

Abstract
Multiciliated cells play critical roles in the airway, reproductive organs, and brain. Generation of multiple cilia requires both activation of a specialized transcriptional program and subsequent massive amplification of centrioles within the cytoplasm. The E2F4 transcription factor is required for both roles and consequently for multiciliogenesis. Here we establish that E2F4 associates with two distinct components of the centriole replication machinery, Deup1 and SAS6, targeting nonhomologous domains in these proteins. We map Deup1 and SAS6 binding to E2F4’s N-terminus and show that this domain is sufficient to mediate E2F4’s cytoplasmic role in multiciliogenesis. This sequence is highly conserved across the E2F family, but the ability to bind Deup1 and SAS6 is specific to E2F4 and E2F5, consistent with their shared roles in multiciliogenesis. By generating E2F4/E2F1 chimeras, we identify a six-residue motif that is critical for Deup1 and SAS6 binding. We propose that the ability of E2F4 and E2F5 to recruit Deup1 and/or SAS6, and enable centriole replication, contributes to their cytoplasmic roles in multiciliogenesis.
INTRODUCTION
Multiciliated cells are terminally differentiated epithelial cells, which assemble numerous motile cilia on their apical surface and function in several organs in many vertebrates (Boutin and Kodjabachian, 2019). Disrupted motile cilia function causes chronic airway diseases and infertility in humans (Brooks and Wallingford, 2014; Spassky and Meunier, 2017). Multiciliogenesis depends on the initiation of a specific transcriptional program in progenitor cells, followed by the massive amplification of centrioles, which are barrel-shaped, microtubule based organelles that convert to the basal bodies of cilia (Boutin and Kodjabachian, 2019; Lewis and Stracker, 2021). In proliferating cells, centriole replication is tightly linked to the cell cycle and only two centrioles are generated per cell cycle through the mother centriole-dependent centriole biogenesis (Nigg and Raff, 2009). In differentiating multiciliated cell progenitors, the mother centriole plays several roles but only a minor role in centriole replication and this, instead, occurs predominately at 200–400 nm diameter ring-shaped structures, called deuterosomes, which are formed exclusively in these multiciliated cell progenitors (Sorokin, 1968; Kubo et al., 1999; Yan, Zhao and Zhu, 2016; Al Jord et al., 2019, Liu et al., 2020).
Deuterosomes and the mother centriole have similar structures and many of the regulators of centriole biogenesis, including centrosomal protein 152 (Cep152), polo-like kinase 4 (Plk4), and centriolar assembly protein 6 (SAS6), function in both (Zhao et al., 2013). However, two paralogues, deuterosome assembly protein 1 (Deup1, also known as Ccdc67) and centrosomal protein 63 (Cep63), participate uniquely in the deuterosome and mother centriole, respectively (Sir et al., 2011; Brown et al., 2013; Zhao et al., 2013). These proteins enable the assembly and maturation of their organelles by recruiting other core components, including Cep152, Plk4, and SAS6 (Sir et al., 2011; Brown et al., 2013; Zhao et al., 2013). Cep63 is broadly expressed, while Deup1 expression is induced as a part of the multiciliogenesis-specific transcriptional program (Zhao et al., 2013; Mori et al., 2017).
Interestingly, a recent study showed that Deup1-deficient cells can still undergo effective multiciliogenesis, even though they lack deuterosomes (Mercey et al., 2019). Additionally, multiciliogenesis is preserved even in the absence of both Deup1 and Cep63, suggesting that the formation of multiple cilia may occur independent of deuterosomes and mother centrioles (Mercey et al., 2019). It is not entirely clear how centriole amplification proceeds in this context, but this is thought to occur at centriolar satellites, formerly known as fibrous granules (Mercey et al., 2019). Notably, although Deup1 and deuterosomes are dispensable for multiciliogenesis, they likely play roles in the normal context that are not well understood, and thus there is rationale for understanding how they function. Additionally, given these results, other pathways for centriole replication must exist that rely on other complexes.
The first indication that multiciliogenesis is dependent on members of the E2F family of transcription factors came from the observation that E2f4–/– mice are susceptible to respiratory failure due to an absence of multiciliated cells from the nasal and airway epithelia (Humbert et al., 2000; Danielian et al., 2007). This and subsequent studies established that E2F4 and E2F5 play functionally redundant roles in multiciliogenesis. Specifically, the defects of the E2f4–/– airway epithelium are recapitulated in E2f4+/–;E2f5–/– compound mutant mice, but not in either E2f4+/– or E2f5–/– mice (Danielian et al., 2007). Moreover, multiciliogenesis is impaired in the efferent ducts by the loss of E2f4 and heterozygosity of E2f5 (in E2f4f/f;E2f5+/–;VilCre mice) but not by E2f4 loss alone (Danielian et al., 2016). Additionally, recent studies showed that the shared role of E2F4 and E2F5 in multiciliogenesis is conserved in zebrafish (Chong et al., 2018). Work in Xenopus and subsequently other organisms revealed that E2F4 or E2F5 activate the multiciliogenesis-specific transcriptional program in association with DP1, the heterodimerization partner for many E2Fs, and two critical transcriptional coregulators of multiciliogenesis, MCIDAS (multicilin) and GEMC1 (Ma et al., 2014; Terré et al., 2016; Lewis and Stracker, 2021).
Previously, we used mouse genetic models and primary air–liquid interface (ALI) cultures to study the role of E2F4 in in vitro differentiation of murine tracheal epithelial cells into the multiciliated cells (Mori et al., 2017). Our results indicated that nuclear E2F4 is crucial to initiate the transcriptional program of multiciliogenesis (Mori et al., 2017), consistent with the prior Xenopus studies (Ma et al., 2014). However, unexpectedly, we found that E2F4 has an additional function in multiciliogenesis at later differentiation stages, in which it relocates to the cytoplasm to promote centriole biogenesis. Specifically, confocal microscopy and superresolution 3D-SIM showed that E2F4 colocalizes with PCM1/centriolar satellites at early stages of multiciliogenesis and also with components of the deuterosome/centriole replication machinery including Deup1, Cep152, Plk4, and SAS6. In reconstitution experiments using E2F4-deficient cells, a solely nuclear form of E2F4 is able to drive the transcriptional program of multiciliogenesis, but the deuterosome components do not assemble appropriately in the cytoplasm and multiciliogenesis fails (Mori et al., 2017). Importantly, when a non-DNA binding, cytoplasmic form of E2F4 is coexpressed with this nuclear E2F4 form, deuterosome assembly and multiciliogenesis are restored (Mori et al., 2017). Finally, using immunoprecipitation (IP) assays, we demonstrated that the endogenous E2F4 and Deup1 physically associate in differentiating progenitor cells (Mori et al., 2017), suggesting a direct role for E2F4 in Deup1’s function during the centriole biogenesis.
These observations raise several key questions. What are the structural requirements for E2F4’s interaction with the centriole biogenesis machinery? What is the structural basis for the selective role of E2F4 and E2F5 in large-scale centriole amplification? In this study, we used site-directed mutagenesis and gene transduction assays in cell lines and primary airway epithelial cultures to address these questions. Our data establish that the N-terminal domain of E2F4 is responsible for interaction with Deup1 as well as a second deuterosome component, SAS6, and accordingly is sufficient to mediate E2F4’s cytoplasmic functions in multiciliogenesis. Although this domain contains a highly conserved region across the E2F family, comparative analysis allowed us to identify a small motif that is critical for Deup1 and SAS6 binding and specific to E2F4 and E2F5. This provides a structural explanation for why the regulation of multiciliogenesis is a unique property of E2F4 and E2F5 and not other E2F family members.
RESULTS
E2F4 binds to the deuterosome-specific protein, Deup1
To dissect interactions between E2F4 and components of the replication machinery, we used a transient coexpression system in 293FT cells. These cells have no endogenous Deup1 but do express various centriolar proteins that are shared components of both deuterosomes and the mother centriole, such as Cep152, Plk4, and SAS6. We coexpressed Flag-tagged Deup1 (Flag-Deup1) and E2F4 and showed by IP-Western blot (IP-Western) experiments that Flag-Deup1 was efficiently recovered in anti-E2F4, but not in control mouse IgG IP (Figure 1A), consistent with our prior report (Mori et al., 2017). This interaction was also observed when the reciprocal experiment of immunoprecipitating Flag-Deup1 and blotting for E2F4 was conducted (Figure 1B). The coexpression of these proteins resulted in unexpected gel mobility shifts for both Deup1 (gaining lower mobility species; Figure 1, A and B) and E2F4 (gaining higher mobility species; Figure 1B). Notably, these novel species were selectively enriched in the IP Flag-Deup1:E2F4 complex, compared with their abundance in the inputs (Figure 1, A and B), suggesting that E2F4 and Deup1 undergo post-translational modifications that result from, or are highly stabilized by, their association. We address these post-translational modifications below.
