

Abstract
New drug production, from target identification to marketing approval, takes over 12 years and can cost around $2.6 billion. Furthermore, the COVID-19 pandemic has unveiled the urgent need for more powerful computational methods for drug discovery. Here, we review the computational approaches to predicting protein–ligand interactions in the context of drug discovery, focusing on methods using artificial intelligence (AI). We begin with a brief introduction to proteins (targets), ligands (e.g. drugs) and their interactions for nonexperts. Next, we review databases that are commonly used in the domain of protein–ligand interactions. Finally, we survey and analyze the machine learning (ML) approaches implemented to predict protein–ligand binding sites, ligand-binding affinity and binding pose (conformation) including both classical ML algorithms and recent deep learning methods. After exploring the correlation between these three aspects of protein–ligand interaction, it has been proposed that they should be studied in unison. We anticipate that our review will aid exploration and development of more accurate ML-based prediction strategies for studying protein–ligand interactions.
Keywords: protein–ligand interaction, drug discovery, binding site, binding affinity, binding pose, machine learning, deep learning
Introduction to protein–ligand interactions
Proteins participate in a wide range of essential intra- and intercellular mechanisms. However, they do not work independently in living organisms. Frequently, they must bind with other molecules (other proteins, nucleic acids, metal ions, organic and inorganic molecules, etc.) to form a specific interaction in order to perform their function [1–4]. Species capable of binding to the protein are known as ligands. As an example, consider the inhibitory drugs peramivir and bosutinib, as demonstrated in Figure 1, which will be used to illustrate protein–ligand interactions in the subsequent sections.
Figure 1.
(A) Sketch of peramivir, an inhibitor of the viral protein neuraminidase from the H1N9 influenza virus. (B) Sketch of human Src kinase inhibitor bosutinib.
Particularly, intermolecular interactions between proteins and ligands occur at specific positions in the protein, known as ligand-binding sites, which has sparked a lot of interest in the domain of molecular docking and drug design. Binding sites, also referred to as binding pockets, are typically concavities on the surface of proteins. Pockets, where small drug-like ligands bind, are typically located in deep cavities. Ligand-binding sites are typically found in large, deep pockets [5, 6] on the protein surface, while some of them may exist in exposed shallow clefts [7, 8]. In medicinal chemistry, there is an emphasis on identifying key proteins whose biochemical functions can be definitively linked to diseases. Such proteins become targets for drug development. In fact, the binding site is considered druggable if the ligand binds with high affinity at the binding site and has an effective therapeutic action [9].
When attempting to predict protein–ligand interactions, a labyrinth of interactions needs to be accounted for to generate an accurate prediction. Biologically, two major factors play into the complexity of protein–ligand interactions, large spectrum of ligand types: small organic molecules, organometallics, nucleic acids, peptides and even other proteins [10]. This paper will primarily focus on small organic compounds as those are immediately relevant to medicinal therapies as this class of ligands is more commonly associated with inhibitor and inactivating ligands than other classes of ligands. The second factor is the resulting intermolecular forces from within the protein–peptide chains, protein-solvent interactions and the binding ligands [10, 11]. Often, these forces are represented by forcefields to simplify computation. However, some estimations resulting from the use of forcefields have been scrutinized as a means of error when generating predictions for ligand interactions and has been shown in the past to significantly misrepresent the potential binding affinity, poses of the suspected ligands [11]. Due to the large variety of ligand types, the defining interactions between protein and ligand can also vary. The most recognizable stabilizing forces are hydrogen bonding and the columbic forces also more commonly referred to as electrostatic interactions. This however is far from the only intermolecular forces driving favorable binding enthalpy. Van der Waals forces including the critical hydrophobic interactions of the London dispersion forces, pi stacking of aromatic compounds and other ion-induced dipole and dipole–dipole interactions also play a role [10, 11]. An example of this can be seen with the human Src kinase inhibited by bosutinib. To better visualize this mode of binding between Src kinase and bosutinib, a hydrophobic surface rendering was generated in chimera as seen in Figure 2. In addition, these images showcase the ligand, bosutinib, drawn in the space fill style to better demonstrate why the ligand is posed in the way that it is as not only do the interactions need to be fulfilled, but they must also satisfy steric requirements to avoid clashes. Another major interaction is the energy involved in overcoming the desolvation of the ligand and the binding residues as they interact with the solvating water molecules. These forces are not static in their accumulative contributions to binding enthalpy. For example, hydrogen bonding can vary greatly ranging from 1 up to 40 kJ/mol due to factors like the donating and accepting species, the type of hydrogen bonding and the distance between the donating and accepting species.
Figure 2.
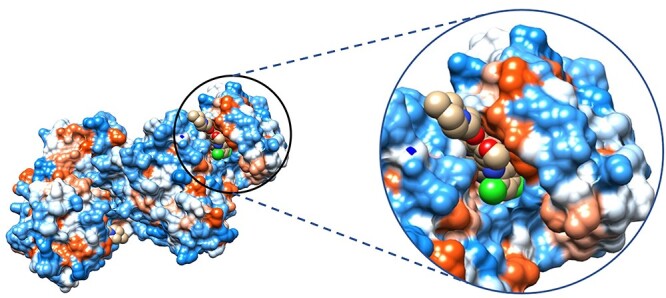
Human Src kinase docked by bosutinib visualized with a hydrophobic surface generated in Chimera, PDB code 4MX0. Most hydrophobic regions colored red; most hydrophilic indicated in blue.
The production of a new drug, from target identification through approval for marketing, can take over 12 years and cost around $2.6 billion [12–14]. The COVID-19 pandemic has unveiled the urgent need for rapid drug development [3]. In most drug design projects, the initial goal is to find ligands that bind to a specific protein target with high affinity and specificity. There is a significant need for expediting the computational process for identifying promising drug candidates for experimental validation [15].
An initial step in the drug discovery pipeline is to identify molecules that bind with high affinity to the target, which can be further developed into drug-like molecules (lead compounds) [16]. Because of our limited understanding of the dynamic relationship between chemical space and genomic space [17–19], identifying novel drugs and their targets remains a difficult task. Experimental methods to identify lead compounds, such as high-throughput screening, can be time-consuming and expensive [16]. In contrast, computational prediction of protein–ligand interaction (PLI) can significantly reduce the resources, time and cost required and reduce the need for physical experimental studies to screen for new therapeutics. Reliable PLI predictive algorithms can thus greatly accelerate the development of new treatments, remove toxic drug candidates and efficiently direct medicinal chemistry [20]. Machine learning (ML) algorithms adopt a different approach from classical virtual screening (VS) [21] approaches. In the case of ligand-based virtual screening (LBVS), it utilizes the active ligand’s information and similarity between candidate ligands and the known active compounds to find new ligands [22]. As a result, these methods are useful when there is no 3-dimensional structure of the target protein available. Likewise, structure-based virtual screening (SBVS) method uses the 3D structure of a target to screen compound libraries [23]. In contrast, ML follows the approach of learning the relation between physicochemical parameters and protein–ligand interactions from the known structures of protein–ligand complex pairs to derive statistical models for predicting the status of other unknown ligands/proteins.
To identify associations between drugs and target proteins (i.e. interaction between them), Yamanishi et al. [24] suggested a kernel regression-based technique to infer protein–ligand interactions by combining the chemical structure information of ligands, sequence information of proteins, as well as the drug–target complex network. Similarly, another published work includes the experiments by Bleakley and Yamanishi [25], called BLM, that employs the supervised learning method. Cao et al. [26] proposed another prediction method based on the random forest (RF) algorithm. Similarly, in the framework of restricted Boltzmann machines, Wang and Zeng [27] introduced the method to predict not just binary contacts between proteins and ligands but also diverse types of interactions, viz how they interact with one other. Readers can explore the review papers [28–31] for the AI-driven drug discovery process including target identification, hit identification, lead optimization, chemical synthesis prediction and drug repositioning.
A simplified illustration of ML technique in protein–ligand interaction prediction is depicted in Figure 3. This figure represents the general workflow of ML architecture. Initially, the features of target protein and ligand are extracted, followed by data preprocessing steps. Normalization (a data preparation technique) is frequently used in machine learning, which converts the values of numeric columns in a dataset to a similar scale without distorting the ranges of values or losing information. Normalized data, thus obtained, are fed to a machine learning model such as neural network in the figure. This step is often performed because data standardization improves accuracy, as shown by empirical evidence [32]. Several processes occur at hidden layers and eventually the output layer outputs the predicted decision. As it does not demand the explicit hard-coded rules curated by human experts to make prediction and potentially yield good prediction accuracy, there has been a lot of interest in using ML and particularly powerful deep learning methods to predict PLIs.
Figure 3.
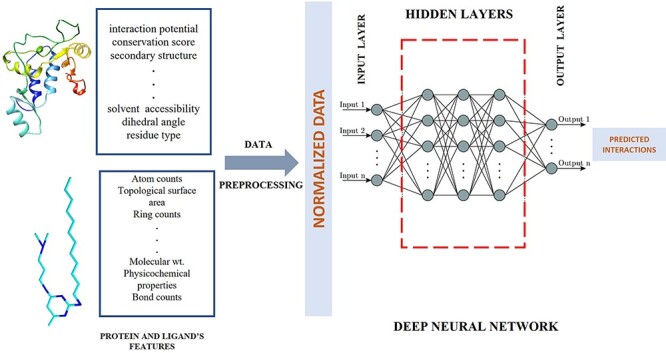
Conceptual workflow of ML pipeline. Inputs are the properties of the target protein and ligands, and output are the predicted interactions.
To achieve highly accurate predictions for new data, a sufficient amount of training data is required. The increase of structural information for protein–ligand complexes, and the cataloging of this information into databases, has enabled researchers to explore artificial intelligence (AI) approaches, mostly ML methods, for virtual screening.
Owing to the advent of many robust AI techniques and the abundance of data in this domain, many surveys have been conducted examining the existing ligand-binding site, binding affinity and binding pose prediction algorithms. A decade ago in 2009, a review group led by Henrich, analyzed various computational methods to identify protein-binding sites for ligand design [33]. Because machine learning applications in this field were still in their infancy years ago, authors focused mostly on the traditional, yet successful, methods. Macari et al. [34] published a review paper focusing on the computational paradigms in the domain of protein-small molecule binding site prediction. Here, they analyzed the approaches from traditional geometrical techniques to recent machine learning strategies and compared the characteristics and performances of the techniques. Similarly, Zhao and his team [35] discussed the expensive computing resources associated with training deep learning algorithms in comparison to traditional machine learning algorithms with an ending note that the prediction problems are still not solved mainly because of presence of some cryptic sites [36].
Focusing on the binding affinity (inhibition constant, dissociation constant and binding energy) prediction models, the team of Heck (in 2017) published a review paper largely concentrating on successful supervised machine learning methods. Authors mention that this holistic credit behind the rapid development of ML strategies in this field goes to the open-source ML libraries and the publicly available data sources, while Yang’s group (in 2020) [37] argue that sufficiently large and unbiased datasets would help training robust AI models more accurately to predict protein–ligand interactions.
In the paper by Ellingson et al. [38], the authors discuss the trends of ML in the domain of drug-binding prediction (binding pose and energy prediction), data sources and potential problems associated with them. Additionally, Chen’s group [39] summarizes the web servers and databases used in drug–target identification and drug discovery. Here, for ML-based approaches, they mostly concentrate on the supervised and semisupervised models. A similar kind of analysis was carried out by Inhester and Rarey [40] describing the publicly available databases containing the affinity data and structural information that plays a vital role in describing interaction geometries and strength of binding.
Recently, Lim’s team [41] published a review paper on compound protein interaction (CPI) prediction models that includes a precise description of the data format used, the techniques associated with model development and emerging methods. They also provide an overview of databases as chemistry-centric, protein-centric and integrated database and analyzed the diversified methods of AI like, tree, neural network, kernel and graph-based methods in the field of CPI. Since widely used human-readable formats, SMILES, generally fail to represent critical information like neighborhood in 3D space, latent vector representation of compounds and proteins is highly recommended. In the same way, since deep learning (DL) methods make black-box decisions (difficult to understand how decisions are made by neural networks), authors are in favor of attention mechanisms to address this issue.
