

Abstract
A major objective of ‘omics’ technologies is to understand genetic causality of complex traits of human diseases. High‐throughput omics technologies and their application to medicine open up remarkable opportunities for realising optimised medical treatment for individuals. Because many major breakthrough and discoveries in this field have been driven by the development of new omics technologies, in this review, the authors aim to provide an in‐depth description of their underlying principles as a foundation of developing another new omics technology, and to introduce their emerging applications for personalised medicine. The systems biology approach is then introduced as a future direction towards actionable personalised medicine.
Inspec keywords: genetics, gene therapy, diseases, reviews, genomics
Other keywords: genetic causality, complex traits, human diseases, high‐throughput omics technologies, optimised medical treatment, review, in‐depth description, systems biology approach, actionable personalised medicine
1 Introduction
Omics technology allows massively parallel analysis of molecules including genes, transcripts, and proteins. Since this technology was created with the aim to generate a massive amount of data in a single experiment, in a short period of time, it featured unprecedented high throughput. Thanks to the omics technology, it is now possible to detect not only qualitative but also quantitative levels of multiple genes, transcripts, and proteins simultaneously. Furthermore, owing to the improved sensitivity, specificity, and accuracy of the technology, key biomarkers for disease prediction can be identified much faster. Most of current association studies of genetic variation and disease phenotypes require a sufficiently large amount of test samples for reliable statistical analysis. High‐throughput omics technology has been playing a key role in accelerating such data‐demanding analyses and now it is rapidly extending its role to molecular diagnosis and personalised medical treatment. In this review, we aim to provide an overview of current omics technologies and their emerging applications for personalised medicine. We will first introduce the key technologies and their underlying principles for analysing a variety of omics data such as genome, epigenome, proteome, and interactome data of a specific tissues or organs in an individual. Finally, we will present personalised approaches to disease diagnosis, medical treatment, and preventive strategies. To conclude, this paper will discuss the limitations of current omics approaches and present future directions.
2 Omics technologies enabling personalised medicine
2.1 Genomics‐related technologies
2.1.1 Whole‐genome (WGS) and whole‐exome sequencing (WES)
WGS technology was first developed during the Human Genome Project (HGP; 1990–2003) and the continuous improvement of this technology has dramatically changed the DNA sequencing industry. At the beginning of this century, sequencing of a single haploid human genome took several years to complete and cost more than 3 billion dollars [1]. These days, it costs merely about a few thousand dollars or less with a few days of labour. Current innovations in sequencing technology such as semiconductor (e.g. Ion S5 sequencer), nanopore (e.g. MinION) and SMRT (single molecule real‐time; e.g. sequel system) sequencing are driving forces decreasing the cost even further (see Table 1, genome sequencing) [2, 3, 4]. Owing to the significantly lower sequencing time and relatively low cost, massive parallel sequencing, also called next generation sequencing (NGS), has gained much popularity as compared with the older Sanger sequencing‐based methods [5]. This technology has been utilised in many different research areas including identification of driver mutations of individuals single‐nucleotide polymorphisms (SNPs), copy number variations, insertions and deletions that are possibly linked to specific diseases.
Table 1a.
Summary of Omics Omics technologies can be largely classified into four categories based on subjects to be analysed: genomic sequencing, transcriptome sequencing, proteome profiling, and interactome profiling. The principle and distinct feature of each technology are here
| Technology | Principle | Features |
|---|---|---|
| Genome sequencing | ||
| pyrosequencing (e.g. GS FLX Titanium XLR70) | SBS: detection of released PPi during DNA synthesis | •Elimination of the use of cloning vectors and library construction |
| •Maximum 500 read length | ||
| illumina sequencing (e.g. HiSeq 2500) | SBS: detection of fluorescent signal that is emitted by each base during DNA synthesis | •Short read assembly |
| •High computation needs | ||
| •Maximum 250 read length | ||
| ion semiconductor sequencing (e.g. Ion torrent) | SBS: detection of released protons (H+) during DNA synthesis | •Removal of optical measurement |
| •Maximum 400 read length | ||
| SMRT sequencing (e.g. PacBio RS II: P6‐C4) | SBS: detection of fluorescent signal that is emitted when base‐paired with immobilised DNA by zero‐mode waveguides (ZMWs) | •Long and highly accurate DNA sequences |
| •Single molecule sequencing (does not need of amplification steps) | ||
| •Provision of epigenetic information through identification of methylated base pairs | ||
| •Maximum 50 kb read length | ||
| nanopore sequencing (e.g. MiniON device from Oxford) | sequencing by translocation: detection of electrical conductivity changes as DNA bases pass through the nanopore (transmembrane protein channels) | •Removal of optical measurement |
| •Single molecule sequencing (does not need of amplification steps) | ||
| •Provision of epigenetic information through identification of methylated base pairs | ||
| •Maximum 48 kb read length | ||
| Transcriptome sequencing | ||
| RNA‐Seq | SBS: Sequencing of the cDNA reverse transcribed from RNA | •High potential for biased results depending on GC content, transcription length, position relative to RNA termini, and priming sequence bias |
| •Needs normalisation of raw data | ||
| GRO‐Seq | SBS: Capturing and SBS of RNA transcript being synthesised tagged with bromouridine enables to identify genes being transcribed at certain time. | •Useful for investigation of the genes that are transcribed at a certain time |
| •High potential for biased results depending on GC content, transcription length, position relative to RNA termini, and priming sequence bias | ||
| •Needs normalisation (through RPKM, FPKM, TPM, spike‐in controls) of raw data to deal with bias | ||
| •Requirement for incubation procedure | ||
| STARR‐Seq | identification of enhancer sequences by making each fragmented DNA self‐transcribe and measuring the amount of the resultant transcribed RNA | •99% detection rate by using pair‐end sequencing |
| •Does not depend on position effects by random genomic integration | ||
| ribosome profiling | capturing global snapshot of transcriptome at a specific time using ribosome foot print and NGS | Capacity to identify |
| •the location of translation start sites | ||
| •the complement of translated ORFs in a cell or tissue | ||
| •the distribution of ribosomes on a messenger RNA | ||
| •the speed of translating ribosomes |
| Technology | Principle | Features |
|---|---|---|
| Proteome profiling | ||
| mass spectrometry (MS) | separation of a protein's fragmented components based on different mass‐to‐charge ratio (m/z) and identification of the protein. | •Qualitative analysis |
| •Useful for identification, structural analysis and interaction between proteins. | ||
| SILAC | non‐radioactive isotopic labeling and quantification of the proteins based on mass shift compared with those of non‐isotope labelled proteins in different cell cultures | •Quantitative analysis |
| •Isotope labeling to proteins in cell culture | ||
| •Identification and quantification of proteins based on mass shift and peak height obtained from MS | ||
| reverse phase protein lysate microarray | quantification of the proteins by microarray with antibodies | •Quantitative analysis |
| •Needs highly specific antibody to detect proteins to be investigated | ||
| CyTOF | metal isotope‐labelled antibody labeling to proteins and quantification of proteins according to antibodies’ m/z in time‐of‐flight (TOF) chambers. | •Quantitative analysis, possible to obtain information up to ∼40 parameters at one time. |
| Interactome profiling | ||
| chromosome conformation capture (3C, 4C, 5C) | analysing DNA‐DNA interaction: identification of interacting DNA fragments by crosslinking physically close DNA strands, ligating the ends of the cross‐linked strands, and sequencing them. | •Analysis of gene function (regulation of gene expression, DNA replication, repair and recombination) |
| •Requires a great number of cells (e.g. ten millions cells on a single microarray of 4c) | ||
| •Limited resolution by random collision | ||
| ChIP‐seq | DNA‐protein interaction: antibody labeling for protein purification and sequencing of DNA fragments attached to the protein | •Profiling of DNA‐binding proteins (transcription factor, RNA polymerase, histone modification, and nucleosome) and their binding sequences |
| ChIA‐PET | analysis of interactions between chromatin and interest proteins: a genome wide version of ChIP‐seq | •ChIP‐based feature |
| •Only identifies interactions between DNA sites bound by the one factor | ||
| high throughput hybrid assay | protein‐protein interaction: yeast two hybrid assay based on the reconstitution of a functional transcription factor (TF) when two proteins of interest interact | •Easy to handle without sophisticated equipment |
| •Possible to screen thousands of proteins |
Note: SBS: sequencing by synthesis, PPi: inorganic pyrophosphate, SMRT: single molecule real time sequencing, MS: mass spectrometry, SILAC: stable isotope labeling by amino acids in cell culture, 3c: chromosome conformation capture, 4c: circularised chromosome conformation capture, 5c: carbon‐copy chromosome conformation, STARR: self‐transcribing active regulatory region, GRO: genomic run‐onm, ChIA‐PET: Chromatin Interaction Analysis by Paired‐End Tag Sequencing.
The NGS technology most frequently used to date is the sequencing‐by‐synthesis (SBS) approach. In SBS, the sequence of a DNA fragment is determined while its complementary strand is being synthesised: each DNA fragment to be sequenced serves as a template. The first SBS based NGS platform was Roche 454 pyrosequencing. It was developed in 1996 and commercialised in 1999. This technology determines DNA sequence by sensing the relative amount of PPi (pyrophosphate), which is released when a complementary dNTP is incorporated onto the sequencing template. PPi is converted into ATP in the presence of a specific enzyme, called ATP sulphurylase and a precursor of ATP — adenosine 5′ phosphosulfate. Because A, C, G, and T nucleotides are sequentially added and removed from the reaction, the sequence can be determined through the luciferase‐catalysed reaction that requires ATP as a substrate to emit light. However, due to the complicated multiple steps and the high error rate for determining homopolymer sequences, longer than 6 bp, pyrosequencing has been phased out and illumina sequencing became an industrial standard. In this section, we will describe the key principles of illumina sequencing, which are largely shared with the pyrosequencing technology.
