

Abstract
Background:
Donation after circulatory death donors (DCD) can expand the donor pool for heart transplantation, which primarily depends on brain death donors. Ischemia and reperfusion injury are inherent to the DCD process. We hypothesize that pharmacologic inhibition of interleukin-1 (IL-1) and/or IL-18 is protective to DCD hearts.
Material and Methods:
Following clinical protocol in-situ ischemia time in control beating-heart donor (CBD) and DCD groups was less than 5 and 40 minutes, respectively. Wild type (WT) C57Bl6/j, IL-1 receptor type I knockout (IL-1RI-KO), and IL-18 KO mice were used. Hearts were reanimated for 90 minutes on a Langendorff system with Krebs-Henseleit buffer at 37°C, to assess physiologic parameters. Recombinant IL-1 receptor antagonist (IL-1Ra) and/or IL-18 binding protein (IL-18BP) were added to the Krebs-Henseleit buffer to inhibit IL-1 and/or the IL-18 signaling, respectively.
Results:
Developed pressure and +/−dP/dt were significantly impaired in the DCD-WT group compared to CBD-WT (p=<0.05). Troponin release was higher in DCD-WT groups. Functional parameters were preserved, and troponin release was significantly less in the DCD knockout groups. Heart function was improved in DCD groups treated with IL-1Ra or IL-18BP compared to the DCD-WT group.
Conclusion:
Heart function was significantly impaired in the DCD-WT group compared to CBD-WT. Genetic deletion or pharmacologic blockage of IL-1 or IL-18 was protective to DCD hearts.
Keywords: Heart Transplantation, Ischemia and reperfusion, Donation after circulatory death, IL-1, IL-18
Introduction:
Advanced heart failure (HF) is a serious morbidity that affects over 6 million patients in the USA alone. The ultimate treatment option for patients with advanced HF is heart transplantation (HTx), which currently has a five-year survival of over 78% and is improving over the past three decades1. This remarkable treatment option is not offered to all patients with advanced HF due to the limited availability of donor hearts. Over 20% of listed patients die waiting for heart transplantation1. Expansion of the donor heart pool is urgently needed. Presently the primary source of donor hearts is a donation after brain death (DBD) donors; however, a potential source of additional heart donors could be donation after circulatory death (DCD) donors. Cardiac ischemia is inherent to the DCD process, and hearts from such donors are selectively utilized for HTx in very few centers2–4. However, other organs such as the liver, kidneys, pancreas, and lungs are utilized from DCD donors with good results. These DCD donor organs now comprise up to 50% of all transplanted organs1. Limiting the ischemic damage and the reperfusion injury in a DCD heart could potentially lead to the clinical use of these organs for HTx.
The nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3), inflammasome pathway is activated in response to ischemia/reperfusion5, 6 and our recent work has shown that the NLRP3 inflammasome inhibition prevented the DCD damage and improved the DCD heart function ex vivo7. The NLRP3 inflammasome amplifies the inflammatory-injury through the rapid production of interleukin-1 beta (IL-1β and IL-18). The IL-1β and IL-18 are known to cause cardiomyocyte damage and dysfunction6, 8, 9. In this study, we sought to determine whether the interleukin-1β and/or the IL-18 signaling mediate the DCD-induced ischemia/reperfusion injury and whether their inhibition could be used to mitigate the DCD induced damage.
Methods:
Ethical aspects
All experimental animals were cared in accordance with institutional guidelines and the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 86–23, revised 2011)10. The study was approved by the institutional animal care and use committee of Virginia Commonwealth University.
Experimental animal model
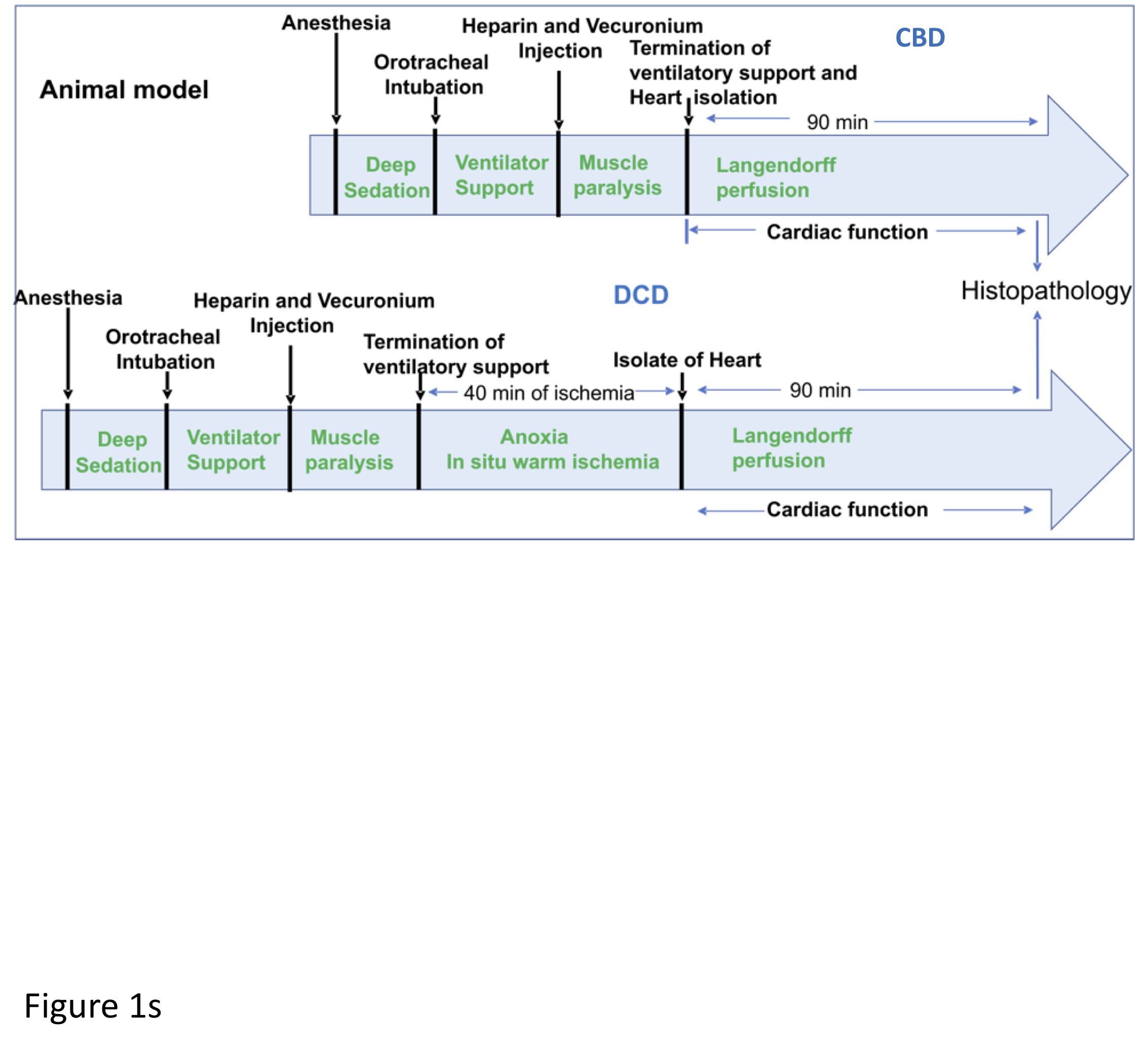
Following clinical protocol, the DCD model was developed. Warm ischemia time was maintained at <5 minutes in control beating-heart donor (CBD) group and at 40 minutes in DCD groups. Figure 1s (supplemental data) shows a schematic of steps and timing for the CBD and DCD hearts. Wild type (WT) C57bl6/j, IL-1 type 1 receptor knock out (IL-RI-KO, strain Il1r1tm1Imx) and IL-18 receptor KO (IL-18-KO, strain Il18tm1Aki) mice were purchased from Jackson Laboratories (Bar Harbor, ME) for this study. Pharmacologic inhibition of IL-1 or IL-18 was obtained with recombinant IL-1 receptor antagonist (IL-1Ra) or IL-18 binding protein (IL-18BP), respectively. IL-1Ra has high specificity as IL-1 inhibitor and has an affinity for the IL-1R of 1.2 μM) (Species-specific binding of IL-1, but not the IL-1 receptor antagonist, by fibroblasts). IL-18BP has a very high affinity for IL-18 (dissociation constant 399 nM)11. IL-18BP can also bind and neutralize the anti-inflammatory cytokine IL-37, which is, however, absent in mice. Adult (10–15 weeks-old) male mice from the 3 strains were randomly assigned to a CBD or a DCD group- CBD-WT, DCD-WT, DCD IL-1RI KO, DCD IL-18 KO (allocation and numbers per group in Table 1). Baseline representative characteristics (body weight, heart weight and rate, fractional shortening) of the 3 strain of mice used are reported in the supplementary table 1s. No differences were noted.
