

Abstract
Transcriptional regulation of gene expression is an essential cellular process that is arranged by transcription factors (TFs), microRNAs (miRNA) and their target genes through a variety of mechanisms. Here, we set out to reconstruct a comprehensive transcriptional regulatory network of Homo sapiens consisting of experimentally verified regulatory information on miRNAs, TFs and their target genes. We have performed topological analyses to elucidate the transcriptional regulatory roles of miRNAs and TFs. When we thoroughly investigated the network motifs, different gene regulatory scenarios were observed; whereas, mutual TF‐miRNA regulation (interactive cooperation) and hierarchical operation where miRNAs were the upstream regulators of TFs came into prominence. Otherwise, biological process specific subnetworks were also constructed and integration of gene and miRNA expression data on ovarian cancer was achieved as a case study to observe dynamic patterns of the gene expression. Meanwhile, both co‐operation and hierarchical operation types were determined in active ovarian cancer and process‐specific subnetworks. In addition, the analysis showed that multiple signals from miRNAs were integrated by TFs. Our results demonstrate new insights on the architecture of the human transcriptional regulatory network, and here we present some lessons we gained from deciphering the reciprocal interplay between miRNAs, TFs and their target genes.
Inspec keywords: RNA, genetics, cellular biophysics, molecular biophysics, cancer
Other keywords: interactive cooperation, microRNA, transcription factor crosstalk, human transcriptional regulatory network, transcriptional regulation, gene expression, cellular process, target genes, regulation mechanism, Homo sapiens, topological analysis, network motifs, miRNA‐TF‐gene regulations, gene regulatory scenarios, mutual TF‐miRNA regulation, biological process, gene integration, miRNA expression data, cooperation operation type, hierarchical operation type, active ovarian cancer, process‐specific subnetworks
1 Introduction
Biological functions are maintained by gene regulatory networks tightly coupled to protein activity and function. All living systems depend on the precise coordinated expression of gene products which interact and specify cellular phenotypes and function. Regulatory modules are required for the coordinated response of distinct cellular processes within an organism, and for responding to the external environment.
One of the most important cellular processes that determine the levels of gene products is transcription. Transcriptional regulation of gene expression is often the prior process due to response of system changes, wherein the information contained in a genome is converted and then ultimately used to produce the proteins required for a given response. It is arranged by transcriptional regulatory proteins and their target genes working in assent through a variety of mechanisms. These regulators, known as transcription factors (TFs), are proteins bind to either enhancer or promoter regions of DNA adjacent to the genes that they regulate. TFs can act as activators or repressors in a tissue‐ or condition‐specific manner. The roles of TF complexes in regulating the levels of protein‐coding genes have been well‐elucidated in many cell types [1, 2].
A recent class of small non‐coding RNAs discovered, microRNAs (miRNAs), also appears to have determining roles in the control of cellular phenotypes, fundamental biological processes and disease [3, 4]. Briefly, miRNAs constitute a unique family of short, with 20–23 nucleotides length, non‐coding endogenous small RNA molecules that have emerged as important modulators of gene expression [5]. They take charge in RNA silencing and post‐transcriptional regulation of gene expression [5, 6]. We are only beginning to understanding the network of individual miRNAs in terms of their regulatory effects on other genes including TFs. However, the networks comprised of TFs and miRNAs are largely disconnected. It has been widely reported that miRNAs and TFs share a common regulatory logic and are capable of controlling hundreds of target genes by binding to distinct cis‐regulatory elements [7, 8]. It is unclear how the transcription of miRNA genes and the feedback of miRNAs into transcription hierarchies are regulated. Since miRNAs constitute an entire layer of the regulatory fabric, it is useful for us to uncover their significance, function and interaction within the hierarchy of other cellular networks.
miRNAs are necessary for many aspects of development, cellular function and cell maintenance. Dysregulation of miRNAs are linked to many diseases, including cancers [9, 10]. They are also implicated in disease by behaving as oncogenes and tumour suppressors [11, 12]. It has been observed that the levels of certain miRNAs are dramatically altered in primary tumours, and global miRNA expression is lower in cancer tissues than in normal tissues.
As regulators of gene expression, miRNAs exert their effects in two modes. miRNAs recognise the target mRNAs by perfect or near‐perfect sequence complementarity, resulting in the cleavage and destruction of mRNA through the RNA interference machinery; or, miRNAs appear to pair with imperfect complementarity to their target mRNAs, and they inhibit protein synthesis through translational inhibition using unknown mechanisms that preserve the mRNA targets [13]. Several studies have provided evidence that translational repression occurs preinitiation or post‐initiation of translation [14, 15]. Also Vasudevan et al. [16], Liu et al. [17] were reported about activator effects of miRNAs on target genes expression. On the other hand, miRNAs can have roles that recapitulate the gain or loss‐of‐function of transcriptional factors. These revelations support the rationale for a deeper investigation into the global and local architecture of miRNA–mRNA–TF interactions.
Construction of biological networks and topological properties tells us a great deal about how they facilitate the cell to respond to its environment and perform complex biological functions such as cancer progression and apoptosis [18]. The topology of the gene regulatory network plays an essential role to understand to the cell response, pathogenesis of genetic diseases and cancer progression. Some repetitive topological units of biological networks, termed network motifs, occur more often than would be expected from a random generated network [19]. Detection of the network motifs in the complex biological networks can yield new mechanistic insights into regulatory system and offer crucial clues into gene expression regulatory mechanisms [20, 21].
Several network based studies were performed to explore the relationships between miRNAs and targeted genes or TFs. These include investigation of the expression patterns between miRNAs and targeted genes [22, 23], prediction of disease‐associated miRNAs and TFs [24, 25, 26], uncovering the local and global architectural features [27, 28], integrative analysis of transcriptional and post‐transcriptional regulatory networks [29, 30], and reconstruction of disease, tissue or process specific transcriptional regulatory subnetworks [31, 32, 33].
In this study, we set out to elucidate the cooperation scenarios of miRNAs and TFs. For this purpose, we reconstructed a generic transcriptional regulatory network of Homo sapiens (H. sapiens) consisting of experimentally verified data on four regulation types among miRNAs, TFs and target genes: miRNAs regulating TFs, TFs regulating miRNAs, TFs regulating target genes and miRNAs regulating target genes. The local and global architectural features of the network were uncovered via topological analyses, determination of hub molecules (TFs and miRNAs), and detection of network motifs. The study was extended via integration of gene and miRNA expression data on ovarian cancer as a case study to observe dynamic patterns of the regulatory mechanisms. Together these findings provide new insights on the architecture of the transcriptional regulatory network, and here we present some lessons we gained from deciphering the reciprocal interplay between miRNAs, TFs and their target genes.
2 Materials and methods
2.1 Reconstruction of transcriptional regulatory network of H. sapiens
The generic transcriptional regulatory network was reconstructed by integrating three distinct regulatory relationships, i.e. miRNA–gene, miRNA–TF and TF–gene interactions. For this purpose, interactome data were compiled from various publicly available databases (Table 1). A comprehensive miRNA–gene interaction dataset was assembled by employing miRDB [34], starBase [35], miRecords [36], miRTarBase [37] and miR2Disease [38] databases. Since two of the databases (miRDB and starBase) store computational predictions, to guarantee that an interaction is experimental validated, miRNA–gene interactions that were reported in at least three distinct databases were incorporated into the dataset. The experimentally validated TF–miRNA interactions were obtained from TransMir (ver.1.2) [39]. The target genes and TFs interactions were extracted from PAZAR database [40].
