finding suggests that alteration in β-arrestin recruitment observed for PTHWT and PTH7d was not caused by defective receptor phosphorylation at the C-terminus, but altered phosphorylation of ICL3 or conformational changes due to alanine substitutions might be the cause of diminished β-arrestin recruitment to PTH7d-bound PTHR.
Because the ICL3 is an immediate indicator of GPCR activation status, we considered that these ligands may uniquely stabilize distinct active PTHR conformations. This hypothesis was investigated experimentally by comparing the capacity of ligands to stabilize the G protein–coupled (RG) and –uncoupled (R0) conformations of PTHR using binding isotherms measured in cell membrane extracts. Compatible with the indistinguishable binding affinity of PTHWT and PTH7d for the receptor in intact cells, these ligands displayed identical affinities for the RG conformation in vitro (Fig. 3A). In contrast, PTH7d had decreased binding affinity for the R0 conformation (Fig. 3A; Table 1), which is usually associated with sustained cAMP originating from endosomes (29), suggesting that PTH7d stabilizes a distinct receptor conformation. We further confirmed that PTHWT and PTH7d stabilized distinct signaling PTHR conformations using PTHRCFP/YFP, an intramolecular FRET-based PTHR sensor (scheme in Fig. 3B) that reports conformational rearrangements as the receptor switches from inactive to active states in response to ligand binding (30). Perfusion of PTHWT to cells expressing this sensor induced a fast increase in the FRET ratio FYFP/FCFP with a time constant τ ≈ 3.4 s (Fig. 3C). In contrast, perfusion of PTH7d induced a FRET change of similar kinetics but substantially lower magnitude than that by PTHWT. Because FRET depends on the distance and the dipole-dipole orientations of fluorophores, these results imply that PTH7d stabilizes a PTHR conformation distinct from that seen in response to PTHWT.
Fig. 3. Molecular changes induced by PTH7d.
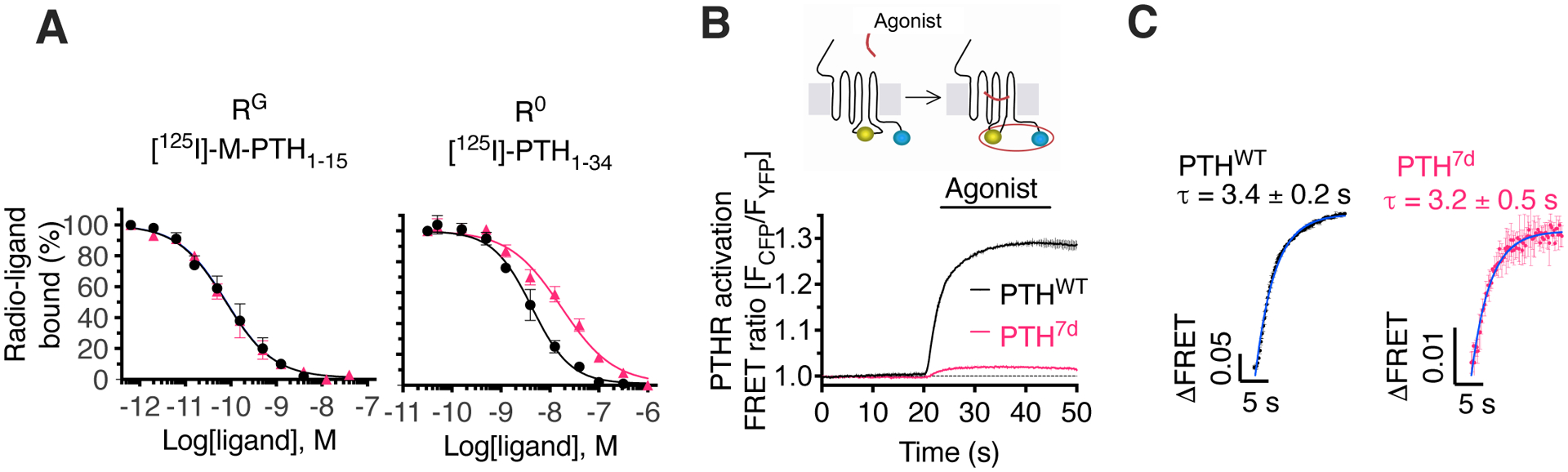
(A) Competition binding at equilibrium with 125I-PTH1–15 and 125I-PTH1–34 as radioligands to detect the RG and R0 states of PTHR, respectively. Data are means ± SEM from N = 5 independent experiments with duplicate wells for each concentration.
(B and C) The inset shows a schematic of the FRET-based PTHR activation sensor (PTHRCFP/YFP) with YFP (yellow) fused to ICL3, and CFP (blue) attached to the receptor C-terminal tail. The graph shows averaged time-courses of PTHR activation by recording changes of the FRET ratio in HEK 293 cells expressing PTHRCFP/YFP with the initial value at t = 0 set to 1 (B), and kinetics of PTHR activation (C). Cells were continuously perfused with control buffer or 1 μM agonist (horizontal bar). Data are means ± SD from N = 2 independent experiments. Note that the FRET data are expressed as FCFP/FYFP, resulting in a positive change upon agonist stimulation.
These observations were further supported by molecular dynamics (MD) simulations. We utilized the recently reported 3.0 Å cryo-EM structure of active state PTHR bound to Gs and LA-PTH (PDB 6NBF) (14) to generate initial models of PTHWT- and PTH7d-bound PTHR for triplicate 200 ns MD simulations. In the initial PTH7d–PTHR model, D-Leu7 is unfavorably close to neighboring receptor transmembrane helix 7 (TM7) residues Trp437 (corresponding to position 7.35 in the Wootten numbering scheme for class B GPCRs (31)) and Met441 (7.39 in the Wootten scheme) (fig. S8A), and simulations revealed that the D-Leu7 side chain shifts to mirror L-Leu7 nonpolar interactions in the PTHWT–PTHR model (fig. S7B). We found that this shift induces a kink in the N-terminal portion of PTH7d helix toward TM6 (fig. S8B), which subsequently permits additional polar interactions between Glu4 of PTH7d and PTHR residues that are not observed for PTHWT (fig. S8C, D; Table S1). In the active-state cryo-EM structure of PTHR, Glu4 promotes an extensive polar interaction network that stabilizes the outward kink of TM6, which is considered prerequisite for coupling to G proteins (12, 14). The increased polar contacts by Glu4 of PTH7d further extends this polar interaction network relative to PTHWT-bound receptor, resulting in a more pronounced TM6 kink and a slight shift of the N-terminal portion of TM6 induced by PTH7d (fig. S8, E and F). While speculative, these MD predictions are consistent with FRET assays indicating different PTHR conformations stabilized by PTH and PTH7d.
Differential pharmacological actions of PTH7d, PTHWT, and LA-PTH in mice
The PTHR is the main regulator of bone remodeling and serum mineral-ion (inorganic phosphate and Ca2+) homeostasis (17). Despite previous reports demonstrating ligand-dependent modulation of these physiological processes, whether the location or the duration of the cAMP response is determinant remains unclear. We thus compared the effects of PTH7d, PTHWT, and LA-PTH on these parameters in mice following four weeks of daily injection (40 μg/kg body weight). Examination of skeletal parameters by micro-computed tomography (Fig. 4A) further highlighted the physiological significance of spatial organization in PTHR-mediated signaling. Whereas all three ligands caused a moderate but significant increase in total bone volume (TV) of the metaphysis at the distal femur (Fig. 4B), only PTHWT and LA-PTH led to a significant increase in trabecular (Tb) bone fractions (Tb bone volume relative to total bond volume, Tb.BV/TV) and thickness (Tb.Th) when compared to vehicle control (Fig. 4B). Despite its ability to prolong the cAMP response at the cell membrane, PTH7d induced a trend of increase in Tb.BV/TV that is comparable to the skeletal response to PTHWT. Additionally, LA-PTH treatment resulted in an increase in trabecular number (Tb.N) that was markedly distinct from that observed for PTHWT and PTH7d (Fig. 4B). These data suggest that endosomal cAMP has a greater impact on skeletal homeostasis than does plasma membrane–associated cAMP.
Fig. 4. Differential pharmacological actions of PTH7d, PTHWT, and LAPTH in mice.