FIGURE 1:
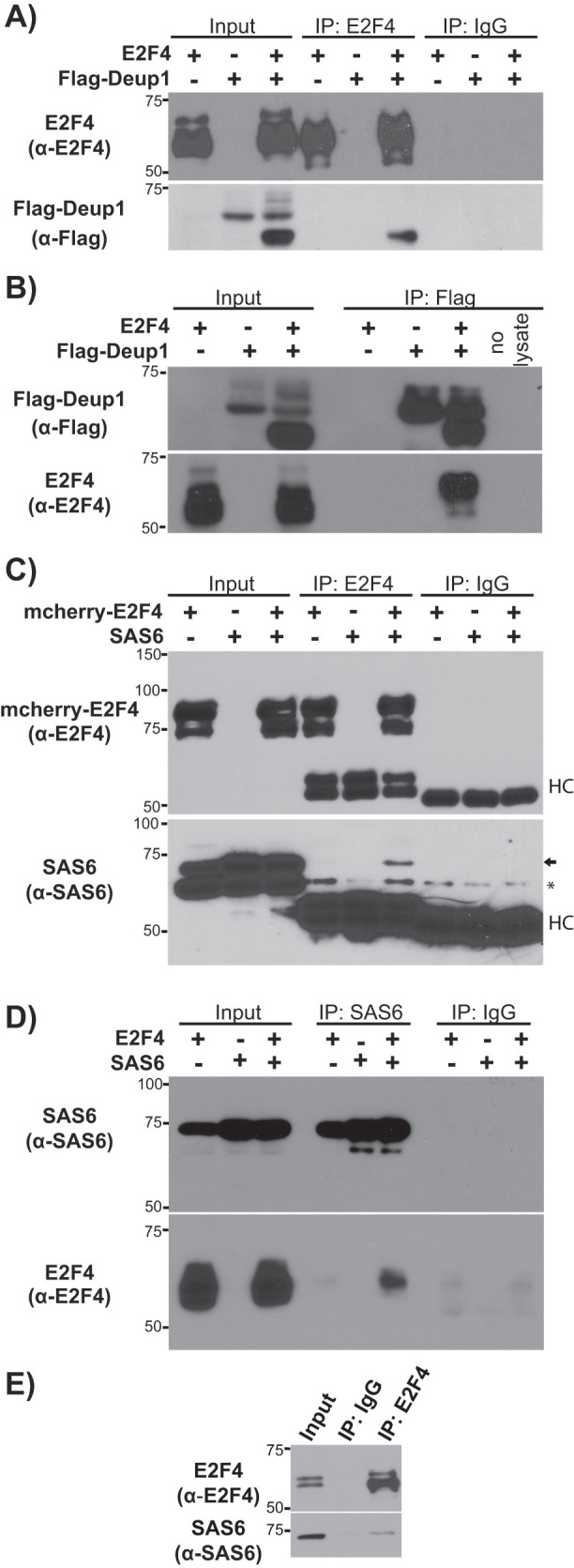
E2F4 associates with Deup1 and SAS6. (A, B) Western blots with α-E2F4 or α-Flag antibodies of input lysates and (A) α-E2F4 or (B) α-Flag immunoprecipitates from 293FT cells overexpressing E2F4 and/or Flag-Deup1. (C, D) Western blots with α-E2F4 or α-SAS6 antibodies of input lysates and (C) α-E2F4 or (D) α-SAS6 immunoprecipitates from 293FT cells overexpressing E2F4 and/or SAS6. Mouse IgG was used as a negative control. For (C), an arrow shows the coimmunoprecipitated SAS6 band, HC denotes the IgG heavy chain, and * denotes a nonspecific band. (E) Western blots with α-E2F4 or α-SAS6 antibodies of input lysate and α-E2F4 immunoprecipitates from 293FT cells verified endogenous interaction of E2F4 and SAS6.
E2F4 also binds to the centriolar protein, SAS6
In addition to deuterosomes, cytoplasmic E2F4 colocalizes with the mother centriole and centriolar satellites in the progenitors of multiciliated cells (Mori et al., 2017). Deup1 is specific to the deuterosome, and in the mother centriole its function is replaced by Deup1 paralogue, Cep63 (Sir et al., 2011; Zhao et al., 2013). Since Deup1 and Cep63 share significant structural and sequence conservation (Supplemental Figures S1 and S2A), we used the transient coexpression system to determine whether E2F4 also binds to Flag-Cep63. Although Flag-Cep63 was expressed at high levels, it did not coprecipitate E2F4 (Supplemental Figure S2B), in clear contrast to Flag-Deup1. Given this finding, we expanded our analyses to consider other centriolar proteins that are shared between the deuterosome and the mother centriole, including Cep152, Plk4, Stil, and SAS6, as well as PCM1, a core component of centriolar satellites (Zhao et al., 2013). We did not observe any specific interaction between E2F4 and Cep152, Plk4, Stil, or PCM1 in our coexpression system. In contrast, E2F4 and SAS6 clearly interacted in our IP-Western blot assay when coexpressed. Specifically, Western blotting with an anti-SAS6 antibody showed that the exogenously expressed SAS6 was recovered in anti-E2F4, but not control anti-IgG IP (Figure 1C). Similarly, in reciprocal IP experiments, blotting with anti-E2F4 antibodies detected E2F4 in SAS6 immunoprecipitates from cells with exogenous SAS6 and E2F4 (Figure 1D). Unlike the E2F4-Deup1 interaction, no change in the mobility of E2F4 and SAS6 was detected following their association. Notably, in both of these experiments, endogenous SAS6 is clearly detected in the total cell lysates, but does not interact with E2F4 (Figure 1, C and D). Given this finding, we wanted to determine whether the endogenous E2F4 and SAS6 proteins actually interact. To address this, we conducted IPs of 293FT cell lysates with anti-E2F4 or control IgG antibodies and then screened for SAS6 by Western blotting. We detected SAS6 specifically in the E2F4, but not in the control IP, validating the in vivo interaction of E2F4 and SAS6 (Figure 1E).
E2F4, like many other E2F family members, functions as a heterodimer with DP proteins (Trimarchi and Lees, 2002). This raised the question of whether DP proteins also associate with Deup1 and SAS6, and thus we coexpressed HA-tagged DP1 and Flag-Deup1 in 293FT cells with or without E2F4. When we immunoprecipitated through Flag-Deup1, Western blotting detected DP1 in cell extracts with cotransfected E2F4, indicating that DP1 is part of the complex (Supplemental Figure S3A). When we conducted the IP via the HA tag on DP1, we were able to co-IP Flag-Deup1 from cells transfected only with HA-tagged DP1 and Flag-Deup1 (Supplemental Figure S3B). It is not clear why exogenous E2F4 is required in the former but not the latter context, but this could reflect differences in complex stability and/or steric hinderance issues with the anti-Flag antibody. Notably, by immunoprecipitating through HA-tagged DP1 and now blotting for SAS6, we also detected an association between cotransfected HA-tagged DP1 and SAS6 (Supplemental Figure S3C). Thus, we conclude that DP1 can participate in both the Deup1 and the SAS6 complexes.
N-terminal regions of Deup1 and SAS6 mediate the interaction with E2F4
Having established that Deup1 and SAS6 both act as binding partners of E2F4, we wanted to identify the regions of Deup1 and SAS6 that mediate these interactions. Coiled-coil domains, which often mediate oligomerization, are present in both Deup1 (four exist in the Deup1 isoform used here) and SAS6 (a single central domain) (Strnad et al., 2007; Zhao et al., 2013). A panel of Flag-tagged deletion mutants based around these structural motifs was tested, beginning with Deup1 (Figure 2A). The N-terminal region of Deup1 (Flag-Deup11–129, residues 1–129), which includes the first coiled-coil domain of Deup1 as well as a portion of the second coiled-coil, immunoprecipitated E2F4 in our assay (Figure 2B). Moreover, this association caused E2F4 to undergo the same mobility shift as induced by its interaction with full-length Deup1 (Figure 2B). In contrast, all other Deup1 mutants, including one with deletion of only the N-terminal 59 residues that comprise the first coiled-coil domain (Flag-Deup160–535), failed to immunoprecipitate E2F4 (Figure 2, B and C). Based on these observations, we conclude that Deup1’s amino-terminal domain (Flag-Deup11–129) is able to complex with E2F4 and induce, or stabilize, E2F4 species with decreased electrophoretic mobility. Since there is significant sequence identity and structural homology between the minimal E2F4 binding region of mouse Deup1 (residues 1–129) and residues 67–192 of mouse Cep63 (Supplemental Figures S1 and S4A), it seemed plausible that an E2F4 binding site might be conserved in Cep63 but occluded in the full-length Cep63. To address this, we tested a Flag-tagged deletion mutant of Cep63 in our interaction assay but again failed to detect any association with E2F4 (Supplemental Figure S4B). Collectively, these data show that Deup1, and not Cep63, interacts specifically with E2F4 and this maps to Deup1’s N-terminal sequences. We believe that subtle amino acid differences between Deup1 and Cep63 within this otherwise conserved region are responsible for the specific interaction with Deup1.
