

Abstract
Adaptation to acute and chronic stress and/or persistent stressors is a subject of wide interest in central nervous system disorders. In this context, stress is an effector of change in organismal homeostasis and the response is generated when the brain perceives a potential threat. Herein, we discuss a nuanced and granular view whereby a wide variety of genotoxic and environmental stressors, including aging, genetic risk factors, environmental exposures, and age- and lifestyle-related changes, act as direct insults to cellular, as opposed to organismal, homeostasis. These two concepts of how stressors impact the central nervous system are not mutually exclusive. We discuss how maladaptive stressor-induced changes in protein connectivity through epichaperomes, disease-associated pathologic scaffolds composed of tightly bound chaperones, co-chaperones, and other factors, impact intracellular protein functionality altering phenotypes, that in turn disrupt and remodel brain networks ranging from intercellular to brain connectome levels. We provide an evidence-based view on how these maladaptive changes ranging from stressor to phenotype provide unique precision medicine opportunities for diagnostic and therapeutic development, especially in the context of neurodegenerative disorders including Alzheimer’s disease where treatment options are currently limited.
Keywords: Alzheimer’s disease, chronic stress and stressors, epichaperomes, maladaptive response to stress, stressor-to-phenotype, synaptic plasticity
Graphical Abstract
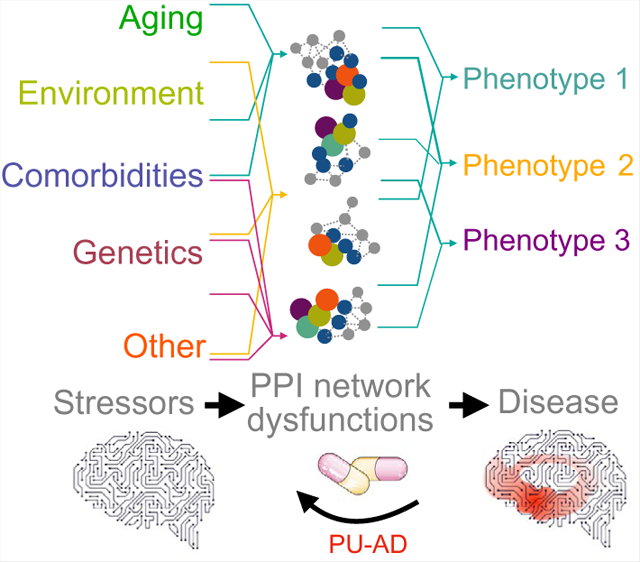
The contents of this page will be used as part of the graphical abstract of html only. It will not be published as part of main article.
We discuss how stressors act as direct insults to cellular homeostasis through epichaperome formation. Epichaperomes are maladaptive disease-associated pathologic scaffolds composed of tightly bound chaperones, co-chaperones, and other factors. They induce proteome-wide changes in protein–protein interaction networks, and thus impact intracellular proteome functionality, altering cellular phenotypes. This disrupts and remodels brain networks ranging from intercellular to brain connectome levels. We provide an evidence-based view on how these maladaptive changes ranging from stressor to phenotype provide unique precision medicine opportunities for diagnostic and therapeutic development, especially in the context of neurodegenerative disorders, such as Alzheimer’s disease, where treatment options are currently limited.
1 |. INTRODUCTION
Established evolutionary biology concepts such as adaptation and maladaptation have been introduced and used by neuroscientists to explain mechanisms related to brain function and plasticity but also to brain dysfunction in numerous disorders ranging from neuropsychiatric disease to neurodevelopmental disorders to neurodegenerative disorders (de Kloet et al., 2005; Vyas et al., 2016). Adaptation in evolutionary biology refers to the process of adjustment for a better fit or more broadly, for survival (Crespi, 2000). For example, an organism could adapt acutely by adjusting to its immediate environment. This could be executed by changing physiological features, such as body temperature or metabolism rate. Adaptation may also refer to a more permanent change so that individuals may have greater success in a specific environmental context. This can be a restructuring in whole-body characteristics (e.g., color, beak shape) and be driven and effected by genetic and epigenetic factors. By the same token, maladaptation is perceived as a deviation from adaptation which at organismal level may imply a state of reduced fitness. Maladaptation however could have different connotations as for example in cancer it may increase fitness at a cellular level yet be detrimental to the organism as a whole (Joshi et al., 2018).
In the discipline of neuroscience, adaptation has been widely studied in the context of response to stress or stressors (de Kloet et al., 2005; McEwen, 2007). Here, stress is broadly defined as a factor that disrupts organism homeostasis. The response to stress is generated when the brain perceives something as an actual or potential threat to generalized well-being. Stress can be beneficial when an acute response is generated to start physiological and behavioral processes that mobilize energy and promote adaptation. For example, the sympathetic nervous system may initiate adrenaline production, thus triggering physical reactions, such as increased heart rate, that prepare the body to respond to threat. This response was coined “fight or flight” by Walter Cannon (Cannon, 1915). As noted by Bruce McEwen (McEwen, 2005), stress can also be processed through the hypothalamic-pituitary-adrenal (HPA) axis that controls adrenal production of glucocorticoids such as cortisol. Cortisol is initially permissive of stress and improves organismal ability to cope with stress (Sapolsky et al., 2000). Successful coping indicates that the response to stress is activated quickly when needed but also efficiently terminated when the stress is removed. However, under conditions where stress occurs repeatedly, persists chronically, or is of such magnitude that an organism fails to re-establish homeostasis, an overload state results that often leads to maladaptive responses and pathological consequences (Godoy et al., 2018; Justice, 2018; McEwen & Gianaros, 2011). Thus, adaptive responses to stress enhance resilience and facilitate coping mechanisms against future stressors, whereas maladaptive responses represent a failure to cope with stress and/or to return to normalcy leading to vulnerability to stress-associated pathology.
As has been described by Novais and coworkers (Novais et al., 2016), the ability to adapt, or conversely maladapt, is modulated by the duration, intensity, controllability, and ability to predict a specific stressor or stressors, as well as the genetic background and life history of the individual. Thus, individuals experience stress and respond to stressors in different ways. Remarkably, as described by Nuno Sousa (Sousa, 2016), stressors trigger in healthy subjects a consistent and reproducible activation of a set of interconnected brain regions. For example, physical stressors (e.g., noxious stimuli, hypovolemia, cytokine exposure, and hypoglycemia) activate brainstem nuclei, which activate corticotropin-releasing hormone and arginine vasopressin releasing neurons in the hypothalamus, ultimately releasing corticosteroids in the adrenal cortex through the HPA axis (Godoy et al., 2018; Herman et al., 2016). Psychosocial stressors on the other hand, may activate limbic brain structures, such as the amygdala, hippocampus, and/or frontal cortex (Bremner, 2006; Godoy et al., 2018). Conversely, as has been described by Sousa (Sousa, 2016), chronic maladaptive stressors engage regions outside the ‘normal’ network, involving a structural and functional reorganization of neural networks. In addition to differences at the brain network level, as has been described by Brivio and coworkers (Brivio et al., 2020) acute and chronic stressors also distinctly impact intracellular networks. Protein pathways associated with synaptic plasticity become activated by acute stress, for example, glucocorticoids signal through serum- and glucocorticoid-regulated kinase (SGK) to increase the trafficking and function of AMPA receptors (AMPAR) leading to altered synaptic potentiation (Krugers et al., 2010; Liu et al., 2010). Conversely, chronic stress reduces these receptors and leads to synaptic dysfunction (Kallarackal et al., 2013; Yuen et al., 2012). Relevant toward understanding the pathobiology of neurodegenerative disorders such as the Alzheimer’s disease (AD) spectrum and tauopathies, chronic stress and high levels of glucocorticoid secretion may trigger misprocessing of amyloid precursor protein (APP), generation of amyloid beta (Aβ), and tau hyperphosphorylation and aggregation (Lopes et al., 2016; Qi et al., 2021; Sotiropoulos et al., 2019). These pathological hallmarks including Aβ and tau also act as cellular stressors to create imbalances in neurotransmission and synaptic plasticity (Costa et al., 2010; Silva et al., 2016; Welikovitch et al., 2020; Zempel et al., 2010) which may serve as nodal points for therapeutic intervention.
Elucidating the cellular, regional, and organism level stress response mechanisms, including alterations under chronic conditions, individual or population differences influencing these processes, and how these factors influence illness susceptibility, has been a dauntingly complex undertaking for neurobiologists. Several aspects of this complexity has been covered in informative reviews over the past two decades (de Kloet et al., 2005; Mattson & Magnus, 2006; McEwen et al., 2015; Saxena & Caroni, 2011; Sousa & Almeida, 2012; Vyas et al., 2016). Herein, we introduce a developing view on stressors and their pathologic impact on the central nervous system (CNS) that has emerged from the cancer field. We perceive stressors as a direct insult onto a cell and its network connections, rather than an indirect manifestation that is perceived first by the brain which then directs the organism, and in turn a cell, to a response. We are cognizant of the vague and/or broad use of the terms ‘stress’ and ‘stressor’ in the scientific community. There are many stressor stimuli of genetic, environmental, and proteotoxic nature that may lead to pathologic responses. Accordingly, we define these stressors throughout the text and figures, as best we could. We present evidence on the involvement and key contribution of pathologic scaffolding platforms termed epichaperomes to the maladaptive stressor-induced rewiring of cellular networks. We strongly believe this ‘bottom up interactome dysfunction’ in addition to a ‘top down brain-initiated stress response’ assessment in the change of directionality of events is of key importance to understand mechanisms driving stressor-to-phenotype responses in brain as well as to develop rational therapeutic approaches to mitigate the impact of specific stressors to CNS disease, especially neurodegenerative disorders.
2 |. CELLULAR RESPONSE TO STRESS
2.1 |. Heat shock response
One of the most widely studied cellular responses to acute stress is the induction of a heat shock response, discovered 60 years ago by Ferruccio Ritossa (1962). As the name implies, the heat shock response is a quick and strong transcriptional activity response in cells when exposed to elevated temperatures (Ritossa, 1962). As noted by Rick Morimoto (1998), it was later recognized that the heat shock response is a general response of cells to acute stress and entails a significant increase in heat shock proteins (HSPs), also referred to as molecular chaperones. The heat shock response is an adaptive response that aims to counteract the negative effects on proteins caused by stressors by helping to prevent or attenuate protein misfolding and provide an environment for proper folding through the increased production of HSPs (Vabulas et al., 2010).
The heat shock response concept was extrapolated to explain several pathologies in the brain and periphery. For example, studies by Brehme and coworkers (Brehme & Voisine, 2016; Brehme et al., 2014) have demonstrated how changes in subnetworks of chaperones are associated with neurodegenerative disorders and may signify a breakdown in the folding capacity of the cell, enabling protein aggregate formation, a hallmark of many neuropathological conditions. Depletion of subsets of molecular chaperones during the progression of disease may also exacerbate protein toxicity and neurodegeneration (Ebrahimi-Fakhari et al., 2011; Muchowski & Wacker, 2005). The concept of heat shock response was also invoked as a potential therapeutic approach, whereby induced HSP production aimed to alleviate proteotoxicity (McAlary et al., 2019; Sweeney et al., 2017). Several excellent reviews are written on this concept whereby a decrease in the folding capacity of the cell is associated with neurodegeneration and accordingly, increasing permissiveness of the folding environment alleviates proteotoxicity (Balch et al., 2008; Boland et al., 2018; Höhn et al., 2020; Kampinga & Bergink, 2016; Lackie et al., 2017; Morimoto, 2008).
2.2 |. Heat shock proteins in relation to chaperones
It cannot be overstated that, albeit widely associated with folding and disaggregation, chaperones have many other functions. For example, under physiological conditions, chaperones assist in the formation of protein complexes (Ellis, 2013). In fact, the term ‘molecular chaperone’ was first used in the context of describing a protein that assists the assembly of folded subunits into oligomeric structures, as evidenced from classic work on bacteriophage lambda, nucleoplasmin, and Rubisco (Barraclough & Ellis, 1980; Goloubinoff et al., 1989; Laskey et al., 1978). Only later was the term extended to include protein folding (Ellis, 2006). Considering all chaperones as being HSPs is incorrect, because only a fraction (i.e., 20%) of human chaperones are actually heat inducible (Finka et al., 2011). Furthermore, a significant number of human heat-induced genes are not chaperones.
Since 1987 when the term ‘molecular chaperone’ was coined by John Ellis (Ellis, 1987), the number of proteins associated with chaperone function continues to increase rapidly. The term ‘chaperome’ was introduced in 2006 to characterize this assembly of chaperones, cochaperones, and related factors (Wang et al., 2006). In 2013, a study categorized 147 members of the human chaperome (Finka & Goloubinoff, 2013). This list included heat shock protein 90s (HSP90s), HSP70s, HSP60, HSP40s (also known as DNAJ proteins), HSP110s, HSP10, the small HSPs, their co-chaperones, as well as peptidylprolyl and protein disulfide isomerases (Finka & Goloubinoff, 2013). HSP family nomenclature comes from molecular mass of the initially identified member. Most HSPs have organelle specific members, including ones localized to the mitochondria and endoplasmic reticulum (ER). For example, cytosolic HSP90α and HSP90β have a mitochondrial paralog, tumor necrosis factor type 1 receptor-associated protein (TRAP1 often termed HSP75 or mortalin) and an ER paralog, glucose regulated protein 94 (GRP94, often termed gp96 or endoplasmin) (Finka & Goloubinoff, 2013). A comprehensive chaperome listing identified 332 chaperones and co-chaperones including 88 chaperones (50 being ATP-dependent), along with 244 co-chaperones (Brehme & Voisine, 2016). A chaperome atlas analysis tool is now accessible on the internet (Hadizadeh Esfahani et al., 2018).
Notably, the chaperome is a highly abundant assembly of proteins. As shown by Finka and Goloubinoff (Finka & Goloubinoff, 2013), the 147 chaperome constituents comprise 7.6% of total polypeptides and 10.3% of total protein mass in HeLa cells. The most abundant chaperome is HSP90, comprising of approximately 2.8% of total protein mass (Finka & Goloubinoff, 2013). HSP90 and HSP70 combine to make up 5.5% of total protein mass (Finka & Goloubinoff, 2013). An additional 1.5% of total protein mass consists of the HSP90 and HSP70 co-chaperones (Finka & Goloubinoff, 2013).
2.3 |. Chaperome heterogeneity following stressor exposure
As noted above the chaperome is an assembly of ubiquitous proteins, including the major chaperones HSP90s and HSP70s which are abundantly expressed and found essentially in all mammalian cells. For a mature human weighing 70 kg, and considering a 16% protein mass, it is estimated there would be ~224–336 g of the major chaperones. What renders this family of great importance for treatment consideration is the relatively recent realization that chaperome members become both structurally and functionally altered in disease (Diezmann et al., 2021; Kamal et al., 2003; Mollapour & Neckers, 2012; Rodina et al., 2016; Wang et al., 2019; Yan et al., 2020). This section will present the chaperome as heterogenous, with stressor-modified entities found in cells undergoing a pathologic process. These entities both distinctly appear and behave differently than ubiquitous folding forms of chaperones and co-chaperones localized to cells under normal physiological conditions (Figure 1a–c).
FIGURE 1.