The unique goal of this review is to illustrate the importance of interconnection of separately discussed topics of PLI: binding site, binding affinity and binding pose prediction beforehand. A systematic search on three aspects of ML-based protein–ligand interactions (i.e. binding site, binding affinity and binding pose prediction) was carried out using Google Scholar. Research methodologies were populated based on the year of publication as well as underlying relevant techniques. Selected literature was analyzed, articles and group of articles were compared, themes identified, and gaps noted, and suggestions recommended for future research. We have tried to provide a comprehensive, organized summary of related databases, recent research trends in AI-guided PLI prediction, their interconnection and prospects so that researchers can fully utilize these resources to develop novel prediction methods.
Existing databases for AI-driven protein–ligand interaction models
The main purpose of ML/AI algorithms is to reveal hidden information/knowledge in data. For ML models to understand how to perform different tasks, training datasets are fed into the algorithm. The model sees and learns from the training data automatically. The model can recognize the underlying, hidden relationships and patterns in the data that are not obvious to human and even experts. The validation dataset, on the other hand, is a different dataset that is often used during training to assess how well the model is performing and used to tune the hyperparameter of the model. After the model has been fully trained and validated, one can run assessment metrics on an independent test dataset not used in training to monitor the performance of the model predictions. As a result, data are critical for such ML applications. Generally, the more data provided to the ML system, the greater its performance of learning and prediction.
Here, we discuss the prevailing datasets in the field of PLIs that serve as sources of training, testing and validating data for ML/AI methods. We studied 63 AI-driven methods that have been published since 2004 and investigated the availability of several databases for training–testing–validating them. In Figure 4A, 32 publications on binding affinity prediction using AI’s most frequently used database is PDBBind [42], followed by CASF benchmarking dataset [42] and BindingDB [43]. In Figure 4B, we observe the recurrence of PDBBind for training AI models for predicting binding pose that signifies its popularity and usefulness. As shown in Figure 4C, out of 25 AI-driven binding site prediction methods, many datasets are mostly self-curated by developers according to the requirement for training–testing–benchmarking purposes. The most frequent datasets used in this domain are BioLip, CASP targets, LigAsite and PDBbind. It is worth noting that many datasets were created using the original protein structure and ligand data in the Protein Data Bank (PDB) [44]. Overall, these datasets would be valuable resources for those who are looking for validating and developing ML-driven PLI prediction methods and for the study of drug design in general.
Figure 4.
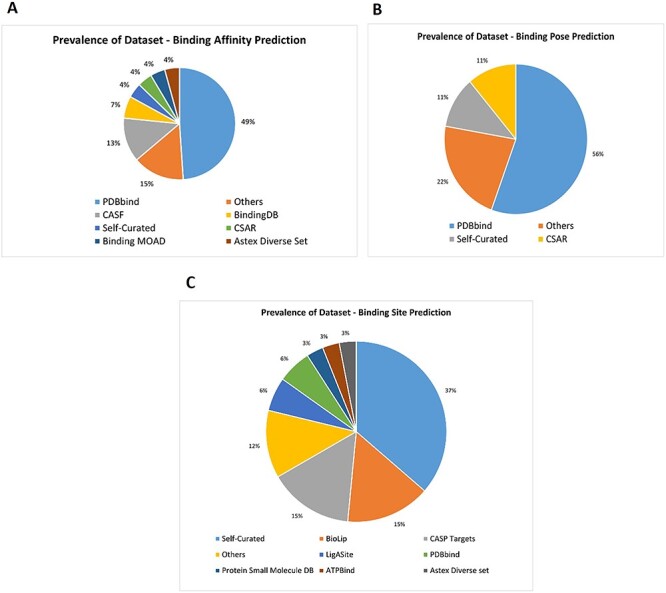
Pie charts showing the distribution of prevailing datasets for the AI-based PLI prediction models. (A) Prevalent dataset for AI based protein-ligand binding affinity prediction models. (B) Prevalent dataset for AI based protein-ligand binding pose prediction models. (C) Prevalent dataset for AI based protein-ligand binding site prediction models.
PDBBind
The PDBbind database was created in 2004 by Wang et al. [42] providing a broad set of binding affinity data that are experimentally determined. The binding affinity data are for all the types of biomolecular complexes that are deposited in the PDB. Originally, PDBbind was limited to complexes formed by proteins and small-molecule ligands. Starting from 2008, other types of biomolecular complexes in PDB were added into PDBbind. Being updated annually, the latest release (i.e. version 2020) contains binding data (Kd, Ki and IC50 values) for 19 443 protein–ligand, 2852 protein–protein, 1052 protein–nucleic acid and 149 nucleic acid–ligand complexes as shown in Figure 5. Here, all binding data are curated by the authors derived from original literatures.
Figure 5.
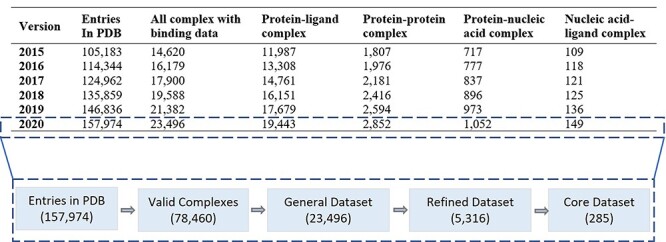
Statistics of PDBBind dataset showing its composition from Version 2015 as well as the basic structure of Version 2020.
The PDBbind version 2020 is based on the contents of PDB officially released at the first week of 2020. It contains 157 974 structures that were experimentally determined. A series of computer programs were implemented to screen the entire PDB to identify four major types of molecular complexes: protein–small ligand, nucleic acid–small ligand, protein–nucleic acid and protein–protein complexes. Version 2020 is the most recent edition at the time of writing this manuscript.
LIGand attachment site (LigASite)
LIGand Attachment SITE (LigASite) is a publicly accessible dataset of biologically relevant binding sites in protein structures. To automatically filter out the biologically irrelevant ligands, an algorithm is used that considers the number of heavy atoms in the ligand and the number of interatomic contacts between protein and ligand [45]. The fact that each protein has both unbound and bound structures ensures that this dataset can be used to benchmark the binding site prediction models. The version 9.7(nr25) contains the list of 391 proteins, while the redundant list contains the information of 816 proteins.
BioLiP
BioLiP is a semimanually curated database of ligand–protein binding interactions that are biologically relevant. After the automated process for determining a ligand’s biological relevance is completed, a thorough manual review is performed to correct any errors. The structure data are mainly obtained from the PDB, with biological insights obtained from literature and other databases. Eventually, the manual check is performed, and possible false-positive entries are verified by reading the original literature and consulting other databases that ensures the completeness and high quality of BioLiP [46]. It is updated on weekly basis and the total number of entries in the current version (17 September 2021: at the time of writing) is 529 047, which includes 109 998 proteins from PDB, 57 059 DNA/RNA ligands, 25 960 peptide ligands, 146 969 metal ligands and 299 051 regular ligands. Out of these entries, 23 492 have binding affinity information.
BindingDB
BindingDB is a public, web-accessible database, extracted from the scientific literature, which consists of binding affinities between protein targets and small, drug-like molecules. The latest version: (September 2021), it has 41 296 entries, containing 2 338 906 binding data with 8617 protein targets and 1 011 134 small molecules [43]. Moreover, the BindingDB website offers a comprehensive collection of tools for querying, analyzing and downloading binding data.
Binding MOAD
Binding MOAD is the largest possible (hence called mother of all databases) deposition of high-quality, protein–ligand complexes available from the PDB, which was augmented with the inclusion of binding data extracted from literature. Binding MOAD’s preference for affinity data is Kd over Ki over IC50 [47–49]. It was designed using the ‘top-down’ approach so that it contained every protein–ligand complex with a 3D structure. Annual updates are done to contain more binding-affinity data as they become available in the PDB. The current release (2019) contains 38 702 protein–ligand structures, 14 324 binding data, 18 939 ligands and 10 500 protein families.
The CASF dataset
Scoring functions are often used to evaluate PLIs in structure-based drug design. Several scoring functions have been established thus far, and hence some open-access benchmarks are required for assessing their strength and weakness. CASF benchmark provides the precalculated prediction results of known scoring functions allowing a fair comparison of the model with existing scoring functions on the same test set. All performance tests enabled in CASF-2016 are based on a set of 285 protein–ligand complexes with high-quality crystal structures and reliable binding data [42]. This test set is selected from the PDBbind refined set (version 2016). In CASF-2016, the evaluation methods have been improved in comparison to its previous version (CASF-2013) and the performance of a scoring function is evaluated by four metrics: (i) scoring power, (ii) ranking power, (iii) docking power and (iv) screening power.
Prediction of the ligand bind sites of proteins
Virtual Screening (VS) requires knowledge of the location of the ligand-binding site (LBS), which in some cases this information is unknown. Accurate prediction of protein–ligand binding sites from a 3D protein structure plays a crucial role in structure-based drug design [50, 51] and can aid in drug side effects prediction [52] as well as understanding a protein’s function [53]. Intermolecular interactions between proteins and ligands occur through amino acid residues at specific positions in the protein, usually located in pocket-like regions. Identification of these key residues is imperative for elucidating protein function, analyzing molecular interactions and facilitating docking computations in virtual screening–based drug design. These specific key amino acid residues in proteins are called the LBSs. Empirical studies show that the actual ligand-binding site correlates to the biggest pocket on the surface of a protein [6, 54]. On a test set of 67 protein structures [55], the SURFNET architecture [56] successfully predicted the ligand-binding site as the largest pocket in 83% of the cases. The findings from LIGSITE [57] also displayed that the ligand-binding site was found in the largest pocket in all 10 proteins tested. Similar was the result yielded from POCKET [58].
Each amino acid (residue) has a distinct impact on the structure and function of a protein. Even if the measured distance between two residues in a protein sequence is long, the spatial distance between them may be short due to protein folding [59–61]. As a result, residues in the sequence that are far from the target residue sequentially, but spatially close, can also have a significant effect on the position of the binding residues. AlphaFold [62] can be considered as one of the major breakthroughs that predicts the tertiary structures of most proteins rather accurately integrating 1D, 2D and 3D protein features. Ultimately, there is a need to consider the spatially neighboring residues for the binding site prediction. Furthermore, the secondary and tertiary structure of the protein also impacts binding, often more significantly than the primary structure.
To further exemplify the concept of ligand-binding site, we take an instance of neuraminidase, an influenza virus protein extensively studied due to its candidacy as a drug target. It is often seen as a good drug target due to the ability to disrupt the life cycle of the virus. Unfortunately, influenza is irritatingly good at manufacturing drug-resistant variations driving the need for constant development of new drugs. One such drug to be developed is peramivir also known as BCX-1812 (sketch shown in Figure 1A. Structurally, peramivir inhibits neuraminidase (interaction shown in Figure 6A) by forming numerous electrostatic interactions including salt bridges and H-bonding [63]. When observing the protein monomer, 11 hydrogen bonds can be found holding peramivir in place greatly increasing the binding affinity for the compound through its enthalpic effects on the system (Figure 6B and C) [6, 54–58]. Although the experimental determination provides the most accurate assignment of the binding locations, it is a time- and labor-intensive process. Computational methods for the detection and characterization of functional sites on proteins have grown in popularity, and as a result, numerous methods have been developed in recent decades attempting to address this issue.
Figure 6.
