

Abstract
Mitophagy is a highly conserved cellular process that maintains the mitochondrial quantity by eliminating dysfunctional or superfluous mitochondria through autophagy machinery. The mitochondrial outer membrane protein BNIP3L/Nix serves as a mitophagy receptor by recognizing autophagosomes. BNIP3L is initially known to clear the mitochondria during the development of reticulocytes. Recent studies indicated it also engages in a variety of physiological and pathological processes. In this review, we provide an overview of how BNIP3L induces mitophagy and discuss the biological functions of BNIP3L and its regulation at the molecular level. We further discuss current evidence indicating the involvement of BNIP3L-mediated mitophagy in human disease, particularly in cancer and neurological disorders.
Subject terms: Cancer, Mitophagy
Facts
BNIP3L promotes mitophagy either via recruiting autophagosomes to target mitochondria or enhancing the formation of autophagosomes.
BNIP3L-mediated mitophagy is regulated at the pre-transcriptional, posttranslational levels, and homodimerization of BNIP3L is required for its mitophagic activity.
BNIP3L either participates in mitophagy independently or interplays with other autophagy/mitophagy pathways.
BNIP3L involves in a variety of human diseases, including cancer, neurological, metabolic, and cardiovascular disorders.
Introduction
Mitochondrial quality control plays a central role in cellular homeostasis. Besides mitochondrial biogenesis, mitochondrial elimination via the autophagosome-lysosome pathway, which is frequently termed mitophagy, plays a crucial role in maintaining the cellular steady state of mitochondrial functions [1, 2]. Mounting evidence indicated the significance of appropriate mitophagy, either in terms of extension and specificity, in cell development and human diseases. To meet the needs of mitochondrial turnover, mammalian cells evolved a variety of molecular machineries. In particular, a multitude of mitophagy receptors connects target mitochondria with autophagosomes for degradation by sensing distinct intracellular or environmental stress [3]. These mitophagy receptors can be regulated at different stages of expression, translation, and post-translation, thus are able to regulate mitochondrial elimination precisely.
BNIP3L/Nix is a mitochondrial protein from the outer membrane that belongs to the BH3-only protein from the BCL2 family. BNIP3L was initially recognized as a proapoptotic protein with milder efficacy in inducing apoptosis compared to other proteins in this family [4, 5]. These phenotypes raised concerns about the molecular function of BNIP3L besides inducing apoptosis [4, 6]. A milestone disclosure in this field was made by the discovery that mice with a Bnip3l gene deletion showed a dysregulated maturation of the reticulocytes, in which massive mitochondria accumulated due to an impaired mitophagic process [7, 8]. BNIP3L has been accepted as a mitophagy receptor that binds to Atg8 proteins [9]. Nevertheless, it is not yet fully understood how BNIP3L-mediated mitophagy is regulated at the molecular level. Hypoxia has been considered as a canonical stress factor that induces BNIP3L-mediated mitophagy since BNIP3L can be transcriptionally upregulated by the hypoxia-inducible factor-1alpha (HIF1A). However, BNIP3L upregulation was not substantial in some cell types despite hypoxic conditions [10–13]. Instead, other transcription factors and non-coding RNAs reportedly regulated the transcription of Bnip3l by sensing distinct environmental fluctuations. More intriguingly, emerging data indicated that posttranslational modification of BNIP3L, in particular phosphorylation and ubiquitination, finely regulates BNIP3L-mediated mitophagy [14–16].
The significance of BNIP3L-mediated mitophagy was initially emphasized in the development of reticulocytes [7]. Since then, accumulating evidence indicated the involvement of BNIP3L-mediated mitophagy in the development process of other cell types. The molecular mechanisms underlying BNIP3L-mediated mitophagy, however, have not yet been fully elucidated. BNIP3L might act as a mitophagy receptor by binding with LC3s; alternatively, it is possible that BNIP3L interplays with some other autophagy/mitophagy pathways, including BECN1, RHEB2, cardiolipin, and PRKN. Furthermore, it is now clear that problems in BNIP3L-mediated mitophagy are associated with a number of human diseases, including neurological disorders, metabolic diseases, and cancer. Unfortunately, due to a poor understanding of the association between BNIP3L-mediated mitophagy and different diseases, few studies addressed potential therapies. Here, we review BNIP3L-mediated mitophagy and discuss how this process is regulated at the molecular level. Furthermore, we update current perspectives on the role of BNIP3L in cell development and human diseases by integrating the findings from novel research studies.
Mitochondria elimination by autophagy
Autophagy maintains intracellular metabolic balance by degrading superfluous and damaged intracellular substances. The process of autophagy has been described elegantly [17]. Briefly, the biogenesis of autophagosomes is initiated by forming phagophores. Autophagosomal membranes are then further extended during maturation and fuse with lysosomes to form autolysosomes, in which the engulfed cargoes are degraded. Autophagy has been widely accepted as a selective, rather than random, process to eliminate cargoes [18]. Mitophagy is essential for mitochondrial quality control. Accumulating evidence indicates mitophagy dysregulation in a variety of human diseases. Mitophagy is restrictively controlled by several proteins, including PINK1/PARKIN, BNIP3L, and FUNDC1, etc. by sensing distinct extracellular signaling, e.g., mitochondrial depolarization, hypoxia, and cellular development (Fig. 1) [19]. Despite the fact that these mitophagy proteins ensure the basal mitophagy activity redundantly in intact cells, dysregulation of a given protein leads to specific disease-related phenotypes [20–23]. Given that most of the mitophagy-related proteins have been elegantly summarized elsewhere [24–26], the regulatory mechanisms of BNIP3L, as a mitophagy receptor, and its association with human disease have not been carefully reviewed.
Fig. 1. An overview of the mitophagy process.
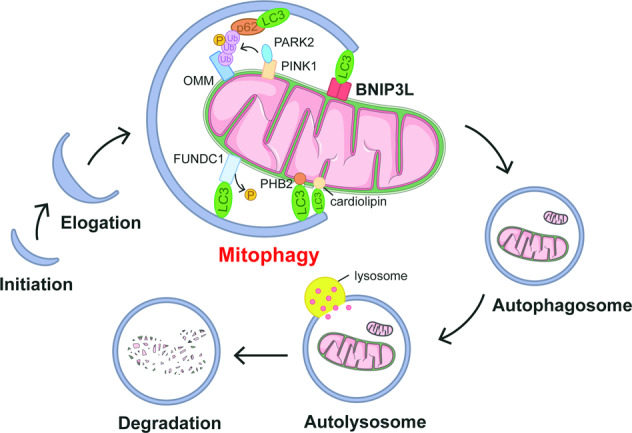
Autophagosomes are generated as phagophores and undergo elongation before ultimately fusing with lysosomes for degradation. Along with this process, mitochondria are recognized with the help from a variety of mitophagy-related proteins. The PINK1/PARK2 pathway senses the depolarization of mitochondria. The accumulation of PINK1 and PARK2 on the mitochondrial outer membrane amplifies the mitophagy signaling by generating phosphorylated ubiquitin, which links the autophagosomal machinery, e.g., P62 and LC3 to engulf the target mitochondria. Alternatively, a variety of mitophagy receptors, including BNIP3L, BNIP3, FUNDC1, PHB2, and cardiolipin, sense distinct extra-/intracellular signaling to recruit autophagosomes by directly binding with Atg8s, like LC3, via their LC3 interacting region (LIR) motif. These pathways ensure the superfluous and/or damaged mitochondria can be eliminated by lysosomes, and thus control the mitochondrial quality.
BNIP3L-mediated mitophagy
BNIP3L has been identified more than two decades ago [27, 28]. Sequencing analysis first recognized BNIP3L as a proapoptotic BCL2 family protein [28, 29] that could induce cell apoptosis or necrosis [30, 31]. Specifically, BNIP3L interacts with the proapoptotic Bcl2 family proteins BAX and BAK to increase the permeability of the mitochondrial outer membrane [32, 33]. This eventually leads to mitochondrial-dependent cell apoptosis. However, either endogenous or ectogenic BNIP3L expression has a poor influence on apoptotic cell death [34–36], compared to other BH3-only proapoptotic proteins [6]. BNIP3L has also been found in the endoplasmic reticulum (ER) [30], increasing its calcium storage through unknown mechanisms and promoting mitochondrial calcium uptake. The disruption of mitochondrial calcium homeostasis increases mitochondrial permeability and ultimately leads to cell demise [30, 37, 38].