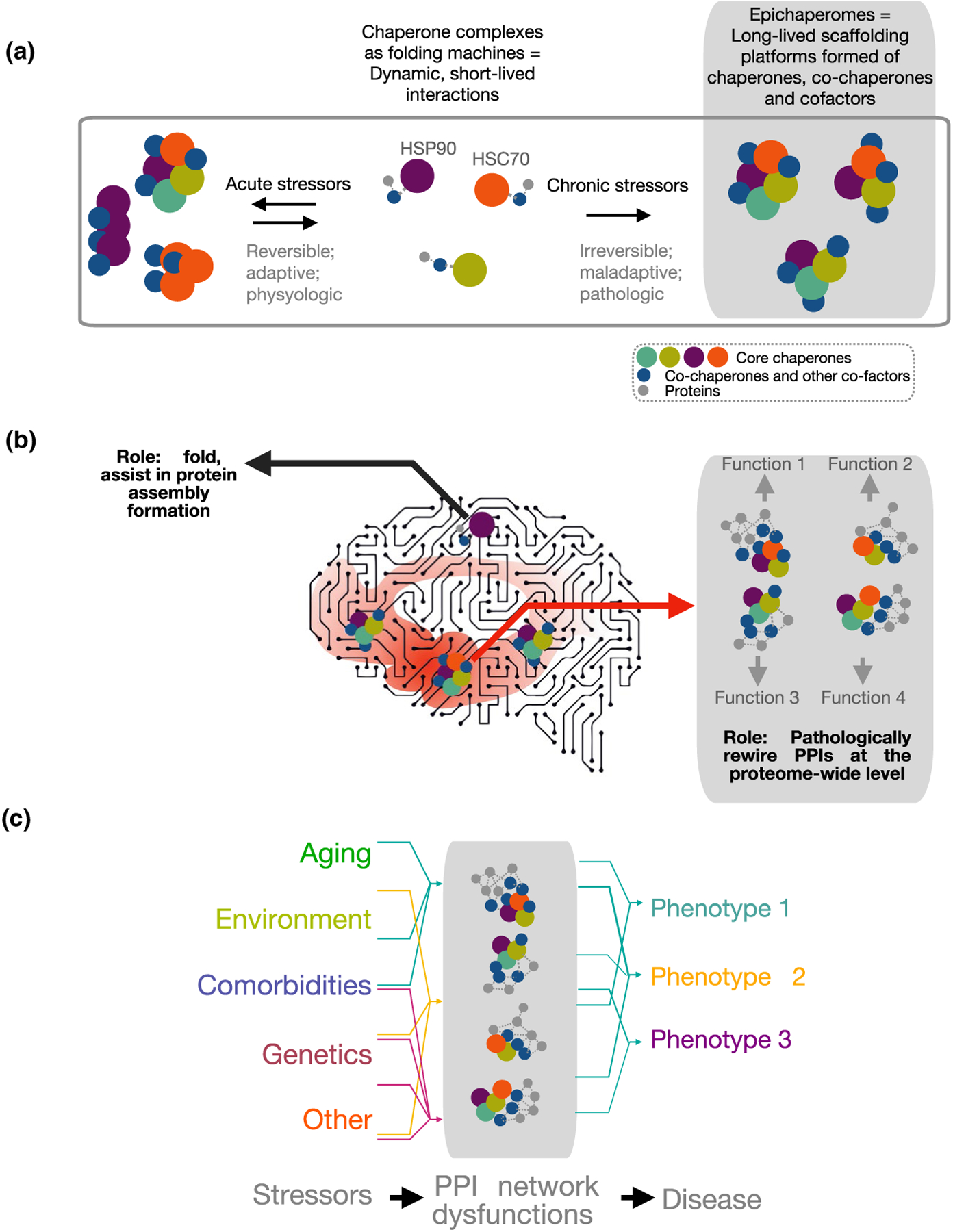
Biochemical (a) and functional (b, c) distinction between chaperones and epichaperomes. There is a fundamental structural, dynamic, and functional difference between HSP90 and HSP90 incorporated into an epichaperome. Epichaperomes are scaffolds. They rewire the connectivity and function of protein networks by remodeling how thousands of proteins interact in conditions of chronic cellular stress. Conversely, HSP90 is a chaperone. Chaperones, co-chaperones, and their complexes have defined functions as protein folders. They interact with a protein to process it through the chaperone folding cycle. Epichaperomes are long-lived oligomers of chaperome members. This differs from chaperones such as HSP90, which interact in a highly dynamic manner with one another and with client proteins on the millisecond to second timescale to make folding versus degradation decisions through transient interactions within the context of the proteostasis network. Epichaperomes are specific to cells exposed to defined stressors and/or combination of stressors. Conversely, chaperones are highly abundant and ubiquitous proteins. Epichaperome composition is stressor specific with distinct epichaperome assemblies impacting specific proteins, and in turn, protein pathways. Therefore, epichaperomes are multimeric, long-lived chaperome structures that act as scaffold for remodeling PPIs and provide a link between stressor-induced protein–protein interaction network perturbations and phenotypes. Abbreviations: HSP90, heat shock protein 90 (HSP90α and HSP90β isoforms, encoded by the HSP90AA1 and HSP90AB1 genes, respectively; HSC70, heat shock cognate 70 protein encoded by the HSPA8 gene; PPIs, protein–protein interactions
As noted by Taldone and coworkers (Taldone et al., 2020), it is now recognized in several disease states, which are effectively embodiments of chronic cellular stress, changes in the interaction strength among chaperome components, alterations in their post-translational modifications, and in the cellular location of chaperome members, in addition to changes in the expression levels of components of the chaperome, are all hallmarks of pathology. These findings are detailed in excellent review articles (Boudesco et al., 2018; Caino & Altieri, 2016; Joshi et al., 2018; Klaips et al., 2018; Mollapour & Neckers, 2012; Wyatt et al., 2013).
2.4 |. The chaperome in disease – a historical perspective
The first chaperone to be given attention as a potential target in cancer was HSP90. A model was built based on the hypothesis that if an oncoprotein or several oncoproteins, termed “client protein(s),” required HSP90 for structural stabilization and/or functional modulation, then an HSP90 inhibitor could facilitate the degradation of such client(s) in tumors, and in turn the tumor will respond to HSP90 therapy (Whitesell & Lindquist, 2005). In this context, HSP90 is a key component of a folding machinery on which certain client proteins, such as kinases with oncogenic activity are transferred over from the HSP70 machinery for folding and maturation (Schopf et al., 2017). If folding were to fail, the protein would be instead directed for proteasomal degradation. In these tumor-specific folding functions, HSP90 was reported to be in high-affinity complexes with co-chaperones such as p23 and HSP-organizing protein (HOP) (Kamal et al., 2003). These complexes characterized tumor cells but not normal cells and provided a rationale for the use of inhibitors of HSP90 as therapeutics to target cancerous cells (Kamal et al., 2003). A role for post-translational modifications in these tumor-specific functions of the chaperome has also been identified (Muller et al., 2013; Walton-Diaz et al., 2013).
This concept was later adapted to neurodegenerative disorders. Several excellent papers from the Petrucelli, Dickey and Gestwicki labs and others reported on the effect of HSP90, HSP70, and other chaperones and co-chaperones, and their complexes, on tau pathology. For example, a 2007 study by the Petrucelli lab presents compelling data showing HSP90 and a number of co-chaperones together in the high-affinity complex directly interact with hyperphosphorylated tau and process it through the chaperone folding pathway (Dickey et al., 2007). Inhibition of HSP90 with EC102 blocked the refolding pathway and promoted hyperphosphorylated tau turnover through degradation (Dickey et al., 2007). A 2012 study from the Gestwicki lab discusses a similar mechanism whereby HSP90 and HSP70 competed for binding to shared sites on tau and an exchange between HSP90 and HSP70 complexes mediates tau degradation (Thompson et al., 2012). The HSP90-bound complex was important in activating tau clearance in cells, with the relative levels of HSP70 and HSP90 determining whether tau was retained or degraded (Thompson et al., 2012). A 2007 study implicated HSP90 also in the regulation of mutant tau forms such as characteristic of frontotemporal dementia (Luo et al., 2007). The structure of the tau-HSP90 complex was identified by the Rüdiger lab to reveal the mechanism of tau regulation by this chaperone (Karagoz et al., 2014). The contribution and implications of several chaperones and co-chaperones in the folding of disease-associated proteins was also studied and reviewed in great detail (Bohush et al., 2019; Carman et al., 2013; Lackie et al., 2017; Lindberg et al., 2015) indicating the generalized importance of this mechanism for disease pathobiology.
3 |. THE EPICHAPEROME AS A PATHOLOGICAL SCAFFOLD AND STRESSOR-TO-PHENOTYPE BIOSENSOR
In this position piece, our goal is to focus on pathologic changes in the chaperome executed by enhancing interaction strength and altering interaction partners. We discuss how these stressor-induced alterations in the chaperome, leading to long-lived assemblies termed epichaperomes, have a negative effect on protein–protein interactions (PPIs; i.e., interactomes) at a proteome-wide level. Epichaperome formation is detrimental to cellular function and cellular interaction networks, perturbing the brain connectome, and in turn brain function. We highlight stressor-related epichaperomes and their functions in several neurodegenerative disorders. Stressors in this context are complex such as aging, genetics, environmental factors, proteotoxic formations, changes in the cellular microenvironment and in the organism as a whole, among others. Since this disease hallmark was first discovered in the context of cancer, we provide a short historical overview.
3.1 |. Epichaperome discovery
Under normative conditions, chaperones and co-chaperones typically interact in a highly dynamic manner with each other and with client proteins on a timescale of milliseconds to a second to execute folding and/or degradation decisions through transient PPIs within the context of the proteostasis network critical to maintaining the cellular proteome (Balchin et al., 2016). For example, pioneering studies on human chaperones in the 1980–1990 era discovered the association of mammalian HSP90 with co-chaperones and client proteins was dynamic (Hutchison et al., 1992). Identification of these multiprotein complexes was enabled through stabilization via oxyanions including molybdate, vanadate, and tungstate (Hutchison et al., 1992). In addition to chemical stabilizers, acute cellular stress can also stabilize chaperone interactions, natively in the cellular environment (Joshi et al., 2018). Chadli and coworkers found that, even though HSP90 purified from chick embryos is observed as single band on SDS-PAGE and as a dimer via native-PAGE, thermal stress generated multiple oligomeric HSP90 forms as observed by native-PAGE (Chadli et al., 1999). HSP90 oligomers were not insoluble aggregates. Rather they were soluble high molecular mass components that maintained binding to co-chaperones including activator of HSP90 ATPase protein 1 (AHA1) (Chadli et al., 1999). AHA1 prefers binding to oligomeric HSP90 compared to the dimer (Lepvrier et al., 2015).
Thus, cellular stress can change the interaction strength between chaperome members. This adaptive “rewiring” of the chaperome by higher order reorganization may enable new functions that are silent under normal conditions but that may be needed to counteract the deleterious effects of acute stress (Figure 1a, acute stress). This mechanism appears to extend beyond the chaperome, as the heat induced “reversible aggregation” or oligomerization of multiple proteins was reported by Wallace and coworkers (Wallace et al., 2015) to reflect an adaptive, autoregulatory process that aids cellular survival under thermal stress.
In addition to providing an adaptive, beneficial effect to cells exposed to stressors, chaperome remodeling results in disease when occurring chronically (Joshi et al., 2018). As noted by Joshi and colleagues (Joshi et al., 2018), this represents a maladaptive response with detrimental effects on the proteome, leading to a pathologic cellular phenotype (Figure 1a, chronic stress). Through the advent of chemical biology tools, the study of the chaperome in native, endogenous cellular states was enabled. In the discipline of chemical biology, chemicals rather than biological systems are “mutated” to interrogate a protein or a cellular state similar to how proteins are mutated in structural biology or cells are engineered in cell biology (Bertozzi et al., 2015). These chemical are referred as ‘probes’, and can be soluble ligands used to inhibit or activate a target, solid-support attached ligands used for affinity capture of the target and its interactors, fluorescently labeled or radiolabeled ligands for target detection and quantification in biological samples and/or whole organisms, among others (Workman & Collins, 2010). In this context, the discovery of several chemical probes that bind HSP90 revealed the complexity both in structure and function of HSP90 in disease that was not appreciated previously through the use of other approaches (Shrestha et al., 2016; Taldone et al., 2020).
By using HSP90 inhibitors immobilized onto agarose beads it was found that each HSP90 probe isolated distinct chaperone pools (Moulick et al., 2011). For example, in K562 chronic myeloid leukemia cells expressing oncogenic BCR-ABL, an aberrant fusion protein between c-ABL and BCR, and the physiologically normative c-ABL, both bind HSP90 (Moulick et al., 2011). Certain chemical probes isolated HSP90 bound to both BCR-ABL and c-ABL HSP90 complexes (Moulick et al., 2011). In contrast, other probes, such as solid support immobilized PU-H71, preferred to associate with the BCR-ABL bound HSP90 pool (Moulick et al., 2011).
Chemical probes that preferred HSP90 bound to BCR-ABL, also showed proclivity for HSP90 bound to oncogenic v-FLIP and v-SRC over the physiological, wild-type c-FLIP and c-SRC respectively (Moulick et al., 2011; Nayar et al., 2013; Ojala, 2013). An interesting observation from these early studies was the HSP90 pool that bound BCR-ABL (the oncogenic form) also tightly bound HSP70 and several co-chaperones that function in concert with HSP70 (Moulick et al., 2011). Conversely, HSP90 that bound c-ABL did not (Moulick et al., 2011). Therefore, HSP90 binds both BCR-ABL (an oncogenic protein) and ABL (its physiologic counterpart), but there is a structural and/or dynamic difference between the two pools, and this difference is sensed by some of the HSP90 chemical probes. As discussed below, these two HSP90 pools also have distinct functions (Figure 1b).
The BCR-ABL-bound HSP90 pool was ultimately distinguished from the c-ABL HSP90 pool by its biochemical signature (Rodina et al., 2016; Yan et al., 2020). This pool contained HSP90 in long-lived heterooligomers along with HSC70, HOP, HSP110, CDC37, AHA1, and other chaperome members (Rodina et al., 2016). Unlike the short-lived complexes consisting of chaperones/co-chaperones which work in a one-on-one, dynamic cyclic fashion to aid in protein folding, degradation, or disaggregation, Rodina and coworkers found these HSP90-containing heterooligomers, dubbed epichaperomes, remain stable under native PAGE, and isoelectric focusing conditions (Rodina et al., 2016). They also had a distinct isoelectric focusing signature. In addition to the HSP90 signature in normal cells, in which the chaperone focuses at an isoelectric point (pI) of 4.9, numerous HSP90 species were evident with a pI >4.9 in the epichaperome pool (Rodina et al., 2016). Unlike dynamic HSP90 complexes which dissociate under native PAGE and appear as a dimer, epichaperomes present as multiple, distinct HSP90 species of variable molecular weights (Bolaender et al., 2021; Inda et al., 2020; Kishinevsky et al., 2018; Pillarsetty et al., 2019; Rodina et al., 2016; Yan et al., 2020), clearly distinguishing them from the normative condition.