Table 1.
Data resources for transcriptional regulatory information
| Interaction type | Database (web‐link) | Data summary | Confidence identifier | References |
|---|---|---|---|---|
| miRNA–gene interactions | MiRDB (http://mirdb.org/miRDB/) | 271, 089 interactions between 2541 miRNAs and 15,788 genes | computational predictions (score ≥80) | Wang [34] |
| miRecords (http://mirecords.biolead.org/) | 1740 interactions between 285 miRNAs and 1110 genes | experimentally validated | Xiao et al. [36] | |
| miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/) | 37, 402 interactions between 576 miRNAs and 12,101 genes | experimentally validated | Hsu et al. [37] | |
| starBase (http://starbase.sysu.edu.cn/) | 423, 405 interactions between 386 miRNAs and 13,801 genes | computational predictions | Yang et al. [35] | |
| miR2Disease (http://www.mir2disease.org/) | 597 interactions between 176 miRNAs and 386 genes | experimentally validated | Jiang et al. [38] | |
| miRNA–TF interactions | TransmiR (http://www.cuilab.cn/transmir) | 670 interactions between 239 miRNAs and 167 TFs | experimentally validated | Wang et al. [39] |
| TF–gene interactions | PAZAR (http://www.pazar.info/cgi‐bin/) | 6071 interactions between 161 TFs and 3690 genes | experimentally validated | Portales‐Casamar et al. [40] |
| miRNA‐disease associations | miR2Disease (http://www.mir2disease.org/) | 81 miRNAs associated with ovarian cancer | experimentally validated | Jiang et al. [38] |
2.2 Topological analysis of transcriptional regulatory networks
Reconstructed networks were visualised and analysed via Cytoscape (v.2.8). Local and global topological features were represented by several metrics, including the degree, betweenness connectivity, shortest path length, heterogeneity, clustering coefficient, and determined via NetworkAnalyzer [41] and Cytohubba [42] plugins. The dual‐metric approach [43] incorporating degree as a local metric and betweenness centrality as a global metric was employed at identification of hub molecules (miRNAs and TFs), and top five molecules in terms of any of the metrics were presented as hubs. Network motifs were determined via FANMOD [44], and significantly enriched motifs with sizes up to five, Z score ≥2, and observation frequency ≥0.001% were considered in further analyses.
2.3 Gene set enrichment analysis
Enrichment analyses of gene sets were performed through DAVID bioinformatics tool [45]. Gene Ontology (GO) terminology was employed for association of genes with biological processes, molecular functions and cellular compartments. Enrichment results with p ‐value <0.05 were considered as statistically significant in all analyses.
2.4 Identification of differentially expressed genes (DEGs) and miRNAs in ovarian cancer
To characterise gene expression in ovarian cancer, two independent transcriptomics datasets (GSE7463 and GSE14407) from the Gene Expression Omnibus (GEO) database [46] were recruited. CEL files were normalised using RMA [47] as implemented in the affy package [48] of R/Bioconductor software (ver. 2.12) [49]. DEGs were identified from the normalised log‐expression values using the multiple testing option of LIMMA (linear models for microarray data) [50]. Benjamini Hochberg's method was used to control the false discovery rate and the 0.01 p ‐value threshold was maintained to identify significant DEGs. Differentially expressed miRNAs (DEmiRs) in ovarian cancer were extracted directly from the miR2disease database. In this database, ovarian cancer associated 81 miRNAs were obtained, 42 of them were represented in transcriptional regulatory network.
2.5 Reconstruction of ovarian cancer specific subnetwork
Considering the DEGs and DEmiRs in ovarian cancer, a disease specific subnetwork of the generic network was constructed. Initially, miRNA–gene and TF–gene associations of DEGs were extracted from the generic network. Then, miRNA–gene and miRNA–TF associations were filtered for DEmiRs. The resultant network was visualised and analysed via Cytoscape (v.2.8).
2.6 Analysis of network performance
We evaluated the performance of the reconstructed generic network via comparison with other combinatorial transcriptional regulatory networks in the literature [24, 51, 52]. For this purpose, gold‐standard datasets were reconstructed. The gold standard positive dataset was composed of 300 interactions, which were randomly selected among the experimentally validated miRNA–TF, TF–gene and miRNA–gene interactions from publicly available databases. The gold standard negative dataset was consisting of randomly generated 300 interactions. Each randomisation was performed 100 times in determination of performance metrics. In comparison of the proposed generic network with the other networks to illustrate the predictive power of the reconstruction process, sensitivity–specificity trade‐off was considered. Sensitivity (recall) and specificity is the ability to identify a true positive and true negative in a data set, respectively. On the other hand, F 1 score, which is the harmonic mean of precision and recall, considers both true positives and true negatives among the total number of cases examined.
2.7 Robustness analysis
Considering the incompleteness of the reconstructed networks, to analyse the robustness of motif analysis results, we performed a systematic analysis on the reconstructed network via removal of 5% true interactions, or addition of 5% false interactions. Interactions were randomly selected or created, and each randomisation was performed 100 times. The statistical significance of z ‐scores (p ‐value) was exhibited using two‐tailed Student's T‐test.
3 Results and discussion
3.1 Generic transcriptional regulatory network of H. sapiens was reconstructed
Although several studies reported conserved features of gene regulatory mechanisms by TFs at network level, very little is known about the features of miRNAs and their interface with the TFs. There is a critical gap in our understanding of how the networks of miRNAs and TFs are connected. Primarily, we reconstructed a generic transcriptional regulatory network of H. sapiens via integrating three distinct regulatory relationships, i.e. miRNA–gene, miRNA–TF and TF–gene interactions. For this purpose, interaction data from various databases were compiled (Table 1). The regulatory mechanisms of miRNAs have made the algorithms for predicting miRNA targets complicated, and the precise determination of the full range of targets of miRNAs is a major bottleneck. To avoid misinterpretations due to false positive interactions arising from incorrect predictions of miRNA target prediction programs, here we employed only the interactions, which were experimentally validated. The miRNA–gene interaction data were taken from five distinct databases, two of which included computational predictions. Therefore, miRNA–gene interactions, which were reported in at least three distinct databases, were incorporated into the generic network in order to guarantee the experimental validation of the interaction. For the other interaction types, data were collected from relevant databases, which provide experimentally validated relationships exclusively. As a whole, the generic network was comprised of 8622 interactions associated with 284 TFs, 345 miRNAs and 4624 target genes (mRNAs) (Table S1).
Combinatorial transcriptional regulatory networks integrating distinct regulatory relationships for H. sapiens are very limited in the literature. Previously, context–specific networks were reconstructed for human cancer [24] and epithelial‐to‐mesenchymal transition in cancer [51]. Data repositories of transcriptional and post‐transcriptional regulatory relationships, such as RegNetwork [52], enabling reconstruction of generic networks is emerging in recent years. Here, we evaluated the performance of the reconstructed generic network via comparison with other combinatorial transcriptional regulatory networks in the literature (Table S2). The reconstructed generic network was more comprehensive (less far away from incompleteness) taking into consideration the number of regulatory elements (miRNAs and TFs) and regulatory interactions associated with miRNAs (miRNA–TF and miRNA–gene interactions). In terms of performance metrics, context‐specific networks indicated high specificity (ability to identify true negatives), but very low sensitivity (ability to identify true positives) due to their limited coverage in terms of regulatory elements and interactions. On the other hand, the performances of the generic networks were comparable to each other, but significantly higher than context‐specific networks, in terms of sensitivity and accuracy. The reconstructed generic network, in this study, represented the highest F 1 score (0.78), which is a measure of accuracy considering both true positives and true negatives.