Latterly the discovery of BNIP3L-promoted mitochondrial elimination by autophagy in developing reticulocytes opened a new horizon in BNIP3L research [7, 8], BNIP3L-induced mitophagy has since been identified in natural killer cells, neurons, retinal ganglion cells, renal cells, and several types of tumor cells [16, 39, 40]. In addition, BNIP3L might be involved in mitophagy in muscle and some cancer cells, even though direct evidence is still absent [41].
One central question is that how BNIP3L modulates mitophagy. BNIP3L disrupts the BCL2-BECN1 complex and releases BECN1, which promotes the formation of autophagosomes [42, 43]. BNIP3L also activates autophagy in epithelial cells from the respiratory tract [44]. Interestingly, BNIP3L-activated autophagy can be mimicked by a BH3 domain-like small molecule, which may serve as a promising drug [45, 46]. Furthermore, BNIP3L can inhibit MTORC1 to induce autophagy [47]. Interestingly, the gene plays a minimal role on the autophagy machinery of developing reticulocytes, ischemic brains, and hypoxic colon carcinoma [7, 16, 48]. Notwithstanding, BNIP3L plays a part in the regulation of autophagy activation, likely relying on the abundance of BECN1 or MTOR within distinct cell types.
The most well-accepted mechanism underlying BNIP3L-mediated mitophagy is through an interaction with proteins from the Atg8 family, by which BNIP3L recruits autophagosomes to targeted mitochondria [9, 49]. Two BNIP3L domains are required for mitophagy, an LIR domain and a transmembrane TM domain (Fig. 2). The LIR domain is composed of four amino acid motifs that are critical for the interaction with LC3s. The BNIP3L genetic sequence contains two LIR motifs, of which the one located at the C-terminus [31–34] is essential for mitophagy. The deletion of the LIR motif in BNIP3L led to a failure to induce mitophagy [9]. However, it was later demonstrated that the deletion of a specific motif known as the minimal essential region (MER), rather than LIR, inhibited BNIP3L-mediated mitophagy [50]. These observations indicated that the MER, but not the LIR motif, is essential for BNIP3L-induced mitophagy. The TM domain located in the C-terminus of BNIP3L allows for its mitochondrial location, and the BNIP3L presence in other organelles leads to organelle-specific autophagy [51]. The TM domain does not participate in the interaction between Atg8 proteins and the LIR motif [49]. Recent studies indicated that BNIP3L may serve as a tag to label the targeted mitochondria for degradation [14, 16, 52], even though the current understanding of the processes regulating BNIP3L mitochondrial distribution remains limited.
Fig. 2. The regions and amino acid sites essential for BNIP3L biological functions.

The LIR (LC3 interaction region) region binds with Atg8 proteins to induce mitophagy. The MER (Minimal essential region) is also essential for mitophagy. BH3 (BCL2 homolog 3) induces apoptosis. The TM (transmembrane domain) domain facilitates BNIP3L mitochondrial location. S34 and S35 phosphorylation promotes mitophagy. S82 phosphorylation is required but not sufficient to induce mitophagy. S117, S118, and S120 are phosphorylated by CK2, but their biological function remains unidentified. S204 and S108 are important for BNIP3L dimer stabilization. S204 and S212 phosphorylation promote BNIP3L homodimerization.
Mitochondria are dynamic organelles undergoing continuous fission, fusion, and transportation. BNIP3L presence leads to the loss of mitochondrial transmembrane potential (ΔΨM) [30, 53–55], which was considered a marker of dysfunctional mitochondria that triggers mitophagy. Hence, BNIP3L seemingly induces ΔΨM collapse and thus initiates mitophagy. However, this assumption was challenged by the observation that BNIP3L does not necessarily cause ΔΨM loss when serving as a mitophagy receptor [7, 56]. Intriguingly, a recent study showed Bnip3l silencing can reduce ΔΨM during programmed mitophagy in cardiac progenitor cells (CPS) [57]. The causal relation among BNIP3L presence, ΔΨM loss and mitophagy should be further investigated. The PRKN/PARK2 can sense ΔΨM loss and ubiquitinate BNIP3L [58]. However, recent studies proposed BNIP3L does not functionally depend on PARK2 for manipulating mitophagy. In cells derived from patients suffering from Parkinson’s disease (PD), BNIP3L seemingly compensates for the loss-of-function mutation of PRKN/PARK2 and restores mitophagy [59]. Accordingly, BNIP3L (and not PRKN/PARK2) is involved in the programmed mitophagy of CPCs [57]. Furthermore, PARK2 deletion did not affect BNIP3L-induced mitophagy in ischemic neurons [16]. Finally, a recent study clearly demonstrated BNIP3L, rather than PARK2, is required for mitophagy clearance of axonal mitochondria [47]. These studies support distinct working mechanisms between BNIP3L and PARK2 and suggest ΔΨM loss may not be the most relevant trigger initiating BNIP3L-mediated mitophagy.
In summary, the role played by BNIP3L as a mitophagy receptor is now consensual, and extends beyond its function as a BH3-only proapoptotic protein. BNIP3L participates in mitophagy by promoting the formation of autophagosomes and, more directly, by recruiting them to target mitochondria. While the mechanistic details behind BNIP3L-mediated mitophagy need further elucidation, current evidence emphasizes BNIP3L is an independent mitophagy-inducing factor from the canonical PARK2.
Regulations of BNIP3L-mediated mitophagy
Distinct mitophagy receptors may sense specific cellular stresses in order to initiate mitophagy. However, the specific environmental cues sensed by BNIP3L to regulate the mitophagic process remain unknown. Hypoxia is known intimately to signal BNIP3L-mediated mitophagy given HIF1A activates the transcription of Bnip3l [48, 60–62]. Paradoxically, this hypothesis originated from the studies in BNIP3, a homologous BNIP3L protein. Both Bnip3 and Bnip3l were transcriptionally upregulated in CHO-K1 cells subjected to hypoxic conditions, but Bnip3l showed a considerably lower sensitivity to hypoxia [10], which was also observed in several tumor cell lines [11]. The assumption that hypoxia activates BNIP3L was further challenged by the observation that BNIP3L expression was neither related with pO2 in tumors [63] nor activated by ischemia in neurons or brain cells [16, 64]. In fact, hypoxia alone might not be sufficient to activate BNIP3L transcription [13, 35]. Some additional transcriptional factors such as TP53, FOXO3, PPARGC1A, E2F1, and FATE1 [34, 65–69], or other important growth factors [70] might be required for hypoxia-induced BNIP3L upregulation. Recent studies proposed that other cellular stresses or environment fluctuations are also involved in BNIP3L regulation. For example, BNIP3L transcription is activated by cytotoxic drugs [41, 71, 72], glucose level perturbations [73], and during the process of cell development [51, 56]. Mounting evidence indicated Bnip3l expression can be regulated by some micro RNAs, including miR-347, miR-30, miR-302/367, miR-133a, etc. [71, 74–76]. The regulation of BNIP3L by miRNAs was reviewed in previous work [77]. Recent data also proposed an epigenetic regulation of Bnip3l through DNA methylation [78, 79]. Therefore, BNIP3L expression can be regulated at both pre- and post-transcriptional levels and triggered by distinct cellular stresses including, but not limited to, hypoxia.
Current knowledge supports mitochondrial BNIP3L recruits autophagosomes needed for mitochondria degradation, which implies BNIP3L abundance is a decisive factor in mitophagy. However, BNIP3L is ubiquitously distributed on most mitochondria but only a portion of the BNIP3L-labeled mitochondria undergoes mitophagy. This paradigm is different from the PRKN/PARK2-induced mitophagy, in which PARK2 selectively accumulates on damaged mitochondria with low transmembrane potential to enable mitophagy. These observations raise the possibility that mechanisms other than location influence BNIP3L-mediated mitophagy.