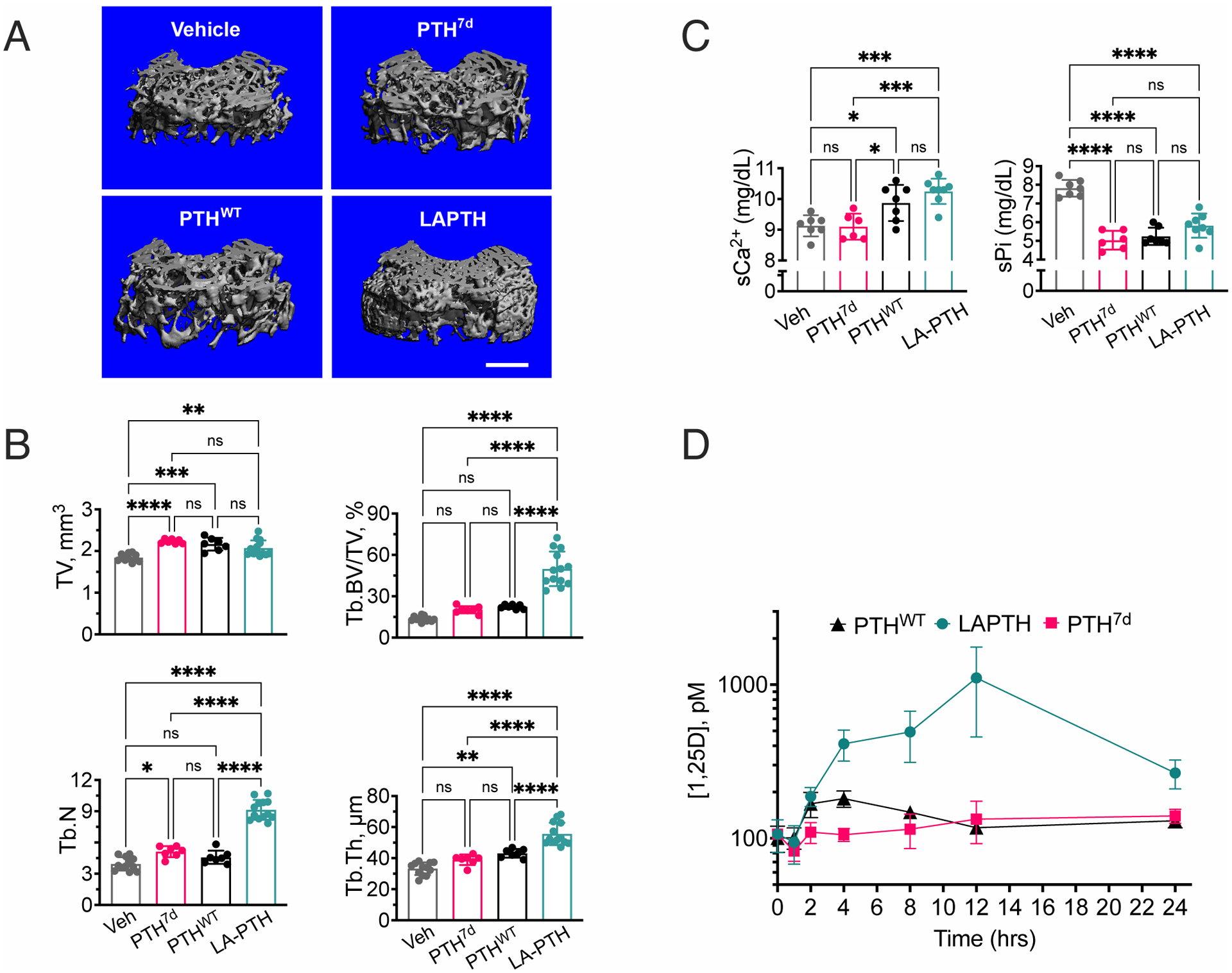
(A) 3-D reconstructed micro-computed tomography (μCT) images of secondary spongia at the distal femurs of mice treated with vehicle, PTH7d, PTHWT (PTH1–34), or LAPTH. Scale bar, 500μm.
(B) Quantifications of skeletal parameters in trabecular (Tb) bone of distal femur, including total bone volume (TV), ratio of Tb bone volume (Tb.BV) to TV (Tb.BV/TV), Tb number (Tb.N), and Tb thickeness (Tb.Th). Parameters were assessed in mice subjected to daily injections of PTH7d, PTHWT, LA-PTH, or vehicle (Veh) for 4 weeks. Data are means ± SD from N = 7 mice/group for PTH7d and PTHWT injections and N = 14 mice/group for LA-PTH and Veh injections. *P < 0.03, ***P < 0.002, ***P < 0.0002 and ****P < 0.0001 vs Veh control mice by one-way ANOVA with Tukey-Kramer post-hoc test.
(C) Quantification of serum Ca2+ (sCa2+) and phosphate (sPi) measured 2 hrs after the last of the 4-week daily injections of PTH7d, PTHWT, LA-PTH (40 μg/kg body weight/injection), or vehicle (Veh). Data are means ± SD from N = 7 mice/group. *P < 0.03, **P < 0.002, ***P < 0.0002 and ****P < 0.0001 vs Veh control mice by one-way ANOVA with Tukey-Kramer post-hoc test.
(D) Quantification of serum 1.25D measured in mice before or 1, 2, 4, 8, 12, or 24 hrs after a single injection of PTH, PTH7d, or LA-PTH. Data are means ± SD from N = 7 mice/time point/drug and 15 mice for time “0” controls. Statistical analysis is shown in the Supplementary Materials (fig. S9).
Analysis of the effects on mineral-ion homeostasis revealed that each ligand caused similar reductions in serum phosphate (sPi) amounts (Fig. 4C), consistent with the previously reported inhibition of phosphate uptake by PKA-dependent phosphorylation of NHERF1 (Na+/H+ exchanger regulatory factor-1) in the cytoplasm (32). However, acute increases in serum Ca2+ (sCa2+) occurred only in response to PTHWT and LA-PTH, but not PTH7d, compared to vehicle control (Fig. 4C). Given that blood samples were taken 2 hrs after the last injection of peptide, the acute actions of the peptide on Ca2+ and Pi were likely due to the responses in the kidney, instead of bone. Previous studies have established that PTHR ligands may indirectly influence these processes by increasing 1,25-dihydroxy vitamin D (1,25D), the active form of vitamin D, in renal cells (33). Consistent with calcemic responses was the acute or sustained increase in circulating 1,25D upon injection of PTHWT or LA-PTH, respectively, into mice that was completely absent in mice treated with PTH7d (Fig. 4D and fig. S9A–D). Pharmacokinetic analysis of serum from mice treated with LA-PTH or PTH7d confirmed that in vivo effects were not influenced by differential degradation of these peptides (fig. S10). The impaired action of PTH7d compared to PTHWT and LA-PTH as it relates to increase in 1,25D may arise from deficient induction of the 25-hydroxyvitamin D 1-α hydroxylase (referred to as 1-α[OH]ase), the rate-limiting enzyme that catalyzes the conversion of 25-hydroxyvitamin D in renal proximal tubule epithelial cells. This premise was supported by the observation that basolateral stimulation with LA-PTH and PTH, but not PTH7d, caused a significant increase in the production of 1-α[OH]ase in polarized MDCK cells (Fig. 5, A and B), despite the ability of both ligands to induce similar cytosolic cAMP responses in this cell line, and with the PTH7d response being restricted to the basolateral membrane (Fig. 5, C and D). Taken together, these findings provide evidence that the location of sustained cAMP signaling serves as a critical regulator for PTHR-mediated effects on serum Ca2+, with increases in 1-α[OH]ase and activation of vitamin D as clear underlying determinants.
Fig. 5. Basolateral actions of PTH and its variants in polarized MDCK cells.