3.2 |. Epichaperome function
Functionally, epichaperomes are also distinct from chaperones as they act as scaffolds for remodeling PPIs at the proteome-wide level, rather than folders of proteins in protein synthesis and degradation pathways (Figure 1b) (Ginsberg et al., 2021; Inda et al., 2020; Joshi et al., 2018; Kishinevsky et al., 2018; Yan et al., 2020). A first glimpse into this role comes from a large-scale unbiased approach conducted by Moulick and coworkers that interrogated the interactome of HSP90-incorporating epichaperomes in the K562 chronic myeloid leukemia cells (Moulick et al., 2011). In addition to BCR-ABL (but not c-ABL) the constellation of proteins interacting with the HSP90-incorporating epichaperomes contained multiple proteins as part of active signaling megacomplexes, as well as adapter proteins which link BCR-ABL to key effectors of multiple aberrantly activated signaling pathways in K562 cells (Moulick et al., 2011). As discussed by Joshi and colleagues (Joshi et al., 2018), these findings indicate a clear presence of HSP90-epichaperomes in assembled—and active—signaling complexes when HSP90 is acting as part of the epichaperome. This observation differs from the one-on-one cyclic, dynamic functions exhibited by HSP90 when acting as a molecular chaperone (Li et al., 2012). Therefore, as noted by Joshi and colleagues (Joshi et al., 2018), HSP90 and other chaperones may use epichaperome formation to trigger functions that are normally silent but become active during stressor conditions. We discuss the constellation of proteins negatively impacted in neurodegenerative disorders and the dysfunctions created by epichaperomes below.
3.3 |. Epichaperome composition
Epichaperome composition is context dependent (Kishinevsky et al., 2018) (Figure 1c). Distinct assemblies have impact on different proteins. For example, HSC70 is an epichaperome constituent along with HSP90 in AD where epichaperomes negatively impact the interactions of proteins important for synaptic plasticity (Inda et al., 2020). HSP60 becomes an epichaperome component in neurons exposed to mitochondrial toxins, such as rotenone, to produce defects in dopamine pathways, whereas HSC70 is co-opted in conditions of genetic stress (e.g., PARKIN mutation), to activate inflammatory pathways (Kishinevsky et al., 2018). GRP94 becomes an epichaperome component in cells where alterations in plasma membrane protein assembly is necessary for phenotypic rewiring (Chaumonnot et al., 2021; Yan et al., 2020). Whereas each of these chaperones is an abundant protein, and found in all cells in the human body, the fraction of a chaperone incorporated into epichaperomes is minor and localized to diseased cells and tissues (Inda et al., 2020; Kishinevsky et al., 2018; Rodina et al., 2016; Yan et al., 2020). Importantly, this critical finding makes the epichaperome optimal for both targeting and imaging (Bolaender et al., 2021; Dunphy et al., 2020; Inda et al., 2020; Jhaveri et al., 2020; Pillarsetty et al., 2019; Sugita et al., 2021).
3.4 |. Epichaperome formation
What triggers and/or enables the switch of a chaperone into epichaperomes? Because HSP90 is stable as a dimer even at high concentrations (Schopf et al., 2017), a conformational change is most likely necessary to drive epichaperome formation, as recently reported for GRP94, the ER resident HSP90 (Yan et al., 2020). This may lead to the unmasking of a multimerization site that is not present in the dimer, generating a new PPI platform with new quaternary structure. Recent studies have shed light on post-translational modifications that may play a role in the switch of a chaperone into epichaperomes. For the chaperone GRP94, N-glycosylation at asparagine 62 is the conformation altering process as it stabilizes a unique GRP94 conformation that facilitates stable interactions with proteins and other chaperones and co-chaperones (Yan et al., 2020). Through such stabilization, the functions of the Glyc62GRP94-epichaperome interacting proteins are altered, and dependent cellular protein networks become remodeled aberrantly (Patel et al., 2013; Yan et al., 2020). The biochemical and structural mechanisms associated with HSP90 switch from chaperone to epichaperome in neurodegenerative disorders remain to be elucidated and exploited for therapeutic intervention.
4 |. EPICHAPEROMES IN THE CONTEXT OF NEURODEGENERATIVE DISORDERS
4.1 |. Parkinson’s disease and related synucleinopathies
Advances in the generation of patient-derived induced pluripotent stem cells (iPSCs) and iPSC-derived neurons enables the possibility to examine how genetic and environmental stressors induce early pathogenic events in neurodegenerative disorders such as Parkinson’s disease (PD). For example, a floor-plate-based differentiation strategy was used to generate midbrain dopaminergic (mDA) neurons that express key markers including transcription factors LMX1A and FOXA2 (Kriks et al., 2011). These neurons efficiently engrafted in vivo and rescued rotation behavior induced by amphetamine delivery and demonstrated improvement in tests of forelimb use and akinesia in several relevant PD animal models (Kriks et al., 2011). mDA neurons derived from patient specific PARK2/Parkin and PINK1 mutant human iPSCs (termed Parkin or PINK PD mDA neurons, respectively) displayed Parkinsonian phenotypes both in vitro and after transplantation in vivo, notably after induction of age-associated stress (Chung et al., 2016). During mitophagy, Parkin and PINK1 interact with each other, and mutations in these two genes are linked to early-onset PD (Ge et al., 2020). Relative to wildtype (WT) mDA neurons, PINK1 and Parkin PD mDA neurons have increased α-synuclein levels at the gene and encoded protein levels, increased vulnerability to mitochondrial toxins including rotenone and carbonyl cyanide m-chlorophenyl hydrazone (CCCP), and challenged with mitochondrial abnormalities (Chung et al., 2016).
Epichaperome formation was evaluated in these neurons, when exposed to genetic stressors (e.g., Parkin mutant) and mitochondrial toxin (e.g., rotenone treatment) (Kishinevsky et al., 2018). Kishinevsky and colleagues (Kishinevsky et al., 2018) found that WT mDA neurons lacked epichaperomes, which was reflected in no multimeric HSP90 pools detected under native PAGE, little interaction with the chemical probe PU-H71 and a refractory profile to PU-H71, where even high concentrations of PU-H71 (≤50 μM) over a 72 h treatment revealed minimal toxicity in day-old 65 mDA neurons (i.e., at 65 days of differentiation). As indicated above, PU-H71 binds HSP90 preferentially when this chaperone reverts to epichaperomes (Inda et al., 2020; Kishinevsky et al., 2018; Rodina et al., 2016). The lack of epichaperomes in unstressed mDA neurons is similar to what was observed for other non-stressed, non-transformed cells and signifies a state of normal cellular proteostasis (Pillarsetty et al., 2019; Rodina et al., 2016; Sugita et al., 2021). Exposure to both genetic and mitochondrial toxin stressors triggered epichaperome formation in mDA neurons, especially with the genetic insult of Parkin mutant introduction (Kishinevsky et al., 2018).
These well-known stressors each led to specific chaperones being recruited into epichaperomes, suggesting that epichaperome composition may represent a stressor-specific fingerprint (Kishinevsky et al., 2018). For example, Kishinevsky and coworkers found that HSP60 recruitment into epichaperomes was enhanced under toxic stress (Kishinevsky et al., 2018). HSP60 is a chaperone with known relevance to PD. Injury of mDA neurons results in increased HSP60 expression, which is believed to create further damage to neighboring neurons after extracellular release and microglial activation (Noelker et al., 2014). Genetic stressors, and to some degree toxic stressors, also led to significant increase in the recruitment of HSC70, HOP, HSP40, and several other co-chaperones to epichaperomes (Kishinevsky et al., 2018).
Interestingly, a study in 2006 conducted by the Trojanowski and Lee lab in postmortem PD brain found both α-synuclein and HSP90 co-localization and high HSP90 expression level intensity, which the authors dubbed intense HSP90 immunoreactivity (iHSP90) along HSC70 and HSP40 in Lewy bodies (LBs) (Uryu et al., 2006). In glial cytoplasmic inclusions (GCIs), iHSP90 was detected along another chaperone, α B-crystallin (Uryu et al., 2006). Conversely, in normal human brain, HSP90 was found homogenously in the neuronal cell body and in proximal dendrites throughout all brain regions examined (Uryu et al., 2006). Assessing numerous synucleinopathies, the authors discovered profuse iHSP90 immunoreactivity within LBs, Lewy neurites (LNs), spheroids in patients with PD, dementia with LBs (DLB) and the LB variant of Alzheimer’s disease (LBVAD), multiple system atrophy (MSA), and frontotemporal dementia with motor neuron disease type (FTD-MND) (Uryu et al., 2006). Interestingly, tau positive neurofibrillary tangles in AD and DLB showed little or no iHSP90 (Dabir et al., 2004; Uryu et al., 2006). Similar findings were reported in α-synuclein (α-syn) transgenic (Tg) mice (Uryu et al., 2006). Age matched A53T mutant α-syn M83 Tg mice (asymptomatic), wild-type α-syn M7 Tg mice, and WT mice had modest, ubiquitous expression of HSP90 in neurons similar to normal human control brains (Uryu et al., 2006). M83 Tg mice displayed α-syn pathology in an age-dependent manner, where α-syn inclusions co-localized with iHSP90 in a subgroup of neurons that contained these inclusions (Uryu et al., 2006). The study did not elucidate the structural and functional relationship between α-syn inclusions and chaperones including iHSP90 and HSC70. However, in the context of the results from PD mDA neurons, we speculate the iHSP90 foci discovered by the Trojanowski and Lee lab in the human brain are epichaperomes and indicate a pathologic end-result of epichaperome formation. A study formally testing the presence of epichaperomes in live and/or postmortem human PD brains and related disorders including DLB, LBVAD, MSA, and FTD-MND has yet to be performed. However, independent identification of the same chaperones in iHSP90 foci that were found to participate along HSP90 in epichaperomes formation in PD mDA neurons and in AD human brains support this hypothesis (Inda et al., 2020; Kishinevsky et al., 2018). Also supportive is the identification of α-synuclein, but not of hyperphosphorylated tau, as a specific epichaperome interactor in human AD brains (Inda et al., 2020). Furthermore the stressor-dependent constituency of epichaperomes (Kishinevsky et al., 2018) is consistent with the distinct chaperone association found for iHSP90 in LBs versus tau-positive GCIs (Uryu et al., 2006).
4.2 |. Alzheimer’s disease and related tauopathies
Epichaperome formation was studied by Inda and colleagues in sporadic AD brains as well as in a variety of cellular and Tg mouse models of AD and related tauopathies where stressors encompassed genetic, environmental, proteotoxic, and other well-established insults (Inda et al., 2020). Results indicated that epichaperomes were detected in human AD (i.e., where stressors are complex such as aging, genetics, environmental factors, and others) but not age-matched no cognitively impaired (NCI) subjects. Similarly, epichaperomes were detected in APP duplication-expressing iPSC-derived neurons (i.e., genetic stressor) but not in WT iPSC-derived neurons, and in PS19 transgenic mouse brains (i.e., proteotoxic stressor produced by mutant tau overexpression) but not in non-transgene containing litter-mates (Inda et al., 2020).
Unbiased mass spectroscopy (MS) analyses of epichaperomes in AD brains determined these formations incorporate HSP90 and a number of other chaperones and co-chaperones as described above, of which HSC70, HOP and CDC37 were also validated by Western blot analysis (Inda et al., 2020). The common epichaperome core, consisting of HSP90 and HSC70, was also found in PD neurons and cancer cells (Kishinevsky et al., 2018; Rodina et al., 2016), suggesting this core may be a nucleator of epichaperomes in both cancer and neurodegenerative disorders, a finding that has important therapeutic significance as it provides a viable solution for inhibitor design (Bolaender et al., 2021).
Interestingly, elevated levels of HOP, a key building block in epichaperome formation in human AD (Inda et al., 2020), was recently reported by Lackie and colleagues in a Tg mouse model of AD and in human AD subjects (Lackie et al., 2020). By overexpressing HOP (also called STI1) in the 5xFAD Tg mouse line, the Prado lab found higher levels of HSP90β, altered expression of Aβ-regulating enzymes (BACE1 and MMP-2) and increased amyloid burden, which amplified neurotoxicity and worsened spatial memory deficits in mice (Lackie et al., 2020). HOP was found to accumulate in dense-core AD plaques in both 5xFAD mice and human brain tissue, implying a role for HOP in Aβ accumulation (Lackie et al., 2020). Thus, despite HOP being protective in C. elegans and in mammalian cultured neurons (Lackie et al., 2020) in vivo, the principal effect of elevated HOP in AD is deleterious and associated with epichaperome formation.
5 |. PROTEINS AND PROTEIN PATHWAYS NEGATIVELY IMPACTED BY EPICHAPEROME FORMATION
The first glimpse into proteins negatively impacted by epichaperome formation comes from cancer where Moulick and coworkers found epichaperome interactomes contained multiple proteins as part of megacomplexes rather than single proteins (Moulick et al., 2011). This finding led to the hypothesis that epichaperome formation may in fact act as a scaffolding platform and lead to formation of maladaptive PPIs in cells exposed to stressors (Ginsberg et al., 2021; Joshi et al., 2018). This postulate was confirmed in a variety of disease settings and disease models (Inda et al., 2020; Kishinevsky et al., 2018). For example, a global analysis of epichaperomes and their interacting proteins performed by Inda and colleagues (Inda et al., 2020) in AD dementia patients and age-matched individuals with NCI identified ~62% of the nodes (in PPI network context, nodes are proteins) and ~98.5% of the edges (the connection between two proteins) were significantly altered in AD through epichaperome formation. The formation of new abnormal PPIs was seen in AD, with 1,191 proteins found to form interactions via epichaperomes. Loss of normal PPIs was also evidenced, with 942 proteins dropping established connections during the progression from NCI to AD (Inda et al., 2020). Although AD pathogenesis is considered heterogeneous in the sporadic cases evaluated to date (i.e., considering pathology, cell composition, underlying disease cause with each patient presenting disease caused by distinct combination of genetic, epigenetic and environmental factors), a distinct commonality was observed, namely each subject displayed significant PPI dysfunctions that were mediated via epichaperomes (Inda et al., 2020).
Functionally, Inda and coworkers found proteins and PPIs impacted by epichaperomes in the human AD brain mediated synaptic plasticity pathways, metabolic processes, inflammation and changes in immune response and adaptation, cell cycle re-entry, cell-cell communication, and other key processes associated with AD (Inda et al., 2020). In addition to AD, epichaperome-mediated changes in inflammation- and cell cycle reentry-related protein pathways were identified by Kishinevsky and colleagues also in mutant Parkin expressing mDA neurons derived from familial PD patients (Kishinevsky et al., 2018).
Synaptic pathways common to AD patients and negatively impacted in the transition from NCI to AD dementia through epichaperome formation were found by Inda and coworkers to include signaling networks such as ‘signaling by second messenger’, ‘Galpha(i) signaling’, ‘signaling by Rho GTPases’, ‘signaling by Wnt’, ‘response to elevated cytosolic Ca2+’, ‘MAPK signaling’, adhesion-regulatory networks such as ‘extracellular matrix organization’ and ‘cell-cell communication’, as well as protein translation-related networks (Inda et al., 2020). Thus, both short-term memory formation, reliant on posttranslational modifications of existing synaptic complexes and associated signaling pathways, and long-term memory preservation, reliant on new protein synthesis (Abraham et al., 2019), are both vulnerable to deleterious PPI network alterations impacted by epichaperome formation (Inda et al., 2020).