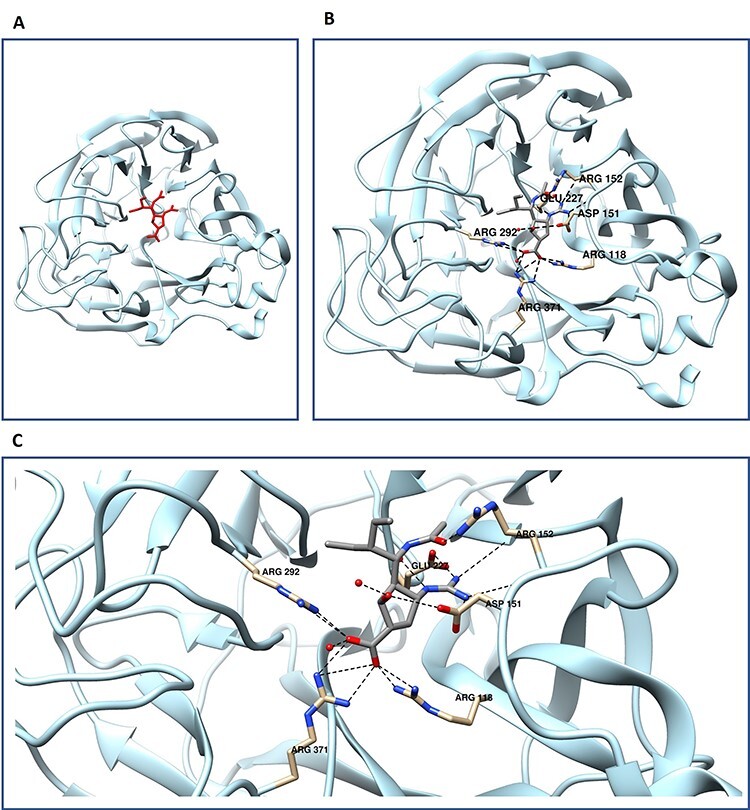
Protein–ligand interactions demonstrated through neuraminidase–peramivir interaction. (A) Neuraminidase monomer with peramivir depicted in red. (B) view of full monomer with peramivir with hydrogen bonding pairs labeled and displayed in canonical atom coloring. Oxygen colored red, and nitrogen in blue. (C) focused view of neuraminidase peramivir hydrogen bonding, PDB code 1L7F.
Binding site prediction methods
Many different approaches to predicting the binding site have been established over the last two decades, based on (i) templates, (ii) energy functions, (iii) geometric considerations and (iv) ML.
The template-based methods attempt to predict the position of binding sites on an input protein using known protein templates. They are based on the assumption that proteins sharing a similar structure can also share a similar function [64]. In comparison to the geometry and energy-based methods, these methods are generally more accurate if a good template can be found [34].
Energy-based approaches rely on the principle to find energetically favorable regions on the protein surface that contain ligand-binding pockets. In most cases, the protein is enclosed in a grid structure, and the interaction energy at each grid point is calculated using one or more probes. The resulting interaction points are then clustered to predict the location of pockets.
In geometry-based methods, the geometry of the molecular surface is analyzed to find surface cavities on the target protein. Based on the algorithm used for cavity localization, Macari et al. [34] have divided it into three subcategories: (i) grid system scanning, (ii) probe sphere filling (iii) alpha shape. In the grid system scanning approach, the protein is enclosed into a three-dimensional grid, and latter, if certain geometric conditions are met, are considered as points belonging to a pocket. The accuracy of this method is dependent on the resolution of the grid. Probe sphere–based approaches are based on directly filling pockets or cavities with specific probe spheres. Furthermore, alpha shape methods rely on the computation of Delaunay triangulation [65] to locate voids on the protein surface. The final step of all these methods involves clustering and a ranking procedure of the pockets identified.
The earlier pioneering binding prediction methods use preexisting templates, employing genetic identity, or molecular geometry to predict the binding pockets. COACH and later COACH-D took advantage of the PDB by using previously solved structures as a template for the predictions of the target complex [66, 67]. Another popular server, LigASite and its upgrade LIGSITEcsc utilizes geometry by scanning the model on a 3D grid map for convolutions by defining grid points to determine protein solvent locations ultimately identifying potential binding locations [68]. Using these concepts of course has its limitations. Accuracy of the predictions begins to decay as the pairwise identity of the target in respect to the template decreases and often become unreliable as the identity approaches the ‘twilight zone’ [69, 70]. The methods based on templates are rather limiting when encountering novel protein structures. As the field matured, FindSite was developed that also took advantage of structural similarity to find templates. Biologically, the rationale behind using structural and genetically similar proteins as templates for the prediction is due to the idea that as proteins accumulating mutations from years of evolution will result in functional regions having a higher degree of conservation within the same protein family due to risking loss of function [67, 70, 71]. However, cautions must be used when using this method as this conservation is not perfect and there can be a high degree of variability among the residues within the family or the function of highly conserved regions of the protein [70].
But how do these non-ML prediction methods compare to ML? Template-based methods perform well if templates having known binding site information can be found, but do not work if there are no good templates. Machine learning methods can learn from existing data and generalize to new data that are not similar to the training data. Energy function–based approaches are calibrated from a small set of known protein–ligand structures using the function designed by human experts, which may not fit a large amount of data best and cannot generalize well to new data. Similarly, geometry-based methods locate surface cavities on the target protein by analyzing the geometry of the molecular surface, whose precision is largely dependent on the resolution of grid [34]. Another limitation to geometry-based method is the sensitiveness to the scanning direction and to the orientation of protein in the grid system [72]. But machine learning, particularly deep learning methods, can directly learn a parameterized function from a large amount of data integrating multiple sources of information, leading to better accuracy of predicting protein–ligand interaction.
The growing availability of high-resolution protein structures in various databases has opened up new possibilities for machine learning (ML) applications. The basic workflow of existing ML methods to predict binding sites can be divided into five main steps: data acquisition and preprocessing, feature engineering, model development, training–testing, hyperparameter tuning and evaluation. At first, several sources of known protein–ligand binding data are aggregated, and several significant features are extracted to represent the protein and ligand, which are then normalized. Then, ML models are designed to use the input features to predict binding sites, including shallow supervised learning algorithms, artificial neural network, convolutional neural network and ensemble methods (different approaches are further described in detail in Sections `Classical ML methods for binding site prediction' and `Deep learning methods for binding site prediction'). A typical ML workflow is illustrated in Figure 3. We group ML methods into two categories: classical ML methods (non–deep learning methods) and modern deep learning methods to be described separately below.
Classical ML methods for binding site prediction
Table 1 contains a summary of a list of the classical methods, their machine learning techniques, input features and training/test data, which are reviewed below.
Table 1.
Classic ML methods to predict protein–ligand binding sites
| SN | Approach | Techniques | Features | Database used | Year |
|---|---|---|---|---|---|
| 1 | Oriented Shell Model [73] | Support vector machine | Developed oriented shell model, utilizing distance and angular position distribution | Self-curated | 2005 |
| 2 | SitePredict [74] | Random forest | Predicted small ligand-binding sites mobilizing backbone structure | Self-curated | 2008 |
| 3 | LIBRUS [75] | Support vector machine | Combined ML and homology information for sequence-based ligand-binding residue prediction | Self-curated + FINDSITE’s database | 2009 |
| 4 | Qiu and Wang’s method [76] | Random forest | Used eight structural properties to train random forest classifiers, latter combined to predict binding residues | Q-SiteFinder’s dataset | 2011 |
| 5 | Wong et al.’s method [77] | Support vector machine + differential evolution | Classified the grid points with the location most likely to contain bound ligands | LigASite | 2012 |
| 6 | DoGSiteScorer [78] | Support vector machine | Web server for binding site prediction, analysis and druggability assessment | Self-curated | 2012 |
| 7 | Wong et al.’s method [79] | Support vector machine | Used SVM to cluster most probable ligand-binding pockets using protein properties | LigASite + self-curated | 2013 |
| 8 | TargetS [80] | Support vector machine + modified AdaBoost | Designed template-free predictor with classifier ensemble and spatial clustering | BioLip | 2013 |
| 9 | Wang et al.’s method [81] | Support vector machine + statistical depth function | SVM model integrating sequence and structural information | PDBbind | 2013 |
| 10 | LigandRFs [82] | Random forest | Applied random forest ensemble to identify ligand-binding residues from sequence information alone | CASP9 targets + CASP8 targets | 2014 |
| 11 | Suresh et al.’s method [83] | Naive Bayes classifier | Trained Naive Bayes classifier using only sequence-based information | Self-curated | 2015 |
| 12 | OSML [84] | Support vector machine | Proposed dynamic learning framework for constructing query-driven prediction models | BioLip + CASP9 targets | 2015 |
| 13 | PRANK [7] | Random forests | Developed mechanism to prioritize the predicted putative pockets | Astex Diverse set + self-curated | 2015 |
| 14 | UTProt Galaxy [85] | Support vector machine + neural network + random forest | Developed pipeline for protein–ligand binding site predictive tools using multiomics big data | Self-curated | 2015 |
| 15 | Chen et al.’s method [86] | Random forest | Proposed dynamic ensemble approach to identify protein–ligand binding residues by using sequence information | ccPDB + CASP9 targets + CASP8 targets | 2016 |
| 16 | Chen et al.’s method [87] | Random forest | Predicted allosteric and functional sites on proteins | PDBbind + allosteric DB + CATH DB | 2016 |
| 17 | TargetCom [88] | Support vector machine + modified AdaBoost algorithm | Designed ligand-specific methods to predict the binding sites of protein–ligand interactions by an ensemble classifier | BioLip | 2016 |
| 18 | P2Rank 2.1 [89] | Bayesian optimization | Improved version of P2Rank | Self-curated | 2017 |
| 19 | P2Rank [90] | Random forest | Built stand-alone template-free tool for prediction of ligand-binding sites | Self-curated | 2018 |
| 20 | PrankWeb [91] | Random forest | Online resource providing an interface to P2Rank | Self-curated | 2019 |
In 2013, Wong et al. [77] proposed a method for predicting protein–ligand binding sites using support vector machines (SVM). SVM was used to cluster the pockets that are most likely to bind ligands based on geometric characteristics (grid values calculated by LIGSITE and SURFNET that can represent binding site), interaction potential (calculated using the PocketFinder method), offset from protein, conservation score (obtained from a residue-level analysis) and properties surrounding the pockets. The dataset (LigASite) used to train the method faces the same issue as most bioinformatics dataset: imbalance, i.e. the number of positive examples (the grid points of binding site) is much less than the negative examples (the other grid points). To mitigate this problem, undersampling of negative examples was used, which resulted in better performance. Likewise, Integrating Data Selection and Extreme Learning Machine for Imbalanced Data (IDELM) [92] can be implemented to overcome the data imbalance problem. IDELM, which was designed by modifying Extreme Learning Machine (ELM) [93], was reported to have a faster learning capacity in comparison to ELM.
In the same year, Yu et al. proposed TargetS [80], a template-free LBS predictor with classifier ensemble and spatial clustering to address the challenge, especially when the target proteins’ 3D structures are unavailable, or no homology models are available in the library. To create discriminative features, protein evolutionary details, predicted protein secondary structure (as determined by PSIPRED [94]) and ligand-specific binding propensities of residues were combined. To address the severe imbalance problem between positive (binding) and negative (nonbinding) samples, an improved AdaBoost classifier ensemble scheme based on random under sampling was used.
Another approach was proposed by Wang et al. [81] introducing the statistical depth function to identify negative samples for predicting binding site using sequence and structural information with SVM. In this study, the statistical depth functions were used to determine the depth of the residues and analyze the protein structure. They chose the half-space depth function to calculate the depth of the residues out of a variety of statistical depth functions because the concept and description of the half-space depth are simple and straightforward. Their research revealed that defining a negative sample in this manner was fair and beneficial to model training.
Inspired by the promising performance of SVM, many other ML approaches have been implemented. In 2005, Guo et al. [73] introduced a new statistical descriptor, named Oriented Shell Model, that considers the distance and angular position distribution of several structural and physicochemical features. Similarly, Kauffman and Karypis developed a sequence-based approach, called LIBRUS [75], based on SVM in 2009. And in 2012, Volkamer et al. published DoGSiteScorer [78], a web-based tool for predicting binding sites and determining druggability. The same SVM technique was applied by Yu et al. in their method: OSML [84].
In Suresh et al.’s method [83], they implemented Naive Bayes classifier with amino acid residue in membrane protein sequence. Here, they predicted whether the given input is a ligand-binding residue or not using only sequence-based information. They opted Bayesian classifiers since they are resistant to real-world noise and missing values [37].
LigandRFs [82], a sequence-based method for identifying protein–ligand binding residues with RF, was developed by Chen et al. In the process of encoding input features, they proposed a hybrid technique to reduce the effects of different sliding residue windows. They also built several balanced datasets, for each of which an RF-based classifier was trained, addressing the high imbalance between ligand-binding sites and non–ligand-binding sites. They discovered that hydrophilic amino acids are more likely to be ligand-binding sites. Besides LigandRFs, RF algorithm was also implemented by Qiu and Wang’s method [76], Bordner [74], PRANK [7], PrankWeb [91] and UTProt Galaxy [85].
Deep learning methods for binding site prediction
Deep learning methods have grown in popularity in recent years due to their potential in capturing complicated relationships hidden within the data. Several deep learning methods for binding site prediction are summarized in Table 2.
