

Abstract
Discovering significant pathways rather than single genes or small gene sets involved in metastasis is becoming more and more important in the study of breast cancer. Many researches have shed light on this problem. However, most of the existing works are relying on some priori biological information, which may bring bias to the models. The authors propose a new method that detects metastasis‐related pathways by identifying and comparing modules in metastasis and non‐metastasis gene co‐expression networks. The gene co‐expression networks are built by Pearson correlation coefficients, and then the modules inferred in these two networks are compared. In metastasis and non‐metastasis networks, 36 and 41 significant modules are identified. Also, 27.8% (metastasis) and 29.3% (non‐metastasis) of the modules are enriched significantly for one or several pathways with p ‐value <0.05. Many breast cancer genes including RB1, CCND1 and TP53 are included in these identified pathways. Five significant pathways are discovered only in metastasis network: glycolysis pathway, cell adhesion molecules, focal adhesion, stathmin and breast cancer resistance to antimicrotubule agents, and cytosolic DNA‐sensing pathway. The first three pathways have been proved to be closely associated with metastasis. The rest two can be taken as a guide for future research in breast cancer metastasis.
Inspec keywords: cancer, genetics, genomics, DNA, molecular biophysics, adhesion, cellular biophysics
Other keywords: breast cancer metastasis, module extraction, gene sets, metastasis‐related pathways, nonmetastasis gene coexpression networks, Pearson correlation coefflcients, breast cancer genes, RB1, CCND1, TP53, glycolysis pathway, cell adhesion molecules, focal adhesion, stathmin, breast cancer resistance, antimicrotubule agents, cytosolic DNA‐sensing pathway, breast cancer metastasis
1 Introduction
Cancer is one of the most daunting worldwide diseases. It causes about 13% of all human death [1]. Distant metastasis is the main cause of death among cancer patients. When a patient is likely to suffer metastasis, he/she may be treated with aggressive adjuvant therapy. However, the poor understanding of the underlying causes and the physiological mechanisms of metastasis may increase the difficulty of diagnosis. In reality, about 70–80% patients receiving such adjuvant therapy would have survived without it [2]. Thus, there is a great need to gain deeper understanding of metastasis so that every patient can receive appropriate therapy. Many efforts have been made to uncover the molecular mechanisms of different cancers.
In the last decade, many disease markers were identified through genome‐wide expression profiles. Differentially expressed genes across different disease states have been identified and their associated functions have been studied to understand the disease mechanisms. In breast cancer, both van't Veer et al. [2] and Wang et al. [3] identified about 70 gene markers that might play a role in the development of metastasis and their prediction accuracy for metastasis is about 0.6–0.7. Similar approaches determined differentially expressed genes for B‐cell lymphoma [4], lung cancer [5] and leukaemia [6]. However, the detected single‐gene markers vary considerably in different data sets for the same disease and thus they may lack universal applicability. Chuang et al. [7] proposed a protein‐network‐based approach for identifying markers of metastasis. The markers are small subnetworks of interacting proteins within protein–protein interaction (PPI) network. It increased the reproducibility across different data sets and the classification accuracy of metastasis. Moreover, Dao et al. [8] addressed the great phenotypical complexity of cancer by employing density‐constrained biclustering to search subnetwork markers. Although the genes and subnetworks they found have provided novel hypotheses for mechanisms of tumour progression, their main idea was to extract a single gene or a small set of genes that were most likely to account for the transition from non‐metastasis to metastasis. Nevertheless, it is widely accepted that all kinds of molecules, including DNAs, RNAs, proteins, and so on, interact with each other and work in concert to influence metastasis of cancer. Focusing only on single genes or small subnetworks tends to restrict the revelation of mechanisms of metastasis.
On the contrary, discovering pathways, processes in which all kinds of molecules interact to perform a specific function, can be a better choice for gaining insight into the mechanisms of metastasis in at least two respects. First, pathway as an aggregate of biomolecules that interact with each other can reveal biological processes involved in metastasis more completely than the existing works did. Second, understanding metastasis in terms of pathway will provide more helpful guidance for cancer therapies. For example, Spek and Arruda [9] provided the evidence on the role of the protein C pathway in cancer metastasis and pointed out the potential of the activated protein C as a novel drug target to reduce cancer progression. Also, it is stated in [10] that the future of cancer drug development may be targeting pathways rather than single genes and their products.
Up to now, considerable pathways have been proved to play an important role in cancer metastasis and many methods have been developed to detect metastasis‐related pathways. Kang et al. [11] provided functional evidence for a switch of Smad pathway, from tumour‐suppressor to prometastatic, in the development of breast cancer bone metastasis. Hu et al. [12] highlighted the evidence for aberration of the Notch signalling pathway in metastasis of tumours such as ostesarcoma, breast cancer, prostate cancer and melanoma. Moreover, Ward et al. [13] revealed a hitherto‐unknown but essential interaction of the RalGFF and ERK pathways to produce a malignant phenotype. And Wang et al. [14] indicated that the overall activity of the cofilin pathway took part in determining the invasive and metastasis phenotype of tumour cells. Several approaches have been put forward to detect metastasis‐related pathways using gene expression data and other biological data. There are at least two types of approaches to identify or discover pathways, that is, knowledge‐based approach, which exploits pathways in public repositories such as the gene ontology (GO) or Kyoto encyclopaedia of genes and genomes (KEGG), and data‐based approach which explores pathways using molecular measurements like gene expression data. The first has been summarised in [15], which reviewed the development of pathway analysis during the last decade. As for the second, some researchers utilised gene expression data coupled with PPI network to discover pathways, including probabilistic model by Segal et al. [16], and colour‐coding methods by Yeh et al. [17]. The knowledge‐based approach suffers from incomplete annotation as indicated in [15], whereas the data‐based approach integrates PPI network with gene expression data to identify pathways and, PPI network, as an incomplete biological database, may cause bias in discovering pathways.