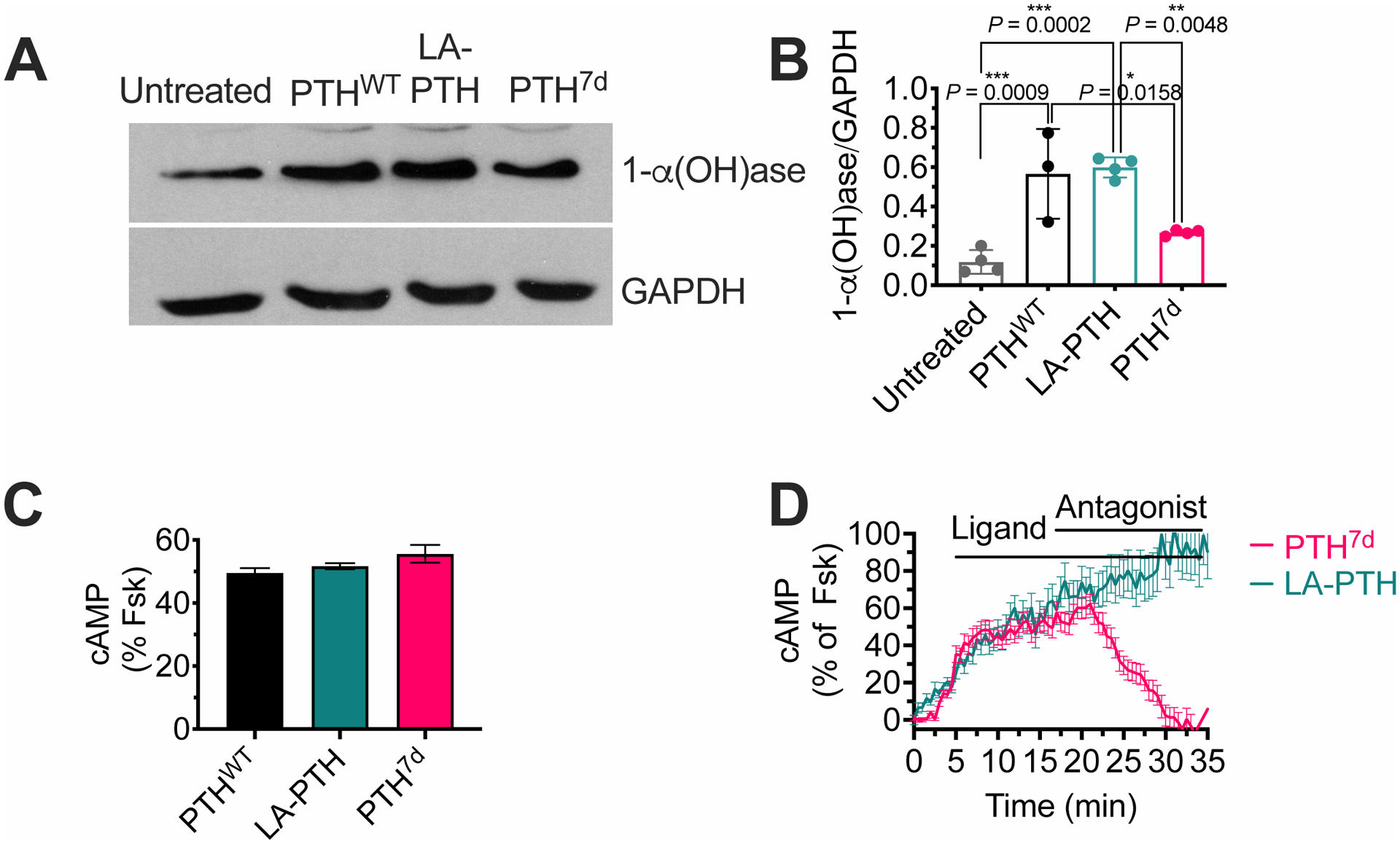
(A and B) Representative western blot (A) and quantification (B) of 1-α(OH)ase relative to GAPDH in polarized PTHR-expressing MDCK cells after basolateral stimulation with either LA-PTH, PTHWT, or PTH7D for 8 hours. Graph shows individual data points and means ± SD. P values were determined by one-way ANOVA with Tukey test. The P value for untreated vs PTH7d is 0.25.
(C) Quantification of cAMP responses in polarized MDCK cells expressing the Green cADDis sensor at t = 30 min after basolateral stimulation with 100 nM PTHWT, LA-PTH, or PTH7d. Percentage of cAMP responses is relative to the response in the presence of forskolin (Fsk). Data are means ± SD from n = 90–107 cells.
(D) cAMP time courses in polarized MDCK cells following addition of 100 nM LA-PTH or PTH7d in the absence or presence of 1 μM D-Trp12,Tyr34-bPTH7–34, a cell-impermeable competitive antagonist at the basolateral membrane. Horizontal bars indicate the application of ligand and antagonist. Data are means ± SEM from n = 48 cells.
DISCUSSION
The present work identifies that amino acid epimerization at a single position in a class B GPCR peptide ligand may provide a means to exquisitely probe ligand-dependent signaling determinants and pharmacological consequences without significantly altering ligand binding affinity or stability in vivo. In the case of PTH7d, L-to-D isomerization at position 7 of PTH revealed significant modulation of PTHR–β-arrestin interactions and thus a shift in the cellular localization of sustained cAMP signaling from endosomes to the plasma membrane. Such negative modulation of PTHR–β-arrestin interaction by PTH7d is likely a result of a distinct receptor conformation triggered by this PTH analog, as indicated by experiments with the PTHR conformational biosensor and further elaborated by our computational analysis. We predict that PTH7d likely stabilizes a receptor conformation with a more pronounced outward shift of TM6. It is well established that β-arrestin–GPCR interaction is a two-step binding process (34), whereby the initial β-arrestin interaction with the receptor’s phosphorylated C-terminus or ICL2/3 is a prerequisite for β-arrestin activation and subsequent engagement to the cytosolic core of a GPCR. Our studies have shown that phosphorylated ICL3 of PTHR is likely a key determinant of initial β-arrestin recruitment to the receptor, whereas the phosphorylated C-terminus is required for stabilization of β-arrestin–PTHR complexes. Our computational investigations suggest that PTH7d binding to PTHR may lead to ICL3 positioning closer to the plasma membrane due to a more pronounced outward movement of TM6, potentially rendering ICL3 inaccessible to both phosphorylation and β-arrestin binding. However, we cannot specifically rule out that alanine substitution in ICL3 could confer a conformational change in this region that also precludes the ability to bind β-arrestins. These studies propose a structural basis of β-arrestin recruitment to PTHR that will be experimentally validated in future structural studies.
Previous studies have proposed a paradigm whereby the duration of the cAMP response serves as the major determinant for PTHR-mediated regulation of serum Ca2+ and bone remodeling, and this model has served as the premise for the development of therapeutics targeting the receptor (9, 35, 36). Our findings reveal that a shift in the spatial bias of cAMP signaling from endosomes to the cell surface markedly reduces PTH-induced effects on serum Ca2+ increases and vitamin D activation without alterating serum Pi reduction. Our study also raises the prospect that the PTHR-generated endosomal cAMP pool ensures increase in serum Vitamin D by inducing the production of the rate-limiting hydroxylase catalyzing the formation of active Vitamin D, which subsequently enhances the increase in serum Ca2+. We propose that the PTHR-generated plasma membrane cAMP pool triggers limited nuclear PKA activity and Vitamin D increases but might contribute to the inhibition of phosphate transport (Fig. 6). Understanding the mechanism by which endosome-generated cAMP leads to nuclear PKA activity, whereas plasma membrane–generated cAMP does not, will require further studies involving, for example, the importance of cAMP diffusion in cells and the role of microdomains in local buffering of cAMP production (37). In addition to the PTHR, several other GPCRs have been reported to engage in cAMP signaling from intracellular compartments, including the vasopressin type 2 receptor (V2R) (38), glucagon-like peptide 1 receptor (GLP-1R) (39), the β1-adrenergic receptor (β1AR) (40), among others, each of which hold potential biological and clinical significance: the V2R is a major regulator of water homeostasis that acts in the kidneys and has been implicated in nephrogenic diabetes insipidus and heart failure (41); the GLP-1R is well-known to modulate insulin secretion from the pancreas and currently serves as a therapeutic target for the treatment of type 2 diabetes (42); and the β1AR contributes to a wide variety of physiological processes, including cardiac function and blood pressure, and currently represents a main therapeutic target for the treatment of hypertension and heart failure (43). This study thus presents implications for other GPCRs that may aid a better understanding of human physiology and facilitate rational drug design in the future.
Fig. 6. Proposed model for location bias in cAMP and PTHR pharmacology.