In a typical illumina sequencing, DNA fragments, typically chopped up from a genome of interest, are first combined with specifically designed adapters (called forked adapters) at both ends of each fragment [6]. About a half of the forked adapter, a part directly attached to the chopped DNA fragment, is fully complemented for each strand, while the other half is not complementary, separating (forking) the top and bottom strands of the half part of the adapter. The two heterogeneous parts result in two different tags that are attached to the 5′ and 3′ ends of the denatured DNA fragment and that serve as (i) complementary binding sites for the two types of nucleotide oligos planted on the surface of a flow cell, where hundreds of millions of DNA fragments can hybridise with one type of the oligos, (ii) binding sites for two primers for illumina sequencing from both ends of the attached DNAs, and (iii) index sequence(s) for identification of each sample. A typical index sequence is a hexamer—theoretically 4096 different indexes can be made, however typically, up to 96 samples (processed in a 96‐well plate) are sequenced simultaneously in a single run. Once the genomic DNA fragments are immobilised on a flow cell, each of the fragments is amplified simultaneously, making up to hundred millions of randomly scattered unique spots of identical DNA fragments, in a process called ‘bridge amplification’ [7]. Engineered dNTPs used for most SBS technologies are commonly tagged with fluorescent dyes so that each nucleotide is identified by its unique colour. For example, in the case of the modified dNTPs for illumina sequencing, 3′‐OH is blocked by adding a specially designed functional group, called a terminating group, or the fluorescent dye itself to it, making the modified dNTP a terminator of DNA polymerisation. After incorporating one of the modified dNTP into a newly being synthesised DNA, DNA polymerase cannot proceed, ensuring that only one nucleotide is added and read in a single cycle of SBS. These modified dNTPs are called reversible terminators because the fluorescent dyes and terminating groups bound to modified dNTP can be easily removed [8]. Because each DNA fragment is sparsely attached and amplified in one spot of the flow cell, fluorescence signals originating from the newly added nucleotides in hundreds of millions spots can be detected simultaneously by a digital image sensor, rendering this a massively parallel technology.
In comparison with WGS, WES is a more cost‐effective and faster technology, in which only the protein‐coding exons, jointly termed the ‘exome’, are sequenced. WES is used to detect very rare genetic variants in the human genome for studying and diagnosing Mendelian diseases. Because exons constitute ∼1% of the entire human genome [9], the cost for exome sequencing is far less than that for WGS. To sequence only the exons, exon‐enrichment strategies such as PCR, molecular inversion probes, and hybrid capture are used [10]. Once DNA fragments that contain exons are highly enriched in a sample, they are processed through high‐throughput DNA sequencing will follow.
2.1.2 RNA sequencing
RNA sequencing (RNA‐seq; see Table 1, transcriptome sequencing) allows quantifying the entire RNA expression in a sample. For RNA‐seq, the target RNAs are first converted to their complementary DNA (cDNA) counterparts through reverse transcription. After fragmentising the cDNAs into smaller pieces and adding synthesised adaptor sequences to both ends, the sequences of the entire DNA fragments are then read in a massively parallel manner. The short reads are then aligned to reconstruct the original RNA sequences based on overlapping sequences of the short reads. If a reference genome sequence is available, this reconstruction process is much more easily completed owing to the additional information [11]. Unlike hybridisation‐based technologies such as microarray, RNA sequencing can be also done without a reference genome. RNA‐seq without a reference genome sequence is called ‘de novo RNA‐seq’. In addition, RNA‐seq measures RNA expression levels more accurately than the hybridisation‐based technology. In the case of microarray, accurate measurement of the concentration is severely limited if the number of a certain RNA fragment is greater than the number of its complementary probe on the array [11].
While the number of genes in the DNA remains constant in most cell types, the quantity of RNAs can vary dramatically among different cell types. Therefore, unlike DNA sequencing, RNA sequencing aims to not only identify but also quantify the transcripts in samples. However, there are obstacles to gaining accurate measurements for the quantity of RNAs of interest. First, with increasing sequencing depth and transcript length, the number of total reads that belongs to a corresponding transcript also increases, hindering a fair comparison of the levels of gene expression among genes or among samples. Various calculations such as read per kilobase per million mapped reads (RPKM), fragments per kilobase per million mapped reads (FPKM) and recently, transcripts per million (TPM) have been introduced as units of RNA expression to address this issue [12]. In addition, despite their usability, there is a growing demand for new ways that can estimate the absolute concentration of each transcript. One option is that synthetic transcripts (spike‐ins) with known sequences and absolute concentrations can be added to the sample pool [13], allowing for better estimation of the absolute concentration of sample RNAs. Horvath et al., [14] for example, was able to establish a precise holistic view of transcriptome of breast cancer: They identified novel expressed variants, allelic prevalence and abundance, co‐expression with other variation, and splicing signature through conducting the spike‐in RNA sequencing of three subtypes of breast cancer. Now RNA sequencing is being extensively used in various applications such as GRO‐seq and STARR‐seq (see Table 1).
2.1.3 Chromatin immunoprecipitation sequencing (ChIP‐seq)
ChIP‐seq is a technique for studying genome‐wide DNA‐protein interactions (see Table 1, interactome profiling) [15]. Brief sonication of cells causes them to lyse and fragments the nuclear DNA. Bead‐attached antibodies are then used to precipitate target proteins and bound DNA fragments from the supernatant of the cell lysate. The co‐precipitated DNA fragments are then purified and sequenced. Through this process, thousands of genomic regions that a certain protein binds in vivo can be identified. Because weakly bound transcription factors and other non‐histone proteins can easily dissociate from their target DNAs before and during ChIP, cross‐linking reagents such as formaldehyde have been used to fix the interaction before sonication. Fixation followed by ChIP is termed ‘XChIP’. However, significant DNA loss can occur during the purification step in which DNA is separated and purified from the cross‐linked protein‐DNA precipitate for high‐throughput sequencing. Hence, typical ChIP‐seq experiments require a high amount of cells for proper analysis. Furthermore, during fixation, epitopes can be masked by cross‐linked molecules or can be denatured by the cross‐linking reagent, which can significantly lower the sensitivity of the ChIP‐seq. As an aside, such fixation technique has also been used in various assays including chromosome conformation capture (see Table 1). In the assay, same cross‐linking is applied to whole genome, which is followed by restriction enzyme treatment and random ligation. Because the restricted DNA fragments are more likely to be ligated to nearby fragments, physical interactions between genomic loci in 3D nuclear space can be measured quantitatively by sequencing the ligated regions.
An alternative ChIP method skips the fixation step and is hence called ‘native ChIP’ (‘NChIP’) [16]. In order to keep DNA‐protein interactions as intact as possible, micrococcal nuclease is used to shear genomic DNA instead of sonication, followed by typical ChIP‐seq procedures. In NChIP, target proteins are naturally intact and co‐precipitated DNA fragments are not cross‐linked; therefore, higher antibody specificity and lower loss of sample DNA during experiments is expected. However, due to the lack of the cross‐linking step, this technique is generally not suitable for weakly bound transcription factors and non‐histone proteins. Both regular and native ChIP‐seq have led to a deeper understanding of the relationship between diseases and various histone modifications. For example, the tumour suppressor genes p16INKA and p14ARF were found to be silenced in T24 bladder cancer cells, and the silencing is correlated with H3K9 hypermethylation and H3K4 hypomethylation in these genes [17].
2.1.4 Bisulphite sequencing
In Eukaryotes, DNA methylation typically occurs at the cytosine of the 5′‐C‐phosphate‐G‐3′ (CpG) region, where guanine follows immediately after cytosine in the 5′ to 3′ direction. DNA methylation in a promoter region can suppress or activate gene expression through two main mechanisms. DNA methylation may directly hinder the binding of transcription factors that are needed for transcriptional activation or repression. In addition, methylated CpGs can recruit chromatin‐modifying enzymes to compact the surrounding chromatin, leading to the repression of gene expression. Such epigenetic modifications can differ from person to person. Therefore, even in case the genomic sequences are identical in two people, their resistance capacity and risk level for certain diseases may significantly vary. Thus, reading the epigenetic landscape of a person will be a key component of personalised medicine.
To check DNA methylation, bisulphite treatment is conducted prior to sequencing. When a DNA sample is treated with bisulphite, normal cytosine bases will change into uracil as they become deaminated, while 5′‐methylcytosines will not change because they do not react with bisulphite. By comparing DNA sequencing results before and after the bisulphite treatment, methylated cytosine residues (the ones do not change in the comparison) can be identified [18]. Thus, epigenetic markers of cancers can be determined by comparing the methylation status of the cancer cells with that of normal cells [19, 20]. Numerous cancer studies have indicated that multiple hypermethylation events in the CpG island within cancer genomes are related to the repression of tumour suppressor genes or activation of oncogenes [21] (see Table 2, epigenomics). For example, upon examination of the methylation profiles of 12 CpG islands in hepatocellular carcinoma (HCC) cells, researchers found hypermethylation in 8 of the CpG islands [22]. Shen et al. identified a connection between colorectal cancer and hypermethylation of the CpG island in a DNA repair protein gene encoding O6‐methylguanine‐DNA methyltransferase (MGMT) and found that a low level of MGMT could be one of the causes of colorectal cancer [23].
Table 2.