In this paper, we assume that the expression of genes in the same pathway is covariant over samples and develops a method for identifying pathways from gene expression data. Our method can be divided into three steps. First, we build the gene co‐expression networks for non‐metastasis group and metastasis group and extract modules in parallel. Second, the pathways that the modules are enriched are identified. Third, we compare the modules from the two groups in terms of pathways and discover metastasis‐related pathways. We apply our method to a breast cancer data set, and meaningful results are discussed.
2 Materials and methods
2.1 Data set
The gene expression data set was downloaded from gene expression omnibus (GEO) with accession number GSE2034: ‘Breast cancer relapse free survival’ [18]. The samples are obtained from primary breast tumours and they are hybridised to Affymetrix Human Genome U133A Array platform to get the gene expression profiles. A total number of 286 breast cancer patients and 22 273 genes are included in the data set. We use the binary logarithm of the original data for our analysis. After computing the variance of each gene over all patients, we finally chose 5292 genes with variances greater than 1.2 to conduct our analysis.
2.2 Network construction
We divided all the patients into two groups, including the metastasis group and non‐metastasis group. Specifically, we assigned 107 patients, with whom metastasis had been detected within 80 months after surgery, into the group of metastasis and the remaining 179 patients into the group of non‐metastasis. Then, for each group, we constructed a weighted gene co‐expression network, where the 5292 nodes represent the 5292 genes and the weights of edges are the absolute values of Pearson correlation coefficients between the two corresponding genes. Apparently, a larger weight of the edges indicates a stronger co‐expression relationship between the two genes.
2.3 Module extraction
A module in a network is a set of nodes that are closely connected with each other and weakly connected to the rest. For our weighted network, the modules are subnetworks that have high weights. The frequently used method for module identification is to divide a network into several modules with each node being in one of the modules. However, in reality, the nodes that have weak connections to all the rest nodes are assigned to a particular module, which may lead to a low enrichment of functions or pathways. Here, we consider extracting the densely connected modules and ignore those sparsely connected genes. We adopted the criterion proposed by Zhao et al. [19] for the module extraction. Let A = [aij ] be the 5292 × 5292 adjacency matrix of our gene co‐expression network, where aij is the absolute value of the Pearson correlation coefficient between gene i and gene j and aii equals 0. Given a particular extracted module S, let S c denote its complement in the network. Then we maximise the following formula over all possible S
| (1) |
where
| (2) |
and |·| denotes the number of genes in a given module.
A great value of criterion (1) is an intuitively reasonable indicator for a module of high quality. The first term of (2) represents the average weight of the edges within S, and the second term is the average connection between S and the rest of the network. So, this criterion can extract modules with a large number of links within itself and a small number of links to the rest of the network [19].
Since (1) is a NP problem in nature, we use tabu search [20], a local optimisation technique, to maximise the criterion. The algorithm is described in Appendix 1 (Fig. 2, Algorithm 1). Owing to the intensive computation of the algorithm and the large size of the network, this algorithm may run slow. Thus before running this algorithm, we first partition our network into a few smaller networks with their module structure being kept. We apply the method proposed in [21]. This method is shown to perform well for module identification and it runs fast compared to most existing methods. Its main idea is to maximise the average degree in each module and minimise the average connections between any two modules. Here, the number of partitions depends on the scale that the module extraction algorithm can handle. After this step, the module extraction algorithm is applied to all the identified subnetworks to extract modules.
Fig. 2.

Find a module in a given network
In our implementation, we first partitioned the network into smaller networks and then carried out module extraction in each of them. The number of nodes in each network varied from 500 to 1000 and the total time of computation turned out to be greatly reduced. We performed Algorithm 1 (Fig. 2) 10 times for 10 initial values of S and adopted the optimal S as the final solution. The parameters were set as
| (3) |
for each network.
After the numerical computation, we determine whether the module should be extracted or not using the permutation test proposed in [19]. Only modules with high significance levels should be extracted. Every time a new module was extracted, 100 permutated versions of the original network were simulated and 100 corresponding W (S) were obtained with the same parameters as (3). We accepted the new module and searched for the next one in the remainder only if there were no more than five permutated networks having larger W (S) than the original one. Otherwise, we rejected the module and stopped searching in the current network since all pairs of genes are nearly equally connected.
2.4 Filtering modules
It is necessary to select modules of high quality from all extracted modules. Since the criterion (1) and the permutation test impose no specific constraints on the sizes of the extracted modules or the strength of links within a module, the method [19] tends to find modules of small sizes in some cases. Gene sets of very small sizes or weak linkage are possible to be extracted inappropriately as modules. We therefore filtered all extracted modules and accepted only those including at least five genes and having average weight of more than 0.5.
3 Results and discussion
3.1 Pathway enrichment analysis
We identified 36 and 41 significant modules in metastasis and non‐metastasis networks (consisting of 338 and 397 genes, respectively). The basic information about the modules (including official gene symbol IDs and average weight) is listed in Appendix 2 (Table 4) and Appendix 3 (Table 5). Since the expression profile was hybridised to Affymetrix Human Genome U133A array platform, we converted the Affymetrix IDs into official gene symbol IDs using the tool ‘Batch Query’ provided by ‘NetAffx Analysis Center’. After a file containing a list of Affymetrix IDs is uploaded, the tool will return the corresponding official gene symbol IDs. However, this conversion is not one‐to‐one. It is possible that one Affymetrix ID corresponds to more than one official gene symbol IDs or that no official gene symbol IDs match an Affymetrix ID, but both situations are rare. We accepted all official gene symbol IDs matching one of Affymetrix IDs of genes in a module, so the number of gene names in a module may not be equal to the size of the module, as presented in Appendix 2 (Table 4) and Appendix 3 (Table 5).