Phosphorylation arguably remains the most carefully studied molecular mechanism behind BNIP3L-mediated mitophagy (Table 1). BNIP3L phosphorylation was initially found in a study identifying the CK2 substrate [80]. The BNIP3L serine residues S120, possibly S117 and S118 (by human sequence) as well, are phosphorylated by CK2, even though the functional significance of this phosphorylation remains unknown. Intriguingly, CK2 phosphorylates another mitophagy receptor FUNDC1 and prevented mitophagy under hypoxic conditions, suggesting an essential role of phosphorylation in receptor-mediated mitophagy [81]. Our previous study identified a serine 81 (S82 in human BNIP3L) phosphorylation is required for BNIP3L-induced mitophagy. The biological significance of S81 phosphorylation was confirmed in ischemic brain cells, in which a BNIP3L S81A mutant failed to eliminate damaged mitochondria and aggravated ischemic neuronal injury [16]. The S81 phosphorylation of BNIP3L was recapitulated in a cadmium-mediated mitophagy in HeLa cells [82]. Crucially, this residue is located in the MER motif region and might serve as an adapter essential for BNIP3L activity. The BNIP3L S81A mutant showed similar mitochondrial localization as the wild-type, but completely eliminated BNIP3L binding with LC3s and showed a limited enhancement of mitophagy induction [16]. This observation implied that unphosphorylated BNIP3L at S81 may act as a “silent” mitophagy receptor to avoid excessive mitophagic activity. The phosphorylation of the other two serine residues (S34 and S35), which are juxtaposed to the LIR motif of BNIP3L, showed a significantly higher affinity to LC3B [14]. In addition, phosphomimetic mutations of BNIP3L (S34, 35D or S34, 35E) reinforced its mitophagic activity. Interestingly, we found the BNIP3L S34, 35D mutation does not promote mitophagy in ischemic neuronal cells (unpublished data). These different pieces of evidence suggest BNIP3L phosphorylation occurring at distinct serine residues may play different functions in different cell types. A recent study by Novak et al. identified the phosphorylation at BNIP3L serine 212 residue promoted the homodimerization of BNIP3L to induce mitophagy [83], which is discussed below. Taken together, these data support a model where phosphorylated BNIP3L serves as a tag on mitochondria targeted for elimination among the BNIP3L-labeled mitochondrial pool. However, the specific phosphatases and kinases involved in controlling BNIP3L phosphorylation remain unknown, but these enzymes may constitute promising drug targets for treating human diseases associated with mitophagy dysfunction.
Table 1.
Posttranslational modification (PTM) of BNIP3L and association with human disease.
| Types | Sites | Biological functions | Related diseases | Ref. |
|---|---|---|---|---|
| Phosphorylation | S34, S35 | S34/35 phosphorylation enhances autophagosome recruitment to mitochondria | Hela cells (Undetermined disease) | [14] |
| S82 | S82 phosphorylation is required for BNIP3L-mediated mitophagy | Cerebral ischemia | [16] | |
| S118, S120 | Undetermined biological function | Undetermined related disease | [80] | |
| S212 | S212 phosphorylation disrupts BNIP3L dimerization | HEK293 and HeLa cells (Undetermined disease) | [83] | |
| Ubiquitylation | Unknown | Ubiquitination of BNIP3L by PARK2 recruits NBR1 to perform mitophagy. | Parkinson’s disease | [52] |
| Unknown | Proteasomal degradation of ubiquitylated BNIP3L leads to mitophagy deficiency | Cerebral ischemia | [15] |
Ubiquitylation is an alternative strategy for the selective elimination of BNIP3L-labeled mitochondria. BNIP3L can act as a PARK2 substrate, which is an E3 ubiquitin ligase. Ubiquitylated BNIP3L recruits the adapter protein NBR1 in the autophagosome to perform mitophagy [58]. This study thus provided an elegant illustration of how BNIP3L cooperates with PARK2 to induce mitophagy. We have also recently revealed an additional contribution of BNIP3L ubiquitylation to mitophagy regulation. Specifically, we identified a proteasomal degradation of ubiquitylated BNIP3L in ischemic brain cells that led to BNIP3L loss and mitophagy deficiency. The prevention of BNIP3L degradation by ubiquitin-proteasome inhibitors restored mitophagic activity and conferred neuroprotection [15]. We thus identified a novel mechanism through which BNIP3L-mediated mitophagy ceases under stressful conditions. We noted that the specific ubiquitylated BNIP3L lysine residues in these two scenarios remain unknown. This is also true for specific E3 ubiquitin ligases responsible for BNIP3L ubiquitylation besides PARK2. Such knowledge would help explore the distribution of E3 ligases within the mitochondria, particularly those associated with the regulation of mitophagy [84–86].
The transmembrane (TM) domain enables BNIP3L mitochondrial distribution, which is essential for its mitophagic activity. Importantly, the deletion of the BNIP3L TM domain does not affect its interaction with BCL2 but hampers the formation of homodimeric BNIP3L forms [27]. BNIP3L homodimers have first been described two decades ago [4, 27], but the biological function of dimeric BNIP3L remains undetermined. We recently showed dimeric BNIP3L is required for mitophagy [15]. Point mutations on the TM domain disrupt BNIP3L dimer formation without affecting its mitochondrial localization. A monomeric BNIP3L mutant failed to induce mitophagy in CCCP-treated HeLa cells and ischemic neuronal cells. Additionally, we found that the dimeric form of BNIP3L is more prone to degradation by the proteasomes in ischemic brains, suggesting an intrinsic mechanism through which neuronal cells tightly limit mitophagy. In accordance with this finding, recent work by Novak et al. supports the essential role of the BNIP3L dimer in mitophagy induction [83]. The same mutation in the TM domain (G204A) led to attenuated LC3A binding and mitophagy defects. Interestingly, the authors provided an elegant mechanistic illustration of the formation of the BNIP3L dimer. The phosphorylated serine 212 prevents BNIP3L dimer formation, indicating phosphorylation of distinct BNIP3L serine residues have opposite effects on mitophagic activity. Of note, the S212 residue is located in the intermembrane space of the mitochondria, suggesting mitochondria-derived signaling regulates mitophagy. These discoveries found an alternative BNIP3L control of mitophagy through the formation of homodimers. However, it is unclear how the BNIP3L dimer triggers mitophagy and the molecular mechanisms underlying BNIP3L dimerization.
BNIP3L-mediated mitophagy is finely controlled by several mechanisms beyond Bnip3l transcription (Fig. 3). Recent evidence indicated posttranslational modifications and homodimerization of BNIP3L are key participants in its mitophagic activity. Further research is necessary to understand how these BNIP3L modifications affect mitophagy as a response to environmental changes and their physiological/pathological significance.
Fig. 3. The regulations of BNIP3L and mitophagic activity.
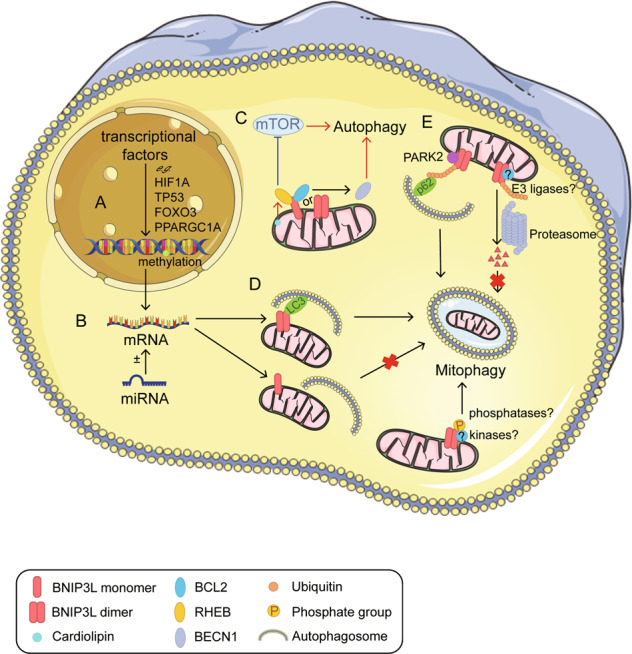
A Regulation of BNIP3L in the nucleus. BNIP3L is transcriptionally upregulated by several transcriptional factors, including hypoxia-inducible factor-1alpha (HIF1A), TP53, and FOXO3. In addition, DNA methylation ensures epigenetic regulation of BNIP3L. B Regulation of BNIP3L by micro RNAs (e.g., miR-347, miR-30, miR-302/367, and miR-133a). C The role played by BNIP3L in modulating autophagy. BNIP3L disrupts the BCL2-BECN1 complex; the released BECN1 subsequently promotes the formation of autophagosomes. Additionally, BNIP3L inhibits MTORC1 by prohibiting RHEB activity and subsequently induces autophagy. We note that the interaction between BNIP3L and RHEB can be enhanced by the accumulation of cardiolipin. D The role played by BNIP3L as a mitophagy receptor. The BNIP3L LIR motif interacts with LC3s to induce mitophagy. The BNIP3L monomer fails to induce mitophagy. BNIP3L phosphorylation at different sites regulates mitophagic activity but the associated kinases and phosphatases have not yet been identified. E BNIP3L dimer is required for mitophagy and is prone to degradation by the proteasome. PARK2 ubiquitinates BNIP3L and promotes mitophagy. Additional E3 ligases participating in BNIP3L degradation have not yet been identified.