FIGURE 2:

E2F4 association is mediated by the N-terminal domains of both Deup1 and SAS6. (A) Schematic representation of Flag-tagged Deup1 deletion mutants and their determined E2F4 association. (B, C) Cell lysates containing E2F4 and the indicated Flag-tagged Deup1 mutants were subjected to Western blotting with α-Flag (upper panel) or α-E2F4 (lower panel) antibodies before (Input) or after IP with α-Flag antibodies. (D) Schematic representation of Flag-tagged SAS6 mutants and their determined E2F4 association. (E) Cell lysates containing E2F4 and Flag-tagged SAS6 mutants were subjected to Western blotting with α-Flag or α-E2F4 antibodies before (Input) or after IP with α-Flag antibodies. (F) Cell lysates overexpressing HA-E2F4 and Flag-tagged WT or point mutants (I62T or F131D) of SAS6 were subjected to Western blotting with α-HA or α-Flag antibodies before (Input) or after IP with α-HA antibodies, showing that Flag-SAS6F131D is unable to associate with E2F4. Numbers indicate amino acid positions. For C and E, * denotes the IgG heavy and light chains, respectively.
We next examined a panel of Flag-tagged SAS6 mutants (Figure 2D) for their ability to interact with E2F4. We found that neither the fragment containing the sole coiled-coil domain (Flag-SAS6160–505) nor the C-terminal region (Flag-SAS6499–656) immunoprecipitated with E2F4 (Figure 2E). Instead, E2F4 interaction mapped to the amino-terminal region of SAS6 (Flag-SAS61–175; Figure 2, D and E). Interestingly, SAS61–175 encompasses the pisa motif (residues 39–91) and a second domain (residues 123–140) named motif II (Figure 2D), which together constitute a continuous, highly conserved patch that is required for head-to-head multimerization of SAS6 dimers (van Breugel et al., 2011). Interestingly, point mutations have been identified within this region that impact SAS6’s biological activity. One, located within the pisa motif (I62T), was identified in members of a consanguineous family afflicted with autosomal recessive primary microcephaly, a classic ciliopathy phenotype (Khan et al., 2014). Another one, which was experimentally generated within motif II (F131D), prevents the multimerization of SAS6 dimers. This multimerization is required to establish the ninefold radially symmetrical cartwheel structure which is implicated in dictating the ninefold symmetry of centrioles (van Breugel et al., 2011; Kitagawa et al., 2011). We therefore generated these point mutants in Flag-tagged SAS6 and asked whether they influenced the E2F4-SAS6 interaction. Flag-SASI62T associated with HA-E2F4 as well as wild-type (WT) Flag-SAS6, indicating that this residue is not critical for interaction with E2F4 (Figure 2F). In contrast, Flag-SAS6F131D failed to co-IP HA-E2F4 (Figure 2F). This raises the possibility that E2F4 binds at this site and/or that multimerization of SAS6 dimers is required for interaction with E2F4.
Partially overlapping domains of E2F4 mediate its association with Deup1 and SAS6
The E2F4 domains necessary for binding to DNA, its DP dimerization partners and other key regulators (e.g., the pocket protein family members), are well characterized. To identify the Deup1 and SAS6 interaction domains, HA-tagged, N- and C-terminal truncation mutants of E2F4 were generated whose boundaries were guided by the location of these known functional domains. Since the interaction between E2F4 and deuterosomes occurs in the cytoplasm (Mori et al., 2017), we verified by immunofluorescence that HA-tagged E2F4 mutants displayed some degree of cytoplasmic localization when expressed alone, or together with Flag-tagged Deup1 or SAS6 (Supplemental Figures S5–S7). We then conducted interaction mapping, beginning with Deup1 (Figure 3A). The C-terminal portion of E2F4 (HA-E2F4198–410) was not detected within the Flag-Deup1 immunoprecipitates, even though it was expressed at similar levels to the other E2F4 species (Figure 3B). In contrast, Flag-Deup1 immunoprecipitated the N-terminal portion of E2F4 (HA-E2F41–197), which includes the DNA binding and dimerization/marked box domains (Figure 3B). The smaller N-terminal mutants (HA-E2F41–100, HA-E2F41–130 and HA-E2F41–158) failed to associate with Flag-Deup1 (Figure 3B). Thus, to further map the minimal interaction domain, internal E2F4 deletion mutants were generated within the context of full-length E2F4: HA-E2F4Δ101–197, HA-E2F4Δ137–197, and HA-E2F4Δ101–137. Notably, we could screen these mutants using an anti-E2F4 antibody, which recognizes sequences within the C-terminal region of E2F4, in addition to the anti-HA antibody. When coexpressed with Flag-Deup1, all three of these internal deletion mutants were detected in the anti-Flag immunoprecipitates with the anti-E2F4 antibody (Figure 3C), but not the anti-HA antibody. We cannot rule out the possibility that E2F4’s carboxy-terminal sequences facilitate folding or provide some additional Deup1 interaction sites, but we believe that this reflects the fact that our anti-E2F4 antibody has a higher avidity for its epitope than the anti-HA antibody. Given these observations, we conclude that E2F41–197 is the minimal region necessary and sufficient for Deup1 association, and that residues 1–100 contain a key Deup1 interaction site since it is sufficient to interact in the presence of the C-terminal domain.
FIGURE 3:

Deup1 and SAS6 association is mediated by the N-terminal domain of E2F4. (A) Schematic representation of HA-tagged E2F4 mutants and their determined association with Deup1 or SAS6. (B, C) Cell lysates containing the indicated HA-tagged E2F4 mutants alone or with Flag-Deup1, either before (Input) or after IP with α-Flag antibodies, were subjected to Western blotting with (B) α-Flag or α-HA antibodies, or (C) α-Deup1 or α-E2F4 antibodies, as indicated. (D, E) Cell lysates from cells expressing SAS6 and the indicated HA-tagged E2F4 mutants were subjected to Western blotting with (D) α-HA or α-SAS6 antibodies, or (E) α-Flag or α-E2F4 antibodies, before (Input) or after IP with either (D) α-HA or (E) α-Flag antibodies. Numbers indicate amino acid positions and Δ denotes the residues deleted from the full-length protein. For B, * denotes the IgG light chain.
We next tested the ability of the HA-tagged E2F4 deletion mutants to bind to SAS6 (Figure 3A). SAS6 associated with HA-E2F41–197, but not any of the smaller N-terminal fragments, HA-E2F41–100, HA-E2F41–130, and HA-E2F41–158 or the C-terminal fragment HA-E2F4198–410 (Figure 3D), essentially mirroring the binding selectivity of Deup1 (Figure 3B). Given this finding, the ability of SAS6 to interact with the E2F4 internal deletion mutants was tested. In contrast to Deup1, we did not detect any interaction between SAS6 and HA-E2F4Δ101–197, HA-E2F4Δ137–197, or HA-E2F4Δ101–137 using either the anti-HA or the anti-E2F4 antibodies (Figure 3E). Thus, we conclude that the N-terminal region of E2F4 (E2F41–197), including the DNA binding and dimerization/marked box domains, is both necessary and sufficient to mediate efficient interaction with both Deup1 and SAS6. However, Deup1 and SAS6 possess subtle differences in either their specific binding site requirements and/or their affinity for E2F4.
E2F4 binding to Deup1 leads to post-translational modifications in both proteins
Our experiments unexpectedly revealed a gel mobility shift of E2F4 when coexpressed with either full-length or the amino-terminal region of Deup1 (Figures 1B and 2B) but not SAS6 (Figure 1D). We wondered whether phosphorylation might account for, or contribute to, E2F4’s mobility shift. To address our hypothesis, we recovered the Flag-Deup1:E2F4 complex through IP with anti-Flag antibodies and then incubated them with lambda protein phosphatase. Subsequent Western blotting showed that the phosphatase treatment eliminated the higher molecular weight E2F4 species and produced two distinct bands of intermediate and lower molecular weight (Supplemental Figure S8A). Phosphatase treatment caused a similar downshift for the higher mobility E2F4 species that was immunoprecipitated with Flag-Deup11–129 (Supplemental Figure S8B). To determine whether this phosphorylation mapped to the amino-terminal 1–197 residues that constituted the Deup1 binding site, we repeated this analysis on immunoprecipitated Flag-Deup1:HA-E2F41–197 complexes. In contrast to full-length E2F4, the mobility of HA-E2F41–197 was not altered by its association with Flag-Deup1, and phosphatase treatment had no detectable effect on its mobility (Supplemental Figure S8C). Together these data argue that coexpression of E2F4 with Deup1 promotes or stabilizes phosphorylation of E2F4, which occurs within the C-terminal domain and/or requires the presence of the C-terminal domain for its deposition. To determine the location of the phosphorylation sites within E2F4, we used mass spectrometry to analyze full-length E2F4 that was immunoprecipitated with Deup1. This identified a single site of phosphorylation, at serine 274, which lies in the C-terminal region that is dispensable for Deup1 interaction (Supplemental Figure S9). Thus, in the context of full-length E2F4, Deup1 preferentially binds to this phospho-E2F4 species, but this phosphorylation event is not required for the N-terminal portion of E2F4 to interact with Deup1.