Topological analysis of the generic network indicated the dominance of TFs as the regulators of transcription, since 39 genes were regulated by a TF, whereas a miRNA regulates 7 genes in average (Figs. 1 a and b, Table S3, S4). This observation may support the hypothesis that TFs play general roles in transcriptional regulatory mechanisms; however, miRNAs take more specific duties, in general. The miRNA–TF associations also constituted an intertwined structure, i.e. 141 miRNAs (41% of all) were found to be regulated by more than one TF, and 106 TFs (63.5% of all) were found to be regulated by at least two miRNAs. When degree distributions were investigated, the generic network follows scale‐free topology which indicated the presence of hubs. For instance, miR‐124‐3p with 139 target genes, and HIF1A with 380 target genes came into prominence as the leading regulators, and the TF MYC was associated with the highest number of miRNAs (Fig. 1 c, Table S5). Hubs play central role in directing the cellular response to stimulus. When hub nodes (TF, miRNA or target gene) were evaluated according to two topological metrics (i.e. degree as the local metric and betweenness centrality as the global metric), network hubs were all TFs, supporting the central role of TFs as key components in the transcriptional regulatory network (Table 2). HIF1A, TP53, FOS, STAT1, RUNX1 and EGR1 were the hub TFs regulating numerous genes. MYC, E2F1, EGR1 and TP53 were the hubs interacting with a wide range of miRNAs. On the other hand, miR‐124‐3p, miR‐16‐5p, miR‐93‐5p, miR‐1, miR‐19b‐3p and miR‐130b‐3p were the hub miRNAs in terms of transcriptional regulation of target genes. In addition, miR‐21 was the leading hub interacting with a wide range of TFs.
Fig. 1.
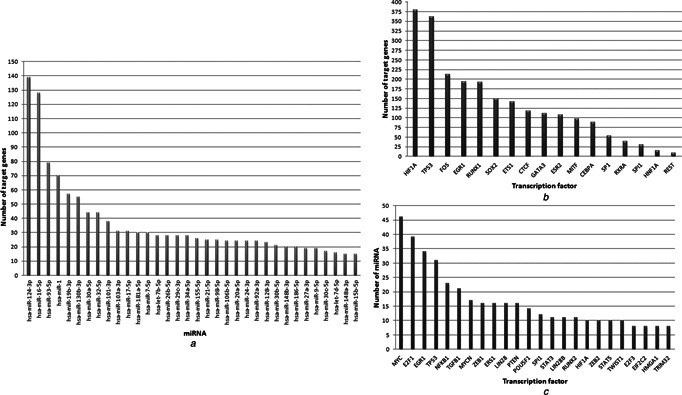
Topological analysis of the transcriptional regulatory network
a Hub miRNAs and their target genes
b Hub TFs of miRNA targeted genes
c Hub TFs and associated miRNAs
Table 2.
Topological features of the generic, core and ovarian cancer specific transcriptional regulatory networks
| Network type | No. of nodes | No. of edges | Hubs | Network heterogeneity | Characteristic path length | Avg. degree |
|---|---|---|---|---|---|---|
| Generic network | ||||||
| TF–miRNA interaction | 409 | 670 | MYC, EGR1, E2F1, TP53, miR‐21 | 1.26 | 5.07 | 3.27 |
| TF–gene interaction | 3801 | 6071 | HIF1A, TP53, FOS, STAT1, RUNX1, EGR1 | 5.78 | 3.87 | 3.19 |
| miRNA–gene interaction | 1469 | 1881 | miR‐124‐3p, miR‐16‐5p, miR‐93‐5p, miR‐1, miR‐19b‐3p, miR‐130b‐3p | 2.73 | 5.35 | 2.56 |
| Whole generic network | 5156 | 8622 | HIF1A, TP53, EGR1, FOS, RUNX1 | 4.50 | 4.55 | 2.84 |
| Core network | ||||||
| TF–miRNA interaction | 215 | 326 | MYC, EGR1, E2F1, NFKB1, TP53 | 1.74 | 4.33 | 2.57 |
| TF–gene interaction | 197 | 239 | HIF1A, TP53, RUNX1, EGR1, FOS, SP1 | 2.12 | 4.51 | 2.29 |
| miRNA–gene interaction | 282 | 272 | miR‐93‐5p, miR‐16‐5p, miR‐124‐3p, miR‐19b‐3p, miR‐29c‐3p, VEGFA | 1.01 | 6.64 | 1.93 |
| Whole core network | 461 | 757 | HIF1A, EGR1, TP53, E2F1, MYC | 1.66 | 4.27 | 3.28 |
| Ovarian cancer specific subnetwork | ||||||
| TF–miRNA interaction | 112 | 225 | MYC, NFKB1, TGFB1, miR‐21, miR‐155, miR‐200c, miR‐200b | 1.07 | 4.08 | 3.16 |
| TF–gene interaction | 1121 | 1291 | HIF1A, TP53, EGR1, ETS1, GATA3, SP1 | 6.9 | 3.74 | 2.3 |
| miRNA–gene interaction | 69 | 65 | miR‐30a‐5p, miR 429, miR‐206, miR142‐3p, miR‐30e‐ 5p, LIMCH1 | 2.78 | 2.06 | 1.88 |
| Whole network | 1277 | 1611 | HIF1A, TP53, EGR1, ETS1, GATA3 | 6.24 | 3.78 | 2.44 |
3.2 Network motifs provide a deeper investigation into the topological architecture
The reconstructed generic transcriptional regulatory network was consisting of different regulatory interaction types, and therefore indicated a complex structure. Uncovering the structural design principles of this complex network might help us to understand the role of transcriptional regulatory components. For this purpose, we utilised the concept of network motifs [19, 53], i.e. the recurrent and statistically significant patterns that occur significantly higher frequencies than expected. These patterns give important clues on the functional abilities of the network, i.e. how particular functions are achieved efficiently, and how networks oversee different tasks.
To investigate the possible regulatory scenarios describing the roles of regulatory elements in regulation of gene transcription, a core network consisting of miRNA–TF‐target gene associations was required. The core network, consisting of 757 interactions associated with 44 TFs, 240 miRNAs and 179 target genes, was reconstructed around the mutual TFs of the TF–gene and miRNA–TF interactions as well as the mutual miRNAs of the miRNA–gene and miRNA–TF interactions (Fig. 2, Table S6). Significantly enriched motifs were determined (Fig. 3) and analysed to provide a deeper investigation into the architecture of miRNA–TF–gene interactions.
Fig. 2.
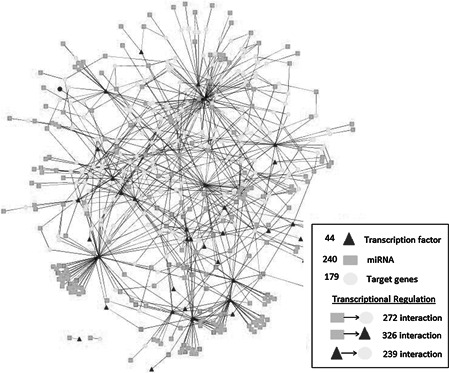
Core transcriptional regulatory network
Fig. 3.