Crosstalk between BNIP3L and other autophagy/mitophagy pathways
Recent data indicated BNIP3L interplays with other autophagy-related pathways [42, 43, 52, 87]. Under hypoxia conditions, BNIP3L disrupts the BCL2–BECN1 complex by competitively binding with BCL2 via its BH3 domain, and the released BECN1 promotes the formation of autophagosomes [42, 43]. In a variety of cancer cell lines, BNIP3L regulation triggers autophagy and decreases cell death. Moreover, BNIP3L was shown to inhibit MTORC1 by preventing RHEB activity and subsequently induced autophagy. A recent study implied that dysfunctioned mitochondria are unable to sustain synaptic homeostasis and increase oxidative damage. The modulating effects of RHEB by BNIP3L in eliminating neuronal axonal mitochondria, showing potential benefits in counteracting Alzheimer’s [47].
Beyond interactions with autophagy-related pathways, BNIP3L also interplays with mitophagy pathways. Cardiolipin is essential for maintaining the structural organization and function of mitochondrial membranes [88], and the accumulation of cardiolipin is accompanied by the upregulation of BNIP3L. PLD6 knockdown prevents BNIP3L-induced MTOR-RPS6KB activation. The crosstalk between BNIP3L and cardiolipin provides a possibility to treat insulin desensitization. BNIP3 and BNIP3L possess common features with the BH3-only protein subgroup of the BCL2 family. Using a stringent, high-throughput yeast two-hybrid system, researchers found that BNIP3 interacts with BNIP3L [89]. However, existing research suggested that BNIP3 and BNIP3L mediate parallel death and autophagy pathways. In patients with breast cancer brain metastases and uveal melanoma, BNIP3 but not BNIP3L may serve as a putative biomarker [90, 91]. Also, previous studies suggested that BNIP3 may be a promising therapeutic target of myocardial infarction, insulin resistance, and osteoarthritis [92–94]. In cases of cell stress, ΔΨM loss can promptly occur and be sensed by the PRKN/PARK2, thus enabling the mitophagic process. BNIP3L was proposed as a PARK2 substrate that recruits adapter proteins in autophagosomes after being ubiquitinated. Briefly, the degradation of BNIP3L attenuates mitophagy and causes injury to DA neurons and PD [58]. Additionally, BNIP3L also interacts with other proteins such as DOK5, EWSR1, RINT1, and ADIPOQ [89, 95]. However, it is still unclear whether these interactions have biological significance.
BNIP3L-mediated mitophagy in human diseases
Mitochondria play key roles in producing energy, buffering calcium, synthesizing steroids, and regulating programmed cell death [96]. The dysregulation of mitochondria is closely related to a variety of human diseases [97]. Neurons have extremely high requirements for energy. Mitochondria dysfunction is intimately associated with neurological disorders [15, 98–102]. Particularly, mitophagy defects may underlie the pathogenesis of Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease [103–107]. Moreover, it has been documented that mitophagy dysregulation plays discrepant role in tumorigenesis [108–110]. Therefore, it comes to a consensus that the disruption of mitophagy is highly involved in a variety of human diseases [97, 111, 112].
BNIP3L has been implicated in a variety of human diseases, including cancer, neurological, metabolic, and cardiovascular disorders. Mounting evidence supports the involvement of BNIP3L-mediated mitophagy in these disorders. Here, we focus on the role of autophagy/mitophagy activity of BNIP3L in human diseases by discussing recent studies in the field that shed light on the potential of targeting BNIP3L for therapeutics.
Cancer
The contribution of BNIP3L to cancer cells remains controversial. BNIP3L is recognized as a tumor suppressor gene and a link between BNIP3L-related autophagy/mitophagy activity and cancer cell death has recently been investigated. BNIP3L reportedly caused autophagic melanoma cell death by recruiting TR3 to mitochondria [55]. The RNA oncomir gene mir-30d promotes tumor progression and impaired the expression of autophagy genes including Bnip3l [113]. Moreover, a study on glioma cells showed BNIP3L is required for the mitophagic cell death caused by the natural compound AT 101 [114]. Finally, another study showed endogenous BNIP3L degradation enabled the survival of Ewing sarcoma cells [68]. These observations showed that BNIP3L-mediated autophagy/mitophagy accelerated cancer cell death. Similarly, higher BNIP3L expression is associated with a better prognosis of acute myeloid leukemia (AML) and sensitized decitabine-induced cell demise without inducing apoptosis [115]. This process likely occurs via mitophagy regulation. In contrast, BNIP3L was considered as an oncogene that disrupts the interaction between BCL2 with BECN1. Accordingly, Bnip3l knockdown led to autophagy defects and increased cell mortality, which means BNIP3L-related autophagy might be related to survival in solid tumors in hypoxic environments [42, 43]. This idea was further supported by a transcriptome analysis indicating BNIP3L upregulation is closely linked with TNF-α resistance in breast cancer cells [116]. Additionally, BNIP3L was found to introduce XIAP into mitochondria through a yet-unidentified mechanism to degrade SMAC. This process can delay mitochondrial-dependent apoptosis in MCF-7 cells [32]. A growing amount of evidence showed that mitochondrial BNIP3L promotes the survival of glioblastoma cells and pancreatic cancer cells. BNIP3L-induced mitophagy alternates either glucose metabolism or mitochondrial OXPHOS status to adapt to the oncogenic environment [117, 118]. In AML cells, Bnip3l silencing leading to increased sensitivity to mitochondria- but not DNA-targeting drugs, also implies a potential link between BNIP3L-mediated mitophagy and cancer cell survival [119].
It must be emphasized that mitophagy reportedly plays a dual role in cancer biology [26, 120]. The level of BNIP3L is high in organoids derived from murine pancreatic intraepithelial neoplasia, thereby delaying progression of pancreatic ductal adenocarcinoma (PDAC) and suggests BNIP3L-mediated mitophagy promotes tumorigenesis [118]. However, a greater amount of evidence from different cancer cell lines and animal models showed BNIP3L-mediated mitophagy alters with the stage of tumor progression.
We also noted that BNIP3L-mediated mitophagy might play a role in anti-cancer treatment by regulating the formation of immunological memory. There is evidence that BNIP3L-mediated mitophagy is able to remove dysfunctional mitochondria and ROS accumulated in natural killer cells when proliferating. This is thought to promote cell survival during the transition to memory cells [39]. In CD8 + T cells, BNIP3L-mediated mitophagy deficiency causes superoxide accumulation in mitochondria and reduction of ATP synthesis, which inhibits the formation of effector memory and recall response [121].
Taken together, BNIP3L-mediated autophagy/mitophagy plays a complex role in the fate of cancer cells (Table 2 and Fig. 4). This complexity can be attributed to the heterogeneity of different types of cancer, the exposure to anti-cancer drugs, the distinct stages and dynamic niches of cancer progression, and the formation of immunological memory. Hence, it is still a pending question whether disturbing BNIP3L-mediated mitophagy represents a safe and efficient strategy in cancer therapy.
Table 2.
The involvement of BNIP3L in human disease.
| Diseases | Evidence | Ref. | |
|---|---|---|---|
| Cancer | Melanoma | BNIP3L causes autophagic melanoma cell death by recruiting TR3 to mitochondria | [55] |
| Glioma | BNIP3L participates in the mitophagic glioma cell death caused by AT 101 | [114] | |
| Ewing sarcoma | Degradation of endogenous BNIP3L is required for the survival of Ewing sarcoma cells | [68] | |
| Acute myeloid leukemia | BNIP3L expression as a useful prognostic marker of acute myeloid leukemia | [115] | |
| Solid tumor (cell lines) | Autophagy via BNIP3L promotes tumors | [42, 43] | |
| Breast cancer | Upregulation of BNIP3L linked with TNF-α resistance | [32, 116] | |
| Upregulation of BNIP3L delays mitophagic apoptosis | |||
| Glioblastoma | BNIP3L-induced mitophagy reduces oxidative damage and promotes glioblastoma cell survival | [117] | |
| Pancreatic cancer | Deletion of Bnip3l delays the progression of pancreatic cancer | [118] | |
| Neurodegeneration | Parkinson’s disease | BNIP3L compensates for mitophagic defects in PD patient-derived cells | [59, 122] |
| Amyotrophic lateral sclerosis | Upregulation of BNIP3L may promote ALS progression via a non-apoptotic function | [127] | |
| Acute brain injury | Cerebral ischemia | BNIP3L protects against cerebral ischemia via mitophagy. The inhibition of BNIP3L degradation by carfilzomib attenuates cerebral ischemia. | [15, 16] |
| Traumatic brain injury | Bnip3l overexpression enhances autophagic flux and attenuates traumatic brain injury | [60] | |
| Intracerebral hemorrhage | BNIP3L is upregulated in intracerebral hemorrhage rats. The increased interaction between BNIP3L and p75 promotes neuronal apoptosis | [137, 138] | |
Fig. 4.