Table 2.
List of deep learning methods to predict protein–ligand binding sites
| SN | Approach | Techniques involved | Feature | Database used | Year |
|---|---|---|---|---|---|
| 1 | DeepCSeqSite [95] | Deep convolutional neural network | Proposed sequence-based approach for ab initio protein–ligand binding residue prediction. | BioLip | 2019 |
| 2 | DELIA [96] | Hybrid Deep neural network + bidirectional long short-term memory network | Designed hybrid deep neural network is to integrate 1D sequence-based features with 2D structure-based amino acid distance matrices. | BioLip + ATPBind | 2020 |
| 3 | Kalasanty [97] | 3D convolutional neural network | Designed model based on U-Net’s architecture. | sc-PDB [98] | 2020 |
| 4 | DeepSurf [99] | Deep convolutional neural network + ResNet | Proposed surface-based deep learning approach for protein–ligand binding residue prediction. | scPDB | 2021 |
| 5 | PUResNet [100] | ResNet | Based on deep ResNet + novel data cleaning process. | scPDB | 2021 |
Cui et al. proposed DeepCSeqSite [95] for predicting protein-ligand binding residues, a sequence-based method based on a deep convolutional neural network (CNN). Several convolutional layers were stacked to obtain hierarchical features from input. Binding residues belonging to any selected ligand class were classified as positive samples in the training sets, whereas the remainder were labeled as negative samples. Seven types of features are used for the protein-ligand binding residue prediction: position-specific score matrix, relative solvent accessibility, secondary structure, dihedral angle (predicted by ANGLOR [101]), conservation scores, residue type and position embeddings, which are purely derived from protein sequences.
In 2020, a 3D fully CNN (based on an architecture called U-Net) was published for finding druggable pockets on protein surface [97]. U-Net [102] is a state-of-the-art neural network architecture that was initially invented to deal with the 2D medical images. In this method, the task of pocket detection was reformulated as a 3D image segmentation problem. Both the input and output are represented as 3D grids of the same dimensions.
Xia et al published a deep learning–based method called DELIA [96], which is a hybrid deep neural network integrating a CNN with a bidirectional long short-term memory network (BiLSTM) to mobilize 1D sequence feature vectors and 2D distance matrices. DELIA’s hybrid neural network architecture is made up of three main modules: (i) feature extractor, (ii) residual neural network (ResNet) and (iii) BiLSTM. To improve the model, oversampling in minibatch, random undersampling and stacking ensemble strategies were used to resolve the problem of the extreme data imbalance between binding and nonbinding residues.
In 2021, Mylonas’ team [99] proposed a binding site prediction method, DeepSurf, based on deep learning architecture. This work is unique in the sense that it mobilizes surface-based representation (implementation of 3D voxelized grids) along with state-of-the-art deep learning architectures to predict potential druggable sites on proteins. After the input features are determined, those grids are imported to a 3D CNN and the resulted ligandability scores of each surface point obtained thus are clustered to create the binding sites.
Recently in late 2021, Kandel et al. published a paper called PUResNet [100], which involves the implementation of Deep ResNet as the backbone of the network in their model for the prediction of protein–ligand binding site. This is unique in the prospect of data cleaning process. Here, 3D protein structure of protein is fed into the model as input and probability of voxel belonging to cavity is given as output. Later, these predictions are saved as mol2 files and visualized using molecular modeling software.
Prediction of protein–ligand binding affinity
In order to be a lead molecule for drug development, a molecule must be able to bind tightly to a target protein; i.e. it must have a high affinity. The degree of attraction between a receptor (e.g. a protein) and its binding partner (e.g. drug or inhibitor) is measured by binding affinity, which can be expressed by the thermodynamic value of dissociation constant (Kd) or in the case of inhibitors (Ki). Table 3 demonstrates a variability of different inhibitors acting upon different proteins. SmCI N23A is a mutant variant of the SmCI inhibitor, demonstrating how small changes to an inhibitor can greatly affect Ki.
Table 3.
Binding affinity (Ki) of the SmCI group of inhibitors on three proteins [161].
| Inhibitor ligand (protein) | Porcine pancreatic elastase | Trypsin | Bovine carboxypeptidase A | Human carboxypeptidase A1 |
|---|---|---|---|---|
| SmCI | 2.66 × 10−8 | 3.81 × 10−8 | 2.83 × 10−8 | – |
| rSmCI | 1.70 × 10−8 | 3.66 × 10−8 | 9.55 × 10−8 | 2.54 × 10−8 |
| SmCI N23A | 1.94 × 10−9 | 4.08 × 10−10 | 4.25 × 10−8 | 1.29 × 10−8 |
Here, – is an indication of no data
Predicting a protein–ligand complex’s binding affinity (such as inhibition constant, dissociation constant and binding energy) is critical for efficient and effective rational drug design. However, experimentally measuring protein–ligand binding affinity is time-consuming and complex, which is one of the major bottlenecks of the drug discovery process.
As discussed earlier, the dissociation constant (Kd) can be used to explain the affinity between a protein and a ligand. The smaller Kd, the stronger the binding. In case of enzymes and their inhibitors, the inhibitory constant Ki is equivalent to Kd. Further information about thermodynamic measurements of PLIs has been reviewed by Perozzo et al. [103].
In computational medicinal chemistry, calculating ligand-binding affinity is an open challenge. The ability to computationally predict binding affinity of small molecules to specific biological targets is extremely useful in the early stages of drug discovery since it allows a mathematical model to determine PLIs. When opposed to conventional experimental methods or computational scoring approaches, ML methods are significantly faster and less expensive.
In the last couple of years, several databases (as discussed in the `Existing databases for AI-driven protein-ligand interaction models' section) have been maintained. The impressive amount of collected experimental data in these datasets can be used to design different deep learning architectures to develop ML-driven ligand-binding affinity prediction methods.
This section is mainly focused on the application of ML in predicting the ligand-binding affinity, which is still an open challenge in computational drug discovery.
Classical ML approaches for binding affinity prediction
While modern ML libraries include many supervised ML techniques, the majority of ML applications for the development of scoring functions have concentrated on three main techniques: SVM, RF and linear regression. Table 4 lists the methods that have been studied using classical ML techniques.
Table 4.
List of classical ML approaches to predict protein–ligand binding affinity
| SN | Approach | Technique involved | Feature | Database used | Year |
|---|---|---|---|---|---|
| 1 | Deng et al.’s method [104] | Kernel partial least squares | Applied knowledge-based QSAR approach + used genetic algorithm-based feature selection method. | Self-curated | 2004 |
| 2 | Ashtawy et al.’s method [105] | KNN + SVM + MLR + MARS + RF + BRT | Explored range of scoring functions employing ML approaches utilizing physicochemical features that characterize protein–ligand complexes. | PDBbind | 2011 |
| 3 | CSCORE [106] | Regression | Developed Cerebellar Model Articulation Controller (CMAC) learning architecture. | PDBbind | 2011 |
| 4 | SFCscoreRF [107] | Random forest | Followed random forest approach to train new regression models. | PDBbind + CSAR | 2013 |
| 5 | B2BScore [108] | Random forest | Predicted binding affinity for protein−ligand complexes based on β contacts and B factor. | PDBBind | 2013 |
| 6 | Li et al.’s method [109] | Random forest + multiple linear regression | Analyzed the importance of structural features to binding affinity prediction using the RF variable importance tool. | PDBbind | 2014 |
| 7 | Wang et al.’s method [110] | Random forest | Predicted the protein–ligand binding affinity based on protein sequence, binding pocket, ligand structure and intermolecular interaction feature set. | PDBbind | 2014 |
| 8 | Cyscore [111] | Linear regression | Improved protein–ligand binding affinity prediction by using a curvature-dependent surface area model. | PDBbind | 2014 |
| 9 | Pred-binding [112] | Random forest + support vector machine | Applied ML algorithms for binding affinity prediction problem based on a large-scale dataset. | PDSP Ki DB + PubChem + DrugBank + ChemSpider | 2016 |
| 10 | Avila et al.’s method [113] | ML methods available in SAnDReS | Applied machine learning box interface of SAnDReS to explore the scoring function virtual space (SFVS). | PDBbind + MOAD + BindingDB | 2017 |
| 11 | Ferreira et al.’s method [114] | ML methods available in SAnDReS | Predicted Gibbs free energy of binding (ΔG) based on the crystallographic structure of complexes. | MOAD + BindingDB + PDBBIND | 2018 |
| 12 | Kundu et al.’s method [115] | GP + LR + MP + SMOR + Kstar + RF | Incorporated Weka 3.6.8 package to select optimum parameters of the ML algorithms. | PDBBind | 2018 |
| 13 | Boyles et al.’s method [116] | Random forest + XGBoost | Used ligand-based features to improve ML scoring functions. | PDBbind + CASF | 2019 |
| 14 | RASPD+ [117] | SVM + LR + KNN + SDN + RF + ERF | Introduced fast prefiltering method for ligand prioritization based on ML models. | PDBbind + DUD-E | 2020 |
| 15 | Amangeldiuly et al.’s method [118] | RF + SVR + XGBOOST + KNN | Designed prediction method for binding kinetics based on the ML analysis of protein−ligand structural features. | BindingDB + self-curated | 2020 |
| 16 | Wee and Xia’s method [119] | Ollivier persistent Ricci curvature-based ML | Persistent attributes were used as molecular descriptors and further combined gradient boosting tree. | PDBbind | 2021 |
In 2014, Li et al. investigated the significance of structural features in binding affinity prediction and discovered that RF can effectively leverage more structural features and more training samples, resulting in better prediction performance than multiple linear regression [109]. Later, in 2016, Shar et al. developed a method called Pred-binding [112], where they compared the performance of RF and SVM, and found that both models (RF and SVM) provide a potent Ki predictability while avoiding overfitting. Similarly, Wang et al. [110] conducted a comparative analysis of affinity prediction for family-specific protein–ligand complex using RF method. Their method predicted the binding affinity using the features like protein sequence, binding pocket, ligand structure and intermolecular interaction.
Inspired by the Cerebellar Model Articulation Controller (CMAC) learning architecture, a method named CSCORE [106] was designed that used a data-driven scoring function for accurate binding affinity prediction. But it had poor interpretability power. It has been noticed that torsion angles play an important role in docking. Despite its significance, it was not considered in this model. In the study reported in B2BScore [108], authors used RF for binding affinity prediction based on β Contacts and B Factor. The key concept of B2Bscore was focused on two physicochemical properties of PLIs: B factor and β contacts, both of which had not previously been used in affinity prediction. Here, the B factor assesses the mobility and flexibility of dynamic atoms in proteins, which is critical in determining the protein’s activity and functions and β contacts are a small fraction of distance-based contacts [120].
Recently, Holderbach’s team published a protein–ligand binding free energy prediction method called RASPD+ [117], which employs the fast prefiltering approach for ligand prioritization, where RF outperforms others. Similarly, Ashtawy et al. [105] investigated a variety of various ML methods in combination with physicochemical features of protein–ligand complexes. Ensemble prediction methods RF and Boosted Regression Trees (BRT) were found to be the most effective in predicting binding affinities of protein–ligand complexes. SFCscoreRF [107] and Boyles et al.’s work [116] followed the RF approach to train new regression models as well.
In 2018, Kandu et al. [115] used the RF and Gaussian process regression algorithms on protein–ligand binding affinity prediction. As part of the feature extraction process, they determined a total of 127 ligand and protein features. For proteins, they used the whole protein rather than just features of pockets and cavities. This is because calculating the features of the cavity necessitates the details of the cavity, which is time-consuming. Similarly, for ligands, all physicochemical properties available in Pubchem [121], as well as a few structural properties measured using a method called Padel Descriptor [122] were included. The Gaussian process, linear regression, multilayer perceptron, sequential minimal optimization (SMO) regression [123], K-star [124] and RF were used to train 2864 instances with 128 features, and they discovered that the RF model was ideally suited to the protein–ligand binding energy prediction problem.
Deep learning methods for binding affinity prediction
Table 5 summarizes a list of the deep learning methods for the binding affinity prediction. Back in 2015, to predict binding affinity, Ashtawy and Mahapatra provided novel scoring functions that used a large ensemble of neural networks. [125]. For accurate predictions, the bagging- and boosting-based ensemble of neural networks scoring functions was used. According to their research, the proposed neural network–based scoring functions BsN-Score and BgN-Score obtained the best results. They also found that the neural network–based ensemble models outperformed RF models.
