

Abstract
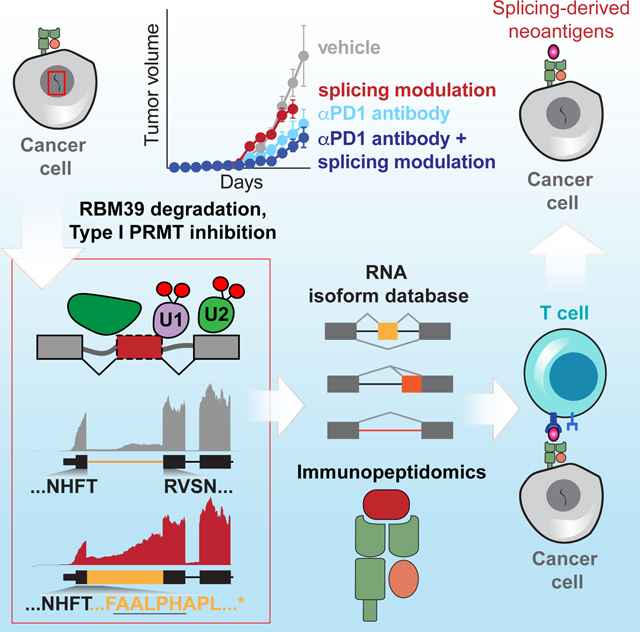
SUMMARY
While mutations in DNA are the best-studied source of neoantigens that determine response to immune checkpoint blockade, alterations in RNA splicing within cancer cells could similarly result in neoepitope production. However, the endogenous antigenicity and clinical potential of such splicing-derived epitopes have not been tested. Here, we demonstrate that pharmacologic modulation of splicing via specific drug classes generates bona fide neoantigens and elicits anti-tumor immunity, augmenting checkpoint immunotherapy. Splicing modulation inhibited tumor growth and enhanced checkpoint blockade in a manner dependent on host T cells and peptides presented on tumor MHC class I. Splicing modulation induced stereotyped splicing changes across tumor types, altering the MHC I-bound immunopeptidome to yield splicing-derived neoepitopes which trigger an anti-tumor T cell response in vivo. These data definitively identify splicing modulation as an untapped source of immunogenic peptides and provide a means to enhance response to checkpoint blockade that is readily translatable to the clinic.
In Brief sentence:
By provoking production of “neoantigens” that are recognized by immune cells, drugs that modulate RNA splicing can enhance cancer immunotherapy.
Original, unedited In Brief (do not use):
Here, we present definitive proof of concept that pharmacologic modulation of RNA splicing enhances anti-tumor immunity via the generation of endogenous, mis-spliced antigenic peptides and augments the efficacy of immune checkpoint blockade.
Keywords: Immune checkpoint blockade, immunotherapy, neoantigens, neoepitopes, immunopeptidome, PD1, PRMTs, RBM39, RNA splicing, splicing
Graphical Abstract
INTRODUCTION
Immune checkpoint blockade has greatly improved outcomes for several malignancies that are challenging to treat. Therapeutic benefit is associated with increased mutational burden (Marabelle et al., 2020; Sha et al., 2020) and mismatch repair deficiency (Le et al., 2017), commonly attributed to high tumor neoantigen load. Coding mutation-derived neoantigens are best studied, but neoantigens can also arise from other processes, such as altered RNA splicing. Two studies analyzing The Cancer Genome Atlas (TCGA) RNA sequencing (RNA-seq) data concluded that tumor-specific alternative splicing is abundant and may generate peptides that contribute to epitope repertoires (Jayasinghe et al., 2018; Kahles et al., 2018). Another study demonstrated that peptides translated from retained introns arising from incomplete RNA splicing can be presented by MHC I in cancer cell lines (Smart et al., 2018). These provided proof of concept that splicing-derived peptides may serve as neoepitopes. However, whether splicing-derived neoepitopes elicit an endogenous immune response remains unknown, in part due to the inherent complexity of RNA processing and corresponding challenges with robustly identifying candidate splicing-derived neoantigens.
Determining whether splicing alterations can generate bona fide neoantigens is particularly important given the development of multiple clinical-grade compounds that alter RNA splicing via non-overlapping mechanisms. Many compounds, such as pladienolide B, GEX1A, E7107, and H3B-8800, inhibit the SF3b splicing complex’s interactions with RNA (Lee et al., 2016; Seiler et al., 2018; Sellin et al., 2019; Yokoi et al., 2011). Recently, a series of “anti-cancer sulfonamides,” including indisulam and E7820, were found to degrade the accessory splicing factor RBM39 by rendering it a neosubstrate for the Ddb1/CUL4 E3 ubiquitin ligase (Han et al., 2017). Additionally, blocking post-translational modifications of splicing factors, which are required for spliceosome assembly and effective splicing catalysis, can robustly perturb splicing. For example, RNA splicing factors are the most heavily arginine-methylated proteins (Musiani et al., 2019), and so drugs which block asymmetric or symmetric arginine dimethylation by inhibiting type I or II protein arginine methyltransferases (PRMTs) potently perturb splicing (Fedoriw et al., 2019; Fong et al., 2019).
Here, we address the central question of whether altered RNA splicing generates immunologically meaningful neoantigens to provoke an effective anti-tumor immune response.
RESULTS
Pharmacologic perturbation of splicing suppresses tumor growth in vivoin a manner dependent on host T cells and tumoral MHC I-presented peptides
We hypothesized that pharmacologic splicing modulation might generate aberrant mRNAs encoding novel proteins, a subset of which could be translated and presented by MHC I as neoepitopes that provoke anti-tumor immunity. We first tested this by treating cancer cell lines with the RBM39-degrader indisulam at doses sub-inhibitory for growth (Fig. 1A). This yielded graded RBM39 degradation but little effect on growth (Fig. 1B and Fig. S1A–B). Across cell lines, ex vivo treatment and drug washout yielded sustained suppression of RBM39 protein following drug removal, with minimal effects on subsequent proliferation or apoptosis (Fig. 1C–D and Fig. S1C–D). Drug treatment also did not notably change MHC I/II, PD-L1, cytokine or death receptor expression (Fig. S1E–F). In contrast, identical cells exhibited strikingly durable growth inhibition following engraftment into mice, despite transient drug exposure (Fig. 1E–F and Fig. S1G). The effect was dose dependent, with increasing drug concentrations causing increased splicing alterations (Fig. S1H–K), reduced tumor growth (Fig. S1L–N), and improved survival (Fig. S1O).
Figure 1. Pharmacologic RNA splicing modulation impairs tumor growth in a manner dependent on immune recognition.

(A) Schema of drug treatment and washout. (B) Western blot of RBM39 in B16-F10 cells after 24h of indisulam treatment. IC50, half-maximal inhibitory concentration. (C) As (B), but with 4 days of 1μM indisulam, then drug washout and continued culture in vitro. (D) Cell growth following 4 days of DMSO or indisulam 1μM, and drug washout (day 0) in vitro. Mean ± sd. (E) In vivo tumor volumes of cells from (D). Each line is an individual tumor (n=10/group, tumors on both flanks). (F) Box-and-whisker plots of tumor volumes from final day of measurement in (E). For box and whiskers plots throughout, bar indicates median, box edges first and third quartiles, and whiskers minimum & maximum. p from Wilcoxon rank-sum test. (G) Schema of drug treatment and engraftment in mice with immune perturbations. (H) Individual B16-F10 tumor volumes following DMSO or indisulam treatment and engraftment in mice with control vs. T cell depletion. Each line is one tumor (n=10/group). (I) Tumor volumes from (H) at day 19; p from Wilcoxon rank-sum test: ***, p = 0.000379; n.s., p > 0.05. (J) H-2Kb/Db expression of control vs. B2m KO B16-F10 cells ± IFNγ. (K) Schema to evaluate requirement of β2M for tumor control in vivo. (L) Individual tumor volumes at day 30 from (K); p from Wilcoxon rank-sum test: ***, p = 0.009; n.s., p > 0.05. See also Figure S1.
The discrepancy between in vitro and in vivo growth suggested non-tumor cell autonomous effects. To assess whether splicing alterations stimulated an anti-cancer immune response, we repeated our experiments but engrafted treated B16-F10 cells into Rag2-deficient C57BL/6 mice and, separately, into mice with T or natural killer (NK) cell depletion (Fig. 1G). In vivo tumor growth inhibition was rescued in lymphocyte-deficient Rag2 recipients and by T cell depletion, but not NK depletion, suggesting a T cell and antigen-dependent mechanism (Fig. 1H–I and Fig. S1P–Y).
To test whether MHC I-bound antigens and CD8+ T cells could be responsible, we evaluated the effects of indisulam versus DMSO pretreatment of isogenic B16-F10 cells with or without CRISPR-mediated knockout (KO) of B2m, encoding β2-microglobulin (Fig. 1J–K). B2m KO rescued the growth inhibition induced by indisulam pretreatment (Fig. 1L). Overall, these results indicate that RBM39 degradation impairs cancer growth in a manner dependent on T cells and tumor MHC I expression.
We similarly observed immune-mediated suppression of tumor growth for MS-023, a splicing modulator which inhibits Type I PRMT enzymes (Eram et al., 2016). Ex vivo MC38 treatment with MS-023 concentrations subinhibitory for in vitro growth resulted in globally reduced asymmetric dimethyl arginines (ADMA), but minimal effects on cell growth in vitro after drug washout (Fig. 2A and Fig. S2A). However, identically treated cells yielded durable suppression of tumor growth in C57BL/6 mice (Fig. 2B–C). These data suggest that splicing modulation can suppress tumor growth in vivo across multiple tumor types and mechanisms of splicing perturbation.
Figure 2. Pharmacologic splicing modulation promotes T cell reactivity without T cell toxicity in vivo.

(A) In vitro growth of MC38 cells following treatment with DMSO or MS-023 for 96 hours and drug washout. Mean ± sd shown. (B) In vivo growth of cells from (A) in C57BL/6 mice; individual tumor volumes shown (n=10/group). (C) Tumor volumes at day 21 from (B). p from Wilcoxon rank-sum test. (D) Percentage of live donor CD8+CFSElo T cells on day 5 of a mixed leukocyte reaction with BMDC from wild-type or B2m KO C57BL/6 mice. Each dot is a technical replicate. Bar represents median. ‘No lysates’: T cells cultured with BMDC and without lysate. p from Wilcoxon rank-sum test. For wild-type BMDCs, DMSO-vs-Ova p = 0.019, vs. indisulam p = 0.001, vs. MS-032 p = 0.032. (E) Representative histograms of CFSE dilution from (D). (F) CFSE-labeled naïve splenic T cells from C57BL/6 mice stimulated with anti-CD3 & CD28 for three days. (G) Wild-type or ovalbumin-expressing B16-F10 cells were cultured alone or with primed OT-1 T cells for 18h and viable tumor cells enumerated. (H) CFSE dilution of donor CD45.1+ T cells adoptively transferred into irradiated Balb/c recipients treated with the indicated compounds. Donor splenic CD4+ T cells on day 3. (I) As (H), but in LP/J recipients; donor splenic T cells on day 5. See also Figure S2.
We next tested whether splicing modulation enhanced tumor immune recognition via drug-induced neoantigen production. To explore this, we compared the ability of professional antigen-presenting cells (APCs) loaded with lysates from control versus drug-treated tumors to stimulate naïve, syngeneic T cells (Fig. S2B). MC38 cells were treated with DMSO, indisulam or MS-023, and used to generate lysates containing potentially immunogenic peptides, but no drug or viable tumor cells. Bone marrow-derived dendritic cells (BMDCs) from wild-type C57BL/6 or B2m knockout mice were pulsed with lysates and incubated with CD45.1+ splenic T cells. BMDCs loaded with lysates from cells treated with indisulam or MS-023 more strongly promoted CD8+ T cell proliferation than did control BMDCs (Fig. 2D–E). This effect was not observed with B2m KO BMDCs, indicating that MHC I peptide presentation was required.
Drug-specific effects of splicing modulators on T cells
Our initial studies did not address in vivo splicing modulator treatment, which can affect immune and hematopoietic compartments as well as tumors. To address this, we evaluated the effects of multiple classes of splicing modulators on T cell function: indisulam, MS-023, pladienolide B, GEX1A, and the PRMT5 inhibitor EPZ015666 (Chan-Penebre et al., 2015). We first assessed the effects of these drugs on the proliferation of CFSE-labeled, naïve splenic T cells upon anti-CD3 and CD8 stimulation (Fig. S2C). With three days of exposure, indisulam and the PRMT inhibitors had minimal effects on proliferation (IC50 ∼1–10 μM), while pladienolide B and GEX1A were markedly inhibitory (IC50 of nMs) (Fig. 2F and Fig. S2D–E). Measurement of apoptosis and activation markers (Fig. S2E) confirmed that these SF3b inhibitors were profoundly immunosuppressive.
We next assessed drug effects on T cell function. Indisulam and MS-023 minimally blunted the in vitro cytotoxicity of primed OT-1 T cells against ovalbumin (OVA)-expressing B16-F10 or MC38 cells (Fig. 2G and Fig. S2F), with little impairment of cell killing at doses <4μM. Exposure to higher drug concentrations did not inhibit OT-1 T cell secretion of IFNγ or TNFα (Fig. S2G–H) or degranulation (Fig. S2I–J). We complemented these functional studies by determining the effects of each compound on gene expression in T cells activated with anti-CD3 and CD28 ex vivo (Fig. S2K). Normally upregulated genes (Fig. S2L) were markedly attenuated by pladienolide B, and to a lesser extent EPZ015666, whereas indisulam and MS-023 caused much milder changes (Fig. S2M).
We then evaluated the effect of each drug on in vivo T cell function in response to antigen. Here, donor CD45.1+ T cells were adoptively transferred into lethally irradiated recipients which were either syngeneic (C57BL/6), mismatched for non-MHC “minor” antigens (LP/J), or major MHC mismatched (Balb/c; H-2b vs. H-2d; Fig. S2N) (Na et al., 2010). In Balb/c mice, donor T cells showed robust activation and proliferation in response to alloantigen (Fig. 2H and Fig. S2O–Q). While in vivo treatment with indisulam, MS-023, or EPZ015666 resulted in minimal impairment of T cell function (Fig. S2P–Q), inhibition of both Type I and II PRMTs blocked T cell proliferation (Fig. 2H–I). Pladienolide B was similarly suppressive (Fig. 2H, Fig. S2O,R). These observations were recapitulated in the C56BL/6 → LP/J model (Fig. 2I). Indisulam, MS-023 and EPZ015666 were permissive to the homeostatic expansion of T cells after syngeneic adoptive transfer (Fig. S2R–S).
Lastly, we assessed the effects of splicing modulators on hematopoiesis in methylcellulose assays. Normal hematopoiesis was intact at even high μM doses of MS-023 and indisulam, whereas EPZ015666 suppressed hematopoiesis (Fig. S2T). Pladienolide B suppressed hematopoiesis at nM concentrations (Fig. S2T). These data indicate that select splicing modulators can be nontoxic at therapeutic concentrations in preclinical models.
Modulating splicing enhances immune checkpoint blockade
We next tested whether perturbing RNA splicing with immune checkpoint blockade promoted control of established tumors. We evaluated in vivo RBM39 degradation alone or with anti-PD1 by engrafting C57BL/6 mice with syngeneic tumors and treated with splicing inhibitors starting on day 3 and anti-PD1 on day 7, the approximate date range at which tumors became measurable (Fig. S3A). Simultaneous indisulam and anti-PD1 led to significantly reduced growth of both B16-F10 and MC38 tumors, which exceeded the effects of either alone (Fig. 3A–F, Fig. S3A) and yielded on-target RBM39 protein reduction (Fig. 3B). We observed similar benefits in LLC tumors, which are resistant to anti-PD1 monotherapy (Bertrand et al., 2017; Lesterhuis et al., 2013) (Fig. 3G–H).
Figure 3. Splicing modulation enhances checkpoint immunotherapy.

