

Abstract
Background & Aims
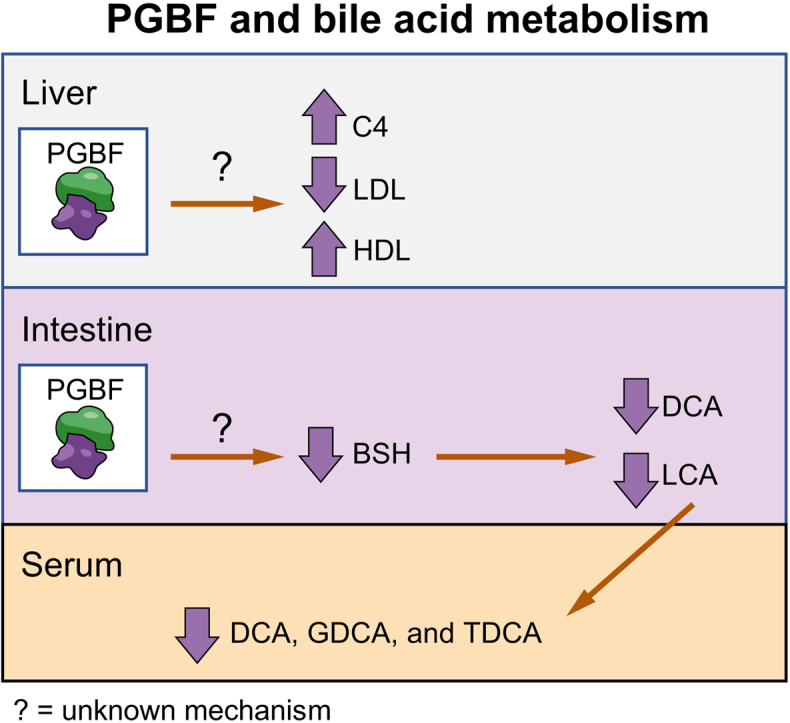
Increased serum bile acids (BAs) have been observed in patients with non-alcoholic steatohepatitis (NASH). Pegbelfermin (PGBF), a polyethylene glycol-modified (PEGylated) analogue of human fibroblast growth factor 21 (FGF21), significantly decreased hepatic steatosis and improved fibrosis biomarkers and metabolic parameters in patients with NASH in a phase IIa trial. This exploratory analysis evaluated the effect of PGBF on serum BAs and explored potential underlying mechanisms.
Methods
Serum BAs and 7α-hydroxy-4-cholesten-3-one (C4) were measured by HPLC-mass spectrometry (MS) using serum collected in studies of patients with NASH (NCT02413372) and in overweight/obese adults (NCT03198182) who received PGBF. Stool samples were collected in NCT03198182 to evaluate faecal BAs by liquid chromatography (LC)-MS and the faecal microbiome by metagenetic and metatranscriptomic analyses.
Results
Significant reductions from baseline in serum concentrations of the secondary BA, deoxycholic acid (DCA), and conjugates, were observed with PGBF, but not placebo, in patients with NASH; primary BA concentrations did not significantly change in any arm. Similar effects of PGBF on BAs were observed in overweight/obese adults, allowing for an evaluation of the effects of PGBF on the faecal microbiome and BAs. Faecal transcriptomic analysis showed that the relative abundance of the gene encoding choloylglycine hydrolase, a critical enzyme for secondary BA synthesis, was reduced after PGBF, but not placebo, administration. Furthermore, a trend of reduction in faecal secondary BAs was observed.
Conclusions
PGBF selectively reduced serum concentrations of DCA and conjugates in patients with NASH and in healthy overweight/obese adults. Reduced choloylglycine hydrolase gene expression and decreased faecal secondary BA levels suggest a potential role for PGBF in modulating secondary BA synthesis by gut microbiome. The clinical significance of DCA reduction post-PGBF treatment warrants further investigation.
Lay summary
Pegbelfermin (PGBF) is a hormone that is currently being studied in clinical trials for the treatment of non-alcoholic fatty liver disease. In this study, we show that PGBF treatment can reduce bile acids that have previously been shown to have toxic effects on the liver. Additional studies to understand how PGBF regulates bile acids may provide additional information about its potential use as a treatment for fatty liver.
Keywords: Microbiome, C4, Biomarkers, Deoxycholic acid, Bile salt hydrolase, FGF21
Abbreviations: ALT, alanine aminotransferase; ApoA1, apolipoprotein A1; AST, aspartate aminotransferase; BA, bile acid; baiCD, 7α-hydroxy-3-oxo-delta4-cholenoic acid oxidoreductase; baiH, 7β-hydroxy-3-oxo-delta4-cholenoic acid oxidoreductase; BSH, bile salt hydrolase; C4, 7α-hydroxy-4-cholesten-3-one; CA, cholic acid; CDCA, chenodeoxycholic acid; CYP7A1, cytochrome P450 7A1; DCA, deoxycholic acid; FGF21, fibroblast growth factor 21; FXR, farnesoid X receptor; GCA, glyco-cholic acid; GCDCA, glyco-chenodeoxycholic acid; GDCA, glyco-deoxycholic acid; GUDCA, glyco-ursodeoxycholic acid; HbA1c, glycated haemoglobin; hdhA, 7-alpha-hydroxysteroid dehydrogenase; HFF, hepatic fat fraction; LCA, lithocholic acid; LC, liquid chromatography; MS, mass spectrometry; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PEGylated, polyethylene glycol-conjugated; PGBF, pegbelfermin; PRO-C3, N-terminal type III collagen propeptide; QD, once daily; QW, once weekly; T2DM, type 2 diabetes mellitus; TCA, tauro-cholic acid; TCDCA, tauro-chenodeoxycholic acid; TDCA, tauro-deoxycholic acid; UDCA, ursodeoxycholic acid
Graphical abstract
Highlights
-
•
Bile acids are elevated in patients with non-alcoholic steatohepatitis.
-
•
Pegbelfermin, a PEGylated human FGF21 analogue, is in phase II trials for non-alcoholic steatohepatitis.
-
•
Pegbelfermin treatment was associated with secondary, but not primary, bile acid reductions.
-
•
Pegbelfermin reduced expression of a gene responsible for secondary bile acid synthesis.