Table 5.
List of deep learning methods to predict protein–ligand binding affinity
| SN | Approach | Technique involved | Feature | Database used | Year |
|---|---|---|---|---|---|
| 1 | BgN-Score and BsN-Score [125] | Ensemble neural networks | Assessed the scoring accuracies of two new ensemble neural network scoring functions based on bagging (BgN-Score) and boosting (BsN-Score). | PDBbind | 2015 |
| 2 | Gomes et al.’s method [126] | Atomic convolution layer | Developed 3D spatial convolution operation for learning atomic-level chemical interactions. | PDBBind | 2017 |
| 3 | KDEEP [127] | 3D convolutional neural networks | Featurized protein and ligand considering eight pharmacophoric-like properties that are used by a three-dimensional CNN model. | PDBbind | 2018 |
| 4 | DeepDTA [128] | Convolutional neural network | Proposed deep learning–based model that uses only sequence information of both targets and drugs to predict drug target interaction binding affinities. | Kinase dataset + KIBA dataset | 2018 |
| 5 | Pafnucy [129] | Deep neural network | Represented molecular complex with a 4D tensor, processed by three convolutional layers and three dense (fully connected) layers. | PDBbind + CASF + Astex Diverse Set | 2018 |
| 6 | OnionNet [130] | Deep convolutional neural network | Constructed modified deep CNN and defined customized loss function to train multiple-layer intermolecular contact features. | PDBbind + CASF | 2019 |
| 7 | Zhu et al.’s method [131] | Neural network | Predicted the binding affinity from a given pose of a 3D protein−ligand complex by pairwise function based on neural network. | PDBbind + CASF-2016 | 2020 |
| 8 | DeepAtom [132] | 3D convolutional neural network | Extracted binding–related atomic interaction patterns automatically from the voxelized complex structure. | PDBbind + Astex Diverse Set | 2020 |
| 9 | Jones et al.’s method [133] | 3D CNN + Spatial Graph-CNN | Developed fusion models to benefit from feature representations of two neural network models to improve the binding affinity prediction. | PDBBind | 2020 |
| 10 | AK-score [134] | 3D CNN ensemble | Used ensemble of multiple independently trained networks that consist of multiple channels of 3D CNN layers. | PDBbind + CASF | 2020 |
| 11 | graphDelta [135] | Graph-convolutional neural network | Designed graph-convolutional neural networks for predicting binding constants of protein−ligand complexes. | PDBbind + CSAR + CASF | 2020 |
| 12 | DeepDTAF [136] | Deep convolutional neural network | Employed dilated convolution to capture multiscale long-range interactions. | PDBbind | 2021 |
| 13 | LigityScore [137] | Convolutional neural network | Designed rotationally invariant scoring functions. | PDBbind + CASF | 2021 |
| 14 | Seo et al.’s method [138] | Deep attention mechanism | Employed deep attention mechanism based on intermolecular interactions. | PDBbind + CSAR | 2021 |
| 15 | DEELIG [139] | Convolutional neural network | CNN was used to learn representations from the features. | Self-curated | 2021 |
| 16 | ResAtom System [140] | ResNet + attention mechanism | Implemented ResNet neural network with added attention mechanism. | PDBbind + CASF | 2021 |
In 2018, Jimenez et al. presented KDEEP [127], a protein–ligand affinity predictor based on 3D convolutional neural networks (CNN), which have shown promising results across a wide range of datasets. In this study, both protein and ligand were featurized via a voxelized 24 Å representation of the binding site considering different pharmacophoric-like properties. These descriptors were used by a 3D CNN model, which learns the binding affinity of the complex given enough training examples. Once trained, the network could predict previously unseen instances. Similarly, in 2020, Mohammad Rezaei’s team published a research method called DeepAtom [132], which utilized 3D CNN to extract the atomic interaction patterns from the voxelized complex structure.
On the other hand, Ozturk et al. proposed a deep learning–based model, DeepDTA [128], which made use of only drug–target’s sequence information. This study introduces a new deep learning–based model for drug–target affinity prediction that utilizes protein and drug character representations.
In 2019, a new method, OnionNet [130], a multiple-layer intermolecular-contact-based CNN was developed for protein−ligand binding affinity prediction. Its input features were based on rotation-free element pair-specific contacts between ligands and protein atoms. Later in 2020, Zhu et al. proposed the binding affinity prediction method by pairwise function based on ANN [131]. Basically, it predicts binding affinity from a given pose of a 3D protein–ligand complex and shows that a simple neural network model based on pairwise interatomic distances performs relatively well for binding affinity prediction.
In addition to these, there have been reports on the works based on the ensemble-based approach. In the study reported in AK-score [134], its model used an ensemble of multiple independently trained networks composed of multiple channels of 3D CNN layers to predict a complex’s binding affinity, which significantly improved prediction quality. The ensemble approach has the advantage of requiring no additional network architecture modifications and being easily applied to most existing models. In 2020, Jones et al. proposed improved protein–ligand binding affinity prediction with structure-based deep fusion inference [133]. In this project, they developed a midlevel fusion model together with 3D CNN and spatial graph CNN to predict protein–ligand binding affinity. graphDelta [135] is a related method that uses graph neural networks to predict binding affinity.
Wang et al. proposed a predictive model called DeepDTAF [136], where the local and global features were generated using only 1D sequence data. (3D structures of proteins, ligands and their complexes were excluded in input representation). It was a successful method for capturing multiscale interactions for protein–ligand binding affinity prediction that merged dilated convolution with traditional convolution. While in the model, developed by Wang’s team, the ResAtom System [140], they implemented ResNet neural network with added attention mechanism. Similar to ResAtom System, Seo et al.’s method [138] also employed attention mechanism to protein–ligand complex binding affinity as attention mechanism was able to capture the ligand-binding sites that contributed to the improvement in the prediction.
The Ahmed group earlier this year in 2021 proposed a deep learning approach called DEELIG [139], which used CNN to extract the spatial relationship information. Docked poses or protein–ligand complexes were not used as input in this research. Another similar CNN application in this domain is LigityScore [137] that includes rotationally invariant scoring functions called LigityScore1D and LigityScore3D.
Predicting and scoring of protein–ligand binding pose (3D structure)
The ligand active conformation is the 3D structure of a ligand when it is coupled to a protein. Binding mode of the ligand/drug is defined as the orientation of a ligand relative to the target in the bound state. Straightforward to grasp, a binding pose is simply a candidate binding mode.
A visual illustration of protein–ligand binding pose can be found in Figure 7 [141]. It depicts the human large GTPase known as dynamin. Biologically dynamin can excise clathrin-coated vesicles anchored to the membrane for endocytosis of the molecules to their targeted destination within the cell. Due to this, some viruses have learned to use this machinery to gain access to their host cell thus making dynamin a promising druggable target. Figure 7 displays an in silico docking model of two pyrimidine analogues docked within the PH domain of the protein. This demonstrates how ligand pose can vary with extremely minor changes even among structurally similar ligands within the same protein further complicating predictions. In molecular docking, many binding poses are computationally generated and then evaluated using a scoring function. A scoring function is a mathematical model that quantifies the binding stability of the pose, which can be used to rank and select binding poses/conformations. The outcome of a docking run, therefore, is a ligand’s top pose selected according to its predicted binding score. Despite some similarities, it is worth noting that the scoring function here is used to measure the binding stability of a pose, which is conceptually different from the scoring function to quantify the experimental binding affinity such as dissociation constant in `Prediction of protein-ligand binding affinity' section.
Figure 7.
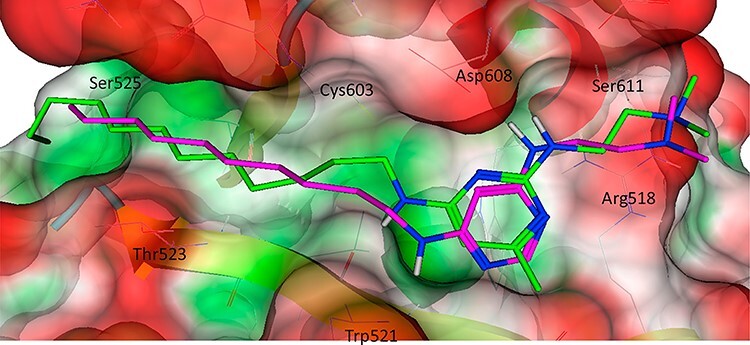
In silico prediction of two similar analogue Inhibitors docked within the binding sight-2 of human dynamin-1 PH domain. (Figure adopted from [141]) Both analogues share very similar intermolecular forces such as H-bonding and yet slight differences in the ligand’s orientation occur.
The current approaches of predicting protein–ligand binding pose typically have two steps: (i) generating protein–ligand binding poses and (ii) evaluating the poses using a scoring function. As the first step is mostly carried out by some standard/mature docking tools such as AutoDock [142], AutoDock Vina [143], Glide [144], GOLD [145] and Internal Coordinate Mechanics (ICM) [146], the recent development is mostly focused on the second step. Therefore, we mostly review the literature on scoring binding poses in the remaining part of this section below.
The scoring function is one of the most critical aspects of molecular docking. It is used for the selection and ranking of the best poses from the potentially wide list of candidates simulated/generated by the docking algorithms. Despite significant progress, designing a good scoring function capable of reliably predicting binding stability for any conformation remains a challenge in molecular docking.
Generally, scoring functions can be divided into four types: forcefield, empirical, knowledge-based and machine learning–based scoring functions. Please refer to these papers [20, 147–151] for in-depth references to first three kinds. The fourth kind ML-based scoring function is reviewed in the following section.
Here, we mainly focus on classical ML-based scoring functions of scoring protein–ligand docking poses, as well as provide insights into recently introduced deep learning (DL) and reinforcement learning–based scoring methods.
Classical ML and DL scoring methods for binding pose prediction
With the rapid expansion of ML techniques, ML-based scoring functions have steadily emerged as a promising alternative for scoring docking poses and virtual screening, with majority of them outperforming a wide range of traditional scoring functions. In recent years, the emergence of more powerful deep learning (DL) methods has inspired interest in the exploitation of more precise scoring functions. Table 6 contains a list of the methods described in the subsequent section.
Table 6.
List of ML methods to predict the binding score of protein–ligand binding pose
| SN | Approach | Technique involved | Feature | Database used | Year |
|---|---|---|---|---|---|
| 1 | Ashtawy et al.’s method [152] | MLR + MARS + KNN + SVM + RF + BRT | Employed ML approaches utilizing physicochemical and geometrical features characterizing protein–ligand complexes | PDBbind | 2015 |
| 2 | Grudininet et al.’s method [153] | Regression | Predicted binding poses and affinities with a statistical parameter estimation | PDBBind + HSP90 dataset + MAP4K dataset | 2016 |
| 3 | Ragoza et al.’s method [154] | Convolutional neural network | Trained CNN scoring function to discriminate binding poses using the differentiable atomic grid format as input | PDBbind | 2017 |
| 4 | Ragoza et al.’s method [155] | Convolutional neural network | Trained and optimized CNN scoring functions to discriminate between correct and incorrect binding poses | CSAR | 2017 |
| 5 | Nguyen1 et al.’s method [156] | Random forest + convolutional neural networks | Used mathematical deep learning for pose and binding affinity prediction | PDBbind | 2018 |
| 6 | Jose et al.’s method [157] | Reinforcement learning | An approach to represent the protein–ligand complex using graph CNN that would help utilize both atomic and spatial features to score protein–ligand poses | PDBbind + self-curated | 2021 |
In 2015, to select the optimal pose of a ligand in protein’s binding site, Ashtawy and Mahapatra utilized a variety of machine learning approaches to estimate the difference (root mean square deviation (RMSD)) between a pose and the true structure of a protein–ligand complex [152]. They used protein–ligand complexes’ physicochemical and geometrical features for their prediction and found that ML models trained to predict RMSD values significantly outperform all traditional scoring functions. The best-known empirical scoring function ASP, which is used in the commercial docking software GOLD [145], had a 70% success rate. On the same test set, their top RMSD-based scoring function, MARS::XARG, had a success rate of 80%, indicating a major improvement in docking performance. They also noticed that increase in training set size and number of features increases the performance of scoring functions.