Table 1.
Group allocation, intervention, and number of hearts per group
| CBD n |
DCD n |
|
|---|---|---|
| Wild type (WT) strain C57bl6/j | 14 | 10 |
| IL-1 type 1 receptor knock out (IL-RI KO) strain Il1r1tm1Imx | - | 7 |
| IL-18 Knock Out (IL-18 KO) strain Il18tm1Aki | - | 7 |
| Wild type (WT) early inhibition with IL-1Ra | - | 13 |
| Wild type (WT) early inhibition with IL-18BP | - | 8 |
| Wild type (WT) late inhibition with IL-1Ra | - | 8 |
| Wild type (WT) late inhibition with IL-18BP | - | 9 |
| Wild type (WT) late inhibition with IL-1Ra and IL-18BP | - | 5 |
Additional DCD-WT groups of mice were assigned to study the pharmacologic inhibition of IL-1, IL-18, a combination of IL-1 and IL-18, as well as early and late inhibition. The mice were anesthetized using sodium pentobarbital (100 mg/Kg) administered intraperitoneally, followed by endotracheal intubation for respiratory support. Heart activity was monitored by EKG. Heparin (1000 U/Kg) was administered through the tail vein, followed by the administration of a paralytic agent (vecuronium bromide, 40 mg/kg) intravenously and allowed to circulate for 5 minutes. Mice in the CBD group underwent a thoracotomy, and hearts were procured while beating. The hearts were then perfused on a Langendorff system primed with KH buffer (115 mM NaCl, 4.0 mM KCl, 2.5 mM CaCl2, 26 mM NaHCO3, 1.1 mM MgSO4, 0.9 mM KH2PO4, 5.5 mM glucose, and 5 IU of insulin/liter, oxygenated with 95% O2 – 5% CO2 to maintain a pH of 7.4) at a constant pressure of 76 mmHg at 37°C for 90 minutes. In the DCD groups, the ventilatory support was terminated, leading to respiratory arrest and cardiac asystole. Hearts were left in situ for approximately 30 minutes from the time of asystole, and then the hearts were procured and reanimated on the Langendorff system at 40 minutes from the termination of ventilation (Figure 1 supplemental data).
In addition to these groups, DCD-WT mice were also allocated to study the protective effect of IL-1Ra, or IL-18BP given 30 minutes prior to induction of DCD (Early inhibition) and at the time of reanimation (diluted in KH buffer) or only at the time of reanimation (diluted in KH buffer- Late inhibition). The doses of IL-1 receptor antagonist and IL-18 binding protein were 10 mg/kg and 1 mg/kg, respectively, given intraperitoneally 30 minutes prior to DCD in the early inhibition group. The IL-1Ra and IL-BP, 1μg/ml, and 1μg/ml, respectively, were added to KH buffer in the late inhibition group. These two inhibitory strategies, pre-treatment (30 minutes prior to DCD induction) and post-treatment, at reanimation only, were designed to test whether the IL-1Ra or IL-18BP blockage before the onset of DCD process offers superior protection to the DCD hearts. Doses of IL-1Ra and IL-18BP were chosen based on the literature and on our prior work12–14.
Measurement of cardiac function
With an intraventricular balloon placed across the mitral valve and connected to a pressure transducer and a Powerlab system myocardial function was assessed with following parameters- heart rate (HR), left ventricle developed pressure (LVDP), rate of contractility (+dP/dt), rate of relaxation (−dP/dt). In addition, to account for differences in HR that may affect the LVDP, we calculated the rate pressure product (RPP) by multiplying the HR and LVDP. Measurements were recorded at 0, 15, 30, 60, 75, and 90 minutes intervals, using Labchart 7 (AD Instruments). The results are reported as average function measured over the 90 minutes period.
Measurement of cardiac damage
Troponin I (cTnI) was measured in the eluate using a commercially available ELISA kit (Life Diagnostic) and expressed as ng/ml. The cTnI values are an average of the cTnI released over 90 minutes. To determine the cTnI concentration, we first determined the relationship between the cTnI concentration (on the X-axis) and the absorbance (on the Y-axis) by interpolating the XY values of the cTnI standard curve in a graph. We have then used the fitting line formula to determine the concentration for each heart using the absorbance values. At the end of perfusion, the hearts were placed in formalin, dehydrated, and embedded in paraffin. DNA fragmentation was measured on 5 μm tissue slides using the TUNEL assay (Clontech/Takara Mountain View CA) and expressed as the percentage of TUNEL positive nuclei.
Statistical Analysis
The Kolgonorov-Smirnov test for normality was used to determine the normality of data distribution. When data were not normally distributed, comparisons between multiple groups were performed using the Kruskal–Wallis test followed by the Dunn’s test for multiple comparisons. Continuous variables are expressed as a Min-Max spread and quartiles (in the figures) and Mean and interquartile ranges (in the tables and text). Data with normal distribution were analyzed using ANOVA and were expressed as mean and standard deviation. Discrete variables are presented as percentages. P values <0.05 were considered statistically significant.
Results:
Damage to the hearts from DCD process:
All parameters of myocardial function were impaired in the DCD WT group compared to CBD WT, with a significant decrease in LVDP, RPP, +/− dp/dt (all p values <0.001) (Figure 1 and Table 2). Troponin I release in the eluate was also very high in the DCD WT group compared to the CBD WT group (Table 1 and Figure 2). The DCD WT group showed significantly increased markers of cell death by TUNEL assay compared to the CBD WT group (table 2).
Figure 1.