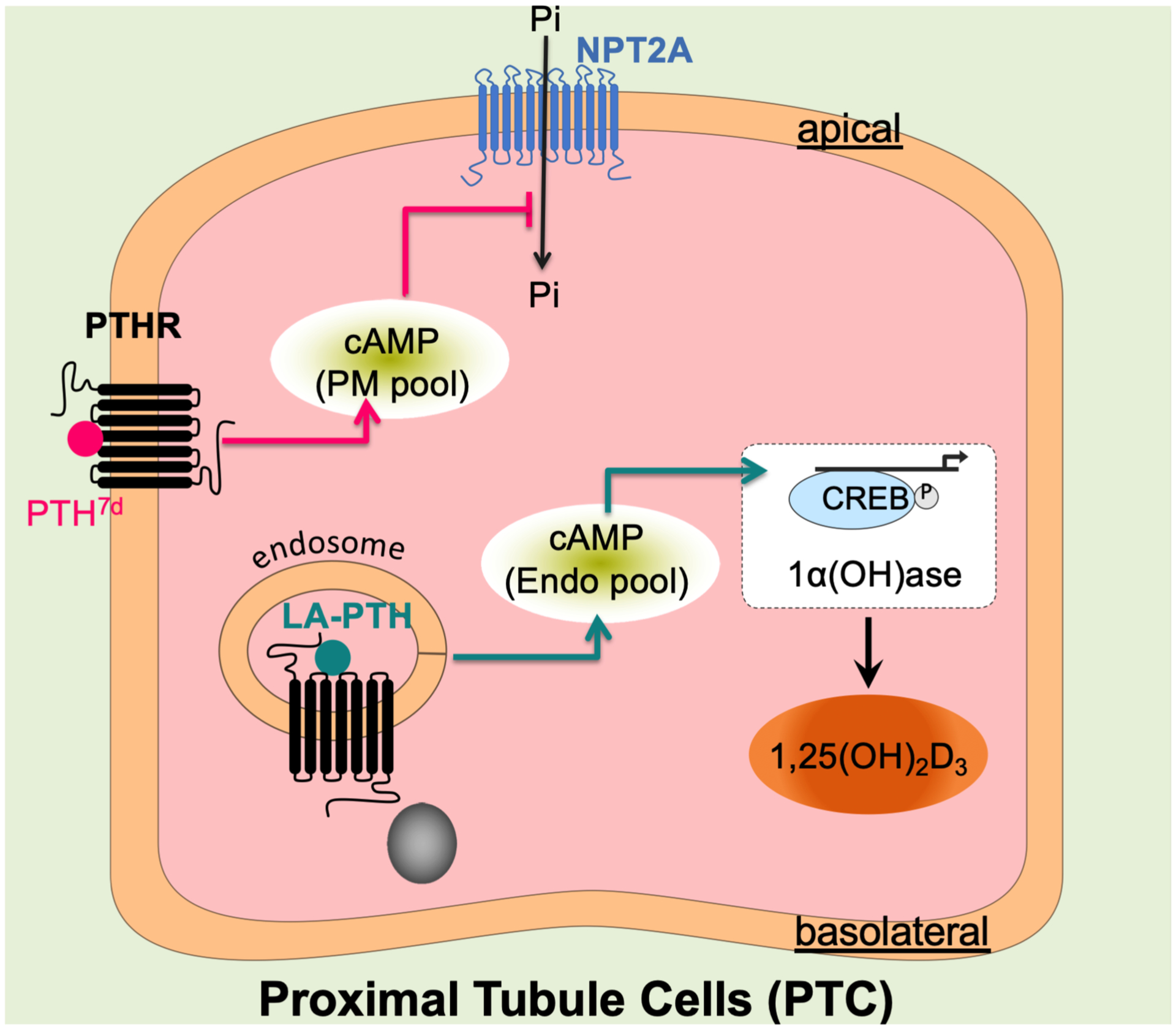
Upon basolateral stimulation of proximal tubule kidney cells, the PTHR-generated endosomal cAMP pool ensures increases in serum Vitamin D by inducing the expression of 1-α(OH)ase, the rate-limiting hydroxylase catalyzing the formation of active Vitamin D, which subsequently stimulates an increase in serum Ca2+. The PTHR-generated plasma membrane cAMP pool might contribute to the inhibition of phosphate import, presumably by increasing endocytosis of the Na+-dependent phosphate cotransporter 2A (NPT2A).
MATERIALS AND METHODS
Cell culture and transfection
Cell culture reagents were obtained from Corning (CellGro). Human embryonic kidney (HEK) cells (HEK293; ATCC, Georgetown, DC) stably expressing the recombinant human PTHR were grown in selection medium (DMEM, 5% FBS, 5% penicillin/streptomycin, 500 μg/ml neomycin) at 37 °C in a humidified atmosphere containing 5% CO2. For transient expression, cells were seeded on glass coverslips coated with poly-D-lysine in six-well plates and cultured for 24 hours prior to transfection with the appropriate cDNAs using Fugene-6 (Promega) or Lipofectamine 3000 (Life Technologies) for 48–72 h before experiments. We optimized expression conditions to ensure the expression of fluorescently labeled proteins was similar in examined cells by performing experiments in cells displaying comparable fluorescence levels. Madin-Darby Canine Kidney (MDCK) cells stably expressing recombinant HA-tagged human PTHR were grown in DMEM/F12 (1:1) supplemented with 5% FBS and 1% penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. For polarization of epithelial monolayers, 9.5×104 cells were plated on 24 mm Transwells (Corning) with 0.4 μm pore size. Cells were used 5 days later, when they reached a transepithelial electric resistance (TER) larger than 500 Ohms*cm2. TER was measured using an EndOhm-24 SNAP chamber with EVOM resistance meter (World Precision Instruments, Inc) following the manufacturer’s instructions.
Chemicals
Forskolin (EMD-Millipore, 344270).
Peptide Synthesis, Purification, and Quantification
PTH1–34, PTH1–34TMR, LA-PTH were synthesized and characterized as previously described (35, 44). (D)-Leu(7)-PTH1–34-NH2 was synthesized by microwave-assisted reactions on NovaPEG Rink amide resin. For each coupling step, the resin was treated with 4 equivalents of protected amino acid, 4 equivalents of HATU, and 8 equivalents of N,N-diisopropylethylamine (DIEA) in DMF. Fmoc deprotection was carried out by using 20% (v/v) piperidine with 0.1 M HOBT in DMF. Upon completion of the synthesis, the peptide was cleaved from the resin using a solution of 94% trifluoroacetic acid (TFA), 2.5 % H2O, 2.5% 1,2-ethanedithiol (EDT), and 1% triisopropylsilane (TIS). After the cleavage, the crude peptide was precipitated by addition of cold diethyl ether. The precipitated material was purified by preparative HPLC. Peptide purity was assessed by UPLC (BEH C18 stationary phase, 2.1 mm × 100 mm, the solvent gradient was 10–60% acetonitrile over 5 min), and peptide mass was checked by MALDI-TOF MS. The peptide reported here was >95% pure as determined by UPLC. Since (D)-Leu(7)-PTH1–34-NH2 contains one tryptophan in its sequence, the peptide concentration was determined by UV spectroscopy using the absorbance at 280 nm (the molecular extinction coefficient of tryptophan at 280 nm is 5690 M−1cm−1). After concentration determination, the peptide was aliquoted, lyophilized to dry powder, and stored at −20 °C. (D)-Ile(5)-PTH1–34 (PTH5d), (D)-Gln(6)-PTH(1–34) (PTH6d), (D)-His(9)-PTH1–34 (PTH9d), and (D)-Asn(10)-PTH1–34 (PTH10d) were synthesized by LifeTein (catalog numbers 829095, 792338, 756617, 829096, respectively). Purity of peptides reported by manufacturer was <98%. Lyophilized peptides were resuspended in 10 mM acetic acid before use.
Plasmids
βarr-2YFP and PTHRSEP were previously described by the Vilardaga lab (5, 21). βarr1Rluc8 was provided by Dr. Zachary Freyberg. Gβ1BiFc (Cer(1–158)-Gβ1) and Gγ2BiFc (CFP(159–238)-Gγ2) were a gift from Dr. Catherine Berlot and can be found in Addgene (55707).
Time-course measurements of cAMP and β-arrestin recruitment by FRET
Cells were transiently transfected with the FRET-based biosensor, Epac1CFP/YFP, for measuring cAMP or PTHRCFP and βarr-2YFP for measuring arrestin recruitment. Cells plated on poly-D-lysine coated glass coverslips were mounted in Attofluor cell chambers (Life Technologies), maintained in Hepes buffer containing 150 mM NaCl, 20 mM Hepes, 2.5 mM KCl and 1 mM CaCl2, 0.1% BSA, pH 7.4, and transferred on an inverted Nikon Ti-E equipped with an oil immersion 40X N.A 1.30 Plan Apo objective and a moving stage (Nikon Corporation). Recordings were performed at 37oC using Nikon A1 confocal unit, through a 60× N.A. = 1.45 objective (Nikon). Fluorescent proteins containing CFP or YFP were excited with 440 and 514 nm lasers (Melles Griot), respectively. Fluorescence data were extracted from single cell using Nikon Element Software (Nikon Corporation). The FRET ratio for single cells was calculated and corrected as previously described (45). Individual cells were perfused with buffer or with the ligand for the time indicated by the horizontal bar.