-
•
Further study is needed to assess the clinical significance of these observations.
Introduction
Non-alcoholic steatohepatitis (NASH) is the advanced, progressive form of non-alcoholic fatty liver disease (NAFLD) and is defined as ≥5% steatosis in the presence of inflammation and hepatocyte injury, with or without fibrosis, on histologic examination of liver biopsy tissue.1 In the general population, the global prevalence of NASH is estimated at 1.5–6.5%2; given that patients with NASH have an increased risk of all-cause and liver-related mortality compared with the NAFLD population,2 there is a large unmet need for pharmacologic treatments that can halt or reverse disease progression. The complexity of NASH pathophysiology has complicated the effort to identify effective treatments for this patient population; however, investigational therapies with diverse mechanisms of action are being evaluated in clinical studies.
Bile acids (BAs) regulate cholesterol homeostasis and lipid metabolism and act as signalling molecules.3 The primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), are synthesised from cholesterol in the liver. Upon conjugation with glycine (glyco-CA [GCA] and glyco-CDCA [GCDCA]) or taurine (tauro-CA [TCA] and tauro-CDCA [TCDCA]), primary BAs are secreted into the intestine and converted into the secondary BAs, deoxycholic acid (DCA) and lithocholic acid (LCA), by intestinal bacteria.4 Gut microbiota initiate primary BA metabolism by deconjugating GCA and GCDCA back to CA and CDCA, a critical step catalysed by choloylglycine hydrolase, also known as bile salt hydrolase (BSH). Although some studies have not demonstrated altered serum BA concentrations in patients with NASH,5 other studies have shown that elevated BA concentrations are associated with the presence and severity of NAFLD6,7 and that DCA concentrations are specifically elevated in patients with NASH relative to healthy adults.6,8 Given the link between BAs and metabolic regulation, there is growing interest in understanding how BA dysregulation may contribute to NASH pathogenesis and progression.
Fibroblast growth factor 21 (FGF21) is a key regulator of energy metabolism and, as such, is a therapeutic target of interest in metabolic diseases, including NASH. Pegbelfermin (PGBF) is a polyethylene glycol-modified (PEGylated) recombinant human FGF21 analogue with a prolonged half-life that supports up to weekly dosing.9 PGBF has been shown to improve the histologic features of NASH and fibrosis in animal models. In a 12-week phase II study of patients with obesity and type 2 diabetes mellitus (T2DM) and a high prevalence of fatty liver, PGBF administration was associated with improvements in insulin sensitivity and lipid profiles and dose-dependent elevation in adiponectin concentrations.9 Further, in a 16-week phase IIa study in patients with NASH and stage 1–3 fibrosis (NCT02413372), PGBF treatment significantly decreased hepatic steatosis and the fibrosis biomarker N-terminal type III collagen propeptide (PRO-C3) and improved metabolic parameters and biomarkers of hepatic injury.10
At present, it has not been reported whether FGF21 mimetics regulate BA metabolism in humans, though FGF21 is a negative regulator of hepatic BA synthesis in animal models.11 BA species are very different in human and mice, and thus far, effects on BA metabolism in mouse models have not faithfully translated to humans.12 This post hoc analysis aimed to evaluate the effects of PGBF on BA metabolism by measuring serum BA concentrations in a phase IIa study in patients with NASH (NCT02413372). To further explore potential mechanisms underlying these observations, the effect of PGBF on the gut microbiome was evaluated using stool samples from a phase I study in healthy overweight or obese adults (NCT03198182).
Patients and methods
Patients
All study protocols were approved by the relevant institutional review boards as noted in the Supplementary information. All patients provided written informed consent before study participation.
Patient samples
NCT02413372 was a randomised, double-blind, placebo-controlled, phase IIa study in which PGBF was administered either 20 mg once weekly (QW) or 10 mg once daily (QD) for 16 weeks to patients with NASH and stage 1–3 fibrosis.10 BA concentrations were evaluated in fasted serum collected on Days 1, 57, and 112. NCT03198182 was a randomised, placebo-controlled, multiple-dose, phase I study of 20 or 40 mg PGBF QW for 14 days in healthy overweight or obese (BMI 25–40 kg/m2) participants.13 Fasted serum samples from Days 1, 15, 22, and 36 were used for the BA analysis, and stool samples collected at Days 1 and 15 were used for the microbiome analysis. Before analysis, the stool samples were collected and stored in Stool Nucleic Acid Collection & Transport Tubes (Second Genome Solutions, South San Francisco, CA, USA) containing a preservative solution (60–99% guanidinium chloride, 1–5% cetrimonium bromide) for DNA/RNA stabilisation.
Sample analysis
Serum BAs
Fasting serum samples from studies NCT02413372 and NCT03198182 were analysed with ultra-HPLC and mass spectrometry (MS) to assess BA levels before and after administration of PGBF or placebo. The BAs with the highest expected concentrations in humans were the focus of this analysis; the primary BAs assayed were CA, GCA, TCA, CDCA, GCDCA, and TCDCA, and the secondary BAs assayed were DCA, glyco-DCA (GDCA), and tauro-DCA (TDCA). All BAs were analysed in a single batch, and a single measurement of serum BAs was performed for each patient at each time point.
Faecal BAs
Faecal samples from study NCT03198182 were analysed by Metabolon (Morrisville, NC, USA). All major human primary and secondary BAs and their respective glycine and taurine conjugates were measured: CA, CDCA, DCA, LCA, ursodeoxycholic acid (UDCA), GCA, GCDCA, GDCA, glyco-UDCA (GUDCA), TCA, TCDCA, TDCA, tauro-LCA (TLCA), tauro-UDCA (TUDCA), and glyco-LCA (GLCA). Further details of this analysis and the stool DNA metagenomic and RNA metatranscriptomic analyses are included in the Supplementary information.
C4 measurement
7α-Hydroxy-4-cholesten-3-one (C4) measurement was performed by the Bristol Myers Squibb laboratory. A 100-μl matrix aliquot was fortified with 25 μl of 800 ng/ml internal standard working solution. Analytes were isolated through protein precipitation using 400 μl of 2% formic acid in acetonitrile. The final extracts were analysed via HPLC and tandem MS detection using positive ion electrospray. The samples were analysed in a single batch.