(A) Treatment schema. (B) Western blot of RBM39 in B16-F10 and immune organs of mice treated with vehicle vs. indisulam for 10 days. (C) B16-F10 tumor volumes in mice treated with vehicle, indisulam, anti-PD1, or both (n=10/group). Mean ± sem. Termination of line before day 28 indicates all animals had reached endpoints. (D) Data from (C) at day 26; p from Wilcoxon rank-sum test vs. PBS; *, p = 0.002; **, p = 0.000581. p indisulam vs. ± PD1 = 0.004. (E) As (C), but for MC38 tumor-bearing mice (n=10/group). (F) Data from (E) at day 31; p as above. *, p = 0.004; **, p = 0.0000682; ***, p = 0.000000101. p indisulam vs. ± PD1 = 0.04. (G) As (C), but for LLC tumor-bearing mice (n=10/group). (H) Data from (G) at day 26. p as above. *, p = 0.048; **, p = 0.004. p indisulam vs. ± PD1 = 0.125. (I) Tumor volumes of B16-F10 tumor-bearing mice treated with vehicle, MS-023, anti-PD1, or both (n=10/group). Mean ± sem. (J) Data from (I) at day 28; p as above. *, p = 0.023; **, p = 0.013; ***, p = 0.0000153. p MS-023 vs. ± PD1 = 0.056 (K) As (I), but for MC38 tumor-bearing mice. (L) Data from (I) at day 31; p as above. *, p = 0.001; **, p = 0.000342; ***, p = 0.00000821. p MS-023 vs. ± PD1 = 0.101. (M) Kaplan-Meier survival from (I). All survivors past day 60 were tumor-free. p from logrank test vs. vehicle. See also Figures S3–4.
We then evaluated MS-023 in vivo, finding that MS-023 treatment significantly improved response to anti-PD1 therapy (Fig. 3I–L). For mice engrafted with MC38 cells, combined MS-023 and anti-PD1 treatment resulted in 50% of mice alive and tumor-free three months after engraftment (p<0.001; Fig. 3M). These survivors demonstrated immune memory: upon rechallenge 6 months later with MC38 cells (with or without MS-023 pretreatment in vitro before engraftment), they exhibited markedly improved tumor control compared to naïve age-matched mice (Fig. S3B–F).
Finally, we assessed potential toxicity in non-tumor tissues. Treatment with indisulam or MS-023 with or without anti-PD1 did not affect blood counts (Fig. S4A). MC38 tumors exhibited increased CD8+ T cells when indisulam or MS-023 were given with anti-PD1, consistent with intra-tumoral T cell expansion (Fig. S4B). Drug-treated animals did not exhibit histologic inflammation or increased CD8+ infiltrates in the skin, lung, gut, or liver (Fig. S4C–F). Concordantly, RNA-seq analyses of lung, colon and splenic T cells from indisulam-treated mice showed mild splicing changes (Fig. S4G–K), and gene pathway analyses did not reveal an inflammatory signature (Fig. S4L–O).
Splicing modulators induce RNA isoforms encoding predicted neoepitopes
We next determined the molecular mechanisms by which splicing modulation enhances anti-tumor immunity. We assessed how splicing modulation altered tumor cell transcriptomes in murine tumor cell lines treated with DMSO, indisulam, or MS-023, performed high-coverage RNA-seq, and quantified differential gene and isoform expression. Both drugs drove dramatic changes in alternative and constitutive splicing, with differential cassette exon inclusion and constitutive intron retention the most common alterations (Fig. 4A, Fig. S5A–C, and Tables S1–2). Identical experiments in human tumor cell lines revealed similar changes (Fig. 4B, Fig. S5D–F, and Tables S3–4). A subset of mis-splicing events were consistently induced across all cell lines in a given species (Fig. S5G–I), and 29.0% (indisulam) and 9.1% (MS-023) of mis-spliced genes were mis-spliced in both species (Fig. S5J), consistent with conserved splicing mechanisms.
Figure 4. Splicing modulation induces widespread potential neoepitope production.

(A) RNA-seq read coverage illustrating shared intron retention (left), cassette exon exclusion (middle), and competing 3’ splice site selection (right) induced by indisulam in mouse and (B) human cancer cell lines. Conditions as in Fig. 1A. (C) Left, stacked bar graph illustrating numbers of differentially retained introns following indisulam treatment. Blue/green, increased/decreased intron retention in indisulam vs. DMSO; percentages shown for blue. Right, heat map illustrating quantitative extent of intron retention for introns significantly mis-spliced in at least one sample. (D) As (C), but for cassette exons. (E) Bar graph of poly(AT) motif enrichment in introns preferentially retained vs. unaffected upon indisulam treatment. Motif enrichment computed relative to a randomly selected group of unaffected introns. Error bars, 95% confidence intervals estimated by bootstrapping. (F) Metagene plot of poly(AT) enrichment across introns that were preferentially retained or unaffected following indisulam treatment. Shading, 95% confidence intervals estimated by bootstrapping. (G) Left, RNA-seq read coverage illustrating Prpf40b intron retention in the cytoplasmic fraction following indisulam treatment of B16-F10. Right, quantification of Prpf40b intron retention in total, nuclear (nuc.), and cytoplasmic (cyto.) fractions. p from unpaired t-test. (H) Predicted 9-mer peptides arising from indisulam-induced Prp40b intron retention. Black/blue, exon/intron-derived amino acids. (I) Filtering strategy to predict potential indisulam-induced, MHC I-bound epitopes. Numbers of unique peptides present at each step are shown for representative MHC I alleles following DMSO or indisulam treatment of B16-F10 and 501-MEL cells. (J) Bar graph illustrating numbers of predicted indisulam-induced 8–14-mer peptides arising from different splicing events following DMSO or indisulam treatment of B16-F10 cells. All analyses performed for n=3 biological replicates for each cell line and treatment unless specified otherwise. See also Figure S5 and Tables S1–5.
Indisulam and MS-023 gave rise to distinct downstream splicing alterations (Fig. S5K). Indisulam-induced splicing alterations were dominated by reduced splicing efficiency: cassette exons were preferentially not included and constitutive introns were preferentially not excised (Fig. 4C–D). MS-023, in contrast, resulted in more balanced changes (Fig. S5C,F). Nonetheless, convergent mis-splicing between drugs was relatively common (∼4–8% of mis-spliced events) (Fig. S5L–M). Constitutive introns preferentially retained following indisulam treatment were significantly depleted for poly(AT) (Fig. 4E–F), consistent with RBM39 binding of poly(AT) motifs (Wang et al., 2019) and on-target RBM39 degradation. For MS-023, we observed no such obvious motif enrichment, as expected given the broad effects of Type I PRMT inhibition.
We next evaluated the cytoplasmic availability of mis-spliced mRNAs for translation. As splicing is linked to cytoplasmic mRNA export (Zhou et al., 2000), splicing alterations could fail to yield novel peptides. We separated nuclear and cytoplasmic RNA pools from DMSO- and indisulam-treated cells (focusing on indisulam, as it led to global decreases in splicing efficiency) (Fig. S5N) and quantified drug-induced isoforms in each subcellular compartment. Indisulam-induced intron retention was readily apparent in the cytoplasmic fraction (Fig. 4G and Fig. S5O–P), where such mRNAs could be translated into potential neoepitopes (Fig. 4H).
We finally estimated the potential consequences of indisulam-induced splicing alterations for neoepitope production. We enumerated all 8–14 amino acid sequences (8–14-mers) arising from mRNA isoforms in the transcriptome and estimated the binding affinity of each to common MHC I alleles with NetMHCPan. We restricted this list to predicted binders, then epitopes arising from differentially spliced genes, and finally epitopes from isoforms promoted by indisulam treatment. This filtering dramatically reduced the space of potential neoepitopes (Table S5), with most arising from cassette exons and constitutive introns (Fig. 4I–J). Many predicted neoepitopes were shared within mouse (5,764) or human (24,378) cell lines (Fig. S5Q), while fewer (1,763) were shared across species (Fig. S5R), presumably reflecting non-conserved splicing alterations and differences in binding preferences of murine and human MHC.
Drug-induced, splicing-derived neoepitopes are presented by MHC I on tumor cells
We augmented transcriptomic analyses with experimental identification of splicing-derived neoepitopes. We purified H-2Kb and H-2Db from DMSO vs. indisulam-treated B16-F10 cells, eluted bound peptides, and performed liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Fig. 5A).
Figure 5. Indisulam-induced neopeptides are presented by MHC I.

(A) Workflow overview. (B) Schematic for RNA isoform and proteome database creation. (C) Histogram of predicted NetMHCpan binding rank of all peptides identified from the H-2Db immunoprecipitation (IP) and full-length proteome. Peptides with rank < 2 are predicted binders. Peptides identified in DMSO-treated (gray, left) and indisulam-treated (red, right) samples are overlaid on a random sample of 1,000 sequences from the full-length proteome (black) for comparison. (D) Sequence logo for 9-mers identified from the H-2Db IP and full-length proteome. (E) Bar plot of numbers of peptides identified from the H-2Db IP using each proteome in (B). (F) Bar plot of numbers of predicted binders and non-binders identified from H-2Db IP using the spiked non-binders proteome, which consists of predicted binders (rank < 2) composing 90% of this proteome, and non-binders (rank > 90), added to constitute 10% of the proteome. (G) Density plots of parent gene expression for peptides identified from the H-2Db IP from DMSO-treated (gray, left) and indisulam-treated (red, right) samples, each compared to the expression of all genes (black) following treatment with DMSO or indisulam, respectively, using the predicted binders proteome. TPM, transcripts per million. (H) Heat map illustrating each peptide identified in at least one replicate (rows) using the predicted binders proteome. Columns are peptides. (I) Bar plot illustrating percentages of indisulam-specific, isoform-specific identified peptides arising from different types of alternative splicing. (J) RNA-seq coverage plots of representative indisulam-induced, candidate splicing-derived neoepitopes generated by intron retention in Hus1 and (K) Zfp512, (L) competing 3’ splice sites in D14Abb1e, and (M) cassette exon skipping in Poldip3. Indisulam-promoted peptide shown in bold, underlined text. (N) Median fluorescence intensities (MFIs) of H-2Db and/or H-2Kb on RMA-S cells following incubation with increasing doses of Hus1, (O) Zfp512, (P) D14Abb1e, and (Q) Poldip3 candidate neoantigenic peptides from (J-M). Mean ± sd shown. For (N-Q), grey lines indicate negative control peptides randomly selected from the predicted non-binder, “spike-in” peptides used in (B). All analyses performed for n=3 biological replicates for each treatment for (A-M) and n=4 biological replicates for (N-Q). For (C-D) and (G), data collated across n=3 replicates per treatment. See also Figure S6 and Table S6.
As MHC I-bound peptide identification from mass spectrometry depends critically upon the search database (proteome) (Murphy et al., 2017), we tested four distinct proteomes. These were “full-length,” i.e. all full-length protein sequences in the transcriptome; “predicted binders,” restricted to 8–14-mers that were predicted MHC I binders; “predicted binders + spiked non-binders”, augmented with decoy predicted non-binders; and “filtered predicted binders,” restricted to predicted binders from differentially expressed or spliced genes (Fig. 5B).
We first evaluated the full-length proteome. Approximately 80% and 86% of identified peptides were predicted binders for H-2Db and H-2Kb versus 0.6% and 0.9% for peptides randomly sampled from the proteome (Fig. 5C and Fig. S6A). Repeating this analysis with MHCflurry (O’Donnell et al., 2018) yielded similar results (not shown). Identified peptides had expected sequence preferences at anchor residues and preferential identification of 9-mers and 8–9-mers for H-2Db and H-2Kb, respectively (Fig. 5D and Fig. S6B–C). We next varied the input proteome to maximize peptide identification. Restricting the search to predicted binders increased recovery ∼2-fold relative to the full-length proteome, while further restricting to differentially expressed or spliced genes decreased recovery ∼3.4-fold for H-2Db (Fig. 5E), with similar results for H-2Kb (Fig. S6D). Restricting to predicted binders did not decrease specificity: we identified only 2 predicted non-binders, versus 2,204 predicted binders, across all six replicates when we queried the spiked non-binder proteome for H-2Db (Fig. 5F), again with similar results for H-2Kb (Fig. S6E). As the predicted binders proteome maximized yield while minimizing false positives, we used it for subsequent analyses. The vast majority of identified peptides arose from genes expressed at moderate to high levels in B16-F10 cells (Fig. 5G and Fig. S6F).
We identified 518 and 366 peptides for H-2Db and H-2Kb that were only recovered from indisulam-treated samples (Fig. 5H). We intersected these with predicted isoform-specific epitopes identified by RNA-seq (Fig. 4J) to obtain 42 and 28 peptides that were bound by H-2Db and H-2Kb, respectively, and arose from mRNA isoforms that were promoted by indisulam treatment (Fig. 5I and Table S6). Due to the known limited sensitivity of mass spectrometry for the MHC I immunopeptidome (Schuster et al., 2018), we also nominated an additional 39 candidate peptides that were supported by RNA-seq alone, but predicted to be high-affinity binders to H-2Db or H-2Kb (Tables S1–2, Fig. S6G). We used this set of 109 (70 from mass spectrometry, 39 from RNA-seq) high-confidence, potentially antigenic peptides in subsequent functional assays.
Splicing-derived neoantigens can trigger an endogenous T cell response
We experimentally assessed binding of each of our 109 candidate neoantigenic peptides, which arose from diverse splicing changes (Fig. 5I–M), to H-2Db or H-2Kb in the RMA-S stabilization assay (Fig. S7A) (De Bruijn et al., 1991; De Silva et al., 1999). Candidate peptides had a range of abilities to stabilize H-2 molecules, with ∼97% (68/70) of peptides identified from both MHC I mass spectrometry and RNA-seq exhibiting some binding, and several exhibiting very strong binding (Fig. 5N–Q, Fig. 6A, and Fig. S7B). Negative control “spike-in” peptides showed no binding.
Figure 6. Splicing-derived neoepitopes are immunogenic in vivo.

(A) Heatmap representing mean MFI of H-2Kb from RMA-S assay. Green, immunogenic controls. (B) Immunization schema. (C) Representative IFNγ ELISpot from CD8+ T cells upon stimulation with syngeneic peptide-loaded splenocytes. Each row is a peptide used for in vivo immunization. Columns, T cells reacted with the indicated stimuli. PMA: Phorbol 12-myristate 13-acetate; Iono: ionomycin. (D) Spots per 105 CD8+ T cells from IFNγ ELISpot for peptides identified as immunogenic. Bar indicates median. SIINFEKL, positive control. Each dot is one technical replicate. (E) Representative IFNγ ELISpot of CD8+ T cells from immunized mice, following stimulation with syngeneic peptide-loaded splenocytes. Each row is one dose. Columns, T cells reacted with the indicated stimuli. Plots on right quantify numbers of dots per well; each dot is one technical replicate. (F) Comparisons of predicted MHC I binding for immunogenic (IFNγ ELISpot-positive) versus nonimmunogenic peptides. (G) As (F), but with RMA-S MFIs. For (F-G), p from two-sided Wilcoxon rank-sum test. See also Figure S7 and Table S7.
As MHC I binding is imperfectly correlated with immunogenicity, we experimentally tested each candidate. We immunized naïve mice with each of the 109 peptides by hock injection, obtained draining lymph nodes (Fig. 6B), and performed IFNγ ELISpot with purified CD8+ T cells incubated with naïve, syngeneic splenocytes loaded with DMSO or cognate peptide (Table S7). ∼43% (30/70) of peptides with both mass spectrometry and RNA-seq support elicited a CD8+ T cell response (Fig. 6C–D and Fig. S7C), and several such peptides were induced by indisulam in all tested mouse cancer cell lines (Fig. S6G). We further confirmed the specificity of these responses by immunizing across a range of peptide doses, revealing a dose-dependent CD8+ T cell response (Fig. 6E and Fig. S7D–E).