Cyclic AMP recordings reported in Figure S4 were performed at room temperature on an inverted Nikon Ti-E epifluorescent microscope in order to allow visualization of RFP-tagged DynaminK44A. CFP and YFP were excited using a mercury lamp. Fluorescence emissions were filtered using a 480 ± 20 nm (CFP) and 535 ± 15 nm (YFP) filter set and collected simultaneously with a LUCAS EMCCD camera (Andor Technology) using a DualView 2 (Photometrics) with a beam splitter dichroic long pass of 505 nm.
cAMP production in polarized HA-PTHR expressing MDCK cells was measured as follows: cells were seeded onto inverted 12 mm Transwells (Corning) with a 0.4 μm pore, as previously described (46). After 5 hours incubation, Transwells were re-inverted with the basolateral membrane facing the bottom of a 12-well plate. Cells were cultured for 5 days until polarization was achieved and measured using transepithelial electric resistance (TER). Two days before imaging, cells were transduced with BacMam containing Green down cADDis sensor (Montana Molecular) following manufacturer’s protocol. Transwells were then transferred to an Attofluor cell chamber (Life Technologies) with a glass coverslip at the bottom. Polarized cells, with the basolateral membrane facing the glass coverslip were imaged on an inverted Nikon Ti-E microscope (confocal Nikon A1) equipped with a Z-driven piezo motor and heated stage. Confocal imaging was performed as described above. Green down cADDis sensor was excited with a 488 nm laser. Cells were stimulated with 100 nM ligand, and images were collected every 30 seconds for 40 minutes. For antagonist assays, the cell-impermeant PTHR antagonist D-Trp12, Tyr34-bPTH7–34 was added 15 min after ligand stimulation at a final concentration of 1 μM. cAMP signals were calculated as (ΔF/F0), corrected for bleaching effect, normalized to the forskolin response, and depicted as the inverse signal because fluorescence intensity of Green cADDis sensor decreases when the cAMP level increases. Data acquisition and image analysis were done using NIS Element Software (Nikon Corporation).
cAMP accumulation assay
HEK293 cells stably expressing the Glosensor cAMP reporter and human PTHR were cultured in DMEM supplemented with 10% FBS. Cells were seeded into 96-well corning plates and used for cAMP assays after forming a confluent monolayer. Upon the removal of culture medium, the intact cells in 96-well plates were incubated in CO2-independent medium containing D-luciferin (0.5 mM) for 20 minutes at room temperature. After this period, cells in each well were treated with peptides at various concentrations, and luminescence resulting from cAMP production was measured for 30 minutes on a BioTek Synergy 2 plate reader. The peak luminescence signaling, which usually appeared 14–20 minutes after peptide addition, were used to generate concentration-response curves. The concentration-response curves were fit to the data by using the sigmoidal dose-response equation in Prism 6.0. Reported EC50 and Emax values represent means ± SEM from N ≥ 3 independent experiments.
Washout assay
For the washout experiments, cells preloaded with D-luciferin were treated with medium (vehicle) or agonists at the concentration of 1 nM for 14 minutes. After this period, the medium in each well was removed and the cells were rinsed twice with CO2-independent medium to remove unbound ligand. After the addition of D-luciferin-containing fresh medium to each well, the luminescence was recorded for an additional 150 minutes. For competitive antagonist-modified washout assays, the experimental protocol was as described above except that 0.5 μM of the competitive antagonist [D-Trp12,Tyr34]bPTH7–34-NH2 (20) was introduced during the ligand-off phase.
β-arrestin recruitment by BRET
CHO-FlpIn cells transfected with PTHRRluc8 and βarr-1venus or βarr-2venus were cultured in DMEM supplemented with 10% FBS and 0.7 mg/mL hygromycin-B. Cells were seeded into 96-well plates at a density of 4×104 cells/well and cultured for 24 hours before the BRET assay. Cells were rinsed twice with DPBS to remove traces of phenol red and incubated in fresh DPBS containing coelenterazine-h (at a final concentration of 5 μM) at 37 °C for 10 minutes. After this period, cells in each well were treated with peptides at various concentrations and BRET readings were collected for 25 minutes on a BioTek Synergy 2 plate reader with 460/40 and 528/20 nm emission filters. The BRET signal was calculated by subtracting the ratio of 528/20 emission over 460/40 emission for vehicle-treated cells from the same ratio for ligand-treated cells. The peak BRET signals (which usually appeared within the initial 5 minutes after the peptide addition) were used to generate concentration-response curves. The concentration-response curves were fit to the data by using sigmoidal dose-response equation in Prism 6.0. Reported EC50 and Emax values represent means ± SEM from N ≥ 3 independent experiments.
Co-immunoprecipitation
HEK293 cells stably expressing hemagglutinin (HA)-tagged PTHR and cultured on a 10-cm dish were stimulated with PTHWT, LA-PTH, or PTH7d at 100 nM for 5 minutes. Cells were then washed with ice-cold PBS prior to cross-linking for 2 hours with DSP (Covachem, 13301) in PBS at 4 °C. The reaction was stopped by addition of 10 mM Tris-HCl for 10 minutes and cell lysates were prepared using lysis buffer (1% Triton X-100, 50 mM Tris-HCl pH 7.4, 140 mM NaCl, 0.5 mM EDTA) containing protease and phosphatase inhibitors (Roche, 11873580001). Protein concentration was determined using BCA protein assay kit (ThermoFisher, 23225), and lysates were incubated with anti-HA agarose antibody beads (Sigma-Aldrich; #A2095 clone HA-7) overnight at 4 °C. Elution was done using LDS loading buffer (Life Technologies) and samples loaded on 10% SDS-PAGE and transferred to nitrocellulose membrane. We used antibodies against HA (Covance, clone 16B12), and βarr-1/2 (Cell Signaling, 4674, clone D24H9). Immunoreactive bands were visualized with Luminata Forte (EMD Millipore) and autoradiography film.
Competition binding at equilibrium in intact live cells
HEK293 cells stably expressing HA-PTHR and seeded in 96-well plates. After 24 hours, cells were incubated in HEPES buffer (137 mM NaCl/5 mM KCl/1 mM MgCl2/20 mM HEPES/0.1% BSA pH 7.4) for 1 h at 4 °C, followed by 2 h incubation at 4 °C in the presence of increasing concentrations of unlabeled PTH or PTH7d with a constant PTHTMR concentration (31.6 nM). Cells were washed twice with the same buffer, and fluorescence intensities were recorded at 580 ± 20 nm using an excitation wavelength of 525 ± 20 nm on a Tecan Spark 20M multimode microplate reader. Nonspecific binding was determined using 1 μM of unlabeled ligand. Data were subsequently analyzed using Graphpad Prism 8.0 (GraphPad Software Inc., La Jolla, CA).
Radioligand binding to the R0 and RG PTHR conformations
Ligand binding to the PTHR in the G protein-coupled and G protein-uncoupled conformation, RG and R0, respectively, was assessed by competition methods using membranes prepared from HEK-293-Gs-null cells (clone CL2) (47) stably transfected with the PTHR. For membranes used in RG reactions, the cells were further transfected transiently with a high-affinity Gs negative dominant (Gs-ND) mutant (48). Binding to R0 was assessed using 125I-PTH1–34 as tracer radioligand and binding to RG was assessed using 125I-M-PTH1–15 as radioligand (48). Binding reactions (100 μl) were assembled at room temperature in membrane binding buffer (20 mM HEPES, pH 7.4, 0.1 M NaCl, 3 mM MgSO4, 20% glycerol, 3 mg/ml bovine serum albumin, protease inhibitors (Sigma-Aldrich Inc., P8849) ~20,000 cpm of radioligand, varying amounts of unlabeled ligand, and were initiated by addition of cell membranes to a membrane protein concentration of 50 ng/μl. Reactions were incubated for 90 minutes at room temperature in 96-well vacuum filtration plates (0.2 μM HPB filter, Millipore Corp.), and terminated by rapid vacuum filtration followed by rinsing with 200 μl buffer, after which the filters were removed and counted for gamma irradiation. Values of non-specific binding, maximum binding, and the 50% inhibition constant (IC50), were determined by curve-fitting the data using Graphpad Prism 8.0 (GraphPad Software Inc., La Jolla, CA) to a sigmoidal dose-response model with variable slope.
Receptor internalization and recycling
Live-imaging trafficking of PTHRSEP was done as described (21) using a Nikon A1 confocal microscope. Briefly, HEK293 cells stably expressing a pH-sensitive GFP variant, superecliptic pHluorin, inserted in the N-terminal domain of the human PTHRSEP were seeded on glass coverslips coated with poly-D-lysine (Sigma, P6407) for 24 hours. Experiments were carried out at 37°C in HEPES buffer used for cAMP experiments. Cells were stimulated by 100 nM ligand for 1 minute then washed out to allow recycling. Images were acquired every 30 s.