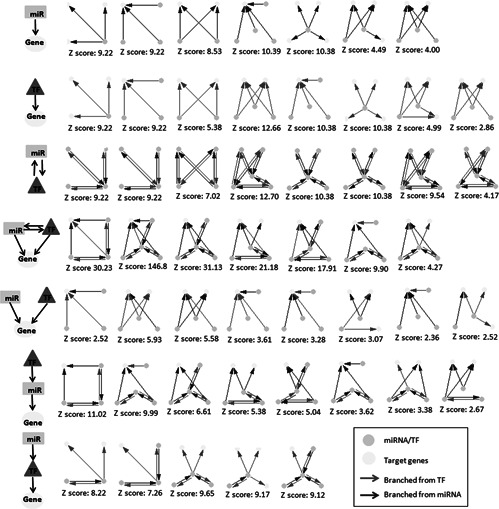
Significantly enriched network motifs
A common limitation of biological network analysis is that currently reported biological networks are far from completeness. Therefore, taking into consideration the hypothesis that the observations may be resulted from data incompleteness, we performed a sensitivity analysis via either removal of 5% true interactions or addition of 5% false interactions in the reconstructed core network to test the robustness of the findings in motif analysis. As a result, the main results remain unchanged (Table S7), i.e. the significantly enriched motifs were also observed within the randomly generated networks with high Z scores (≥2) and observation frequencies (≥0.001%), and no statistically significant changes were observed in their Z scores (p ‐value >0.05).
When significantly enriched motifs were investigated, in general, the number of regulations originated from miRNAs was pretty much the same with that from TFs (161 and 152, respectively). Motifs describing gene regulation by either TFs or miRNAs were describing a well‐balanced distribution, i.e. the upstream regulators of 28 cases were miRNAs, whereas 29 regulations were originated from TFs. On the other hand, in motifs describing cooperative regulation of target genes by miRNAs and TFs, the number of regulations originated from miRNAs was higher than that from TFs (48 and 36, respectively). These results suggest that mutual TF–miRNA regulation come into prominence in transcriptional regulatory network. Also, regulation of target genes by miRNAs has got a crucial ratio in transcriptional regulation mechanisms.
Several features observed in transcriptional factor networks were repeated in the miRNA landscape (Fig. 3). Fifteen motifs were describing gene regulation by either TFs or miRNAs (with observation frequencies between 2.86 and 12.66), and eight motifs were consisting of miRNA–TF interactions (with observation frequencies between 4.17 and 12.7). These motifs represented three different topologies (Fig. 4 a): (i) pleiotropic structure (single regulatory element regulating several targets), (ii) multi‐input structure (several regulatory elements regulating single target) and (iii) super structure (several regulatory elements regulating several targets). On the other hand, four different gene regulatory scenarios were observed (Fig. 4 b) when we thoroughly investigated the network motifs including miRNA–TF–gene regulations: (i) interactive cooperation of miRNAs and TFs (7 motifs with significantly high observation frequencies up to 146.77), (ii) non‐interactive cooperation of miRNAs and TFs (8 motifs with observation frequencies up to 5.93), (iii) hierarchical operation of miRNAs and TFs where miRNAs were the upstream regulators of TFs (5 motifs with observation frequencies around 8.7) and (iv) hierarchical operation of miRNAs and TFs where TFs were the upstream regulators of miRNAs (8 motifs with observation frequencies between 2.67 and 11.02).
Fig. 4.
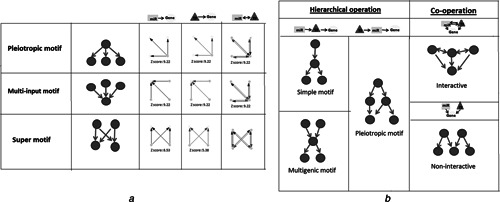
Different gene regulatory scenarios of motifs
A (i) Pleiotropic structure, (ii) multi‐input structure and (iii) super structure of miRNA‐gene and TF‐gene interactions
B (i) Hierarchical operation of miRNAs and TFs where miRNAs were the upstream regulators of TFs, (ii) hierarchical operation of miRNAs and TFs where TFs were the upstream regulators of miRNAs, (iii) interactive cooperation of miRNAs and TFs, (iv) non‐interactive cooperation of miRNAs and TFs
3.3 Core network topology endorses the previous findings on miRNA and gene interactions
Genome‐wide studies (i.e. ChIP‐on‐Chip and ChIP‐seq) on human TFs show that they bind to a remarkably widespread, overlapping set of genomic regions [54] and transcriptional regulation of many genes by a single TF is the prominent motif of TF networks [19]. One observation was that, similar to TFs, a single miRNA has the potential to target many genes (pleiotropic motif in Fig. 4 a). For example, miR‐124‐3p and miR‐16‐5p are targeting 139 and 128 genes, respectively. Also it was determined that 135 miRNAs (75.8% of all) regulate two or more genes. The coordinated control of many genes by a miRNA should be explained by the observation that mammalian miRNAs do not require perfect base pair complementarity with cognate mRNA sites [55]; thereby one miRNA can target many genes with no overall sequence homology.
Analogous to transcription regulation where several TFs tend to work in synergism through assembly as a protein complex to regulate the promoters and enhancers of target genes, we observed multi‐input motifs of miRNAs, i.e. a single gene was targeted by several miRNAs (Fig. 4 a). For instance, vascular endothelial growth factor (VEGFA), which is responsible for programmed angiogenic processes during menstrual cycle, is regulated by 12 miRNAs. However, 378 genes (29.3% of all) are found to be regulated by more than one miRNA and a gene is regulated by 2.6 miRNAs, in average. Since the recognition sites of mRNAs vary considerably in length and sequence, they carry numerous potential binding sites for the same or different miRNAs. Just as TFs recognise short sequences, miRNAs likewise utilise sequence complementarity to its target for recognition. This specificity‐determining region is often short, comprising seven or eight nucleotides [56].
Another recapitulated feature of TFs is the interwind structure of their subnetworks. A miRNA subnetwork was often linked and interlocked with several subnetworks centred upon other miRNAs (Fig. 2). This was mainly due to sharing common target genes (i.e. multi‐input and super motifs, as represented in Fig. 4 a). This result presents the challenging task of understanding how the targeting of multiple genes by a single miRNA, and how the intersection of networks can lead to a cellular effect.
3.4 Target genes may be regulated in cooperation of regulators
The simplest motif of co‐regulation was observed when a common gene is coordinately targeted by a TF and a miRNA (Fig. 4 b). For instance, a common downstream target gene may be regulated by an upstream TF and repressed by a miRNA, which represents a frequently reported regulatory strategy in the literature [29]. If the TF is a repressor, the overall function of the motif is to silence the target, and this represents a coherent motif since both upstream regulators act synergistically in the same direction. In contrast, the TF may be an activator, and the negative interaction between the transcription activator and miRNA creates an incoherent motif [57].
The cooperation of miRNAs and TFs may occur in an interactive manner where miRNAs and TFs form interconnected loops whereby each mutually regulates the other, in addition to common downstream targets (Fig. 4 b). For instance, this type of a motif may comprise of a miRNA which represses target genes, and at the same time, silences a transcription activator for these common downstream targets. In contrast, there may not be an interaction between upstream regulators, the target genes of miRNAs and TFs may be mutual. The interactive cooperation of miRNAs and TFs came into prominence in the human network with seven motifs having the highest observation frequencies, when compared to other cases (p ‐value < 0.05).
Another important observation on cooperative motifs was that a TF was interacting with at least two miRNAs in gene regulation. Multiple inputs as exemplified by TFs provide positive signals for ensuring consistent activity in regulating a common gene. This renders it insensitive to transient changes in individual input strength [58]. Similar to TF based feed‐forward loops comprised of activators and repressors; the integration of miRNAs may provide a negative input thus enabling the circuit to behave as a sensor that can respond to the balance of signals [59].