The implications of BNIP3L in human diseases.
Neurodegeneration
Mitophagy has been recognized as essential for neuronal survival during neurodegeneration. The discovery that Parkinson’s disease (PD)-related genes Pink1 and Park2 participate in mitophagy highlighted how this process maintains the steady state of striatum dopaminergic neurons. However, Park2 deletion failed to recapitulate PD phenotypes in mice, implying other compensatory mechanisms exist to ensure mitophagy [122, 123]. While it is still debatable whether PARK2 is involved in BNIP3L-mediated mitophagy, recent evidence implied BNIP3L may compensate for mitophagy in PD. Accordingly, an MPTP-induced parkinsonism model BNIP3L was downregulated, but this could be reversed through neuroprotectant rescuing dopamine neuron death [124]. In a murine model carrying a GBA mutant, which is a genetic risk factor for PD, the BNIP3L dimer was reduced and this was accompanied by mitophagy dysfunction. Of note, the GBA mutation did not lead to PARK2 and ubiquitin alternations, implying less involvement of PARK2 in mitophagy defects [125]. In a murine model carrying a GBA mutant, which is a genetic risk factor for PD, the BNIP3L dimer was reduced and this was accompanied by mitophagy dysfunction. Of note, the GBA mutation did not lead to PARK2 and ubiquitin alternations, implying less involvement of PARK2 in mitophagy defects [59]. Even though these data suggested BNIP3L acts as an alternative molecular pathway restoring mitophagy in PD, it is still unclear whether BNIP3L is able to rescue dopaminergic neuronal death and PD symptoms, especially in the sporadic cases of PD.
BNIP3L upregulation was found in the cerebrospinal fluid of patients suffering from amyotrophic lateral sclerosis (ALS), a degenerative disorder of the motor neurons [126]. Likewise, BNIP3L upregulation was documented in the astrocytes of SOD1 mutant mice, a widely used model for ALS [127]. It is unraveled that whether upregulated BNIP3L can increase mitophagy activity, although some papers denoted that mitophagy defects might underlie ALS pathology [128, 129]. Besides PD and ALS, other studies indicated the potential involvement of BNIP3L-mediated autophagy/mitophagy in other neurodegenerative conditions, but the evidence is not conclusive [47, 130, 131]. Finally, there is no evidence indicating an association between Bnip3l mutation and neurodegeneration, although an emerging preliminary study implied its involvement in schizophrenia [132]. As compensation for mitophagy defects, it is likely that enhancing BNIP3L-mediated mitophagy offers a potential therapeutic strategy to treat neurodegenerative disorders.
Acute brain injury
Current evidence indicates BNIP3L is involved in acute brain injury disorder including cerebral ischemia, intracerebral hemorrhage (ICH), and traumatic brain injury, while most evidence indicates a proapoptotic role of BNIP3L. However, recent studies shed light on BNIP3L-mediated mitophagy in these diseases. The first report indicating BNIP3L upregulation and neuronal apoptosis in ischemic brain cells dates from 2005, before the gens was recognized as a mitophagy receptor [133]. Enhanced binding of BNIP3L with LC3 was observed in a delayed manner (72 h) after acute brain ischemia, suggesting excessive mitophagy may be correlated with delayed neuronal death [134]. Nevertheless, the contribution of BNIP3L-mediated mitophagy to brain ischemia was not determined by silencing or deleting the Bnip3l gene in animal cells. Our previous work revealed a neuroprotective role of mitophagy in acute brain ischemia in Bnip3l knockout mice [135, 136]. We demonstrated BNIP3L is required for ischemia-reperfusion-induced neuronal mitophagy and BNIP3L-mediated mitophagy offers protection against ischemic stroke by reinforcing clearance of damaged mitochondria [16]. In line with these findings, BNIP3L was found to be degraded by the proteasomes in permanent ischemic mice brains, which further leads to mitophagy defects. Blocking BNIP3L loss with the proteasomal inhibitor drug carfilzomib restored mitophagy and attenuated ischemic stroke [15]. These data emphasized the benefits of BNIP3L-mediated mitophagy in acute brain ischemia. Similarly, in a model evaluating traumatic brain injury in rats, neuronal BNIP3L reduction was found within 24 h after trauma. The overexpression of Bnip3l enhanced autophagic flux and attenuated traumatic brain injury, but it is still unclear whether mitophagy was also activated [60].
In contrast to studies on ischemic and traumatic brain injuries, there is a paucity of data indicating the involvement of BNIP3L-mediated mitophagy in intracerebral hemorrhage. It was shown that BNIP3L undergoes transient upregulation after brain hemorrhage in rats. However, the BNIP3L binding with BCL2 remained intact, suggesting minimal impacts on autophagy induction [137]. Alternatively, the bond between BNIP3L and p75 might stabilize BNIP3L on the outer mitochondrial membrane and promote neuronal apoptosis in the ICH brain [138]. However, these findings were argued since only a few neuronal cells showed features of apoptotic or autophagic death after ICH [139].
Overall, there is no strong evidence demonstrating a fundamental role of BNIP3L in any specific disorders, and there are no existing clinic approaches targeting BNIP3L. However, current evidence supports BNIP3L is involved in a variety of human diseases, from cancer to neurological disorders. Besides, BNIP3L-mediated mitophagy also involves in retinal ganglion cell development, intestinal inflammation, adult cardiac progenitor cells differentiation, and myocyte insulin resistance [57, 140–142]. Interestingly, an emerging study revealed BNIP3L mutations in human schizophrenia [132]. These studies highlighted the potential value of BNIP3L as a promising biomarker and drug target for specific human diseases.
Perspectives
It is increasingly clear that BNIP3L induces mitophagy by either activating autophagy or serving as a mitophagy receptor. Several studies showed BNIP3L is involved in diseases such as cancer, neurodegenerative disorders, and acute brain injury. BNIP3L plays a dual role in different kinds of cancer progression. In solid tumors (cell lines), breast cancer, glioblastoma, and pancreatic cancer, BNIP3L exacerbate cancer progression. However, it has an inverse role in the case of acute myeloid leukemia, melanoma, glioma, and Ewing sarcoma [32, 42, 43, 55, 68, 114–119]. These contrasting roles might be related to the unique metabolic environments of different tumors, but remained unclear. Additionally, it was reported that mitophagy also plays dual roles at different stages of cancer progression, despite a lack of studies addressing this specific question. In the case of neurodegenerative diseases such as Parkinson’s and ALS, BNIP3L accelerated disease progression [59, 122, 127]. However, due to the complexity of neurodegeneration and current technical limitations, it is not yet possible to thoroughly understand the underlying mechanisms. In the case of acute brain injury, BNIP3L protects the brain from damage. We found this is primarily related to BNIP3L-mediated mitophagy and independent of the PRKN/PARK2 [16]. We also showed BNIP3L dimer mediates mitophagy activity [15]. However, there are several important and outstanding questions in this case, including (but not limited to): (1) What are the key extra- and intracellular stimuli that trigger BNIP3L-mediated mitophagy? (2) How does BNIP3L regulate the extension and selectivity of mitophagy by sensing these stimuli? (It is possible BNIP3L responses to stimuli during phosphorylation and other posttranslational modifications). Nevertheless, the key amino acid residues and enzymes responsible for phosphorylation remain elusive. The identification of these enzymes might bring forward promising drug targets for BNIP3L-related human diseases, particularly neurological disorders. Unlike the case of PRKN/PARK2 and Parkinson’s disease, there is a paucity of data supporting the association between BNIP3L mutations and specific diseases. Given the known involvement of BNIP3L-mediated mitophagy in development and diseases, it is essential to identify key BNIP3L mutations to understand the biological function of this gene and provide the rationale for discovering novel therapeutic strategies.
Acknowledgements
Figures 1, 3, and 4 were modified from the Servier Medical Art website (http://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License (https://creativecommons.org/licenses/by/3.0/).
Author contributions
X.Z., X.W., Y.Y., Z.S., and Y.Z. conceived and designed the study; X.Z., Y.Li., and W.Z. performed the literature investigation and wrote the manuscript; and Y.Li., Y.Lu., L.P., S.M., J.W., and Z.C. edited and revised the paper. All authors read and approved the final manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (81822044 and 81773703) and Zhejiang Provincial Natural Science Foundation (Z21H310002).