We also examined the nature of the downward mobility shift of Deup1 that exists within the Flag-Deup1:E2F4 complex. In this case, the lower molecular weight Deup1 species was largely unaffected by phosphatase treatment. Given this, and the magnitude of the reduction in Deup1’s molecular weight, we hypothesized that protein cleavage might be responsible for the shift. To address our hypothesis, a Deup1 construct with distinct tags at the N- (Flag) and C- (HA) termini was generated and coexpressed with E2F4. In the whole cell lysate, anti-Flag and anti-HA antibodies both efficiently detected a common higher mobility Flag-Deup1-HA species. However, the lower mobility Deup1 species was only detected by the anti-Flag and not the anti-HA, antibody in E2F4-immunoprecipates, suggesting the loss of the C-terminus (Supplemental Figure S10A). These data argue that the formation of a lower molecular weight Deup1 species in the presence of E2F4 reflects removal of C-terminal Deup1 sequences. The molecular weight shift suggests that approximately 11 kDa are lost. To more precisely map the cleavage sequence, we generated several double-tagged C-terminal Deup1 truncation mutants (1–491, 1–461, and 1–432) and screened for the recovery of these Flag- and HA-tagged species following IP via E2F4. All three constructs retained the HA-tag and showed no downward mobility shift, indicating that cleavage requires sequences within the last 45 amino acids of Deup1 (Supplemental Figure S10B). Collectively, our experiments show that coexpression of E2F4 with Deup1 promotes or stabilizes E2F4 phosphorylation, likely within its C-terminal domain, and induces removal of Deup1’s C-terminal sequences. Notably, while these modifications are enriched in the E2F4:Deup1 complex, they both fall outside of the minimal interaction domains.
E2F4 1–197ΔDBD is sufficient for the cytoplasmic function of E2F4 in multiciliogenesis
We previously established the essential cytoplasmic function of E2F4 in multiciliogenesis using an ALI assay, in which primary murine tracheal cells undergo differentiation such that a subset becomes multiciliated, with others adopting other lineage fates. Using mouse tracheal progenitors isolated from E2f4f/f;R26CreERT/+ mice and treating them with 4-hydroxytamoxifen to knock out the endogenous E2f4, we showed that E2F4-deficient cells fail to undergo multiciliogenesis, and that this defect can be rescued by lentivirus-mediated expression of WT E2F4 (Mori et al., 2017). Most importantly, a nuclear E2F4 variant, which lacks the nuclear export signal (E2F4ΔNES) but retains transcriptional activity, was unable to induce multiciliogenesis but the coexpression of E2F4ΔNES and a cytoplasmic, non-DNA binding (DBD), E2F4 variant (E2F4ΔDBD) effectively restored multiciliogenesis (Mori et al., 2017). Having established that E2F41–197 is sufficient to associate with both Deup1 and SAS6, we used this reconstitution assay to determine whether this domain was sufficient to perform the cytoplasmic function of E2F4 in multiciliogenesis. A non-DNA binding variant of the N-terminal fragment, E2F41–197ΔDBD, was therefore expressed with or without HA-E2F4ΔNES in adult mouse E2f4f/f;R26CreERT/+tracheal progenitors. In parallel, we generated cells expressing full-length HA-E2F4ΔDBD and HA-E2F4ΔNES as a positive control. The cells were then treated with 4-hydroxytamoxifen, to knock out the endogenous E2f4, and subjected to the ALI differentiation protocol. Immunofluorescence with anti-HA antibodies, to detect HA-E2F4 variants, and antibodies against acetylated-α-tubulin, to detect the cilia of multiciliated cells, was used to quantify the fraction of multiciliated HA-positive cells. Consistent with our prior report (Mori et al., 2017), coexpression of full-length HA-E2F4ΔDBD and HA-E2F4ΔNES induced the differentiation of multiciliated cells in E2F4-deficient cultures, as verified by staining for cilia (Figure 4A, upper panels). The fraction of cells undergoing multiciliogenesis was within the range expected for the ALI assay since some cells adopt alternate fates. In contrast, E2F41–197ΔDBD alone was insufficient to rescue multiciliated cell differentiation, as expected (Figure 4A, middle panels). Importantly, coexpression of HA-E2F41–197ΔDBD and HA-E2F4ΔNES induced multicilia formation in E2f4 null precursor cells (Figure 4A, lower panels). Moreover, the quantification showed that HA-E2F41–197ΔDBD rescued multiciliogenesis as effectively as full-length HA-E2F4ΔDBD when coexpressed with HA-E2F4ΔNES (Figure 4B). Thus, we conclude that E2F41–197 is sufficient to perform E2F4’s cytoplasmic role in multiciliogenesis, presumably via interaction with Deup1 and/or SAS6.
FIGURE 4:

E2F41–197ΔDBD is sufficient for the cytoplasmic function of E2F4 in multiciliogenesis. E2F4f/f;R26CreERT2/+ airway epithelial progenitors were transduced with E2F4ΔDBD, E2F41–197ΔDBD and/or E2F4ΔNES as indicated, treated with 4-hydroxytamoxifen to delete the endogenous E2F4, and then subjected to the ALI differentiation assay. (A) Expression of E2F4 was visualized by α-HA tag staining (red signal) and multiciliated cells were detected by staining with the cilia marker α-tubulin (green signal) by confocal imaging. (B) Quantitation of the frequency of HA positive cells in ALI cultures that are multiciliated; n = 2 experiments and 200 cells counted per condition. The bar represents the mean of each population. Two-tailed unpaired t test is used to identify designated significant differences. *p < 0.05, ****p < 0.0001, n.s. = not significant. These data shows that E2F41–197ΔDBD rescues multiciliogenesis as efficiently as the full-length E2F4ΔDBD.
Deup1 and SAS6 bind to E2F4 and E2F5, but not E2F1, via a unique shared motif
We wanted to extend our analysis to consider other members of the E2F family based on two observations. First, E2F4’s role in multiciliogenesis is known to be shared by E2F5, which is its closest relative, but has not been linked to any other E2F family member (Danielian et al., 2007; Ma et al., 2014; Danielian et al., 2016; Terré et al., 2016). Second, the Deup1 and SAS6 interaction maps to the region of E2F4 that contains structural and functional domains that are highly conserved across E2F family members, especially E2F1-E2F6, although some sequence differences do exist. Since Deup1 and SAS6 binding is important for multiciliogenesis, we hypothesized that this function would be conserved between E2F4 and E2F5, but not in other E2Fs and such selectivity could be used to home in on sequences that could be interaction sites. To address these questions, we conducted transfection and IP experiments using HA-tagged E2F5 or, as an alternative E2F, HA-tagged E2F1 (Figure 5A). These experiments showed that Flag-Deup1 (Figure 5B) and Flag-SAS6 (Figure 5C) both specifically immunoprecipitated HA-E2F5, but not HA-E2F1 (Figure 5, B and C). Thus, the interaction between E2F4/5 and these two components of the centriole replication machinery is specific and conserved.
FIGURE 5:

Association of Deup1 and SAS6 with other E2F family members. (A) Schematic representation of the full-length E2F4, E2F5, and E2F1 and their determined Deup1 and SAS6 binding abilities. (B) Cell lysates containing the indicated HA-tagged E2F species with or without Flag-Deup1 were subjected to Western blotting with the indicated α-Flag and α-HA antibodies before (Input) or after IP of Flag-Deup1. (C) Cell lysates overexpressing indicated HA-tagged E2F species with or without SAS6 were subjected to Western blotting with the indicated α-HA or α-SAS6 antibodies before (Input) or after IP with α-HA antibodies.
Having established this specificity, we used a chimeric strategy to narrow down the interacting region(s) of E2F4. Specifically, we generated chimeras between E2F41–197 and the equivalent regions of E2F1 using published 3D structures (Zheng et al., 1999; Liban et al., 2017) to position the junctions in the linker regions between the key structural domains (Figure 6A; Supplemental Figure S11). Chimera 1-4-4, which carries the DBD of E2F1 (residues 121–198) and the dimerization/marked box domains of E2F4 (residues 90–197), did not bind to Deup1, while chimeras 4-1-1 and 4-4-1 showed partial or strong binding, respectively (Figure 6B). These data argue that E2F41–89 is the main interaction region of E2F4, in agreement with the earlier internal E2F4 deletion mutant analyses (Figure 3C).