Statistical analysis
Treatment effects on BAs in the NASH study
Linear mixed models were fit to predict log (CA, CDCA, GCA, GCDCA, TCA, TCDCA, and TDCA) or square-root (DCA and GDCA) transformed BA levels from treatment and time, adjusting for sex, stratum (T2DM vs. no T2DM), and age, and including a random intercept for each patient. The model included an interaction between treatment and time, allowing the slopes for the predicted BA levels to differ over time across the different treatments. The full model including each interaction term is as follows:
| BileAcid ∼ (1|SUBJID) + B0 + B1 ∗ Sex + B2 ∗ Stratum + B3 ∗ Age + B4 ∗ Day57 + B5 ∗ Day112 + B6 ∗ Dose10mg + B7 ∗ Dose20mg + γ46 ∗ Day57:Dose10mg + γ47 ∗ Day57:Dose20mg + γ56 ∗ Day112:Dose10mg + γ57 ∗ Day112:Dose20mg |
To determine whether there were significant differences between treatment arms over time, we assessed whether the model coefficients for the interaction terms were equal to 0 (γ46 = γ47 = γ56 = γ57 = 0). Under the null hypothesis, the slopes for the predicted BA levels over time would be the same for each dose level as for placebo, with all of the trajectories being parallel. Rejection of the null hypothesis implies that the change in BA levels for the placebo arm differs significantly from at least 1 of the PGBF-treated arms.
Treatment effects on microbial gene expression in the study with healthy participants
For feature significance, functional gene, and pathway significance testing, the Wilcoxon rank sum test was used to identify differentially abundant genes or pathways. Where samples could be paired across categories, a paired Wilcoxon signed rank test was employed. Values of p were adjusted by the Benjamini–Hochberg procedure to control for false discovery rates from multiple testing.
Results
PGBF treatment significantly reduced serum levels of secondary BAs but not primary BAs in patients with NASH
Serum samples from 72 of 75 patients with NASH and stage 1–3 fibrosis were available for BA concentration analysis. Baseline characteristics for these patients are shown in Table 1. The mean age of patients in the PGBF treatment arms (52 years) was greater than those in the placebo arm (47 years), but there were no clear differences in metabolic or clinical parameters between arms.
Table 1.
Baseline demographics and disease characteristics for patients with NASH.
| Parameters | Placebo (n = 25) |
20 mg QW PGBF (n = 24) |
10 mg QD PGBF (n = 23) |
|---|---|---|---|
| Age, mean (SD), years | 47 (11) | 51 (12) | 52 (10) |
| Sex, n (%) | |||
| Male | 9 (36) | 7 (29) | 9 (39) |
| Female | 16 (64) | 17 (71) | 14 (61) |
| BMI, mean (SD), mg/kg2 | 36 (7) | 35 (6) | 34 (4) |
| NAFLD activity score, mean (SD) | 4 (1) | 4 (1) | 5 (1) |
| ALT, mean (SD), IU/L | 76 (53) | 72 (33) | 67 (46) |
| AST, mean (SD), IU/L | 49 (30) | 53 (23) | 50 (26) |
| HFF, mean (SD), % | 21 (8) | 20 (6) | 18 (8) |
| Triglycerides, mean (SD), mg/dl | 205 (134) | 192 (56) | 195 (89) |
| Total cholesterol, mean (SD), mg/dl | 197 (59) | 199 (49) | 196 (42) |
| HDL, mean (SD), mg/dl | 49 (10) | 45 (12) | 47 (11) |
| LDL, mean (SD), mg/dl | 122 (56) | 128 (44) | 122 (35) |
| HbA1c, mean (SD), % | 6.1 (1.1) | 6.3 (1.2) | 6.1 (1.0) |
| T2DM, n (%) | 11 (44) | 9 (38) | 9 (39) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; HbA1c, glycated haemoglobin; HFF, hepatic fat fraction; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PGBF, pegbelfermin; QD, once daily; QW, once weekly; T2DM, type 2 diabetes mellitus.
Median primary and secondary BA concentrations for all arms at baseline were not significantly different, although the median GCA concentration for the PGBF 20 mg QW arm was numerically higher than those for the placebo and PGBF 10 mg QD arms (Fig. S1A and B). Significant positive correlations were observed between the baseline concentrations of DCA and its conjugates GDCA and TDCA. Concentrations of conjugated BAs, especially conjugated primary BAs (TCA, GCA, TCDCA, and GCDCA), were highly correlated with each other (Fig. S1C). Serum concentrations of DCA, GDCA, and TDCA were significantly lower at Days 57 and 112 than at baseline in the PGBF arms, whereas no decrease was observed in patients who received placebo (Fig. S1A). DCA and conjugates were significantly decreased in the PGBF arms compared with the placebo arm in a mixed-model analysis (10 mg PGBF QD vs. placebo: p <0.001; 20 mg PGBF QW vs. placebo: p ≤0.01; Fig. 1 and Fig. S1A). Concentrations of DCA and conjugates at Day 112 were decreased by approximately 60–80% in both PGBF treatment arms with no significant difference between those who received 10 mg QD or 20 mg QW (Fig. 1). In contrast to secondary BA concentrations, there were no significant differences in primary BA concentrations from baseline to Day 112 following PGBF treatment compared with placebo (Fig. 2 and Figs. S1B and S2).
Fig. 1.

Change in serum concentration of secondary BAs in patients with NASH.
(A) Percentage and (B) absolute change in serum concentration of DCA + conjugates in patients from study NCT02413372. In (A), error = bootstrapped 95% CI; in (B), whiskers extend to most extreme data point that is ≤1.5× the IQR from the box. BA, bile acid; D, day; DCA, deoxycholic acid; GDCA, glyco-deoxycholic acid; NASH, non-alcoholic steatohepatitis; PGBF, pegbelfermin; QD, once daily; QW, once week; TDCA, tauro-deoxycholic acid.
Fig. 2.

Percentage change in primary BA concentrations in patients with NASH.