Anisotropic network model (ANM) analysis
ANM data and methods were originally presented in Clark, et al. (15). In brief, we generated a model of PTH-bound receptor using the 3.0 Å cryo-EM structure of active PTHR–Gs complex (PDB 6NBF) (14). This model was inserted into a membrane model consisting of an SC lattice using the Orientations of Proteins in Membranes (OPM) database (49). In the ANM, the protein system (receptor and ligand) is represented as a network where residues (α-carbons) serve as the nodes (489 nodes). The membrane accounted for 3,953 additional nodes. The overall potential is represented as the sum of harmonic potentials between pairs of nodes within an interaction range (Cα-Cα distance < 15 Å). Normal mode 14 was used for our analysis, and cross-correlations between PTHR Thr392 and PTH residues in this mode are shown in figure S1. All computations were performed using the ProDy API (50, 51).
MD system preparation
The 3.0 Å cryo-EM structure of LA-PTH–PTHR–Gs–Nb35 complex (PDB 6NBF) (14) was used to generate initial PTHR models. I-TASSER was then used to model flexible loops absent in the cryo-EM structure: ECD residues 56–104, ECL1 residues 247–275, and ICL3 residues 394–398 (52–54). In PyMOL (Version 2.1, Schrödinger), structures of Gs, Nb35, palmitic acid, and cholesterol were removed. LA-PTH residues were mutated to the corresponding PTHWT residues, as necessary, using the PyMOL Mutagenesis Wizard. The chirality of Leu7 in the PTHWT model was changed in PyMOL to generate the PTH7d model. Each initial model was oriented in a model membrane using the Orientations of Proteins in Membranes (OPM) PPM Server (49). Using oriented models, inputs for Nanoscale Molecular Dynamics (NAMD) were generated using CHARMM-GUI Membrane Builder (55, 56). Disulfide bonds Cys48-Cys117, Cys108-Cys148, Cys131-Cys170, and Cys281-Cys351 were specified to ensure correct formation. A heterogeneous lipid bilayer consisting of 75% POPC and 25% cholesterol was assembled around the receptor model using the Replacement Method. The system was solvated in a box of TIP3P waters, and ions were added to a concentration of 150 mM NaCl using Monte Carlo sampling.
MD simulations
All-atom simulations were performed in triplicate for PTHWT–PTHR and PTH7d–PTHR simulations using NAMD with the CHARMM36m force field (57, 58). Prior to production simulations, 10,000 steps of conjugate gradient energy minimization were performed followed by 0.675 ns equilibration in which restraints were applied and then slowly released over six steps following the protocol established by the CHARMM-GUI group (58). Next, 200 ns production simulation with 2 fs timestep was performed. Non-bonded interactions were cut off at 12.0 Å, and van der Waals force switching was applied between 10.0 Å and 12.0 Å. Langevin dynamics and Langevin piston were used to maintain temperature at 303.15 K and pressure at 1 atm.
MD trajectory analysis
MD trajectories were analyzed in Visual Molecular Dynamics (VMD) and PyMOL (59). PTHR snapshots were aligned using receptor TM helices (PTHRTMD without ECLs or ICLs: residues 180–211, 218–246, 280–311, 317–343, 359–392, 399–425, 435–460). PTH Glu4 hydrogen bond analysis was performed using HBonds Plugin in VMD. Number of hydrogen bonds throughout the trajectories was plotted in GraphPad Prism.
Photo-crosslinking in cells
We previously reported a detailed description of this experiment in Clark et al., (15). In brief, we used a β-arrestin mutant with the photo-activatable amino acid p-Benzoyl-L-phenylalanine (Bpa) incorporated into position 75 of a HA-tagged β-arrestin-1. Photo-crosslinking experiments are performed in 10 cm dishes of HEK293 cells expressing a PTHR and the βarr1 mutant. Twenty-four hours after transfection, cells were stimulated with 200 nM PTHWT or PTH7d for 10 min, washed twice and irradiated with UV light (365 nm) for 15 min in cold PBS using UVP crosslinker (Analytik Jena) with 2000× 100 microjoules per cm2 energy exposure at a 2.5 cm distance. For co-immunoprecipitation experiments, 150 μL of anti-HA agarose was added to the supernatant and incubated with gentle end-over-end mixing overnight at 4 °C. Using centrifugation at 1000×g for 5 min at 4 °C and aspiration of the supernatant, the beads were washed with RIPA buffer six times. The protein complex was eluted from the beads by incubating with 3 mg/mL HA peptide solution at for 30 min at 25 °C. After centrifugation at 1000×g for 5 min at 4 °C, the supernatant was collected for subsequent in-gel digestion.
1-α(OH)ase protein expression
MDCK cells stably expressing HA-PTHR were polarized on 24 mm Transwells (Corning) with 0.4 μm pore size. Polarized cells were basolaterally stimulated with 30 nM PTHWT, LA-PTH or 30 nM PTH7D diluted in growth media, with or without FBS, for 8 hours or left untreated. After stimulation, cells were lysed in lysis buffer containing 50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 10% glycerol, and 1% IGEPAL®CA-630 (Sigma-Aldrich), supplemented with protease and phosphatase inhibitors, as described before (60). Briefly, Transwells were washed in cold PBS and then membranes were cut out of their plastic supports and put in a microcentrifuge tube with 300 μL lysis buffer. Membranes were vortexed and incubated in rotation at 4 °C for 30 minutes. Lysates were then centrifuged at maximum speed for 10 min in a benchtop refrigerated centrifuge. Supernatants were transferred to a clean tube. Protein concentration was measured using BCA protein assay kit (ThermoFisher, 23225). For Western blot analysis, 10 μg of lysates were loaded per well of a 10% SDS-PAGE. Gels were transferred to a nitrocellulose membrane and blocked in TBS-T with 5% milk. For detection of 1-α(OH)ase, membranes were blotted with anti-CYP27B1 polyclonal antibody (Novus Biologicals, NBP2–29942) at 1:1000 dilution. Anti-GAPDH monoclonal antibody (Santa Cruz Biotechnologies, sc-32233) at 1:1000 dilution was used as loading control. HRP-conjugated antibodies (Dako Denmark) were used as secondaries antibodies. Immunoreactive bands were visualized with immobilon forte western HRP substrate (EMD Millipore, WBLUF0100). Band intensity quantification was performed using ImageJ software (NIH).
Mouse studies
To test the impact of PTH7d, LAPTH, and PTHTWT (PTH1–34) (Bachem, Torrance, CA, Cat# H-5460) on serum and skeletal parameters, 3-month-old male C57BL/6J (C57/B6) mice (Jackson Laboratory, Stock No: 000664) were ear-tagged for identification, randomly assigned to groups, and injected daily with the drugs (40 μg/kg bogy weight/injection) for 4 weeks after the mice were acclimated with housing environment for 2 weeks. In this study, blood was sampled through the retroorbital route under isoflurane anesthesia after 2 hrs of the last drug injection. The mice were allowed to recover before another blood collection by cardiac puncture and bone collection from the mice euthanized by isoflurane overdose 24 hrs after the last drug injection. Serum Ca2+ and phosphate ion (Pi) levels were assessed by an ACE Axcel bioanalyzer (Alfa Wassermann, West Coldwell, NJ) and skeletal parameters of distal femurs were assessed by a SCANCO μCT 50 scanner and analytic software (Scanco USA, Inc., Wayne, PA, USA) as detailed previously (61). In a separate time-course study, different groups of mice were injected with a single dose of PTHWT, PTH7d, or LAPTH for different time points before blood collections by cardiac puncture. Their serum 1,25D levels were assessed by an ELISA kit (AC-62F1; Immunodiagnostic systems, Tyne & Wear, United Kingdom) according to the manufacturer instructions. All mice were kept in a climate-controlled room (22°C; 45–54% relative humidity) with a 12-hour light/12-hour dark cycle. Water and standard chow (1.3% calcium and 1.03% phosphate) were given ad libitum. All animal experiments (Protocol #18–013) were approved and performed according to guidelines of the Institutional Animal Care and Use Committee at the San Francisco Department of Veterans Affairs Medical Center.
Pharmacokinetic analysis
Bioactive LA-PTH or PTH7d content of blood plasma was assessed by applying 5 μL of plasma (supplemented with protease inhibitors) to the HEK293-derived cell line GP2.3 in which human PTHR and the luciferase-derived Glosensor cAMP reporter were stably expressed. Cells were seeded into 96-well white plates and assayed 24 hours later in CO2-independent culture medium (Life Technologies, Carlsbad, CA, USA) containing 0.1% BSA. Cells were pre-incubated with D-luciferin (0.5 mM) for 2 hours, then blood plasma samples were added, and cAMP-dependent luminescence was measured at 2-minute intervals. For each well, the maximum luminescence observed, which typically occurred 10–20 minutes after sample addition, was used to establish relative bioactive peptide.