3.5 Target gene may be regulated by multiple upstream effectors in a hierarchical operation
In regulation of a target gene, in 13 motifs, the upstream regulators also have an impact on each other, which is reminiscent of a hierarchical operation. The control of a downstream target may occur through the synergism of upstream factors, where one of the factors is also the upstream of the other.
The hierarchical operation of miRNAs and TFs where TFs were the upstream regulators of miRNAs was represented by eight motifs (Fig. 3). For example, the upstream TF could activate the production of a miRNA and simultaneously repress the transcription of a common target gene, or the upstream factor could repress the production of a miRNA, whilst activating a target gene which is repressed by the miRNA. This design could minimise the effects of leaky gene transcription when their expression is not desired [57]. On the other hand, the hierarchical operation where miRNAs were the upstream regulators of TFs was also represented with five enriched motifs having significantly higher observation frequencies when compared to the hierarchical operation with TFs as the upstream regulators (p ‐value <0.05). These motifs may represent the silencing of a transcription activator by miRNAs. This result presents the challenging task of understanding the regulatory roles of miRNAs, especially in regulation of TFs.
A considerable observation was that the hierarchical operation of miRNAs and TFs where TFs were the upstream regulators of miRNAs was represented by only pleiotropic motifs, employing single TF (Fig. 4 b). On the other hand, the hierarchical operation where miRNAs were the upstream regulators of TFs was represented by either simple or multi‐input motifs. The abundance of multi‐input motifs pointed out the combinatorial interaction among miRNAs, which is likely to allow for more accurate control of translation rates. As observed with TFs, combinatorial control allows multiple inputs, represented by individual miRNAs, to be integrated into the post‐transcriptional regulation of target mRNAs. Understanding the extent of combinatorial interactions among miRNAs will provide clues toward the differential control of protein expression.
3.6 Process‐specific subnetworks were also dominated by hierarchical operation of regulators
To understand how regulatory networks were rewired in a process specific manner, gene set enrichment analysis was performed for the target genes in the generic network and process‐specific subnetworks were reconstructed for the top five enriched processes, namely metabolic process, regulation of transcription, cell death, cell cycle and reproductive process (Fig. 5, Table S8). The abundance of miRNAs around a small number of TFs was remarkable in all subnetworks. For instance, VEGFA, which is responsible for programmed angiogenic processes during menstrual cycle [60, 61] was one of the hubs in reproductive process‐specific subnetwork and interacting with 12 miRNAs (Fig. 5 e). A deeper investigation into the topological architecture of subnetworks pointed out the dominance of multi‐input motifs representing the hierarchical operation where miRNAs were the upstream regulators of TFs.
Fig. 5.
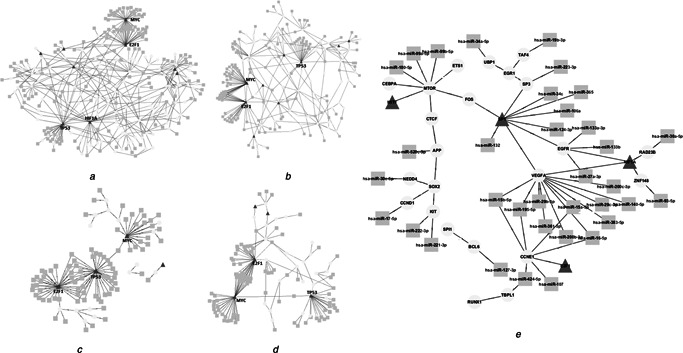
Process‐specific subnetworks for the significantly enriched processes
a Metabolic process
b Regulation of transcription
c Cell death
d Cell cycle and
e Reproductive process
3.7 Case study: ovarian cancer
Integration of functional genomics datasets with the reconstructed network allows us to understand dynamic aspects of transcriptional regulation mechanisms. Here, transcriptomics datasets associated with ovarian cancer were employed as a case study to elucidate the active mechanisms of transcriptional regulation in ovarian cancer based on gene and miRNA expression levels. We determined DEGs and DEmiRs in ovarian cancer, and constructed an ovarian cancer specific subnetwork consisting of 1611 interactions among 34 miRNAs, 90 TFs and 1153 DEG targets (Fig. 6 a). As the regulators, the TFs HIF1A, TP53, EGR1, ETS1 and GATA3 as well as the miRNAs miR‐30a‐5p, miR‐429, miR‐206, miR‐142‐3p and miR‐30e‐5p dominated the disease‐specific subnetwork regulating the excessive number of genes. The hub TFs HIF1A, TP53, EGR1, ETS1 and GATA3 have been implicated in tumorigenesis [62, 63]. Among them, EGR1 and HIF1A [64, 65] were TFs specifically associated with ovarian cancer. In addition, miR‐21, miR‐155, miR‐200c and miR‐200b came into prominence as co‐regulators of TFs. miR‐21 and miR‐200 induce mesenchymal‐to‐epithelial transition, in other words, they have important regulatory functions at metastasis of cancer cells [66, 67]. Upregulated expression of miR‐155 at tumour initiating cells was shown and this miRNA was associated with cancer promotion [68]. These miRNAs and TFs have critical impact on cancer progression and to an extent, cancer phenotype as a result of a failure on cell cycle mechanisms.
Fig. 6.
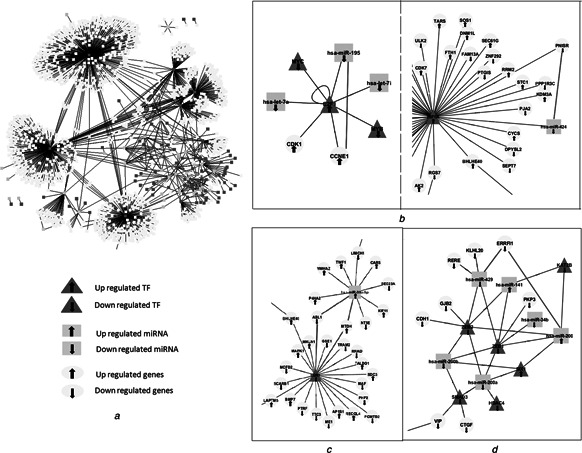
Case study: ovarian cancer
a Ovarian cancer specific subnetwork
b Interactive cooperation examples
c Non‐interactive manner examples
d Hierarchical operations examples on the lessons derived from motif analysis
The subgraphs of this network represent us several examples on the lessons derived from motif analysis. For instance, the regulation of cyclin E1 (CCNE1) gene in ovarian cancer was mediated via the interactive cooperation of miR‐195 and E2F TF 1 (E2F1). The TF (E2F1) and target gene (CCNE1) were both up‐regulated, whereas the expression of the miR‐195 was down‐regulated. This result suggests that TF may positively regulate the transcription of target gene; however, miRNA may regulate gene expression as a repressor. This subgraph, named as feed forwards loop, was detected in previous studies [22, 63]. On the other hand, the regulation of PNISR gene encoding PNN‐interacting serine/arginine‐rich protein was also mediated through an interactive cooperation, but this time the gene and its upstream regulator miR‐424 were down‐regulated whereas the TF HIF1A was up‐regulated (Fig. 6 b). In this scenario, preference of positive correlation between miRNA‐target genes was shown and TF may function as a repressor in gene regulation. The transcriptional regulator EGR1 and miR‐30a‐5p cooperate in a non‐interactive manner in regulation of ABL1 and MTDH genes (Fig. 6 c). Here, EGR1 and ABL1 were down‐regulated, whereas miR‐30a‐5p and MTDH were up‐regulated in ovarian cancer. It was observed that positive and negative correlations occur in the active subnetwork, and therefore the expression profiles of regulatory components should be evaluated to understand the mechanism. The miRNA–TF–gene hierarchy was the dominated strategy in gene regulatory network of ovarian cancer (Fig. 6 d). For example, the regulation of PKP3 gene in ovarian cancer was mediated via hierarchical operation where several miRNAs were the upstream regulators of the TF ZEB1. In addition, almost all transcriptional regulatory components as well as target genes were down regulated in this active subnetwork.