Diagnosis markers based on genetic or epigenetic profiles. Previous diagnostic methods are mainly based on physiological symptoms and histological patterns. However, the development of omics technologies makes it possible to investigate correlations between specific genetic or epigenetic variants and the probability of disease occurrence. Based on their genomic profiles obtained from sequencing, people can be told how much they are likely to have Alzheimers disease, hereditary breast and ovarian cancer, Mucolipidosis IV, Parkinson, hypertrophic cardiomyopathy, and Canavan disease. Moreover, epigenomic profiles, which are especially correlated with cancer occurrence, can let patients know how risky they are to have diverse cancers such as colon cancer, liver cancer, lung cancer, oesophageal cancer and gastric cancer
| Bioinformatics used for diagnosis | Disease | Marker gene | Variant |
|---|---|---|---|
| genomics | Alzheimer's disease | APOE | ε4 status |
| hereditary breast and ovarian cancer | BRCA1 |
5382insC 185delAG |
|
| BRCA2 | 6174delT | ||
| mucolipidosis IV | MCOLN1 |
IVS3‐2A > G 511del6464 |
|
| Parkinson | LRRK | G2019S | |
| GBA | N370S | ||
| hypertrophic cardiomyopathy | MYBPC3 | 25bpdel | |
| canavan disease | ASPA |
E285A A305E Y231X |
|
| epigenomics | colon cancer | CpG island of ESR1 | hypermethylation |
| CpG island of CDKN2A | |||
| CpG island of THBS1 | |||
| CpG island of MGMT | |||
| liver cancer | CpG island of ESR1 | hypermethylation | |
| CpG island of CDKN2A | |||
| CpG island of PTGS2 | |||
| CpG island of MINT31 | |||
| CpG island of MINT1 | |||
| CpG island of MINT27 | |||
| CpG island of MINT2 | |||
| lung cancer | CpG island of CDKN2A | hypermethylation | |
| CpG island of MGMT | |||
| CpG island of DAPK | |||
| CpG island of SOCS1 | |||
| CpG island of RASSF1A | |||
| CpG island PTGS2 | |||
| CpG island of RARB | |||
| esophageal cancer | CpG island of APC | hypermethylation | |
| CpG island of CDH1 | |||
| CpG island of ESR1 | |||
| CpG island of CDKN2A | |||
| gastric cancer | CpG island of CDKN2A | hypermethylation | |
| CpG island of hMLH1 |
Previous diagnostic methods are mainly based on physiological symptoms and histological patterns. However, the development of omics technologies makes it possible to investigate correlations between specific genetic or epigenetic variants and the probability of disease occurrence. Based on their genomic profiles obtained from sequencing, people can be told how much they are likely to have Alzheimer's disease, Hereditary breast and ovarian cancer, Mucolipidosis IV, Parkinson, Hypertrophic cardiomyopathy, and Canavan disease. Moreover, epigenomic profiles, which are especially correlated with cancer occurrence, can let patients know how risky they are to have diverse cancers such as colon cancer, liver cancer, Lung cancer, oesophageal cancer and Gastric cancer. Here are acronyms of terms in Table 2: APOE : Apolipoprotein E, BRCA : Breast cancer, MCOLN1 : Mucolipin1, LRRK : Leucine‐rich repeat kinase, GBA : Glucosidase beta acid, MYBPC3 : Myosin binding protein C, Cardiac, ASPA : Aspartoacylase, ESR1 : Estrogen receptor 1, CDKN2A : Cyclin‐dependent kinase inhibitor 2A, THBS1 : Thrombospondin 1, MGMT : O‐6‐methylguanine‐DNA methyltransferase, PTGS2 : Prostaglandin‐endoperoxide synthase 2, MINT: Methylated in tumour, CpG, DAPK : Death‐associated protein kinase, SOCS1 : Suppressor of cytokine signalling 1, RASSF1A: Ras association domain family 1 isoform A, RARB : Retinoic acid receptor, Beta, APC : Adenomatous polyposis coli, CDH1 : Cadherin‐1, CDKN2A : Cyclin‐dependent kinase inhibitor 2A, hMLH1 : Human mutL homologue 1.
2.1.5 Single‐cell sequencing
For precise and effective cancer treatment, it is important to know the composition and progressive development of the cancer cell subpopulation. With the development of technologies for single cell isolation and single DNA‐based amplification, it has become possible to conduct cancer research at single‐cell resolution. In single‐cell sequencing, individual cells are isolated through micromanipulation methods such as fluorescence‐activated cell sorting (FACS), laser‐capture micro‐dissection, or microfluidics device. Then, lysis is conducted in individual reaction chambers. Finally, the gene of interest is amplified and sequenced [24]. Previously, if the subpopulation to be analysed was small, accurate sequencing and subsequent analysis were extremely difficult. Degradation, loss, or contamination of the sample may occur [25] and further, conducting PCR on single DNA strand requires a significant amount of skill and luck.
There are two major methods for amplifying a single DNA strand or an extremely small amount of DNA: multiple displacement amplification (MDA) and multiple annealing and looping‐based amplification (MALBAC). In MDA, random hexamer primers are simultaneously bound to multiple locations of a long, stretched DNA: the use of different primers ensures their binding to every 4 kb of DNA on average. 29 DNA polymerase, used in MDA, can proceed polymerisation even when it encounters a newly synthesised DNA from an upstream primer by tearing off (debranching) the DNA strand from its template. Therefore, MDA generates multiple elongating DNA fragments that are branched out from the template DNA. The branched DNA serves as a new template for another round of DNA synthesis, giving rise to heavy exponential amplification of a single DNA molecule at multiple positions. 29 amplifies template DNA isothermally at and is a high‐fidelity polymerase that can synthesise DNA fragments longer than 70 kb; therefore, the DNA must be fragmented before sequencing. In addition, the very low error rate (1 out of 10−6 to 10−7 nucleotides) of the 29 DNA polymerase makes MDA a reliable method for single‐cell sequencing. However, due to the massive amplification in random branches, sequencing bias can occur. In MALBAC, using special primers, only an input DNA serves as a template for amplification: DNA amplification occurs linearly on the template, removing uneven biased sequencing depth. These innovative technologies have made single‐cell sequencing possible.
Navin et al. [26] investigated the tumour population of breast cancer through single‐cell sequencing and discovered three distinct subpopulations. Through single‐cell exome sequencing of clear cell renal cell carcinoma, researchers were able to describe a detailed intra‐tumoural genetic landscape [27]. These studies are significant because the clinical response towards the same type of cancer may vary depending on the composition of heterogeneous subpopulations [28]. Furthermore, if single‐cell sequencing is combined with a protocol such as fluorescent in situ sequencing(FISSEQ), spatially resolved genetic information of the copy number variation, existence of circular DNA, tumour heterogeneity, and RNA localisation in a sample tissue can be obtained [29]. When compared with normal healthy samples, this allows researchers to analyse the tumour development and the connectivity of individual cell activities. In addition, it allows determining which part of the tissue the disease originated from. By checking the dominant subpopulation in the malignant cancer tissue of a patient, the subtype of the cancer can be determined and the most effective and relevant medical treatment for the patient can be suggested.
2.2 Proteomics‐related technologies
2.2.1 Mass spectroscopy (MS)‐based technologies
MS technology (see Table 1, Proteome Profiling) has revolutionised the field of proteomics. MS can identify proteins, characterise their structures and post‐translational modifications, and even analyse protein‐protein interactions in a high‐throughput manner. In addition, it can be used to conduct de novo peptide sequencing of a sample. This technology has led to the identification of numerous protein biomarkers and the characterisation of their activities for accurate diagnosis of various diseases such as Alzheimer's, Parkinson's, and prion diseases [30].
When electrically charged molecules (ions) are accelerated and linearly subjected to a magnetic field, the trajectories of the molecules are deflected under the influence of the field. The degree of deflection depends on the mass‐to‐charge (m/z) ratio of the ion and the magnetic field strength. The mass spectrometer measures the degree of the deflection, which allows calculating the m/z ratio of the ion, and generates a mass spectrum as a function of the m/z ratios. The bigger the m/z ratio, the less the deflection. Instead of the degree of deflection, the time of flight (TOF) of accelerated molecular ions can also be used for calculating the m/z ratio — TOF and m/z are inversely proportional. The height of the spectral peak indicates the amount of ions with the same m/z ratio. If the charge numbers (z) are controlled to be constant for ions, the positional difference of the MS peaks is directly proportional to the difference of their molecular weights. Using these principles, MS can accurately measure absolute masses of molecules in a sample.
In MS, there are two main methods for protein identification. Once the mass spectrum of cleaved peptides of a protein is obtained, it can be compared with a database of documented mass spectra of known proteins to find the best match. However, such database search cannot be used for novel, unregistered proteins. Such cases require de novo peptide sequencing through tandem MS (MS/MS). In tandem MS, the first MS step (MS1) measures the m/z ratio of the proteolytic peptide ions (also called ‘precursor ions’). The mass spectrum from MS1 can be used for database searches. Each precursor ions generated in MS1 can be further broken into small pieces for peptide sequencing by peptide fragmentation techniques such as collision‐induced dissociation (CID). Note that isobaric (e.g. Ile and Leu) amino acids are not distinguishable in MS/MS with low‐energy CID. Near‐isobaric (e.g. Lys and Glu) amino acids may not be distinguishable in a low‐resolution MS spectrum.
2.2.2 High throughput protein quantification: SILAC, iTRAQ and CyTOF
Mass spectroscopy is used not only for qualitative analysis such as protein identification and peptide sequencing, but also for quantitative analysis of proteins and their post‐translational modifications. To compare the expression levels of proteins between two or more samples, they undergo sample‐specific molecular labelling. Frequently used molecular tags are isotope‐labelled amino acids, isotope‐labelled chemical reagents, and metal isotope‐labelled antibodies. These 3 tags are used in three different techniques — SILAC (Stable Isotope Labelling with Amino Acids in Cell Culture), iTRAQ (Isobaric Tags for Relative and Absolute Quantitation), and cytometry by time of flight (CyTOF), respectively, and they have their own benefits and disadvantages (see Table 1, interactome profiling).
SILAC is a simple and one of the most accurate approaches for comparing protein concentrations between samples [31]. Typically, two amino acids, non‐radioactive (stable) isotope‐labelled ‐lysine and ‐arginine are fed during the culturing of cells, which results in the labelling of every peptide before trypsin digestion and LC‐MS analysis. In another cell culture, only natural amino acids are fed. The two samples are combined and processed for protein quantification, which makes SILAC simple and accurate; no technical variation between samples occurs. Because the heavy and light (natural) amino acids are chemically identical, HPLC does not separate isotope‐labelled peptides from their chemically identical counterparts. Therefore, they are loaded together in MS and scanned together, resulting in a single spectrum plot. Owing to the differential molecular weight, however, the peaks of peptides from the different samples are clearly distinguishable. By comparing the median values of the heights of the distinguishable peaks, the relative concentrations of peptides, and subsequently, the concentration of each protein can be determined. However, this technique is applicable only to culturable cells and it takes a relatively long period of time; multiple generations of cell culture are required for full incorporation of the heavy amino acids. In addition, only up to three samples are directly comparable by utilising additional heavy amino acids‐ ‐lysine and ‐arginine. In an alternative approach, called ‘spike‐in SILAC’, the sample labelled with heavy amino acids is generated separately and used as an internal standard for other experimental samples. Peptide ratios between the sample and the heavy spike‐in standard are then determined for each of the samples. Because the spike‐in standard is identical in all experimental samples, relative concentration changes between the experimental samples can be inferred by normalising them with the spike‐in signal in the spectrum. This spike‐in SILAC allows a theoretically unlimited number of sample comparisons and extends its application range to dead cell extracts, unculturable cells, and even tissues. As a trade‐off, however, spike‐in SILAC carries a risk of significant technical variation because each experimental sample, after being combined with the heavy spike‐in, is processed separately.