The D3R grand challenges hosted by the Drug Design Data Resource (D3R) has also provided opportunities for computer scientists and bioinformatics researchers to explore the recent advancement in this field. In 2015 D3R Challenge, Grudinin et al. evaluated their procedure to score binding poses for protein–ligand complexes using a regression method [153]. They used the affinity and structural data from the PDBBind database to train the model’s free parameters with a regularized regression.
Similarly, in the following D3R Grand Challenge in 2018, Nguyen et al. developed a ML-based scoring function [156] to select the poses generated by GOLD [145], GLIDE [144] and Autodock Vina [158]. They created a training dataset of complexes from the PDB after being given a ligand target. Then, using docking software, they re-docked ligands to proteins in those selected complexes. They implemented RF to learn the biomolecular structure and used CNNs to capture topological features. The consensus of the energy values predicted by these two ML strategies was the final predictions for this method.
In 2017, Ragoza et al. developed a method using CNN scoring functions to take a detailed 3D representation of a PLI as input automatically learning the main features of PLIs that correlate with binding [155]. Their CNN scoring functions were trained and optimized to distinguish between correct and incorrect binding poses, as well as known binders and nonbinders. The CSAR-NRC HiQ dataset was used as the pose prediction training set, with the addition of the CSAR HiQ Update. They discretized a protein–ligand structure into a grid that is 24 Å on each side and centered around the binding site with a default resolution of 0.5 to handle the 3D structural data as input. Their CNN models were defined and trained using the Caffe deep learning framework [159]. The classes were balanced by sampling the same number of positive and negative examples in each batch after shuffling training data. The CNN model outperformed the Autodock Vina scoring function significantly in terms of intertarget ranking of CSAR poses. They did, however, perform worse in terms of intratarget pose ranking.
Recently, a reinforcement learning–based method for predicting the score of ligand pose has been put forward by the team of Jose [157]. In this method, the agent optimizes the correct pose and can also be trained to locate the binding site. The hypothesis is that training on protein–ligand complexes with known binding poses would aid the reinforcement learning algorithm in approximating the underlying molecular interactions, using the input atomic and spatial features provided as molecule fingerprints. Based on the atomic, spatial and molecular features, this would result in an optimized pose in the desired binding site of the protein of interest. Here, the overall network for reinforcement learning–based protein–ligand docking consists of a GraphCNN layer that represents atomic and molecular properties as a feature vector, as well as an optimization mechanism that approximates the docking scoring function.
Interconnection of protein–ligand binding site, binding affinity and binding pose
Like all matter, proteins are influenced and shaped by thermodynamic principles that include protein–ligand interactions. Often when characterizing these interactions, intermolecular forces such as hydrogen bonding, van der Waals forces, ion-induced dipoles, desolvation and electrostatic forces are described. All of which directly impact the enthalpy ΔH of the system. Interactions such as H-bonding can net approximately 20 kJ/mol assuming optimal geometry and distance. The intermolecular interactions of the ligand can also dictate the pose of the ligand within the binding pocket. Poses can vary greatly from ligand to ligand due to adjustments of the ligand to bend or twist to accommodate the attractive and repulsive forces involved. It is important to remember molecules are dynamic in nature with varying degrees of flexibility and not static and stiff. Ligands will attempt to orient themselves in the lowest energy conformation possible. Of course, entropy also plays a big role as hydrophobicity of the compounds and the environment in which it is docking can greatly shift what kinds of ligands can bind and how the compound can be accommodated within the binding pocket. The hydrophobicity of the pocket and the ligand can drastically alter the pose of the prospective ligand or outright prevent docking even if the compound is capable of accommodating some of the intermolecular forces needed for specific binding. The entropic penalty of the protein–ligand complex must also not be ignored as it will require most of the free energy that would result from the stabilizing interactions. Because system enthalpy and entropy are so critical in determining the likeliness and orientation of ligand binding, it is commonplace for Gibbs free energy, ΔG to be used as it incorporates both thermodynamic parameters along with accounting for temperature and pressure of the system, which can be valuable when investigating kinetics and molecular dynamics. ΔG is also useful when describing proteins, allowing for equilibrium constants to be derived, in this case, Kd or Ki. The relationship and the effects of the thermodynamic parameters have been reviewed, and it is easy to see how ignoring either thermodynamic principle can greatly impact binding prediction accuracy.
To demonstrate these principles in a more tangible manner, we will use four peptidases with two inhibitors as an example; see Table 3. In this example, we can note that the inhibitor in this case is a family of proteins identified as SmCIs. Biologically, these proteases are regulated through the inhibitory action of its ligand, SmCI proteins. Within Table 3, we see two inhibitors targeting four different peptidases plus one mutant. When comparing SmCI with rSMCI in bovine carboxypeptidase A, we can find an almost 3-fold difference in their Ki. When looking at trypsin, we find the introduction of somewhat conservative mutation of Asn to Ala causes a drastic magnitudinous change within the Ki. The substantial change in Ki highlights the significance of small thermodynamic changes within the system can have upon ligand binding. This mutation also demonstrates how changes in the system may be counterintuitive if the entire system is not considered.
It is important to be mindful of the system as a whole. Isolating individual parameters such as binding affinity from binding pose will lead to a lackluster prediction, an accurate prediction can only be obtained by accurately representing the controlling natural phenomenon as close as possible. In this case, thermodynamics is the physical link that dictates what and how a ligand will bind to a protein. Therefore, it is essential to integrate the three traditionally separated tasks of predicting binding sites, binding affinity and binding pose together in one comprehensive machine learning system.
Conclusion and future direction’
In conclusion, based on the findings presented in studies in the above sections, it appears that ML-based PLI prediction methods can reach a higher level of accuracy if we incorporate the use of a large number of physicochemical properties and implement state-of-the-art deep learning techniques.
In the case of binding site prediction, there exists severe data imbalance in the benchmark datasets, making it an imbalanced learning problem, in which the number of samples in different classes (binding or nonbinding) differs significantly. It has been found that applying conventional ML algorithms to imbalanced problems, which presume that samples in different classes are balanced, often results in poor performance. To address this problem, the random undersampling technique can be used to alter the size of the majority class by randomly removing samples from the majority class. Since random undersampling eliminates samples from the original dataset, it provides a sparse training dataset. Moreover, a part of the vital information buried in the removed samples may also be lost simultaneously. Hence, the method of combing multiple random undersampling with classifier ensemble is exploited to balance the sample distribution and at the same time reduce the information loss caused by undersampling. The powerful deep learning approaches have been applied to predict ligand binding sites recently. However, the existing deep learning methods are based on conventional convolutional/recurrent network architectures. Next-generation deep learning architectures based on the attention that has achieved success in protein structure prediction and interpretation [62, 160] shall be developed to further improve the accuracy of LBS prediction.
Protein–ligand binding affinity prediction is still an open challenge in computational drug discovery since it is a highly selective process. It depends on the shape, size, constitutional makeup and physicochemical properties of both drug and its target. Hence, feature selection must be performed with extreme caution, as training a machine is heavily reliant on features. Upon study, it has been found that RF can effectively leverage more structural features and more training samples, resulting in better prediction performance than multiple linear regression. Moreover, RF-based scoring functions are supposed to capture the nonlinear nature of the data more comprehensively than multivariate linear regression (MLR)-based scoring functions. Moreover, among a variety of novel scoring functions using various ML methods in combination with physicochemical features of protein–ligand complexes, ensemble prediction methods RF and Boosted Regression Trees are found to be effective in predicting binding affinities. Regardless of the merits of classical ML algorithms, most of them heavily rely on biological feature engineering to extract explicit fingerprints. Since it is focused on expert information, it is supposed to be biased. Deep learning models, on the other hand, which fall into the descriptor-based category that can automatically extract features from raw data, tend to reduce the bias. It is expected that deep learning methods will play an increasingly significant role in this area.
For achieving a good binding pose, it has been found that picking the right receptor template and reducing the binding pocket size (and hence the size of the search space) as much as possible are critical. Furthermore, research has shown that the flexibility of protein side chains within the binding pocket has no effect in improving the quality of docking poses. Since ML can express nonlinear dependencies between chemical features, it has become an increasingly popular approach for scoring docking poses. RFs, SVMs and neural networks are the algorithms that have been used to solve scoring problems in a number of situations, and they are said to provide more flexibility and expressiveness than traditional empirical scoring methods because they learn both parameters and model structure from data. Furthermore, we see a promising prospect of deep learning and reinforcement learning in the domain of binding pose prediction. It is likely more such methods will be developed in the future. Moreover, as the tasks of predicting binding sites, binding affinity and binding poses are related, advanced deep learning methods of predicting the three simultaneously via multitasking are worth exploring.
Key Points
There is a significant need for improving the prediction of protein–ligand binding site, binding affinity and binding pose to aid drug discovery, drug design and protein function study.
Artificial intelligence, particularly data-driven machine learning, can significantly advance the prediction of protein–ligand interaction.
The growing amount of valuable structural and functional data of protein–ligand complexes makes it possible to train highly sophisticated deep learning architectures to predict protein–ligand interactions.
There is a promising potential of integrating the three traditionally separated tasks of predicting binding site, binding affinity and binding pose together in one comprehensive deep learning system via multitask learning.
Ashwin Dhakal is a Computer Science graduate student at the University of Missouri-Columbia. His research is focused on analysis and design of machine learning models for predicting protein–ligand interactions.
Cole McKay is a Biochemistry graduate student with the University of Missouri-Columbia. His research is focused on structural and biological function of uncharacterized protein domains.
John J. Tanner is a professor in the Departments of Biochemistry and Chemistry at the University of Missouri-Columbia. His research interest includes structural biology, X-ray crystallography and enzymes.
Jianlin Cheng is a professor at the Department of Electrical Engineering and Computer Science, University of Missouri-Columbia. His research is focused on bioinformatics and machine learning.
Contributor Information
Ashwin Dhakal, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, MO, 65211, USA.
Cole McKay, Department of Biochemistry, University of Missouri, Columbia, MO, 65211, USA.
John J Tanner, Department of Biochemistry, University of Missouri, Columbia, MO, 65211, USA; Department of Chemistry, University of Missouri, Columbia, MO, 65211, USA.
Jianlin Cheng, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, MO, 65211, USA.
Funding
This work was supported by the Department of Energy (DE-AR0001213, DE-SC0020400 and DE-SC0021303); National Science Foundation (DBI1759934 and IIS1763246); and National Institute of Health (R01GM093123).