Both cooperation and hierarchical operation types were determined in active subnetworks. Overall results provided that TFs and miRNAs may mostly tend to co‐regulate their target genes (in an interactive manner) and transcriptional regulations of target genes may occur through hierarchical mechanisms where miRNAs were the upstream regulators of TFs. The important discovery from this study is that TFs pick up signals from multiple inputs (miRNAs) and provide positive signals for ensuring consistent activity in regulating a common gene like a sensor. TFs and miRNAs in the transcriptional regulation mechanisms are relevant components and their regulatory effect (activator or supressor) are complicated depend on the mechanism robustness.
Our results provide a new perspective on analysing the possible functions of miRNAs, as well as deeper understanding of transcriptional regulatory mechanisms. Yu et al. [69] previously reported two classes of miRNAs with distinct network topological properties. The first class of miRNAs is regulated by a large number of TFs, whereas the second is regulated by only a few TFs. Recently, it is presented that extensive context dependency are widespread in miRNA‐mediated gene regulation, similar to TF‐mediated regulation, implying a much more complex regulatory network than is currently known [70]. Also, studies based on computational predictions mostly presented an overestimation of the number of predicted miRNA regulators so these perspectives may be added to the increasing complexity of miRNA‐mediated regulation. In this study, human transcriptional regulatory network was curated with experimentally known data. In addition, the necessary tools to illuminate transcriptional regulation mechanisms need to be much more involved, and much more experimentally validated data will be needed in order to understand possible miRNA–TF regulation mechanism and post‐transcriptional regulatory network.
4 Conclusions
In this work, we aimed to examine the cooperation scenarios of miRNAs and TFs, and for this purpose we mapped a comprehensive transcription regulatory network in H. sapiens comprised of interactome data concerning to TFs, miRNAs and their target genes. The essential findings of this study may be summarised as follows: (i) miRNA is an inseparable component of the overall gene regulatory fabric; (ii) most of the TFs play general roles in transcriptional regulatory mechanisms; however, miRNAs take more specific duties; (iii) mutual TF–miRNA regulation (interactive cooperation) and hierarchical operation where miRNAs were the upstream regulators of TFs come into prominence in transcriptional regulatory network; (iv) multiple inputs as exemplified by TFs provide positive signals for ensuring consistent activity in regulating a common gene; and (v) the hierarchical operation where TFs were the upstream regulators of miRNAs was represented by only pleiotropic motifs, employing single TF.
Whole results ensured that TFs and miRNAs may mostly regulate their target genes in an interactive manner and gene expression regulations of target genes may occur through hierarchical mechanisms where miRNAs were the upstream regulators of TFs. These findings may assist researchers in understanding complex gene expression mechanism and miRNA functions toward system biology studies. In the future work, it will be investigated the various tissues or conditions specific transcriptional regulatory networks depend on expression data and establishment of TFs and miRNAs precise regulatory roles.
Supporting information
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
5 References
- 1. Mercer T.R. Mattick J.S.: ‘Understanding the regulatory and transcriptional complexity of the genome through structure ’, Genome Res., 2013, 23, (7), pp. 1081–1088 (doi: 10.1101/gr.156612.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yu H. Gerstein M.: ‘Genomic analysis of the hierarchical structure of regulatory networks ’, Proc. Natl. Acad. Sci., 2006, 103, (40), pp. 14724–14731 (doi: 10.1073/pnas.0508637103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang Y. Shen X.J. Zou Q. et al.: ‘Biological functions of microRNAs: a review ’, J. Stress Physiol. Biochem., 2011, 67, (1), pp. 129–139 (doi: 10.1007/s13105-010-0050-6) [DOI] [PubMed] [Google Scholar]
- 4. Croce C.M.: ‘Causes and consequences of microRNA dysregulation in cancer ’, Nat. Rev. Genet., 2009, 10, (10), pp. 704–714 (doi: 10.1038/nrg2634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bartel D.P.: ‘MicroRNAs: genomics, biogenesis, mechanism, and function ’, Cell, 2004, 116, (2), pp. 281–297 (doi: 10.1016/S0092-8674(04)00045-5) [DOI] [PubMed] [Google Scholar]
- 6. Filipowicz W. Jaskiewicz L. Kolb F.A. et al.: ‘Post‐transcriptional gene silencing by siRNAs and miRNAs ’, Curr. Opin. Struct. Biol., 2005, 15, (3), pp. 331–341 (doi: 10.1016/j.sbi.2005.05.006) [DOI] [PubMed] [Google Scholar]
- 7. Hobert O.: ‘Gene regulation by transcription factors and microRNAs ’, Science, 2008, 319, (5871), pp. 1785–1786 (doi: 10.1126/science.1151651) [DOI] [PubMed] [Google Scholar]
- 8. Liang C. Li Y. Luo J. et al.: ‘A novel motif‐discovery algorithm to identify co‐regulatory motifs in large transcription factor and microRNA co‐regulatory networks in human ’, Bioinformatics, 2015, 31, (14), pp. 2348–2355 (doi: 10.1093/bioinformatics/btv159) [DOI] [PubMed] [Google Scholar]
- 9. Calin G.A. Sevignani C. Dumitru C.D. et al.: ‘Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers ’, PNAS, 2004, 101, (9), pp. 2999–3004 (doi: 10.1073/pnas.0307323101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iorio M.V. Croce C.M.: ‘MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review ’, EMBO Mol. Med., 2012, 4, (3), pp. 143–159 (doi: 10.1002/emmm.201100209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang B. Pan X. Cobb G.P. et al.: ‘microRNAs as oncogenes and tumor suppressors ’, Dev. Biol., 2007, 302, (1), pp. 1–12 (doi: 10.1016/j.ydbio.2006.08.028) [DOI] [PubMed] [Google Scholar]
- 12. Shenouda S.K. Alahari S.K.: ‘MicroRNA function in cancer: oncogene or a tumor suppressor? ’, Cancer Metastasis Rev., 2009, 28, (3), pp. 369–378 (doi: 10.1007/s10555-009-9188-5) [DOI] [PubMed] [Google Scholar]
- 13. Ambros V.: ‘The functions of animal microRNAs ’, Nature, 2004, 431, pp. 350–355 (doi: 10.1038/nature02871) [DOI] [PubMed] [Google Scholar]
- 14. Wang B. Love T.M. Call M.E. et al.: ‘Recapitulation of short RNA‐directed translational gene silencing in vitro ’, Mol. Cell., 2006, 22, (4), pp. 553–560 (doi: 10.1016/j.molcel.2006.03.034) [DOI] [PubMed] [Google Scholar]