FIGURE 6:

Efficient Deup1 and SAS6 binding requires a short motif (E2F448–53) that is conserved in E2F4 and E2F5 but not other E2Fs. (A) Schematic representation of E2F41–197, E2F1121–301 and the various E2F4/E2F1 chimeras. Red lines show the positions of the M1 and M2 mutations. (B) Cell lysates containing HA-tagged E2F4/E2F1 chimeras (HA-E2F4/1), alone or with Flag-Deup1 were subjected to Western blotting with the indicated α-Deup1 or α-HA antibodies before (Input) or after IP with α-Flag antibodies. (C) Amino acid alignments of E2F4, E2F5 and E2F1 with the red boxes indicating the M1 and M2 motifs, which were swapped from E2F4 to E2F1 sequences in the M1 and M2 mutants. (D) Crystal structure of the E2F4 DBD illustrating the exterior surface position of the M1 site (in red). The M2 mutation site is C-terminal to the structured DBD region and was unresolved in the crystal structure (Zheng et al., 1999). (E–G) Cell lysates with the indicated WT, M1, M2 or M1+2 versions of (F, G) HA-tagged full-length E2F4 or (E) HA-E2F41–197, expressed alone or with (E, F) Flag-Deup1 or (G) Flag-SAS6, were subjected to Western blotting with the indicated (E, F) α-Deup1, (G) α-Flag, (E) α-HA, and (F, G) α-E2F4 antibodies before (Input) or after IP with α-Flag antibodies. For F and G, HC denotes the heavy chain of IgG.
To further delineate the interaction site, we compared the sequence of residues 1–89 of E2F4 with the equivalent regions of E2F5, versus E2F1, E2F2, and E2F3, across multiple species (Supplemental Figure S12). Regions that were shared between E2F4 and E2F5 and diverged from E2F1, E2F2, and E2F3 were then mapped in silico onto the structure of the E2F4-DP heterodimer to assess whether they were potential interaction sites on the surface of the protein that would not be occluded or interfere with DP binding (Zheng et al., 1999). This analysis identified two candidate motifs: motif 1, which corresponded to E2F448–53 versus E2F1158–162, and motif 2, which corresponded to E2F484–92 versus E2F1193–198 (Figure 6, C and D). Based on this analysis, we introduced three E2F4 mutations (M1, M2, and M1+2) in which we swapped one, or both, of these motifs in the context of either the N-terminal E2F41–197 fragment or the full-length E2F4. As a control for folding, we established that these small sequence swaps had no detectable effect on E2F4’s ability to heterodimerize with DP1 (Supplemental Figure S13), confirming that these mutants retained significant 3D structure and also ascertained that they displayed some degree of colocalization with Deup1 and SAS6, as judged by immunofluorescence (Supplemental Figures S14–S16). We then conducted co-IP experiments to determine their ability to bind to Deup1 and SAS6. The M2 mutant retained the ability to bind to both Deup1 (Figure 6, E and F) and SAS6 (Figure 6G). In contrast, the M1 mutation alone abolished the ability of E2F41–197 to bind Deup1 (Figure 6E) and greatly impaired association of full-length E2F4 to Deup1 (Figure 6F). Moreover, it also prevented full-length E2F4 from binding to SAS6 (Figure 6G). The combined M1 and M2 mutations abrogated all interaction between full-length E2F4 to Deup1 (Figure 6F). Thus, these experiments identify specific residues of E2F4, particularly 48–53, as being critical for efficient binding to Deup1 and SAS6.
E2F4 and E2F5 are not essential for primary ciliogenesis
Given that E2F4 and E2F5 interact with SAS6, which functions in both deuterosome and the mother centriole of primary cilia, and that endogenous E2F4 localizes to the mother centriole in vivo (Mori et al., 2017), we wondered whether E2F4 and E2F5 are important for the generation of primary cilia. In our prior analyses of E2f4 and E2f5 mutant mouse models, we detected multiciliogenesis and not primary cilia defects, but these studies were conducted with single and compound mutants (E2f4–/–;E2f5+/– or E2f4+/–;E2f5–/–), never double homozygous mutants. Thus, we used two approaches to generate cells that completely lacked both E2F4 and E2F5. First, we generated mouse embryo fibroblasts (MEFs) from E2f4f/f;E2f5–/– embryos, or E2f4f/f;E2f5+/+ as controls, and infected these with lentivirus expressing Cre recombinase to ablate E2f4. Following drug selection for infected cells, these populations were transferred into reduced serum media to induce quiescence and the formation of primary cilia. Staining for Arl13b, a primary cilia marker, showed that the E2f4 and E2f5 double mutant cells continued to produce primary cilia (Figure 7A) and the frequency of cilia-bearing cells was not significantly different from the control (Figure 7B). For the second approach, we examined the skin sections of e14.5 E2f4f/f;E2f5–/–;Meox2-Cre double mutant mouse embryos relative to the control, E2f4f/+;E2f5+/–;Meox2-Cre embryos. We initially established that E2F4 expression was absent from E2f4f/f;E2f5–/–;Meox2-Cre embryos by immunohistochemistry (Figure 7C). Staining for Arl13b again showed the combined loss of E2F4 and E2F5 did not abolish the formation of primary cilia (Figure 7D). Accordingly, gross examination of E2f4f/f;E2f5–/–;Meox2-Cre embryos did not reveal phenotypes typical of loss of primary cilia function, such as an open neural tube (Goetz, Ocbina and Anderson, 2009). These data therefore show that E2F4 and E2F5 play specific roles in multiciliogenesis and are not essential for primary ciliogenesis.
FIGURE 7:

E2F4 and E2F5 are not essential for primary cilia formation. (A) E2f4f/f;E2f5+/+ and E2f4f/f;E2f5–/– MEFs were transduced with lentivirus expressing Cre recombinase to ablate E2F4 and then stained via immunofluorescence for primary cilia (Arl13b, green signal) and nuclei (DAPI, blue signal). Inset is 2× main figure. Scale bar represents 15 μm. (B) Quantification shows that the percentage of cells carrying primary cilia is not significantly reduced in the double mutant MEFs relative to the E2f4f/f;E2f5+/+ controls. (C) Immunohistochemistry for E2F4 (brown signal) shows the efficient loss of E2F4 protein in the skin of e14.5 E2f4f/f;E2f5–/–;Meox2-Cre double mutant mouse embryos vs. the control E2f4f/+;E2f5+/–;Meox2-Cre embryos. Nuclei are counterstained (blue). (D) Adjacent skin sections from control (E2f4f/+;E2f5+/–;Meox2-Cre) and double knockout embryos (E2f4f/f;E2f5–/–; Meox2-Cre) stained by immunofluorescence for primary cilia (Arl13b, green signal) and nuclei (DAPI, blue signal) show that both form primary cilia.
DISCUSSION
This study shows that in a transfected cell line, E2F4 can physically associate with Deup1 and SAS6, but not with other core deuterosome components which colocalize with E2F4 during multiciliated cell differentiation in tracheal cultures, including Cep152, Plk4, and Centrin1. These results do not preclude the participation of these proteins in higher order cytoplasmic E2F4 complexes. Although we were unable to isolate any, we suspect that their formation requires multiple components, post-translational modifications, and/or other intracellular conditions that are not present in the transfected cell line. By focusing on the most robust interactions identified, we determined that Deup1 and SAS6 use structurally distinct amino- terminal domains to bind E2F4. For Deup1, E2F4 binding maps to residues 1–129. This region includes the full sequence of the first coiled-coil domain. In contrast, SAS6 uses residues 1–175, which contains a structurally unique pisa motif (residues 39–91) and motif II (residues 123–140) and excludes its single coiled-coil domain. Together, pisa and motif II constitute a continuous, highly conserved patch that is required for SAS6 homodimers to multimerize, via head-to-head contacts, to form a cartwheel structure with ninefold radial symmetry (van Breugel et al., 2011). A synthetic single amino acid mutation (F131D) within motif II that prevents the multimerization of SAS6 dimers (van Breugel et al., 2011; Kitagawa et al., 2011) was sufficient to abolish SAS6’s binding to E2F4. In contrast, the I62T mutation, found in members of a consanguineous family afflicted with a classic ciliopathy syndrome (Khan et al., 2014), did not alter SAS6’s binding to E2F4. Based on the SAS6 crystal structure (van Breugel et al., 2011), I62 is not involved in the multimerization of SAS6 dimers. Together, the differential phenotypes of these two SAS6 point mutants point to a high degree of specificity for the SAS6-E2F4 interaction and raises the possibility that E2F4 actively facilitates SAS6 multimerization and/or binds specifically to the cartwheel assembly of SAS6 dimers.
During these studies, both E2F4 and Deup1 were observed to undergo mobility-shifts on coexpression and these new species were specifically enriched in E2F4-Deup1 complexes. In the case of Deup1, we observe the loss of carboxy-terminal sequences, suggesting that cleavage of Deup1 occurs as a consequence of complex formation. For E2F4, we see the coexpression of full-length Deup1 or Deup11–129, but not SAS6, promotes phosphorylation, showing that it is Deup1-specific. Interestingly, this phosphorylation is only observed with the full-length E2F4, not the E2F41–197 region that mediates the interaction with Deup1. Moreover, mobility shifts of E2F5 were not observed following IP with Deup1. Thus, it is unclear whether these modifications are essential for multiciliogenesis, or even act to modulate this process.