Remarkably, a commonality of distinct stressors was a profound impact on PPIs related to synaptic plasticity including a genetic stressor (APP duplication) in iPSC-derived neurons, and proteotoxic stressors via human tau overexpression in N2a cells and in PS19 transgenic mice (Ginsberg et al., 2021; Inda et al., 2020). As noted by Inda and coworkers (Inda et al., 2020), although the identity of individual synaptic plasticity-related proteins impacted by epichaperome formation often differed between stressor conditions, epichaperome-mediated changes in the interaction of these proteins all had functional output imbalances in protein pathways integral for synaptic plasticity. Taken together, defects in synaptic plasticity is a pervasive epichaperome-mediated vulnerability of brain cells in response to various genetic, toxic, and environmental stressors (Inda et al., 2020). The observed vulnerability of synaptic function to stressors is not surprising because of the critical involvement of this pathway in a number of neurological conditions, such as neurodegenerative diseases (e.g., AD, PD, and Huntington’s disease, among others), neurodevelopmental disorders (e.g., autism and Down syndrome), as well as neuropsychiatric disorders (e.g., bipolar disorder, major depressive disorder, and schizophrenia) (Compta & Revesz, 2021; Counts et al., 2014; Eltokhi et al., 2021; Garner & Wetmore, 2012; Ginsberg et al., 2012; Henstridge et al., 2016). Each of these distinct disorders may arise because of perturbation in, and via a combination of, a multiplicity of genetic, epigenetic, and environmental stressors, which we posit can be interrogated by epichaperomics (Ginsberg et al., 2021).
Linking epichaperome formation to dysfunction in synaptic plasticity induced by a variety of stressors is highly relevant translationally as it provides a final common path for therapeutically rectifying synaptic plasticity defects (Ginsberg et al., 2021; Inda et al., 2020). PU-AD, a small molecule that dismantles epichaperomes into normal folding chaperome components (Bolaender et al., 2021; Inda et al., 2020), reverted the pathologic effects of several genetic and toxic stressors on synaptic protein pathways (Inda et al., 2020). For example, Inda and colleagues found that introduction of human tau into N2a cells was sufficient to rewire a fraction of the cellular chaperome into epichaperomes resulting in functional imbalances within synaptic protein networks detected postmortem in AD brains (Inda et al., 2020).
Epichaperome formation by these stressors was associated with aberrant changes in the function of several protein pathways involved in cell adhesion, actin remodeling and translation initiation pathways (Ben Zablah et al., 2020; Chesnokova et al., 2017; Clevers, 2006). Epichaperome-mediated defects were also seen in signaling such as by second messengers and MAPK pathways (Inda et al., 2020) that converge on CREB phosphorylation and activation of its transcriptional activity (Saura & Valero, 2011). These pathways regulate phosphorylation of the GluA1 subunit of AMPAR on S845, which leads to subsequent increases in GluA1 surface expression during synaptic plasticity, and thus are critical for proper long-term potentiation (LTP) execution (Huganir & Nicoll, 2013). In particular, during LTP and synaptic up-scaling, AMPARs are accumulated at synapses to increase synaptic strength. Inda and coworkers found PU-AD rebalanced the activity of these networks to their pre-tau state suggesting a causal link between pathologic changes in synaptic plasticity related neuronal pathways and epichaperome formation (Inda et al., 2020).
Similar restorative effects were also observed by Inda and colleagues in Tg mice (Inda et al., 2020). For example, in the PS19 mouse model, mutant tau overexpression is one of the major stressors that imbalances synaptic protein networks also detected in human AD brains (Inda et al., 2020). Treatment of PS19 mice with PU-AD resulted in a significant rebalance in the activity of synaptic protein networks to levels observed in the WT mice, as determined by Western blot monitoring of the levels of the above-mentioned functional effector proteins (e.g., p-cofilin, active β-catenin, p-CREB, p-eIF4E, and p-AMPAR) (Inda et al., 2020). PU-AD treatment restored postsynaptic responsiveness and repaired a largely compromised synaptic plasticity, as recorded using slice electrophysiology (Inda et al., 2020). Functionally, epichaperome inhibition resulted in the reversal of cognitive decline, as measured by Barnes maze. Studies conducted in both the preventive and the interventional treatment settings in PS19 mice demonstrated cognitive improvements with measured parameters equaling those in WT mice. Similar results on memory testing were found in the 3xTg AD model expressing human APPswe, PS1M146V, and tauP301L (Inda et al., 2020).
To appreciate how distinct stressors effect synaptic plasticity through epichaperome formation, we reanalyzed published datasets of proteins impacted by epichaperome formation in the transition from NCI to AD (Figure 2a) (Inda et al., 2020). Subnetwork classification of the datasets (n = 5 AD and n = 3 NCI human brains) via unsupervised clustering analyses (Alashwal et al., 2019) led to the identification of three major subtypes (Figure 2b). Dysfunctions in synaptic transmission (i.e., AMPA receptor trafficking, CREB phosphorylation, and presynaptic polarization) and metabolic processes (i.e., glucose and pyruvate metabolism, energy integration, and TCA cycle) were shared among all analyzed AD samples (see heatmap, Figure 2b and Reactome functional mapping of proteins, red circles, Figure 2c).
FIGURE 2.
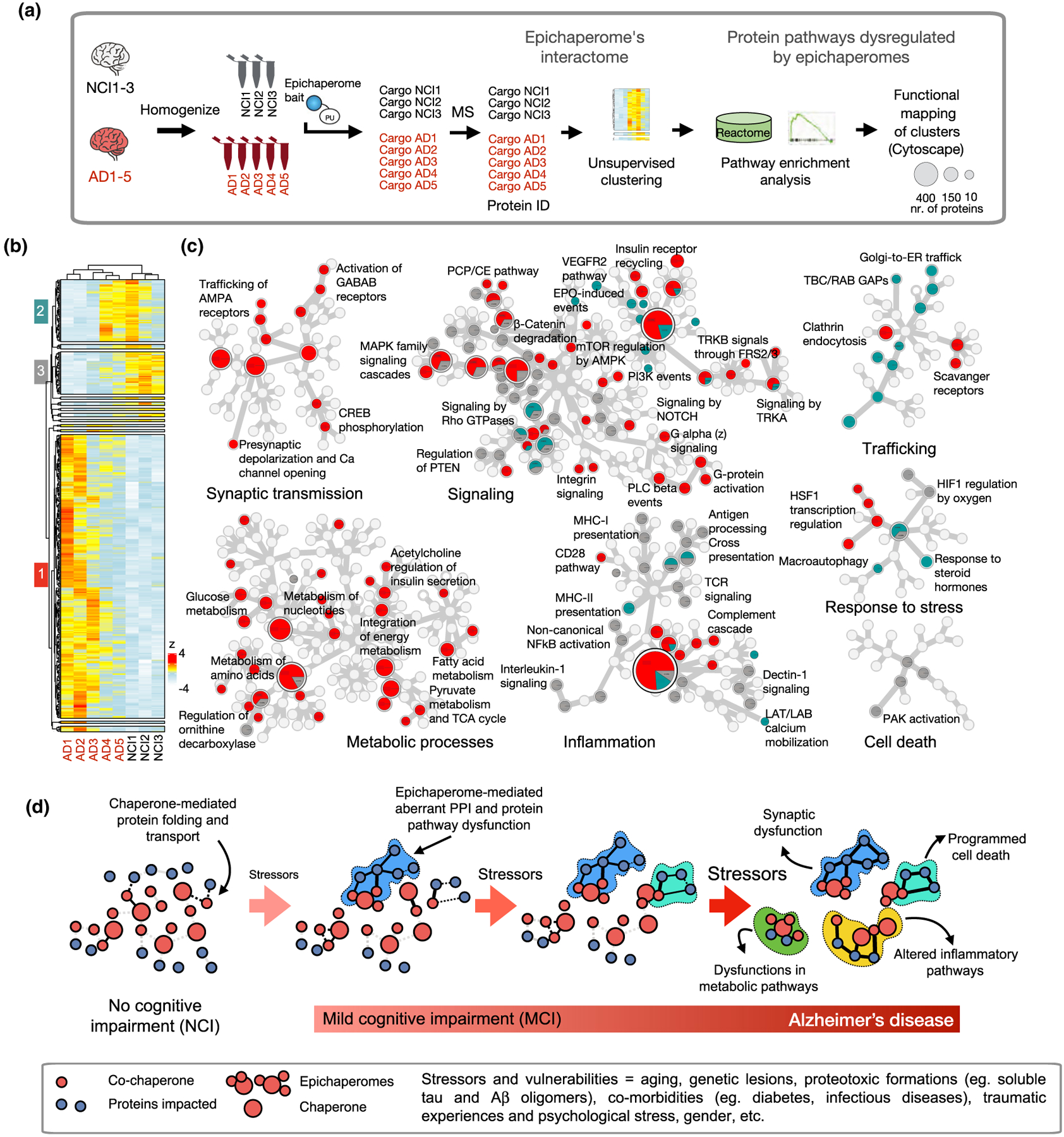
Stressor-to-phenotype assessments via epichaperomics, the omics method that uses epichaperomes to identify and study aberrant PPIs in disease. (a) Schematic showing epichaperomics sample analysis and data processing. (b) Unsupervised clustering of datasets from Inda et al. (2020). Human brains (sporadic late-onset AD, n = 5 vs NCI = 3). (c) Reactome mapping of proteins in each of the three major clusters. In the Reactome map, generated in Cytoscape, each circle represents a function (i.e., a protein pathway). If the circle is divided among gray, red, and teal, it means the dysfunction in the particular protein pathway is characteristic of each cluster. (d) Schematic showing how the accumulation of stressors during the disease spectrum can negatively impact PPI to cascade in impaired cellular activity, intra-cellular communication, brain region connectivity, ultimately leading to decline seen in these disorders, such as AD. Abbreviations: AD, Alzheimer’s disease; NCI, no cognitive impairment
This indicates that these pathways are vulnerable to most stressors and cellular insults associated with AD. These findings are concordant with observations that neurons at higher risk in AD are particularly vulnerable to energy deprivation (Fu et al., 2018; Mattson & Magnus, 2006; Saxena & Caroni, 2011). In early-onset AD cases, mild cognitive deficit conditions, which frequently progress to AD, correlate with reduced glucose utilization in the brain (Johnson et al., 2012, 2020; Mattson & Arumugam, 2018). However, more proteins were negatively affected in AD cases AD1-AD3 than in AD cases AD4 and AD5 (see heatmap, red cluster), suggesting some stressors are more likely to impact these key pathways than others. Perhaps the magnitude of impact on these pathways is stressor dependent on an individual basis. Accordingly, assessing epichaperomes may prove to be a biosensor of the degree of dysfunction in a particular cell type and brain region (Figure 2d and (Ginsberg et al., 2021)). Specifically, the higher the epichaperome levels, the higher the number of proteins negatively impacted upon, and in turn, the higher the severity of perturbation to the complex network of molecular interactions in affected cells (Kishinevsky et al., 2018; Rodina et al., 2016). Importantly, dismantling epichaperomes may be envisioned as a viable and potentially transformative therapeutic approach to rewire network dysfunctions and revert brain cells, and in turn brain connectomes, to the pre-stressor, non-pathological states (Inda et al., 2020).
Other key protein pathways such as signaling, inflammatory/immune processes, trafficking, and stress response are likely stressor specific, as we find in each cluster different proteins to map to these pathways (see color coding of circles, Figure 2). Clustering also points to another interesting observation, in that patients AD4 and AD5 share a subset of proteins with the NCI subset (see heatmap, teal/green cluster). Also intriguing is that ‘Response to steroid hormones’, which encompasses the classical hormone-mediated response of the brain to stressors conditions (McEwen, 2007), is another epichaperome-mediated network dysfunction also found in the teal/green protein subset. Together with the low representation of the AD4 and AD5 interactomes in the AD-specific cluster (i.e., red), the teal/green protein subset may represent an intermediate and/or transitional state, as it relates to interactome and phenotype dysfunction and its magnitude, in the transition from NCI to AD, a hypothesis that merits further investigation. This postulate agrees with the hypothesis put forward by Pico Caroni and others in the field (Mattson & Magnus, 2006; McEwen et al., 2015; Saxena & Caroni, 2011; De Strooper & Karran, 2016), where the gradual accumulation of stressors (e.g., genetic, environmental, lifestyle, among others) may reach a critical mass stressor threshold that tips the balance in vulnerable neuron from function to dysfunction, leading to subsequent pathology. In this scenario, neurodegenerative disorders are seen as a spectrum and/or continuum in the accumulation of stressors that can negatively impact cellular activity, intra-cellular communication, brain region connectivity, ultimately leading to decline seen in these disorders (Figure 2d).
6 |. SPATIO-TEMPORAL TRAJECTORY OF EPICHAPEROME FORMATION
iPSC-derived neurons represent a model and embodiment of alterations that occur early in the disease spectrum before the formation of pathologic aggregates. Epichaperome detection in these cells (Inda et al., 2020; Kishinevsky et al., 2018) is supportive of epichaperome formation as an early hallmark across the disease spectrum. Employing PS19 mice which express a mutant tau gene, abundant epichaperome formation was observed as early as 3 months of age (MO) in forebrain structures implicated in AD, but little in other brain regions relatively spared in AD (Inda et al., 2020; Robinson, 2020). For example, epichaperomes were found within the hippocampal formation, including the entorhinal cortex, hippocampus, and subiculum, as well as the amygdala. The dentate gyrus and hippocampal CA3 layer had highest presence at this age (Inda et al., 2020). In neocortex, epichaperome expression was highest in temporal lobe areas adjacent to the hippocampal formation and amygdala, with a pattern of spreading toward somatosensory areas (Inda et al., 2020).
Epichaperome abundance was age-dependent with formation in the PS19 hippocampus displaying a linear increase between 3 MO and 8 MO, followed by a plateau. With increasing age, the presence of epichaperome staining became evident in most brain areas (Inda et al., 2020). Epichaperome expression reached a maximal value earlier in the hippocampus when compared to other areas and also retained the highest level of epichaperomes at all ages, commensurate with the hypothesized nidus of human AD pathology (Inda et al., 2020).
These studies in mice, together with proof-of-principle epichaperome imaging in human patients (Inda et al., 2020), indicate that epichaperome-sensitive brain areas largely coincide with those obtained through systematic mapping of stress-responsive circuits in the brain (Sousa & Almeida, 2012). For example, these stress-responsive areas in the brain are organized into functionally integrated networks of the hippocampus, amygdala, brainstem nuclei (locus coeruleus and raphe), hypothalamus, orbitofrontal cortex, prefrontal cortex, and striatum (Sousa & Almeida, 2012). This complex stress-related connectivity may explain why disruptive effects of stressors are not restricted to a single brain function and likely involve numerous interactomes with functional consequences. For example, stressors may initially impede hippocampus-dependent declarative memory but progressively interferes with corticocortical-dependent executive functions (e.g., behavioral flexibility, decision-making, and working memory), as well as behavioral domains (e.g., anxiety, fear, and mood, among others) that are regulated by multiple limbic and affective brain circuits (Godoy et al., 2018; Goldfarb et al., 2020; McEwen & Gianaros, 2010).
In contrast with Tg mice, no or little epichaperomes were detected by Inda and colleagues in age-paired WT mice or in the brains of NCI human subjects (Inda et al., 2020). Importantly, individual chaperones and co-chaperones including HSP90, HSP70, and HSP110, key building blocks for the epichaperomes (Inda et al., 2020; Rodina et al., 2016), were expressed at similar levels throughout the brain, and not significantly different between PS19 and WT mice (Inda et al., 2020).