Unlike SILAC that uses in vivo, metabolic labelling, iTRAQ uses in vitro, chemical labelling. It directly adds isobaric (same‐weight) tags to the N termini of the peptides, which significantly reduces labelling time and can be used to quantify any protein sample from any source, including unculturable cell types. Eight different isobaric tags are currently available, implying that proteins in up to eight samples can be directly compared. However, in iTRAQ, until the chemical labelling, all experimental steps (protein extraction, fractionation, trypsin treatment, and peptide labelling) are performed separately, increasing technical variability. Peptides labelled with different isobaric tags are combined and transferred to LS‐MS/MS. Each isobaric tag consists of two parts; a reporter and a balance group. Reporter groups, which are well ionisable, have the same chemical structure but different molecular weights by controlling the number of heavy isotopes in the group. Balance groups also have the same structure but different weights, which compensate for the weight differences of the reporter groups, hence the term ‘isobaric tag’. This is the key feature of iTRAQ that makes the ‘differentially labelled’ proteins from all different sources indistinguishable until MS2, allowing strong signal amplification of the peak of each peptide (precursor) in MS1 and the peak of each fragment ion in MS2, with exception of the reporter ions. In the MS2 spectrum, peaks of different reporter ions do not co‐localise and thus are distinguishable owing to their differential weights. By comparing the intensity of the reporter peaks, the concentration of each peptide and therefore, each protein, can be determined. The main advantage of iTRAQ is that sequencing and quantification of peptides originating from up to 8 different samples can be done together in a single MS2 spectrum.
CyTOF is a single cell‐based protein quantification technique. It can simultaneously measure expression levels of more than 60 different proteins in an individual cell [32]. CyTOF is a type of cytometry, in which antibodies are labelled with heavy‐metal isotope tags rather than with fluorescent molecules used in conventional flow cytometry (e.g. FACS). Unlike SILAC and iTRAQ, CyTOF can only measure the abundance of antibody‐available proteins within a cell. As a first step in the CyTOF device, testing cells treated with the heavy‐metal labelled antibodies are nebulised into single‐cell droplets and individually introduced into a plasma. The high temperature plasma breaks down each whole cell into atomic ions. Among the atomic ion mixture, the heavy metal isotope ions from the antibody tags can be quantified via TOF—MS: the metal ions are much heavier than the other ions originating from the cells. Owing to the high sensitivity of MS that can determine the mass of a single neutron, theoretically, the maximum number of proteins to be measured is equivalent to the number of non‐biological heavy metal isotopes — about a hundred. This approach overcomes limitations of broad spectral overlap of fluorescent molecules, which limits the number of proteins that can be measured simultaneously in a fluorescence flow‐cytometric experiment. In addition, compared with SILAC and iTRAQ, in which only the same peptides from different samples can be compared in a single spectrum, CyTOF can directly compare the amounts of up to hundred different proteins simultaneously from a single cell in a single spectrum. This technology offers unprecedented opportunity in the analysis of heterogeneous cancer tissues. By simultaneously scanning cell‐surface and cytoplasmic protein markers, it can characterise all of the subpopulations and their key signalling pathways in a tumour sample [33, 34]. Tumour subpopulation data of a patient, combined with pharmacogenomics, can dramatically improve patient care and the treatment of cancers and other diseases. However, CyTOF destroys the cells while scanning target proteins, precluding further analysis of the individual cells. Further, it currently requires more than 100 molecules per protein per cell for detection [35].
3 Prenatal screening
In the following sections, we will review various applications of the omics technologies described previously, along with the biography of a person from pre‐womb to tomb (see Fig. 1). The health of a baby is one of the most important matters for parents. Genetic defects inherited from parents can be a serious threat to the health of a baby. To address this issue, omics technologies are increasingly involved in a person's life even before birth. Prenatal screening has become popular in many countries for examining not only deformities of a foetus but also the potential risks of diseases or abnormal conditions after birth. If there is a genetic predisposition for disorders such as cystic fibrosis or muscular dystrophy, a couple can consult a medical doctor or a direct‐to‐consumer genetic diagnosis company to plan a non‐invasive genetic diagnosis for their baby. The following sections will describe personalised prenatal screening, enabled by omics technologies.
Fig. 1.
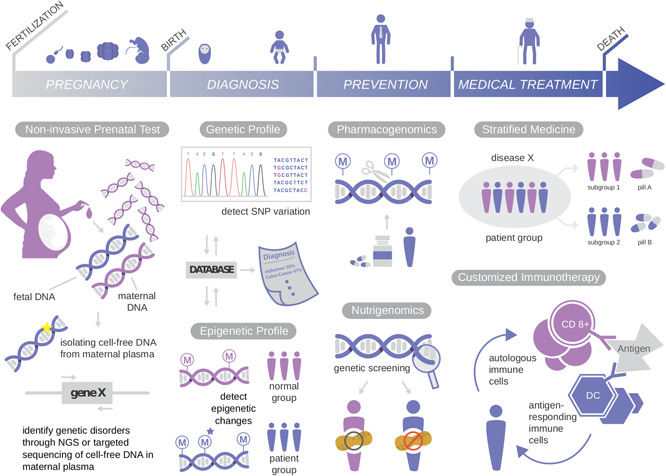
Personalised medicine throughout the lifespan
3.1 Technologies for prenatal diagnosis
The advances in molecular and cellular biology in the late 20th century allowed various genetic diagnoses for foetuses (see Fig. 1, PREGNANCY and DIAGNOSIS). In order to check the genetic abnormalities in a foetus, foetal genetic materials must be obtained from the biological mother. Till date, invasive technologies such as amniocentesis and chorionic villus sampling have been used for prenatal diagnosis. In these methods, foetal genetic samples are extracted directly from foetal tissue in amniotic fluid and chorionic villus, respectively, using specialised needles and tubes. Even though the methods have been improved, there is still risk of miscarriage and serious side effects such as foetal limb reduction and placental mosaicism [36, 37]. To overcome these problems, attempts have been made for decades to develop non‐invasive prenatal screening technologies.
3.1.1 Foetal DNA‐based diagnosis: sequencing using nucleated cells vs. cell‐free DNA
Currently, the most common non‐invasive technology for prenatal screening is ultrasound scanning. However, it can only detect morphological defects, not the genetic constitution of an embryo or foetus. One of the new approaches to evade the side effects of conventional invasive technologies is to isolate and analyse foetal nucleated cells from maternal blood. However, the number of foetal cells that can be isolated is extremely low; 3–5 cells per 30 ml of maternal blood [38]. In addition, this method requires the isolation of the foetal cells among a huge number of maternal cells. Multiple techniques including FACS, magnetic‐activated cell sorting, micro‐dissection, and size separation have been applied, but the detection sensitivity for foetal cells remains to be improved. For example, Bianchi et al. reported a detection sensitivity of male foetal DNA in cells of 41.4% and a false‐positive rate of 11.1% [39].
Recent omics technologies, combined with various experiments, have enabled a huge leap in the field of non‐invasive prenatal testing (NIPT). In 1997, a research group discovered that cell‐free foetal DNA exists in maternal blood. [40]. This finding opened a new door for non‐invasive prenatal diagnosis. The use of DNA amplification technology combined with NGS enabled the direct detection of the foetal DNA in maternal blood [41]. Currently, non‐invasive cell‐free foetal DNA sequencing allows to determine the sex and Rh blood type, and to analyse chromosomal aneuploidies [42, 43, 44, 45, 46]. The cell‐free foetal DNA is typically released in maternal blood during the process of apoptosis of placental cells. The fragmented cell‐free foetal DNA is ∼200 bp long and has a half‐life of ∼16 min. It is currently detectable in maternal plasma from early pregnancy (5–7 weeks) and diminishes significantly after birth [47, 48]. Due to its short length and relatively low amount (10%) compared with maternal cell‐free DNA (90%), identifying the detailed physical and biological characteristics of the foetal genome remains challenging. However, decades of research on cell‐free foetal DNA in maternal plasma and the rapid development of related omics technologies make future prospects for NIPT look bright.
3.1.2 Foetal RNA and epigenetics‐based diagnosis
In addition to foetal DNA, foetal RNAs can be detected and used for NIPT [49]. For example, foetal trisomy 21 can be diagnosed by checking the allelic ratio of PLAC4 mRNA, produced from chromosome 21 in the placenta [49]. If a baby inherits heterogeneous alleles from the parents, an unequal allelic ratio (2 : 1) indicates trisomy. Foetal epigenetic profiles can be used for identifying foetus‐specific DNA in maternal blood. It has been reported that the RASSF1A tumour suppressor gene is hypermethylated in the cell‐free foetal DNA, while it is hypomethylated in the maternal DNA [50]. Thus, the methylation status allows identifying and isolating foetal DNAs from maternal plasma through methylated DNA immunoprecipitation or restriction enzyme treatment, followed by NGS.
3.1.3 Future of prenatal diagnosis
Integrating massive amounts of data from various types of experiments in various institutions and research labs will help pushing the prenatal genetic diagnostic technology forward. Prenatal diagnosis based on multiple sources such as foetal genomic, transcriptomic, proteomic, metabolomic, and epigenetic data will be a powerful predictive tool in the near future. The ultimate goal of prenatal diagnosis would be to non‐invasively identify any genetic disorder in the early phase of pregnancy, to attain health information on the foetus, and to treat any genetic defects before birth. However, the early prenatal diagnosis technology could raise ethical issues with regard to the right of choice and decision‐making — whether or not to perform NIPT and abandon pregnancy if the NIPT result indicates serious genetic defects.