We used DAVID to do pathway‐enrichment analysis for each module. 27.8% (metastasis) and 29.3% (non‐metastasis) of the modules are enriched for one or several pathways when setting the threshold of p ‐value to be 0.05. The pathways we found in each module are listed in Tables 1 and 2.
Table 1.
Pathways in the metastasis group
| Metastasis | Pathways | Included genes | p ‐value |
|---|---|---|---|
| 1 | glycolysis pathway | GAPDH, PGK1 | 0.015 |
| 5 | cell adhesion molecules (CAMs) | ITGA6, HLA‐G, HLA‐DRA | 0.017 |
| 15 | PPAR signalling pathway | ADIPOQ, FABP4, LPL | 0.001,1 |
| 20 | stathmin and breast cancer resistance to antimicrotubule agents | CD2, CD247 | 0.027 |
| T cell receptor signalling pathway | CD247, CD3D, LCK | 0.001,3 | |
| primary immunodeficiency | CD3D, LCK | 0.021 | |
| 21 | cytokine‐cytokine receptor interaction | CD27, CXCL9, LTB | 0.047 |
| 24 | chemokine signalling pathway | XCL1, XCL2, CCL19, PRKCB | 0.013 |
| 25 | toll‐like receptor signalling pathway | CCL5, CXCL10, CXCL11 | 0.001,2 |
| chemokine signalling pathway | CCL5, CXCL10, CXCL11 | 0.003,9 | |
| cytokine–cytokine receptor interaction | CCL5, CXCL10, CXCL11 | 0.007,7 | |
| cytosolic DNA‐sensing pathway | CCL5, CXCL10 | 0.032 | |
| 30 | signalling in immune system | IGK@, IGKC, CKAP2, IGLC1, IGLV1‐44 | 0.000,049 |
| 32 | Lck and Fyn tyrosine kinases in the initiation of TCR activation | HLA‐DRA, PTPRC | 0.007,7 |
| B lymphocyte cell surface molecules | HLA‐DRA, PTPRC | 0.007,7 | |
| activation of Csk by cAMP‐dependent protein kinase inhibits signalling through the T ‐cell receptor | HLA‐DRA, PTPRC | 0.012 | |
| 33 | focal adhesion | COL1A1, COL6A1, FLNA, MYL9 | 0.000,061 |
| 33 | ECM‐receptor interaction | COL1A1, COL6A1 | 0.049 |
First column represents the index of a metastasis module. Column ‘Pathways’ represents pathway(s) for which the module is enriched. The p ‐values are obtained from KEGG or Biocarta
Table 2.
Pathways in the non‐metastasis group
| Non‐metastasis | Pathways | Included genes | p ‐value |
|---|---|---|---|
| 1 | signalling in immune system | CKAP2, IGLC1, IGLV1‐44, IGK@, IGKC, LOC642838, LOC100130100 | 0.000,59 |
| 7 | Lck and Fyn tyrosine kinases in initiation of TCR activation | HLA‐DRA, PTPRC | 0.007,7 |
| B lymphocyte cell surface molecules | HLA‐DRA, PTPRC | 0.007,7 | |
| activation of Csk by cAMP‐dependent protein kinase inhibits signalling through the T ‐cell receptor | HLA‐DRA, PTPRC | 0.012 | |
| 10 | vascular smooth muscle contraction | MYLK, MYH11 | 0.022 |
| 16 | ECM‐receptor interaction | COL6A1, COL6A2 | 0.033 |
| 17 | insulin signalling pathway | SHC1, TSC2, CRK | 0.022 |
| 18 | chemokine signalling pathway | ARRB2, CXCR4, RAC2, STAT1 | 0.000,46 |
| leukocyte transendothelial migration | CXCR4, CYBA, RAC2 | 0.005,1 | |
| endocytosis | ARRB2, CXCR4, HLA‐G | 0.012 | |
| 23 | primary immunodeficiency | CD3D, LCK | 0.021 |
| 24 | T ‐cell receptor signalling pathway | CD247, CD3G, PTPRC, ZAP70 | 0.001,4 |
| cytokine–cytokine receptor interaction | CD27, XCL1, XCL2, CCL5, CXCL9, LTB | 0.001,7 | |
| 24 | chemokine signalling pathway | XCL1, XCL2, CCL5, CXCL9, PRKCB | 0.006,5 |
| natural killer cell‐mediated cytotoxicity | CD247, PRKCB, ZAP70 | 0.032 | |
| 29 | PPAR signalling pathway | PLIN1, CD36, ADIPOQ | 0.000,54 |
| adipocytokine signalling pathway | CD36, ADIPOQ | 0.039 | |
| 31 | toll‐like receptor signalling pathway | CXCL10, STAT1, CXCL11 | 0.001,2 |
| chemokine signalling pathway | CXCL10, STAT1, CXCL11 | 0.003,9 | |
| 36 | cell cycle | CDC20, TTK | 0.049 |
| 39 | B cell receptor signalling pathway | CR2, CD19 | 0.044 |
First column represents the index of a non‐metastasis module. The column ‘Pathways’ represents pathway(s) for which the module is enriched. The p ‐values are obtained from KEGG or Biocarta
Many known breast cancer susceptibility genes are in the same pathways of metastasis modules. CDH1 is in cell adhesion molecules (CAM) pathway. ERBB2, PTEN and CCND1 are in focal adhesion pathway. RB1, CCND1, TP53 and MYC are involved in cell cycle pathway.
3.2 Significant pathways in breast cancer metastasis
There are five significant pathways that are only identified in metastasis modules. They are glycolysis pathway, CAMs, focal adhesion, stathmin and breast cancer resistance to antimicrotubule agents, and cytosolic DNA‐sensing pathway. Distributions of weights within those five modules are plotted in Fig. 1. The first three pathways are shown to be closely related with metastasis in previous research.
Fig. 1.