Epichaperome formation in PS19 mice preceded the appearance of hyperphosphorylated tau, as determined by Inda and colleagues through AT8 staining (Inda et al., 2020) a marker for tau pathology, and as evaluated by Maruyuma and colleagues using tau ligands for visualizing multiple tau inclusions (Maruyama et al., 2013). Notably, epichaperome formation preceded microglial activation (imaged with [11C]-AC-5216, TSPO tracer as a surrogate) (Maeda et al., 2011), MRI-measurable regional atrophy (Yoshiyama et al., 2007), and impairments of cerebral glucose metabolism (imaged with [18F]-FDG (Maeda et al., 2011) in PS19 mice (Inda et al., 2020). Taken together, these preclinical studies support the hypothesis that epichaperomes and associated dysfunctions in protein pathways, may precede morphometric changes, pathological deposits, synaptic loss, and metabolic dysfunction in the context of AD, proposing a role for epichaperomes as a biomarker for early detection of stressor-induced pathologic changes in neuronal cells and brain regions that may foretell disease.
Another key question is whether aging itself is a sufficiently powerful stressor to tip the balance in vulnerable neurons and/or other cells from function to dysfunction through epichaperome formation, ultimately leading to pathological and functional deficits. Initial assessments indicate significantly higher levels in the expression of epichaperomes within postmortem AD brains compared to age-matched NCI subjects (Inda et al., 2020). The spatio-temporal analysis of epichaperome expression in the PS19 mouse model of tauopathy showed its specific presence in disease-relevant regions of the brain but no expression in age-matched mice even at advanced age and despite similar overall expression of chaperones in both the Tg and the WT mice (Inda et al., 2020). Whole-body epichaperome imaging data by positron emission tomography (PET) is now emerging from both mice and humans to indicate disease specific presentation irrespective of age (Bolaender et al., 2021; Jhaveri et al., 2020; Pillarsetty et al., 2019). This body of evidence indicates that, if epichaperomes form in the organism undergoing normal aging, they are either transient in nature and/or form at an expression level that is undetectable by sensitive techniques such as positron emission tomography. Therefore, a reasonable hypothesis to put forth and test is that additional stressors and/or vulnerabilities in tandem with aging may be needed to initiate pathologic changes seen in neurodegenerative conditions and related phenotypes in animal and cellular models.
7 |. TARGETING AND IMAGING THE EPICHAPEROME FOR THERAPEUTIC INTERVENTION IN NEURODEGENERATIVE DISORDERS
7.1 |. Targeting epichaperomes
How is it possible to target a chaperone when incorporated into epichaperomes specifically over the chaperone itself? Considering the high abundance and ubiquity of chaperones in the human body when compared to the small fraction in the epichaperome it would appear as an impossible task. However, it is crucial to consider when incorporated into an epichaperome, a chaperone is both structurally and dynamically distinct (Bolaender et al., 2021; Inda et al., 2020; Joshi et al., 2018; Kishinevsky et al., 2018; Rodina et al., 2016; Yan et al., 2020). An epichaperome is a unit as distinct from a chaperone as another protein altogether. A short overview on how small molecules that bind and dismantle epichaperomes were discovered is presented.
As described by Taldone and coworkers (Taldone et al., 2020), the availability of small molecule chemical probes, including PU-H71 (Figure 3a) made the discovery of epichaperomes possible. While initially discovered as a potential HSP90 inhibitor (He et al., 2006), it was soon realized that the PU-H71 probe preferred and acted on a subset or a pool of HSP90 as found in cancer cells, and that this pool was absent in non-transformed, non-cancerous cells (Goldstein et al., 2015; Kucine et al., 2015; Moulick et al., 2011; Zong et al., 2015). This HSP90 pool contained tightly bound chaperones and co-chaperones as opposed to the transient HSP90-co-chaperone folding complexes (Moulick et al., 2011). Subsequent investigations revealed this pool to be the fraction of HSP90 incorporated into epichaperomes (Inda et al., 2020; Kishinevsky et al., 2018; Rodina et al., 2016).
FIGURE 3.
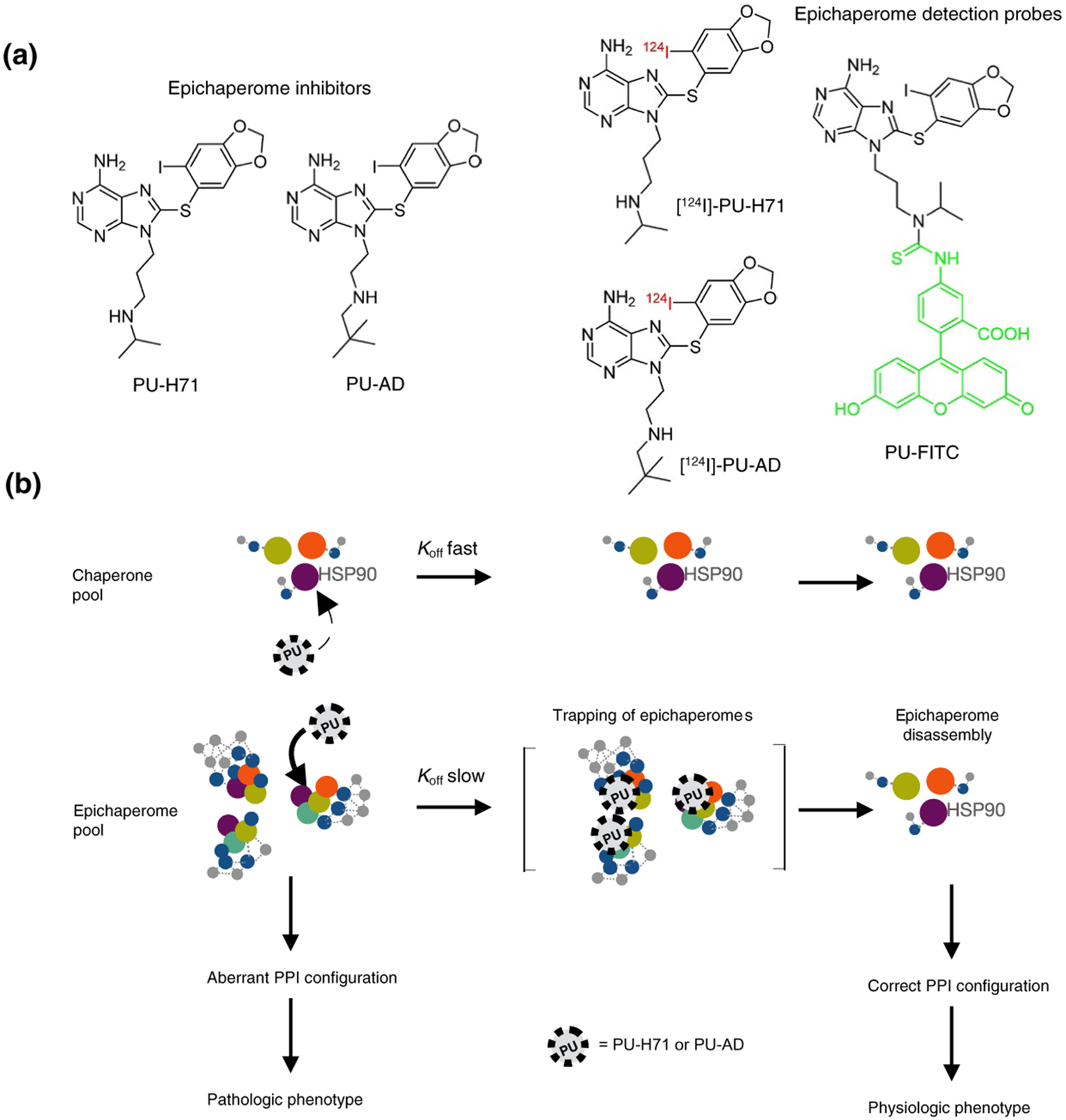
Chemical structure of epichaperome inhibitors and epichaperome detection probes (a) and the biochemical mechanism for their selective binding to HSP90-incorporated into epichaperomes over the abundant and ubiquitously expressed HSP90 chaperone pool, and for their restorative effect on cellular function (b). Abbreviations: Koff, dissociation rate constant
When the crystal structure of PU-H71 bound to HSP90 was first revealed by the Gewirth laboratory (Immormino et al., 2006), it indicated a binding mode that was distinct from classical HSP90 inhibitors. Albeit most such inhibitors occupy the ATP-binding pocket of HSP90, they do so in distinct ways as discussed by Wang and colleagues (Wang et al., 2019). For example, the ATP-binding pocket of HSP90 can be subdivided into three major sub-pockets (referred to as A, B and C) (Taldone et al., 2013). The binding of geldanamycin, the classical HSP90 inhibitor (Neckers, 2006), was restricted to sub-pockets A and C (Taldone et al., 2013). Conversely, PU-H71 inserted also into sub-pocket B, creating a new binding channel (Immormino et al., 2006; Taldone et al., 2013). This binding mode is of importance as ligands that bind the pocket configuration unveiled by PU-H71, termed the “helical” conformation of HSP90 by Amaral and colleagues (Amaral et al., 2017), display much slower dissociation rates when compared to similar ligands which bind in the configuration of geldanamycin bound HSP90, termed the “loop” conformation of HSP90 (Amaral et al., 2017). The conformation adapted by HSP90 can therefore greatly influence the residence time (i.e., the dissociation rate constant) of an inhibitor on HSP90. These analyses were made with recombinant HSP90, but one can envision a similar situation in cells where the epichaperome-permissive and PU-H71 preferred conformation of HSP90 is enriched. Concordantly, PU-H71 was found to dissociate much slower from epichaperome-positive cell homogenates than from epichaperome-negative cell homogenates despite equally high levels of HSP90 in both homogenates (Rodina et al., 2016). Thus, PU-H71 prefers epichaperomes over HSP90 by kinetically selecting for the epichaperome-HSP90 pool even if its affinity (i.e., Kd) for other intracellular HSP90 pools may (or may not) be the same (Figure 3b).
As discussed by Taldone and colleagues (Taldone et al., 2020), a classic example of kinetic selectivity in discerning seemingly similar protein targets comes from drugs that target muscarinic receptors (MRs) for the treatment of asthma (Moulton & Fryer, 2011). Inhibitors of MRs, such as atropine and scopolamine, naturally occurring in several nightshade family plants, have been in medicinal use since ancient times. The pharmacologically effective dose range for atropine and other MR inhibitors is close to the toxic range, limiting treatment use (Moulton & Fryer, 2011). There are several MR receptors, with the M3 being responsible for the anti-asthma activity of these inhibitors and the M2 subtype for toxicity (Moulton & Fryer, 2011). The muscarinic antagonists developed to date, including Atropium, Ipatropium, Clidinium, and Tiotropium, are characterized by similar Kds, (i.e., equal affinity for the three MR receptor subtypes) (Moulton & Fryer, 2011). Yet, tiotropium, despite having equal affinity for the M1, M2, and M3 receptors, is kinetically, and in turn functionally selective, for M3 receptors (Moulton & Fryer, 2011). This selectivity is provided by the ability of tiotropium to dissociate from M2 receptors 10 times faster than it does from M3 receptors (T1/2 = 3.6 h for M2 vs. T1/2 = 34.7 h for M3) (Moulton & Fryer, 2011). This kinetic selectivity for the M3 receptor provides tiotropium with a superior safety profile over the other inhibitors and supports its clinical application for chronic obstructive pulmonary disease.
To put PU-H71 in this context, its dissociation from HSP90 occurs in minutes whereas its off-rate from epichaperomes is in contrast measured in days (Jhaveri et al., 2020; Pillarsetty et al., 2019; Rodina et al., 2016). Since PU-H71 kinetically discriminates the small fraction of HSP90 in epichaperome from the abundant HSP90 pools found along epichaperomes in the same cell and throughout the body, radiolabeled or fluorescently-labeled versions of PU-H71 can be used as in vivo epichaperome detection reagents (Figure 3a) (Bolaender et al., 2021; Inda et al., 2020; Jhaveri et al., 2020; Merugu et al., 2020; Pillarsetty et al., 2019; Sugita et al., 2021). If the in vivo off-target of PU-H71 (and PU-AD, see further) was the chaperone HSP90, high nonspecific uptake would occur not only in all epichaperome-positive tumors but also in epichaperome-negative tumors and in all tissues and organs. This would be evident because HSP90 is one of the most abundantly expressed proteins in a cell (see Section 2.3 above). This is decidedly not the case, indicating that uptake is highly specific for epichaperomes (Bolaender et al., 2021; Inda et al., 2020; Jhaveri et al., 2020; Merugu et al., 2020; Pillarsetty et al., 2019; Sugita et al., 2021).
The PU-H71 and PU-AD agents and their radiolabeled versions rapidly clear from the human body and normal cells but stay lodged in epichaperome-positive cells proving the favorable signal-to-noise ratio needed for diagnostic purposes, such as for target engagement and/or imaging (Bolaender et al., 2021; Dunphy et al., 2020; Inda et al., 2020; Pillarsetty et al., 2019). This property of PU-H71 can also provide a favorable therapeutic index. For example, in poor prognosis malignancies such as metastatic triple negative breast cancers (Jhaveri et al., 2020) and myeloproliferative neoplasms transformed to acute myeloid leukemia (Sugita et al., 2021), clinical studies with PU-H71 demonstrated that epichaperome inhibition leads to durable responses and is safe.
Unfortunately, PU-H71 is not an effective probe or treatment for neurodegenerative disorder or other CNS maladies as it poorly crosses the blood-brain barrier (BBB). Through medicinal chemistry modifications, this liability of PU-H71 was rectified by Bolaender and coworkers (Bolaender et al., 2021) resulting in the BBB-permeable agent PU-AD (Figure 3a). PU-AD is structurally very close to PU-H71 and retains its epichaperome selectivity as well as its safety profile (Bolaender et al., 2021; Inda et al., 2020). For example, Inda and coworkers (Inda et al., 2020) conducted a standard safety study of PU-AD administered chronically to healthy C57/BL6 mice with mice dosed three times per week for 90 to 180 days (40–70 PU-AD doses) at a dose of 75 mg kg−1 (i.e., target saturating dose under intraperitoneal (i.p.) administration as determined through co-injection with radiolabeled PU-AD). No mortality or morbidity was observed during treatment or observation periods and no significant weight loss was found (Inda et al., 2020). Hematological and clinical chemistry findings were normal. Histopathology of major organs showed no toxic changes induced by PU-AD treatment (Inda et al., 2020). PU-AD was also evaluated when given over the entire adulthood of PS19 mice (>250 PU-AD doses; 3×per week 75 mg kg−1 target saturating under i.p. administration). In PS19 mice, tau pathology typically results in decreased survival because of severe paralysis (Yoshiyama et al., 2007). Inda and coworkers (Inda et al., 2020) found that PU-AD administration from 3 MO until death was well tolerated and significantly increased survival, with 50% of treated male mice having a 100–150 day delay in the terminal paralysis phenotype (p = 0.0022).
Importantly, PU-AD has transitioned to clinical investigation in human patients. A single and multiple ascending dose study of PU-AD given orally was initiated to evaluate the safety and pharmacokinetics of PU-AD in healthy subjects (ClinicalTrials.gov, 2019 [NCT03935568]). At the 2020 AAIC meeting in San Diego it was reported that there were no serious adverse events or deaths, and no subject experienced an adverse events leading to discontinuation from the study (Silverman et al., 2020). The majority of adverse events were reported as Grade 1 and self-limited (Silverman et al., 2020). All doses were well-tolerated in clinical subjects with no dose limiting adverse events observed (Silverman et al., 2020). PU-AD was detected in CSF samples providing evidence of brain penetration (Silverman et al., 2020). Based on these studies, a Phase 2a in AD with mild AD dementia was initiated planning for a recruitment of 150 patients under a 6 months of treatment paradigm. The study is designed as a classic, randomized, double blind, placebo controlled, parallel group study including PU-AD and matching placebo, designed to assess safety, tolerability, and pharmacological effects of oral PU-AD (dihydrochloride salt) in subjects with mild AD (ClinicalTrials.gov, 2020 [NCT04311515]).