- 15. Petersen C.P. Bordeleau M.E. Pelletier J. et al.: ‘Short RNAs repress translation after initiation in mammalian cells ’, Mol. Cell., 2006, 21, (4), pp. 533–542 (doi: 10.1016/j.molcel.2006.01.031) [DOI] [PubMed] [Google Scholar]
- 16. Vasudevan S. Tong Y. Steitz J.A.: ‘Switching from repression to activation: microRNAs can up‐regulate translation ’, Science, 2007, 318, (5858), pp. 1931–1934 (doi: 10.1126/science.1149460) [DOI] [PubMed] [Google Scholar]
- 17. Liu N. Williams A.H. Kim Y. et al.: ‘An intragenic MEF2‐dependent enhancer directs muscle‐specific expression of microRNAs 1 and 133 ’, PNAS, 2007, 104, (52), pp. 20844–20849 (doi: 10.1073/pnas.0710558105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barabasi A.L. Oltvai Z.N.: ‘Network biology: understanding the cell's functional organization ’, Nat. Rev. Genet., 2004, 5, (2), pp. 101–113 (doi: 10.1038/nrg1272) [DOI] [PubMed] [Google Scholar]
- 19. Milo R. Shen‐Orr S. Itzkovitz S. et al.: ‘Network motifs: simple building blocks of complex networks ’, Science, 2002, 298, (5594), pp. 824–827 (doi: 10.1126/science.298.5594.824) [DOI] [PubMed] [Google Scholar]
- 20. Grochow J.A. Kellis M.: ‘Network motif discovery using subgraph enumeration and symmetry‐breaking ’, RECOMB, Springer‐Verlag, Heidelberg, 2007, pp. 92–106 [Google Scholar]
- 21. Martinez N.J. Walhout A.J.: ‘The interplay between transcription factors and microRNAs in genome‐scale regulatory networks ’, Bioessays, 2009, 31, (4), pp. 435–445 (doi: 10.1002/bies.200800212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsang J. Zhu J. van Oudenaarden A.: ‘MicroRNA‐mediated feedback and feedforward loops are recurrent network motifs in mammals ’, Mol. Cell., 2007, 26, (5), pp. 753–767 (doi: 10.1016/j.molcel.2007.05.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gennarino V.A. D'Angelo G. Dharmalingam G. et al.: ‘Identification of microRNA‐regulated gene networks by expression analysis of target genes ’, Genome Res., 2012, 22, pp. 1163–1172 (doi: 10.1101/gr.130435.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu H. Tu K. Wang Y.J. et al.: ‘Combinatorial network of transcriptional regulation and microRNA regulation in human cancer ’, BMC Syst. Biol., 2012, 6, (1), p. 61 (doi: 10.1186/1752-0509-6-61) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sengupta D. Bandyopadhyay S.: ‘Topological patterns in microRNA–gene regulatory network: studies in colorectal and breast cancer ’, Mol. BioSyst., 2013, 9, (6), pp. 1360–1371 (doi: 10.1039/c3mb25518b) [DOI] [PubMed] [Google Scholar]
- 26. Calimlioglu B. Karagoz K. Sevimoglu T. et al.: ‘Tissue‐specific molecular biomarker signatures of Type 2 diabetes: an integrative analysis of transcriptomics and protein–protein interaction data ’, Omics, 2015, 19, (9), pp. 563–573 (doi: 10.1089/omi.2015.0088) [DOI] [PubMed] [Google Scholar]
- 27. Shalgi R. Lieber D. Oren M. et al.: ‘Global and local architecture of the mammalian microRNA‐transcription factor regulatory network ’, PLoS Comput. Biol., 2007, 3, (7), p. e131 (doi: 10.1371/journal.pcbi.0030131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin C.C. Chen Y.J. Chen C.Y. et al.: ‘Crosstalk between transcription factors and microRNAs in human protein interaction network ’, BMC Syst. Biol., 2012, 6, p. 18 (doi: 10.1186/1752-0509-6-18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y. Ferguson J. Chang J.T. et al.: ‘Inter‐and intra‐combinatorial regulation by transcription factors and microRNAs ’, BMC Genomics, 2007, 8, (1), p. 396 (doi: 10.1186/1471-2164-8-396) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qiu C. Wang J. Yao P. et al.: ‘microRNA evolution in a human transcription factor and microRNA regulatory network ’, BMC Syst. Biol., 2010, 4, p. 90 (doi: 10.1186/1752-0509-4-90) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo A.Y. Sun J. Jia P. et al.: ‘A novel microRNA and transcription factor mediated regulatory network in schizophrenia ’, BMC Syst. Biol., 2010, 4, p. 10 (doi: 10.1186/1752-0509-4-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen C.Y. Chen S.T. Fuh C.S. et al.: ‘Coregulation of transcription factors and microRNAs in human transcriptional regulatory network ’, BMC Bioinf., 2011, 12, (Suppl 1), p. S41 (doi: 10.1186/1471-2105-12-S1-S41) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun J. Gong X. Purow B. et al.: ‘Uncovering microRNA and transcription factor mediated regulatory networks in glioblastoma ’, PLoS Comput. Biol., 2012, 8, p. e1002488 (doi: 10.1371/journal.pcbi.1002488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang X.: ‘miRDB: a microRNA target prediction and functional annotation database with a wiki interface ’, RNA, 2008, 14, pp. 1012–1017 (doi: 10.1261/rna.965408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang J.H. Li J.H. Shao P. et al.: ‘starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute CLIP‐Seq and Degradome‐Seq data ’, Nucleic Acids Res., 2011, 39, (Suppl 1), pp. D202–D209 (doi: 10.1093/nar/gkq1056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao F. Zuo Z. Cai G. et al.: ‘miRecords: an integrated resource for microRNA‐target interactions ’, Nucleic Acids Res., 2009, 37, (Suppl 1), pp. D105–D110 (doi: 10.1093/nar/gkn851) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsu S.D. Tseng Y.T. Shrestha S. et al.: ‘miRTarBase update 2014: an information resource for experimentally validated miRNA‐target interactions ’, Nucleic Acids Res., 2014, 42, (D1), pp. D78–D85 (doi: 10.1093/nar/gkt1266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang Q. Wang Y. Hao Y. et al.: ‘miR2Disease: a manually curated database for microRNA deregulation in human disease ’, Nucleic Acids Res., 2009, 37, (Suppl 1), pp. D98–D104 (doi: 10.1093/nar/gkn714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang J. Lu M. Qiu C. et al.: ‘TransmiR: a transcription factor–microRNA regulation database ’, Nucleic Acids Res., 2010, 38, (Suppl 1), pp. D119–D122 (doi: 10.1093/nar/gkp803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Portales‐Casamar E. Arenillas D. Lim J. et al.: ‘The PAZAR database of gene regulatory information coupled to the ORCA toolkit for the study of regulatory sequences ’, Nucleic Acids Res., 2009, 37, (Suppl 1), pp. D54–D60 (doi: 10.1093/nar/gkn783) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Assenov Y. Ramírez F. Schelhorn S.E. et al.: ‘Computing topological parameters of biological networks ’, Bioinformatics, 2008, 24, (2), pp. 282–284 (doi: 10.1093/bioinformatics/btm554) [DOI] [PubMed] [Google Scholar]