All 39 candidate peptides selected based solely on RNA-seq analyses and MHC I binding predictions exhibited some H-2 binding (Fig. S7F), and 28% (11/39) were immunogenic in vivo (Fig. S7G). Our in silico analyses thereby identified a reasonable proportion of splicing-derived, potentially immunogenic peptides, but nonetheless a number of candidate peptides with verified MHC I binding failed to elicit CD8+ T cell activation in vivo (Fig. S7H–J). To understand the basis for this differential response, we interrogated potential distinctions between immunogenic and nonimmunogenic splicing-derived peptides. Analyses of predicted binding affinity (NetMHCpan), experimental ability to stabilize MHC I (RMA-S assay), parent gene expression, type and magnitude of splicing alteration, and predicted induction of NMD revealed that only binding to MHC I (NetMHCpan or RMA-S assay) differed significantly between immunogenic and nonimmunogenic peptides (Fig. 6F–G).
We next tested the ability of CD8+ T cells from peptide-immunized mice to kill tumor cells presenting the cognate peptide. While DMSO-immunized CD8+ T cells exerted no cytotoxic activity regardless of the peptide presented, CD8+ T cells from mice immunized with an immunogenic peptide selectively killed B16-F10 cells presenting that same peptide (Fig. 7A–B).
Figure 7. Splicing-derived neoantigens trigger an endogenous T cell response.

(A) Schema of co-culture of CD8+ T cells from peptide-immunized C57BL/6 mice with peptide-loaded B16-F10 cells for cytotoxicity. (B) Bar plot of live B16-F10 cells from (A). Each dot is a technical replicate. p from Wilcoxon rank-sum test. (C) Schema of CD8+ T cells from peptide-immunized C57BL/6 mice, stimulated with B16-F10 cells treated with DMSO or indisulam for IFNγ ELISpot. (D) Representative IFNγ ELISpot from CD8+ T cells following stimulation with DMSO or indisulam-treated B16-F10 cells, or B16-F10 cells overexpressing ovalbumin. Rows, peptides used for immunization. (E) Bubble plot of data from (C-D); size of bubble indicates number of spots. p from Wilcoxon rank-sum test. (F-H) Box-and-whisker plots for representative peptides from (E). Each dot is one technical replicate. p from Wilcoxon rank-sum test. (I) RNA-seq coverage plots demonstrating mis-splicing of Eif4g3 (left) and Stat2 (right) upon indisulam exposure and the resulting neoantigenic peptides. (J) Representative plots of peptide:MHC I tetramer staining of CD8+ T cells from tumor-draining lymph nodes of B16-F10 tumor-bearing mice treated with vehicle, anti-PD1, indisulam, or the combination and analyzed at day 14, gated on CD3+ T cells. Each row is one neoantigenic peptide, and columns indicate treatment condition. (K) Quantification of (J); each dot is one mouse. p from Kruskal-Wallis ANOVA. See also Figure S7 and Table S7.
Finally, we assessed the endogenous consequences of splicing-derived peptide production by testing whether drug-treated tumors generated neoantigenic peptides at concentrations which activated CD8+ T cells. We repeated the above experiments, but used B16-F10 cells treated with indisulam as APCs (Fig. 7C). This demonstrated that indisulam treatment of tumor cells indeed stimulates endogenous generation of specific splicing-derived neoantigens that trigger antigen-specific T cell activation (Fig. 7D–H). We therefore tested whether indisulam treatment drove the expansion of antigen-specific CD8+ T cells that recognized neoantigenic peptides in vivo (Fig. 7I). We generated H-2Kb tetramers loaded with peptides which elicited strong IFNγ secretion and cytotoxicity in the above experiments. These were used to stain tumor-draining lymph nodes of B16-F10 tumor-bearing mice treated with vehicle, indisulam, anti-PD1, or the combination (Fig. 7J, Fig. S7K). This revealed increased frequencies of CD8+ T cells capable of recognizing splicing-derived peptides in mice receiving indisulam or the combination of indisulam and anti-PD1 (Fig. 7K). Together, these data demonstrate that splicing modulation triggers the production of specific splicing-derived neoantigens at levels sufficient to drive expansion of CD8+ T cells recognizing those antigens.
DISCUSSION
Mutation-derived neoantigen burden is an established determinant of response to immune checkpoint blockade. Here, we describe a distinct and abundant source of immunogenic peptides derived from novel mRNA species. We demonstrated that multiple clinical-grade compounds, acting via distinct mechanisms, can thereby enhance immune checkpoint blockade by inducing such MHC I-presented neopeptides. These studies thus identify a means to acutely and reversibly induce tumor neoantigens without genomic changes, and demonstrate the antigenic potential of splicing-derived neoepitopes in vivo.
Splicing modulation generates many novel mRNAs derived from large-scale events, including inclusion of intronic sequence into mature mRNA, juxtaposition of exons not normally spliced together, and exons with abnormal 5’ or 3’ ends. Each can result in the production of peptides containing wholly novel sequences—confirmed by our cytoplasmic RNA sequencing and MHC I mass spectrometry—potentially contributing to the many immunogenic peptides that we identified.
While direct comparisons of the frequencies of neoantigens derived from aberrant splicing to those derived from single-nucleotide variants is challenging due to differences in experimental methodologies, the frequency of antigenic peptides derived from splicing may be high. For example, of candidate neoantigenic peptides derived from intersecting RNA-seq and MHC I proteomics, we found that 30 of 70 (∼43%) could elicit a CD8+ T cell immune response in naïve C57BL/6 mice by IFNγ ELISpot. Predicted neoantigenic peptides derived from RNA-seq alone exhibited a positivity rate of 11 / 39 (∼28%). Of these neoantigenic peptides, we then demonstrated that four were associated with the expansion of antigen-specific CD8+ T cells following splicing modulator treatment in vivo. In comparison, an early seminal study of MC38 cells reported that out of ∼1,300 coding variations, ∼13% resulted in peptides predicted to bind MHC I, 0.5% of which were identified by mass spectrometry, and ∼0.25% of which were immunogenic in vivo (Yadav et al., 2014). A recent consortium effort evaluating human melanoma and non-small cell lung cancer neoantigens predicted to bind MHC reported an immunogenicity rate of 6% (Wells et al., 2020). Overall, our results highlight the immunological relevance and clinical potential of splicing modulation.
Limitations of the Study
Although we identified many splicing-derived, potentially immunogenic peptides produced upon exposure to splicing inhibitors, some of which triggered reactive T cell expansion in vivo, it is unclear which are most important for controlling tumor growth. However, as splicing modulation yields diverse such peptides, multiple peptides may contribute. Our work also highlights outstanding questions. For example, does splicing modulation affect CD4+ T cells and MHC II-presented neoantigens? Are there MHC-independent anti-tumor B cell and antibody responses elicited by neoantigenic proteins on the cell surface? Do cancer-associated mutations in RNA splicing factors (Dvinge et al., 2016) alter the response to checkpoint immunotherapy? Finally, does pharmacologically altering RNA metabolic processes beyond splicing, such as intronic polyadenylation or NMD, affect tumor immunogenicity? Our finding that multiple modes of splicing modulation promote tumor immunogenicity will hopefully motivate further efforts to develop novel means of therapeutically modulating splicing and other RNA metabolic processes.
STAR Methods
Resource Availability
Lead Contact:
Omar Abdel-Wahab; abdelwao@mskcc.org
Materials Availability:
Unique reagents were not generated for this study.
Data and Code Availability:
RNA-seq data generated as part of this study were deposited at the Gene Expression Omnibus (accession GSE162818). Software RRIDs (also available in the Key Resources Table) are: Bowtie RRID: SCR_005476, RSEM RRID: SCR_013027, TopHat RRID: SCR_013035, MISO RRID: SCR_003124, Samtools RRID: SCR_002105, Bioconductor RRID: SCR_006442, dplyr RRID: SCR016708, ggplot2 RRID: SCR_014601, Proteome Discoverer RRID: SCR_014477.
Experimental Model and Subject Details
Mice:
All in vivo experiments were approved by the Institutional Animal Care and Use Committees (IACUC) of Memorial Sloan-Kettering Cancer Center and/or Fred Hutchinson Cancer Research Center and conducted in accordance with ARRIVE guidelines. All animals were housed in the respective specific pathogen-free (SPF) barrier facilities and maintained under standard husbandry conditions. B6(Cg)-Rag2tm1.1Cgn/J (RAG2 KO) mice, C57BL/6-Tg (TcraTcrb)1100Mjb/J (OT-1) mice and B6.129P2-B2mtm1Unc/DcrJ (B2M KO) mice were obtained from Jackson Laboratories (Cat. 008440, 003831, 002087 respectively). C57BL/6 mice, congenic B6.SJL-Ptprca Pepcb/BoyJ (CD45.1) mice, Balb/c and LP/J mice were also obtained from Jackson Laboratories (Cat. 000664, 002014, 000651 and 000676). CD45.1, RAG2 KO, B2M KO, and OT-1 mice are all fully backcrossed onto the C57BL/6 genetic background. Unless otherwise noted in the text, females from 6–8 weeks old were used for all experiments. Animals within each genotype and treatment condition were randomly allocated to experimental groups; we did not exclude any allocated animals from our analyses. We minimized nuisance variables associated with the vivarium by checking on each cage of animals at least daily, and randomizing the location of cages when animals were placed back in their housing. Multiple authors (S.X.L., D.C., H.C., M.S., C.E.) were assigned to each experiment to minimize bias due to animal handling techniques. Blinding was not feasible for animal tumor challenge, drug treatment, or tumor measurements as the same authors performed all of the above. Animal group sample sizes (indicated in Figure Legends) were determined based on prior published literature on the same tumor models, with differences in tumor growth as the primary outcome measure. Individual study designs, outcome measures and corresponding statistical tests for animal experiments and ex vivo analyses of primary murine tissues are otherwise described in the text and figure legends.
Cell lines:
B16-F10, CT26.WT (CT26), and LLC cells were obtained from ATCC (Cat. CRL-6475, CRL-2638, and CRL-1642 respectively). MB49 cells were obtained from MilliporeSigma (Cat. SCC148, Burlington, MA); MC38 cells were obtained from Kerafast (Cat. ENH204-FP, Boston, MA). B16-F10 and MC38 cells expressing chicken ovalbumin (B16ova and MC38ova) were a kind gift of Jeff Ravetch (Rockefeller University, New York, NY). To generate β2 microglobulin-deficient cell lines for in vitro experiments, four candidate sgRNAs for mouse β2 microglobulin (#1 AGTATACTCACGCCACCCACCGG, #2 TCACGCCACCCACCGGAGAATGG, #3 GGCGTATGTATCAGTCTCAGTGG, #4 TCGGCTTCCCATTCTCCGGTGGG) or non-targeting control (GGAGCGCACCATCTTCTTCA) were cloned into pSpCas9(BB)-2A-Puro (PX459) as previously described (Ran et al., 2013) and used to engineer deficient MC38, B16-F10, and CT26 cell lines via transfection using XtremeGene 9 reagent as per manufacturer’s instructions (MilliporeSigma Cat. 6365809001) followed by puromycin selection at 10 μg/mL for three days. Polyclonal cell populations were obtained by flow sorting for H-2Kb/Db and β2 microglobulin double-negative cells, and gene knockout further confirmed by stimulating a culture of these sorted cells for 48 hours with 10U/mL mouse IFNγ and analyzing for the same markers. For in vivo experiments, lentiCas9-Blast was used to generate Cas9-expressing B16-F10 cells. B2m gRNAs (GAGGGGTTTCTGAGGGCCAC, AGTATACTCACGCCACCCAC) and non-targeting control gRNAs (AAAAAGTCCGCGATTACGTC, ACCCATCCCCGCGTCCGAGA) were cloned into lentiGuide-Puro and introduced into Cas9-expressing B16-F10 cells via lentiviral transduction as previously described (Thomas et al., 2020) and underwent similar selection. PX459, lentiCas9-blast, and lentiGuide-Puro were kind gifts of Feng Zhang (Addgene Cat. 62988, 52962, 52963).
Quantification and Statistical Analysis
All data summarization, visualization, and statistical analysis were performed using GraphPad Prism v9.0 (GraphPad Software, San Diego, CA) or in the R programming language. Details for statistical procedures, including statistical test used, number of replicates, definition of center, and definition of error bars are found within the figure legends. Unless otherwise noted in the text, n represents biological replicates of the sample type (e.g., individual tumors, independent cell cultures, etc.) indicated in the figure legend. The normality of data was assessed using a Shapiro-Wilk test. If data were normally distributed, a parametric test (e.g., unpaired, two-sided t-test) was used to test for significant differences between groups; otherwise, a non-parametric test (e.g., Wilcoxon rank-sum test) was used, as indicated in the figure legends. Differences between groups were considered significant if p was less than 0.05. No methods were used for sample randomization or sample size estimation and no data were excluded from analysis.
Method Details:
Pretreatment with splicing inhibitors
Unless otherwise specified, cell lines were treated with splicing inhibitors at the indicated concentrations for 96 hours in vitro, harvested and washed three times with PBS in excess to remove all drug, and then used for downstream analyses and/or subsequent studies, including phenotyping, RNA-seq analyses, continued growth in vitro, or tumor challenge in vivo into syngeneic animals.
In vivo tumor challenge
Unless otherwise specified, syngeneic B6 or Balb/c mice were engrafted subcutaneously on bilateral flanks with MC38, B16-F10, CT26 or LLC tumor cells at the following doses: MC38 106 cells, B16-F10 0.5×106 cells, CT26 0.25×106 cells, LLC 0.25×106 cells. Tumors were measured serially twice or three times weekly and tumor volumes were estimated by length x width x height. Animals were monitored daily for survival and weighed twice weekly. Experimental endpoints mandating euthanasia were approved by the IACUC and included: animal lethargy, severe kyphosis or evidence of pain, difficulty with ambulation or feeding, tumor ulceration > 1 cm or bleeding tumor, evidence of infected tumor, tumor volumes exceeding 2.5 cm3, or animal total body weight loss > 10% from baseline.
Determination of cell growth, Annexin V, and activation marker IC50 values
Cell lines were grown with half-log10 concentrations of the indicated drug in 4 to 8 technical replicates under standard conditions until the control condition (DMSO or vehicle) was confluent by microscopy. For tumor cell lines, viable cells were quantified via the CellTiter-Glo® assay (Promega Cat. G7573) as per manufacturer instructions. For the ex vivo proliferation of T cells, viable cells were instead quantified via flow cytometry using counting beads. The percentage or number of viable cells with drug treatment was calculated relative to DMSO control (as 100%). These data were log10 transformed and a three-parameter nonlinear fit of log(inhibitor) vs. response was performed in GraphPad Prism v9.0 (GraphPad Software, San Diego, CA) to determine IC50 values. For absolute cell number, Annexin V+, CD25+, and PD1+ flow cytometry data presented in Figure S4, dose-response models and IC50 values were computed using the R language’s drc package(Ritz et al., 2015).
OT-1 cytotoxicity assay
Bulk splenocytes from OT-1 animals were cultured for three days with 100 U/mL murine IL-2 and 100 μg/mL SIINFEKL peptide to activate CD8+ T cells. Cultures were subsequently washed thoroughly to remove ova peptide and rested for at least 24 hours prior to use. OT-1 cells were passaged in T cell media with 50 U/mL IL-2 for no more than seven days from animal sacrifice prior to use. For the cytotoxicity assay, tumor cells alone or OT-1 + tumor cells (1:1 ratio) were incubated in T cell media for 18 hours under standard conditions with the indicated concentrations of splicing drugs and analyzed by flow cytometry to quantify killing. OT-1 cells and other hematopoietic cells were excluded with the use of CD45, CD3, and CD8 staining. Tumor cell viability was measured using DAPI.
LAMP-1 T cell degranulation assay
OT-1 cells were generated as described for the cytotoxicity assay and incubated with ovalbumin-expressing tumor cell lines (pre-treated overnight with IFNγ 100U/mL to upregulate cell-surface MHC I) in the presence of DMSO or varying concentrations of splicing modulator drugs as indicated, in the presence of LAMP-1 antibody for 5–6 hours under standard incubator conditions. After the first hour of incubation, BD GolgiPlug™ (brefeldin A) and BD GolgiStop™ (monensin) was added at 1:1,1000 + 1:1,500 respectively into cells. At the end of incubation, cells were washed and stained for cell surface markers prior to standard flow cytometry.