In our opinion, epichaperome inhibitor treatment should be dictated by diagnostic identification both of epichaperome formation and abundance (i.e. where and how much), which as proposed by data presented in Section 6, would occur presymtomatically and increase longitudinally as disease progresses. Probes for epichaperome PET imaging, thus amenable for both epichaperome detection and quantitation, have been developed using I-124 radiotracers and are currently optimized with F-18 isotope incorporation (Bolaender et al., 2021; Inda et al., 2020; Pillarsetty et al., 2019), as we detail on below. It is also important that introduction of epichaperome inhibitors to treatment is guided by adequately chosen dose and schedule regimens as dictated by the specific nature of the target and pharmacokinetic and pharmacodynamic profile of the inhibitor, as discussed previously (Bolaender et al., 2021; Pillarsetty et al., 2019; Sugita et al., 2021).
7.2 |. Imaging, tracking, and quantifying epichaperomes
Leveraging the kinetic selectivity of PU-H71 for the epichaperome over HSP90, an I-124 radiolabeled version, [124I]-PU-H71, was made by Pillarsetty and coworkers (Pillarsetty et al., 2019) for positron emission tomography (PET) imaging (Figure 3a). Over 60 patients were imaged using [124I]-PU-H71 in cancer to show the feasibility of detecting epichaperomes in live human beings through imaging (Dunphy et al., 2020; Jhaveri et al., 2020; Pillarsetty et al., 2019). Combined with therapeutic trials of PU-H71, this imaging agent has been used to demonstrate its use in patient selection (e.g., identify epichaperome-positive tumors and evaluate target expression levels in individual tumors), in evaluating target engagement by a specific administered dose of PU-H71 and in determining real-time molar concentrations of PU-H71 at the site of action (Dunphy et al., 2020; Jhaveri et al., 2020; Pillarsetty et al., 2019). Thus, the combination of drug (e.g., PU-H71), companion diagnostic (e.g., [124I]-PU-H71 for solid tumors (Pillarsetty et al., 2019; Jhaveri et al., 2020) and PU-FITC for hematologic malignancies (Merugu et al., 2020; Sugita et al., 2021) and biomarker (e.g., epichaperome levels (Rodina et al., 2016)) enables a precision medicine approach in epichaperome therapy that is based of stressor-induced molecular dysfunctions rather than genetics or pathology.
Taking lessons from oncology paradigms, a similar platform was also designed for neurodegenerative disorders, and a radiolabeled PU-AD was developed – [124I]-PU-AD (Figure 3a) was developed by Bolaender and colleagues (Bolaender et al., 2021). Because the therapeutic agent PU-AD has an endogenous iodide, [124I]-PU-AD was created by Bolaender and colleagues (Bolaender et al., 2021) by exchanging this naturally occurring iodine-127 isotope with iodine-124. In addition to studies in mice where [124I]-PU-AD was used to determine the spatio-temporal trajectory of epichaperomes in transgenic mice (Inda et al., 2020), the [124I]-PU-AD probe was used to provide proof-of-principle demonstration of its brain permeability in human patients (n = 3) via imaging (Bolaender et al., 2021). PET imaging with [124I]-PU-AD confirmed the feasibility of epichaperomes detection and quantitation, in addition to demonstrating their localization in AD relevant regions but not in relatively spared regions (Bolaender et al., 2021; Inda et al., 2020), enabling the possibility of diagnostic detection for therapeutic intervention.
While first-generation CNS epichaperome PET probes were instrumental in providing proof-of-principle, the next step is to make imaging probes with wider acceptability by the neuroscience and neurology communities. As noted by Cascini and colleagues (Cascini et al., 2014), I-124 labeled PET probes are less practical for routine clinical use in patients because of several disadvantages: (i). decay properties (long half-life, low positron ratio, high energy positron, prompt gammas) and (ii). production challenges (requires solid enriched target, low production yields leading to high costs). Therefore, a logical next step is to develop probes radiolabeled with isotopes, such as 18F, that have shorter half-life, high β+ emission efficiency, lower β+ energy and easier production (Jacobson et al., 2015). This is expected to significantly improve sensitivity and spatial and temporal image quality, enable dynamic imaging, reduce radiation burden and imaging times, and increase acceptance and availability for widespread clinical application. This type of companion diagnostic would be invaluable in the development of epichaperome drugs with use in patient selection, in monitoring target engagement by inhibitor in real time, as well as for quantitative evaluations that can be used to optimize dose and schedule selection, as has been successful for the non-CNS drug PU-H71 (Pillarsetty et al., 2019).
A practical and innovative aspect for a probe such as [18F]-PU-AD is that it could be added to the currently available imaging biomarker portfolio for CNS disorders such as AD, as the biology and preliminary imaging in mice predict that the resulting epichaperome imaging may capture the anticipated spatio-temporal progression of the disease from very early stage. To our knowledge, there is no other probe to detect pathologic stressor-induced molecular dysfunction that foretells AD onset and precedes morphometric changes (as measured by MRI), pathological deposits (as measured by tau PET), inflammation (as measured by microglia activation), and metabolic dysfunction (as measured by fluorodeoxyglucose PET). Early stage detection may facilitate early clinical intervention. While it is true [18F]-PU-AD imaging will not differentiate AD pathologic events from those of another underlying pathology, for diagnosis in AD and other neurodegenerative disorders, there is undoubtedly a need for composite neuroimaging biomarkers that combine information about molecular alterations; amyloid and tau aggregation; structural and functional alterations; and synaptic, cellular, and molecular dysfunction (Khoury & Ghossoub, 2019; Young et al., 2020). Different imaging modalities offer complementary information, and the spatial distribution of these obtained measurements can generate valuable information that can be used for tracking and staging within individuals and groups that is currently unavailable and extremely important for rational therapeutic intervention moving forward. By this token, pathologic tau formations, inflammation and synaptic loss are a hallmark of AD but also of many other diseases of the CNS – yet several imaging probes detecting such disease hallmarks are of wide interest for development in AD diagnosis that can be used in combination/association with epichaperome detectors/inhibitors.
8 |. SUMMARY AND CONCLUSIONS
In this piece, we discuss how the maladaptation to stressors, ranging from genotoxic to environmental, negatively impacts an organism holistically as well as being deleterious to intracellular and intercellular networks, which we posit is especially detrimental to the critical cellular interactomes and brain connectomes that underlie normative function. This is of particular interest in the context of the etiology of CNS diseases, especially late-onset neurodegenerative disorders, where stressors are likely to aggregate over time exacerbating vulnerabilities in forebrain circuits which are decimated in AD.
We present supporting evidence suggesting neurological disorders can be viewed as protein connectivity dysfunctions through aberrant functional scaffolds (e.g., epichaperomes) induced by a combination of stressors in vulnerable brain cells and circuits, which causes faulty intercellular communication leading to rewiring of brain regions, and ultimately compromised function. These endophenotypes are shaped by a variety of stressors, including aging, genetic risk alleles, environmental exposures, and lifestyle choices, with disease being a manifestation of the combined influence of several ‘hits’ to an organism. Therefore, individuals likely damage vulnerable cells and brain circuitry to varying degrees over the course of the lifespan. Disease occurs when any combination of stressors damages enough PPIs, and in turn proteome functions, in vulnerable cells in specific tissues and organs beyond a tolerable threshold. This realization is of importance as it supports epichaperomics, the ‘omics method that uses epichaperomes to identify and study stressor-induced PPI alterations at the proteome-wide level (Ginsberg et al., 2021), is ideally poised to provide mechanistically and therapeutically relevant insights into context-dependent proteome-wide functional changes in CNS disorders.
Stress and stressors are able to influence the organism to alter its phenotype through global ‘top down’ brain-initiated actions as well as ‘bottom up’ alterations in protein–protein connectivity (i.e. interactomes) that can facilitate disease pathogenesis. We provide evidence for the importance of ‘bottom up interactome dysfunction’ which differs from the conventional stress biology concept of ‘top down’ in how stressor-to-phenotype alterations cause disease. We reiterate that these two concepts of stressor-driven disease mechanisms within the CNS are not mutually exclusive, and likely are complementary in terms of disease generation within the individual over the course of a lifetime.
With the development of chemical biology probes to interrogate epichaperomes in cancer (for proof-of-concept) and recently in neurodegenerative disorders including AD and PD and relevant animal/cellular models of these disorders, we provide an evidence-based view on how aberrant stressor-to-phenotype alterations through epichaperomes actually provide a unique precision medicine intervention roadmap for diagnostic and therapeutic development. This exciting new lineage of translational and potentially transformative research is especially prescient in the context of neurodegenerative disorders such as the AD spectrum where treatment options are limited, and early diagnosis is becoming increasingly more important. Assessment of epichaperome formation in neurodegenerative conditions, such as Huntington’s disease, motor neuron disease, or frontotemporal dementias, among others remain to be fully investigated and reported.
In conclusion, understanding mechanisms driving the stressor-to-phenotype phenomena and controlling these maladaptive responses in the wake of stressor presentation may lead to the development of rational therapeutic approaches to abrogate dysfunction from interactomes to connectomes which are now being increasingly appreciated as endemic to neurodegenerative disorders. Thus, detecting and modifying stressors-induced maladaptive responses through the epichaperome platform – from chemical probes to ‘omics approaches – may provide a previously unavailable treatment approach to disorders, including AD, that are currently awaiting viable interventions.
ACKNOWLEDGEMENTS
This work was supported by the U01 AG032969, R56 AG061869, R56 AG072599, R01 AG067598, R01 AG067598, P01 AG014449, P01 AG017617, Coins for Alzheimer’s Research, ADDF, the Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of the Memorial Sloan Kettering Cancer Center, P30 CA008748 (NCI Core Facility Grant).
Funding information
National Institute on Aging, Grant/Award Number: U01 AG032969, R56 AG061869, R56 AG072599, R01 AG067598 and R01 AG067598
Abbreviations:
- AD
Alzheimer’s disease
- AHA1
activator of HSP90 ATPase protein 1
- AMPAR
AMPA receptors
- APP
amyloid precursor protein
- Aβ
amyloid beta
- BBB
blood-brain barrier
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- CNS
central nervous system
- DLB
dementia with LBs
- ER
endoplasmic reticulum
- FTD-MND
frontotemporal dementia with motor neuron disease type
- GCIs
glial cytoplasmic inclusions
- GRP94
glucose regulated protein 94
- HOP
HSP-organizing protein
- HPA
hypothalamic-pituitary-adrenal
- HSP90s
heat shock protein 90s
- HSPs
heat shock proteins
- i.p.