Percentage change in serum primary BA concentrations in patients from study NCT02413372; error = bootstrapped 95% CI. BA, bile acid; CA, cholic acid; CDCA, chenodeoxycholic acid; D, day; GCA, glyco-cholic acid; GCDCA, glyco-chenodeoxycholic acid; NASH, non-alcoholic steatohepatitis; PGBF, pegbelfermin; QD, once daily; QW, once weekly; TCA, tauro-cholic acid; TCDCA, tauro-chenodeoxycholic acid.
The decrease in total DCA + conjugates over time in both PGBF arms, but not in the placebo arm, corresponded with a decrease in the percentage of total DCA + conjugates in the total serum BA pool (Fig. 3A and B). Accordingly, the concentration of total BAs was decreased in the PGBF vs. placebo arms; median reductions of 24.7 and 30.8% were observed in the 10 mg PGBF QD and 20 mg PGBF QW arms, respectively, at Day 112 (Fig. 3C). The median concentration of total BAs in the placebo arm at baseline was numerically lower than what was observed in the PGBF arms, but the differences were not significant because of large inter-patient variability (Table S1).
Fig. 3.

Effects of PGBF on total DCA + conjugates, total primary BAs, and total BAs in patients with NASH.
(A) Percentage change from baseline of DCA + conjugates and total primary BAs; error = bootstrapped 95% CI. (B) Fraction of DCA + conjugates and total primary BAs relative to total serum BAs. (C) Percentage change from baseline total serum BAs from study NCT02413372. Whiskers extend to most extreme data point that is ≤1.5× the IQR from the box. BA, bile acid; D, day; DCA, deoxycholic acid; NASH, non-alcoholic steatohepatitis; PGBF, pegbelfermin; QD, once daily; QW, once weekly.
To assess the generalisability of the findings observed in patients with NASH from the phase IIa study, BA data were extracted from a metabolomics analysis performed on patient samples from a phase II study of PGBF treatment in overweight or obese patients with T2DM (NCT02097277).9 Samples were available from 109 patients assigned to receive the following: placebo QD (n = 22), 1 mg PGBF QD (n = 22), 5 mg PGBF QD (n = 22), 20 mg PGBF QD (n = 20), and 20 mg PGBF QW (n = 23). In this semiquantitative assessment of BA concentrations (Supplemental methods), GDCA concentration was markedly decreased at Day 84 in the PGBF 20 mg QD and 20 mg QW arms by approximately 46 and 50%, respectively, relative to baseline. DCA concentration was similarly reduced in the PGBF arms compared with baseline, although the extent of the reduction was smaller in the PGBF 20 mg QW arm than in the PGBF 20 mg QD arm (Fig. S3). TDCA concentrations were not detectable in this analysis. Similar to the results observed in patients with NASH, no changes in primary BAs or their conjugates were observed in patients who received PGBF vs. placebo (Fig. S4).
PGBF increased levels of C4, a biomarker of active BA synthesis, and decreased the LDL/HDL ratio
To further investigate the effect of PGBF on BA metabolism, C4, a stable intermediate in BA biosynthesis from cholesterol and a biomarker of primary BA synthesis in the liver, was measured in plasma of the patients with NASH from study NCT02413372. A significant increase in C4 levels was observed in the PGBF arms compared with the placebo arm (p <0.001; Fig. 4A and B). Spearman correlation analysis demonstrated that at Day 112, a moderate but significant negative correlation was observed for percent change in C4 concentration with percent change in DCA concentration (ρ = −0.45; p = 0.0002), with percent change in GDCA concentration (ρ = −0.33; p = 0.0098), and with percent change in TDCA concentration (ρ = −0.46; p = 0.0043). No correlation between changes in concentrations of C4 and primary BAs was observed. Median levels of LDL cholesterol were lower in the PGBF 10 mg QD arm, and median HDL cholesterol levels were increased in both PGBF arms compared with baseline, resulting in lower median LDL/HDL ratios in both PGBF arms at Days 57 and 112 (Fig. S5). As a result, the LDL/HDL ratio was reduced by approximately 20% from baseline in the PGBF 10 mg QD arm at Days 57 and 112; minimal change from baseline in the LDL/HDL ratio was observed in the placebo arm (Fig. 4C).
Fig. 4.

Effects of PGBF on C4 concentration and LDL/HDL ratio in patients with NASH.
(A) Percentage and (B) absolute change in C4. (C) Percentage change in LDL/HDL ratio. (D) Correlation between percentage change in C4 from baseline to Day 112 and percentage change in LDL/HDL ratio from baseline to Day 112 from study NCT02413372. In (A), error = bootstrapped 95% CI; in (B and C), whiskers extend to most extreme data point that is ≤1.5× the IQR from the box. C4, 7α-hydroxy-4-cholesten-3-one; D, day; NASH, non-alcoholic steatohepatitis; PGBF, pegbelfermin; QD, once daily; QW, once weekly.
Correlations between serum concentrations of cholesterol parameters and C4 were then investigated. At baseline, C4 concentrations were moderately but significantly correlated with concentrations of apolipoprotein A1 (ApoA1), the major component of HDL particles, and HDL cholesterol and were modestly correlated with total cholesterol concentration (Table 2). The percentage change from baseline to Day 112 in C4 concentration was negatively correlated with percentage change in LDL/HDL ratio (Fig. 4D) and was positively correlated with percentage change in concentrations of ApoA1 and HDL cholesterol (Table 2). The percentage changes from baseline to Day 112 in concentrations of DCA, TDCA, and, to a lesser extent, GDCA also were positively correlated with percentage change in LDL/HDL ratio.
Table 2.