Generation and use of peptide:H-2Kb tetramers
Peptide:MHC I tetramers with neoantigenic peptides and murine H-2Kb were generated using the QuickSwitch™ Quant Tetramer Kit-PE (Cat. TB-7400-K1, MBL International) per manufacturer instructions. Briefly, 10 μg of peptide together with 50 μL of the tetramer reagent and 1 μL of peptide exchange factor were incubated at room temperature for 5–6 hours and used to stain cell populations of interest. Clone KT15 of an anti-CD8 antibody (Cat. D271-A64, MBL International) was used to identify CD8+ T cells of interest as this clone does not interfere with tetramer binding.
Intracellular cytokine staining
OT-1 cells were prepared and incubated with ovalbumin-expressing tumors as described above in the LAMP-1 assay. For some experiments OT-1 cells were instead left unstimulated (DMSO) or treated with PMA 1 μg/mL + ionomycin 1 μM as a supraphysiologic stimulus. In all cases, T cells underwent a 5–6 hour incubation period in the presence of DMSO or splicing modulators at the indicated concentrations, and with brefeldin A and monensin present for the entire duration. Cells were subsequently washed, stained for surface markers, and then fixed/permeabilized for intracellular staining of the indicated cytokines according to manufacturer instructions (BD Biosciences).
Western blotting
Western blotting was performed as per standard techniques. Anti-RBM39 (Atlas Antibodies, Cat. HPA001591 or Bethyl laboratories, Cat. A300–291A) were used to detect RBM39 degradation. ADMA and SDMA levels were determined using antibodies from Cell Signaling Technologies (Cat. 13222S and Cat. 13522S). Actin antibody (clone AC-15) was obtained from MilliporeSigma (Cat. A5441-.2ML). Densitometry of RBM39 and actin loading control was performed using ImageJ software in order to calculate RBM39 degradation IC50 values.
Therapeutic treatment with splicing compounds and anti-PD1
Animals were subcutaneously engrafted on bilateral flanks with tumor cells (MC38 1×106, B16-F10 0.5×106 and LLC 0.25×106 cells unless otherwise specified) on day 0, and treated continuously with splicing inhibitors (MS-023 50 mg/kg i.p., indisulam 25 mg/kg i.v. or vehicle) daily for 5 of 7 weekly days starting from day +3 of tumor challenge. Indisulam was obtained from MilliporeSigma (Cat. SML1225–25MG) and MS-023 in sufficient quantities for in vivo studies was synthesized by the authors as previously described (Eram et al., 2016). For in vivo formulation, indisulam was dissolved in sterile DMSO at 50 mg/mL and this was combined in a 1:20 ratio with 15% 2-Hydroxypropyl-β-cyclodextrin (Sigma. Cat. H107–100G) in sterile water (w/v) and filtered through a 0.45 μM filter to yield a final solution of 2.5 mg/mL. For in vivo formulation, 62.5 mg of MS-023 was dissolved in 563 microliters of 1-methyl-2-pyrrolidinone (NMP, Sigma. 328634–1L), diluted with 2.257 mL of 20% Captisol in sterile water (w/v, SelleckChem Cat. S4592) and further combined with 2.257 mg of polyethylene glycol 400 (PEG-400, Sigma Cat. PX1286B-2), and 6.21 mL of PBS, mixed by vortexing and sterile filtered to yield a solution of 5.5 mg/mL. Mice were weighed weekly for weight-based drug dosing. Animals were treated with 250μg of anti-PD1 flat dose (clone RMP1–14, BioXCell Cat. BE0146) or PBS i.p. starting on day +7 and twice weekly thereafter for a total of five doses.
In vivoT cell or NK cell depletion
For depleting T cells, mice were treated with simultaneous anti-CD4 (clone GK1.5, BioXCell Cat. BE0003–1) together with anti-CD8 (clone 2.43, BioXCell Cat. BE0061) versus PBS control, at days −7, −4, +4, and +7 relative to tumor challenge on day 0. Each depleting antibody was administered i.p. at 0.5 mg per dose. 0.5×106 B16-F10 which were treated in vitro with indisulam at 1μM or DMSO for 96 hours were engrafted subcutaneously on the flanks of animals receiving T cell depletion or PBS control. For NK cell depletion, an identical experimental schedule and dose using clone PK136 (BioXCell Cat. BE0036) was utilized. To verify T cell depletion, CD4 clone H129.19 (Biolegend Cat. 130310), CD8 clone 53–5.8 (Biolegend Cat. 140410) were used. NKp46 (Biolegend Cat. 137608) was used to verify NK cell depletion.
CFSE adoptive T cell transfer and splicing modulator treatment
Splenic T cells were obtained from naïve B6 or CD45.1 donors by CD5 positive selection (Miltenyi Biotec, Cat. 130–049-301), labeled with CellTrace CFSE (ThermoFisher Cat. C34570) at 10μM, and adoptively transferred by tail vein injection into lethally irradiated B6, Balb/c, or LP/J recipients, with 107 labeled donor T cells transferred per recipient. All recipients were irradiated on day −1 prior to adoptive T cell transfer with 7 Gy as a single fraction and continuously received splicing inhibitor drugs or vehicle control at the indicated doses, from day −1 until day of sacrifice, with the initial dose of drug at least 4 hours after lethal irradiation. Recipients were treated with each splicing modulatory compound at doses used in prior studies that result in target engagement in vivo (Fong et al., 2019; Wang et al., 2019). Indisulam and MS-023 were solubilized for in vivo administration and animals were treated daily as above. Pladienolide B (Tocris, Cat. 6070) and GEX1A (Cayman Chemicals, Cat. 25136) were both dissolved in vehicle (10% ethanol and 4% Tween-80 in sterile PBS) and administered i.p., with pladienolide B dosed at 10 mg/kg every other day, and GEX1A dosed at 1.25 mg/kg every four days. For in vivo use, EPZ015666 was dissolved in DMSO and solubilized in 0.5% methylcellulose in water to 20 mg/mL; animals were treated daily with 200mg/kg by oral gavage.
Anti-CD3/CD28 T cell activation
Plates were coated with 10μg/mL anti-CD3 (clone 145–2C11, Biolegend Cat. 100302) and 2 μg/mL anti-CD28 (clone 37.51, Biolegend Cat. 102102) in PBS overnight at 4°C and washed twice with cold PBS prior to use. CFSE-labeled CD5-selected splenic T cells from naïve C57BL/6J mice were obtained identically as for adoptive cell transfer, and 5×104 cells incubated with coated plates in the presence of splicing inhibitor drugs at the indicated concentrations, followed by analysis by standard flow cytometry on day 3. Of note, for RNA-seq analyses, T cells were not labeled with CFSE, and underwent activation for 4 days (96 hours) in the presence of various splicing modulator drugs to harmonize experimental conditions with RNA-seq analyses of tumors treated with splicing inhibitors. For the RNA-seq experiments only, T cells in all conditions were also incubated with IL-2 at 50U/mL to maximize viability and yield.
Mixed leukocyte reaction
RBC lysed bone marrow obtained from the femurs and tibias of C57BL/6 or β2 microglobulin deficient mice (Jackson Laboratories Cat. 2087) were cultured with mouse IL-3 (PeproTech Cat. 213–13) and mouse FLT3 ligand (PeproTech Cat. 250–31L) both at 10 ng/mL each in RPMI + 10% FCS for 7 days to generate bone marrow derived dendritic cells. Separately, 107 MC38 treated with splicing inhibitors vs. DMSO or expressing chicken ovalbumin were harvested, washed and resuspended in sterile PBS, and subjected to five cycles of rapid freeze-thaw (alternating between 37°C and dry ice/acetone) to generate a cell lysate. After brief centrifugation at 100xg, the soluble fraction in PBS was added to bone marrow derived DCs and left to incubate overnight for antigen phagocytosis in the presence of LPS (ThermoFisher Cat. 00–4976-93). DCs were subsequently washed three times to remove cell-free lysates and LPS and incubated in a 1:1 ratio with CFSE-labeled B6 splenic T cells (105 stimulators with 105 responders) as described above. The MLR was analyzed at day 5 by flow cytometry.
M3434 methylcellulose colony assay
25,000 red blood cell-lysed bone marrow mononuclear cells from C57BL/6 mice were plated in duplicates or triplicates in each well of a non tissue-culture treated 6 well plate with M3434 methylcellulose media in the presence of splicing drugs at the indicated concentrations as per manufacturer’s instructions (StemCell Technologies, Cat. 03434) and incubated for seven days prior to quantification of colonies by manual microscopy.
Intracellular flow cytometry
Cells were fixed with 2.1% formaldehyde in PBS for 10 minutes at 37°C, washed and permeabilized with ice-cold 90% methanol for 30 minutes, and washed prior to staining. If required, cell surface staining was performed after fixation but prior to permeabilization. For some experiments, intracellular staining was performed using the eBioscience™ Foxp3 transcription factor staining buffer set (ThermoFisher Cat. 00–5523-00) or reagents for intracellular cytokine staining (BD Cytofix/Cytoperm™, Cat. 554714, and BD Perm/Wash™, Cat. 554723) as per manufacturer’s instructions.
Histology
Animal tissues were fixed in 4% paraformaldehyde, decalcified (for bone), dehydrated and paraffin embedded. Blocks were sectioned and stained with hematoxylin and eosin or anti-CD8. Images were acquired using an Axio Observer A1 microscope (Carl Zeiss, Oberkochen, Germany) or scanned using an Aperio AT slide scanner (Leica Biosystems, Buffalo Grove, IL). Automated quantification of infiltrating CD8+ T cells was performed using HALO software (Indica labs, Albuquerque, NM). Pathologic evaluation of immune-related tissue toxicities was performed in a blinded fashion by one of the authors who is a trained pathologist (Ben Durham, MD).
Cellular fractionation for RNA sequencing
Nuclear and cytoplasmic cellular fractions were isolated from B16-F10 cells using reagents from Active Motif (Cat. 25501) as per manufacturers’ instructions, with the exception of RNA isolation and purification from each fraction using the QIAgen RNeasy Mini kit.
RNA sequencing
Bulk lung and colon were homogenized using a Qiagen TissueRuptor. For all tissues and cell types, RNA was extracted using an RNeasy kit (Qiagen, Frederick, MD) and quantified using a NanoDrop 8000 (ThermoFisher Scientific). A minimum of 500 ng of high-quality RNA (as determined by Agilent Bioanalyzer) per sample or replicate was used for library preparation. Poly(A)-selected, strand-specific (dUTP method) Illumina libraries were prepared with a modified TruSeq protocol and sequenced on the Illumina HiSeq 2000 (∼100M 2×101 bp paired-end reads per sample or replicate).
RNA-seq data analysis
RNA-seq analysis was performed as previously described (Dvinge et al., 2014). Briefly, FASTQ files were mapped using RSEM version 1.2.4 (Li and Dewey, 2011) (modified to call Bowtie (Langmead et al., 2009) with option ‘-v 2’) to mouse or human transcriptome annotations built using transcript information from Ensembl v71.1 (Flicek et al., 2013), UCSC knownGene (Meyer et al., 2013), and MISO v2.0 (Katz et al., 2010). Reads that did not align at this step were then mapped using TopHat version 2.0.8b (Trapnell et al., 2009) to the mouse (GRCm38/mm10) or human (GRCh37/hg19) genome assemblies, as well as to a database of annotated splice junctions as well as all possible new junctions consisting of linkage between each co-linear annotated 5’ and 3’ splice sites within individual genes. Aligned reads from these two mapping steps were merged to generate final BAM files for all subsequent analyses.
Gene expression estimates were computed using RSEM (performed concordantly with the RNA-seq read mapping procedure described above). Significantly differentially expressed genes were defined as those meeting the follow criteria: minimum expression of 1 transcript per million (TPM); minimum fold-change of 1.5 (log2 scale); p ≤ 0.05 (computed using an unpaired, two-sided t-test comparing replicate groups for a given treatment and cell line) or a minimum Bayes factor of 100 (computed using Wagenmakers’s Bayesian framework (Wagenmakers et al., 2010) for the median of gene expression and associated read counts over replicates for a given treatment and cell line). Splice junction-spanning reads were filtered to require a minimum overhang of 6 nt.
MISO v2.0 was used to quantify all expression of isoforms arising from exon skipping (cassette exons), competing 5’ splice site selection, competing 3’ splice site selection, and annotated intron retention. Quantification of constitutive intron retention, where constitutive introns were defined as those whose 5’ and 3’ splice sites were never joined to other splice sites in the knownGene annotation, was calculated as previously described (Hubert et al., 2013) using reads with a minimum of 6 nt overhang in both the exon and intron. Events were considered significantly differentially spliced if they met the following criteria: a minimum of 20 identifying reads (reads which align only to one, but not both, isoforms constituting a given splicing event) in each sample; a minimum of 10% change (absolute scale) in isoform ratio or minimum fold-change of 2 (log2 scale) in absolute isoform ratio; p ≤ 0.05 (computed using an unpaired, two-sided t-test comparing replicate groups for a given treatment and cell line) or a minimum Bayes factor of 5 (computed using Wagenmakers’s Bayesian framework (Wagenmakers et al., 2010) for the median of isoform ratios and distinguishing read counts over replicates for a given treatment and cell line). All data parsing, statistical analyses, and data visualization were performed using the R programming environment with Bioconductor (Huber et al., 2015).
MHC I immunoprecipitation, peptide purification, and mass spectrometry
Peptide-MHC complexes were isolated as previously described (Abelin et al., 2017), with the following modifications: anti-mouse H-2Db (clone B22–249.R1, CedarLane laboratories, Cat. CL9001AP) or H-2Kb (clone Y-3, BioXCell Cat. BE0172) non-covalently linked to GammaBind Plus Sepharose beads were co-incubated with soluble lysates overnight. After washing with lysis buffer twice, 10mM Tris pH 8 twice, and dH2O twice, the peptides were desalted on C18 StaGE tips (Ishihama et al., 2006) (Pierce, Cat. 87784) and eluted using a 20%−35%−50% acetonitrile stepwise gradient. Eluted fractions were dried using a SpeedVac™ vacuum concentrator and stored until mass spectrometry. For B16-F10, cells in all experimental conditions were treated with 10U/mL mouse IFNγ (PeproTech Cat. 315–05) for 48 hours prior to cell harvest and immunoprecipitation to upregulate surface MHC I expression.
Mass spectrometry
Desalted, dried samples enriched for MHC peptides were resolubilized in 8uL 0.1% TFA and 3uL were loaded onto a packed-in-emitter 12cm/75um ID/3um C18 particles column (Nikkyo Technos Co., Ltd. Japan). Peptides were eluted using a gradient delivered at 300nL/min increasing from 2% Buffer B (0.1% formic acid in 80% acetonitrile) / 99% Buffer A (0.1% formic acid) to 30% Buffer B / 70% Buffer A, over 70 minutes (EasyLC 1200, Thermo Scientific). All solvents were LCMS grade (Optima, Fisher Scientific). MS and MS/MS (HCD type fragmentation) experiments were performed in data dependent mode with lock mass (m/z 445.12003) using Fusion Lumos (Thermo Scientific). Precursor mass spectra were recorded from m/z 300–1500 m/z range at 60,000 resolution. 1, 2 and 3 positive charges were selected for fragmentation experiments. MS/MS spectra were recorded at 30,000 resolution and lowest mass set at m/z 110. For MS/MS acquisition, injection time was set to maximum 100 milliseconds with an Auto Gain Control setting of 5e4. Normalized collision energy was set to 30. All experiments were recorded in FT-mode.