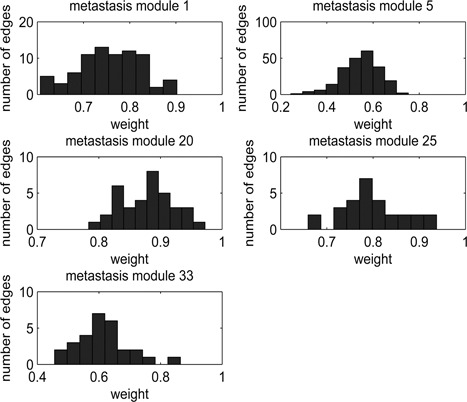
Distribution of weights of edges within each of five modules corresponding to five exclusive pathways in the metastasis group
Up regulation of glycolysis is a near‐universal property in metastasis cancers, including breast cancer. It represents adaptations to a hypoxic microenvironment that is associated with tumour invasion, metastasis and lethality [22]. And many researches around glycolysis pathway has been conducted. Gatenby and Gillies [23] proposed an explanation about high aerobic glycolysis in cancer. Zhong et al. [22] pointed out that HIF‐1α plays an important role in human cancer progression. In their study, HIF‐1α overexpression was detected in only 29% of primary breast cancers but in 69% of breast cancer metastasis [22]. Sun et al. [24] discovered that DCA has anti‐proliferative properties and can be effective against highly metastatic diseases.
CAMs is also a signal of cancer metastasis. Alterations in the expression and function of CAMs correlate with the progression to tumour malignancy. There are many researches about the relationship between CAMs and different cancers. For example, Christofori and Semb [25] pointed out that the loss of the CAM E ‐cadherin is involved in the formation of epithelial cancer. He also discussed in his review [26] that a possible signalling role of CAMs, and with it, tumour malignancy. Paschos et al. [27] suggested that CAMs may also mediate the selection of the host organ, for the development of distant colorectal metastasis.
When we talk about focal adhesion pathway, much attention has been paid on focal adhesion kinase (FAK). For example, Kornberg [28] pointed out that FAK may have two roles: over‐expression of FAK leads to (i) increased cell migration and (ii) increased cell survival under anchorage‐independent conditions. Planas‐Silva et al. [29] showed that FAK is likely to be biomarkers to predict the risk of recurrence in ER‐positive breast cancer. Since FAK is closely associated with focal adhesion pathway, deeper research in the pathway can shed light on mechanism of breast cancer metastasis.
For the other two significant pathways, stathmin and breast cancer resistance to antimicrotubule agents, and cytosolic DNA‐sensing pathway, few relative work is found. However, these pathways may be viewed as putative markers for breast cancer metastasis which may serve as a guide for future research.
3.3 Significance test for pathway enrichment
To test the significance of the difference in pathways between metastasis and non‐metastasis groups in the background of genes we studied, we conducted a significance test on three pathways: glycolysis pathway, CAMs and focal adhesion, which have been proved metastasis‐related above. Such a test is necessary because there is no guarantee that these three pathways result from the difference between the structures of metastasis and non‐metastasis networks.
First, we define the similarity between two modules. Let G be the set of all genes involved in our analysis, and A, B ⊂ G be the two modules. We measure their similarity by
| (4) |
where |·| represents the number of genes a module contains. A great value of (4) indicates a large proportion of genes which A and B share.
For each of the three pathways, we use M 1 to denote its corresponding module of the metastasis group, and let M 2 be the module of the non‐metastasis group that maximises S (M 1, M 2). The difference in gene composition between these two modules can be viewed as a physiological change from non‐metastasis to metastasis. In order to prove it is the change in pathway activity from non‐metastasis to metastasis that most likely accounts for the change from M 1 to M 2, we have designed a significance test for M 1 and M 2. The null hypothesis is that all genes are equally possible to be extracted.
Assuming we randomly extract two modules R 1, R 2 ⊂ G under the condition that
| (5) |
the p ‐value is computed as
| (6) |
| (7) |
| (8) |
| (9) |
where
| (10) |
| (11) |
Here, P denotes the set of all human genes involved in the pathway that are obtained from KEGG. More specifically, here the p ‐value is the probability that
| (12) |
under the condition (5).
In addition, to demonstrate that these three pathways are much more significant in the metastasis network than in the non‐metastasis one, we compare the significant levels of each pathway in two networks in the background G. Take the pathway glycolysis as an example. For the metastasis network, we define the p ‐value as the probability that we obtain no less than k 1 = 2 genes in glycolysis when randomly selecting |M 1 | = 13 genes from G. The p ‐value of glycolysis in the non‐metastasis network is defined in the same fashion with k 2 = 1 and = |M 2 | = 17. Then the ratio of the non‐metastasis p ‐value to the metastasis one is calculated.
The p ‐values, ratios and other information are listed in Table 3. The large ratios indicate that the significant levels of pathway in metastasis network are many times higher than those in non‐metastasis network. This means there is a large gap with regard to glycolysis between metastasis and non‐metastasis networks, although there is only a discrepancy of 1 in the number of glycolysis genes contained in their modules (2 and 1, respectively). The small p ‐values demonstrate the significance of difference in the pathway enrichment and prove the validity of our method.
Table 3.