Mapping within E2F4 identified the amino-terminal region (1–197) as necessary and sufficient to bind both Deup1 and SAS6. Moreover, our ALI in vitro differentiation experiments showed that E2F41–197 is able to mediate the cytoplasmic functions of E2F4 in multiciliogenesis as efficiently as the full-length E2F4 protein. This provides strong support for the idea that E2F4’s cytoplasmic role in multiciliogenesis is mediated through these interactions. Considering that E2F4 and E2F5 are important for multiciliogenesis in mammals and fish (Stracker, 2019), we hypothesized that the interaction domain would be conserved between these two E2Fs and not other E2Fs. Consistent with this hypothesis, we found that Deup1 and SAS6 can both associate with either E2F4 or E2F5, but not E2F1. Moreover, this specificity led us to identify two motifs, M1 and M2, both within the DBD, which differ between these two E2F subgroups. Mutation of these motifs suggests that M2 may play a supporting role in enabling the interaction between Deup1 and the full-length E2F4 protein, since mutation of M1 alone does not fully abrogate the interaction while mutation of M1+2 does. Most importantly, the data strongly argue that the binding of both Deup1 and SAS6 to E2F4 is primarily dependent on M1, which corresponded to residues 48–53 of E2F4, and is conserved in E2F5 but not E2F1. The fact that Deup1 and SAS6 both rely on the same sequence in E2F4 for interaction raises three possibilities: the existence of a trimolecular E2F4-Deup1-SAS6 complex; the existence of, as yet unidentified, bridging proteins; or independent, and thus presumably mutually exclusive, binding of SAS6 or Deup1 to E2F4. We are not aware of any reports of an interaction between SAS6 and Deup1 and we were unable to detect this ourselves. Moreover, in our assays, SAS6 is clearly able to bind E2F4 in cells that lack Deup1, arguing that SAS6 or another bridging protein directly contact E2F4. Additionally, we find that Deup1 is able to bind internal E2F4 deletion mutants (HA-E2F4Δ101–197, HA-E2F4Δ137–197, and HA-E2F4Δ101–137) that SAS6 cannot. Given these findings, we favor the notion that E2F4 binds independently to SAS6 and Deup1 using the M1 site, suggesting that these interactions are mutually exclusive and potentially could occur in a spatiotemporally regulated manner. During multiciliogenesis, E2F4/5 also interacts with MCIDAS or GEMC1 to activate the transcription of genes required for multiciliogenesis, including Deup1 (Ma et al., 2014). This MCIDAS/GEMC1 interaction does not require the DBD of E2F4 (Ma et al., 2014); thus, it is quite distinct from E2F4’s interaction with Deup1 and SAS6. Presumably E2F4’s interactions with MCIDAS and GEMC1 occur predominantly within the nucleus and are not concurrent with E2F4/5 binding to Deup1/SAS6 in the cytoplasm; however, this remains to be established. Interestingly, the nucleo-cytoplasmic shuttling of other proteins, such as the splicing factor SRSF1, is also important for multiciliogenesis (Haward et al., 2021).
We had previously found that E2F4 colocalizes with the Cep63 at the mother centriole at early stages of the multiciliogenesis differentiation process (Mori et al., 2017). Despite the conservation between Deup1 and Cep63, E2F4 did not associate with Cep63 in our transfection assay, suggesting that E2F4 localization to the mother centriole is mediated by other centriole components, with SAS6 being a good candidate. We wondered whether this SAS6 association and mother centriole localization might reflect a role for E2F4 and E2F5 in the formation of primary cilia. However, our in vivo analyses of E2f4 and E2f5 double mutant cells showed that these proteins are specifically required for multiciliogenesis and not primary cilia formation. This specific requirement may reflect the sheer extent of centriole replication required to generate a multiciliated cell in terms of both the levels of transcription of genes encoding the centriole replication machinery complex and the role of E2F4/5 in enabling massive centriole replication in the cytoplasm.
As described earlier, multiciliogenesis can occur in cells lacking Deup1 and Cep63 and thus without both deuterosomes and mother centrioles (Mercey et al., 2019). We suggest that this does not negate the potential importance of the E2F4-Deup1 interaction because, in the context of WT cells, the deuterosome is the predominant site of centriole amplification. However, it is intriguing to note that, in cells lacking Deup1 and Cep63, centriole replication occurs in centriolar satellites, a site where we have previously shown that E2F4 colocalizes with PCM1 (Mori et al., 2017). This raises the possibility that E2F4 could facilitate centriole replication, at this site, potentially via interaction with SAS6, even in the absence of Deup1 and Cep63. Taking all these data together, we propose that following their transcriptional role in multiciliogenesis, E2F4 and E2F5 play a scaffolding or kinetic function in promoting the assembly or efficacy of centriole replication via their interaction with Deup1 and/or SAS6.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Molecular cloning
HA-tagged E2F4 truncation mutants HA-E2F41–100, HA-E2F41–130, HA-E2F41–158, HA-E2F41–197, and HA-E2F4198–410 and HA-tagged internal E2F4 deletion mutants HA-E2F4Δ101–197 (retains residues 1–100 plus 198–410), HA-E2F4Δ137–197 (retains residues 1-136 plus 198–410), and E2F4Δ101–137 (retains residues 1–100 plus 138–410) were generated using standard PCR cloning techniques starting with the pCMVSPORT2 (Invitrogen) murine E2F4 cDNA plasmid. Plasmids expressing Flag-tagged Deup1 full-length and its truncation mutants (Flag-Deup11–129, Flag-Deup1309–535, Flag-Deup1401–535, and Flag-Deup1130–309), Flag-Cep63, Flag-Cep152, and Flag-Plk4 were kind gifts of Xueliang Zhu. pEGFP-hPCM1 and hPCM1-EGFP plasmids were kind gifts of Song-Hai Shi. CMV-HA-DP1 is previously described (Wu et al., 1995). Flag-tagged Deup60–535, Deup186–535, and Cep6367–192 plasmids were cloned into pcDNA3.1(+) (Thermo Fisher V79020). A kinase dead version of Plk4 was a gift from Erich Nigg (Addgene 41164). Amplification of Deup1 using forward primer encoding Flag tag and reverse primer encoding HA tag allowed generation of the double-tagged Deup1 construct (pcDNA3.1-Flag-Deup1-HA). C-terminal truncation mutants of double-tagged Deup1 constructs (Flag-Deup11–491-HA, Flag-Deup11–461-HA, and Flag-Deup11–432-HA) were generated using standard PCR-based cloning techniques. pENTR-age-Hs-SAS6 was a kind gift from Pierre Gonczy (Addgene 46381). Amino-terminus Flag-tagged SAS6 truncation mutants (Flag-SAS61–175, Flag-SAS6160–505, and Flag-SAS6499–656) were generated by PCR amplification of human SAS6 gene from pENTR-age-Hs-SAS6 plasmid using primers including Flag tag sequences and cloning into pcDNA3.1(+) plasmid. Flag-tagged SAS6 point mutants (SAS6I62T and SAS6F131D) were cloned using QuikChange II XL-site-directed mutagenesis kit (Agilent Technologies, 200521). We generated pcDNA3.1-SAS6 I62T using the following primer pair:
Forward 5′-CCATTTTTTTTATATAACCTTGTTACATCTGAGGAAGATTTTCAAAG
Reverse 5′-CTTTGAAAATCTTCCTCAGATGTAACAAGGTTATATAAAAAAAATGG.
We generated pcDNA3.1-SAS6 F131D using the following primer pair:
Forward 5′-GGTAGAGACAAATCCTGATAAGCATCTTACACACC
Reverse 5′-GGTGTGTAAGATGCTTATCAGGATTTGTCTCTACC.
pCMV-Neo-Bam-HA-E2F1 plasmid was previously generated in our lab (Moberg et al., 1996) and HA-tagged E2F5 was a kind gift from Kristian Helin (Addgene 24213). Plasmids expressing HA-tagged E2F4/E2F1 chimeras were generated using the Gibson assembly cloning kit (New England Biolabs E5510S). The oligonucleotides used in Gibson cloning are listed in Supplemental Table S1. All sequences were verified by DNA sequencing; details are available on request.
Cell culture
The 293FT (Invitrogen) cells were maintained in DMEM (Corning 10-017-CV) supplemented with heat-inactivated, 10% fetal bovine serum (FBS, HyClone SH30910.03) and 1% penicillin/streptomycin (P/S, Corning MT30002Cl). U2OS cells (ATCC HTB-96) were maintained in DMEM supplemented with 10% FBS, 1% P/S, and 2 mM L-glutamine (GE Healthcare SH30034). Phosphate-buffered saline (PBS) was obtained from Corning 21-03-1CV. Cells lines were tested yearly for mycoplasma.