Correlation between serum concentrations of C4 and cholesterol parameters in patients with NASH treated with PGBF.
|
Baseline concentrations |
ρ∗ |
p value† |
| C4 vs. ApoA1 | 0.40 | 0.000621 |
| C4 vs. HDL cholesterol | 0.31 | 0.00998 |
| C4 vs. total cholesterol | 0.29 | 0.0164 |
| C4 vs. LDL cholesterol | 0.19 | 0.110 |
| C4 vs. LDL/HDL ratio |
-0.04 |
0.687 |
|
Percentage change from baseline to Day 112 |
ρ∗ |
p value† |
| % C4 vs. % LDL/HDL ratio | -0.41 | 0.000858 |
| % C4 vs. % ApoA1 | 0.40 | 0.002070 |
| % C4 vs. % HDL | 0.30 | 0.016500 |
| % C4 vs. % LDL | -0.08 | 0.541000 |
| % C4 vs. % total cholesterol | 0.07 | 0.590000 |
| % DCA vs. % LDL/HDL ratio | 0.44 | 0.000328 |
| % TDCA vs. % LDL/HDL ratio | 0.39 | 0.001900 |
| % GDCA vs. % LDL/HDL ratio | 0.31 | 0.015100 |
| % total DCA + conjugates vs. % LDL/HDL ratio | 0.41 | 0.001050 |
ApoA1, apolipoprotein A1; C4, 7α-hydroxy-4-cholesten-3-one; DCA, deoxycholic acid; GDCA, glyco-deoxycholic acid; NASH, non-alcoholic steatohepatitis; PGBF, pegbelfermin; TDCA, tauro-deoxycholic acid.
Spearman correlation was calculated using Spotfire software (TIBCO Software Inc., Palo Alto, CA, USA). Values >0.3 were considered significant.
Unadjusted p values were calculated using Spotfire software. Values <0.05 were considered significant.
PGBF affects secondary BA synthesis in healthy overweight or obese adults
As secondary BAs are produced by gut microbes, we evaluated whether PGBF treatment could modulate the gut microbiome. Matched serum and stool samples from healthy overweight or obese participants were collected during a phase I study (NCT03198182), which allowed for paired analysis of BA composition and gut microbiome activity before and after PGBF treatment. Complete BA data for all arms at all time points are shown in Table S2. BA levels were variable in this cohort; however, the median percentage change from baseline in serum DCA concentrations in both PGBF arms at Day 15 was lower than that in the placebo arm, and decreased median concentrations of GDCA and TDCA were observed in the PGBF 40 mg QW arm compared with placebo at Days 15 and 22 (Fig. S6). The changes in levels of DCA and its conjugates did not demonstrate a clear dose-dependent pattern. There was not a consistent change from baseline in serum concentrations of primary BAs and conjugates in the PGBF arms compared with the placebo arm (Fig. S7).
Fig. 5 shows the comparison between the percentage of DCA and conjugates and the percentage of primary BAs and conjugates in the total BA pool. The median percentages of total DCA and conjugates were variable across the 3 arms at baseline, although the differences were not significant because of the small sample size. In the PGBF arms, the percentage of total DCA and conjugates in the total BA pool was decreased at Day 15 and reduced further at Day 22 compared with baseline, but a clear dose-dependent response was not observed. This progressive decrease in the percentage of total DCA over time was not observed for the placebo arm.
Fig. 5.

Percentage of primary and secondary BA concentrations in relation to the total BA pool in healthy overweight or obese adults.
Total DCA + conjugates (DCA, GDCA, and TDCA) and total primary BAs (CA, GCA, TCA, CDCA, GCDCA, and TCDCA) as a percentage of the total BA pool from study NCT03198182. Median percentages for each arm at each time point are presented. Formal statistical analyses were not performed on these data because of the small sample size. BA, bile acid; CA, cholic acid; CDCA, chenodeoxycholic acid; D, day; DCA, deoxycholic acid; GCA, glyco-cholic acid; GCDCA, glyco-chenodeoxycholic acid; GDCA, glyco-deoxycholic acid; PGBF, pegbelfermin; QW, once weekly; TCA, tauro-cholic acid; TCDCA, tauro-chenodeoxycholic acid; TDCA, tauro-deoxycholic acid.
To evaluate whether PGBF-mediated changes in the gut microbiome might provide a mechanism to explain the selective effects on secondary BAs, a metagenomics analysis evaluating bacterial taxonomy was performed on stool samples collected pre- (Day 1) and post-PGBF treatment (Day 15). In total, 355 bacterial species were detected in all participants, and there was no PGBF-related difference in alpha diversity (microbial richness and evenness) or beta diversity (differences among samples) for any arms. Relative abundance of the phylum Actinobacteria was decreased at Day 15 compared with baseline in all arms, and there was no difference observed between the PGBF and placebo arms (data not shown). At the family level, relative abundance of Bifidobacteriaceae was also decreased at Day 15 compared with baseline in all arms (data not shown). Thus, after administration of 3 weekly doses, PGBF treatment had no discernable effect on faecal microbiome taxonomy in this analysis.
To evaluate whether PGBF treatment affects gut microbiome activity by altering microbial gene expression, metatranscriptomics analysis by RNA sequencing was performed. In all, 396 unique pathways and 9,168 unique genes were identified. In an analysis of pathway abundance, relative abundance of the secondary BA synthesis pathway decreased at Day 15 from baseline in a dose-dependent manner after PGBF, but not placebo, administration (Fig. S8). Furthermore, the median percentage change from baseline in secondary BA pathway gene abundance was lower in the PGBF arms (−41 and −66% for 20 and 40 mg QW, respectively) than in the placebo arm (+68%); the baseline median secondary BA pathway abundance was also lower in the placebo arm than in the PGBF arms.
Expression of individual genes related to the secondary BA synthesis pathway was analysed. The gene encoding choloylglycine hydrolase (BSH; EC 3.5.1.24) had detectable counts/abundance at baseline in all 24 samples tested (Table S3). In addition, genes encoding 7-alpha-hydroxysteroid dehydrogenase (hdhA), NAD+-dependent 7alpha-hydroxy-3-oxo bile acid-CoA-ester 4-dehydrogenase (baiCD) and NAD+-dependent 7beta-hydroxy-3-oxo bile acid-CoA-ester 4-dehydrogenase (baiH) had detectable abundance in the majority of the baseline samples. Of these genes, relative levels of the BSH gene, calculated as gene counts/total gene counts in the same sample, were the only ones that decreased after PGBF, but not placebo, administration (Fig. 6A and B). Median percent change in relative BSH gene counts from baseline to Day 15 was 46% for the placebo arm, -34% for the 20 mg QW PGBF arm, and -71% for the 40 mg QW PGBF arm. The magnitude of the BSH gene changes was similar to the changes in secondary BA pathway abundance (Fig. S8). To explore whether the changes in BSH gene expression were associated with the changes in serum DCA and conjugates, Spearman correlation was performed. Percentage change in GDCA at Day 15 had a moderate but significant positive correlation with percentage changes in BSH gene expression at Day 15 (ρ = 0.46; p = 0.025), whereas percentage change in total DCA and conjugates showed a non-significant, moderate correlation with percentage change in BSH gene expression (ρ = 0.40; p = 0.07).