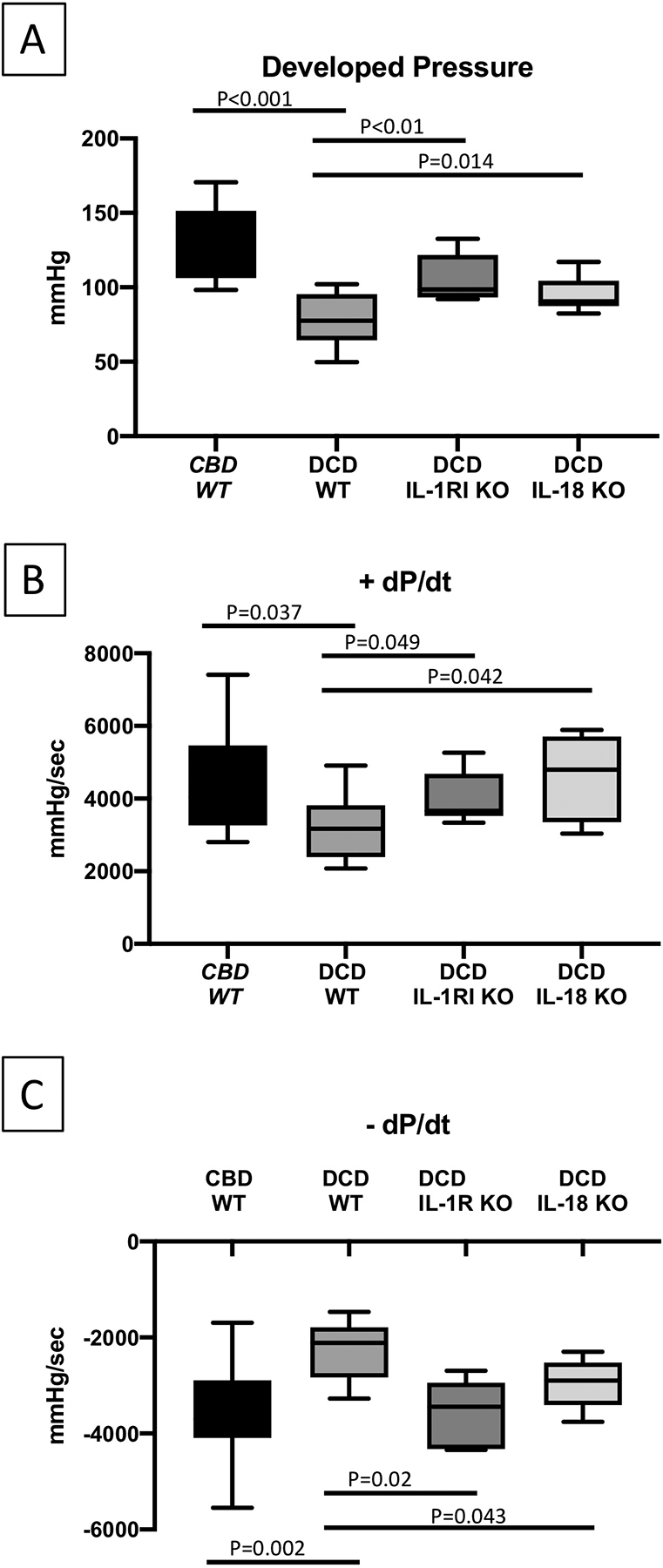
Heart function of reanimated CBD WT, DCD WT, IL-1RI KO DCD and IL-18 KO DCD mice hearts on a Langendorff system. Mean developed pressure (A), +dP/dt (B) and −dP/dt (C), measured with balloon tip catheter placed in left ventricle. CBD=Control Beating-heart Donor; DCD=Donation after Circulatory Death; WT=wild type, IL-1RI KO=Interleukin-1 Receptor type I knock out; IL-18 KO=Interleukin-18 knock-out. Values are expressed as Min-Max spread with quartiles.
Table 2.
Heart function and markers of cell injury in CBD and DCD mice hearts by genetic modification
| CBD-WT Control |
DCD-WT Control |
DCD- IL-1RI KO | DCD- IL-18 KO | |
|---|---|---|---|---|
| N =14 | N=10 | N = 7 | N=7 | |
| Functional Data | ||||
| Heart Rate b/min; Median (Q1-Q3) | 324(308–325) | 399(381–409) | 447(428–465) | 427(383–443) |
| Perfusion rate ml/min; Median (Q1-Q3) | 2.1(2.0–2.3) | 1.9(1.7–2.0) | 2.1(1.8–2.7) | 2.0(1.8–2.4) |
| Developed pressure mmHg; Median (Q1-Q3) | 115(106–145) | 77(74–93)a | 99(94–120)b | 91(81–103)b |
| Rate Pressure Product (HR X LVDP); Median (Q1-Q3) | 44207(32899–51054) | 30744(23091–38180)a | 45239(41382–55474)b | 39439(35486–43182) |
| Rate of positive Developed pressure dt/dp mmHg/ms; Median (Q1-Q3) | 4038(3319–5279) | 3167(2477–3415)a | 3661(3569–4428)b | 4799(3774–5418)b |
| Rate of negative Developed pressure −dt/dp mmHg/ms; Median (Q1-Q3) | −3697(−3925–−2927 | −2113(−2790–−1884)a | −3444(−3972–−3023)b | −2897(−3307–−2605)b |
| Makers of myocyte damage | ||||
| Eluate troponin level ng/ml; Median (Q1-Q3) | 0.49(0.30–0.73) | 7.74(5.67–9.65)a | 0.72(0.51–0.87)b | 5.16(3.27–5.69) |
| TUNEL assay (% of TUNEL+ cells); Median (Q1-Q3) | 0.44(0.16–1.10) | 1.49(1.35–2.79)a | 0.91(0.79–1.35) | 1.48(1.06–2.00) |
P<0.05 vs CBD WT control,
P<0.05 vs DCD WT control.
Figure 2.