Co-IP and Western blotting
Plasmids were cotransfected into 293FT cells, 10-cm dishes (8 µg per plasmid, per dish) using Lipofectamine 2000 (Thermo Fisher Scientific 11668019). After 48 h, transfected cells were lysed in NP40-based lysis buffer: 20 mM HEPES, pH 7.8 (VWR EM-5310), 15% glycerol (VWR 117240), 250 mM potassium chloride (KCL, Malinckrodt 6858), 0.2 mM EDTA (EDTA, VWR MK4931-04), 1% IGEPAL (Sigma-Aldrich CA-630), 50 mM L-glutamic acid (Sigma-Aldrich G1251), 50 mM L-arginine (Sigma-Aldrich RPI A50010), 1 mM dithiothreitol (DTT, VWR 97061-338), 50 mM sodium fluoride (NaF, Sigma-Aldrich S7920), 1 mM sodium orthovanadate (NaVO3, Sigma-Aldrich 13721-39-6), 0.1 mM phenylmethanesulfonyl fluoride (PMSF, Sigma-Aldrich P7626), 1 µg/ml leupeptin (Sigma-Aldrich L2884), and 1 µg/ml aprotinin (Sigma-Aldrich A1153). For the endogenous E2F4-SAS6 interaction, 293FT cells were lysed in a milder lysis buffer: 50 mM Tris-HCl, pH7.5 (Macron fine chemicals, 7544-06), 0.5 mM EDTA, 150 mM sodium chloride (NaCl, VWR 0497), 0.5% IGEPAL, 1 mM NaVO3, 0.1 mM PMSF, 1 µg/ml leupeptin, and 1 µg/ml aprotinin. For all IP experiments, cell lysates were centrifuged at 1000 rpm for 10 min at 4°C. Proteins were quantified using a BCA protein assay kit (Thermo Fisher Scientific 23250), and equal amounts (0.8 mg for overexpressed or 1.5 mg endogenous protein lysates) of each sample were precleared by incubation with protein A/G agarose beads (Santa Cruz sc-2003, Thermo Fisher Scientific 20421) for 30 min at 4°C and subsequently incubated for 2 h at 4°C with α-E2F4 agarose beads (Santa Cruz sc-866 AC), α-HA magnetic beads (Thermo Fisher Scientific 88836), α-Flag M2 magnetic beads (Sigma-Aldrich M8823), α-SAS6 agarose beads (Santa Cruz 81451), or anti-mouse or anti-rabbit IgG agarose beads as appropriate (Santa Cruz sc-2343, Santa Cruz sc-2345). The beads were washed 5 times in IP buffer and boiled in 90 µl of 2× SDS loading buffer containing 20% DTT for 10 min. Total lysates and immunoprecipitates were resolved in SDS–PAGE gels, transferred to PVDF membranes (Sigma-Aldrich IPVH00010), blocked in 8% nonfat milk powder (Biorad 1706404XTU), and probed with relevant primary antibodies for 2 h at room temperature or overnight at 4°C: α-E2F4 (LLF4.2 (Moberg et al., 1996), 1:500), α-Deup1 (provided by Xueliang Zhu, 1:3000), α-SAS6 (Santa Cruz sc-81431, 1:1000), α-Flag (Sigma-Aldrich F1804, 1:1000; Sigma-Aldrich F3165, 1:2000), α-HA (12CA5, Roche 11583816001, 1:2000), and native secondary antibodies for 1 h at room temperature: HRP–conjugated mouse (Rockland 18-8817-33, 1:5000) or rabbit (Rockland 18-8816-33, 1:5000), washing in TBST (Tween 20, Sigma-Aldrich P2287) three rounds following both primary and secondary antibody incubation. Western blot experiments were conducted as described (Moberg et al., 1996). The specificity of antibodies was verified by blotting extracts containing untagged proteins or untransfected cells. Western blots were developed using Amersham ECL prime Western blotting detection reagent (GE Healthcare, RPN2232) and exposed to autoradiography film (Thomas Scientific 1159T41). All IP and Western blotting experiments were repeated independently three times.
Protein phosphatase treatment
Flag-tagged beads were incubated with lysates of cells that overexpress either full-length or N-terminal domain Flag-tagged Deup1 and E2F4. Beads were washed three times with lysis buffer and then treated with lambda protein phosphatase according to the NEB Lambda PP kit (New England Biolabs P07535). These experiments were repeated independently three times.
Mass spectrometry and data analysis
Lysates of cells overexpressing Flag-Deup1 and E2F4 were immunoprecipitated with Flag-tagged beads and analyzed by mass spectrometry by the Biopolymer & Proteomics core facility. Samples were reduced with 5 mM DTT and alkylated with 10 mM iodoacetamide before digesting with either 0.5 µg of trypsin, or chymotrypsin, or 0.5 µg of GluC followed by overnight digestion with 0.5 µg of trypsin. Samples were acidified by adding formic acid to 2% final. Acidified peptides were loaded with the help of an autosampler directly onto a 50-cm EASY-Spray C18 column (ES803a, Thermo Scientific). A Thermo Easy nanoLC system was used to elute the peptides with a 3-min gradient from 0% buffer B to 3% buffer B (100% acetonitrile, 0.1% formic acid), followed by a 55-min gradient to 23% and a 5-min gradient to 100% B (buffer B: 80% acetonitrile in 0.1% formic acid) and held constant for 3 min. Finally, the gradient was changed from 100% buffer B to 100% buffer A (100% water, 0.1% formic acid) over 0.5 mins and then held constant at 100% buffer A for 13.5 more minutes. Peptides were sprayed into the Thermo QExactive HFX by applying voltage of 2.0 kV. Mass spectrometer-scanning functions and HPLC gradients were controlled by the Xcalibur data system (Thermo Scientific). For the data-dependent acquisition (DDA) experiments, MS1 scan parameters were 60,000 resolution, scan range m/z 375–1500, AGC at 3e6, IT at 50 ms. MS2 scan parameters were at 60,000 resolution, isolation width at m/z 1.2, HCD collision energy at 28%, AGC target at 1e6%, maximum IT at 118ms, TopN at 10, with an intensity threshold at 3.4e4. Charge states 2–5 were considered for MS2. An inclusion list comprising all possible peptides created by a double digest with GluC and trypsin, with one missed cleavage and possible phosphorylation of proteins Deup1 and E2F4, was also added. For parallel reaction monitoring (PRM) experiments, instrument settings were changed to 60,000 resolution, 1e6 AGC target with maximum IT of 118 ms, isolation window of m/z 1.4 containing nine potential phosphorylated peptides in the inclusion list. DDA data was analyzed by ProteomeDiscoverer 2.3.0.523 as well as Byonic v3.5.0. Masses of potential phosphorylated peptides were used in the inclusion list for the PRM experiments. Data from the PRM experiments were manually analyzed.
Immunofluorescence
U2OS cells were plated at low density on gelatin (Sigma-Aldrich G7041)-coated glass coverslips (VWR 48380-046). The next day, cells were transiently transfected with HA-tagged truncation mutants alone or with Flag-tagged Deup1 or SAS6; 48 h later, the cells were washed with PBS, fixed in 4% paraformaldehyde (Electron Microscopy Sciences 15710) for 10 min, incubated in PBS 50 mM ammonium chloride (Sigma-Aldrich A0171) for 10 min, then washed and permeabilized with 0.25% Triton X-100 (Sigma-Aldrich T9284) in PBS for 4 min. After washing in PBS 10 mg/ml bovine serum albumin (BSA) (Sigma-Aldrich A7906) and blocking in 5% goat serum (Vector Laboratories S-1000) in PBS for 30 min at 37°C, the cells were incubated with α-HA (Cell Signaling 3724, 1/1600 dilution) or α-Flag (Sigma-Aldrich F1804, 1/1000 dilution) in PBS 10 mg/ml BSA for 1 h, washed three times in the same buffer, and then incubated with Alexa Fluor 488 (1/1000, Thermo Fisher A11001) or Texas Red (1/1000, Thermo Fisher T6391)–conjugated secondary antibodies in PBS 10 mg/ml BSA containing 1 µg/ml DAPI (BD Pharmingen 564907) for 1 h in the dark and then washed three times. Following the last wash, coverslips were washed with water, placed on mounting media (Vector Labs Vectashield Plus H1900), and sealed with nail polish. All experiments were repeated three times. Images were captured using a Zeiss AxioPlan II upright microscope or a Nikon Spinning-Disk Confocal microscope. Lentiviral transduction and staining for Arl13b on cells or tissue sections were performed as described (Guen et al., 2017). The lentiviral backbone plasmid containing Cre recombinase was a gift from Tyler Jacks. Images were analyzed using Fiji software (https://imagej.net).
ALI cultures of airway epithelial progenitors
Culture experiments and microscopy were performed as described (Mori et al., 2017). Quantification of the HA-positive multiciliated cell populations was performed blind. Prism8 (GraphPad) was used to generate the graph and statistical significance was determined using a two-tailed unpaired t test (https://www.graphpad.com/scientific-software/prism/).
Transgenic mouse strains and immunohistochemistry
Animal studies were approved by the Committee for Animal Care and conducted in compliance with the Animal Welfare Act Regulations and other federal statutes relating to experiments involving animals and adhered to the principles set forth in the Guide for the Care and Use of Laboratory Animals (8th ed., National Research Council, 2011 [PHS institutional animal welfare assurance # A3125–01]). The E2f4f/f, R26CreERT2/+, and E2f5+/– transgenic strains and the genotyping protocols are as described previously (Danielian et al., 2016; Mori et al., 2017). The Meox2-Cre allele was purchased from the Jackson Laboratory (#003755). Cre recombinase expression from the Meox2 promoter is largely ubiquitous in the embryo proper by e6 (Hayashi et al., 2002; Tallquist and Soriano, 2000). Embryos at e14.5 were collected, fixed in formalin, and processed in paraffin for sectioning and subsequent immunohistochemistry for E2F4, as described (Danielian et al., 2007). Four embryos of each genotype were analyzed by immunohistochemistry and immunofluorescence.