Fig. 6.

Effects of PGBF on faecal levels of choloylglycine hydrolase (BSH) gene, DCA, and LCA in healthy overweight or obese adults.
(A) Relative faecal levels of choloylglycine hydrolase gene. (B) Percentage change from baseline to Day 15 in relative faecal choloylglycine hydrolase gene levels. (C) Faecal DCA and LCA levels at baseline and Day 15. (D) Percentage change from baseline to Day 15 in faecal DCA and LCA levels from study NCT03198182. Whiskers extend to most extreme data point that is ≤1.5× the IQR from the box. BSH, bile salt hydrolase; D, day; DCA, deoxycholic acid; LCA, lithocholic acid; PGBF, pegbelfermin; QD, once daily; QW, once weekly.
BSH catalyses the critical first step of secondary BA synthesis by deconjugating conjugated BAs. Therefore, decreases in BSH suggest the possibility of a corresponding decrease in secondary BA synthesis. Of the 15 BAs measured in faecal samples, the secondary BAs DCA and LCA constituted the major faecal BA species (80–99%; data not shown). Weight-corrected faecal levels of DCA and LCA were generally lower at Day 15 than at baseline for most PGBF-treated study participants (7 of 12 in the 20 mg PGBF QW arm; 5 of 6 in the 40 mg PGBF QW arm); however, only 2 of 6 participants who received placebo had a decrease in DCA and LCA levels at Day 15 relative to baseline (Fig. 6C). The median percentage decreases in faecal DCA and LCA levels at Day 15 tended to be greater in PGBF groups than in placebo, and the highest reductions were observed in the 40 mg PGBF QW arm (Fig. 6D). No other treatment-related changes were consistently observed for other BAs that are naturally present at very low levels in stool.
Discussion
In these exploratory post hoc analyses, serum concentrations of the secondary BAs DCA and conjugates were selectively decreased following PGBF treatment in patients with NASH, obese patients with T2DM, and healthy overweight or obese adults. The selective reduction of DCA and conjugates led to changes in the total BA pool, namely, a decreased percentage of DCA and conjugates and an increase in the percentage of primary BAs relative to the total BA pool. Furthermore, the data suggest that the decrease in total DCA concentration led to a lower serum concentration of total BAs after PGBF administration compared with placebo. To our knowledge, this is the first demonstration that treatment with a FGF21 mimetic modulates serum BA concentrations by selectively regulating gut microbiome-produced DCA in humans.
Of all BAs, the secondary BAs DCA and LCA have the highest hydrophobicity, a property that is thought to be linked to hepatotoxicity. Increased concentrations of DCA and its conjugates GDCA and TDCA are associated with drug-induced hepatic injury14 and are thought to promote hepatocellular carcinoma15,16 and colon cancer.17,18 Additionally, elevated DCA concentrations have been observed in patients with NASH relative to healthy adults.6,8 Thus, by selectively reducing serum concentration of DCA, treatment with PGBF may result in an overall reduction in the hydrophobicity of the total BA pool and lower the risk of further hepatotoxicity.
In this analysis, the decrease in total DCA following PGBF treatment led to an approximate 20–30% reduction in total BAs, which is similar to what has been observed after treatment with farnesoid X receptor (FXR) agonists.19 The selective effect of PGBF on secondary BAs may have advantages over other clinical-stage therapeutics that target primary BA synthesis, because reducing primary BA synthesis can decrease cholesterol removal and increase LDL cholesterol, which has been observed in clinical studies of FXR agonists19,20 and an FGF19 mimetic.21 Conversely, treatment with PGBF in patients with NASH was associated with a decrease in LDL cholesterol and an increase in HDL cholesterol,10 resulting in a decrease in LDL/HDL ratio, which estimates cardiovascular disease risk with better predictive value than LDL cholesterol alone.22 The PGBF-associated decreases in concentrations of DCA and conjugates were not associated with decreases in liver BA synthesis in patients with NASH. In contrast, elevated plasma C4 was observed in patients with NASH treated with PGBF, suggesting that primary BA synthesis may be increased in the liver. However, serum primary BA concentrations were not elevated after PGBF treatment, which may be indicative of increased turnover or excretion of BAs into bile. The underlying mechanisms that link PGBF administration with increased C4 levels are not known. The increase in C4 may be secondary to the decrease in DCA and conjugates, which is suggested by a moderate negative correlation for percentage changes from baseline in C4 and total DCA and conjugates (data not shown, ρ = −0.44, p = 0.00005). Interestingly, at baseline, C4 levels were moderately correlated with HDL cholesterol and ApoA1 levels, whereas increases in C4 levels after PGBF treatment were correlated with increases in ApoA1 and decreases in LDL/HDL ratio, suggesting that PGBF-associated changes in BA metabolism may contribute to an improvement in lipoprotein profiles that could possibly lead to a reduction of cardiovascular risk in patients with NASH. The decrease in C4 levels associated with FXR agonist treatment was associated with decreased HDL and increased LDL cholesterol in patients with NASH,20,23 suggesting that the correlation observed in this study between C4, HDL, and LDL/HDL may reflect the biological relationship of these parameters. Therefore, PGBF may reduce total BA levels to a similar degree as does FXR agonist treatment but may uniquely selectively decrease the most hydrophobic BAs without elevating LDL cholesterol as an unintended consequence. However, it is not known whether the effects on lipoproteins are caused by direct effects of PGBF on hepatocyte gene expression or caused by altered lipoprotein metabolism outside the liver. Although decreasing secondary BA concentrations appears to be a worthy therapeutic goal, it is presently unknown whether reduction of the secondary BA pool has a clinically meaningful impact on patients with NASH, specifically in terms of reducing or reversing fibrosis or reducing the risk of hepatocellular carcinoma or colon cancer.[15], [16], [17], [18] Future studies to address these questions are warranted.