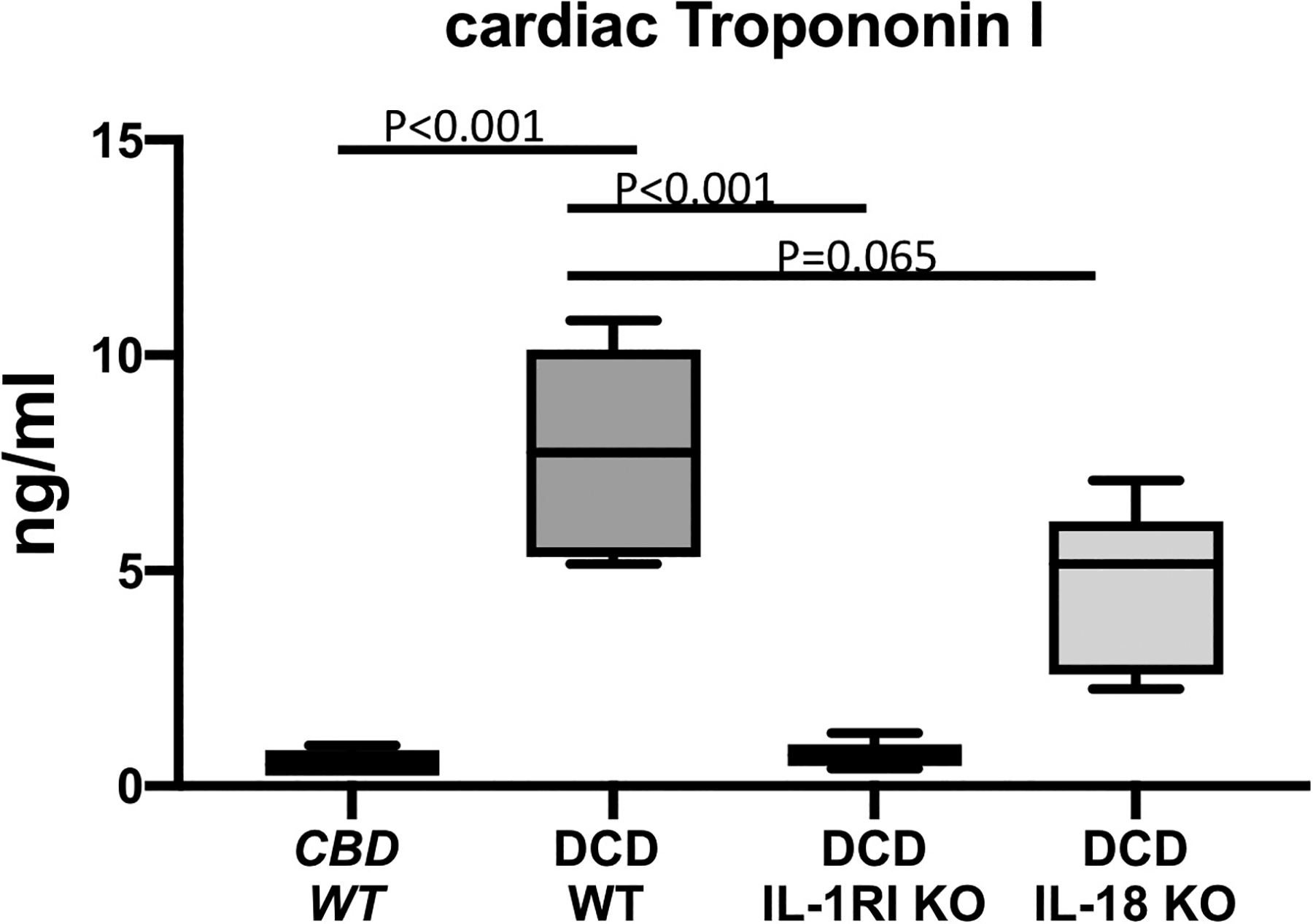
Cardiac troponin I measured in the coronary sinus samples of CBD WT, DCD WT, IL-1RI KO DCD and IL-18 KO DCD mice hearts reanimated on a Langendorff system. CBD=Control Beating-heart Donor; DCD=Donation after Circulatory Death; WT=wild type, IL-1RI KO=Interleukin-1 Receptor type I knock out; IL-18 KO=Interleukin-18 knock out. Values are expressed as Min-Max spread with quartiles.
Genetic deletion of IL-1RI or IL-18 preserves myocardial function:
The DCD IL-1RI KO and DCD IL-18 KO groups had preserved LVDP, which were significantly better compared to the DCD WT group, and were comparable to the CBD group (Figure 1 and Table 2). There was no significant difference in RPP between the IL-1RI KO, IL-18 KO and the CBD WT groups. We measured the cTnI levels in coronary sinus samples as a marker of cardiomyocyte damage (Figure 2). The average cTnI in the eluate collected from DCD WT hearts was significantly elevated compared to the CBD WT hearts, 7.74 (5.67–9.65) vs. 0.49 (0.30–0.73) ng/ml, p<0.05. Genetic deletion of IL-1RI and IL-18 protected myocytes from DCD damage with significantly less troponin release compared to the DCD WT group (Figure 2 and Table 2).
Pharmacological inhibition of IL-1 and IL-18:
Interleukin-1 and IL-18 blockage at reperfusion (Late inhibition groups)- separate groups of DCD WT mice hearts were reanimated on the Langendorff system primed with KH buffer at a constant pressure of 76 mmHg at 37°C for 90 minutes. Added to KH buffer, one group received IL-1Ra, second group IL-18BP and third group received both IL-1Ra and IL-18BP. In these groups of DCD hearts, we noticed an improvement in myocardial protection (less troponin release) with the inhibitors, especially with IL-18BP and combined IL-1Ra and IL-18BP, where the differences attained statistical significance, p<0.05 (Figure 3). We also noticed a trend in improved myocardial function in these groups of hearts, especially with IL-18BP and combined IL-1Ra and IL-18BP, where the differences were statistically significant (Figure 4). No additional improvement was observed between the individual IL-18BP or IL-1Ra treatment and the combined IL-1Ra and IL-18BP treatment (Figure 4, Table 3).
Figure 3.
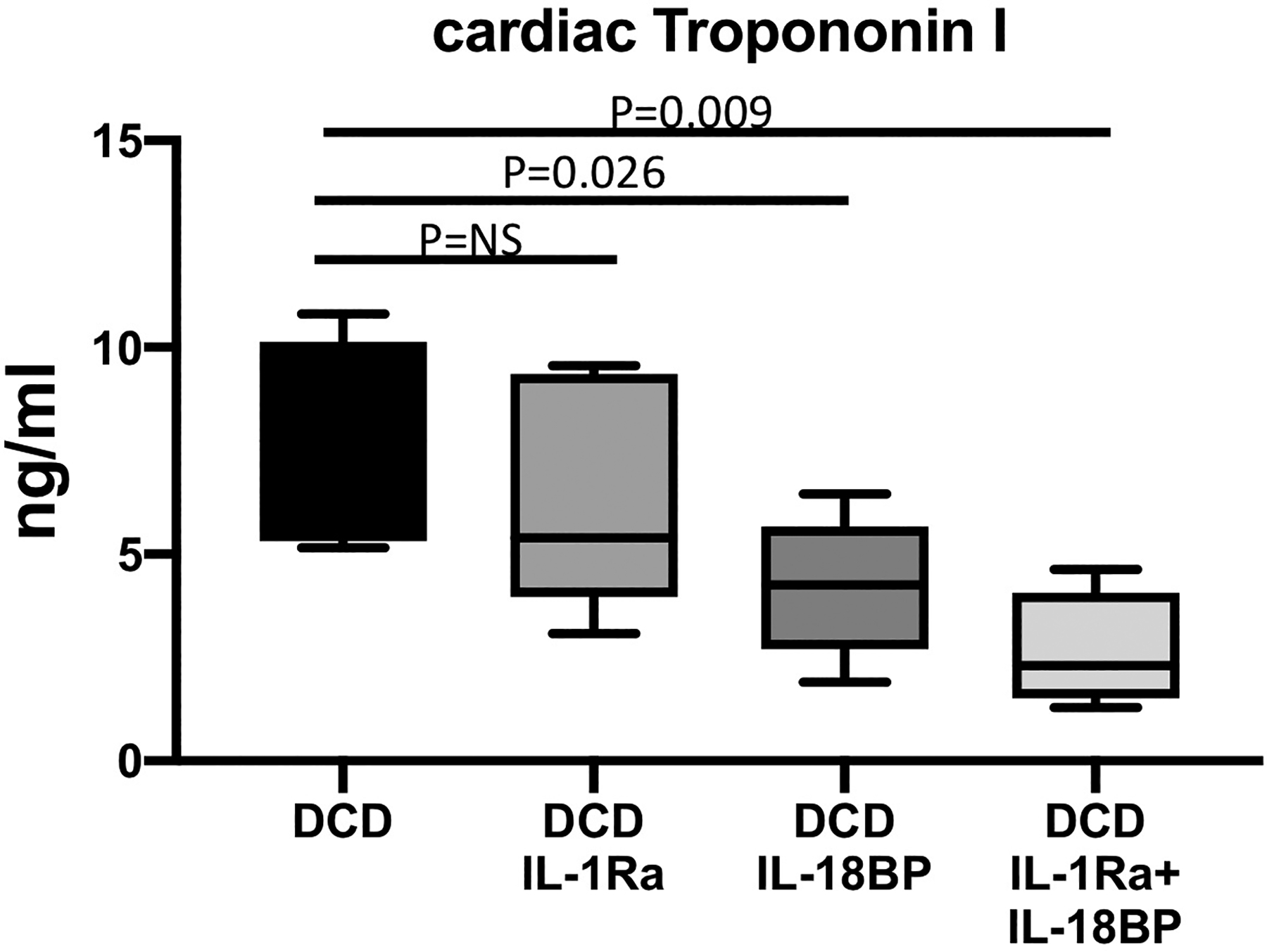
Cardiac troponin I measured in the coronary sinus samples of DCD WT hearts reanimated with regular Krebs Henseleit (KH) buffer or KH buffer supplemented with IL-1 Receptor antagonist (IL-1Ra), IL-18 binding protein (IL-18BP) or the combination of IL-1Ra and IL-18BP. CBD=Control Beating-heart Donor; DCD=Donation after Circulatory Death; WT=wild type. Values are expressed as Min-Max spread with quartiles.