Results of the significance test for three pathways; number of studied genes N = 5292
| Pathway | M 1 | M 2 | |M 1 | | |M 2 | | k 1 | k 2 | c | n | p ‐value | Ratio |
|---|---|---|---|---|---|---|---|---|---|---|
| glycolysis | 1 | 14 | 13 | 17 | 2 | 1 | 6 | 31 | 1.9 × 10−3 | 38 |
| CAMs | 5 | 19 | 22 | 10 | 3 | 0 | 6 | 78 | 1.3 × 10−3 | 256 |
| focal adhesion | 33 | 21 | 8 | 6 | 4 | 1 | 3 | 127 | 9.6 × 10−6 | 6667 |
Column ‘M 1 ’ denotes the index of the corresponding module in the metastasis group, while the column ‘M 2 ’ denotes the index of the module in the non‐metastasis group that maximises S (M 1, M 2). The column ‘k 1 (k 2)’ denotes the number of genes in both M 1 (M 2) and the pathway. ‘c ’ denotes the number of genes in both M 1 and M 2, and ‘n ’ denotes the number of genes that the pathway includes and that we studied
4 Concluding remarks
In many previous researches on discovering the pathological characteristics of cancer, people focused on extracting a single gene or subnetwork differentially expressed across different cancer states. However, single‐gene identification lacks reproducibility across different data sets and subnetwork extraction may be biased due to incomplete priori biological information and relatively small size of subnetwork. Here, we detect the pathways involved in cancer metastasis via a module extraction approach without any preceding biological knowledge in a completely quantitative way. Five exclusive cancer‐related pathways are discovered in the metastasis group and the significance test proved that our method is able to detect the differences in topological structures of networks.
Although we have obtained a basically satisfying result, it is possible for us to improve the method in two respects. First, the extraction criterion (1) proposed by [19] tends to find relatively small modules, so it is likely that only some key genes of a complete module are extracted, which may impair the completeness of our modules. Had some extraction method producing more complete modules been adopted, more information of metastasis‐related pathways would have been mined. Secondly, computation intensity for module search increases rapidly with the size of network increasing. A more computationally efficient method is desired.
5 Acknowledgments
This work was supported in part by NSFC grants 10901042, 91130032, Shanghai Natural Science Foundation 13ZR1403600 and Fudan's Undergraduate Research Opportunities Program.
See Fig. 2.
See Table 4.
Table 4.
Modules of metastasis group
| Index | Size | Gene name | AW |
|---|---|---|---|
| 1 | 13 | HSPD1, PSMC3, LAMP1, TMED2, ABCF2, HSPA8, AKAP1, MSH6, HSP90AB1, PGK1, TMEM230, ACTB, GAPDH | 0.78 |
| 2 | 12 | P4HB, CALR, UBE2M, APOE, PTP4A3, CDC37, JUND, TSC2, FARSA, ELOVL1, SH3BGRL3 | 0.73 |
| 3 | 19 | CRK, TMED2, ARF1, STAT3, CDC5 L, MPDU1, YWHAE, NOTCH2, TAGLN2, HLA‐G, ARPC4, NFYC, POLR2E, PICALM, CDK11A, CDK11B, C14orf1, ENO1, EXOC5 | 0.71 |
| 4 | 7 | FBN1, FBLN1, CDH11, COL6A2, VCAN, DCN | 0.76 |
| 5 | 22 | SNX17, PGRMC1, PPIF, SPTLC1, CDYL, SMARCA2, MED6, HLA‐DRA, KLF6, PRPF4, WTAP, ACLY, HLA‐G, DIMT1, PRPF4B, USP34, ADAM10, TRRAP, ITGA6, RPS2, TMCO3, STAT1 | 0.60 |
| 6 | 8 | PRKCD, CDK5, CPT2, MAD1L1, MED8, LTBP3, CCNL2, INO80B | 0.61 |
| 7 | 14 | PRPF31, MMP11, CLDN3, TFAP2A, HPCAL1, CRIP2, HMG20B, PPAP2C, CD46, MUC1, TP53I11, PLXNB1, LPPR2 | 0.60 |
| 8 | 7 | TPSAB1, TPSB2 | 0.82 |
| 9 | 7 | SRRT, NPRL3, MAZ, STIP1, NUMA1, PLEC, ELMO2 | 0.64 |
| 10 | 6 | PKP4, GOLGA2, SRSF10, H2AFY, UBE2H, AKIRIN1 | 0.60 |
| 11 | 11 | NUCB1, CYBA, ARRB2, NUP62, TSPAN4, AP3D1, COL4A2, ALOX5, FADS3, MBTPS1, EHBP1L1 | 0.59 |
| 12 | 5 | MEFV, SPN, SCD5, SNW1 | 0.58 |
| 13 | 5 | HBA1, HBA2 | 0.92 |
| 14 | 10 | AEBP1, FBN1, COL10A1, INHBA, MFAP5, ADAM12, LRRC15, COL10A1, COL8A2 | 0.76 |
| 15 | 8 | FHL1, LPL, FABP4, LEP, ADIPOQ, G0S2, GPD1, RBP4 | 0.74 |
| 16 | 7 | FBLN1, SFRP4, DCN, COL14A1, DPT, SPON1 | 0.73 |
| 17 | 7 | ITM2A, ABCA8, ENPP2, IGF1, ADH1B, CHRDL1 | 0.71 |
| 18 | 5 | MYH11, LMOD1, SVEP1 | 0.69 |