intraperitoneal
- iHSP90
intense HSP90 immunoreactivity
- iPSCs
induced pluripotent stem cells
- LBs
Lewy bodies
- LBVAD
LB variant of Alzheimer’s disease
- LNs
Lewy neurites
- LTP
long-term potentiation
- mDA
midbrain dopaminergic
- MO
months of age
- MRs
muscarinic receptors
- MS
mass spectrometry
- MSA
multiple system atrophy
- PD
Parkinson’s disease
- PET
positron emission tomography
- pl
isoelectric point
- PPIs
protein-protein interactions
- SGK
serum- and glucocorticoid-regulated kinase
- TRAP1
tumor necrosis factor type 1 receptor-associated protein
- WT
wild-type
Key terms
- Acute stress or stressor
An immediate insult to a cell or organism that leads to a rapid reaction that dissipates quickly once the stressor event has passed
- Adaptive response
A rapid response of a cell or organism that protects from damage by external stressor agents and events and facilitates coping mechanisms against future stressors
- Chaperome
The collective name for chaperones, co-chaperones, and other factors that in a cell ensure that proteins have the correct structure and assembly for their function
- Chaperone
Protein that through one-on-one dynamic interactions assists in the conformational folding or unfolding as well as the assembly or disassembly of other macromolecular structures so they can work correctly
- Chronic stress or stressor
A prolonged and constant exposure of a cell or organism to a stressor or a combination of stressors that has detrimental effects, leading to a pathologic cellular phenotype and disease
- Connectome
An interaction network, from intercellular to brain regional to circuit level, that drives specific brain functions
- Epichaperomes
Malfunctioning long-lived assemblies of chaperome members that act as aberrant scaffolding structures leading to misorganized proteins including loss of normal protein–protein interactions and gain of abnormal protein–protein interactions within a vulnerable cell, exacerbating disease onset and progression
- Interactome
The complex network of protein–protein interactions in the cell
- Maladaptive response
A failure to cope with stress(ors) and/or to return to normalcy leading to vulnerability to stress(or)-associated pathology
- Stressor
An insult that can be of genetic, proteotoxic, or environmental in nature
Footnotes
CONFLICT OF INTEREST
Memorial Sloan Kettering Cancer Center holds the intellectual rights to PU-H71 and PU-AD. Samus Therapeutics Inc, of which G.C. has partial ownership, and is a member of its board of directors, has licensed this portfolio. G.C. is an inventor on the licensed intellectual property. All other authors declare no competing interests.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available at DOI: https://doi.org/10.1038/s41467-019-14082-5. These data were derived from the following resources available in the public domain: https://www.nature.com/articles/s41467-019-14082-5
REFERENCES
- Abraham WC, Jones OD, & Glanzman DL (2019). Is plasticity of synapses the mechanism of long-term memory storage? NPJ Science of Learning, 4, 9. 10.1038/s41539-019-0048-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaalm L, Crunden JL, Butcher M, Obst U, Whealy R, Williamson CE, O’Brien HE, Schaffitzel C, Ramage G, Spencer J, & Diezmann S (2021). Identification and phenotypic characterization of Hsp90 phosphorylation sites that modulate virulence traits in the major human fungal pathogen Candida albicans. Frontiers in Cellular and Infection Microbiology, 10.3389/fcimb.2021.637836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alashwal H, El Halaby M, Crouse JJ, Abdalla A, & Moustafa AA (2019). The application of unsupervised clustering methods to Alzheimer’s disease. Frontiers in Computational Neuroscience, 13, 31. 10.3389/fncom.2019.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M, Kokh DB, Bomke J, Wegener A, Buchstaller HP, Eggenweiler HM, Matias P, Sirrenberg C, Wade RC, & Frech M (2017). Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nature Communications, 8, 2276. 10.1038/s41467-017-02258-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, & Kelly JW (2008). Adapting proteostasis for disease intervention. Science, 319, 916–919. 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, & Hartl FU (2016). In vivo aspects of protein folding and quality control. Science, 353, aac4354. 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Barraclough R, & Ellis RJ (1980). Protein synthesis in chloroplasts. IX. Assembly of newly-synthesized large subunits into ribulose bisphosphate carboxylase in isolated intact pea chloroplasts. Biochimica Et Biophysica Acta, 608, 19–31. [DOI] [PubMed] [Google Scholar]
- Ben Zablah Y, Merovitch N, & Jia Z (2020). The role of ADF/Cofilin in synaptic physiology and Alzheimer’s disease. Frontiers in Cell and Developmental Biology, 8, 594998. 10.3389/fcell.2020.594998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertozzi CR, Stockwell BR, Kubicek S et al. (2015). Voices of chemical biology. Nature Chemical Biology, 11, 378–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohush A, Bieganowski P, & Filipek A (2019). Hsp90 and its co-chaperones in neurodegenerative diseases. International Journal of Molecular Sciences, 20, 4976. 10.3390/ijms20204976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolaender A, Zatorska D, He H, Joshi S, Sharma S, Digwal CS, Patel HJ, Sun W, Imber BS, Ochiana SO, Patel MR, Shrestha L, Shah SK, Wang S, Karimov R, Tao H, Patel PD, Martin AR, Yan P, … Chiosis G (2021). Chemical tools for epichaperome-mediated interactome dysfunctions of the central nervous system. Nature Communications, 12, 4669. 10.1038/s41467-021-24821-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, Pastores GM, Rubinsztein DC, Nixon RA, Duchen MR, Mallucci GR, Kroemer G, Levine B, Eskelinen E-L, Mochel F, Spedding M, Louis C, Martin OR, & Millan MJ (2018). Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nature Reviews Drug Discovery, 17, 660–688. 10.1038/nrd.2018.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudesco C, Cause S, Jego G, & Garrido C (2018). Hsp70: A cancer target inside and outside the cell. Methods in Molecular Biology, 1709, 371–396. [DOI] [PubMed] [Google Scholar]
- Brehme M, & Voisine C (2016). Model systems of protein-misfolding diseases reveal chaperone modifiers of proteotoxicity. Disease Models & Mechanisms, 9, 823–838. 10.1242/dmm.024703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, & Morimoto RI (2014). A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Reports, 9, 1135–1150. 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD (2006). Traumatic stress: Effects on the brain. Dialogues in Clinical Neuroscience, 8, 445–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brivio E, Lopez JP, & Chen A (2020). Sex differences: Transcriptional signatures of stress exposure in male and female brains. Genes, Brain, and Behavior, 19, e12643. 10.1111/gbb.12643. [DOI] [PubMed] [Google Scholar]
- Caino MC, & Altieri DC (2016). Molecular pathways: Mitochondrial reprogramming in tumor progression and therapy. Clinical Cancer Research, 22, 540–545. 10.1158/1078-0432.CCR-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon WB (1915). Bodily changes in pain, hunger, fear and rage: An account of recent researches into the function of emotional excitement: Bodily changes in pain, hunger, fear and rage: An account of recent researches into the function of emotional excitement. D Appleton & Company, New York, NY, US. [Google Scholar]
- Carman A, Kishinevsky S, Koren J 3rd, Lou W, & Chiosis G (2013). Chaperone-dependent neurodegeneration: A molecular perspective on therapeutic intervention. Journal of Alzheimers Disease and Parkinsonism, 2013, 007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascini GL, Niccoli Asabella A, Notaristefano A, Restuccia A, Ferrari C, Rubini D, Altini C, & Rubini G (2014). 124Iodine: A longer-life positron emitter isotope—new opportunities in molecular imaging. BioMed Research International, 2014, 672094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadli A, Ladjimi MM, Baulieu EE, & Catelli MG (1999). Heat-induced oligomerization of the molecular chaperone Hsp90. Inhibition by ATP and geldanamycin and activation by transition metal oxyanions. Journal of Biological Chemistry, 274, 4133–4139. 10.1074/jbc.274.7.4133. [DOI] [PubMed] [Google Scholar]
- Chaumonnot K, Masson S, Sikner H, Bouchard A, Baverel V, Bellaye PS, Collin B, Garrido C, & Kohli E (2021). The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death & Disease, 12, 114. 10.1038/s41419-020-03288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnokova E, Bal N, & Kolosov P (2017). Kinases of eIF2a switch translation of mRNA subset during neuronal plasticity. International Journal of Molecular Sciences, 18, 2213. 10.3390/ijms18102213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, Puspita L, Valiulahi P, Sulzer D, Milner TA, Taldone T, Krainc D, Studer L, & Shim J-W (2016). Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and α-synuclein accumulation. Stem Cell Reports, 7, 664–677. 10.1016/j.stemcr.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta-catenin signaling in development and disease. Cell, 127, 469–480. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov. (2019) A Single and Multiple Ascending Dose Study to Evaluate the Safety and Pharmacokinetics of PU-AD in Healthy Subjects. ClinicalTrials.gov Identifier: NCT03935568, https://clinicaltrials.gov/ct2/show/record/NCT03935568. [Google Scholar]
- ClinicalTrials.gov. (2020) To Evaluate Safety, Tolerability and Pharmacological Effects of PU AD in Subjects with Mild AD Dementia. ClinicalTrials.gov Identifier: NCT04311515, https://clinicaltrials.gov/ct2/show/NCT04311515. [Google Scholar]
- Compta Y, & Revesz T (2021). Neuropathological and biomarker findings in Parkinson’s disease and alzheimer’s disease: From protein aggregates to synaptic dysfunction. Journal of Parkinson’s Disease, 11, 107–121. 10.3233/JPD-202323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RO, Ferreiro E, Cardoso SM, Oliveira CR, & Pereira CM (2010). ER stress-mediated apoptotic pathway induced by Abeta peptide requires the presence of functional mitochondria. Journal of Alzheimer’s Disease, 20, 625–636. [DOI] [PubMed] [Google Scholar]
- Counts SE, Alldred MJ, Che S, Ginsberg SD, & Mufson EJ (2014). Synaptic gene dysregulation within hippocampal CA1 pyramidal neurons in mild cognitive impairment. Neuropharmacology, 79, 172–179. 10.1016/j.neuropharm.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi BJ (2000). The evolution of maladaptation. Heredity (Edinb.), 84, 623–629. [DOI] [PubMed] [Google Scholar]
- Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM, & Forman MS (2004). Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. American Journal of Pathology, 164, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Joels M, & Holsboer F (2005). Stress and the brain: From adaptation to disease. Nature Reviews Neuroscience, 6, 463–475. 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- De Strooper B, & Karran E (2016). The Cellular phase of Alzheimer’s disease. Cell, 164, 603–615. 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Hutton M, Burrows F, & Petrucelli L (2007). The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. Journal of Clinical Investigation, 117, 648–658. 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy MPS, Pressl C, Pillarsetty N, Grkovski M, Modi S, Jhaveri K, Norton L, Beattie BJ, Zanzonico PB, Zatorska D, Taldone T, Ochiana SO, Uddin MM, Burnazi EM, Lyashchenko SK, Hudis CA, Bromberg J, Schöder HM, Fox JJ, … Larson SM (2020). First-in-human trial of epichaperome-targeted PET in patients with cancer. Clinical Cancer Research, 26, 5178–5187. 10.1158/1078-0432.CCR-19-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Wahlster L, & McLean PJ (2011). Molecular chaperones in Parkinson’s disease–present and future. Journal of Parkinson’s Disease, 1, 299–320. 10.3233/JPD-2011-11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J (1987). Proteins as molecular chaperones. Nature, 328, 378–379. 10.1038/328378a0. [DOI] [PubMed] [Google Scholar]
- Ellis RJ (2006). Molecular chaperones: Assisting assembly in addition to folding. Trends in Biochemical Sciences, 31, 395–401. 10.1016/j.tibs.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Ellis RJ (2013). Assembly chaperones: A perspective. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 368, 20110398. 10.1098/rstb.2011.0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltokhi A, Santuy A, Merchan-Perez A, & Sprengel R (2021). Glutamatergic dysfunction and synaptic ultrastructural alterations in schizophrenia and autism spectrum disorder: Evidence from human and rodent studies. International Journal of Molecular Sciences, 22, 59. 10.3390/ijms22010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finka A, & Goloubinoff P (2013). Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress and Chaperones, 18, 591–605. 10.1007/s12192-013-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finka A, Mattoo RU, & Goloubinoff P (2011). Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress and Chaperones, 16, 15–31. 10.1007/s12192-010-0216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Hardy J, & Duff KE (2018). Selective vulnerability in neurodegenerative diseases. Nature Neuroscience, 21, 1350–1358. 10.1038/s41593-018-0221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner CC, & Wetmore DZ (2012). Synaptic pathology of down syndrome. Advances in Experimental Medicine and Biology, 970, 451–468. [DOI] [PubMed] [Google Scholar]
- Ge P, Dawson VL, & Dawson TM (2020). PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Molecular Neurodegeneration, 15, 20. 10.1186/s13024-020-00367-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Hemby SE, & Smiley JF (2012). Expression profiling in neuropsychiatric disorders: Emphasis on glutamate receptors in bipolar disorder. Pharmacology, Biochemistry and Behavior, 100, 705–711. 10.1016/j.pbb.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Neubert TA, Sharma S, Digwal CS, Yan P, Timbus C, Wang T, & Chiosis G (2021). Disease-specific interactome alterations via epichaperomics: The case for Alzheimer’s disease. FEBS Journal, 10.1111/febs.16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy LD, Rossignoli MT, Delfino-Pereira P, Garcia-Cairasco N, & de Lima Umeoka EH (2018). A comprehensive overview on stress neurobiology: Basic concepts and clinical implications. Frontiers in Behavioural Neurosciences, 12, 127. 10.3389/fnbeh.2018.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb EV, Rosenberg MD, Seo D, Constable RT, & Sinha R (2020). Hippocampal seed connectome-based modeling predicts the feeling of stress. Nature Communications, 11, 2650. 10.1038/s41467-020-16492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RL, Yang SN, Taldone T, Chang B, Gerecitano J, Elenitoba-Johnson K, Shaknovich R, Tam W, Leonard JP, Chiosis G, Cerchietti L, & Melnick A (2015). Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. Journal of Clinical Investigation, 125, 4559–4571. 10.1172/JCI80714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P, Gatenby AA, & Lorimer GH (1989). GroE heat-shock proteins promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase oligomers in Escherichia coli. Nature, 337, 44–47. 10.1038/337044a0. [DOI] [PubMed] [Google Scholar]
- Hadizadeh Esfahani A, Sverchkova A, Saez-Rodriguez J, Schuppert AA, & Brehme M (2018). A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLoS Computational Biology, 14, e1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, Wu N, Immormino RM, Gewirth DT, & Chiosis G (2006). Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. Journal of Medicinal Chemistry, 49, 381–390. 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- Henstridge CM, Pickett E, & Spires-Jones TL (2016). Synaptic pathology: A shared mechanism in neurological disease. Ageing Research Reviews, 28, 72–84. 10.1016/j.arr.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, Scheimann J, & Myers B (2016). Regulation of the hypothalamic-pituitary-adrenocortical stress response. Comprehensive Physiology, 6, 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höhn A, Tramutola A, & Cascella R (2020). Proteostasis failure in neurodegenerative diseases: Focus on oxidative stress. Oxidative Medicine and Cellular Longevity, 2020, 1–21. 10.1155/2020/5497046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, & Nicoll RA (2013). AMPARs and synaptic plasticity: The last 25 years. Neuron, 80, 704–717. 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison KA, Stancato LF, Jove R, & Pratt WB (1992). The protein-protein complex between pp60v-src and hsp90 is stabilized by molybdate, vanadate, tungstate, and an endogenous cytosolic metal. Journal of Biological Chemistry, 267, 13952–13957. 10.1016/S0021-9258(19)49662-2. [DOI] [PubMed] [Google Scholar]
- Immormino RM, Kang Y, Chiosis G, & Gewirth DT (2006). Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. Journal of Medicinal Chemistry, 49, 4953–4960. 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- Inda MC, Joshi S, Wang T, Bolaender A, Gandu S, Koren J III, Che AY, Taldone T, Yan P, Sun W, Uddin M, Panchal P, Riolo M, Shah S, Barlas A, Xu KE, Chan LYL, Gruzinova A, Kishinevsky S, … Chiosis G (2020). The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nature Communications, 11, 319. 10.1038/s41467-019-14082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson O, Kiesewetter DO, & Chen X (2015). Fluorine-18 radio-chemistry, labeling strategies and synthetic routes. Bioconjugate Chemistry, 26, 1–18. 10.1021/bc500475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri KL, Dos Anjos CH, Taldone T et al. (2020). Measuring tumor epichaperome expression using [(124)I] PU-H71 positron emission tomography as a biomarker of response for PU-H71 plus nab-paclitaxel in HER2-negative metastatic breast cancer. JCO Precision Oncology, 4, 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, Thambisetty M, Montine TJ, Lee EB, Trojanowski JQ, Beach TG, Reiman EM, Haroutunian V, Wang M, Schadt E, … Seyfried NT (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nature Medicine, 26, 769–780. 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Fox NC, Sperling RA, & Klunk WE (2012). Brain imaging in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 2, a006213. 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL, & Chiosis G (2018). Adapting to stress – chaperome networks in cancer. Nature Reviews Cancer, 18, 562–575. 10.1038/s41568-018-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice NJ (2018). The relationship between stress and Alzheimer’s disease. Neurobiol Stress, 8, 127–133. 10.1016/j.ynstr.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallarackal AJ, Kvarta MD, Cammarata E, Jaberi L, Cai X, Bailey AM, & Thompson SM (2013). Chronic stress induces a selective decrease in AMPA receptor-mediated synaptic excitation at hippocampal temporoammonic-CA1 synapses. Journal of Neuroscience, 33, 15669–15674. 10.1523/JNEUROSCI.2588-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, & Burrows FJ (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature, 425, 407–410. 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kampinga HH, & Bergink S (2016). Heat shock proteins as potential targets for protective strategies in neurodegeneration. The Lancet Neurology, 15, 748–759. 10.1016/S1474-4422(16)00099-5. [DOI] [PubMed] [Google Scholar]