Figure 4.
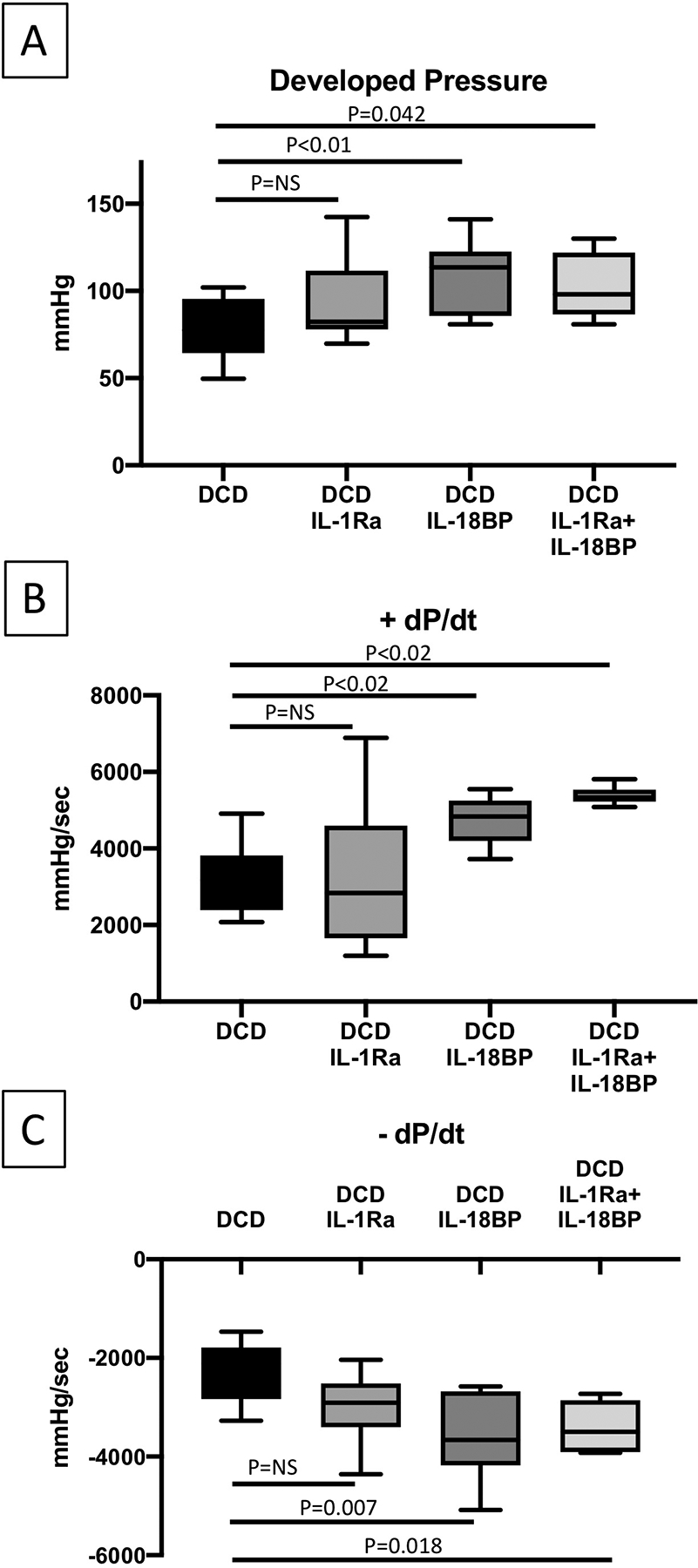
Heart function of DCD WT hearts reanimated with regular Krebs Henseleit (KH) buffer or KH buffer supplemented with IL-1 Receptor antagonist (IL-1Ra), IL-18 binding protein (IL-18BP) or the combination of IL-1Ra and IL-18BP. DCD=Donation after Circulatory Death; WT=wild type. Values are expressed as Min-Max spread with quartiles.
Table 3.