Supplementary Material
Acknowledgments
We thank the Koch Institute Swanson Biotechnology Center for technical support, particularly the Microscopy, Biopolymers, and Proteomics and Hope Babette Tang Histology core facilities. We also thank mem¡bers of the Lees and Yaffe laboratories at MIT for helpful advice and discussions and Song-Hai Shi, Pierre Gonczy, Kristian Helin, Tyler Jacks, David Livingston, Erich Nigg, and Xueliang Zhu for plasmids, mouse strains, and reagents. Funding support was from the National Cancer Institute for the Koch Institute Support (core) Grants P30-CA14051 and PO1 CA42063 (to J.A.L.); from the National Institutes of Health; the National Heart, Lung, and Body Institute R35 HL135834-01 (to W.V.C); and from the National Institute of General Medical Science R01 GM124148 (to S.M.R.). R.H. was recipient of a David H. Koch Fellowship for Cancer Research. J.A.L. is the Virginia and D.K. Ludwig Professor for Cancer Research at the Koch Institute at MIT.
Abbreviations used:
- ALI
air–liquid interface
- BSA
bovine serum albumin
- Cep63
centrosomal protein 63
- DBD
DNA binding domain
- DDA
data-dependent acquisition
- Deup1
deuterosome assembly protein 1
- DTT
dithiothreitol
- FBS
fetal bovine serum
- IP
immunoprecipitation
- MEF
mouse embryo fibroblast
- NES
nuclear export signal
- PBS
phosphate-buffered saline
- Plk4
polo-like kinase 4
- PMSF
phenylmethanesulfonyl fluoride
- PRM
parallel reaction monitoring
- P/S
penicillin/streptomycin
- SAS6
spindle assembly homolog 6
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-01-0039).
REFERENCES
- Al Jord A, Spassky N, Meunier A (2019). Motile ciliogenesis and the mitotic prism. Biol Cell 111, 199–212. [DOI] [PubMed] [Google Scholar]
- Boutin C, Kodjabachian L (2019). Biology of multiciliated cells. Curr Opin Genet Dev 56, 1–7. [DOI] [PubMed] [Google Scholar]
- van Breugel M, Hirono M, Andreeva A, Yanagisawa H, Yamaguchi S, Nakazawa Y, Morgner N, Petrovich M, Ebong IO, Robinson CV, et al. (2011). Structures of SAS-6 suggest its organization in centrioles. Science 331, 1196–1199. [DOI] [PubMed] [Google Scholar]
- Brooks ER, Wallingford JB (2014). Multiciliated cells: a review. Curr Biol R973–R982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NJ, Marjanovic M, Luders J, Stracker TH, Costanzo V (2013). Cep63 and cep152 cooperate to ensure centriole duplication. PLoS One 8, e69986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YL, Zhang Y, Zhou F, Roy S (2018). Distinct requirements of E2f4 versus E2f5 activity for multiciliated cell development in the zebrafish embryo. Dev Biol 443, 165–172. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Bender Kim CF, Caron AM, Vasile E, Bronson RT, Lees JA (2007). E2f4 is required for normal development of the airway epithelium. Dev Biol 305, 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, Hess RA, Lees JA (2016). E2f4 and E2f5 are essential for the development of the male reproductive system. Cell Cycle 15, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Ocbina PJR, Anderson KV (2009). The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol 94, 199–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guen VJ, Chavarria TE, Kröger C, Ye X, Weinberg RA, Lees JA (2017). EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc Natl Acad Sci USA 114, E10532–E10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haward F, Malson MM, Yeyati PL, Bellora N, Hansen JN, Aitken S, Lawson J, Kriegsheim A, Wachten D, Mill P, et al. (2021). Nucleo-cytoplasmic shuttling of splicing factor SRSF1 is required for development and cilia function. eLife 10, e65104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP (2002). Efficient gene modulation in mouse epiblast using Sox2Cre transgenic mouse strain. Mech Dev 119(Supp. 1), S97–S101. [DOI] [PubMed] [Google Scholar]
- Humbert PO, Rogers C, Ganiatsas S, Landsberg RL, Trimarchi JM, Dandapani S, Brugnara C, Erdman S, Schrenzel M, Bronson RT, et al. (2000). E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell 6, 281–291. [DOI] [PubMed] [Google Scholar]
- Khan MA, Rupp VM, Orpinell M, Hussain MS, Altmüller J, Steinmetz MO, Enzinger C, Thiele H, Höhne W, Nürnberg G, et al. (2014). A missense mutation in the PISA domain of HsSAS-6 causes autosomal recessive primary microcephaly in a large consanguineous Pakistani family. Hum Mol Genet 23, 5940–5949. [DOI] [PubMed] [Google Scholar]
- Kitagawa D, Vakonakis I, Olieric N, Hilbert M, Keller D, Olieric V, Bortfeld M, Erat MC, Flückiger I, Gönczy P, et al. (2011). Structural basis of the 9-fold symmetry of centrioles. Cell 144, 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo A, Sasaki H, Yuba-Kubo A, Tsukita S, Shiina N (1999). Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J Cell Biol 147, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M, Stracker TH (2021). Transcriptional regulation of multiciliated cell differentiation. Seminars Cell Dev Biol 110, 51–60. [DOI] [PubMed] [Google Scholar]
- Liban TJ, Medina EM, Tripathi S, Sengupta S, Henry RW, Buchler NE, Rubin SM (2017). Conservation and divergence of C-terminal domain structure in the retinoblastoma protein family. Proc Natl Acad Sci USA 114, 4942–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Nguyen QPH, Nanjundappa R, Delgehyr N, Megherbi A, Doherty R, Thompson J, Jackson C, Abulescu A, Heng YM, et al. (2020). Super-resolution microscopy and FIB-SEM imaging reveal parental centriole-derived, hybrid ciliun in mammalian multiciliated cells. Dev Cell 55, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Quigley I, Omran H, Kintner C (2014). Multicilin drives centriole biogenesis via E2f proteins. Genes Dev 28, 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercey O, Levine MS, LoMastro GM, Rostaing P, Brotslaw E, Gomez V, Kumar A, Spassky N, Mitchell BJ, Meunier A, et al. (2019). Massive centriole production can occur in the absence of deuterosomes in multiciliated cell. Nat Cell Biol 21, 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg K, Starz MA, Lees JA (1996). E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol 16, 1436–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Hazan R, Danielian PS, Mahoney JE, Li H, Lu J, Miller ES, Zhu X, Lees JA, Cardoso WV (2017). Cytoplasmic E2f4 forms organizing centres for initiation of centriole amplification during multiciliogenesis. Nat Commun 8, 15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW (2009). Centrioles, centrosomes, and cilia in health and disease. Cell 139, 663–678. [DOI] [PubMed] [Google Scholar]
- Sir JH, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D’Santos C, Woods CG, Gergely F (2011). A primary microcephaly protein complex forms a ring around parental centrioles, Nat Genet 43, 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin SP (1968). Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci 3, 207–230. [DOI] [PubMed] [Google Scholar]
- Spassky N, Meunier A (2017). The development and functions of multiciliated epithelia. Nat Rev Mol Cell Biol 18, 423–436. [DOI] [PubMed] [Google Scholar]
- Stracker TH (2019). E2F4/5-mediated transcriptional control of multiciliated cell differentiation: redundancy or fine-tuning? Dev Biol 446, 20–21. [DOI] [PubMed] [Google Scholar]
- Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gonczy P (2007). Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell 13, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P (2000). Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26, 113–115. [DOI] [PubMed] [Google Scholar]
- Terré B, Piergiovanni G, Segura-Bayona S, Gil-Gómez G, Youssef SA, Attolini CS, Wilsch-Bräuninger M, Jung C, Rojas AM, Marjanović M, et al. (2016). GEMC1 is a critical regulator of multiciliated cell differentiation. EMBO J 35, 942–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3, 11–20. [DOI] [PubMed] [Google Scholar]
- Yan X, Zhao H, Zhu X (2016). Production of Basal Bodies in bulk for dense multicilia formation. F1000Research 5, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CL, Zukerberg LR, Ngwu C, Harlow E, Lees. JA (1995) In vivo association of E2F and DP family proteins. Mol Cell Biol 15, 2536–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhu L, Zhu Y, Cao J, Li S, Huang Q, Xu T, Huang X, Yan X, Zhu X (2013). The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Nat Cell Biol 15, 1434. [DOI] [PubMed] [Google Scholar]
- Zheng N, Fraenkel E, Pabo CO, Pavletich NP (1999). Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev 13, 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.