Many scientists have been concerned that the outcome of the HGP and advances in recent genomic technology would raise serious ethical issues in the near future. To assess and anticipate these issues, National Institute of Health launched a research program called ethical, legal, and social implications (ELSI) — as an integral part of the HGP in 1990. Under ELSI, many researchers have studied the impact of new technologies on society, such as possible discrimination based on genetic information, undetermined harmful effects of nanotechnologies on humans and nature, and copyright infringements due to the development of information technology. Now the scope of ELSI has grown to include all types of research dealing with the relationships between technologies and society.
4 Postnatal diagnosis
4.1 Before the onset of disease: genome sequencing‐based diagnosis
Identifying genetic markers such as mutations in genomic DNA that are associated with a specific disease trait is key for estimating the disease risk (see Table 2, genomics). Because the genetic information of a person remains largely unchanged, the mutated sequences can be reliable indicators, regardless of when diagnosis is conducted, for certain diseases if they are significantly associated. One of the applications of omics technology in this regard is genome‐wide association studies (GWASs), which identify common genetic variants, generally SNPs, associated with certain disease traits, e.g. Alzheimer's disease and hereditary cancers. In general, SNP information is collected from two groups, a patient group and a healthy control group, and then statistically analysed to identify SNPs significantly associated with the specific disease trait of the patient group. SNP array chips that have been used in multiple GWASs commonly can scan 5 million or more SNPs [51, 52]. Even though more than 85 million SNPs have been identified in the human genome [53], many of them are likely to be inherited together — those SNPs, typically clustered in a stretch of DNA, are called a haplotype. It is, therefore, possible to perform a GWAS using a few SNP arrays that scan only representative SNPs for individual haplotypes [52]. Some of examples of GWAS study and other omics based approaches will be reviewed in the following sections.
4.1.1 Neurodegenerative diseases: Alzheimer's disease
Apolipoprotein E (APOE) gene is a well‐known genetic factor that is associated with the late‐onset Alzheimer's disease (LOAD). Strittmatter et al. [54] histochemically observed that APOE is bound to ‐amyloid with high avidity in LOAD patients. They also found that patients with LOAD have three times higher frequency of having APOE 4 allele than control group. APOE binding to hydrophobic ‐amyloid peptide is thought to initiate neurodegeneration in AD [55]. Since GWAS for Alzheimer's disease has been started, it has pinpointed SNPs and genes that have a high association with LOAD. Coon et al. [56] assessed 502,627 SNPs in 1086 AD patients and demonstrated that rs4420638 on chromosome 19 is the clearest marker among 502,627 SNPs distinguishing AD patients from the control group. Moreover, Takei et al. discovered three additional genes other than APOE, namely (PVRL2, TOMM40 and APOC1), which all have a particular connection with LOAD [57]. This indicates the importance of omics technology and the resultant data for genetic disease screening.
4.1.2 Hereditary cancers: hereditary breast cancer
BRCA1 and BRCA2 genes are one of the most prominent genetic markers for hereditary breast and ovarian cancers [58] (see Table 2, genomics). Association between BRCA genes and breast cancer was first reported by genetic linkage analysis using microsatellite markers [59, 60]. These genes produce tumour suppressor proteins, which help repairing damaged DNA through homology directed repair [61, 62, 63]. Some mutations in BRCA1 and/or BRCA2 greatly increase the risk of developing breast and/or ovarian cancers. For BRCA1, two mutations, 185delAG and 5382insC, are well known cancer markers [64]. ‘Del’ and ‘ins’ indicate ‘deletion’ and ‘insertion’, respectively, and the numbers indicate the distance from the transcription start site. For BRCA2, 617delT is the best‐known marker. Approximately 55–65% of women who inherit one of the harmful BRCA1 mutations and around 45% of women who carry the BRCA2 mutation have developed breast cancer by the age of 70 [65, 66] while only 12% of general population have developed breast cancer in the same time span [67]. Sanger sequencing has been a standard procedure for the molecular diagnosis of the BRCA genes [68]. However, for large genes, such as BRCA1 and BRCA2 with lengths of 127.1 kb and 85.9 kb, respectively, pre‐sequencing steps such as denaturing HPLC and high‐resolution melting are typically conducted prior to sanger sequencing, increasing the cost of diagnosis [69]. NGS has become a faster, less expressive, and most importantly, more accurate alternative to conventional procedures in molecular diagnostics including BRCA gene scanning. For example, it has been reported that NGS‐based diagnosis shows higher sensitivity (99.99%) than the traditional method (67.6%), and it allowed detecting additional mutations including a causative BRCA1 mutation missed by the traditional screening [70]. Note that NGS‐based diagnosis can be conducted easily for 96 patients simultaneously with a typical NGS machine.
4.1.3 Other disease traits
Various other disease traits such as autosomal‐dominant colorectal cancer, Li—Fraumeni multi‐cancer syndrome, mucolipidosis IV, and hypertrophic cardiomyopathy, have been successfully identified using NGS [71, 72, 73, 74, 75, 76, 77] (see Table 2, genomics). As genomics‐related technologies, analytic tools, and resulting datasets continue to be improved, more genetic markers for specific disease traits will be registered and clinically applied. However, a growing body of evidence indicates that genetic markers generally explain only a small part of the total trait variation. For example, only 5–10% of breast cancers in the United States are linked to an inherited gene mutation [78]. In the case of human body length, genetic variations identified by GWAS explained only about 5% of the variation [79]. Together, these indicate the insufficiency of genetic screening alone (known as missing heritability) in predicting the risks for a certain disease or non‐disease trait. Such missing heritability is accounted for, in part, by rare variants, which only can be detected through individual whole exome or genome sequencing, highlighting the value of NGS in predictive medicine.
4.2 At the early phase of disease development
Epigenetic variations are also considered a part of the missing heritability. Epigenetic changes can be stably inherited from a biological parent or occur as an effect of gene‐environment interaction. Various epigenetic changes have been reported to be correlated with the early stage of various cancers, mostly before neoplastic transformation (abnormal growth of a tumour) [19] (see Table 2, epigenomics). About a half of the known suppressor genes are inactivated due to epigenetic changes rather than genetic variations [19]. Because epigenetic profiles in early‐stage tumour cells in many cases can be stably and irreversibly transferred to the daughter cells, certain epigenetic profiles can be relevant markers for diagnosis at the early phase of cancers, even though the tumours are not histologically detectable, reducing the cost for effective medical treatment and increasing the recovery rate.
It is well known that colorectal cancer is more frequently detected in men than in women, and it has been suggested that oestrogen may help suppress colorectal cancer [80]. It was found that a form of colorectal cancer is correlated with frequent methylation in the oestrogen receptor (ESR) gene, which leads to silencing of the gene [81]. In addition, by utilising bisulphite DNA sequencing, Shen et al. investigated the epigenetic profiles of tumour, adjacent mucosa, and non‐adjacent mucosa samples of 95 colorectal cancer patients, which revealed that the promoter of the MGMT, a DNA repair gene, was frequently methylated [23]. In addition, the analysis of 85 HCC samples revealed significant hypermethylation in 8 CpG islands in ESR, CDKN2A, PTGS2, MINT31, MINT1, MINT27 and MINT2 genes [22]. For instance, 62 and 42% of the samples possessed hypermethylation in the ESR and CDKN2A genes, respectively, and all of the 8 CpG islands were positively correlated, indicating that the CpG island methylator phenotype might be a distinctive marker for HCC.
5 Disease treatment
5.1 Stratified medicine
A major goal of personalised (precision) medicine is to select the most effective treatment option for an individual (see Fig. 1, medical Treatment) (see Table 3, stratified medicine). For example, currently, breast cancer can be categorised into three distinctive subtypes based on the molecular characteristics: HER2 Amp (amplified human epidermal growth factor receptor 2), HR + (hormone receptor‐positive), and triple‐negative breast cancer (TNBC) [82]. Effective treatment options for the first two subtypes consist of HER2‐targeted monoclonal antibody and HR antagonists, respectively [83, 84, 85]. In the case of the TNBC subtype, which features low expression of both HER2 and HR, conventional cytotoxic chemotherapy is generally used [86]. A recent study utilised enzyme‐linked immunosorbent assay to measure the amount of surface receptors and downstream signalling kinases and their phosphorylation states to estimate the sensitivity of 23 targeted therapeutics for individual patients [87]. Simultaneous quantification of multiple protein markers will play an important role in subtyping tumours and selecting optimised therapies for each subtype. High‐throughput, single cell‐based protein quantification techniques, such as CyTOF, can be excellent alternatives for immunohistochemistry‐based methods and have been paving the way for simultaneously measuring various molecular characters of an array of cancers in a short time [88]. This stratified (subtyping) approach marks the early stage of personalised medicine. More personalised approaches involving the patient's immune system will be discussed in the next section.
Table 3.