Stable Isotope Labeling with Amino acids in Cell culture (SILAC)
We used the HEK-293 cell line stably expressing the human HA-tagged PTHR previously generated in our lab (62). Three pools of HA-PTHR/HEK-293 cells were maintained side-by-side in Stable Isotope Labeling with Amino acids in Cell (SILAC) “Light”, “Medium” and “Heavy” media (fig. S5A). The SILAC media were prepared from custom-ordered DMEM powder without arginine, lysine, and leucine (Gibco, formula # 03–5080EB) (Gibco/Invitrogen). [2H4]-L-lysine (50 mg/liter) and [13C6]-L-arginine (25 mg/liter) (Cambridge Isotope Laboratories) were added to the “Medium” culture medium; and [13C6, 15N2]-L-lysine (50 mg/liter) and [13C6,15N4]-L-arginine (25 mg/liter) (Cambridge Isotope Laboratories) were added to the “Heavy” culture medium, whereas equal concentrations of conventional (i.e., natural) lysine and arginine were added to the “Light” culture medium. All “Light”, “Medium” and “Heavy” media were supplemented with L-leucine (104 mg/liter), L-proline (10 mg/liter), 10% dialyzed FBS (Hyclone) (Thermo Scientific), 1% penicillin/streptomycin, and G418 (150 mg/ml). The SILAC cells were cultured for more than six doublings until the isotope incorporation rates in “Medium” and “Heavy” cells were higher than 95%. The SILAC cells were then expanded. When the cells reached ~80% confluence, they were serum-starved for 4 hours. To map the phosphorylation sites on the PTHR induced by PTHWTor PTH7d, the “Light” labeled cells cultured in “Light” medium were treated with 30 nM PTHWT for 5 min before harvesting; the “Medium” labeled cells cultured in “Medium” medium were treated with 30 nM for 5 min before harvesting; the “Heavy” labeled cells cultured in “Heavy” medium served as control without any treatment. Equal numbers of “Light”, “Medium” and “Heavy” labeled cells (generally six 150-mm culture dishes for each) were mixed, flash-frozen in liquid nitrogen, and stored at −80 °C. The SILAC experiments were repeated three times.
HA-PTHR isolation, digestion and peptide desalting
The HA-PTHRs were isolated from SILAC cells over-expressing HA-PTHR using Pierce™ Anti-HA Agarose beads (Catalog number: 26181, Thermo Fisher Scientific). Briefly, crude membrane fractions were prepared from the equally mixed (“Light”:“Medium:“Heavy”=1:1:1) SILAC cells as previously described (27, 63). The HA-PTHRs were then extracted from crude membrane preparations with 1× buffer [20 mM tris-HCl (pH 8.0), 100 mM NaCl, and 2 mM EDTA] containing 1% DDM (n-dodecyl b-D-maltoside), protease inhibitors and phosphatase inhibitors. HA-PTHRs were isolated from the extraction solution by incubating with 200 μL Pierce™ Anti-HA Agarose beads with rotation at 4 °C for 4 hours. The HA-PTHR bound anti-HA Agarose beads were washed with 1× buffer containing 0.1% DDM five times and then eluted with 100 μL 2X SDS-PAGE buffer (containing 10 mM dithiothreitol (DTT)) by incubating at 37 °C for 1 hour. The receptor proteins were alkylated with 30 mM iodoacetamide (IAA) in the dark at room temperature for 30 min. The samples were then subjected to SDS-PAGE separation. The protein bands corresponding to HA-PTHRs were excised from SDS-PAGE gel for in-gel protein digestion.
Tryptic digestion of HA-PTHRs was performed as previously described (64, 65). In brief, the excised gel bands were chopped into small pieces and destained by distaining solution (50 mM ammonium bicarbonate in 50% acetonitrile). Sequence grade trypsin (10 ng/μL, modified, Promega) in 50 mM NH4HCO3 (pH 8.0) was then added to the tubes to cover the destained gel pieces. The tryptic digestion reactions were incubated at 37 °C for overnight. An equal volume of 100% acetonitrile (CH3CN) was added to the digested gel samples for peptide extraction and repeated for three times. The extracted peptides were pooled into a pre-washed protein LoBind tube (Catalog number: 13698793, Fisher) and then dried under vacuum on a SpeedVac evaporator. The peptide samples were desalted with handmade Stage Tips as previously described (64). The desalted peptides were lyophilized with a SpeedVac evaporator, reconstituted in 0.1% trifluoroacetic acid, 2% acetonitrile, and 25 mM citrate, and subjected to LC-MS/MS analysis.
MS and data analyses
LC-MS/MS analyses were performed on a Thermo Scientific LTQ Orbitrap Velos mass spectrometer (Thermo Fisher) with a Finnigan Nanospray II electrospray ionization source. The peptide samples were loaded onto a nanoViper Compatible PicoChip Column (Catalog number: 1PCH7515–105H354-NV, New Objective) and separated with a Waters nanoACQUITY UPLC System. Instrument control and primary data processing were done with the Xcalibur software package. The LTQ Orbitrap Velos was operated in data-dependent mode using a TOP10 strategy (66). MS/MS spectra were searched with the SEQUEST algorithm against a composite database containing the human HA-PTHR sequence or HA-PTHR with its interacting proteins, as well as their reverse sequences. Search parameters allowed for three missed tryptic cleavages, a mass tolerance of ±80 parts per million (ppm), a static modification of 57.02146 daltons (carboxyamidomethylation) on cysteine, and up to eight total dynamic modifications: 79.96633 daltons (phosphorylation) on serine, threonine, and tyrosine; 15.99491 daltons (oxidation) on methionine; 6.02012 daltons or 10.00827 daltons on arginine; and 4.00709 daltons or 8.01420 daltons on lysine. Search results were filtered to include <1% matches to reverse sequences by restricting the mass tolerance window, and setting thresholds for Xcorr and dCn′ (defined as the normalized difference between Xcorr values of the top-ranked candidate peptide and the next candidate with a different amino acid sequence). Matches for phosphopeptides were validated manually with special consideration of intense fragment ions formed through cleavage N-terminal to proline residues and neutral losses of phosphoric acid. Peptide quantification was performed with the Vista program (67) as well as by manual calculation with Qual Browser (version 3.0.63). In brief, the theoretical mass of “Light”, “Medium” and “Heavy” variants of each peptide was calculated and used to identify ion peaks in the high mass accuracy precursor scans for each. The intensity of the peaks was used to construct ion chromatograms. For each isotopic variant, the peak height and background-subtracted area under the curve were used to calculate the “Light”-to-“Heavy”(PTHWT: nonstimulation) and “Medium”-to-“Heavy” (PTH7d: nonstimulation) abundance ratios.
Statistics
Data were processed using GraphPad Prism 9.2 and expressed as mean ± SEM or SD. Paired data were statistically compared by using Student’s t-test assuming unequal variances for the two stets. Multiple comparisons were analyzed using one-way or two-way ANOVA with Turkey post-hoc test.
Supplementary Material
Acknowledgments:
We are grateful to Asuka Inoue (U. Tohoku) for advice in using Gs-null cells.
Funding:
Research reported in this publication was supported by grants from the US National Institutes of Health (NIH) (R01-DK111427 and R01-DK116780 to J.-P.V., R01-DK122259 to J.-P.V. and W.C., R01-AR067291 to W.C., DK11794 and AR066261 to T.J.G., R01-GM-056414 to S.H.G., P41-GM103712 and R01-GM139297 to I. B., and F31-AR074843 to L.J.C.), and the Department of Veterans Affairs (I0-BX003453-03 and IK6BX004835 to W.C.).
Footnotes
Competing interests: S.L. and S.H.G. are co-inventors on a patent application that includes PTH7d. S.H.G. is a cofounder of and has a secondary affiliation with Longevity Biotech, Inc. The other authors declare that they have no competing interests.
Data and materials availability:
The mass spectrometry data have been deposited with the ProteomeXchange Consortium with the dataset identifier (MSV000087510/PXD026256). All other data needed to evaluate the conclusions in the paper are present in the paper or in the Supplementary Materials. All materials, data, animal models, and associated protocols will be made available upon reasonable request.