Proteome creation
Gene and isoform annotations were created as described in RNA-seq data analysis. This merged transcript annotation, as well as the RefSeq annotations of the human and mouse genomes, was used to create the four distinct proteomes described in the main text as follows. Isoforms were computationally translated into proteins and digested into unique 8–14-mers. Isoforms were translated into proteins “conservatively,” in the sense that the translation was performed assuming that the annotated start codons were used and no stop codon readthrough or internal translation initiation occurred (e.g., generally only the first portion of a retained intron would be translated until an in-frame premature termination codon was encountered, after which translation was assumed to halt). The binding affinity for each resulting peptide to the relevant MHC alleles was then predicted using NetMHCpan v4.0 (Jurtz et al., 2017). Each peptide was annotated with relevant information about its encoding transcript, including parent gene, parent isoform(s), differential gene and/or isoform expression (if relevant), position within parent transcript, unique assignment to one versus two or more isoforms of the originating splicing event (if relevant), etc.
Four distinct, custom proteomes for subsequent spectra mapping were created (illustrated in Fig. 5B). (1) “full-length proteome”, created using peptides arising from all unique full-length isoforms. (2) “predicted binders”, created by further restricting to unique 8–14-mers that had a NetMHCpan 4.0 percentile rank < 2 (the recommended cutoff for binders from NetMHCpan 4.0). Two versions of this proteome were created, one including only those isoforms derived from differentially retained constitutive introns based on the RNA-seq data, and one including all isoforms derived from constitutive intron retention (constituting an increase in unique 8–14-mers of ∼28%). Analyses used the complete (latter) proteome unless otherwise indicated. (3) “predicted binders + spiked non-binders”, created by augmenting the “predicted binders” proteome with peptides that were predicted to not bind the relevant MHC alleles with high-confidence, defined as having NetMHCpan percentile rank > 90, with the number of such non-binders chosen such that they comprised 10% of the final proteome after adding to the “predicted binders” proteome. (4) “filtered predicted binders”, created by further filtering the “predicted binders” proteome by restricting to peptides arising from genes that were significantly differentially expressed or isoforms that were significantly differentially spliced in indisulam-treated versus DMSO-treated samples, defined based on the RNA-seq analysis for the corresponding cell lines.
Peptide identification from mass spectrometry data
Mass spectra from all MHC immunoprecipitations were analyzed using Proteome Discoverer v2.4.1.15, with the following workflow. Spectra from each replicate were searched against each distinct proteome (described above) as follows. For each proteome, searches were performed with no enzyme specificity, precursor mass tolerance of 10 ppm, and fragment mass tolerance of 0.6 Da. Oxidation (+15.995 Da), phosphorylation (+79.966 Da), and deamidated (+0.984 Da) dynamic modifications were included, in addition to N-terminal glutamate to pyro-glutamate (−17.027 Da). False discovery rate (FDR) estimation was performed computationally using the Percolator software. Peptides reaching the 5% FDR threshold were retained for downstream analyses. For the “full-length” proteome, identified peptides were further restricted to those of length 8–14 amino acids before being used as input for subsequent analyses. For the “predicted binders”, “predicted binders + spiked non-binders”, and “filtered predicted binders” proteomes, peptides corresponding to subsequences of the sequences in the input proteomes were removed before the identified peptides were used for subsequent analyses.
Candidate neoepitope identification
As described in the main text, two distinct groups of candidate neoepitopes were selected for subsequent immunization experiments. The first group was based on the intersection between mass spectrometry analyses and RNA-seq analyses. Peptides were first identified using the mass spectrometry analysis described above. These peptides were then restricted to the set of indisulam-specific peptides, where an indisulam-specific peptide was defined as a peptide that was identified in one or more indisulam-treated samples, but not recovered in any DMSO-treated samples. These indisulam-specific peptides were then filtered to retain only those peptides arising from alternative isoforms that were significantly differentially spliced in indisulam-treated versus DMSO-treated cells, and subsequently additionally filtered to require (1) isoform specificity and (2) appropriate direction of differential splicing, with those two criteria defined as follows. (1) An isoform-specific peptide was defined as a peptide which arose exclusively from one isoform associated with a given splicing event (e.g., a peptide from a retained intron event is isoform-specific if it arises from translation of the intronic portion of the unspliced mRNA, or if it arises from translation of the exon-exon junction within the spliced mRNA). This definition means that differential splicing of a given event is predicted to alter levels of the isoform encoding an isoform-specific peptide, and therefore likely similarly alter abundance of the isoform-specific peptide itself. (2) Peptides that exhibit appropriate direction of differential splicing are those isoform-specific peptides which are specifically encoded by differentially spliced isoforms that are promoted by indisulam treatment (e.g., the encoding isoform is present at higher levels in indisulam-treated versus DMSO-treated cells). Isoform-specific peptides were only used for subsequent immunization experiments if their parent isoform was more prevalent in the indisulam treatment, signifying that the peptide is expected to be more abundant in indisulam-treated cells. These criteria yielded 72 peptides, which were subsequently tested in immunization experiments.
The second group of peptides used for immunization experiments was derived by combining evidence from RNA-seq analyses and MHC I binding predictions. This set of peptides was defined using the same criteria described above for the first set (derived by intersecting predictions from mass spectrometry analyses as well as RNA-seq analyses), but without the requirement that peptides be detected as indisulam-specific epitopes via MHC I mass spectrometry. To compensate for the fact that direct protein-level detection was not required, a stringent predicted MHC I binding threshold of rank < 0.5 (the NetMHCpan recommended threshold for strong binders) for one or more relevant alleles was applied (versus the more lenient threshold of rank < 2 used for other, mass spectrometry-based predictions and analyses). Peptides were additionally restricted to those of lengths between 8 and 11 amino acids, as such lengths are preferred by the studied alleles. The final set of peptides used for subsequent immunization experiments was then derived by additionally requiring that peptides be isoform-specific; arise from genes with expression >5 TPM in corresponding indisulam-treated samples (in order to favor peptides from relatively highly expressed genes); and have a difference in isoform ratio >20% in indisulam-treated versus DMSO-treated samples, and isoform ratio <25% in DMSO-treated samples (in order to restrict to peptides that were associated with more dramatic splicing changes). These criteria yielded 39 peptides, which were subsequently tested in immunization experiments.
Peptide Synthesis
Experimental peptides were individually custom synthesized via the solid-phase method by GenScript (Piscataway, NJ), with standard removal of trifluoracetic acid and replacement with hydrochloride, purified to >98% by HPLC, and lyophilized for storage. Peptides were reconstituted in DMSO at 10 mg/mL and frozen at −80C until use.
RMA-S peptide H-2 stabilization assay
RMA-S cells were maintained under standard conditions in RPMI + 7.5% FCS for expansion. H-2 stabilization experiments were performed as previously described (Ross et al., 2012). Briefly, RMA-S were exposed to 31°C and 5% CO2 conditions overnight, incubated with peptides of interest for 30 minutes at 31°C, and then returned to 37°C and 5% CO2 for three hours prior to cell surface staining for H-2Kb (clone AF6–88.5) and H-2Db molecules (clone KH95) and standard flow cytometry analysis. For Figure 5N–Q), spike-in negative control peptides were as follows for each experimental peptide – Hus1: PPSGRALLW; Zpf512: QKPKGSQRG; D14Abb1e: LKPQAKRSK; Poldip3: GESWQEKER
TiterMax immunization
Unless otherwise specified, 10 μg of peptide was emulsified with TiterMax Classic (TiterMax Corp., Norcross, GA) and injected into the hocks of anesthetized animals. On day +7 after challenge, draining lymph nodes were collected and CD8+ T cells purified by magnetic selection (Miltenyi Biotec, Cat. 130–117-044).
IFNγ ELISpot
CD8+ T cells from TiterMax immunized animals were cultured overnight with 20 U/mL mouse IL-2 (PeproTech, Cat. 212–12) and plated at 105 per well in combination with 3×105 T cell depleted syngeneic splenocytes which had been loaded with 100 μg/mL of peptides of interest for 18 hours. PMA 1 μg/mL + ionomycin 500 ng/mL stimulation of T cells served as positive control. In some experiments, in lieu of peptide-loaded splenocytes, instead ovalbumin-expressing B16-F10 cells or B16-F10 cells treated with DMSO or indisulam 1 μM for 96 hours were stimulated overnight with IFNγ 100U/mL for the last 24 hours of cell culture. Such cells were then non-enzymatically harvested, washed repeatedly to remove IFNγ, and irradiated to 60 Gy from a 60Co source to inhibit growth and further upregulate MHC I. Tumor cells thus generated were counted and incubated with CD8+ T cells at identical ratios as for splenocytes (105 CD8+ T cells + 3×105 melanoma cells). IFNγ ELISpot was performed as per manufacturer’s instructions (BD Biosciences, Cat. 551083). Spots were imaged and quantified on an Immunospot® analyzer (Cellular Technology Limited, Cleveland, OH).
B16-F10 co-culture cytotoxicity assay
B16-F10 cells were harvested, counted, and plated at 104 per well in the presence of 100U/mL IFNγ overnight to upregulate MHC I. After washing, peptides were loaded onto tumor cells at 100 μg/mL, and 106 CD8+ T cells from TiterMax immunized animals were added to the tumor cells. 50 U/mL mouse IL-2 was added to this co-culture of tumor cells + CD8+ T cells, which was incubated for three days. After washing to remove free (detached) B16-F10 and T cells, viable B16-F10 were harvested, stained (to exclude T and other hematopoietic cells) and absolute cell numbers enumerated via flow cytometry using counting beads according to the manufacturers’ instructions (ThermoFisher Cat. C36950).
Supplementary Material
Figure S1 —related to Figure 1. Pharmacologic perturbation of RNA splicing impairs tumor growth in vivo, in a dose-dependent fashion dependent on adaptive immunity. (A) Left, Western blot of RBM39 after in vitroexposure to the indicated concentrations of indisulam for 24 hours in MC38 and CT26 cells. Right, half-maximal inhibitory concentration (IC50, computed using the Hill equation) values for cell growth inhibition and RBM39 degradation. (B) Dose-response curves for RBM39 abundance (normalized to β-actin) across the indicated concentrations of indisulam treatment as measured by densitometry. IC50 values are shown. (C) Western blot of RBM39 after MC38 or CT26 cells were exposed to 1μM indisulam for 96 hours, drug was washed off, and RBM39 levels were allowed to recover. (D) Annexin-V and DAPI analysis of MC38, B16-F10, CT26, and MB49 cells after in vitro treatment with indisulam 1μM or DMSO for 96 hours. (E) Heatmap of MFI values above isotype control of cell surface levels of H-2K/H-2D, I-A/I-E (MHC II), or PD-L1 after 96 hours of treatment with DMSO, indisulam 1μM, MS-023 5μM, or EPZ015666 1μM, or the same drugs in the presence of mouse IFNγ 10U/mL. CRISPR-mediated β2-microglobulin knockout cells served as biologic control for H-2K/H-2D staining. B16-F10 cells shown on top (blue). CT26 cells on bottom (red). Representative histograms of B16-F10 data are shown on right. (F) Cell surface levels of cytokine and death receptors in B16-F10 cells after treatment with conditions identical to (D). (G) Sample photos of B16-F10 (top), MC38 (middle), CT26 (bottom) bearing mice treated with indisulam or DMSO and engrafted into syngeneic animals. (H) Experimental schema. MC38 or CT26 cells were treated with DMSO or indisulam at the indicated concentrations for 96 hours in technical triplicate; these were then subjected to RNA-seq analyses or used for biological experiments as in (L-O). (I) numbers of constitutive U2-type introns retained in MC38 cells treated with the indicated doses of indisulam compared with DMSO. (J) Total number of splicing alterations in CT26 and MC38 tumors exposed to the indicated doses of indisulam, as compared with DMSO. (K) RNA-seq coverage plot depicting two representative intron retention events in Agxt2l2 and Hmgxb4 with increasing doses of indisulam. (L) experimental schema showing the engraftment of CT26 tumors treated with indisulam in vitro into syngeneic Balb/c mice. (M) tumor volumes of CT26 bearing mice from (L) over time (n=15 mice/group; tumors engrafted on bilateral flanks of mice). Mean ± sem. (N) tumor volumes from (M) at day 24. P-values were calculated for the indicated group compared to DMSO control using the Wilcoxon rank-sum test: *, p = 0.048; ***, p = 0.000798; ****, p = 0.000363. (O) Kaplan-Meier survival curve of animals from (L). (P) Individual tumor growth curves for DMSO-treated and indisulam-treated B16-F10 cells in Rag2 knockout mice. n=10 mice per group; tumors engrafted on bilateral flanks. (Q) Box-and-whisker plots of day 28 individual tumor volumes from (P). (R) Schema of CD4 & CD8 T cell depletion or NK cell depletion versus control. (S) Representative flow cytometry confirmation of T cell depletion using anti-CD4 and anti-CD8 antibodies, gated for CD3ε in the spleens of animals sacrificed at day 19 post-tumor challenge. Remaining double-negative population are NK1.1+ NKT cells (not shown). (T) Representative flow cytometry confirmation of NK cell depletion via NKp46 antibody; peripheral blood on day 14 after tumor challenge. (U-V) Box-and-whisker plot quantification of (S) and (T). Each dot represents an individual animal. (W) Sample photos of tumor-bearing animals treated with CD4/CD8 T cell depletion and challenged with B16-F10 cells pre-treated with indisulam or control. (X) Individual tumor growth curves for NK1.1 depletion experiment. n=10 per group; tumors engrafted on bilateral flanks. (Y) Box-and-whisker plots of day 21 tumor volumes for B16-F10 treated as in Fig. 1A, with or without NK cell depletion. *p<0.05; **p<0.01. ANOVA calculated using the Kruskal-Wallis test and multiple comparisons vs. DMSO alone calculated using the Dunn’s test.
Figure S2 —related to Figure 2. Class-specific effects of splicing modulator drugs on hematopoiesis, T cell gene expression and function in vitro and in vivo. (A) Left, western blot showing dose-dependent reduction in asymmetric dimethyl arginine (ADMA) with MS-023 treatment in MC38 tumor cells; right, corresponding flow cytometry analysis for intracellular ADMA. (B) Schema for the mixed leukocyte reaction, depicting generation of tumor lysates, generation of bone marrow derived dendritic cells, process of pulsing dendritic cells with lysates and lipopolysaccharide (LPS) for maturation and antigen presentation, and mixed leukocyte reaction with congenic CD45.1 CFSE-labeled naïve splenic T cells. (C) Schema depicting purification of splenic T cells, CFSE labeling, and in vitro stimulation with plate-bound anti-CD3 and anti-CD28 antibody in the presence of splicing drugs. (D) Full dose range of splicing drugs tested in (C) demonstrating dose-dependent inhibition of CFSE dye dilution and T cell proliferations at higher drug concentrations; representative histogram from N=8 technical replicates each. (E) absolute numbers of CD4 (left) and CD8 (right) T cells from experiment in (C) for each drug and concentration were quantified to calculate IC50 for cell proliferation (table). Each dot is the average from 3–8 technical replicates. Curves and IC50 values for the activation markers CD25 and PD1 are shown (bottom panels), and EC50 for induction of apoptosis as measured by annexin-V staining is shown (middle panel). (F) OT-1 killing assay against wildtype or ovalbumin-expressing MC38 tumors; experiment performed as in Fig. 2G. (G) representative contour plots showing the staining of OT-1 cells for TNFα and IFNγ or isotype control, after exposure to DMSO (left) or PMA/Iono (Phorbol 12-myristate 13-acetate and Iono: ionomycin, right). Percentage of positive cells are quantified in the associated bar plots. Each dot is one technical replicate. (H) As in (G) but OT-1 cells were stimulated with PMA/Iono (top) or incubated with MC38 cells overexpressing ovalbumin at a 1:1 ratio (bottom), at the indicated concentrations of indisulam or MS-023 on the x-axis. Graph shows mean ± sem. N=4 technical replicates per condition. (I) Experimental schema demonstrating normal intracellular localization of the LAMP1 protein on cytotoxic granules in resting cytotoxic T cells, and appearance of LAMP1 on the plasma membrane upon T cell degranulation, rendering the protein stainable for flow cytometry. Bar plots at right show LAMP1 MFI of OT-1 cells remaining unstimulated, exposed to PMA/ionomycin, or incubated at a 1:1 ratio with MC38 overexpressing ovalbumin. Each dot indicates a technical replicate. P-values were calculated using the Wilcoxon rank-sum test. (J) experiment as in (I) except in the presence of the indicated doses of indisulam or MS-023 on the x-axis. N=4 technical replicates per condition. Graph shows mean ± SD. P-values were calculated using the Kruskal-Wallis non-parametric ANOVA test. (K) experimental design: naïve C56BL/6 splenic T cells were isolated and cultured ex vivo for 96 hours in media with IL-2 50U/mL. Cells were either left unstimulated, or activated with plate-bound anti-CD3 + CD28 antibody in the presence of DMSO or the indicated splicing compounds: indisulam 1μM, MS-023 1μM, EPZ015666 5μM, or pladienolide B 2nM. At the end of 96 hours, cells were subjected to RNA-seq. N=3 replicates for each condition. (L) Differential gene expression from (K) of unstimulated T cells versus those stimulated in the presence of DMSO. (M) Box plots of fold changes of the top 100 upregulated genes in T cells undergoing activation from (K) in the presence of the indicated drugs. Each dot is one replicate. (N) Schema of adoptive transfer of C57BL/6 CD45.1 CFSE labeled T cells into syngeneic, minor antigen mismatched, or major antigen mismatched lethally irradiated recipients to assess T cell activation and expansion. (O) Histograms of CFSE dilution of donor adoptively transferred CD45.1+ CD8+ T cells into Balb/c mice on day 3 to assess for alloreactivity. (P) Representative flow cytometry plots of T cell activation and death markers in adoptively transferreddonor CFSE-labeled CD45.1 splenic CD4+ or CD8+ T cells into lethally irradiated Balb/c recipients with or without daily indisulam (25 mg/kg) treatment. Spleens were analyzed on day 3. Two animals from each condition are shown. (Q) as in (P) but with daily MS-023 treatment, 50mg/kg daily. (R-S) Histograms of CFSE dilution in donor adoptively transferred CD45.1+ T cells into syngeneic C57BL/6 mice to assess for homeostatic proliferation. Cells in the spleen on day 7 after transfer are shown. (T) Numbers of colonies in M3434 methylcellulose media on day 7. 25,000 red blood cell-lysed bone marrow hematopoietic stem and precursor cells (HSPC) from C57BL/6 mice were grown in methylcellulose with continuous exposure to splicing compounds at the indicated concentrations.