References
- 1. Chen K, Mizianty MJ, Kurgan L. ATPsite: sequence-based prediction of ATP- binding residues. IEEE Int Conf Bioinforma Biomed 2010;2011(9):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ma B, Shatsky M, Wolfson HJ, et al. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci 2009;11:184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller DW, Dill KA. Ligand binding to proteins: the binding landscape model. Protein Sci 1997;6:2166–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Whitesides GM, Krishnamurthy VM. Designing ligands to bind proteins. Q Rev Biophys 2005;38:385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weisel M, Proschak E, Schneider G. PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem Cent J 2007;1:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sotriffer C, Klebe G. Identification and mapping of small-molecule binding sites in proteins: computational tools for structure-based drug design. Farmacia 2002;57:243–51. [DOI] [PubMed] [Google Scholar]
- 7. Krivák R, Hoksza D. Improving protein-ligand binding site prediction accuracy by classification of inner pocket points using local features. J Chem 2015;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghersi D, Sanchez R. EasyMIFs and SiteHound: a toolkit for the identification of ligand-binding sites in protein structures. Bioinformatics 2009;25:3185–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov 2002;1:727–30. [DOI] [PubMed] [Google Scholar]
- 10. Bronowska AK. Thermodynamics of ligand-protein interactions: implications for molecular design. Thermodyn Interact Stud Solids Liq Gases 2011;1–49. [Google Scholar]
- 11. Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct 2007;36:21–42. [DOI] [PubMed] [Google Scholar]
- 12. Mohs RC, Greig NH. Drug discovery and development: role of basic biological research. Alzheimer’s Dement Transl Res Clin Interv 2017;3:651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dimasi JA, Feldman L, Seckler A, et al. Trends in risks associated with new drug development: success rates for investigational drugs. Clin Pharmacol Ther 2010;87:272–7. [DOI] [PubMed] [Google Scholar]
- 14. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 2016;47:20–33. [DOI] [PubMed] [Google Scholar]
- 15. Asselah T, Durantel D, Pasmant E, et al. COVID-19: discovery, diagnostics and drug development Tarik. J Hepatol 2020;74:168–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hughes JP, Rees SS, Kalindjian SB, et al. Principles of early drug discovery. Br J Pharmacol 2011;162:1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Odling-smee L, Dobson CM. Chemical space and biology. Nature 2004;432:824–8. [DOI] [PubMed] [Google Scholar]
- 18. Kanehisa M, Goto S, Hattori M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 2006;34:D354–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stockwell BR. Chemical genetics: ligand-based discovery of gene function. Nat Rev Genet 2000;1:116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kitchen DB, Decornez H, Furr JR, et al. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 2004;3:935–49. [DOI] [PubMed] [Google Scholar]
- 21. Sliwoski G, Kothiwale S, Meiler J, et al. Computational methods in drug discovery. Pharmacol Rev 2014;66:334–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tresadern G, Bemporad D, Howe T. A comparison of ligand based virtual screening methods and application to corticotropin releasing factor 1 receptor. J Mol Graph Model 2009;27:860–70. [DOI] [PubMed] [Google Scholar]
- 23. Lyne PD. Structure-based virtual screening: an overview. Drug Discov Today 2002;7:1047–55. [DOI] [PubMed] [Google Scholar]
- 24. Yamanishi Y, Araki M, Gutteridge A, et al. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008;24:232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bleakley K, Yamanishi Y. Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 2009;25:2397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao DS, Zhang LX, Tan GS, et al. Computational prediction of Drug_Target interactions using chemical, biological, and network features. Mol Inform 2014;33:669–81. [DOI] [PubMed] [Google Scholar]
- 27. Wang Y, Zeng J. Predicting drug-target interactions using restricted Boltzmann machines. Bioinformatics 2013;29:126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim H, Kim E, Lee I, et al. Artificial intelligence in drug discovery: a comprehensive review of data-driven and machine learning approaches. Biotechnol Bioprocess Eng 2020;25:895–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiménez-Luna J, Grisoni F, Weskamp N, et al. Artificial intelligence in drug discovery: recent advances and future perspectives. Expert Opin Drug Discovery 2021;16:1–11. [DOI] [PubMed] [Google Scholar]
- 30. Hessler G, Baringhaus KH. Artificial intelligence in drug design. Molecules 2018;23:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abbasi K, Razzaghi P, Poso A, et al. Deep learning in drug target interaction prediction: current and future perspectives. Curr Med Chem 2021;28:2100–2113. [DOI] [PubMed] [Google Scholar]
- 32. Shanker MS, Hu MY, Hung MS. Effect of data standardization on neural network training. Omega 1996;24:385–97. [Google Scholar]
- 33. Henrich S, Salo-Ahen OMH, Huang B, et al. Computational approaches to identifying and characterizing protein binding sites for ligand design. J Mol Recognit 2010;23:209–19. [DOI] [PubMed] [Google Scholar]
- 34. Macari G, Toti D, Polticelli F. Computational methods and tools for binding site recognition between proteins and small molecules: from classical geometrical approaches to modern machine learning strategies. J Comput Aided Mol Des 2019;33:887–903. [DOI] [PubMed] [Google Scholar]
- 35. Zhao J, Cao Y, Zhang L. Exploring the computational methods for protein-ligand binding site prediction. Comput Struct Biotechnol J 2020;18:417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cimermancica P, Weinkama P, Frasera JS, et al. CryptoSite: expanding the druggable proteome by characterization and prediction of cryptic binding sites. J Mol Biol 2016;428:709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang J, Shen C, Huang N. Predicting or pretending: artificial intelligence for protein-ligand interactions lack of sufficiently large and unbiased datasets. Front Pharmacol 2020;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ellingson SR, Davis B, Allen J. Machine learning and ligand binding predictions: a review of data, methods, and obstacles. Biochim Biophys Acta Gen Subj 2020;1864:129545. [DOI] [PubMed] [Google Scholar]
- 39. Chen X, Yan CC, Zhang X, et al. Drug-target interaction prediction: databases, web servers and computational models. Brief Bioinform 2016;17:696–712. [DOI] [PubMed] [Google Scholar]
- 40. Inhester T, Rarey M. Protein-ligand interaction databases: advanced tools to mine activity data and interactions on a structural level. Wiley Interdiscip Rev Comput Mol Sci 2014;4:562–75. [Google Scholar]
- 41. Lim S, Lu Y, Cho CY, et al. A review on compound-protein interaction prediction methods: data, format, representation and model. Comput Struct Biotechnol J 2021;19:1541–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang R, Fang X, Lu Y, et al. The PDBbind database: methodologies and updates. J Med Chem 2005;48:4111–9. [DOI] [PubMed] [Google Scholar]
- 43. Liu T, Lin Y, Wen X, et al. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res 2007;35:198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berman HM, Battistuz T, Bhat TN, et al. The protein data bank. Acta Crystallogr Sect D Biol Crystallogr 2000;28:235–42. [Google Scholar]
- 45. Dessailly BH, Lensink MF, Orengo CA, et al. LigASite—a database of biologically relevant binding sites in proteins with known apo-structures. Nucleic Acids Res 2008;36:667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang J, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res 2013;41:1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benson ML, Smith RD, Khazanov NA, et al. Binding MOAD, a high-quality protein-ligand database. Nucleic Acids Res 2008;36:674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu L, Benson ML, Smith RD, et al. Binding MOAD (mother of all databases). Proteins Struct Funct Bioinforma 2005;60:333–40. [DOI] [PubMed] [Google Scholar]
- 49. Ahmed A, Smith RD, Clark JJ, et al. Recent improvements to binding MOAD: a resource for protein-ligand binding affinities and structures. Nucleic Acids Res 2015;43:D465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng X, Gan L, Wang E, et al. Pocket-based drug design: exploring pocket space. AAPS J 2013;15:228–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pérot S, Sperandio O, Miteva MA, et al. Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today 2010;15:656–67. [DOI] [PubMed] [Google Scholar]
- 52. Xie L, Xie L, Bourne PE. Structure-based systems biology for analyzing off-target binding. Curr Opin Struct Biol 2011;21:189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Konc J, Janežič D. Binding site comparison for function prediction and pharmaceutical discovery. Curr Opin Struct Biol 2014;25:34–9. [DOI] [PubMed] [Google Scholar]
- 54. Campbell SJ, Gold ND, Jackson RM, et al. Ligand binding: functional site location, similarity and docking. Curr Opin Struct Biol 2003;13:389–95. [DOI] [PubMed] [Google Scholar]
- 55. Laskowski RA, Luscombe NM, Swindells MB, et al. Protein clefts in molecular recognition and function. Protein Sci 1996;5:2438–24352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Laskowski RA. SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J Mol Graph 1995;13:323–30. [DOI] [PubMed] [Google Scholar]
- 57. Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model 1997;15:359–63. [DOI] [PubMed] [Google Scholar]
- 58. Levitt DG, Banaszak LJ. POCKET: a computer graphies method for identifying and displaying protein cavities and their surrounding amino acids. J Mol Graph 1992;10:229–34. [DOI] [PubMed] [Google Scholar]
- 59. Hu X, Dong Q, Yang J, et al. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 2016;32:3260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin CT, Lin KL, Yang CH, et al. Protein metal binding residue prediction based on neural networks. Int J Neural Syst 2005;15:71–84. [DOI] [PubMed] [Google Scholar]
- 61. Chauhan JS, Mishra NK, Raghava GPS. Identification of ATP binding residues of a protein from its primary sequence. BMC Bioinformatics 2009;10:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Smith BJ, McKimm-Breshkin JL, McDonald M, et al. Structural studies of the resistance of influenza. Virus neuramindase to inhibitors. J Med Chem 2002;45:2207–12. [DOI] [PubMed] [Google Scholar]
- 64. Dey F, Zhang QC, Petrey D, et al. Toward a ‘structural BLAST’: using structural relationships to infer function. Protein Sci 2013;22:359–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee DT, Schachter BJ. Two algorithms for constructing a Delaunay triangulation. Int J Comput Inf Sci 1980;9:219–42. [Google Scholar]
- 66. Yang J, Roy A, Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013;29:2588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Skolnick J, Brylinski M. FINDSITE: a combined evolution/structure-based approach to protein function prediction. Brief Bioinform 2009;10:378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Huang B, Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct Biol 2006;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rost B. Twilight zone of protein sequence alignments. Protein Eng 1999;12:85–94. [DOI] [PubMed] [Google Scholar]
- 70. Wu Q, Peng Z, Zhang Y, et al. COACH-D: improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res 2018;46:W438–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brylinski M, Skolnick J. A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci USA 2008;105:129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yu J, Zhou Y, Tanaka I, et al. Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2009;26:46–52. [DOI] [PubMed] [Google Scholar]
- 73. Guo T, Shi Y, Sun Z. A novel statistical ligand-binding site predictor: application to ATP-binding sites. Protein Eng Des Sel 2005;18:65–70. [DOI] [PubMed] [Google Scholar]
- 74. Bordner AJ. Predicting small ligand binding sites in proteins using backbone structure. Bioinformatics 2008;24:2865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kauffman C, Karypis G. LIBRUS: combined machine learning and homology information for sequence-based ligand-binding residue prediction. Bioinformatics 2009;25:3099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Qiu Z, Wang X. Improved prediction of protein ligand-binding sites using random forests. Protein Pept Lett 2011;18:1212–8. [DOI] [PubMed] [Google Scholar]
- 77. Wong GY, Leung FHF, Ling SH. Predicting protein-ligand binding site with differential evolution and support vector machine. Proc 2012 Int Jt Conf Neural Networks 2012;10–5. [Google Scholar]
- 78. Volkamer A, Kuhn D, Rippmann F, et al. DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012;28:2074–5. [DOI] [PubMed] [Google Scholar]
- 79. Wong GY, Leung FHF, Ling SH. Predicting protein-ligand binding site using support vector machine with protein properties. IEEE/ACM Trans Comput Biol Bioinforma 2013;10:1517–29. [DOI] [PubMed] [Google Scholar]
- 80. Yu DJ, Hu J, Yang J, et al. Designing template-free predictor for targeting protein-ligand binding sites with classifier ensemble and spatial clustering. IEEE/ACM Trans Comput Biol Bioinforma 2013;10:994–1008. [DOI] [PubMed] [Google Scholar]
- 81. Wang K, Gao J, Shen S, et al. An accurate method for prediction of protein-ligand binding site on protein surface using SVM and statistical depth function. Biomed Res Int 2013;2013:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen P, Huang JZ, Gao X. LigandRFs: random forest ensemble to identify ligand-binding residues from sequence information alone. BMC Bioinformatics 2014;15:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Suresh MX, Gromiha MM, Suwa M. Development of a machine learning method to predict membrane protein-ligand binding residues using basic sequence information. Adv Bioinformatics 2015;2015: Article ID 843030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yu DJ, Hu J, Li QM, et al. Constructing query-driven dynamic machine learning model with application to protein-ligand binding sites prediction. IEEE Trans Nanobiosci 2015;14:45–58. [DOI] [PubMed] [Google Scholar]