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Competing interests
The authors declared no competing interests. We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Footnotes
Edited by Professor Paolo Pinton
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Zhong Chen, Email: chenzhong@zju.edu.cn.
Xiangnan Zhang, Email: xiangnan_zhang@zju.edu.cn.
References
- 1.Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705. doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20:1013–22. doi: 10.1038/s41556-018-0176-2. [DOI] [PubMed] [Google Scholar]
- 3.Xian H, Liou YC. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2020;28:827–42. doi: 10.1038/s41418-020-00657-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohi N, Tokunaga A, Tsunoda H, Nakano K, Haraguchi K, Oda K, et al. A novel adenovirus E1B19K-binding protein B5 inhibits apoptosis induced by Nip3 by forming a heterodimer through the C-terminal hydrophobic region. Cell Death Differ. 1999;6:314–25. doi: 10.1038/sj.cdd.4400493. [DOI] [PubMed] [Google Scholar]
- 5.Yasuda M, Han JW, Dionne CA, Boyd JM, Chinnadurai G. BNIP3alpha: a human homolog of mitochondrial proapoptotic protein BNIP3. Cancer Res. 1999;59:533–7. [PubMed] [Google Scholar]
- 6.Zhang J, Ney PA. Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid Redox Signal. 2011;14:1959–69. doi: 10.1089/ars.2010.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–5. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. 2000;97:9082–7. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–73. [PubMed] [Google Scholar]
- 12.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, et al. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem. 2006;281:1442–8. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 13.Bacon AL, Fox S, Turley H, Harris AL. Selective silencing of the hypoxia-inducible factor 1 target gene BNIP3 by histone deacetylation and methylation in colorectal cancer. Oncogene. 2007;26:132–41. doi: 10.1038/sj.onc.1209761. [DOI] [PubMed] [Google Scholar]
- 14.Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7:1131. doi: 10.1038/s41598-017-01258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Zheng Y, Liu M, Li Y, Ma S, Tang W, et al. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy. 2020;17:1–13. doi: 10.1080/15548627.2020.1802089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13:1754–66. doi: 10.1080/15548627.2017.1357792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10:322–37. doi: 10.1038/nrendo.2014.35. [DOI] [PubMed] [Google Scholar]
- 19.Yoo SM, Jung YK. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells. 2018;41:18–26. doi: 10.14348/molcells.2018.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–7. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–46. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Y, Chiang WC, Sumpter R, Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–38. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–73. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W. The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): a promising biomarker and potential therapeutic target of human diseases. Genes Dis. 2021;8:640–54. doi: 10.1016/j.gendis.2020.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Y, Liu J, Kang R, Tang D. Mitophagy receptors in tumor biology. Front Cell Dev Biol. 2020;8:594203. doi: 10.3389/fcell.2020.594203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imazu T, Shimizu S, Tagami S, Matsushima M, Nakamura Y, Miki T, et al. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene. 1999;18:4523–9. doi: 10.1038/sj.onc.1202722. [DOI] [PubMed] [Google Scholar]
- 28.Matsushima M, Fujiwara T, Takahashi E, Minaguchi T, Eguchi Y, Tsujimoto Y, et al. Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer. 1998;21:230–5. [PubMed] [Google Scholar]
- 29.Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, et al. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999;274:7–10. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- 30.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Investig. 2009;119:203–12. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–68. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamacher-Brady A, Choe SC, Krijnse-Locker J, Brady NR. Intramitochondrial recruitment of endolysosomes mediates Smac degradation and constitutes a novel intrinsic apoptosis antagonizing function of XIAP E3 ligase. Cell Death Differ. 2014;21:1862–76. doi: 10.1038/cdd.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19:555–66. doi: 10.1007/s10495-014-0967-2. [DOI] [PubMed] [Google Scholar]
- 34.Real PJ, Benito A, Cuevas J, Berciano MT, de Juan A, Coffer P, et al. Blockade of epidermal growth factor receptors chemosensitizes breast cancer cells through up-regulation of Bnip3L. Cancer Res. 2005;65:8151–7. doi: 10.1158/0008-5472.CAN-05-1134. [DOI] [PubMed] [Google Scholar]
- 35.Papandreou I, Krishna C, Kaper F, Cai D, Giaccia AJ, Denko NC. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res. 2005;65:3171–8. doi: 10.1158/0008-5472.CAN-04-3395. [DOI] [PubMed] [Google Scholar]
- 36.Guscetti F, Nath N, Denko N. Functional characterization of human proapoptotic molecules in yeast S. cerevisiae. FASEB J. 2005;19:464–6. doi: 10.1096/fj.04-2316fje. [DOI] [PubMed] [Google Scholar]
- 37.Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. doi: 10.1016/j.ceca.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Kowaltowski AJ, Menezes-Filho SL, Assali EA, Goncalves IG, Cabral-Costa JV, Abreu P, et al. Mitochondrial morphology regulates organellar Ca(2 + ) uptake and changes cellular Ca(2 + ) homeostasis. FASEB J. 2019;33:13176–88. doi: 10.1096/fj.201901136R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. 2015;43:331–42. doi: 10.1016/j.immuni.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu D, Chen P, Wang B, Wang Y, Miao N, Yin F, et al. NIX-mediated mitophagy protects against proteinuria-induced tubular cell apoptosis and renal injury. Am J Physiol Ren Physiol. 2019;316:F382–395. doi: 10.1152/ajprenal.00360.2018. [DOI] [PubMed] [Google Scholar]
- 41.Yan C, Luo L, Guo CY, Goto S, Urata Y, Shao JH, et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017;388:34–42. doi: 10.1016/j.canlet.2016.11.018. [DOI] [PubMed] [Google Scholar]
- 42.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–81. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazure NM, Pouyssegur J. Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy. 2009;5:868–9. doi: 10.4161/auto.9042. [DOI] [PubMed] [Google Scholar]
- 44.Zhang M, Shi R, Zhang Y, Shan H, Zhang Q, Yang X, et al. Nix/BNIP3L-dependent mitophagy accounts for airway epithelial cell injury induced by cigarette smoke. J Cell Physiol. 2019;234:14210–20. doi: 10.1002/jcp.28117. [DOI] [PubMed] [Google Scholar]
- 45.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 46.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han S, Jeong YY, Sheshadri P, Su X, Cai Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020;21:e49801.. doi: 10.15252/embr.201949801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–81. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 49.Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–8. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- 50.Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy. 2012;8:1325–32. doi: 10.4161/auto.20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brennan LA, McGreal-Estrada R, Logan CM, Cvekl A, Menko AS, Kantorow M. BNIP3L/NIX is required for elimination of mitochondria, endoplasmic reticulum and Golgi apparatus during eye lens organelle-free zone formation. Exp Eye Res. 2018;174:173–84. doi: 10.1016/j.exer.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, et al. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. 2015;24:2528–38. doi: 10.1093/hmg/ddv017. [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA. 2010;107:9035–42. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujimoto K, Ford EL, Tran H, Wice BM, Crosby SD, Dorn GW, 2nd, et al. Loss of Nix in Pdx1-deficient mice prevents apoptotic and necrotic beta cell death and diabetes. J Clin Invest. 2010;120:4031–39. doi: 10.1172/JCI44011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang WJ, Wang Y, Chen HZ, Xing YZ, Li FW, Zhang Q, et al. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat Chem Biol. 2014;10:133–40. doi: 10.1038/nchembio.1406. [DOI] [PubMed] [Google Scholar]
- 56.Xiang G, Yang L, Long Q, Chen K, Tang H, Wu Y, et al. BNIP3L-dependent mitophagy accounts for mitochondrial clearance during 3 factors-induced somatic cell reprogramming. Autophagy. 2017;13:1543–55. doi: 10.1080/15548627.2017.1338545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15:1182–98. doi: 10.1080/15548627.2019.1580095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, et al. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. 2015;24:2528–38. doi: 10.1093/hmg/ddv017. [DOI] [PubMed] [Google Scholar]