Figure S3 —related to Figure 3. Treatment of tumor-bearing animals with splicing modulator compounds in vivo enhances tumor control and can elicit memory. (A) Growth curves of individual B16-F10 tumors from C57BL/6 mice exposed to the indicated treatment, as summarized in Figure 3C. Each line represents one tumor; termination of lines prior to the end of the experiment indicates animal mortality or euthanasia according to predetermined endpoints, as described in the Methods section. (B) Experimental design: MC38 cells were treated with DMSO or MS-023 in vitro for 96 hours, and then 106 cells engrafted onto the bilateral flanks of either naïve C57BL/6 mice, or animals which had successfully rejected MC38 tumors previously after treatment with anti-PD1 and MS-023 in vivo (from Figure 3K–M). (C) Growth curves of individual tumors from (B). Pink and purple arrows indicate tumors which were implanted but rejected. (D) Line graphs summarizing growth curves from (C), showing mean ± sem. (E) Violin plots showing tumor volumes at day 28. P-values (calculated using the Wilcoxon rank-sum test) for the “DMSO survivor” -vs “DMSO naïve” and “MS-023 survivor” -vs- “MS-023 naïve” comparisons are 0.044 and 0.011, respectively. (F) Representative animals from (E).
Figure S4 —related to Figure 3. Lack of toxicity and absence of inflammatory signatures or increased CD8+ T cell infiltrates into healthy, non-tumor tissues in animals treated with splicing modulator compounds with or without anti-PD1. (A) Peripheral blood counts of tumor-bearing mice treated for 3 weeks with vehicle, anti-PD1 (250μg flat dose), indisulam (25 mg/kg), MS-023 (50 mg/kg), or combined anti-PD1 with MS-023 or indisulam. ALC: absolute lymphocyte count; ANC: absolute neutrophil count; Hgb: hemoglobin; Plt: platelet count; WBC: white blood cell count. (B) Percentage of CD8+cells amongst CD45+ cells (left) and proportion of CD8+/CD11b+ cells (right) in tumors at day 21 following treatment of animals bearing MC38 tumors with the treatments indicated on the x-axis. n=10 mice/group. ANOVA calculated using the Kruskal-Wallis test. (C) H&E stained tissue sections from mice treated with indisulam, MS-023, anti-PD1, or combination at day 21. N=5 animals per treatment group per tissue were assessed. Review of the necropsy tissues shows no overt evidence of toxicity, and the tongue shows typical squamous epithelium overlying bundles of skeletal muscle. The lung demonstrates typical alveolar spaces, alveolar septa, and bronchioles with respiratory epithelium. The large intestine shows typical colonic mucosa, submucosa, and muscularis propria. The skin from the ear demonstrates typical epidermis, dermis, and subcutaneous tissues overlying cartilage. The liver shows typical portal tracts and hepatocytes, and the small intestine demonstrates normal villi with typical mucosa, submucosa, and muscularis propria. Representative micrographs of the findings are depicted. (H&E; Magnification 400x; Scale bar: 50 microns). (D) Liver and pancreas enzymes and markers in the blood from these same animals at the time of sacrifice (n=3 mice/group). ALT: alanine aminotransferase. AST: aspartate aminotransferase. Mean ± sd shown. (E) B16-F10-tumor bearing C57BL/6 mice received the indicated treatment; tissues were collected on day 21. Paraformaldehyde-fixed and paraffin-embedded tissues were sectioned and stained for CD8 by immunohistochemistry. Representative sections of the tongue (muscle), liver, ear (skin), small and large bowel and lung are shown. 20x magnification. Scale bar: 100 microns. (F) Violin plots depicting the automated enumeration of CD8+ T cell infiltrates in the corresponding organs are shown. N=3 mice per tissue per treatment were analyzed, and N=5 fields per mouse were analyzed. Fold-change in CD8+ T cell infiltrates versus the vehicle condition are shown. p-values were calculated using the Wilcoxon rank-sum test. (G) Experimental design: C57BL/6 mice were engrafted with 106 MC38 tumor cells on each flank and subjected to treatment with vehicle, indisulam 25mg/kg beginning day +3, anti-PD1 250 μg twice weekly beginning day +7, or the combination. Lung, colon, and splenic T cells were obtained on day 14 after tumor challenge for RNA-seq. (H) Stacked bar graphs indicating the absolute number and type of splicing alterations in the lungs, colon and T cells of animals receiving the indicated therapy. Splicing alterations were defined as a 5% change in splicing with a p-value threshold of 0.05 (I-K) Scatter plots indicating constitutive intron retention in tissues of animals treated with indisulam versus vehicle. (L-N) Scatter plots indicating differentially expressed genes in the colon, lung and T cells of animals treated with vehicle versus combination of indisulam and anti-PD1. (O) Pathway analyses of differential gene expression from (L-M) in the colon and lung. No significantly enriched gene ontology pathways were identified in the T cell RNA-seq dataset.
Figure S5 —related to Figure 4. Splicing alterations generated by MS-023 and indisulam treatment. (A) Left, scatter plot comparing constitutive intron splicing in DMSO- versus MS-023-treated B16-F10 cells. Right, RNA-seq read coverage illustrating increased intron retention following MS-023 treatment. (B) As (A), but for cassette exons. (C) Bar graphs representing numbers of differentially spliced events of each indicated type in DMSO- versus MS-023-treated B16-F10 cells. Blue/green, mis-splicing events indicative of reduced/increased splicing efficiency (for example, decreased/increased cassette exon inclusion). (D) As (A), but for human SK-MEL-239 cells. (E) As (D), but for cassette exons. (F) As (C), but for human SK-MEL-239 cells. (G) representative example of a 5’ alternative splice site event induced with indisulam treatment in all of the murine cell lines studied. (H-I) two representative examples of intron retention events observed in all murine cell line studied. (J) Venn diagram depicting the number of genes mis-spliced in common across mouse versus human cell lines analyzed, upon exposure to either indisulam or MS-023. (K) Left, heat map illustrating Pearson correlations for all retained introns, constitutive introns, and cassette exons that are differentially spliced following indisulam or MS-023 treatment in at least one sample from the indicated mouse cell lines. Pearson correlations computed using the absolute isoform expression values (0–100%) for each splicing event. Right, identical analysis, but for human data. Dendrograms, unsupervised clustering by the Euclidean distance method. (L) Venn diagrams depicting shared mis-splicing events upon exposure to indisulam or MS-023 in the indicated murine cell lines. (M) as for (L) but for the indicated human cell lines studied. (N) Bar graph of Malat1 RNA levels in the cytoplasmic (cyto.) and nuclear (nuc.) fractions of DMSO-treated B16-F10 cells. As Malat1 RNA is restricted to the nucleus(Hutchinson et al., 2007), these data confirm the specificity of our fractionation. (O) RNA-seq read coverage illustrating that increased Prpf40b intron retention following indisulam treatment of B16-F10 cells is evident in total (left), nuclear (middle), and cytoplasmic (right) fractions. Gray, DMSO treatment; red, indisulam treatment. (P) Scatter plot comparing constitutive intron splicing in the cytoplasmic fractions of DMSO- versus indisulam-treated B16-F10 cells. Increased intron retention is readily evident in the cytoplasm. (Q) Venn diagram indicating shared and unique predicted neoantigens among mouse and human cell lines, treated with either indisulam or MS-023. (R) Shared and unique predicted neoantigens, across mouse and human cell lines upon treatment with splicing modulator compounds.
Figure S6 —related to Figure 5. Analysis of mass spectrometry results for H-2Db and H-2Kb immunoprecipitations. (A) As Fig. 5A, but for H-2Kb immunoprecipitation. (B) As Fig. 5D, but for H-2Kb immunoprecipitation. (C) Histogram of lengths of identified peptides using the predicted binders proteome. The majority of identified peptides are 8–9-mers, as expected. (D) As Fig. 5E, but for H-2Kb immunoprecipitation. (E) As Fig. 5F, but for H-2Kb immunoprecipitation. (F) As Fig. 5G, but for H-2Kb immunoprecipitation. (G) Table showing selected candidate neoantigenic peptides induced by indisulam exposure and shared across MC38, B16-F10, CT26 and MB49 mouse tumor cell lines, which were subjected to experimental analysis by TiterMax immunization of C57BL/6 mice followed by evaluation of reactive CD8+ T cells by IFNγ ELISpot.
Figure S7 —related to Figures 6 and 7. Functional evaluation of candidate splicing-derived, neoantigenic peptides. (A) Schema of MHC I peptide stabilization assay on TAP-deficient RMA-S cells. (B) Heatmap of mean MFI values of H-2Db levels from RMA-S peptide stabilization experiments across 30 peptides identified as binding to H-2Db in the RMA-S assay. Green text indicates control known immunogenic peptide with H-2Db binding (gp100). (C) Number of spots per 105 CD8+ T cells from IFNγ ELISpot quantified across 70 peptides derived from mass spectrometry analyses and tested in vivo which did not elicit an immune response. Bar indicates median. Each dot represents a technical replicate. (D) Diagram of experiment immunizing C57BL/6 mice with graded doses of candidate peptides at 0.1, 1, 10, or 100 μg followed by analysis of CD8+ T cells in IFNγ ELISpot. (E) Results from experiment described in (D), with graded doses of the indicated peptide on the x-axis. Each dot indicates one technical replicate. (F) As (B), but for H-2Kb and H-2Db expression levels from the RMA-S assay for candidate splicing-derived neoantigenic peptides induced by indisulam that were identified based on RNA-seq data and MHC I binding predictions alone. Some peptides appear twice as they bind to both H-2Kb and H-2Db. (G) As (C), but for the 39 candidate splicing-derived neoantigenic peptides induced by indisulam identified based on RNA-seq data and MHC I binding predictions alone. (H) RNA-seq coverage plots of representative indisulam-induced, candidate splicing-derived neoantigens generated by intron retention events in Kdm4b and H2–Ke6, cassette exon inclusion in Mon2, and competing 3’ splice sites in Zpf637. Indisulam-promoted peptide shown in bold, underlined text. (I) Median fluorescence intensities (MFIs) of H-2Db and/or H-2Kb on RMA-S cells following incubation with increasing doses of candidate nonantigenic peptide from (H). Mean ± sd shown. (J) Representative ELISpot images for the peptides from (H). (K) Schema of peptide:MHC I tetramer studies. B16-F10 tumor-bearing animals were treated with vehicle, anti-PD1, indisulam or the combination and were sacrificed at day 14 post-tumor challenge. Tumor-draining lymph nodes were analyzed for the frequency of tetramer-positive CD8+ T cells.
Table S1 —related to Figure 4. Differentially spliced events in DMSO- versus indisulam-treated mouse cell lines. Each row corresponds to isoform 1 of a splicing event, where isoform 1 is defined for each splicing event type as follows: “se”, inclusion isoform of cassette exons; “a3ss” and “a5ss”, intron-proximal isoform for competing 3’ or 5’ splice site events; “ri”, spliced isoform for annotated intron retention events; “ci”, spliced isoform for constitutive intron retention events. The table columns are defined as: “event”, event ID (encoding description of event type and coordinates of identifying splice junctions); “coordinates”, genomic coordinates of splicing event; “gene”, Ensembl gene ID; “geneName”, name of gene; “type”, type of splicing event; “dpsi_X-vs-dmso”, difference in isoform ratio between the indicated indisulam-treated cell line and DMSO-treated control, computed as (indisulam - DMSO); “pval_X-vs-dmso”, p (computed using unpaired, two-sided t-test) associated with difference in isoform ratio between indisulam-treated cells and DMSO-treated control cells for the indicated cell line. Table is restricted to events that were differentially spliced in at least one comparison of indisulam- or MS-023-treated mouse cell lines, where differential splicing is defined as |difference in isoform ratio| ≥ 0.10 and p ≤ 0.05.
Table S2 —related to Figure 4. Differentially spliced events in DMSO- versus MS-023-treated mouse cell lines. As Table S1, but for MS-023-treated mouse cell lines.
Table S3 — related to Figure 4. Differentially spliced events in DMSO- versus indisulam-treated human cell lines. As Table S1, but for indisulam-treated human cell lines.
Table S4 —related to Figure 4. Differentially spliced events in DMSO- versus MS-023-treated human cell lines. As Table S1, but for MS-023-treated human cell lines.
Table S5 —related to Figure 4. Potential indisulam-induced, MHC I-bound epitopes for H-2Db, H-2Kb and HLA-A*02:01. Table lists all 8–14-mers that met the filtering criteria in Fig. 4I with H-2Db, H-2Kb, and HLA-A*02:01 binding predictions.
Table S6 —related to Figure 5. Peptide identifications from MHC I mass spectrometry data. Table lists Proteome Discoverer Peptide Groups output for all samples using each of the four distinct proteomes, in both H-2Kb and H-2Db immunoprecipitations. Proteomes are specified as follows: “as_ci_full” is the “full-length” proteome; “as_ci_binders” is the “predicted binders” proteome; “as_ci_binders_spiked10” is the “predicted binders + spiked non-binders” proteome; “as_ci_diff” is the “filtered predicted binders” proteome.
Table S7 —related to Figure 6. Candidate peptides that were tested experimentally. Table lists all candidate splicing-derived, MHC I-bound, putative neoantigenic peptides tested in RMA-S assays and in vivo immunization.
Highlights:
Pharmacologic modulation of splicing enhances immune checkpoint blockade efficacy.
RBM39 degradation and Type I PRMT inhibition are well tolerated by immune cells.
Splicing perturbation induces splicing-derived neoepitopes which are immunogenic.
Rigorous framework for the identification of splicing-derived antigenic peptides.