- 85. Komiyama Y, Banno M, Ueki K, et al. Automatic generation of bioinformatics tools for predicting protein-ligand binding sites. Bioinformatics 2016;32:901–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chen P, Hu S, Zhang J, et al. A sequence-based dynamic ensemble learning system for protein ligand-binding site prediction. IEEE/ACM Trans Comput Biol Bioinforma 2016;13:901–12. [DOI] [PubMed] [Google Scholar]
- 87. Chen ASY, Westwood NJ, Brear P, et al. A random Forest model for predicting allosteric and functional sites on proteins. Mol Inform 2016;35:125–35. [DOI] [PubMed] [Google Scholar]
- 88. Hu X, Wang K, Dong Q. Protein ligand-specific binding residue predictions by an ensemble classifier. BMC Bioinformatics 2016;17:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Krivak R, Hoksza D, Skoda P. Improving quality of ligand-binding site prediction with Bayesian optimization. 2017 IEEE Int Conf Bioinforma Biomed BIBM 2017;2017:2278–9. [Google Scholar]
- 90. Krivák R, Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J Chem 2018;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jendele L, Krivak R, Skoda P, et al. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res 2019;47:W345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mahdiyah U, Imah EM, Irawan MI. Integrating data selection and extreme learning machine to predict protein-ligand binding site. Contemp Eng Sci 2016;9:791–7. [Google Scholar]
- 93. Bin HG, Zhu QY, Siew CK. Extreme learning machine: theory and applications. Neurocomputing 2006;70:489–501. [Google Scholar]
- 94. Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999;292:195–202. [DOI] [PubMed] [Google Scholar]
- 95. Cui Y, Dong Q, Hong D, et al. Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinformatics 2019;20:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Xia CQ, Pan X, Shen H-B. Protein-ligand binding residue prediction enhancement through hybrid deep heterogeneous learning of sequence and structure data. Bioinformatics 2020;36:3018–27. [DOI] [PubMed] [Google Scholar]
- 97. Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. Improving detection of protein-ligand binding sites with 3D segmentation. Sci Rep 2020;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Desaphy J, Bret G, Rognan D, et al. Sc-PDB: a 3D-database of ligandable binding sites-10 years on. Nucleic Acids Res 2015;43:D399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mylonas SK, Axenopoulos A, Daras P. DeepSurf: a surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics 2021;37:1681–90. [DOI] [PubMed] [Google Scholar]
- 100. Kandel J, Tayara H, Chong KT. PUResNet: prediction of protein-ligand binding sites using deep residual neural network. J Chem 2021;13:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wu S, Zhang Y. ANGLOR: a composite machine-learning algorithm for protein backbone torsion angle prediction. PLoS One 2008;3:e3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ronneberger O, Fischer P, Brox T. U-net: convolutional networks for biomedical image segmentation. Int Conf Med image Comput Comput Interv 2015;234–41. [Google Scholar]
- 103. Perozzo R, Folkers G, Scapozza L. Thermodynamics of protein-ligand interactions: history, presence, and future aspects. J Recept Signal Transduct 2004;24:1–52. [DOI] [PubMed] [Google Scholar]
- 104. Deng W, Breneman C, Embrechts MJ. Predicting protein - ligand binding affinities using novel geometrical descriptors and machine-learning methods. J Chem Inf Comput Sci 2004;44:699–703. [DOI] [PubMed] [Google Scholar]
- 105. Ashtawy HM, Mahapatra NR. A comparative assessment of conventional and machine-learning-based scoring functions in predicting binding affinities of protein-ligand complexes. 2011 IEEE Int. Conf. Bioinforma. Biomed. BIBM 2011 2011; 627–30
- 106. Ouyang X, Handoko SD, Kwoh CK. CScore:a simple yet effective scoring function for protein ligand binding affinity prediction using modified cmac learning architecture. J Bioinform Comput Biol 2011;9:1–14. [DOI] [PubMed] [Google Scholar]
- 107. Zilian D, Sotriffer CA. SFCscoreRF: a random forest-based scoring function for improved affinity prediction of protein-ligand complexes. J Chem Inf Model 2013;53:1923–33. [DOI] [PubMed] [Google Scholar]
- 108. Liu Q, Kwoh CK, Li J. Binding affinity prediction for protein-ligand complexes based on β contacts and B factor. J Chem Inf Model 2013;53:3076–85. [DOI] [PubMed] [Google Scholar]
- 109. Li H, Leung KS, Wong MH, et al. Substituting random forest for multiple linear regression improves binding affinity prediction of scoring functions: Cyscore as a case study. BMC Bioinformatics 2014;15:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang Y, Guo Y, Kuang Q, et al. A comparative study of family-specific protein-ligand complex affinity prediction based on random forest approach. J Comput Aided Mol Des 2015;29:349–60. [DOI] [PubMed] [Google Scholar]
- 111. Cao Y, Li L. Improved protein-ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics 2014;30:1674–80. [DOI] [PubMed] [Google Scholar]
- 112. Shar PA, Tao W, Gao S, et al. Pred-binding: large-scale protein–ligand binding affinity prediction. J Enzyme Inhib Med Chem 2016;31:1443–50. [DOI] [PubMed] [Google Scholar]
- 113. Ávila MB, Xavier MM, Pintro VO, et al. Supervised machine learning techniques to predict binding affinity. A study for cyclin-dependent kinase 2. Biochem Biophys Res Commun 2017;494:305–10. [DOI] [PubMed] [Google Scholar]
- 114. Bitencourt-Ferreira G, Azevedo WF. Development of a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes. Biophys Chem 2018;240:63–9. [DOI] [PubMed] [Google Scholar]
- 115. Kundu I, Paul G, Banerjee R. A machine learning approach towards the prediction of protein-ligand binding affinity based on fundamental molecular properties. RSC Adv 2018;8:12127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Boyles F, Deane CM, Morris GM. Learning from the ligand: using ligand-based features to improve binding affinity prediction. Bioinformatics 2020;36:758–64. [DOI] [PubMed] [Google Scholar]
- 117. Holderbach S, Adam L, Jayaram B, et al. RASPD+: fast protein-ligand binding free energy prediction using simplified physicochemical features. Front Mol Biosci 2020;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Amangeldiuly N, Karlov D, Fedorov MV. Baseline model for predicting protein-ligand unbinding kinetics through machine learning. J Chem Inf Model 2020;60:5946–56. [DOI] [PubMed] [Google Scholar]
- 119. Wee J, Xia K. Ollivier persistent Ricci curvature-based machine learning for the protein-ligand binding affinity prediction. J Chem Inf Model 2021;61:1617–26. [DOI] [PubMed] [Google Scholar]
- 120. Liu Q, Kwoh CK, Hoi SCH. Beta atomic contacts: identifying critical specific contacts in protein binding interfaces. PLoS One 2013;8:e59737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res 2016;44:D1202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Allouche A. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 2011;32:1466–74. [DOI] [PubMed] [Google Scholar]
- 123. Keerthi SS, Shevade SK, Bhattacharyya C, et al. Improvements to Platt’s SMO algorithm for SVM classifier design. Neural Comput 2001;13:637–49. [Google Scholar]
- 124. Cleary JG, Trigg LE. K*: an instance-based learner using an entropic distance measure. Mach Learn Proc 1995;1995:108–114. [Google Scholar]
- 125. Ashtawy HM, Mahapatra NR. BgN-score and BsN-score: bagging and boosting based ensemble neural networks scoring functions for accurate binding affinity prediction of protein-ligand complexes. BMC Bioinformatics 2015;16:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gomes J, Ramsundar B, Feinberg EN, et al. Atomic convolutional networks for predicting protein-ligand binding affinity arXiv Prepr. arXiv. 2017;1–17.
- 127. Jiménez J, Škalič M, Martínez-Rosell G, et al. KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J Chem Inf Model 2018;58:287–96. [DOI] [PubMed] [Google Scholar]
- 128. Öztürk H, Özgür A, Ozkirimli E. DeepDTA: deep drug-target binding affinity prediction. Bioinformatics 2018;34:i821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018;34:3666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zheng L, Fan J, Mu Y. OnionNet: a multiple-layer intermolecular-contact-based convolutional neural network for protein-ligand binding affinity prediction. ACS Omega 2019;4:15956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Zhu F, Zhang X, Allen JE, et al. Binding affinity prediction by pairwise function based on neural network. J Chem Inf Model 2020;60:2766–72. [DOI] [PubMed] [Google Scholar]
- 132. Rezaei MA, Li Y, Wu DO, et al. Deep learning in drug design: protein-ligand binding affinity prediction. IEEE/ACM Trans Comput Biol Bioinform 2020;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jones D, Kim H, Zhang X, et al. Improved protein-ligand binding affinity prediction with structure-based deep fusion inference. J Chem Inf Model 2021;61:1583–92. [DOI] [PubMed] [Google Scholar]
- 134. Kwon Y, Shin WH, Ko J, et al. AK-score: accurate protein-ligand binding affinity prediction using an ensemble of 3D-convolutional neural networks. Int J Mol Sci 2020;21:8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Karlov DS, Sosnin S, Fedorov MV, et al. GraphDelta: MPNN scoring function for the affinity prediction of protein-ligand complexes. ACS Omega 2020;5:5150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wang K, Zhou R, Li Y, et al. DeepDTAF: a deep learning method to predict protein–ligand binding affinity. Brief Bioinform 2021;22:1–15. [DOI] [PubMed] [Google Scholar]
- 137. Azzopardi J, Ebejer JP. LigityScore: convolutional neural network for binding-affinity predictions. Bioinformatics 2021;3:38–49. [Google Scholar]
- 138. Seo S, Choi J, Park S, et al. Binding affinity prediction for protein-ligand complex using deep attention mechanism based on intermolecular interactions. bioRxiv 2021; 2021.03.18.436020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ahmed A, Mam B, Sowdhamini R. DEELIG: A deep learning approach to predict protein-ligand binding affinity. Bioinform Biol Insights 2021;15:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wang Y, Wu S, Duan Y, et al. ResAtom system: protein and ligand affinity prediction model based on deep learning. arXiv 2021. [Google Scholar]
- 141. Odell LR, Abdel-Hamid MK, Hill TA, et al. Pyrimidine-based inhibitors of dynamin I GTPase activity: competitive inhibition at the Pleckstrin homology domain. J Med Chem 2017;60:349–61. [DOI] [PubMed] [Google Scholar]
- 142. Cosconati S, Forli S, Perryman AL, et al. Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discovery 2010;5:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;32:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Friesner RA, Banks JL, Halgren TA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1739–49. [DOI] [PubMed] [Google Scholar]
- 145. Verdonk ML, Cole JC, Hartshorn MJ, et al. Improved protein-ligand docking using GOLD. Proteins Struct Funct Genet 2003;52:609–23. [DOI] [PubMed] [Google Scholar]
- 146. Neves MAC, Totrov M, Abagyan R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J Comput Aided Mol Des 2012;26:675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophys Rev 2017;9:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. McGann M. FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des 2012;26:897–906. [DOI] [PubMed] [Google Scholar]
- 149. Huang N, Kalyanaraman C, Irwin JJ, et al. Physics-based scoring of protein–ligand complexes: enrichment of known inhibitors in large-scale virtual screening. J Chem Inf Model 2006;46:243–53. [DOI] [PubMed] [Google Scholar]
- 150. Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des 2002;16:11–26. [DOI] [PubMed] [Google Scholar]
- 151. Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein–ligand interactions. J Mol Biol 2000;295:337–56. [DOI] [PubMed] [Google Scholar]
- 152. Ashtawy HM, Mahapatra NR. Machine-learning scoring functions for identifying native poses of ligands docked to known and novel proteins. BMC Bioinformatics 2015;16:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Grudinin S, Kadukova M, Eisenbarth A, et al. Predicting binding poses and affinities for protein–ligand complexes in the 2015 D3R grand challenge using a physical model with a statistical parameter estimation. J Comput Aided Mol Des 2016;30:791–804. [DOI] [PubMed] [Google Scholar]
- 154. Ragoza M, Turner L, Koes DR. Ligand pose optimization with atomic grid-based convolutional neural networks arXiv Prepr. arXiv. 2017; 1710.07400.
- 155. Ragoza M, Hochuli J, Idrobo E, et al. Protein-ligand scoring with convolutional neural networks. J Chem Inf Model 2017;57:942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Nguyen DD, Cang Z, Wu K, et al. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R grand challenges. J Comput Aided Mol Des 2019;33:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Jose J, Gupta K, Alam U, et al. Reinforcement learning based approach for ligand pose prediction. bioRxiv 2021. [Google Scholar]
- 158. Allouche A. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2012;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Jia Y, Shelhamer E, Donahue J, et al. Caffe: convolutional architecture for fast feature embedding. Proc ACM Conf Multimed 2014;2014:675–8. [Google Scholar]
- 160. Chen C, Wu T, Guo Z, et al. Combination of deep neural network with attention mechanism enhances the explainability of protein contact prediction. Proteins Struct Funct Bioinforma 2021;89:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Reytor Gonzalez ML, Alonso del Rivero Antigua M. Reviewing the experimental and mathematical factors involved in tight binding inhibitors Ki values determination: The bi-functional protease inhibitor SmCI as a test model. Biochimie 2021;181:86–95. [DOI] [PubMed] [Google Scholar]