Conflicting observations of the regulation of BA synthesis by FGF21 or FGF21 mimetics have been reported previously in animal models. Treatment with FGF21 or a long-acting FGF21 mimetic reduced cytochrome P450 7A1 (CYP7A1) and BA concentrations in the mouse liver and reduced plasma C4 concentrations in monkeys.11 However, chronic hepatic overexpression of FGF21 (via adeno-associated virus) increased BA synthesis by increasing expression of Cyp7a1 in diet-induced obese mice. FGF21 overexpression may increase Cyp7a1 by antagonising the function of FGF15/19 in the liver.24 Selective regulation of secondary BAs has not been reported in animal models.
Because production of DCA is mediated by gut microbes, we hypothesised that the changes in serum DCA concentrations may be associated with changes in microbiome activity. In a small cohort of adults who were overweight or obese but otherwise healthy, a reduction in serum concentrations of DCA and conjugates was observed as early as 2 weeks after initiation of PGBF treatment. Interestingly, dose-related decreases in the relative levels of microbial BSH gene expression in matched faecal samples were observed, whereas no PGBF-related changes in microbiome abundance were detected. BSH mediates the critical process of deconjugating conjugated BAs and serves as a gatekeeper to provide unconjugated BAs as substrates for subsequent BA transformations.25 The decrease in serum concentration of total DCA was moderately correlated with the decrease in BSH gene expression, suggesting that the decrease in BSH may contribute to the decreased serum concentration of DCA. Furthermore, decreased levels of DCA and LCA in stool would be consistent with the decrease in BSH, which would lead to the decrease in the synthesis of secondary BAs. Several published studies support the idea that DCA concentration may be regulated by BSH. In 1 study, pharmacologic activation of the pregnane X receptor and the constitutive androstane receptor led to reduction of Bifidobacterium, with reduction of BSH and a reduction in hepatic DCA concentration.26 Data from a 6-month randomised clinical trial demonstrated a significant increase in serum concentration of DCA and conjugates in participants who were fed a high-fat diet compared with those fed a low-fat diet; the increase in DCA was accompanied by an increase in BSH-expressing gut microbiota.27 Although PGBF-related changes in microbiota abundance were not observed in overweight or obese participants in this 2-week study, we cannot exclude the possibility that longer-term PGBF treatment may exert effects on the abundance of microbiota involved in secondary BA synthesis in patients with NASH.
It is encouraging that similar patterns of decreased secondary, but not primary, BAs in samples from 3 independent study populations treated with PGBF were observed in this post hoc analysis; however, larger, prospectively designed studies are needed to confirm these results and evaluate the clinical significance of the selective reduction of secondary BAs. Additionally, there are several other important limitations of this study that should be taken into consideration. Although decreased secondary BA concentrations were observed following PGBF treatment, the studies were not designed or powered to detect BA changes over time; further, patients were not stratified by baseline BA levels to ensure balancing across all study arms. Similarly, a caveat to the microbial gene expression analysis was that there were differences in secondary BA pathway abundance in the placebo vs. PGBF arms (Fig. S8). The sample size of the study of healthy overweight or obese adults was very small and, thus, did not permit statistical testing for the analyses of microbial activity; additionally, the PGBF treatment duration was only 2 weeks. Because of the inter-study differences and limitations of each study, data from a study of short-term PGBF treatment in healthy overweight or obese adults cannot necessarily be extrapolated to predict what might be observed in patients with NASH who receive long-term PGBF treatment.
In conclusion, this post hoc analysis demonstrates that treatment with PGBF in patients with NASH selectively decreased serum concentrations of the secondary BAs DCA and conjugates and reduced the percentage of secondary BAs in the total BA pool, the properties of which may be altered as a result of these changes. Preliminary data from healthy overweight or obese adults who received PGBF indicated that faecal secondary BAs and microbial BSH gene expression were reduced, suggesting that reduced secondary BA biosynthesis may be a potential mechanism that could account for our observations of selective reduction in serum DCA concentration following PGBF treatment. However, the molecular mechanisms that mediate the effect of PGBF on BSH gene expression and secondary BA synthesis remain elusive and warrant further investigation.
Financial support
This study was fully funded by Bristol Myers Squibb (Princeton, NJ, USA).
Authors’ contributions
Yi Luo: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Supervision, Validation, Writing - original draft, Writing - review & editing ; Benjamin E. Decato: Data curation, Formal analysis, Methodology, Validation, Visualization; Edgar D. Charles: Conceptualization, Investigation, Supervision, Writing - review & editing; Diane E. Shevell: Investigation, Resources, Writing - review & editing; Colleen McNaney and Petia Shipkova: Data curation, Methodology, Resources; Abraham Apfel: Data curation, Methodology; Giridhar S. Tirucherai: Investigation, Resources, Writing - review & editing; Arun J. Sanyal: Conceptualization, Supervision, Writing - original draft; Writing - review & editing.
Data availability statement
BMS policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
Conflicts of interest
YL, EDC, DES, CM, PS, AA, and GST are employees of Bristol Myers Squibb and may own company stock. BED was an employee of Bristol Myers Squibb at the time of the study. AJS has been a consultant for and received grants from Allergan, AstraZeneca, Bristol Myers Squibb, Intercept Pharmaceuticals, and Viking Therapeutics; has consulted for AbbVie, Affyimmune Therapeutics, Ardelyx, Chemomab, Conatus Pharmaceuticals, Echosens, Fractyl, Galectin Therapeutics, Immuron, Nitto Denko, Nimbus Therapeutics, Nordic Bioscience, Novo Nordisk, and Synlogic Therapeutics; has received grants from Cumberland Pharmaceuticals, Gilead Sciences, Merck, Mallinckrodt Pharmaceuticals, Novartis, Salix Pharmaceuticals, and Shire; owns stock in Akarna Therapeutics, Durect, Genfit, and Tiziana Life Sciences; and is employed by Sanyal Bioscience.
Please refer to the accompanying ICMJE disclosure forms for further details.
Acknowledgements
The authors thank Michael Reily (formerly of Bristol Myers Squibb) for his valuable contributions as a member of the research team that carried out this analysis. Medical writing support was provided by Amanda Martin, PhD, of Medical Expressions (Chicago, IL), and was funded by Bristol Myers Squibb.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhepr.2021.100392.