ACKNOWLEDGMENTS
We thank Phil Gafken and Lisa Jones for mass spectrometry advice. SXL is supported by a Parker Institute for Cancer Immunotherapy (PICI) Bridge Scholar award, the NIH/NCI (K08 CA245242–01), and the Leukemia & Lymphoma Society (5492–20). EDN was supported in part by the NIH/NCI (T32 CA009657). JDT is a Washington Research Foundation Postdoctoral Fellow.JDW is funded through NIH/NCI Cancer Center P30 CA008748, Ludwig Collaborative and Swim Across America Laboratory, and PICI. OA-W and RKB were supported by the Edward P. Evans Foundation, NIH/NCI (R01 CA251138), and NIH/NHLBI (R01 HL128239). OA-W is supported in part by the NIH/NCI (R01 CA242020) and Henry and Marilyn Taub Foundation for MDS Research. RKB was supported in part by the NIH/NIDDK (R01 DK103854) and NIH/NHLBI (R01 HL151651). RKB is a Scholar of The Leukemia & Lymphoma Society (1344–18). RET is supported by NIH R01GM123741 and the Walther Cancer Foundation through the Harper Cancer Research Institute at Notre Dame. Computational studies were supported by FHCRC’s Scientific Computing Infrastructure (ORIP S10 OD028685).
DECLARATION OF INTERESTS
OA-W has served as a consultant for H3B Biomedicine, Foundation Medicine, Merck, Prelude Therapeutics, and Janssen, and is on the Scientific Advisory Board of Envisagenics, Pfizer Boulder, and AIChemy; OA-W has received prior research funding from Loxo Oncology and H3 Biomedicine unrelated to this work. SXL has served as a consultant (uncompensated) for PTC Therapeutics. BR has served as a consultant for Bayer and Roche. DZ reports clinical research support to his institution from Astra Zeneca, Plexxikon, and Genentech; personal/consultancy fees from Merck, Synlogic Therapeutics, Tesaro, Bristol Myers Squibb (BMS), Genentech, Xencor, Memgen, Calidi Biotherapeutics, and Agenus. LAD is a member of the board of directors of Personal Genome Diagnostics (PGDx) and Jounce Therapeutics. LAD is a compensated consultant to PGDx, 4Paws (PetDx), Innovatus CP, Se’er, Delfi, Kinnate and Neophore. LAD is an uncompensated consultant for, but has received clinical trial support from Merck. LAD holds equity in PGDx, Jounce, Se’er, Delfi, Kinnate and Neophore, and divested equity in Thrive Earlier Detection in 2021. His spouse holds equity in Amgen. JDW is a consultant for: Amgen; Apricity; Arsenal IO; Ascentage Pharma; AstraZeneca; Astellas; Boehringer Ingelheim; BMS; Chugai; Dragonfly; F Star; Eli Lilly; Georgiamune; IMVAQ; Merck; Polynoma; Psioxus, Recepta; Trieza; Truvax; Sellas, Werewolf Therapeutics. JDW has Grant/Research Support from: BMS; Sephora. JDW reports equity in: Tizona Pharma.; Imvaq; Beigene; Linneaus, Apricity, Arsenal IO; Georgiamune. TM is an inventor on patents involving the use of anti-PD-1 antibodies. TM is a consultant for Immunos Therapeutics and Pfizer. TM is a cofounder of and equity holder in IMVAQ. TM receives research funding from BMS, Surface Oncology, Kyn Therapeutics, Infinity Pharma., Peregrine Pharma., Adaptive Biotech., Leap Therapeutics, and Aprea Therapeutics. SXL, OA-W, and RKB are inventors on a patent application submitted by FHCRC related to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, Stevens J, Lane W, Zhang GL, Eisenhaure TM, et al. (2017). Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 46, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, Rochaix P, Andrieu-Abadie N, Levade T, Meyer N, et al. (2017). TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun 8, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 432–437. [DOI] [PubMed] [Google Scholar]
- De Bruijn ML, Schumacher TN, Nieland JD, Ploegh HL, Kast WM, and Melief CJ (1991). Peptide loading of empty major histocompatibility complex molecules on RMA-S cells allows the induction of primary cytotoxic T lymphocyte responses. Eur J Immunol 21, 2963–2970. [DOI] [PubMed] [Google Scholar]
- De Silva AD, Boesteanu A, Song R, Nagy N, Harhaj E, Harding CV, and Joyce S. (1999). Thermolabile H-2Kb molecules expressed by transporter associated with antigen processing-deficient RMA-S cells are occupied by low-affinity peptides. J Immunol 163, 4413–4420. [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Ries RE, Ilagan JO, Stirewalt DL, Meshinchi S, and Bradley RK (2014). Sample processing obscures cancer-specific alterations in leukemic transcriptomes. Proc Natl Acad Sci U S A 111, 16802–16807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram MS, Shen Y, Szewczyk M, Wu H, Senisterra G, Li F, Butler KV, Kaniskan HU, Speed BA, Dela Sena C, et al. (2016). A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem Biol 11, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, Rioux N, Lingaraj T, Ribich SA, Pappalardi MB, et al. (2019). Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 36, 100–114 e125. [DOI] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. (2013). Ensembl 2013. Nucleic Acids Res 41, D48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, Musiani D, Massignani E, Kotini AG, Penson A, et al. (2019). Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 36, 194–209 e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, and Nijhawan D. (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert CG, Bradley RK, Ding Y, Toledo CM, Herman J, Skutt-Kakaria K, Girard EJ, Davison J, Berndt J, Corrin P, et al. (2013). Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev 27, 1032–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, and Chess A. (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, Rappsilber J, and Mann M. (2006). Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J Proteome Res 5, 988–994. [DOI] [PubMed] [Google Scholar]
- Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, Zhao Y, Climente-Gonzalez H, Chai S, Wang F, et al. (2018). Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell reports 23, 270–281 e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, and Nielsen M. (2017). NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research, N., et al. (2018). Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 34, 211–224 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, Harken JA, Robinson BW, and Lake RA (2013). Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One 8, e61895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, Chung HC, Kindler HL, Lopez-Martin JA, Miller WH Jr., et al. (2020). Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 21, 1353–1365. [DOI] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. (2013). The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41, D64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JP, Konda P, Kowalewski DJ, Schuster H, Clements D, Kim Y, Cohen AM, Sharif T, Nielsen M, Stevanovic S, et al. (2017). MHC-I Ligand Discovery Using Targeted Database Searches of Mass Spectrometry Data: Implications for T-Cell Immunotherapies. J Proteome Res 16, 1806–1816. [DOI] [PubMed] [Google Scholar]
- Musiani D, Bok J, Massignani E, Wu L, Tabaglio T, Ippolito MR, Cuomo A, Ozbek U, Zorgati H, Ghoshdastider U, et al. (2019). Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci Signal 12. [DOI] [PubMed] [Google Scholar]
- Na IK, Lu SX, Yim NL, Goldberg GL, Tsai J, Rao U, Smith OM, King CG, Suh D, Hirschhorn-Cymerman D, et al. (2010). The cytolytic molecules Fas ligand and TRAIL are required for murine thymic graft-versus-host disease. J Clin Invest 120, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, and Hammerbacher J. (2018). MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst 7, 129–132 e124. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D. (2015). Dose-Response Analysis Using R. PLoS One 10, e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross P, Holmes JC, Gojanovich GS, and Hess PR (2012). A cell-based MHC stabilization assay for the detection of peptide binding to the canine classical class I molecule, DLA-88. Vet Immunol Immunopathol 150, 206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster H, Shao W, Weiss T, Pedrioli PGA, Roth P, Weller M, Campbell DS, Deutsch EW, Moritz RL, Planz O, et al. (2018). A tissue-based draft map of the murine MHC class I immunopeptidome. Sci Data 5, 180157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. (2018). H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin M, Mack R, Rhodes MC, Zhang L, Wei W, Breslin P, Berg S, Huang S, Taylor R, and Zhang J. (2019). The Splicing Modulator GEX1A Exhibits Potent Anti-Leukemic Activity Both in Vitro and In Vivo through Inducing an MCL1 Splice-Switch in Pre-Clinical Models of Acute Myeloid Leukemia. Blood 134, 2666–2666. [Google Scholar]
- Sha D, Jin Z, Budczies J, Kluck K, Stenzinger A, and Sinicrope FA (2020). Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, and Van Allen EM (2018). Intron retention is a source of neoepitopes in cancer. Nature biotechnology 36, 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Polaski JT, Feng Q, De Neef EJ, Hoppe ER, McSharry MV, Pangallo J, Gabel AM, Belleville AE, Watson J, et al. (2020). RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat Genet 52, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers EJ, Lodewyckx T, Kuriyal H, and Grasman R. (2010). Bayesian hypothesis testing for psychologists: a tutorial on the Savage-Dickey method. Cogn Psychol 60, 158–189. [DOI] [PubMed] [Google Scholar]
- Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, Hockemeyer K, Ghebrechristos YE, Yoshimi A, Inoue D, et al. (2019). Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 35, 369–384 e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. (2020). Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818–834 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. [DOI] [PubMed] [Google Scholar]
- Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane K, Hamaguchi M, Iwata M, et al. (2011). Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J 278, 4870–4880. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Luo MJ, Straesser K, Katahira J, Hurt E, and Reed R. (2000). The protein Aly links pre-messenger-RNA splicing to nuclear export in metazoans. Nature 407, 401–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 —related to Figure 1. Pharmacologic perturbation of RNA splicing impairs tumor growth in vivo, in a dose-dependent fashion dependent on adaptive immunity. (A) Left, Western blot of RBM39 after in vitroexposure to the indicated concentrations of indisulam for 24 hours in MC38 and CT26 cells. Right, half-maximal inhibitory concentration (IC50, computed using the Hill equation) values for cell growth inhibition and RBM39 degradation. (B) Dose-response curves for RBM39 abundance (normalized to β-actin) across the indicated concentrations of indisulam treatment as measured by densitometry. IC50 values are shown. (C) Western blot of RBM39 after MC38 or CT26 cells were exposed to 1μM indisulam for 96 hours, drug was washed off, and RBM39 levels were allowed to recover. (D) Annexin-V and DAPI analysis of MC38, B16-F10, CT26, and MB49 cells after in vitro treatment with indisulam 1μM or DMSO for 96 hours. (E) Heatmap of MFI values above isotype control of cell surface levels of H-2K/H-2D, I-A/I-E (MHC II), or PD-L1 after 96 hours of treatment with DMSO, indisulam 1μM, MS-023 5μM, or EPZ015666 1μM, or the same drugs in the presence of mouse IFNγ 10U/mL. CRISPR-mediated β2-microglobulin knockout cells served as biologic control for H-2K/H-2D staining. B16-F10 cells shown on top (blue). CT26 cells on bottom (red). Representative histograms of B16-F10 data are shown on right. (F) Cell surface levels of cytokine and death receptors in B16-F10 cells after treatment with conditions identical to (D). (G) Sample photos of B16-F10 (top), MC38 (middle), CT26 (bottom) bearing mice treated with indisulam or DMSO and engrafted into syngeneic animals. (H) Experimental schema. MC38 or CT26 cells were treated with DMSO or indisulam at the indicated concentrations for 96 hours in technical triplicate; these were then subjected to RNA-seq analyses or used for biological experiments as in (L-O). (I) numbers of constitutive U2-type introns retained in MC38 cells treated with the indicated doses of indisulam compared with DMSO. (J) Total number of splicing alterations in CT26 and MC38 tumors exposed to the indicated doses of indisulam, as compared with DMSO. (K) RNA-seq coverage plot depicting two representative intron retention events in Agxt2l2 and Hmgxb4 with increasing doses of indisulam. (L) experimental schema showing the engraftment of CT26 tumors treated with indisulam in vitro into syngeneic Balb/c mice. (M) tumor volumes of CT26 bearing mice from (L) over time (n=15 mice/group; tumors engrafted on bilateral flanks of mice). Mean ± sem. (N) tumor volumes from (M) at day 24. P-values were calculated for the indicated group compared to DMSO control using the Wilcoxon rank-sum test: *, p = 0.048; ***, p = 0.000798; ****, p = 0.000363. (O) Kaplan-Meier survival curve of animals from (L). (P) Individual tumor growth curves for DMSO-treated and indisulam-treated B16-F10 cells in Rag2 knockout mice. n=10 mice per group; tumors engrafted on bilateral flanks. (Q) Box-and-whisker plots of day 28 individual tumor volumes from (P). (R) Schema of CD4 & CD8 T cell depletion or NK cell depletion versus control. (S) Representative flow cytometry confirmation of T cell depletion using anti-CD4 and anti-CD8 antibodies, gated for CD3ε in the spleens of animals sacrificed at day 19 post-tumor challenge. Remaining double-negative population are NK1.1+ NKT cells (not shown). (T) Representative flow cytometry confirmation of NK cell depletion via NKp46 antibody; peripheral blood on day 14 after tumor challenge. (U-V) Box-and-whisker plot quantification of (S) and (T). Each dot represents an individual animal. (W) Sample photos of tumor-bearing animals treated with CD4/CD8 T cell depletion and challenged with B16-F10 cells pre-treated with indisulam or control. (X) Individual tumor growth curves for NK1.1 depletion experiment. n=10 per group; tumors engrafted on bilateral flanks. (Y) Box-and-whisker plots of day 21 tumor volumes for B16-F10 treated as in Fig. 1A, with or without NK cell depletion. *p<0.05; **p<0.01. ANOVA calculated using the Kruskal-Wallis test and multiple comparisons vs. DMSO alone calculated using the Dunn’s test.
Figure S2 —related to Figure 2. Class-specific effects of splicing modulator drugs on hematopoiesis, T cell gene expression and function in vitro and in vivo. (A) Left, western blot showing dose-dependent reduction in asymmetric dimethyl arginine (ADMA) with MS-023 treatment in MC38 tumor cells; right, corresponding flow cytometry analysis for intracellular ADMA. (B) Schema for the mixed leukocyte reaction, depicting generation of tumor lysates, generation of bone marrow derived dendritic cells, process of pulsing dendritic cells with lysates and lipopolysaccharide (LPS) for maturation and antigen presentation, and mixed leukocyte reaction with congenic CD45.1 CFSE-labeled naïve splenic T cells. (C) Schema depicting purification of splenic T cells, CFSE labeling, and in vitro stimulation with plate-bound anti-CD3 and anti-CD28 antibody in the presence of splicing drugs. (D) Full dose range of splicing drugs tested in (C) demonstrating dose-dependent inhibition of CFSE dye dilution and T cell proliferations at higher drug concentrations; representative histogram from N=8 technical replicates each. (E) absolute numbers of CD4 (left) and CD8 (right) T cells from experiment in (C) for each drug and concentration were quantified to calculate IC50 for cell proliferation (table). Each dot is the average from 3–8 technical replicates. Curves and IC50 values for the activation markers CD25 and PD1 are shown (bottom panels), and EC50 for induction of apoptosis as measured by annexin-V staining is shown (middle panel). (F) OT-1 killing assay against wildtype or ovalbumin-expressing MC38 tumors; experiment performed as in Fig. 2G. (G) representative contour plots showing the staining of OT-1 cells for TNFα and IFNγ or isotype control, after exposure to DMSO (left) or PMA/Iono (Phorbol 12-myristate 13-acetate and Iono: ionomycin, right). Percentage of positive cells are quantified in the associated bar plots. Each dot is one technical replicate. (H) As in (G) but OT-1 cells were stimulated with PMA/Iono (top) or incubated with MC38 cells overexpressing ovalbumin at a 1:1 ratio (bottom), at the indicated concentrations of indisulam or MS-023 on the x-axis. Graph shows mean ± sem. N=4 technical replicates per condition. (I) Experimental schema demonstrating normal intracellular localization of the LAMP1 protein on cytotoxic granules in resting cytotoxic T cells, and appearance of LAMP1 on the plasma membrane upon T cell degranulation, rendering the protein stainable for flow cytometry. Bar plots at right show LAMP1 MFI of OT-1 cells remaining unstimulated, exposed to PMA/ionomycin, or incubated at a 1:1 ratio with MC38 overexpressing ovalbumin. Each dot indicates a technical replicate. P-values were calculated using the Wilcoxon rank-sum test. (J) experiment as in (I) except in the presence of the indicated doses of indisulam or MS-023 on the x-axis. N=4 technical replicates per condition. Graph shows mean ± SD. P-values were calculated using the Kruskal-Wallis non-parametric ANOVA test. (K) experimental design: naïve C56BL/6 splenic T cells were isolated and cultured ex vivo for 96 hours in media with IL-2 50U/mL. Cells were either left unstimulated, or activated with plate-bound anti-CD3 + CD28 antibody in the presence of DMSO or the indicated splicing compounds: indisulam 1μM, MS-023 1μM, EPZ015666 5μM, or pladienolide B 2nM. At the end of 96 hours, cells were subjected to RNA-seq. N=3 replicates for each condition. (L) Differential gene expression from (K) of unstimulated T cells versus those stimulated in the presence of DMSO. (M) Box plots of fold changes of the top 100 upregulated genes in T cells undergoing activation from (K) in the presence of the indicated drugs. Each dot is one replicate. (N) Schema of adoptive transfer of C57BL/6 CD45.1 CFSE labeled T cells into syngeneic, minor antigen mismatched, or major antigen mismatched lethally irradiated recipients to assess T cell activation and expansion. (O) Histograms of CFSE dilution of donor adoptively transferred CD45.1+ CD8+ T cells into Balb/c mice on day 3 to assess for alloreactivity. (P) Representative flow cytometry plots of T cell activation and death markers in adoptively transferreddonor CFSE-labeled CD45.1 splenic CD4+ or CD8+ T cells into lethally irradiated Balb/c recipients with or without daily indisulam (25 mg/kg) treatment. Spleens were analyzed on day 3. Two animals from each condition are shown. (Q) as in (P) but with daily MS-023 treatment, 50mg/kg daily. (R-S) Histograms of CFSE dilution in donor adoptively transferred CD45.1+ T cells into syngeneic C57BL/6 mice to assess for homeostatic proliferation. Cells in the spleen on day 7 after transfer are shown. (T) Numbers of colonies in M3434 methylcellulose media on day 7. 25,000 red blood cell-lysed bone marrow hematopoietic stem and precursor cells (HSPC) from C57BL/6 mice were grown in methylcellulose with continuous exposure to splicing compounds at the indicated concentrations.