Personalised medical treatments
| Classification of medicine | Disease | Variant/subtype* | Recommended Medical treatment | |
|---|---|---|---|---|
| stratified medicine | cancer | diverse cancer (colorectal, breast, oesophageal, stomach cancer) | D949V | 5‐FU should be avoided |
| colorectal cancer | rs2233921 | 5‐FU chemotherapy | ||
| DLBCL | GCB‐DLBCL | R‐CHOP treatment | ||
| ABC‐DLBCL | Ibrutinib treatment | |||
| thyroid nodule | expression of miR‐222, miR‐328, miR‐197, miR‐21 | physical dissection | ||
| breast cancer |
high expression of HER2 receptor (HERamp subtype) |
trastuzumab treatment, pertuzumab treatment | ||
| high expression of ER, PR (HR + subtype) | tamoxifen treatment | |||
| low expression of HER2, ER, PR (TNBC subtype) | cytotoxic chemotherapy | |||
| cardiovascular disease | general disease | CYP2C9 mutation | warfarin treatment quantity should be different | |
| LQTS | KCNQ1 mutation (LQTS1) | propranolol treatment, nadolol treatment | ||
| KCNH2 mutation (LQTS2) | amiloride treatment | |||
| SCN5A mutation (LQTS3) | sodium channel blocker treatment | |||
| AIDS | presence of HLA‐DR7, HLA‐DQ3 | abcavir should be avoided | ||
| customised immunotherapy medicine | glioma | individual‐specific | DC‐based immunotherapy, Cancer vaccine | |
| leukaemia | DC‐based immunotherapy | |||
| AIDS | CD4 + T lymphocyte‐based immunotherapy, DC‐based immunotherapy |
Though a group of patients are diagnosed to have the same disease, their effective medical treatments can be different, depending on bioinformation each patient has. Stratified medicine is, after grouping patients into several subgroups, to adapt different medicines to each subgroup. Customised immunotherapy is to treat individuals using each person's distinctive bioinformation. Here are acronyms of terms in Table 4: DPYD: Dihydropyrimidine dehydrogenase, EGFR: Epidermal growth factor receptor, DLBCL: Diffuse large B‐cell lymphoma, GCB‐DLBCL: Germinal centre B‐cell such as DLBCL, ABC‐DLBCL: Activated B‐cell such as DLBCL, 5‐FU: 5‐fluorouracil, R‐CHOP: The initials of the five drugs(rituximab, cyclophosphamide, doxorubicin (hydroxydaunomycin), vincrinstine (Oncovin®), prednisolone), HER2: Human epidermal growth factor receptor 2, ER: Estrogen receptor, PR: Progesterone receptor, CYP2C9 : Cytochrome P450 2C9, VKORC : Vitamin K epoxide reductase complex, LQTS: Long QT syndrome, KCNQ1 : Potassium channel, voltage gated KQT‐like subfamily Q, member 1, KCNH2 : Potassium channel, voltage gated eag related subfamily H, member 2, SCN5A : Sodium channel, voltage‐gated, type V alpha subunit, HLA‐B: Human leukocyte antigen Major histocompatibility complex, class I, B, HLA‐DR: Human leukocyte antigen – antigen D Related, HLA‐DQ: Human Lymphocyte Antigen‐DQ, DC: Dendritic cell, AIDS: Acquired immune deficiency syndrome.
5.2 Customised immunotherapy
A new approach for treating cancers — letting the patient's immune system combat his or her cancer — has become the fifth major cancer treatment, following surgery, chemotherapy, radiation therapy, and antibody‐based treatment. This approach is called as adoptive cell transfer (ACT), has demonstrated its bright potential (see Table 3, customised immunotherapy medicine). For example, glioblastoma multiforme (GBM) is the most common and aggressive type of malignant primary brain tumour. Although GBM has been treated with surgical removal, radiation treatment, and chemotherapy methods, there have been no noticeable effects [89], primarily because the glioma cells invade distant brain tissues, and it is almost impossible to remove each one of them [90]. Over the past 20 years, numerous clinical trials have been conducted to use the immune system to treat cancers such as GBM. Among various ACT methods, particular focus has been given to dendritic cell (DC)‐ and T cell‐based vaccines: these vaccines can penetrate and induce anti‐tumoural response in the brain, an immunologically privileged site [91, 92], and show great potential for effectively killing various cancers including the malignant and advanced types.
Autologous DCs, a type of antigen‐presenting cells, obtained from tumour tissue or in the form of a tumour lysate of a patient, can be grown in vitro in the presence of tumour‐associated antigens (TAAs), a technique called ‘pulsing’. The pulsed, autologous DCs are reintroduced into the patient to stimulate specific, cell‐mediated, anti‐tumoural cytotoxicity against GBM and other malignant cancers. Autologous DCs can also be fused with the patient's tumour cells in vitro. The DC‐tumour cell hybrids, consistently expressing tumour antigen on the surface of the fused DCs, may stimulate a strong anti‐tumoural immune response. Despite the intensive studies conducted on DC‐based vaccines, there is a need for further research in patients with early‐stage cancer, who do not have strong immunosuppression [93]. More radical approaches, e.g. engineering specific immune cells using zinc‐finger nucleases (ZFNs) and CRISPR technology, have also been tried. Leukaemia tumour‐specific lymphocytes were successfully created in a mouse model by removing the endogenous T cell receptor (TCR) ‐ and ‐ chains in autologous lymphocytes using ZFNs. An exogenous TCR gene able to recognise a specific tumour was then incorporated into the genome of the lymphocytes by using lentivirus [94]. Another remarkable example is the chimeric antigen receptor (CAR) T‐cell treatment. It engineers the patient's own immune cells by adding a CAR that recognises and helps attack its target tumour cells. A part of anti‐TAA antibody is covalently linked to an engineered T‐cell receptor (a CAR part) located on the surface of the engineered T cells. Surprisingly, one single CAR T cell can kill more than 1000 cancer cells, and once in contact with tumour cells, the CAR T cells are significantly amplified [95, 96]. CAR T cells behave such as a ‘living drugs’ that keep patrolling and destroying cancer cells upon detection in the body. This approach, however, is currently restricted to small clinical trials, and needs further study before application to personalised medicine.
6 Disease prevention and future prospects
One of the ultimate goals of personalised medicine is to prevent the development of various diseases before their onset (see Fig. 1, prevention). However, there are serious obstacles to be overcome before reaching this goal. Current preventive treatments are limited to surgery — excising a tissue or an organ if a person possesses noxious cancer markers such as BRCA mutations. However, this approach cannot be applied to several diseases, such as Alzheimer's disease, Huntington's disease, and congenital muscular dystrophy, as the target tissues are not excisable. The ability to not only accurately diagnose, but also to prevent disease onset without surgery, will be a major feat in the field of personalised medicine. One of the recent approaches to address this issue is the nutrigenomics‐based diet treatment. Nutrigenomics assesses dietary influence at the levels of gene and protein expression, as well as epigenetic and metabolic profiles in various tissues and cell types. An impressive example of nutrigenomics studies reported that Genistein, the most abundant natural isoflavone found in the soybean, induces gene expression of tumour suppressor genes, p21WAF1/CIP1 and p16INK4a in prostate cancer cells [97]. This study showed that the treatment of Genistein increased acetylated histones at the p21 and p16 transcription start sites and induced apoptosis in cancer cells, suggesting the possibility of an dietary preventive method.
We are, however, still at the very early stage of non‐invasive or minimally invasive preventive medicine. One of the major hurdles is that predicting disease traits itself is very challenging. Common variant genetic markers, assessed by GWAS, have little explanatory power for most of the complex, polygenic disease traits. Rare variants, which cannot be detected by GWAS, may also play an important role in certain diseases. Epistatic effects of combinations of multiple variants and environmental effects must also be taken into account. Furthermore, we currently have a very limited number of actionable gene variants for which medical actions can be taken to prevent the associated disease from occurring. For example, APOE is known to significantly increase the risk of late onset Alzheimer's disease, yet, there is no proven method to prevent the disease [29]. In order to understand how genetic variations result in distinctive disease traits and to discover novel molecular markers or actionable molecular targets, the causal variants and their influence on the molecular network of the disease‐related cells or tissues must be elucidated. One of the main limitations of current statistical approaches is that it is very difficult to identify and validate causal variants [98]. Furthermore, because the complex disease traits are orchestrated by simultaneously operating molecular interactions altered by causal variants, it is currently impossible to determine the full effects of such complex interactions by contemporary experimental and statistical approaches alone. However, recent advances in computational modelling of various molecular networks have provided a new avenue into the personalised medicine. For example, Choi et al. [99] were able to build a disease network model of breast cancer in silico and successfully identified novel actionable therapeutic targets, WIP1 and MDM2. By treating WIP1 inhibitor and Nutlin‐3 together with the breast cancer cell line, MCF7, they induced p53‐mediated apoptosis as computationally predicted, indicating the importance of the systems biology approach to advance current personalised medicine research to the next level.
7 Acknowledgments
This work was supported by Basic Science Research Program through the National Research Foundation of Korea(NRF) funded by the Ministry of Education (2016940619), and No. 20150091 of Handong Global University Research Grants.