| 19 | 10 | IGHG1, IGHG2, IGHM, IGHV4‐31, GUSBP11, IGLC1, IGK@, IGKC, LOC100294406, CYAT1, IGLV1‐44, IGLL3P | 0.92 |
| 20 | 9 | LCK, CD2, GZMK, TRAC, CD247, TRBC1, CD3D | 0.85 |
| 21 | 15 | CXCL9, PTPRCAP, ACAP1, CD27, MAP4K1, CD3G, LTB, SH2D1A, GPR18, ICOS, PPP1R16B, NKG7, MAP4K1, CD52 | 0.67 |
| 22 | 7 | KRT5, TRIM29, S100A2, SERPINB5, JUP, KRT17, KRT14 | 0.73 |
| 23 | 9 | CA12, ESR1, GATA3, TBC1D9 | 0.84 |
| 24 | 15 | ARHGAP25, POU2AF1, TRAF3IP3, CD1C, GPR171, PRKCB, LGALS2, CCL19, GZMB, PTGDS, CLIC2, XCL1, XCL2, TRAT1 | 0.61 |
| 25 | 8 | CCL5, GBP1, TAP1, CXCL10, CXCL11 | 0.77 |
| 26 | 8 | SFRP1, GABRP, MIA, ID4, SOX10, SYNM | 0.74 |
| 27 | 6 | XBP1, FOXA1, AGR2, SPDEF, MLPH | 0.83 |
| 28 | 8 | NNMT, PDGFRA, FBLN2, GAS1, C1S, RARRES2, CXCL12, PDPN | 0.68 |
| 29 | 6 | MYLK, CNN1, DST, LOC100652766, ID4, LOC100287705, PTN | 0.56 |
| 30 | 14 | IGK@, IGKC, LOC642838, CKAP2, IGLC1, IGLV1‐44, LOC100130100, LOC100287723 | 0.87 |
| 31 | 15 | IGHM, IGH@, IGHA1, IGHA2, IGHD, IGHG1, IGHG3, IGHG4, IGHV3‐23, IGHV4‐31, IGLJ3, IGHG2, LOC100291917, IGKC, IGLC1, IGLV1‐44, LOC100294406, IGLL5 | 0.71 |
| 32 | 6 | SRGN, CTSS, PTPRC, HLA‐DRA, HLA‐DQA1, HLA‐DQA2, LOC100507718, LOC100509457, HLA‐DRB1, HLA‐DRB3, HLA‐DRB4, LOC100507709, LOC100507714 | 0.81 |
| 33 | 8 | MYL9, COL1A1, MFAP2, EMILIN1, TAGLN, BMP1, COL6A1, FLNA | 0.69 |
| 34 | 5 | DCN, SLIT3, SFRP4, COPZ2 | 0.72 |
| 35 | 8 | BIRC5, FOXM1, NDC80, NEK2, TTK, CENPE, MKI67, NCAPH | 0.62 |
| 36 | 8 | STK10, CD52, LCP2, PSMB8, HLA‐DQB1, LOC100293977, CASP1, FYN, SLC15A3 | 0.57 |
‘Size’ denotes the number of Affymetrix IDs in a module, whereas ‘AW’ represents the average weight within a module
See Table 5.
Table 5.
Modules of non‐metastasis group; ‘Size’ denotes the number of Affymetrix IDs in a module, whereas ‘AW’ represents the average weight within a module
| Index | Size | Gene name | AW |
|---|---|---|---|
| 1 | 13 | IGLJ3, IGKC, LOC642838, CKAP2, IGLC1, IGLV1‐44, CKAP2, IGK@, IGH@, IGHA1, IGHA2, IGHG1, IGHG2, IGHG3, IGHM, IGHV4‐31, LOC100130100, LOC100287723 | 0.88 |
| 2 | 13 | IGHM, IGH@, IGHA1, IGHA2, IGHD, IGHG1, IGHG3, IGHG4, IGHV3‐23, IGHV4‐31, IGHV4‐31, IGHG2, LOC100291917, IGK@, IGLV1‐44, IGLL5 | 0.79 |
| 3 | 7 | AEBP1, COL11A1, INHBA, LRRC15, COL10A1, COL8A2 | 0.78 |
| 4 | 5 | DCN, COL1A1, FBLN1 | 0.83 |
| 5 | 10 | C7, ABCA8, MEOX1, TNXA, TNXB, DARC, IGF1, CHRDL1, COL14A1 | 0.75 |
| 6 | 12 | NNMT, FBN1, FBLN1, PDGFRA, FBLN2, GAS1, DCN, RARRES2, DPT, CFH, SPON1 | 0.71 |
| 7 | 8 | SRGN, CTSS, LCP2, PTPRC, HLA‐DRA, HLA‐DQA1, HLA‐DQA2, LOC100507718, LOC100509457, HLA‐DRB1, HLA‐DRB3, HLA‐DRB4, LOC100507709, LOC100507714 | 0.81 |
| 8 | 10 | IGHA1, IGHA2, IGHD, IGHG1, IGHG3, IGHG4, IGHM, IGHV4‐31, IGH@, LOC100291917, LOC100653245, IGLV1‐44, IGLC1, IGLJ3, IGHV3‐48 | 0.66 |
| 9 | 6 | FHL1, FABP4, LEP, G0S2, GPD1, ITGA7 | 0.75 |
| 10 | 5 | MYH11, MYLK, LMOD1, CNN1 | 0.71 |
| 11 | 5 | TPSAB1, TPSB2 | 0.86 |
| 12 | 5 | MYL9, TAGLN, SERPINH1, CTGF, ADAM12 | 0.65 |
| 13 | 6 | CD52, CD79A, ACAP1, PTGDS, CD7, MS4A1 | 0.60 |
| 14 | 17 | P4HB, HSPD1, CALR, PSMC3, LAMP1, UBE2M, APOE, ARF1, MIR3620, TAGLN2, COL4A2, POLR2E, HSP90AB1, FARSA, ELOVL1, SH3BGRL3, ACTB, LOC100505829, GAPDH | 0.77 |
| 15 | 16 | PGRMC1, SPTLC1, TMED2, YWHAE, HSPA8, SNORD14C, SNORD14D, NOTCH2, PRPF4B, MSH6, CCNG2, NFYC, RBFOX2, ADAM10, PICALM, TMEM230 | 0.71 |
| 16 | 7 | FBN1, EMILIN1, CDH11, COL6A2, VCAN, COL6A1 | 0.70 |
| 17 | 29 | NUCB1, SHC1, CRK, PRPF31, JUND, CLDN3, TFAP2A, EIF2S3, HPCAL1, PTP4A3, CBX4, BTBD2, CRIP2, RALGDS, HMG20B, PRPF4, TSPAN4, HNRNPUL1, CDC37, AKAP1, ARPC4, TP53I11, TSC2, ENO1, MBTPS1, PTMS, INO80B, INO80B‐WBP1, EHBP1L1 | 0.65 |
| 18 | 7 | CYBA, ARRB2, RAC2, CXCR4, HLA‐G, STAT1 | 0.64 |
| 19 | 10 | SNX17, PPIF, CDYL, MED6, ABCF2, STAT3, CDC5L, WTAP, ACLY, LOC100652805, LOC100653302, PGK1 | 0.62 |
| 20 | 18 | MAT2A, BHLHE40, SLC29A1, ATP6V1A, NFYA, MPDU1, FOXO3, FOXO3B, TP53, CD46, STIP1, USP34, MTUS1, TRRAP, PLXNB1, SEC23IP, C14orf1, EXOC5, TMCO3 | 0.57 |
| 21 | 6 | MMP14, MFAP2, MMP11, BMP1, FLNA | 0.61 |
| 22 | 12 | IGLC1, IGHG1, IGHG2, IGHM, IGHV4‐31, IGK@, IGKC, GUSBP11, LOC100294406, CYAT1, IGLV1‐44, IGLL3P | 0.90 |