- 42. Smoot M.E. Ono K. Ruscheinski J. et al.: ‘Cytoscape 2.8: new features for data integration and network visualization ’, Bioinformatics, 2011, 27, (3), pp. 431–432 (doi: 10.1093/bioinformatics/btq675) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karagoz K. Sinha R. Arga K.Y.: ‘Triple negative breast cancer: a multi‐omics network discovery strategy for candidate targets and driving pathways ’, Omics, 2015, 19, (2), pp. 115–130 (doi: 10.1089/omi.2014.0135) [DOI] [PubMed] [Google Scholar]
- 44. Wernicke S.: ‘Efficient detection of network motifs ’, IEEE/ACM Trans. Comput. Biol., 2006, 3, (4), pp. 347–359 (doi: 10.1109/TCBB.2006.51) [DOI] [PubMed] [Google Scholar]
- 45. Huang D.W. Sherman B.T. Lempicki R.A.: ‘Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists ’, Nucleic Acids Res., 2009, 37, (1), pp. 1–13 (doi: 10.1093/nar/gkn923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barrett T. Wilhite S.E. Ledoux P. et al.: ‘NCBI GEO: archive for functional genomics data sets‐update ’, Nucleic Acids Res., 2013, 41, (Suppl 1), pp. D991–D995 (doi: 10.1093/nar/gks1193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bolstad B.M. Irizarry R.A. Åstrand M. et al.: ‘A comparison of normalization methods for high density oligonucleotide array data based on variance and bias ’, Bioinformatics, 2003, 19, (2), pp. 185–193 (doi: 10.1093/bioinformatics/19.2.185) [DOI] [PubMed] [Google Scholar]
- 48. Gautier L. Cope L. Bolstad B.M. et al.: ‘affy – analysis of Affymetrix GeneChip data at the probe level ’, Bioinformatics, 2004, 20, (3), pp. 307–315 (doi: 10.1093/bioinformatics/btg405) [DOI] [PubMed] [Google Scholar]
- 49. Gentleman R.C. Carey V.J. Bates D.M. et al.: ‘Bioconductor: open software development for computational biology and bioinformatics ’, Genome Biol., 2004, 5, (10), p. R80 (doi: 10.1186/gb-2004-5-10-r80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smyth G.K.: ‘Limma: linear models for microarray in bioinformatics and computational biology solutions using R and bioconductor ’ (Springer, 2005), pp. 397–420 [Google Scholar]
- 51. Le T.D. Liu L. Liu B. et al.: ‘Inferring microRNA and transcription factor regulatory networks in heterogeneous data ’, BMC Bioinf., 2013, 14, (92), doi: 10.1186/1471‐2105‐14‐92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu Z.‐P. Wu C. Miao H. et al.: ‘RegNetwork: an integrated database of transcriptional and post‐transcriptional regulatory networks in human and mouse ’, 2015, Database (Oxford), 2015, 2015, pp. 1–12, doi: 10.1093/database/bav095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shen‐Orr S.S. Milo R. Mangan S. et al.: ‘Network motifs in the transcriptional regulation network of Escherichia coli ’, Nat. Genet., 2002, 31, (1), pp. 64–68 (doi: 10.1038/ng881) [DOI] [PubMed] [Google Scholar]
- 54. Todeschini A.L. Georges A. Veitia R.A.: ‘Transcription factors: specific DNA binding and specific gene regulation ’, Trends Genet., 2014, 30, (6), pp. 211–219 (doi: 10.1016/j.tig.2014.04.002) [DOI] [PubMed] [Google Scholar]
- 55. Grosswendt S. Filipchyk A. Manzano M. et al.: ‘Unambiguous identification of miRNA: target site interactions by different types of ligation reactions ’, Mol. Cell., 2014, 54, (6), pp. 1042–1054 (doi: 10.1016/j.molcel.2014.03.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lewis B.P. Shih I.H. Jones‐Rhoades M.W. et al.: ‘Prediction of mammalian microRNA targets ’, Cell, 2003, 115, (7), pp. 787–798 (doi: 10.1016/S0092-8674(03)01018-3) [DOI] [PubMed] [Google Scholar]
- 57. Liu E.T. Lauffenburger D.A.: ‘Systems biomedicine: concepts and perspectives ’ (Academic Press, 2009) [Google Scholar]
- 58. Mangan S. Alon U.: ‘Structure and function of the feed‐forward loop network motif ’, PNAS, 2003, 100, (21), pp. 11980–11985 (doi: 10.1073/pnas.2133841100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hornstein E. Shomron N.: ‘Canalization of development by microRNAs ’, Nat Genet., 2006, 38, pp. S20–S24 (doi: 10.1038/ng1803) [DOI] [PubMed] [Google Scholar]
- 60. Gutman G. Barak V. Maslovitz S. et al.: ‘Regulation of vascular endothelial growth factor‐A and its soluble receptor sFlt‐1 by luteinizing hormone in vivo: implication for ovarian follicle angiogenesis ’, Fertil. Steril., 2008, 89, (4), pp. 922–926 (doi: 10.1016/j.fertnstert.2007.03.097) [DOI] [PubMed] [Google Scholar]
- 61. Panda H. Pelakh L. Chuang T.D. et al.: ‘Endometrial miR‐200c is altered during transformation into cancerous states and targets the expression of ZEBs, VEGFA, FLT1, IKKβ, KLF9, and FBLN5 ’, Reprod. Sci., 2012, 19, (8), pp. 786–796 (doi: 10.1177/1933719112438448) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Muller P.A. Vousden K.H.: ‘Mutant p53 in cancer: new functions and therapeutic opportunities ’, Cancer Cell, 2014, 25, (3), pp. 304–317 (doi: 10.1016/j.ccr.2014.01.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lu G. Zhang Q. Huang Y. et al.: ‘Phosphorylation of ETS1 by Src family kinases prevents its recognition by the COP1 tumor suppressor ’, Cancer Cell, 2014, 26, (2), pp. 222–234 (doi: 10.1016/j.ccr.2014.06.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheng J.C. Klausen C. Leung P.C.: ‘Hypoxia‐inducible factor 1 alpha mediates epidermal growth factor‐induced down‐regulation of E‐cadherin expression and cell invasion in human ovarian cancer cells ’, Cancer Lett., 2013, 329, (2), pp. 197–206 (doi: 10.1016/j.canlet.2012.10.029) [DOI] [PubMed] [Google Scholar]
- 65. Cheng J. Chang H. Leung P.: ‘Egr‐1 mediates epidermal growth factor‐induced downregulation of E‐cadherin expression via slug in human ovarian cancer cells ’, Oncogene, 2013, 32, (8), pp. 1041–1049 (doi: 10.1038/onc.2012.127) [DOI] [PubMed] [Google Scholar]
- 66. Han M. Liu M. Wang Y. et al.: ‘Antagonism of miR‐21 reverses epithelial‐mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN ’, PLoS One, 2012, 7, (6), p. e39520 (doi: 10.1371/journal.pone.0039520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McDonald J. Bowen N.J. Wang L.: ‘MIR‐200 family induces mesenchymal‐to‐epithelial transition (MET) in ovarian cancer cells ’. Google Patents, 2014.
- 68. Farooqi A.A. Qureshi M.Z. Coskunpinar E. et al.: ‘MiR‐421, miR‐155 and miR‐650: emerging trends of regulation of cancer and apoptosis ’, Asian Pac. J. Cancer Prev., 2014, 15, pp. 1909–1912 (doi: 10.7314/APJCP.2014.15.5.1909) [DOI] [PubMed] [Google Scholar]
- 69. Yu X. Lin J. Zack D.J. et al.: ‘Analysis of regulatory network topology reveals functionally distinct classes of microRNAs ’, Nucleic Acids Res., 2008, 36, (20), pp. 6494–6503 (doi: 10.1093/nar/gkn712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Erhard F. Haas J. Lieber D. et al.: ‘Widespread context dependency of microRNA‐mediated regulation ’, Genome Res., 2014, 24, (6), pp. 906–919 (doi: 10.1101/gr.166702.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data
Supplementary Data