- 59.Koentjoro B, Park JS, Sue CM. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci Rep. 2017;7:44373. doi: 10.1038/srep44373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma J, Ni H, Rui Q, Liu H, Jiang F, Gao R, et al. Potential roles of NIX/BNIP3L pathway in rat traumatic brain injury. Cell Transpl. 2019;28:585–95. doi: 10.1177/0963689719840353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abaci HE, Truitt R, Luong E, Drazer G, Gerecht S. Adaptation to oxygen deprivation in cultures of human pluripotent stem cells, endothelial progenitor cells, and umbilical vein endothelial cells. Am J Physiol Cell Physiol. 2010;298:C1527–37. doi: 10.1152/ajpcell.00484.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, Zhang J, Liu H, Wang J, Xin J, Deng M. Changes in levels of hypoxia-induced mediators in rat hippocampus during chronic cerebral hypoperfusion. Neurochem Res. 2013;38:2433–39. doi: 10.1007/s11064-013-1158-1. [DOI] [PubMed] [Google Scholar]
- 63.Le QT, Kong C, Lavori PW, O’Byrne K, Erler JT, Huang X, et al. Expression and prognostic significance of a panel of tissue hypoxia markers in head-and-neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys. 2007;69:167–75. doi: 10.1016/j.ijrobp.2007.01.071. [DOI] [PubMed] [Google Scholar]
- 64.Cho B, Choi SY, Park OH, Sun W, Geum D. Differential expression of BNIP family members of BH3-only proteins during the development and after axotomy in the rat. Mol Cells. 2012;33:605–10. doi: 10.1007/s10059-012-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer cell. 2004;6:597–609. doi: 10.1016/j.ccr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 66.Yurkova N, Shaw J, Blackie K, Weidman D, Jayas R, Flynn B, et al. The cell cycle factor E2F-1 activates Bnip3 and the intrinsic death pathway in ventricular myocytes. Circ Res. 2008;102:472–9. doi: 10.1161/CIRCRESAHA.107.164731. [DOI] [PubMed] [Google Scholar]
- 67.Yu D, Li M, Ni B, Kong J, Zhang Z. Induction of neuronal mitophagy in acute spinal cord injury in rats. Neurotox Res. 2013;24:512–22. doi: 10.1007/s12640-013-9397-0. [DOI] [PubMed] [Google Scholar]
- 68.Gallegos ZR, Taus P, Gibbs ZA, McGlynn K, Gomez NC, Davis I, et al. EWSR1-FLI1 Activation of the Cancer/Testis Antigen FATE1 Promotes Ewing Sarcoma Survival. Molecular and cellular biology. 2019;39:e00138–19. doi: 10.1128/MCB.00138-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi GE, Lee HJ, Chae CW, Cho JH, Jung YH, Kim JS, et al. BNIP3L/NIX-mediated mitophagy protects against glucocorticoid-induced synapse defects. Nat Commun. 2021;12:487. doi: 10.1038/s41467-020-20679-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilkinson S, Ryan KM. Growth factor signaling permits hypoxia-induced autophagy by a HIF1alpha-dependent, BNIP3/3L-independent transcriptional program in human cancer cells. Autophagy. 2009;5:1068–69. doi: 10.4161/auto.5.7.9821. [DOI] [PubMed] [Google Scholar]
- 71.Roca-Alonso L, Castellano L, Mills A, Dabrowska AF, Sikkel MB, Pellegrino L, et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in beta-adrenergic signaling and enhances apoptosis. Cell Death Dis. 2015;6:e1754. doi: 10.1038/cddis.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Du B, Dai XM, Li S, Qi GL, Cao GX, Zhong Y, et al. MiR-30c regulates cisplatin-induced apoptosis of renal tubular epithelial cells by targeting Bnip3L and Hspa5. Cell Death Dis. 2017;8:e2987. doi: 10.1038/cddis.2017.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang C, Lu H, Xu J, Yu H, Wang X, Zhang X. Protective roles of autophagy in retinal pigment epithelium under high glucose condition via regulating PINK1/Parkin pathway and BNIP3L. Biol Res. 2018;51:22. doi: 10.1186/s40659-018-0169-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gubern C, Camos S, Ballesteros I, Rodriguez R, Romera VG, Canadas R, et al. miRNA expression is modulated over time after focal ischaemia: up-regulation of miR-347 promotes neuronal apoptosis. FEBS J. 2013;280:6233–46. doi: 10.1111/febs.12546. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Z, Hong Y, Xiang D, Zhu P, Wu E, Li W, et al. MicroRNA-302/367 cluster governs hESC self-renewal by dually regulating cell cycle and apoptosis pathways. Stem Cell Rep. 2015;4:645–57. doi: 10.1016/j.stemcr.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mughal W, Martens M, Field J, Chapman D, Huang J, Rattan S, et al. Myocardin regulates mitochondrial calcium homeostasis and prevents permeability transition. Cell Death Differ. 2018;25:1732–48. doi: 10.1038/s41418-018-0073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li W, Chen H, Li S, Lin G, Feng D. Exploring microRNAs on NIX-dependent mitophagy. Methods Mol Biol. 2018;1759:111–21. doi: 10.1007/7651_2017_15. [DOI] [PubMed] [Google Scholar]
- 78.Veron V, Marandel L, Liu J, Velez EJ, Lepais O, Panserat S, et al. DNA methylation of the promoter region of bnip3 and bnip3l genes induced by metabolic programming. BMC Genomics. 2018;19:677. doi: 10.1186/s12864-018-5048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pedanou VE, Gobeil S, Tabaries S, Simone TM, Zhu LJ, Siegel PM, et al. The histone H3K9 demethylase KDM3A promotes anoikis by transcriptionally activating pro-apoptotic genes BNIP3 and BNIP3L. eLife. 2016;5:e16844.. doi: 10.7554/eLife.16844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta. 2009;1793:847–59. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 81.Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–77. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 82.Naeem S, Qi Y, Tian Y, Zhang Y. NIX compensates lost role of parkin in cd-induced mitophagy in HeLa cells through phosphorylation. Toxicol Lett. 2020;326:1–10. doi: 10.1016/j.toxlet.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 83.Marinkovic M, Sprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy. 2020;17:1232–43. doi: 10.1080/15548627.2020.1755120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18:495–509. doi: 10.15252/embr.201643309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li J, Qi W, Chen G, Feng D, Liu J, Ma B, et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy. 2015;11:1216–29. doi: 10.1080/15548627.2015.1017180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Di Rita A, Peschiaroli A, P DA, Strobbe D, Hu Z, Gruber J, et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat Commun. 2018;9:3755. doi: 10.1038/s41467-018-05722-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han S, Jeong YY, Sheshadri P, Su X, Cai Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020;21:e49801. doi: 10.15252/embr.201949801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ren M, Phoon CK, Schlame M. Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. 2014;55:1–16. doi: 10.1016/j.plipres.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 89.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–78. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 90.Schulten HJ, Bangash M, Karim S, Dallol A, Hussein D, Merdad A, et al. Comprehensive molecular biomarker identification in breast cancer brain metastases. J Transl Med. 2017;15:269. doi: 10.1186/s12967-017-1370-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang Z, Yu F, Li M. Upregulation of BCL2 19 kD protein-interacting protein 3 (BNIP3) is predictive of unfavorable prognosis in uveal melanoma. Med Sci Monit. 2018;24:4711–17. doi: 10.12659/MSM.907679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–33. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tol MJ, Ottenhoff R, van Eijk M, Zelcer N, Aten J, Houten SM, et al. A PPARgamma-Bnip3 axis couples adipose mitochondrial fusion-fission balance to systemic insulin sensitivity. Diabetes. 2016;65:2591–605. doi: 10.2337/db16-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ma Z, Wang D, Weng J, Zhang S, Zhang Y. BNIP3 decreases the LPS-induced inflammation and apoptosis of chondrocytes by promoting the development of autophagy. J Orthop Surg Res. 2020;15:284. doi: 10.1186/s13018-020-01791-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161:647–60. doi: 10.1016/j.cell.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Annesley SJ, Fisher PR. Mitochondria in health and disease. Cells. 2019;8:680.. doi: 10.3390/cells8070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–59. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maruszak A, Safranow K, Branicki W, Gaweda-Walerych K, Pospiech E, Gabryelewicz T, et al. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J Alzheimers Dis. 2011;27:197–210. doi: 10.3233/JAD-2011-110710. [DOI] [PubMed] [Google Scholar]