- Karagöz GE, Duarte AMS, Akoury E, Ippel H, Biernat J, Morán Luengo T, Radli M, Didenko T, Nordhues BA, Veprintsev DB, Dickey CA, Mandelkow E, Zweckstetter M, Boelens R, Madl T, & Rüdiger SGD (2014). Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell, 156, 963–974. 10.1016/j.cell.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury R, & Ghossoub E (2019). Diagnostic biomarkers of Alzheimer’s disease: A state-of-the-art review. Biomarkers in Neuropsychiatry, 1, 100005. 10.1016/j.bionps.2019.100005. [DOI] [Google Scholar]
- Kishinevsky S, Wang T, Rodina A, Chung SY, Xu C, Philip J, Taldone T, Joshi S, Alpaugh ML, Bolaender A, Gutbier S, Sandhu D, Fattahi F, Zimmer B, Shah SK, Chang E, Inda C, Koren J, Saurat NG, … Studer L (2018). HSP90-incorporating chaperome networks as biosensor for disease-related pathways in patient-specific midbrain dopamine neurons. Nature Communications, 9, 4345. 10.1038/s41467-018-06486-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips CL, Jayaraj GG, & Hartl FU (2018). Pathways of cellular proteostasis in aging and disease. Journal of Cell Biology, 217, 51–63. 10.1083/jcb.201709072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim J-W, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, & Studer L (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature, 480, 547–551. 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugers HJ, Hoogenraad CC, & Groc L (2010). Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nature Reviews Neuroscience, 11, 675–681. 10.1038/nrn2913. [DOI] [PubMed] [Google Scholar]
- Kucine N, Marubayashi S, Bhagwat N, Papalexi E, Koppikar P, Sanchez Martin M, Dong L, Tallman MS, Paietta E, Wang K, He J, Lipson D, Stephens P, Miller V, Rowe JM, Teruya-Feldstein J, Mullighan CG, Ferrando AA, Krivtsov A, … Kleppe M (2015). Tumor-specific HSP90 inhibition as a therapeutic approach in JAK-mutant acute lymphoblastic leukemias. Blood, 126, 2479–2483. 10.1182/blood-2015-03-635821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy WY, Duennwald ML, Prado VF, & Prado MAM (2017). The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Frontiers in Neuroscience, 11, 254. 10.3389/fnins.2017.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackie RE, Marques-Lopes J, Ostapchenko VG, Good S, Choy WY, van Oosten-Hawle P, Pasternak SH, Prado VF, & Prado MAM (2020). Increased levels of stress-inducible phosphoprotein-1 accelerates amyloid-beta deposition in a mouse model of Alzheimer’s disease. Acta Neuropathologica Communications, 8, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskey RA, Honda BM, Mills AD, & Finch JT (1978). Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature, 275, 416–420. 10.1038/275416a0. [DOI] [PubMed] [Google Scholar]
- Lepvrier E, Moullintraffort L, Nigen M, Goude R, Allegro D, Barbier P, Peyrot V, Thomas D, Nazabal A, & Garnier C (2015). Hsp90 oligomers interacting with the Aha1 cochaperone: An outlook for the Hsp90 chaperone machineries. Analytical Chemistry, 87, 7043–7051. 10.1021/acs.analchem.5b00051. [DOI] [PubMed] [Google Scholar]
- Li J, Soroka J, & Buchner J (2012). The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochimica Et Biophysica Acta, 1823, 624–635. 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Lindberg I, Shorter J, Wiseman RL, Chiti F, Dickey CA, & McLean PJ (2015). Chaperones in Neurodegeneration. Journal of Neuroscience, 35, 13853–13859. 10.1523/JNEUROSCI.2600-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yuen EY, & Yan Z (2010). The stress hormone corticosterone increases synaptic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors via serum-and glucocorticoid-inducible kinase (SGK) regulation of the GDI-Rab4 complex. Journal of Biological Chemistry, 285, 6101–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes S, Vaz-Silva J, Pinto V, Dalla C, Kokras N, Bedenk B, Mack N, Czisch M, Almeida OFX, Sousa N, & Sotiropoulos I (2016). Tau protein is essential for stress-induced brain pathology. Proceedings of the National Academy of Sciences of the United States of America, 113, E3755–3763. 10.1073/pnas.1600953113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, & Chiosis G (2007). Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proceedings of the National Academy of Sciences of the United States of America, 104, 9511–9516. 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Zhang MR, Okauchi T et al. (2011). In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. Journal of Neuroscience, 31, 4720–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang M-R, Trojanowski JQ, Lee V-Y, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, Kudo Y, Chang Q, Saido TC, … Higuchi M (2013). Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron, 79, 1094–1108. 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, & Arumugam TV (2018). Hallmarks of brain aging: Adaptive and pathological modification by metabolic states. Cell Metabolism, 27, 1176–1199. 10.1016/j.cmet.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, & Magnus T (2006). Ageing and neuronal vulnerability. Nature Reviews Neuroscience, 7, 278–294. 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlary L, Plotkin SS, & Cashman NR (2019). Emerging developments in targeting proteotoxicity in neurodegenerative diseases. CNS Drugs, 33, 883–904. 10.1007/s40263-019-00657-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS (2005). Stressed or stressed out: What is the difference? Journal of Psychiatry and Neuroscience, 30, 315–318. [PMC free article] [PubMed] [Google Scholar]
- McEwen BS (2007). Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiological Reviews, 87, 873–904. 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, & Nasca C (2015). Mechanisms of stress in the brain. Nature Neuroscience, 18, 1353–1363. 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, & Gianaros PJ (2010). Central role of the brain in stress and adaptation: Links to socioeconomic status, health, and disease. Annals of the New York Academy of Sciences, 1186, 190–222. 10.1111/j.1749-6632.2009.05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, & Gianaros PJ (2011). Stress-and allostasis-induced brain plasticity. Annual Review of Medicine, 62, 431–445. 10.1146/annurev-med-052209-100430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merugu S, Sharma S, Kaner J, Digwal C, Sugita M, Joshi S, Taldone T, Guzman ML, & Chiosis G (2020). Chemical probes and methods for single-cell detection and quantification of epichaperomes in hematologic malignancies. Methods in Enzymology, 639, 289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, & Neckers L (2012). Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochimica Et Biophysica Acta, 1823, 648–655. 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI (1998). Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes & Development, 12, 3788–3796. 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morimoto RI (2008). Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & Development, 22, 1427–1438. 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, Vu LP, Zhao X, Zatorska D, Taldone T, Smith-Jones P, Alpaugh M, Gross SS, Pillarsetty N, Ku T, … Chiosis G (2011). Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nature Chemical Biology, 7, 818–826. 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton BC, & Fryer AD (2011). Muscarinic receptor antagonists, from folklore to pharmacology; finding drugs that actually work in asthma and COPD. British Journal of Pharmacology, 163, 44–52. 10.1111/j.1476-5381.2010.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski PJ, & Wacker JL (2005). Modulation of neurodegeneration by molecular chaperones. Nature Reviews Neuroscience, 6, 11–22. 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, & Vojtesek B (2013). C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene, 32, 3101–3110. 10.1038/onc.2012.314. [DOI] [PubMed] [Google Scholar]
- Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, Melnick A, Cerchietti L, Chiosis G, Wang YL, & Cesarman E (2013). Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood, 122, 2837–2847. 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L (2006). Chaperoning oncogenes: Hsp90 as a target of Geldanamycin. In Starke K, & Gaestel M (Eds.), Molecular chaperones in health and disease (pp. 259–277). Springer. [DOI] [PubMed] [Google Scholar]
- Noelker C, Morel L, Osterloh A, Alvarez-Fischer D, Lescot T, Breloer M, Gold M, Oertel WH, Henze C, Michel PP, Dodel RC, Lu L, Hirsch EC, Hunot S, & Hartmann A (2014). Heat shock protein 60: An endogenous inducer of dopaminergic cell death in Parkinson disease. Journal of Neuroinflammation, 11, 86. 10.1186/1742-2094-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novais A, Monteiro S, Roque S, Correia-Neves M, & Sousa N (2016). How age, sex and genotype shape the stress response. Neurobiol Stress, 6, 44–56. 10.1016/j.ynstr.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala PM (2013). Naughty chaperone as a target for viral cancer. Blood, 122, 2767–2768. 10.1182/blood-2013-08-522425. [DOI] [PubMed] [Google Scholar]
- Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, & Chiosis G (2013). Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nature Chemical Biology, 9, 677–684. 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillarsetty N, Jhaveri K, Taldone T, Caldas-Lopes E, Punzalan B, Joshi S, Bolaender A, Uddin MM, Rodina A, Yan P, Ku A, Ku T, Shah SK, Lyashchenko S, Burnazi E, Wang T, Lecomte N, Janjigian Y, Younes A, … Dunphy MPS (2019). Paradigms for precision medicine in epichaperome cancer therapy. Cancer Cell, 36, 559–573.e557. 10.1016/j.ccell.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Klyubin I, Ondrejcak T, Hu NW, & Rowan MJ (2021). Enduring glucocorticoid-evoked exacerbation of synaptic plasticity disruption in male rats modelling early Alzheimer’s disease amyloidosis. Neuropsychopharmacology, 46(12), 2170–2179. 10.1038/s41386-021-01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritossa F (1962). A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia, 18, 571–573. 10.1007/BF02172188. [DOI] [Google Scholar]
- Robinson R (2020). The epichaperome appears early in Alzheimer’s disease models and may drive tau pathology. NeurologyToday, 20, 18–13. [Google Scholar]
- Rodina A, Wang T, Yan P et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature, 538, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, & Munck AU (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocrine Reviews, 21, 55–89. [DOI] [PubMed] [Google Scholar]
- Saura CA, & Valero J (2011). The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Reviews in the Neurosciences, 22, 153–169. 10.1515/rns.2011.018. [DOI] [PubMed] [Google Scholar]
- Saxena S, & Caroni P (2011). Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron, 71, 35–48. 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- Schopf FH, Biebl MM, & Buchner J (2017). The HSP90 chaperone machinery. Nature Reviews Molecular Cell Biology, 18, 345–360. 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- Shrestha L, Patel HJ, & Chiosis G (2016). Chemical tools to investigate mechanisms associated with HSP90 and HSP70 in disease. Cell Chemical Biology, 23, 158–172. 10.1016/j.chembiol.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, Zhang Z, Huang Y, Temple S, Coppola G, Geschwind DH, Karydas A, Miller BL, Kosik KS, Gao F-B, Steen JA, & Haggarty SJ (2016). Human iPSC-derived neuronal model of Tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Reports, 7, 325–340. 10.1016/j.stemcr.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman M, Wallner B, Key C, Duggan SM, & Reynolds L (2020). Poster -A Single-and Multiple-Ascending Dose Study to Evaluate the Safety and Pharmacokinetics of Oral PU-AD, an Epichaperome Inhibitor to Treat Alzheimer’s Disease. Alzheimer’s Association International Conference (AAIC 2020). Poster #: 41144. Session: P3 [Posters: Drug Development] Human / Trial design. Date and Time WednesdayJuly 29, 2020: 12:00 AM – 11:59 AM [Google Scholar]
- Sotiropoulos I, Silva JM, Gomes P, Sousa N, & Almeida OFX (2019). Stress and the etiopathogenesis of Alzheimer’s disease and depression. Advances in Experimental Medicine and Biology, 1184, 241–257. [DOI] [PubMed] [Google Scholar]
- Sousa N (2016). The dynamics of the stress neuromatrix. Molecular Psychiatry, 21, 302–312. 10.1038/mp.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa N, & Almeida OF (2012). Disconnection and reconnection: The morphological basis of (mal)adaptation to stress. Trends in Neurosciences, 35, 742–751. 10.1016/j.tins.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Sugita M, Wilkes DC, Bareja R, Eng KW, Nataraj S, Jimenez-Flores RA, Yan LB, De Leon JP, Croyle JA, Kaner J, Merugu S, Sharma S, MacDonald TY, Noorzad Z, Panchal P, Pancirer D, Cheng S, Xiang JZ, Olson L, … Guzman ML (2021). Targeting the epichaperome as an effective precision medicine approach in a novel PML-SYK fusion acute myeloid leukemia. NPJ Precision Oncology, 5, 44. 10.1038/s41698-021-00183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R, McCampbell A, Leblanc G, Venkateswaran A, Nurmi A, & Hodgson R (2017). Protein misfolding in neurodegenerative diseases: Implications and strategies. Translational Neurodegeneration, 6, 6. 10.1186/s40035-017-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Patel PD, Patel M, Patel HJ, Evans CE, Rodina A, Ochiana S, Shah SK, Uddin M, Gewirth D, & Chiosis G (2013). Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. Journal of Medicinal Chemistry, 56, 6803–6818. 10.1021/jm400619b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Wang T, Rodina A et al. (2020). A Chemical biology approach to the chaperome in cancer-HSP90 and beyond. Cold Spring Harbor Perspectives in Biology, 12, a034116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AD, Scaglione KM, Prensner J, Gillies AT, Chinnaiyan A, Paulson HL, Jinwal UK, Dickey CA, & Gestwicki JE (2012). Analysis of the tau-associated proteome reveals that exchange of Hsp70 for Hsp90 is involved in tau degradation. ACS Chemical Biology, 7, 1677–1686. 10.1021/cb3002599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Richter-Landsberg C, Welch W et al. (2006). Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. American Journal of Pathology, 168, 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M, & Hartl FU (2010). Protein folding in the cytoplasm and the heat shock response. Cold Spring Harbor Perspectives in Biology, 2, a004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Rodrigues AJ, Silva JM, Tronche F, Almeida OF, Sousa N, & Sotiropoulos I (2016). Chronic stress and glucocorticoids: From neuronal plasticity to neurodegeneration. Neural Plasticity, 2016, 6391686. 10.1155/2016/6391686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace EWJ, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, Airoldi EM, Pan T, Budnik BA, & Drummond DA (2015). Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell, 162, 1286–1298. 10.1016/j.cell.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton-Diaz A, Khan S, Bourboulia D, Trepel JB, Neckers L, & Mollapour M (2013). Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Medicinal Chemistry, 5, 1059–1071. 10.4155/fmc.13.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Rodina A, Dunphy MP, Corben A, Modi S, Guzman ML, Gewirth DT, & Chiosis G (2019). Chaperome heterogeneity and its implications for cancer study and treatment. Journal of Biological Chemistry, 294, 2162–2179. 10.1074/jbc.REV118.002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR, & Balch WE (2006). Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell, 127, 803–815. 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Welikovitch LA, Do Carmo S, Maglóczky Z, Malcolm JC, Lőke J, Klein WL, Freund T, & Cuello AC (2020). Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain. Proceedings of the National Academy of Sciences of the United States of America, 117, 6844–6854. 10.1073/pnas.1914593117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, & Lindquist SL (2005). HSP90 and the chaperoning of cancer. Nature Reviews Cancer, 5, 761–772. 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Workman P, & Collins I (2010). Probing the probes: Fitness factors for small molecule tools. Chemistry & Biology, 17, 561–577. 10.1016/j.chembiol.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt AR, Yerbury JJ, Ecroyd H, & Wilson MR (2013). Extracellular chaperones and proteostasis. Annual Review of Biochemistry, 82, 295–322. 10.1146/annurev-biochem-072711-163904. [DOI] [PubMed] [Google Scholar]
- Yan P, Patel HJ, Sharma S, Corben A, Wang T, Panchal P, Yang C, Sun W, Araujo TL, Rodina A, Joshi S, Robzyk K, Gandu S, White JR, de Stanchina E, Modi S, Janjigian YY, Hill EG, Liu B, … Chiosis G (2020). Molecular stressors engender protein connectivity dysfunction through aberrant n-glycosylation of a chaperone. Cell Reports, 31, 107840. 10.1016/j.celrep.2020.107840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, & Lee V-Y (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron, 53, 337–351. 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Young PNE, Estarellas M, Coomans E, Srikrishna M, Beaumont H, Maass A, Venkataraman AV, Lissaman R, Jiménez D, Betts MJ, McGlinchey E, Berron D, O’Connor A, Fox NC, Pereira JB, Jagust W, Carter SF, Paterson RW, & Schöll M (2020). Imaging biomarkers in neurodegeneration: Current and future practices. Alzheimer’s Research & Therapy, 12, 49. 10.1186/s13195-020-00612-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Wei J, Liu W, Zhong P, Li X, & Yan Z (2012). Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron, 73, 962–977. 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, & Mandelkow EM (2010). Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. Journal of Neuroscience, 30, 11938–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Gozman A, Caldas-Lopes E, Taldone T, Sturgill E, Brennan S, Ochiana SO, Gomes-DaGama EM, Sen S, Rodina A, Koren J, Becker MW, Rudin CM, Melnick A, Levine RL, Roboz GJ, Nimer SD, Chiosis G, & Guzman ML (2015). A hyperactive signalosome in acute myeloid leukemia drives addiction to a tumor-specific Hsp90 species. Cell Reports, 13, 2159–2173. 10.1016/j.celrep.2015.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available at DOI: https://doi.org/10.1038/s41467-019-14082-5. These data were derived from the following resources available in the public domain: https://www.nature.com/articles/s41467-019-14082-5