Heart function and markers of cell injury in DCD hearts by treatment received
| DCD Control |
DCD Inhibition Prior to DCD Early Intervention |
DCD Inhibition at Reperfusion Late Intervention |
||||
|---|---|---|---|---|---|---|
| N=10 | IL-1Ra N = 13 |
IL-18BP N = 8 |
IL-1Ra N = 8 |
IL-18BP N = 9 |
IL-1Ra AND IL-18BP N = 5 |
|
| Functional Data | ||||||
| Heart Rate b/min; Median (Q1-Q3) | 399(381–409) | 421(398–447) | 447(419–469) | 416(393–427) | 441(426–457) | 420(367–434) |
| Perfusion rate ml/min; Median (Q1-Q3) | 1.9(1.7–2.0) | 2.1(1.8–2.4) | 2.8(2.4–3.2) | 2.2(2.0–2.4) | 2.5(2.4–2.7) | 2.0(1.6–2.2) |
| Developed pressure mmHg; Median (Q1-Q3) | 77(74–93)a | 101(86–120)a | 89(86–115)a | 82(80–107) | 114(90–119)a | 98(92–114)a |
| Rate Pressure Product; Median (Q1-Q3) | 30744(23091–38180) | 41203(32553–48944) | 38309(37064–51247)a | 34832(31666–43698) | 46072(32613–50139)a | 32375(26415–39335) |
| Rate of positive developed pressure dt/dp mmHg/ms; Median (Q1-Q3) | 3167(2477–3415) | 3186(2233–5578) | 4634(4039–5776)a | 2839(2126–3736) | 4836(4281–5150)a | 5357(5266–5447)a |
| Rate of negative developed pressure −dt/dp mmHg/ms; Median (Q1-Q3) | −2113(−2790–−1884) | −3070(−3402–−2442)a | −2938(−3657–−2784)a | −2909(−3197–−2600) | −3664(−4117–−2728)a | −3496(−3890–−2988)a |
| Makers of myocyte damage | ||||||
| Eluate troponin level ng/ml; Median (Q1-Q3) | 7.74(5.67–9.65) | 8.37(5.80–9.02) | 3.87(2.89–4.54)a | 5.39(4.29–8.58) | 4.25(3.23–5.17)a | 2.30(1.99–2.94)a |
| TUNEL assay; Median (Q1-Q3) | 1.49(1.35–2.79) | 1.24(1.09–1.88) | 0.55(0.44–0.72) | 1.99(1.28–2.82) | 0.86(0.75–0.87)a | 1.03(0.66–1.37) |
P<0.05 vs DCD control
Pharmacological early inhibition of IL-1 and IL-18:
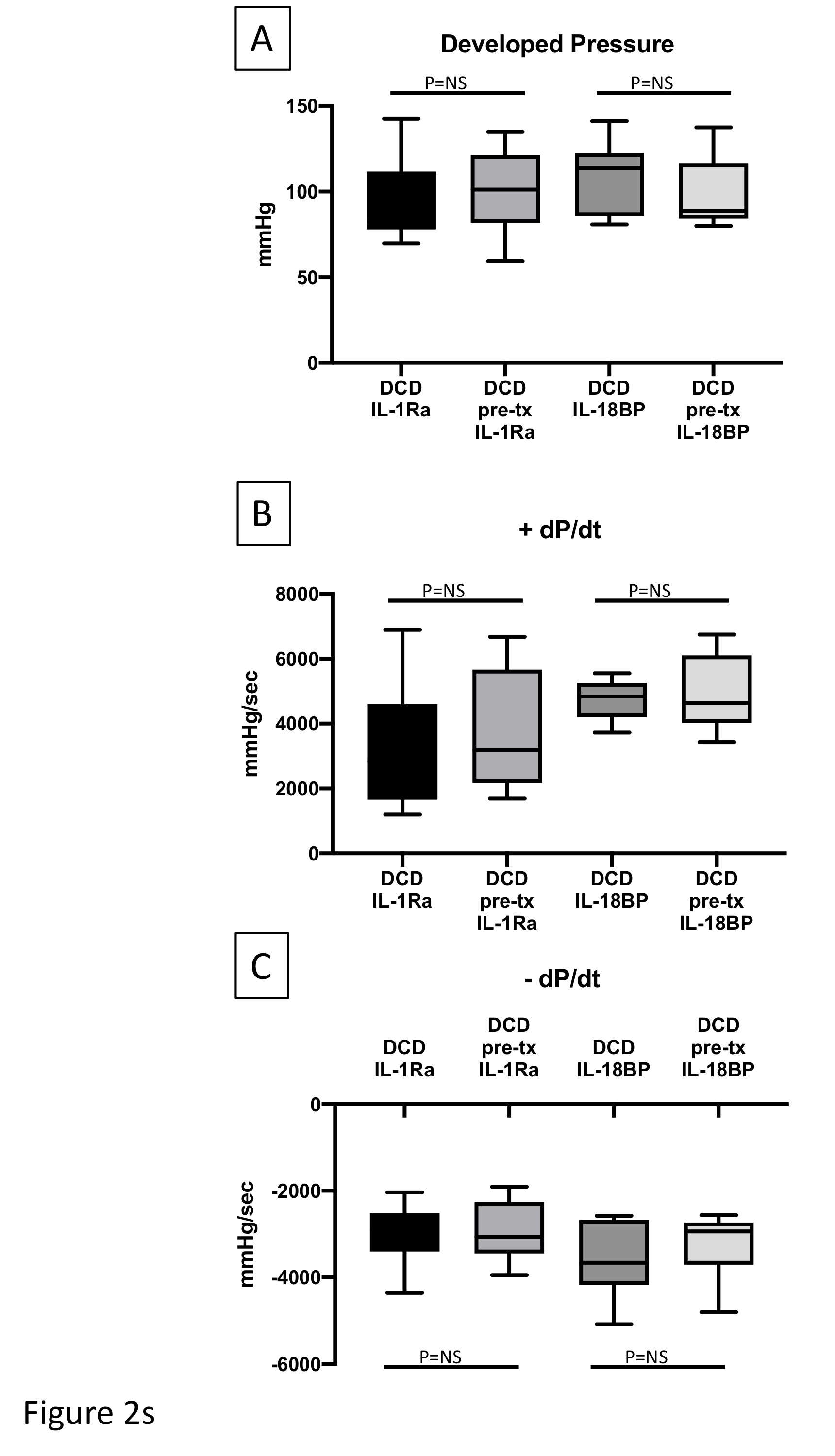
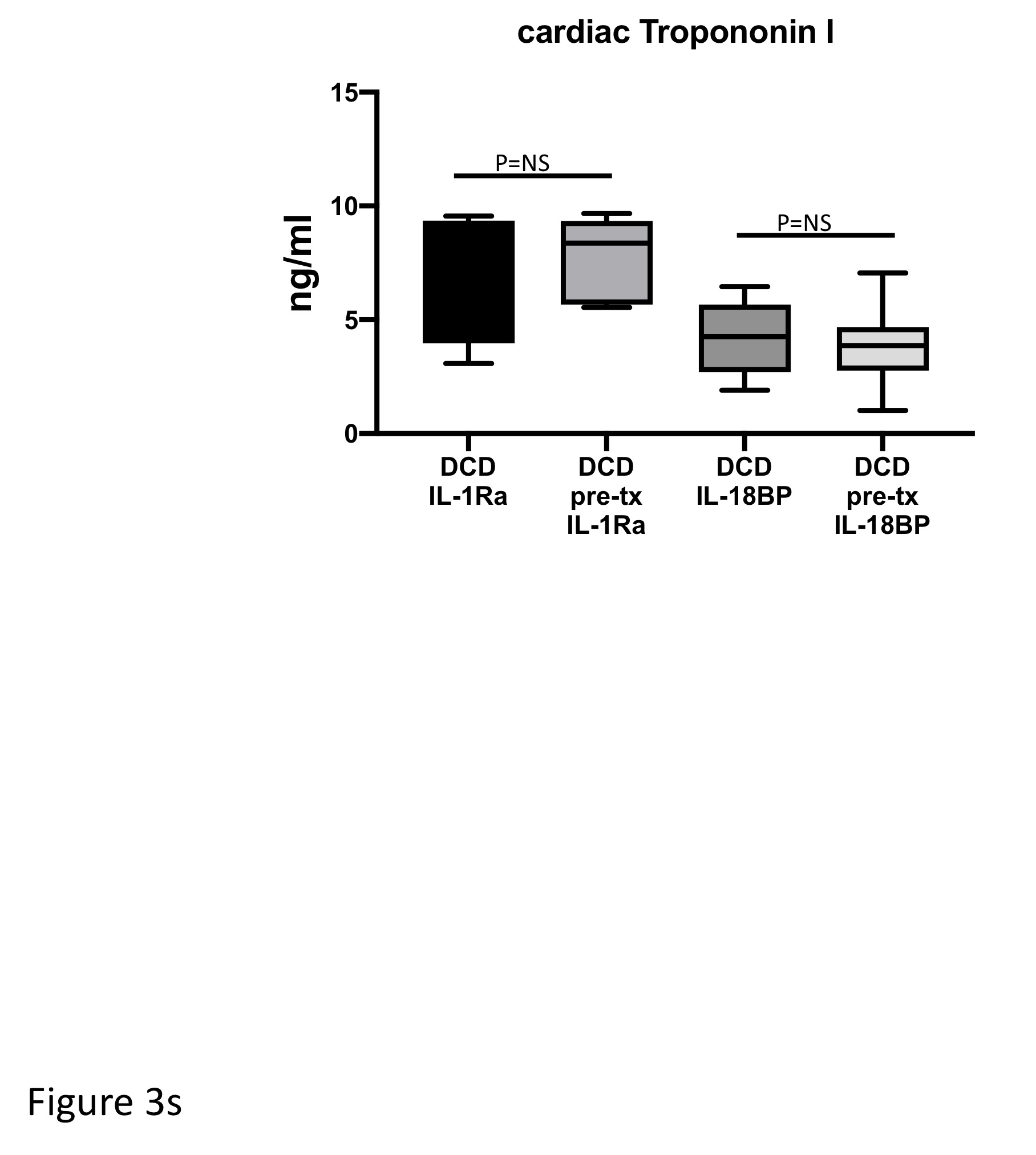
To determine whether the blockade of IL-1 or IL-18 before the DCD induction could protect myocardial damage, two groups of mice were allocated to receive IL-1Ra (10 mg/kg) or IL-18BP (1 mg/kg) administered intraperitoneally 30 minutes before induction of the DCD protocol. Although pre-treatments are not allowed in clinical DCD practice, we wanted to explore whether pre-treatment could augment the protection that we observed with the inhibition of IL-1 or IL-18 at reperfusion (late protection-as above). In addition, these DCD hearts also were treated with IL-1Ra (1μg/ml) or IL-18BP (1μg/ml) mixed in KH buffer when they were reanimated on the Langendorff system. Compared to the late treatment with IL-1Ra or IL-18BP, the early treatment with IL-1Ra or IL-18BP did not afford any additional benefits as measured with myocardial function or markers of cell damage (Table-3 and Figures 2s, 3s supplemental data).