| 23 | 10 | LCK, CD2, GZMK, TRAC, SH2D1A, TRBC1, CD3D, CD52 | 0.88 |
| 24 | 20 | CCL5, CXCL9, ARHGAP25, PTPRCAP, POU2AF1, CD27, MAP4K1, CD3G, LTB, GPR171, PRKCB, CD247, PTPRC, PPP1R16B, ZAP70, CTSW, XCL1, XCL2 | 0.69 |
| 25 | 9 | IGHM, IGK@, IGKC, IGLJ3, IGLC1, IGLV1‐44 | 0.80 |
| 26 | 11 | CA12, FOXA1, ESR1, GATA3, TBC1D9, MLPH | 0.82 |
| 27 | 7 | SFRP1, GABRP, MIA, SOX10, SYNM | 0.78 |
| 28 | 8 | LRMP, LCK, IKZF1, CCL19, PTGDS, KLRB1, YME1L1 | 0.67 |
| 29 | 6 | PLIN1, CD36, ADIPOQ, ADH1B, RBP4 | 0.80 |
| 30 | 6 | MX1, IFI27, IFI44L, ISG15, RSAD2, IFI44 | 0.78 |
| 31 | 7 | GBP1, TAP1, CXCL10, STAT1, CXCL11 | 0.81 |
| 32 | 6 | KRT5, TRIM29, SERPINB5, JUP, KRT17, KRT6B | 0.71 |
| 33 | 7 | XBP1, SLC44A4, AGR2, AR, SPDEF, SIDT1, SPDEF | 0.71 |
| 34 | 8 | SFRP4, ITGBL1, COMP, OMD, COL10A1, MFAP5, ASPN, COPZ2 | 0.69 |
| 35 | 8 | HBB, HBA1, HBA2 | 0.87 |
| 36 | 10 | TOP2A, BIRC5, FOXM1, CDC20, NDC80, NEK2, TTK, CENPE, NCAPH, CDCA8 | 0.71 |
| 37 | 14 | RUNX3, LAMP3, CD38, ITGB7, ADAMDEC1, XCL1, NCF4, GZMB, ICOS, NKG7, LYZ, BIN2, SLAMF8, IL21R | 0.65 |
| 38 | 6 | CLDN5, CCL14, CCL14‐CCL15, CCL15, IGF1, MFAP4, SVEP1 | 0.70 |
| 39 | 10 | SIT1, CR2, HLA‐DOB, CD19, GPR18, P2RX5, IGHM, BANK1, STAP1, LRMP | 0.58 |
| 40 | 12 | CCND2, IL32, NCF1, NCF1B, NCF1C, GNLY, GABBR1, UBD, PTPN22, LAG3, CCR5, GZMH, BIRC3, KLRD1 | 0.56 |
| 41 | 5 | RARRES1, CHI3L1 | 0.77 |
‘Size’ denotes the number of Affymetrix IDs in a module, whereas ‘AW’ represents the average weight within a module
6 References
- 1.‘Cancer’. http://en.wikipedia.org/wiki/Cancer, accessed October 2013
- 2. van't Veer L.J. Dai H., and Van De Vijver M.J. et al.: ‘Gene expression profiling predicts clinical outcome of breast cancer’, Nature, 2002, 415, (6871), pp. 530–536 (doi: 10.1038/415530a) [DOI] [PubMed] [Google Scholar]
- 3. Wang Y. Klijn J.G.M., and Zhang Y. et al.: ‘Gene‐expression profiles to predict distant metastasis of lymph‐node‐negative primary breast cancer’, The Lancet, 2005, 365, (9460), pp. 671–679 (doi: 10.1016/S0140-6736(05)17947-1) [DOI] [PubMed] [Google Scholar]
- 4. Alizadeh A.A. Eisen M.B., and Eric Davis R. et al.: ‘Distinct types of diffuse large b‐cell lymphoma identified by gene expression profiling’, Nature, 2000, 403, (6769), pp. 503–511 (doi: 10.1038/35000501) [DOI] [PubMed] [Google Scholar]
- 5. Beer D.G. Kardia S.L.R., and Huang C.‐C. et al.: ‘Gene‐ expression profiles predict survival of patients with lung adenocarcinoma’, Nat. Med., 2002, 8, (8), pp. 816–824 [DOI] [PubMed] [Google Scholar]
- 6. Golub T.R. Slonim D.K., and Tamayo P. et al.: ‘Molecular classification of cancer: class discovery and class prediction by gene expression monitoring’, Science, 1999, 286, (5439), pp. 531–537 (doi: 10.1126/science.286.5439.531) [DOI] [PubMed] [Google Scholar]
- 7. Chuang H.‐Y. Lee E. Liu Y.‐T. Lee D., and Ideker T.: ‘Network‐based classification of breast cancer metastasis’, Mol. Syst. Biol., 2007, 3, (1) article 140; doi:10.1038/msb4100180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dao P. Colak R., and Salari R. et al.: ‘Inferring cancer subnet‐work markers using density‐constrained biclustering’, Bioinformatics, 2010, 26, (18), pp. i625–i631 (doi: 10.1093/bioinformatics/btq393) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arnold Spek C., and Arruda V.R.: ‘The protein c pathway in cancer metastasis’, Thrombosis Res., 2012, 129, pp. S80–S84 (doi: 10.1016/S0049-3848(12)70022-1) [DOI] [PubMed] [Google Scholar]
- 10.‘A new pathway for cancer research’. http://www.wired.com/wiredscience/2008/09/war‐on‐cancer‐g/, accessed October 2013
- 11. Kang Y. He W., and Tulley S. et al.: ‘Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway’. Proc. National Academy of Sciences of the United States of America, 2005, vol. 102, no. 39, pp. 13909–13914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu Y.‐Y. Zheng M.‐h. Zhang R. Liang Y.‐M., and Han H.: ‘Notch signaling pathway and cancer metastasis’. Notch Signaling in Embryology and Cancer (Springer, 2012), pp. 186–198 [DOI] [PubMed] [Google Scholar]