8 References
- 1. Venter J.C. Adams M.D. Myers E.W. et al.: ‘The sequence of the human genome ’, Science, 2001, 291, (5507), pp. 1304–1351 [DOI] [PubMed] [Google Scholar]
- 2. Rothberg J.M. Hinz W. Rearick T.M. et al.: ‘An integrated semiconductor device enabling non‐optical genome sequencing ’, Nature, 2011, 475, (7356), pp. 348–352 [DOI] [PubMed] [Google Scholar]
- 3. Kasianowicz J.J. Brandin E. Branton D. et al.: ‘Characterization of individual polynucleotide molecules using a membrane channel ’, Proc. Natl. Acad. Sci., 1996, 93, (24), pp. 13770–13773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eid J. Fehr A. Gray J. et al.: ‘Real‐time dna sequencing from single polymerase molecules ’, Science, 2009, 323, (5910), pp. 133–138 [DOI] [PubMed] [Google Scholar]
- 5. Sanger F. Coulson A.R.: ‘A rapid method for determining sequences in dna by primed synthesis with dna polymerase ’, J. Mol. Biol., 1975, 94, (3), pp. 441–448 [DOI] [PubMed] [Google Scholar]
- 6. Bentley D.R. Balasubramanian S. Swerdlow H.P. et al.: ‘Accurate whole human genome sequencing using reversible terminator chemistry ’, Nature, 2008, 456, (7218), pp. 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fedurco M. Romieu A. Williams S. et al.: ‘Bta, a novel reagent for dna attachment on glass and efficient generation of solid‐phase amplified dna colonies ’, Nucleic Acids Res., 2006, 34, (3), pp. e22–e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Metzker M.L.: ‘Sequencing technologiesthe next generation ’, Nat. Rev. Genet., 2010, 11, (1), pp. 31–46 [DOI] [PubMed] [Google Scholar]
- 9. Ng P.C. Murray S.S. Levy S. et al.: ‘An agenda for personalized medicine ’, Nature, 2009, 461, pp. 724–726 [DOI] [PubMed] [Google Scholar]
- 10. Turner E.H. Lee C. Ng S.B. et al.: ‘Massively parallel exon capture and library‐free resequencing across 16 genomes ’, Nat. Methods, 2009, 6, (5), pp. 315–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Z. Gerstein M. Snyder M.: ‘Rna‐seq: a revolutionary tool for transcriptomics ’, Nat. Rev. Genet., 2009, 10, (1), pp. 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li B. Ruotti V. Stewart R.M. et al.: ‘Rna‐seq gene expression estimation with read mapping uncertainty ’, Bioinformatics, 2010, 26, (4), pp. 493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang L. Schlesinger F. Davis C.A. et al.: ‘Synthetic spike‐in standards for RNA‐seq experiments ’, Genome Res., 2011, 21, pp. 1543–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horvath A. Pakala S.B. Mudvari P. et al.: ‘Novel insights into breast cancer genetic variance through RNA sequencing ’, Sci. Rep., 2013, 3, p. 2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park P.J.: ‘ChIP–seq: advantages and challenges of a maturing technology ’, Nat. Rev. Genet., 2009, 10, pp. 669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. ONeill L.P. Turner B.M.: ‘Immunoprecipitation of native chromatin: Nchip ’, Methods, 2003, 31, (1), pp. 76–82 [DOI] [PubMed] [Google Scholar]
- 17. Nguyen C.T. Weisenberger D.J. Velicescu M. et al.: ‘Histone H3‐Lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5‐aza‐2 ‐deoxycytidine ’, Cancer Res., 2002, 62, pp. 6456–6461 [PubMed] [Google Scholar]
- 18. Grunau C. Clark S.J. Rosenthal A.: ‘Bisulfite genomic sequencing: systematic investigation of critical experimental parameters ’, Nucleic Acids Res., 2001, 29, pp. E65–E65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Issa J.‐P.: ‘Cancer prevention: epigenetics steps up to the plate ’, Cancer Prev. Res., 2008, 1, pp. 219–222 [DOI] [PubMed] [Google Scholar]
- 20. Landan G. Cohen N.M. Mukamel Z. et al.: ‘Epigenetic polymorphi1sm and the stochastic formation of differentially methylated regions in normal and cancerous tissues ’, Nat. Genet., 2012, 44, pp. 1207–1214 [DOI] [PubMed] [Google Scholar]
- 21. Esteller M.: ‘CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future ’, Oncogene, 2002, 21, pp. 5427–5440 [DOI] [PubMed] [Google Scholar]
- 22. Shen L. Ahuja N. Shen Y. et al.: ‘Dna methylation and environmental exposures in human hepatocellular carcinoma ’, J. Natl. Cancer Inst., 2002, 94, (10), pp. 755–761 [DOI] [PubMed] [Google Scholar]
- 23. Shen L. Kondo Y. Rosner G.L. et al.: ‘Mgmt promoter methylation and field defect in sporadic colorectal cancer ’, J. Natl. Cancer Inst., 2005, 97, (18), pp. 1330–1338 [DOI] [PubMed] [Google Scholar]
- 24. Shapiro E. Biezuner T. Linnarsson S.: ‘Single‐cell sequencing‐based technologies will revolutionize whole‐organism science ’, Nat. Rev. Genet., 2013, 14, pp. 618–630 [DOI] [PubMed] [Google Scholar]
- 25. Nawy T.: ‘Single‐cell sequencing ’, Nat. Methods, 2014, 11, p. 18 [DOI] [PubMed] [Google Scholar]
- 26. Navin N. Kendall J. Troge J. et al.: ‘Tumour evolution inferred by single‐cell sequencing ’, Nature, 2011, 472, pp. 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu X. Hou Y. Yin X. et al.: ‘Single‐cell exome sequencing reveals single‐nucleotide mutation characteristics of a kidney tumor ’, Cell, 2012, 148, pp. 886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Du W. Elemento O.: ‘Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies ’, Oncogene, 2015, 34, pp. 3215–3225 [DOI] [PubMed] [Google Scholar]
- 29. Topol E.J.: ‘Individualized medicine from prewomb to tomb ’, Cell, 2014, 157, pp. 241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei X. Li L.: ‘Mass spectrometry‐based proteomics and peptidomics for biomarker discovery in neurodegenerative diseases ’, Int. J. Clin. Exp. Pathol., 2009, 2, (2), pp. 132–148 [PMC free article] [PubMed] [Google Scholar]
- 31. Ong S.‐E. Blagoev B. Kratchmarova I. et al.: ‘Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics ’, Mol. Cell. Proteomics, 2002, 1, (5), pp. 376–386 [DOI] [PubMed] [Google Scholar]
- 32. Bandura D.R. Baranov V.I. Ornatsky O.I. et al.: ‘Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time‐of‐flight mass spectrometry ’, Anal. Chem., 2009, 81, (16), pp. 6813–6822 [DOI] [PubMed] [Google Scholar]
- 33. Qiu P. Simonds E.F. Bendall S.C. et al.: ‘Extracting a cellular hierarchy from highdimensional cytometry data with spade ’, Nat. Biotechnol., 2011, 29, (10), pp. 886–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amir E.‐a.D. Davis K.L. Tadmor M.D. et al.: ‘visne enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia ’, Nat. Biotechnol., 2013, 31, (6), pp. 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Behbehani G.K. Bendall S.C. Clutter M.R. et al.: ‘Single‐cell mass cytometry adapted to measurements of the cell cycle ’, Cytometry A, 2012, 81, (7), pp. 552–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cederholm M. Haglund B. Axelsson O.: ‘Infant morbidity following amniocentesis and chorionic villus sampling for prenatal karyotyping ’, BJOG: Int. J. Obstet. Gynaecol., 2005, 112, (4), pp. 394–402 [DOI] [PubMed] [Google Scholar]
- 37. Grati F.R.: ‘Chromosomal mosaicism in human feto‐placental development: implications for prenatal diagnosis ’, J. Clin. Med., 2014, 3, (3), pp. 809–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schlütter J.M. Kirkegaard I. Petersen O.B. et al.: ‘Fetal gender and several cytokines are associated with the number of fetal cells in maternal blood–an observational study ’, PloS One, 2014, 9, (9), p. e106934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bianchi D.W. Simpson J. Jackson L. et al.: ‘Fetal gender and aneuploidy detection using fetal cells in maternal blood: analysis of nifty i data ’, Prenat. Diagn., 2002, 22, (7), pp. 609–615 [DOI] [PubMed] [Google Scholar]
- 40. Lo Y.M. Corbetta N. Chamberlain P.F. et al.: ‘Presence of fetal DNA in maternal plasma and serum ’, Lancet, 1997, 350, pp. 485–487 [DOI] [PubMed] [Google Scholar]
- 41. Chiu R.W. Chan K.A. Gao Y. et al.: ‘Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of dna in maternal plasma ’, Proc. Natl. Acad. Sci., 2008, 105, (51), pp. 20458–20463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Norwitz E.R. Levy B.: ‘Noninvasive prenatal testing: the future is now ’, Rev. Obstet. Gynecol., 2013, 6, (2), pp. 48–62 [PMC free article] [PubMed] [Google Scholar]
- 43. Bianchi D.W. Parsa S. Bhatt S. et al.: ‘Fetal sex chromosome testing by maternal plasma dna sequencing: clinical laboratory experience and biology ’, Obstet. Gynecol., 2015, 125, (2), pp. 375–382 [DOI] [PubMed] [Google Scholar]
- 44. Sparks A.B. Wang E.T. Struble C.A. et al.: ‘Selective analysis of cell‐free dna in maternal blood for evaluation of fetal trisomy ’, Prenat. Diagn., 2012, 32, (1), pp. 3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kitzman J.O. Snyder M.W. Ventura M. et al.: ‘Noninvasive whole‐genome sequencing of a human fetus ’, Sci. Transl. Med., 2012, 4, (137), pp. 137ra76–137ra76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hill M. Barrett A.N. White H. et al.: ‘Uses of cell free fetal dna in maternal circulation ’, Best Pract. Res. Clin. Obstet. Gynaecol., 2012, 26, (5), pp. 639–654 [DOI] [PubMed] [Google Scholar]
- 47. Chan K.C.A. Zhang J. Hui A.B.Y. et al.: ‘Size distributions of maternal and fetal DNA in maternal plasma ’, Clin. Chem., 2004, 50, pp. 88–92 [DOI] [PubMed] [Google Scholar]
- 48. Lo Y.M. Zhang J. Leung T.N. et al.: ‘Rapid clearance of fetal DNA from maternal plasma ’, Am. J. Hum. Genet., 1999, 64, pp. 218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lo Y.M.D. Tsui N.B.Y. Chiu R.W.K. et al.: ‘Plasma placental RNA allelic ratio permits noninvasive prenatal chromosomal aneuploidy detection ’, Nat. Med., 2007, 13, pp. 218–223 [DOI] [PubMed] [Google Scholar]
- 50. Chan K.A. Ding C. Gerovassili A. et al.: ‘Hypermethylated rassf1a in maternal plasma: a universal fetal dna marker that improves the reliability of noninvasive prenatal diagnosis ’, Clin. Chem., 2006, 52, (12), pp. 2211–2218 [DOI] [PubMed] [Google Scholar]