Discussion:
Our study demonstrated several fundamental findings as they pertain to the DCD heart injury in a mouse model. Compared to the CBD controls, DCD hearts sustained warm ischemia that manifested as higher levels of cardiac troponin I release in the eluate, increased markers of cell injury on the histopathologic exam, and a significant decrease in physiologic functions of hearts. As we hypothesized, IL-1 and IL-18 were contributory to the DCD mediated injury to the myocardium. Genetically modified mice without the IL-1RI or the IL-18 gene were less susceptible to DCD induced injury and were able to maintain significantly better heart function compared to the wild type DCD mice. Pharmacologic inhibition of IL-1 or IL-18 either individually or in combination was beneficial in decreasing IL-1- and IL-18-mediated injury with a consistent trend towards better heart function preservation. Pre-treatment with the specific cytokine blockers (early treatment) did not afford additional benefit to what is achieved with the blockers administered at reperfusion alone (late treatment).
Presently most of the heart transplantation procedures are done utilizing DBD donors, where after thorough evaluation in a controlled condition, hearts are procured following administration of cardioplegia1. Ischemia is limited, and a finite time is allowed before the donor heart is implanted into a recipient. DCD process is less controlled, and significant myocardial damage is inevitable2, 15; hence DCD hearts are not utilized for heart transplantation except for small case series from few centers2, 16. Interventions that would limit myocardial damage in DCD hearts, especially during reperfusion, could expand heart transplantation numbers. Our study is designed to further this objective.
Our previous work on DCD induced ischemia has substantiated the role of NLRP3 in activating the NLRP3 inflammasome and its deleterious effects7. Since the IL-1β and IL-18 are the main down-stream mediators of inflammasome-mediated inflammation, we elected to study them further. While there is compelling evidence for the role of IL-1 in mediating regional myocardial ischemia and remodeling17–20, the literature on its role in DCD induced injury is lacking. To understand the role of IL-1 in mediating DCD myocardial injury, we studied seven mice with genetic deletion of the interleukin-1 receptor type I. These IL-1RI KO mice were protected from the deleterious effects of DCD process. Pharmacologic inhibition of IL-1 receptor has been shown to be beneficial in the setting of regional myocardial ischemia to prevent both short-term adverse outcomes and in limiting long-term adverse remodeling21–24. In our study, pharmacologic inhibition with recombinant IL-1Ra administered at the time of reperfusion to DCD hearts showed a consistent trend towards a decrease in cardiac troponin I release and improved heart function; however, all functional parameters did not attain a statistical significance. The possible reasons could be a concurrent injury from other pathways or other cytokines that were not blocked25 or insufficient treatment time. These concerns merit further studies in the future.
The second down-stream effector cytokine in DCD hearts is IL-187. The literature on the role of IL-18 in the global ischemia model, such as with DCD, is very limited. In IL-18 KO mice, the troponin I release was significantly less compared to DCD-WT and heart function parameters were significantly preserved. While there are several studies exploring and demonstrating the benefits of inhibiting IL-1 in acute myocardial ischemia, there is a paucity of data on modulating IL-18. A recombinant IL-18 binding protein is available, and its role in regulating connective tissue diseases is speculated26, but to date, the studies exploring its role in diminishing myocardial ischemia and reperfusion injury are limited27, 28. In our study, we treated a group of DCD mice with IL-18 inhibitor at reperfusion. The troponin I release was significantly less in the treated group as well as all the parameters of heart function were better compared to DCD controls.
It is noteworthy that the beneficial effect of concomitant pharmacologic blockage of IL-1 and IL-18 is marginally better in protecting DCD induced damage than the individual blockade with IL-18BP or IL-Ra, suggesting the potential interdependent nature of downstream effects of IL-1 and IL-18. In addition, the early pharmacologic inhibition of these cytokines (given prior to initiation of the DCD process) did not afford additional myocardial protection compared to what is observed with treatment at reperfusion. A possible explanation for the lack of additional myocardial protection could be that the activation of inflammasome from the DCD process is a pre-requisite to the enhanced production of IL-1 and IL-18, the target of our pharmacologic intervention.
The intracellular signal of IL-1 and IL-18 is complex and involves several intracellular adaptors, kinases and effectors. Myeloid differentiation factor 88 (MyD88) is the first signaling protein activated by the IL-18R or the IL-1RI26. Inhibition of MyD88 can, therefore, work as a combined inhibition of the two pathways. Despite sharing signaling proteins, like MyD88, the intracellular effects of the IL-1 and IL-18 are different. In fact, IL-1 preferentially promotes nuclear factor-kappa B (NF-κB) signaling and IL-18 favors the p38 mitogen-activated protein (MAP) kinase13, 29. In addition, NF-κB and p38 can be activated by other cytokines30, 31. IL-1 and IL-18 may not be the only cytokines increased during the DCD process. Therefore, NF-κB and p38 inhibitors could be other potential protective treatments to test against the DCD damage in future studies.
Our study findings support the role of IL-1 and IL-18 in mediating DCD induced myocardial injury and provides a mechanistic approach to modulate it. Future studies should focus on the appropriate time and dosing of IL-1 and IL-18 inhibitors, on the inhibition of downstream signaling effectors, like MyD88, NF-κB and p38, and perhaps in conjunction with other known factors that influence reperfusion injury such as Na/K channel blockers, cyclosporine, erythropoietin, etc. in a larger animal model where the outcomes could be more compelling to move towards a phase I clinical trials.