Supplementary data
The following are the supplementary data to this article:
References
- 1.Chalasani N., Younossi Z., Lavine J.E., Charlton M., Cusi K., Rinella M., et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–357. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L., Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Asgharpour A., Kumar D., Sanyal A. Bile acids: emerging role in management of liver diseases. Hepatol Int. 2015;9:527–533. doi: 10.1007/s12072-015-9656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arab J.P., Karpen S.J., Dawson P.A., Arrese M., Trauner M. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology. 2017;65:350–362. doi: 10.1002/hep.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Legry V., Francque S., Haas J.T., Verrijken A., Caron S., Chávez-Talavera O., et al. Bile acid alterations are associated with insulin resistance, but not with NASH, in obese subjects. J Clin Endocrinol Metab. 2017;102:3783–3794. doi: 10.1210/jc.2017-01397. [DOI] [PubMed] [Google Scholar]
- 6.Puri P., Daita K., Joyce A., Mirshahi F., Santhekadur P.K., Cazanave S., et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67:534–548. doi: 10.1002/hep.29359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiao N., Baker S.S., Chapa-Rodriguez A., Liu W., Nugent C.A., Tsompana M., et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut. 2018;67:1881–1891. doi: 10.1136/gutjnl-2017-314307. [DOI] [PubMed] [Google Scholar]
- 8.Ferslew B.C., Xie G., Johnston C.K., Su M., Stewart P.W., Jia W., et al. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig Dis Sci. 2015;60:3318–3328. doi: 10.1007/s10620-015-3776-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charles E.D., Neuschwander-Tetri B.A., Pablo Frias J., Kundu S., Luo Y., Tirucherai G.S., et al. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: results from a randomized phase 2 study. Obesity. 2019;27:41–49. doi: 10.1002/oby.22344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanyal A., Charles E.D., Neuschwander-Tetri B.A., Loomba R., Harrison S.A., Abdelmalek M.F., et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2018;392:2705–2717. doi: 10.1016/S0140-6736(18)31785-9. [DOI] [PubMed] [Google Scholar]
- 11.Chen M.M., Hale C., Stanislaus S., Xu J., Véniant M.M. FGF21 acts as a negative regulator of bile acid synthesis. J Endocrinol. 2018;237:139–152. doi: 10.1530/JOE-17-0727. [DOI] [PubMed] [Google Scholar]
- 12.Li J., Dawson P.A. Animal models to study bile acid metabolism. Biochim Biophys Acta Mol Basis Dis. 2019;1865:895–911. doi: 10.1016/j.bbadis.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tirucherai G.S., Ide T., Shevell D., Mora J., Charles E.D. Safety and pharmacokinetics of multiple-dose BMS-986036 in healthy Japanese and non-Japanese adults [abstract] Hepatology. 2018;68:756A–757A. [Google Scholar]
- 14.Woolbright B.L., McGill M.R., Staggs V.S., Winefield R.D., Gholami P., Olyaee M., et al. Glycodeoxycholic acid levels as prognostic biomarker in acetaminophen-induced acute liver failure patients. Toxicol Sci. 2014;142:436–444. doi: 10.1093/toxsci/kfu195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshimoto S., Loo T.M., Atarashi K., Kanda H., Sato S., Oyadomari S., et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 16.Yamada S., Takashina Y., Watanabe M., Nagamine R., Saito Y., Kamada N., et al. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget. 2018;9:9925–9939. doi: 10.18632/oncotarget.24066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayerdörffer E., Mannes G.A., Richter W.O., Ochsenkühn T., Wiebecke B., Köpcke W., et al. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology. 1993;104:145–151. doi: 10.1016/0016-5085(93)90846-5. [DOI] [PubMed] [Google Scholar]
- 18.Ochsenkuhn T., Bayerdorffer E., Meining A., Schinkel M., Thiede C., Nussler V., et al. Colonic mucosal proliferation is related to serum deoxycholic acid levels. Cancer. 1999;85:1664–1669. [PubMed] [Google Scholar]
- 19.Neuschwander-Tetri B.A., Loomba R., Sanyal A.J., Lavine J.E., Van Natta M.L., Abdelmalek M.F., et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel K., Harrison S.A., Elkhashab M., Trotter J.F., Herring R., Rojter S.E., et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58–71. doi: 10.1002/hep.31205. [DOI] [PubMed] [Google Scholar]
- 21.Harrison S.A., Rinella M.E., Abdelmalek M.F., Trotter J.F., Paredes A.H., Arnold H.L., et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2018;391:1174–1185. doi: 10.1016/S0140-6736(18)30474-4. [DOI] [PubMed] [Google Scholar]
- 22.Millán J., Pintó X., Muñoz A., Zúñiga M., Rubiés-Prat J., Pallardo L.F., et al. Lipoprotein ratios: physiological significance and clinical usefulness in cardiovascular prevention. Vasc Health Risk Manag. 2009;5:757–765. [PMC free article] [PubMed] [Google Scholar]
- 23.Younossi Z.M., Ratziu V., Loomba R., Rinella M., Anstee Q.M., Goodman Z., et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–2196. doi: 10.1016/S0140-6736(19)33041-7. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J., Gupte J., Gong Y., Weiszmann J., Zhang Y., Lee K.J., et al. Chronic over-expression of fibroblast growth factor 21 increases bile acid biosynthesis by opposing FGF15/19 action. EBioMedicine. 2017;15:173–183. doi: 10.1016/j.ebiom.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foley M.H., O'Flaherty S., Barrangou R., Theriot C.M. Bile salt hydrolases: gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dempsey J.L., Wang D., Siginir G., Fei Q., Raftery D., Gu H., et al. Pharmacological activation of PXR and CAR downregulates distinct bile acid-metabolizing intestinal bacteria and alters bile acid homeostasis. Toxicol Sci. 2019;168:40–60. doi: 10.1093/toxsci/kfy271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan Y., Yuan J., Li J., Li H., Zhang J., Tang J., et al. Unconjugated and secondary bile acid profiles in response to higher-fat, lower-carbohydrate diet and associated with related gut microbiota: a 6-month randomized controlled-feeding trial. Clin Nutr. 2019;39:395–404. doi: 10.1016/j.clnu.2019.02.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BMS policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.