- 51. Grygalewicz B. Woroniecka R. Rygier J. et al.: ‘Monoallelic and biallelic deletions of 13q14 in a group of cll/sll patients investigated by cgh haematological cancer and snp array (8 × 60k) ’, Mol. Cytogenet., 2016, 9, (1), p. 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Atlija M. Arranz J.‐J. Martinez‐Valladares M. et al.: ‘Detection and replication of qtl underlying resistance to gastrointestinal nematodes in adult sheep using the ovine 50k snp array ’, Genet. Sel. Evol., 2016, 48, (1), pp. 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sherry S.T. Ward M.‐H. Kholodov M. et al.: ‘dbsnp: the ncbi database of genetic variation ’, Nucleic Acids Res., 2001, 29, (1), pp. 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strittmatter W.J. Saunders A.M. Schmechel D. et al.: ‘Apolipoprotein e: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial alzheimer disease ’, Proc. Natl. Acad. Sci., 1993, 90, (5), pp. 1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu C.‐C. Kanekiyo T. Xu H. et al.: ‘Apolipoprotein e and alzheimer disease: risk, mechanisms and therapy ’, Nat. Rev. Neurol., 2013, 9, (2), pp. 106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Coon K.D. Myers A.J. Craig D.W. et al.: ‘A high‐density whole‐genome association study reveals that APOE is the major susceptibility gene for sporadic late‐onset alzheimer's disease ’, J. Clin. Psychiatry, 2007, 68, pp. 613–618 [DOI] [PubMed] [Google Scholar]
- 57. Takei N. Miyashita A. Tsukie T. et al.: ‘Genetic association study on in and around the APOE in late‐onset alzheimer disease in Japanese ’, Genomics, 2009, 93, pp. 441–448 [DOI] [PubMed] [Google Scholar]
- 58. King M.‐C. Marks J.H. Mandell J.B. et al.: ‘Breast and ovarian cancer risks due to inherited mutations in brca1 and brca2 ’, Science, 2003, 302, (5645), pp. 643–646 [DOI] [PubMed] [Google Scholar]
- 59. Hall J. Lee M. Newman B. et al.: ‘Linkage of early‐onset familial breast cancer to chromosome 17q21 ’, Science, 1990, 250, (4988), pp. 1684–1689 [DOI] [PubMed] [Google Scholar]
- 60. Wooster R. Neuhausen S.L. Mangion J. et al.: ‘Localization of a breast cancer susceptibility gene, brca2, to chromosome 13q12‐13 ’, Science, 1994, 265, (5181), pp. 2088–2090 [DOI] [PubMed] [Google Scholar]
- 61. Easton D.F. et al.: ‘How many more breast cancer predisposition genes are there ’, Breast Cancer Res., 1999, 1, (1), pp. 14–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Holloman W.K.: ‘Unraveling the mechanism of brca2 in homologous recombination ’, Nat. Struct. Mol. Biol., 2011, 18, (7), pp. 748–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Silver D.P. Livingston D.M.: ‘Mechanisms of brca1 tumor suppression ’, Cancer Discov., 2012, 2, (8), pp. 679–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Antoniou A.C. Pharoah P.D.P. Narod S. et al.: ‘Breast and ovarian cancer risks to carriers of the BRCA1 5382insc and 185delAG and BRCA2 6174delt mutations: a combined analysis of 22 population based studies ’, J. Med. Genet., 2005, 42, pp. 602–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Antoniou A. Pharoah P. Narod S. et al.: ‘Average risks of breast and ovarian cancer associated with brca1 or brca2 mutations detected in case series unselected for family history: a combined analysis of 22 studies ’, Am. J. Human Genet., 2003, 72, (5), pp. 1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen S. Parmigiani G.: ‘Meta‐analysis of brca1 and brca2 penetrance ’, J. Clin. Oncol., 2007, 25, (11), pp. 1329–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Howlader N. Noone A. Krapcho M. et al.: ‘Cancer statistics review, 1975–2011 ’ (National Cancer Institute, Bethesda, MD, 2014) [Google Scholar]
- 68. Gerhardus A. Schleberger H. Schlegelberger B. et al.: ‘Diagnostic accuracy of methods for the detection of brca1 and brca2 mutations: a systematic review ’, Eur. J. Hum. Genet., 2007, 15, (6), pp. 619–627 [DOI] [PubMed] [Google Scholar]
- 69. Vuttariello E. Borra M. Calise C. et al.: ‘A new rapid methodological strategy to assess brca mutational status ’, Mol. Biotechnol., 2013, 54, (3), pp. 954–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. D'Argenio V. Esposito M.V. Telese A. et al.: ‘The molecular analysis of brca1 and brca2: Next‐generation sequencing supersedes conventional approaches ’, Clin. Chim. Acta, 2015, 446, pp. 221–225 [DOI] [PubMed] [Google Scholar]
- 71. Frebourg T. Barbier N. Yan Y. et al.: ‘Germ‐line p53 mutations in 15 families with li‐fraumeni syndrome. ’, Am J. Hum. Genet., 1995, 56, (3), p. 608 [PMC free article] [PubMed] [Google Scholar]
- 72. Lang G.A. Iwakuma T. Suh Y.‐A. et al.: ‘Gain of function of a p53 hot spot mutation in a mouse model of li‐fraumeni syndrome ’, Cell, 2004, 119, (6), pp. 861–872 [DOI] [PubMed] [Google Scholar]
- 73. Bach G.: ‘Mucolipidosis type IV ’, Mol. Genet. Metab., 2001, 73, pp. 197–203 [DOI] [PubMed] [Google Scholar]
- 74. Hantash F.M. Olson S.C. Anderson B. et al.: ‘Rapid one‐step carrier detection assay of mucolipidosis IV mutations in the Ashkenazi Jewish population ’, J. Mol. Diagn., 2006, 8, pp. 282–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Waldmüller S. Sakthivel S. Saadi A.V. et al.: ‘Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy ’, J. Mol. Cell. Cardiol., 2003, 35, pp. 623–636 [DOI] [PubMed] [Google Scholar]
- 76. Richard P. Charron P. Carrier L. et al.: ‘Hypertrophic cardiomyopathy distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy ’, Circulation, 2003, 107, (17), pp. 2227–2232 [DOI] [PubMed] [Google Scholar]
- 77. Deprez R.L. Muurling‐Vlietman J.J. Hruda J. et al.: ‘Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the mybpc3 gene ’, J. Med. Genet., 2006, 43, (10), pp. 829–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. A.C. Society : ‘American cancer society breast cancer facts & figures ’. Breast Cancer Facts and Figures, 2015, pp. 1–38
- 79. Manolio T.A. Collins F.S. Cox N.J. et al.: ‘Finding the missing heritability of complex diseases ’, Nature, 2009, 461, pp. 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Barzi A. Lenz A.M. Labonte M.J. et al.: ‘Molecular pathways: Estrogen pathway in colorectal cancer ’, Clin. Cancer Res., 2013, 19, pp. 5842–5848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Issa J.‐P.J. Ottaviano Y.L. Celano P. et al.: ‘Methylation of the oestrogen receptor cpg island links ageing and neoplasia in human colon ’, Nat. Genet., 1994, 7, (4), pp. 536–540 [DOI] [PubMed] [Google Scholar]
- 82. Schnitt S.J.: ‘Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy ’, Mod. Pathol., 2010, 23, pp. S60–S64 [DOI] [PubMed] [Google Scholar]
- 83. Nielsen D.L. Kümler I. Palshof J.A. et al.: ‘Efficacy of her2‐targeted therapy in metastatic breast cancer. monoclonal antibodies and tyrosine kinase inhibitors ’, Breast, 2013, 22, (1), pp. 1–12 [DOI] [PubMed] [Google Scholar]
- 84. Klijn J.G. Setyono‐Han B. Foekens J.A.: ‘Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer ’, Steroids, 2000, 65, (10), pp. 825–830 [DOI] [PubMed] [Google Scholar]
- 85. Sommer S. Fuqua S.A.: ‘Estrogen receptor and breast cancer ’, in ‘Seminars in cancer biology ’ (Elsevier, 2001), vol. 11, pp. 339–352 [DOI] [PubMed] [Google Scholar]
- 86. Isakoff S.J.: ‘Triple negative breast cancer: role of specific chemotherapy agents ’, Cancers J. (Sudbury, Mass.), 2010, 16, (1), p. 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Niepel M. Hafner M. Pace E.A. et al.: ‘Profiles of basal and stimulated receptor signaling networks predict drug response in breast cancer lines ’, Sci. Signal., 2013, 6, p. ra84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vallejos C.S. Gómez H.L. Cruz W.R. et al.: ‘Breast cancer classification according to immunohistochemistry markers: subtypes and association with clinicopathologic variables in a peruvian hospital database ’, Clin. Breast Cancer, 2010, 10, (4), pp. 294–300 [DOI] [PubMed] [Google Scholar]
- 89. G. M.‐a. T. G. Group : ‘Chemotherapy in adult high‐grade glioma: a systematic review and meta‐analysis of individual patient data from 12 randomised trials ’, Lancet, 2002, 359, (9311), pp. 1011–1018 [DOI] [PubMed] [Google Scholar]
- 90. Yamanaka R.: ‘Cell‐ and peptide‐based immunotherapeutic approaches for glioma ’, Trends Mol. Med., 2008, 14, pp. 228–235 [DOI] [PubMed] [Google Scholar]
- 91. Yu J.S. Wheeler C.J. Zeltzer P.M. et al.: ‘Vaccination of malignant glioma patients with peptide‐pulsed dendritic cells elicits systemic cytotoxicity and intracranial t‐cell infiltration ’, Cancer Res., 2001, 61, (3), pp. 842–847 [PubMed] [Google Scholar]
- 92. Schneider T. Gerhards R. Kirches E. et al.: ‘Preliminary results of active specific immunization with modified tumor cell vaccine in glioblastoma multiforme ’, J. Neurooncol., 2001, 53, pp. 39–46 [DOI] [PubMed] [Google Scholar]
- 93. Palucka K. Banchereau J.: ‘Dendritic‐cell‐based therapeutic cancer vaccines ’, Immunity, 2013, 39, pp. 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Provasi E. Genovese P. Lombardo A. et al.: ‘Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer ’, Nat. Med., 2012, 18, pp. 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brentjens R.J. Davila M.L. Riviere I. et al.: ‘Cd19‐targeted t cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia ’, Sci. Transl. Med., 2013, 5, (177), pp. 177ra38–177ra38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grupp S.A. Kalos M. Barrett D. et al.: ‘Chimeric antigen receptor‐modified t cells for acute lymphoid leukemia ’, New Engl. J. Med., 2013, 368, (16), pp. 1509–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Majid S. Kikuno N. Nelles J. et al.: ‘Genistein induces the p21waf1/cip1 and p16ink4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification ’, Cancer Res., 2008, 68, (8), pp. 2736–2744 [DOI] [PubMed] [Google Scholar]
- 98. Stitziel N.O. Kiezun A. Sunyaev S.: ‘Computational and statistical approaches to analyzing variants identified by exome sequencing ’, Genome Biol., 2011, 12, (9), pp. 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Choi M. Shi J. Jung S.H. et al.: ‘Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to dna damage ’, Sci. Signal, 2012, 5, (251), p. ra83 [DOI] [PubMed] [Google Scholar]