Study limitations:
Murine heart model while practical, and reproducible, does not mirror the clinical scenario where several patient variables confound the results. However, the murine DCD model has the advantage of studying targeted pathway inhibition with two complementary strategies (genetic and pharmacological approach). This approach strengthens our study findings. The use of KH buffer instead of a blood-based perfusate is a limitation of our model because crystalloid solutions such as KH display lower oncotic pressure. However, in our case, the use of KH buffer has shown the effect of the IL-1 and IL-18 produced by the myocardium, without the confounding effect of cytokines produced by immune cells. The study has been designed to test whether the treatment of the heart ex-vivo is sufficient to reduce the damage induced by the DCD process followed by the reperfusion ex-vivo. Treatments to reduce the reperfusion injury in vivo will likely be achieved with the dosage currently approved for the recombinant human IL-1Ra or the investigational dose of recombinant human IL-18BP. The functional data obtained with a balloon tip catheter placed in the left ventricle simulates the working heart condition but is not a substitute for the physiologic working heart model.
Conclusion:
With increasing numbers of patients suffering from advanced heart failure, there is an urgency in finding more donor hearts for transplantation. Donation after circulatory death donors can expand the donor pool for heart transplantation, provided that we develop strategies to reduce cardiac injury in DCD hearts. Inflammasome mediated cytokines (IL-1, IL-18) play a downstream effector role in ischemia/reperfusion injury in DCD hearts. Targeted inhibition of IL-1 and IL-18 is possible and results in significant preservation of the DCD heart function.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments:
American Heart Association SDG (award number- 16SDG31080002) and Veterans Administration Merit Review grants awarded to Mohammed Quader (Grant ID CARA-015-17S, Award No. I01 BX003859), National Institute of Health grant awarded to Stefano Toldo (R01HL150115) and funds from the Pauley Heart Center to Mohammed Quader and Stefano Toldo (Grant No. 50408).
Footnotes
Disclosure: Dr. Toldo is PI of investigator-initiated studies from Olatec and Kiniksa. All other authors have no conflict of interest to disclose.
References:
- 1.Colvin M, Smith JM, Hadley N, et al. OPTN/SRTR 2016 Annual Data Report: Heart. Am J Transplant. 2018;18 Suppl 1:291–362. [DOI] [PubMed] [Google Scholar]
- 2.Dhital KK, Iyer A, Connellan M, et al. Adult heart transplantation with distant procurement and ex-vivo preservation of donor hearts after circulatory death: a case series. Lancet. 2015;385(9987):2585–91. [DOI] [PubMed] [Google Scholar]
- 3.Chew HC, Iyer A, Connellan M, et al. Outcomes of Donation After Circulatory Death Heart Transplantation in Australia. Journal of the American College of Cardiology. 2019;73(12):1447–59. [DOI] [PubMed] [Google Scholar]
- 4.Quader M, Toldo S, Chen Q, Hundley G, Kasirajan V. Heart transplantation from donation after circulatory death donors: Present and future. Journal of cardiac surgery. 2020. [DOI] [PubMed] [Google Scholar]
- 5.Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123(6):594–604. [DOI] [PubMed] [Google Scholar]
- 6.Mezzaroma E, Toldo S, Farkas D, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108(49):19725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quader M, Mezzaroma E, Kenning K, Toldo S. Modulation of Inflammasome Mediated Ischemia and Reperfusion Injury in Donation After Circulatory Death Heart. Circulation. 2018;138 Suppl_1 A10771(A10771). [Google Scholar]
- 8.Toldo S, Mezzaroma E, Mauro AG, et al. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22(13):1146–61. [DOI] [PubMed] [Google Scholar]
- 9.Van Tassell BW, Seropian IM, Toldo S, Mezzaroma E, Abbate A. Interleukin-1beta induces a reversible cardiomyopathy in the mouse. Inflamm Res. 2013;62(7):637–40. [DOI] [PubMed] [Google Scholar]
- 10.Council NR. Guide for the Care and Use of Laboratory Animals. In: th, editor. Guide for the Care and Use of Laboratory Animals. The National Academies Collection: Reports funded by National Institutes of Health. Washington (DC) 2011. [Google Scholar]
- 11.Kim SH, Eisenstein M, Reznikov L, et al. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A. 2000;97(3):1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbate A, Salloum FN, Van Tassell BW, et al. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PloS one. 2011;6(11):e27923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toldo S, Mezzaroma E, O’Brien L, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014;306(7):H1025–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novick D, Kim SH, Fantuzzi G, et al. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10(1):127–36. [DOI] [PubMed] [Google Scholar]
- 15.Rosenfeldt F, Ou R, Salamonsen R, et al. A novel combination technique of cold crystalloid perfusion but not cold storage facilitates transplantation of canine hearts donated after circulatory death. J Heart Lung Transplant. 2016;35(11):1358–64. [DOI] [PubMed] [Google Scholar]
- 16.Messer SJ, Axell RG, Colah S, et al. Functional assessment and transplantation of the donor heart after circulatory death. J Heart Lung Transplant. 2016;35(12):1443–52. [DOI] [PubMed] [Google Scholar]
- 17.Bageghni SA, Hemmings KE, Yuldasheva NY, et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pascual-Figal DA, Bayes-Genis A, Asensio-Lopez MC, et al. The Interleukin-1 Axis and Risk of Death in Patients With Acutely Decompensated Heart Failure. Journal of the American College of Cardiology. 2019;73(9):1016–25. [DOI] [PubMed] [Google Scholar]
- 19.Schofer N, Zeller T. Response to: Soluble regulators of Interleukin-1 signaling: Novel biomarkers for early acute myocardial infarction diagnosis and to predict ischemia/reperfusion injury? Int J Cardiol. 2019;279:32. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, Wan J. Soluble regulators of Interleukin-1 signaling: Novel biomarkers for early acute myocardial infarction diagnosis and to predict ischemia/reperfusion injury? Int J Cardiol. 2019;274:357. [DOI] [PubMed] [Google Scholar]
- 21.Abbate A, Salloum FN, Vecile E, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117(20):2670–83. [DOI] [PubMed] [Google Scholar]
- 22.Zheng ZH, Zeng X, Nie XY, et al. Interleukin-1 blockade treatment decreasing cardiovascular risk. Clin Cardiol. 2019;42(10):942–51. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine. 2017;377(12):1119–31. [DOI] [PubMed] [Google Scholar]
- 24.Ridker PM, MacFadyen JG, Glynn RJ, et al. Inhibition of Interleukin-1beta by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. Journal of the American College of Cardiology. 2018;71(21):2405–14. [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1beta inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. European heart journal. 2019. [DOI] [PubMed] [Google Scholar]
- 26.Dinarello CA. Targeting interleukin 18 with interleukin 18 binding protein. Ann Rheum Dis. 2000;59 Suppl 1:i17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A. 2001;98(5):2871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M, Tan J, Wang Y, et al. IL-18 binding protein-expressing mesenchymal stem cells improve myocardial protection after ischemia or infarction. Proc Natl Acad Sci U S A. 2009;106(41):17499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JK, Kim SH, Lewis EC, et al. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A. 2004;101(23):8815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.