Figure S3 —related to Figure 3. Treatment of tumor-bearing animals with splicing modulator compounds in vivo enhances tumor control and can elicit memory. (A) Growth curves of individual B16-F10 tumors from C57BL/6 mice exposed to the indicated treatment, as summarized in Figure 3C. Each line represents one tumor; termination of lines prior to the end of the experiment indicates animal mortality or euthanasia according to predetermined endpoints, as described in the Methods section. (B) Experimental design: MC38 cells were treated with DMSO or MS-023 in vitro for 96 hours, and then 106 cells engrafted onto the bilateral flanks of either naïve C57BL/6 mice, or animals which had successfully rejected MC38 tumors previously after treatment with anti-PD1 and MS-023 in vivo (from Figure 3K–M). (C) Growth curves of individual tumors from (B). Pink and purple arrows indicate tumors which were implanted but rejected. (D) Line graphs summarizing growth curves from (C), showing mean ± sem. (E) Violin plots showing tumor volumes at day 28. P-values (calculated using the Wilcoxon rank-sum test) for the “DMSO survivor” -vs “DMSO naïve” and “MS-023 survivor” -vs- “MS-023 naïve” comparisons are 0.044 and 0.011, respectively. (F) Representative animals from (E).
Figure S4 —related to Figure 3. Lack of toxicity and absence of inflammatory signatures or increased CD8+ T cell infiltrates into healthy, non-tumor tissues in animals treated with splicing modulator compounds with or without anti-PD1. (A) Peripheral blood counts of tumor-bearing mice treated for 3 weeks with vehicle, anti-PD1 (250μg flat dose), indisulam (25 mg/kg), MS-023 (50 mg/kg), or combined anti-PD1 with MS-023 or indisulam. ALC: absolute lymphocyte count; ANC: absolute neutrophil count; Hgb: hemoglobin; Plt: platelet count; WBC: white blood cell count. (B) Percentage of CD8+cells amongst CD45+ cells (left) and proportion of CD8+/CD11b+ cells (right) in tumors at day 21 following treatment of animals bearing MC38 tumors with the treatments indicated on the x-axis. n=10 mice/group. ANOVA calculated using the Kruskal-Wallis test. (C) H&E stained tissue sections from mice treated with indisulam, MS-023, anti-PD1, or combination at day 21. N=5 animals per treatment group per tissue were assessed. Review of the necropsy tissues shows no overt evidence of toxicity, and the tongue shows typical squamous epithelium overlying bundles of skeletal muscle. The lung demonstrates typical alveolar spaces, alveolar septa, and bronchioles with respiratory epithelium. The large intestine shows typical colonic mucosa, submucosa, and muscularis propria. The skin from the ear demonstrates typical epidermis, dermis, and subcutaneous tissues overlying cartilage. The liver shows typical portal tracts and hepatocytes, and the small intestine demonstrates normal villi with typical mucosa, submucosa, and muscularis propria. Representative micrographs of the findings are depicted. (H&E; Magnification 400x; Scale bar: 50 microns). (D) Liver and pancreas enzymes and markers in the blood from these same animals at the time of sacrifice (n=3 mice/group). ALT: alanine aminotransferase. AST: aspartate aminotransferase. Mean ± sd shown. (E) B16-F10-tumor bearing C57BL/6 mice received the indicated treatment; tissues were collected on day 21. Paraformaldehyde-fixed and paraffin-embedded tissues were sectioned and stained for CD8 by immunohistochemistry. Representative sections of the tongue (muscle), liver, ear (skin), small and large bowel and lung are shown. 20x magnification. Scale bar: 100 microns. (F) Violin plots depicting the automated enumeration of CD8+ T cell infiltrates in the corresponding organs are shown. N=3 mice per tissue per treatment were analyzed, and N=5 fields per mouse were analyzed. Fold-change in CD8+ T cell infiltrates versus the vehicle condition are shown. p-values were calculated using the Wilcoxon rank-sum test. (G) Experimental design: C57BL/6 mice were engrafted with 106 MC38 tumor cells on each flank and subjected to treatment with vehicle, indisulam 25mg/kg beginning day +3, anti-PD1 250 μg twice weekly beginning day +7, or the combination. Lung, colon, and splenic T cells were obtained on day 14 after tumor challenge for RNA-seq. (H) Stacked bar graphs indicating the absolute number and type of splicing alterations in the lungs, colon and T cells of animals receiving the indicated therapy. Splicing alterations were defined as a 5% change in splicing with a p-value threshold of 0.05 (I-K) Scatter plots indicating constitutive intron retention in tissues of animals treated with indisulam versus vehicle. (L-N) Scatter plots indicating differentially expressed genes in the colon, lung and T cells of animals treated with vehicle versus combination of indisulam and anti-PD1. (O) Pathway analyses of differential gene expression from (L-M) in the colon and lung. No significantly enriched gene ontology pathways were identified in the T cell RNA-seq dataset.
Figure S5 —related to Figure 4. Splicing alterations generated by MS-023 and indisulam treatment. (A) Left, scatter plot comparing constitutive intron splicing in DMSO- versus MS-023-treated B16-F10 cells. Right, RNA-seq read coverage illustrating increased intron retention following MS-023 treatment. (B) As (A), but for cassette exons. (C) Bar graphs representing numbers of differentially spliced events of each indicated type in DMSO- versus MS-023-treated B16-F10 cells. Blue/green, mis-splicing events indicative of reduced/increased splicing efficiency (for example, decreased/increased cassette exon inclusion). (D) As (A), but for human SK-MEL-239 cells. (E) As (D), but for cassette exons. (F) As (C), but for human SK-MEL-239 cells. (G) representative example of a 5’ alternative splice site event induced with indisulam treatment in all of the murine cell lines studied. (H-I) two representative examples of intron retention events observed in all murine cell line studied. (J) Venn diagram depicting the number of genes mis-spliced in common across mouse versus human cell lines analyzed, upon exposure to either indisulam or MS-023. (K) Left, heat map illustrating Pearson correlations for all retained introns, constitutive introns, and cassette exons that are differentially spliced following indisulam or MS-023 treatment in at least one sample from the indicated mouse cell lines. Pearson correlations computed using the absolute isoform expression values (0–100%) for each splicing event. Right, identical analysis, but for human data. Dendrograms, unsupervised clustering by the Euclidean distance method. (L) Venn diagrams depicting shared mis-splicing events upon exposure to indisulam or MS-023 in the indicated murine cell lines. (M) as for (L) but for the indicated human cell lines studied. (N) Bar graph of Malat1 RNA levels in the cytoplasmic (cyto.) and nuclear (nuc.) fractions of DMSO-treated B16-F10 cells. As Malat1 RNA is restricted to the nucleus(Hutchinson et al., 2007), these data confirm the specificity of our fractionation. (O) RNA-seq read coverage illustrating that increased Prpf40b intron retention following indisulam treatment of B16-F10 cells is evident in total (left), nuclear (middle), and cytoplasmic (right) fractions. Gray, DMSO treatment; red, indisulam treatment. (P) Scatter plot comparing constitutive intron splicing in the cytoplasmic fractions of DMSO- versus indisulam-treated B16-F10 cells. Increased intron retention is readily evident in the cytoplasm. (Q) Venn diagram indicating shared and unique predicted neoantigens among mouse and human cell lines, treated with either indisulam or MS-023. (R) Shared and unique predicted neoantigens, across mouse and human cell lines upon treatment with splicing modulator compounds.
Figure S6 —related to Figure 5. Analysis of mass spectrometry results for H-2Db and H-2Kb immunoprecipitations. (A) As Fig. 5A, but for H-2Kb immunoprecipitation. (B) As Fig. 5D, but for H-2Kb immunoprecipitation. (C) Histogram of lengths of identified peptides using the predicted binders proteome. The majority of identified peptides are 8–9-mers, as expected. (D) As Fig. 5E, but for H-2Kb immunoprecipitation. (E) As Fig. 5F, but for H-2Kb immunoprecipitation. (F) As Fig. 5G, but for H-2Kb immunoprecipitation. (G) Table showing selected candidate neoantigenic peptides induced by indisulam exposure and shared across MC38, B16-F10, CT26 and MB49 mouse tumor cell lines, which were subjected to experimental analysis by TiterMax immunization of C57BL/6 mice followed by evaluation of reactive CD8+ T cells by IFNγ ELISpot.
Figure S7 —related to Figures 6 and 7. Functional evaluation of candidate splicing-derived, neoantigenic peptides. (A) Schema of MHC I peptide stabilization assay on TAP-deficient RMA-S cells. (B) Heatmap of mean MFI values of H-2Db levels from RMA-S peptide stabilization experiments across 30 peptides identified as binding to H-2Db in the RMA-S assay. Green text indicates control known immunogenic peptide with H-2Db binding (gp100). (C) Number of spots per 105 CD8+ T cells from IFNγ ELISpot quantified across 70 peptides derived from mass spectrometry analyses and tested in vivo which did not elicit an immune response. Bar indicates median. Each dot represents a technical replicate. (D) Diagram of experiment immunizing C57BL/6 mice with graded doses of candidate peptides at 0.1, 1, 10, or 100 μg followed by analysis of CD8+ T cells in IFNγ ELISpot. (E) Results from experiment described in (D), with graded doses of the indicated peptide on the x-axis. Each dot indicates one technical replicate. (F) As (B), but for H-2Kb and H-2Db expression levels from the RMA-S assay for candidate splicing-derived neoantigenic peptides induced by indisulam that were identified based on RNA-seq data and MHC I binding predictions alone. Some peptides appear twice as they bind to both H-2Kb and H-2Db. (G) As (C), but for the 39 candidate splicing-derived neoantigenic peptides induced by indisulam identified based on RNA-seq data and MHC I binding predictions alone. (H) RNA-seq coverage plots of representative indisulam-induced, candidate splicing-derived neoantigens generated by intron retention events in Kdm4b and H2–Ke6, cassette exon inclusion in Mon2, and competing 3’ splice sites in Zpf637. Indisulam-promoted peptide shown in bold, underlined text. (I) Median fluorescence intensities (MFIs) of H-2Db and/or H-2Kb on RMA-S cells following incubation with increasing doses of candidate nonantigenic peptide from (H). Mean ± sd shown. (J) Representative ELISpot images for the peptides from (H). (K) Schema of peptide:MHC I tetramer studies. B16-F10 tumor-bearing animals were treated with vehicle, anti-PD1, indisulam or the combination and were sacrificed at day 14 post-tumor challenge. Tumor-draining lymph nodes were analyzed for the frequency of tetramer-positive CD8+ T cells.
Table S1 —related to Figure 4. Differentially spliced events in DMSO- versus indisulam-treated mouse cell lines. Each row corresponds to isoform 1 of a splicing event, where isoform 1 is defined for each splicing event type as follows: “se”, inclusion isoform of cassette exons; “a3ss” and “a5ss”, intron-proximal isoform for competing 3’ or 5’ splice site events; “ri”, spliced isoform for annotated intron retention events; “ci”, spliced isoform for constitutive intron retention events. The table columns are defined as: “event”, event ID (encoding description of event type and coordinates of identifying splice junctions); “coordinates”, genomic coordinates of splicing event; “gene”, Ensembl gene ID; “geneName”, name of gene; “type”, type of splicing event; “dpsi_X-vs-dmso”, difference in isoform ratio between the indicated indisulam-treated cell line and DMSO-treated control, computed as (indisulam - DMSO); “pval_X-vs-dmso”, p (computed using unpaired, two-sided t-test) associated with difference in isoform ratio between indisulam-treated cells and DMSO-treated control cells for the indicated cell line. Table is restricted to events that were differentially spliced in at least one comparison of indisulam- or MS-023-treated mouse cell lines, where differential splicing is defined as |difference in isoform ratio| ≥ 0.10 and p ≤ 0.05.
Table S2 —related to Figure 4. Differentially spliced events in DMSO- versus MS-023-treated mouse cell lines. As Table S1, but for MS-023-treated mouse cell lines.
Table S3 — related to Figure 4. Differentially spliced events in DMSO- versus indisulam-treated human cell lines. As Table S1, but for indisulam-treated human cell lines.
Table S4 —related to Figure 4. Differentially spliced events in DMSO- versus MS-023-treated human cell lines. As Table S1, but for MS-023-treated human cell lines.
Table S5 —related to Figure 4. Potential indisulam-induced, MHC I-bound epitopes for H-2Db, H-2Kb and HLA-A*02:01. Table lists all 8–14-mers that met the filtering criteria in Fig. 4I with H-2Db, H-2Kb, and HLA-A*02:01 binding predictions.
Table S6 —related to Figure 5. Peptide identifications from MHC I mass spectrometry data. Table lists Proteome Discoverer Peptide Groups output for all samples using each of the four distinct proteomes, in both H-2Kb and H-2Db immunoprecipitations. Proteomes are specified as follows: “as_ci_full” is the “full-length” proteome; “as_ci_binders” is the “predicted binders” proteome; “as_ci_binders_spiked10” is the “predicted binders + spiked non-binders” proteome; “as_ci_diff” is the “filtered predicted binders” proteome.
Table S7 —related to Figure 6. Candidate peptides that were tested experimentally. Table lists all candidate splicing-derived, MHC I-bound, putative neoantigenic peptides tested in RMA-S assays and in vivo immunization.
Data Availability Statement
RNA-seq data generated as part of this study were deposited at the Gene Expression Omnibus (accession GSE162818). Software RRIDs (also available in the Key Resources Table) are: Bowtie RRID: SCR_005476, RSEM RRID: SCR_013027, TopHat RRID: SCR_013035, MISO RRID: SCR_003124, Samtools RRID: SCR_002105, Bioconductor RRID: SCR_006442, dplyr RRID: SCR016708, ggplot2 RRID: SCR_014601, Proteome Discoverer RRID: SCR_014477.