REFERENCES AND NOTES
- 1.Fredriksson R, Schioth HB, The repertoire of G-protein-coupled receptors in fully sequenced genomes. Molecular pharmacology 67, 1414–1425 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Bockaert J, Pin JP, Molecular tinkering of G protein-coupled receptors: an evolutionary success. The EMBO journal 18, 1723–1729 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galandrin S, Oligny-Longpre G, Bouvier M, The evasive nature of drug efficacy: implications for drug discovery. Trends in pharmacological sciences 28, 423–430 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Maudsley S, Martin B, Luttrell LM, The origins of diversity and specificity in g protein-coupled receptor signaling. The Journal of pharmacology and experimental therapeutics 314, 485–494 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrandon S et al. , Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 5, 734–742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calebiro D et al. , Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 7, e1000172 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutkeviciute I, Vilardaga JP, Structural insights into emergent signaling modes of G protein-coupled receptors. J Biol Chem 295, 11626–11642 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dean T, Vilardaga JP, Potts JT Jr., Gardella TJ, Altered selectivity of parathyroid hormone (PTH) and PTH-related protein (PTHrP) for distinct conformations of the PTH/PTHrP receptor. Molecular endocrinology 22, 156–166 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okazaki M et al. , Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation. Proceedings of the National Academy of Sciences of the United States of America 105, 16525–16530 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wehbi VL et al. , Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gbetagamma complex. Proceedings of the National Academy of Sciences of the United States of America 110, 1530–1535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feinstein TN et al. , Retromer terminates the generation of cAMP by internalized PTH receptors. Nature chemical biology 7, 278–284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutkeviciute I, Clark LJ, White AD, Gardella TJ, Vilardaga JP, PTH/PTHrP Receptor Signaling, Allostery, and Structures. Trends Endocrinol Metab 30, 860–874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horwitz MJ et al. , Continuous PTH and PTHrP infusion causes suppression of bone formation and discordant effects on 1,25(OH)2 vitamin D. J Bone Miner Res 20, 1792–1803 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Zhao LH et al. , Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark LJ et al. , Allosteric interactions in the parathyroid hormone GPCR-arrestin complex formation. Nat Chem Biol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bahar I, Lezon TR, Bakan A, Shrivastava IH, Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem Rev 110, 1463–1497 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardella TJ, Vilardaga JP, International Union of Basic and Clinical Pharmacology. XCIII. The parathyroid hormone receptors--family B G protein-coupled receptors. Pharmacol Rev 67, 310–337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vilardaga JP, Jean-Alphonse FG, Gardella TJ, Endosomal generation of cAMP in GPCR signaling. Nature chemical biology 10, 700–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardella TJ, Juppner H, Molecular properties of the PTH/PTHrP receptor. Trends Endocrinol Metab 12, 210–217 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Goldman ME et al. , A new highly potent parathyroid hormone antagonist: [D-Trp12,Tyr34]bPTH-(7–34)NH2. Endocrinology 123, 2597–2599 (1988). [DOI] [PubMed] [Google Scholar]
- 21.McGarvey JC et al. , Actin-Sorting Nexin 27 (SNX27)-Retromer Complex Mediates Rapid Parathyroid Hormone Receptor Recycling. The Journal of biological chemistry 291, 10986–11002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jean-Alphonse FG et al. , beta2-adrenergic receptor control of endosomal PTH receptor signaling via Gbetagamma. Nature chemical biology 13, 259–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sample V et al. , Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nature chemical biology 8, 375–382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer D et al. , Distinct G protein-coupled receptor phosphorylation motifs modulate arrestin affinity and activation and global conformation. Nature communications 10, 1261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liggett SB, Phosphorylation Barcoding as a Mechanism of Directing GPCR Signaling. Science signaling 4, pe36 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Staus DP et al. , Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in beta-arrestin coupling. Proc Natl Acad Sci U S A 115, 3834–3839 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobles KN et al. , Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Science signaling 4, ra51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vilardaga JP et al. , Internalization determinants of the parathyroid hormone receptor differentially regulate beta-arrestin/receptor association. The Journal of biological chemistry 277, 8121–8129 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Vilardaga JP, Gardella TJ, Wehbi VL, Feinstein TN, Non-canonical signaling of the PTH receptor. Trends Pharmacol Sci 33, 423–431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vilardaga JP, Bunemann M, Krasel C, Castro M, Lohse MJ, Measurement of the millisecond activation switch of G protein-coupled receptors in living cells. Nat Biotechnol 21, 807–812 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM, Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proceedings of the National Academy of Sciences of the United States of America 110, 5211–5216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang B et al. , Ezrin-anchored protein kinase A coordinates phosphorylation-dependent disassembly of a NHERF1 ternary complex to regulate hormone-sensitive phosphate transport. The Journal of biological chemistry 287, 24148–24163 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenza HL et al. , Parathyroid hormone activation of the 25-hydroxyvitamin D3–1alpha-hydroxylase gene promoter. Proc Natl Acad Sci U S A 95, 1387–1391 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheerer P, Sommer ME, Structural mechanism of arrestin activation. Curr Opin Struct Biol 45, 160–169 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Shimizu M et al. , Pharmacodynamic Actions of a Long-Acting PTH Analog (LA-PTH) in Thyroparathyroidectomized (TPTX) Rats and Normal Monkeys. J Bone Miner Res 31, 1405–1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noda H et al. , Optimization of PTH/PTHrP Hybrid Peptides to Derive a Long-Acting PTH Analog (LA-PTH). JBMR Plus 4, e10367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bock A et al. , Optical Mapping of cAMP Signaling at the Nanometer Scale. Cell 182, 1519–1530 e1517 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinstein TN et al. , Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem 288, 27849–27860 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuna RS et al. , Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic beta-cells. American journal of physiology. Endocrinology and metabolism 305, E161–170 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Irannejad R et al. , Functional selectivity of GPCR-directed drug action through location bias. Nature chemical biology 13, 799–806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boone M, Deen PM, Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch 456, 1005–1024 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacDonald PE et al. , The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 51 Suppl 3, S434–442 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Lymperopoulos A, Rengo G, Koch WJ, Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113, 739–753 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gidon A et al. , Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nature chemical biology 10, 707–709 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gidon A, Feinstein TN, Xiao K, Vilardaga JP, Studying the regulation of endosomal cAMP production in GPCR signaling. Methods Cell Biol 132, 109–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wakabayashi Y, Chua J, Larkin JM, Lippincott-Schwartz J, Arias IM, Four-dimensional imaging of filter-grown polarized epithelial cells. Histochem Cell Biol 127, 463–472 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Stallaert W et al. , Purinergic Receptor Transactivation by the beta2-Adrenergic Receptor Increases Intracellular Ca(2+) in Nonexcitable Cells. Mol Pharmacol 91, 533–544 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Dean T et al. , Mechanisms of ligand binding to the parathyroid hormone (PTH)/PTH-related protein receptor: selectivity of a modified PTH(1–15) radioligand for GalphaS-coupled receptor conformations. Molecular endocrinology 20, 931–943 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL, OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40, D370–376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bakan A et al. , Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 30, 2681–2683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakan A, Meireles LM, Bahar I, ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J et al. , The I-TASSER Suite: protein structure and function prediction. Nat Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy A, Kucukural A, Zhang Y, I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J et al. , CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput 12, 405–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu EL et al. , CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J Comput Chem 35, 1997–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phillips JC et al. , Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang J et al. , CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humphrey W, Dalke A, Schulten K, VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Oztan A et al. , Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Molecular biology of the cell 18, 3978–3992 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McElderry JD et al. , Tracking circadian rhythms of bone mineral deposition in murine calvarial organ cultures. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 28, 1846–1854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White AD et al. , Ca(2+) allostery in PTH-receptor signaling. Proc Natl Acad Sci U S A 116, 3294–3299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao K, Shenoy SK, Beta2-adrenergic receptor lysosomal trafficking is regulated by ubiquitination of lysyl residues in two distinct receptor domains. The Journal of biological chemistry 286, 12785–12795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kahsai AW, Rajagopal S, Sun J, Xiao K, Monitoring protein conformational changes and dynamics using stable-isotope labeling and mass spectrometry. Nat Protoc 9, 1301–1319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao K et al. , Revealing the architecture of protein complexes by an orthogonal approach combining HDXMS, CXMS, and disulfide trapping. Nat Protoc 13, 1403–1428 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Haas W et al. , Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Molecular & cellular proteomics : MCP 5, 1326–1337 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Bakalarski CE et al. , The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. Journal of proteome research 7, 4756–4765 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry data have been deposited with the ProteomeXchange Consortium with the dataset identifier (MSV000087510/PXD026256). All other data needed to evaluate the conclusions in the paper are present in the paper or in the Supplementary Materials. All materials, data, animal models, and associated protocols will be made available upon reasonable request.