- 99.Mkrtchyan GV, Ucal M, Mullebner A, Dumitrescu S, Kames M, Moldzio R, et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim Biophys Acta Bioenerg. 2018;1859:925–31. doi: 10.1016/j.bbabio.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 100.Cha MY, Chen H, Chan D. Removal of the Mitochondrial Fission Factor Mff Exacerbates Neuronal Loss and Neurological Phenotypes in a Huntingtonas Disease Mouse Model. PLoS Curr. 2018;10:ecurrents.hd.a4e15b80c4915c828d39754942c6631f.. doi: 10.1371/currents.hd.a4e15b80c4915c828d39754942c6631f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chun BY, Rizzo JF., 3rd Dominant optic atrophy and Leber’s hereditary optic neuropathy: update on clinical features and current therapeutic approaches. Semin Pediatr Neurol. 2017;24:129–34. doi: 10.1016/j.spen.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 102.Zsurka G, Kunz WS. Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol. 2015;14:956–966. doi: 10.1016/S1474-4422(15)00148-9. [DOI] [PubMed] [Google Scholar]
- 103.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22:401–12. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chu CT. Multiple pathways for mitophagy: a neurodegenerative conundrum for Parkinson’s disease. Neurosci Lett. 2019;697:66–71. doi: 10.1016/j.neulet.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palomo GM, Granatiero V, Kawamata H, Konrad C, Kim M, Arreguin AJ, et al. Parkin is a disease modifier in the mutant SOD1 mouse model of ALS. EMBO Mol Med. 2018;10:e8888.. doi: 10.15252/emmm.201808888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rogers RS, Tungtur S, Tanaka T, Nadeau LL, Badawi Y, Wang H, et al. Impaired mitophagy plays a role in denervation of neuromuscular junctions in ALS mice. Front Neurosci. 2017;11:473. doi: 10.3389/fnins.2017.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hwang S, Disatnik MH, Mochly-Rosen D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol Med. 2015;7:1307–26. doi: 10.15252/emmm.201505256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chourasia AH, Boland ML, Macleod KF. Mitophagy and cancer. Cancer Metab. 2015;3:4. doi: 10.1186/s40170-015-0130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Drake LE, Springer MZ, Poole LP, Kim CJ, Macleod KF. Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol. 2017;47:110–24. doi: 10.1016/j.semcancer.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang JY, Yi HS, Kim HW, Shong M. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim Biophys Acta Bioenerg. 2017;1858:633–40. doi: 10.1016/j.bbabio.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 111.Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16:3–17. doi: 10.1080/15548627.2019.1603547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun K, Jing X, Guo J, Yao X, Guo F. Mitophagy in degenerative joint diseases. Autophagy. 2021;17:2082–92. doi: 10.1080/15548627.2020.1822097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang X, Zhong X, Tanyi JL, Shen J, Xu C, Gao P, et al. mir-30d Regulates multiple genes in the autophagy pathway and impairs autophagy process in human cancer cells. Biochem Biophys Res Commun. 2013;431:617–22. doi: 10.1016/j.bbrc.2012.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Meyer N, Zielke S, Michaelis JB, Linder B, Warnsmann V, Rakel S, et al. AT 101 induces early mitochondrial dysfunction and HMOX1 (heme oxygenase 1) to trigger mitophagic cell death in glioma cells. Autophagy. 2018;14:1693–709. doi: 10.1080/15548627.2018.1476812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lazarini M, Machado-Neto JA, Duarte AD, Pericole FV, Vieira KP, Niemann FS, et al. BNIP3L in myelodysplastic syndromes and acute myeloid leukemia: impact on disease outcome and cellular response to decitabine. Haematologica. 2016;101:e445–8. doi: 10.3324/haematol.2016.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moussay E, Kaoma T, Baginska J, Muller A, Van Moer K, Nicot N, et al. The acquisition of resistance to TNFalpha in breast cancer cells is associated with constitutive activation of autophagy as revealed by a transcriptome analysis using a custom microarray. Autophagy. 2011;7:760–70. doi: 10.4161/auto.7.7.15454. [DOI] [PubMed] [Google Scholar]
- 117.Jung J, Zhang Y, Celiku O, Zhang W, Song H, Williams BJ, et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 2019;79:5218–32. doi: 10.1158/0008-5472.CAN-19-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Disco. 2019;9:1268–87. doi: 10.1158/2159-8290.CD-18-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rodrigo R, Mendis N, Ibrahim M, Ma C, Kreinin E, Roma A, et al. Knockdown of BNIP3L or SQSTM1 alters cellular response to mitochondria target drugs. Autophagy. 2019;15:900–7. doi: 10.1080/15548627.2018.1558002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miller DR, Thorburn A. Autophagy and organelle homeostasis in cancer. Developmental cell. 2021;56:906–18. doi: 10.1016/j.devcel.2021.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gupta SS, Sharp R, Hofferek C, Kuai L, Dorn GW, 2nd, Wang J, et al. NIX-mediated mitophagy promotes effector memory formation in antigen-specific CD8( + ) T cells. Cell Rep. 2019;29:1862–77. doi: 10.1016/j.celrep.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–35. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 123.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–62. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ren ZL, Wang CD, Wang T, Ding H, Zhou M, Yang N, et al. Ganoderma lucidum extract ameliorates MPTP-induced parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis. Acta Pharm Sin. 2019;40:441–450. doi: 10.1038/s41401-018-0077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15:113–130. doi: 10.1080/15548627.2018.1509818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sharma A, Varghese AM, Vijaylakshmi K, Sumitha R, Prasanna VK, Shruthi S, et al. Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem Res. 2016;41:965–984. doi: 10.1007/s11064-015-1779-7. [DOI] [PubMed] [Google Scholar]
- 127.Duval N, Sumner WA, Andrianakos AG, Gray JJ, Bouchard RJ, Wilkins HM, et al. The Bcl-2 homology-3 domain (BH3)-only proteins, Bid, DP5/Hrk, and BNip3L, are upregulated in reactive astrocytes of end-stage mutant SOD1 mouse spinal cord. Front Cell Neurosci. 2018;12:15. doi: 10.3389/fncel.2018.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Evans CS, Holzbaur ELF. Autophagy and mitophagy in ALS. Neurobiol Dis. 2019;122:35–40. doi: 10.1016/j.nbd.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USA. 2014;111:E4439–E4448. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kodron A, Hajieva P, Kulicka A, Paterczyk B, Jankauskaite E, Bartnik E. Analysis of BNIP3 and BNIP3L/Nix expression in cybrid cell lines harboring two LHON-associated mutations. Acta Biochim Pol. 2019;66:427–435. doi: 10.18388/abp.2019_2837. [DOI] [PubMed] [Google Scholar]
- 131.Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 2020;17:1–18. doi: 10.1080/15548627.2020.1728096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou J, Ma C, Wang K, Li X, Jian X, Zhang H, et al. Identification of rare and common variants in BNIP3L: a schizophrenia susceptibility gene. Hum Genomics. 2020;14:16. doi: 10.1186/s40246-020-00266-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Birse-Archbold JL, Kerr LE, Jones PA, McCulloch J, Sharkey J. Differential profile of Nix upregulation and translocation during hypoxia/ischaemia in vivo versus in vitro. J Cereb Blood Flow Metab. 2005;25:1356–1365. doi: 10.1038/sj.jcbfm.9600133. [DOI] [PubMed] [Google Scholar]
- 134.Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther. 2014;20:1045–1055. doi: 10.1111/cns.12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321–1333. doi: 10.4161/auto.25132. [DOI] [PubMed] [Google Scholar]
- 136.Yuan Y, Zhang X, Zheng Y, Chen Z. Regulation of mitophagy in ischemic brain injury. Neurosci Bull. 2015;31:395–406. doi: 10.1007/s12264-015-1544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rui Y, Ke K, Li L, Zheng H, Xu W, Tan X, et al. Up-regulated expression of Bnip3L after intracerebral hemorrhage in adult rats. J Mol Histol. 2013;44:497–505. doi: 10.1007/s10735-013-9506-7. [DOI] [PubMed] [Google Scholar]
- 138.Shen J, Chen X, Li H, Wang Y, Huo K, Ke K. p75 neurotrophin receptor and its novel interaction partner, NIX, are involved in neuronal apoptosis after intracerebral hemorrhage. Cell Tissue Res. 2017;368:13–27. doi: 10.1007/s00441-016-2510-y. [DOI] [PubMed] [Google Scholar]
- 139.Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48:1033–1043. doi: 10.1161/STROKEAHA.116.015609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Esteban-Martinez L, Boya P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy. 2018;14:915–917. doi: 10.1080/15548627.2017.1332567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Vincent G, Novak EA, Siow VS, Cunningham KE, Griffith BD, Comerford TE, et al. Nix-mediated mitophagy modulates mitochondrial damage during intestinal inflammation. Antioxid Redox Signal. 2020;33:1–19. doi: 10.1089/ars.2018.7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.da Silva Rosa SC, Martens MD, Field JT, Nguyen L, Kereliuk SM, Hai Y, et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy. 2021;17:2257–2272. doi: 10.1080/15548627.2020.1821548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.