- 13. Ward Y. Wang W. Woodhouse E. Linnoila I. Liotta L., and Kelly K.: ‘Signal pathways which promote invasion and metastasis: critical and distinct contributions of extracellular signal‐regulated kinase and Ral‐specific guanine exchange factor pathways’, Mol. Cell. Biol., 2001, 21, (17), pp. 5958–5969 (doi: 10.1128/MCB.21.17.5958-5969.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang W. Eddy R., and Condeelis J.: ‘The cofilin pathway in breast cancer invasion and metastasis’, Nat. Rev. Cancer, 2007, 7, (6), pp. 429–440 (doi: 10.1038/nrc2148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khatri P. Sirota M., and Butte A.J.: ‘Ten years of pathway analysis: current approaches and outstanding challenges’, PLoS Comput. Biol., 2012, 8, (2), p. e1002375 (doi: 10.1371/journal.pcbi.1002375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Segal E. Wang H., and Koller D.: ‘Discovering molecular pathways from protein interaction and gene expression data’, Bioinformatics, 2003, 19, (Suppl 1), pp. i264–i272 (doi: 10.1093/bioinformatics/btg1037) [DOI] [PubMed] [Google Scholar]
- 17. Yeh C.‐Y. Yeh H.‐Y. Arias C.R., and Soo V.‐W.: ‘Pathway detection from protein interaction networks and gene expression data using color‐coding methods and a* search algorithms’, Sci. World J., 2012, 2012, article ID 315797 (doi: 10.1100/2012/315797) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.‘Breast cancer relapse free survival’. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2034, accessed September 2013
- 19. Zhao Y. Levina E., and Zhu J.: ‘Community extraction for social networks’, Proc. Natl Acad. Sci., 2011, 108, (18), pp. 7321–7326 (doi: 10.1073/pnas.1006642108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glover F.: ‘Tabu search: a tutorial’, Interfaces, 1990, 20, (4), pp. 74–94 (doi: 10.1287/inte.20.4.74) [DOI] [Google Scholar]
- 21. Zhang S., and Zhao H.: ‘Community identification in networks with unbalanced structure’, Phys. Rev. E, 2012, 85, p. 066114 (doi: 10.1103/PhysRevE.85.066114) [DOI] [PubMed] [Google Scholar]
- 22. Zhong H. De Marzo A.M., and Laughner E. et al.: ‘Overexpression of hypoxia‐inducible factor 1α in common human cancers and their metastases’, Cancer Res., 1999, 59, (22), pp. 5830–5835 [PubMed] [Google Scholar]
- 23. Gatenby R.A., and Gillies R.J.: ‘Why do cancers have high aerobic glycolysis?’, Nat. Rev. Cancer, 2004, 4, (11), pp. 891–899 (doi: 10.1038/nrc1478) [DOI] [PubMed] [Google Scholar]
- 24. Sun R.C. Fadia M. Dahlstrom J.E. Parish C.R. Board P.G., and Blackburn A.C.: ‘Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo’, Breast Cancer Res. Treat., 2010, 120, (1), pp. 253–260 (doi: 10.1007/s10549-009-0435-9) [DOI] [PubMed] [Google Scholar]
- 25. Christofori G., and Semb H.: ‘The role of the cell‐adhesion molecule e‐cadherin as a tumour‐suppressor gene’, Trends Biochem. Sci., 1999, 24, (2), pp. 73–76 (doi: 10.1016/S0968-0004(98)01343-7) [DOI] [PubMed] [Google Scholar]
- 26. Christofori G.: ‘Changing neighbours, changing behaviour: cell adhesion molecule‐mediated signalling during tumour progression’, EMBO J., 2003, 22, (10), pp. 2318–2323 (doi: 10.1093/emboj/cdg228) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paschos K.A. Canovas D., and Bird N.C.: ‘The role of cell adhesion molecules in the progression of colorectal cancer and the development of liver metastasis’, Cell. Signal., 2009, 21, (5), pp. 665–674 (doi: 10.1016/j.cellsig.2009.01.006) [DOI] [PubMed] [Google Scholar]
- 28. Kornberg L.J.: ‘Focal adhesion kinase and its potential involvement in tumor invasion and metastasis’, Head Neck, 1998, 20, (8), pp. 745–752 (doi: 10.1002/(SICI)1097-0347(199812)20:8<745::AID‐HED14>3.0.CO;2‐Z) [DOI] [PubMed] [Google Scholar]
- 29. Planas‐Silva M.D. Bruggeman R.D. Grenko R.T., and Stanley Smith J.: ‘Role of c‐SRC and focal adhesion kinase in progression and metastasis of estrogen receptor‐positive breast cancer’, Biochem. Bio‐Phys. Res. Commun., 2006, 341, (1), pp. 73–81 (doi: 10.1016/j.bbrc.2005.12.164) [DOI] [PubMed] [Google Scholar]