

Abstract
Pantothenate kinase (PANK) is the critical regulator of intracellular levels of coenzyme A and has emerged as an attractive target for treating neurological and metabolic disorders. This report describes the optimization, synthesis, and full structure–activity relationships of a new chemical series of pantothenate competitive PANK inhibitors. Potent drug-like molecules were obtained by optimizing a high throughput screening hit, using lipophilic ligand efficiency (LipE) derived from human PANK3 IC50 values to guide ligand development. X-ray crystal structures of PANK3 with index inhibitors from the optimization were determined to rationalize the emerging structure activity relationships. The analysis revealed a key bidentate hydrogen bonding interaction between pyridazine and R306′ as a major contributor to the LipE gain observed in the optimization. A tractable series of PANK3 modulators with nanomolar potency, excellent LipE values, desirable physicochemical properties, and a well-defined structural binding mode was produced from this study.
Keywords: Pantothenate Kinase, Lipophilic ligand efficiency, Pyridazine, Hit-to-lead
1. Introduction
Coenzyme A is an essential metabolic cofactor required for mitochondrial function, fatty acid, and folate metabolism.1 Pantothenate Kinase (PANK) catalyzes the first and rate-limiting step in Coenzyme A biosynthesis, regulating intracellular concentrations of CoA.1–2 Human cells express four closely related isoforms of PANKs (PANK1α, PANK1β, PANK2, and PANK3) encoded by three genes in tissue-dependent ratios.3–5 PANK2 is the major isoform in neural cells, and disabling genetic lesions in the PANK2 gene result in a devastating neurological disorder – pantothenate kinase associated neurodegeneration (PKAN).3,6,7 Recently, we have demonstrated that the loss of PANK2 function can be compensated for by pharmacological activation of PANK, as a novel strategy to treat PKAN.8 We have also shown that pharmacological activation of PANK can be used as novel method to treat coenzyme A sequestration and improve mitochondrial function in mice with propionic acidemia.9 Inhibition of PANK may also be therapeutically desirable in some instances, such as for the treatment of diabetes where Pank1 gene deletion produces mild hypoglycemia in the fasted state in mice,10 or as an antimicrobial drug discovery target due to kingdom-specific differences in PanK function and structure.11–12
Given the documented association of PANK with diseases like PKAN and other metabolic diseases, we aimed to develop chemical probes that target PANK and are capable of modulating CoA levels to assess the potential of such compounds as therapeutics to treat these disorders. We performed a high throughput screen towards this goal.13 However, the most potent hits identified suffered from poor solubility and a flat structure–activity relationship in preliminary follow-up studies. In response, we adjusted our hit prioritization strategy by applying both molecular weight (<350) and lipophilic ligand efficiency (LipE > 2) restrictions to our original hit list, to identify initial leads with improved biophysical properties (Fig 1A).14 This reanalysis identified piperazine urea compound 1 (Fig 1B), with promising potency (IC50 = 0.51 μM) against PANK3 and a reasonable ligand efficiency (LipE = 2.8), but limited solubility (1.4 μM). In a recent communication, we described the use of the preclinical lead, 57, as a therapeutic to treat pantothenate kinase associated neurodegeneration and briefly described the rationale behind its development.8 Here, we report the synthesis of the 58 new molecules and the structure–activity relationships (SAR) responsible for the advancement of screening hit 1 to lead 57. Optimization was guided by PANK3 IC50s and crystal structures of the PANK3•ATP•pantazine complexes, but advancement of individual molecules was primarily driven by the LipE score to ensure the development of biologically useful PANK3 probes.
Figure 1.
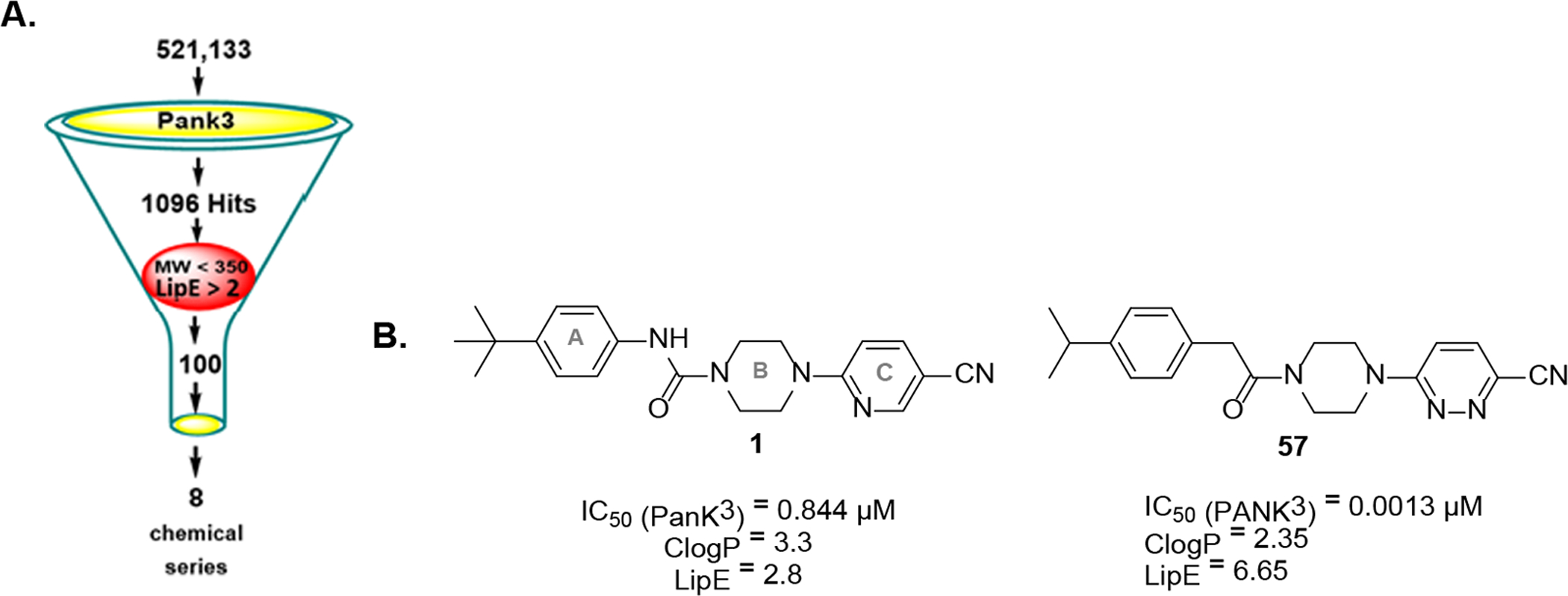
A. Process of hit-triage using MW (<350) and LipE (>2) leading to 8 chemotypes. B. Chemical structure of screening hit (1) with piperazine ureas scaffold with ring labeling and optimized lead 57.
2. Results and discussion
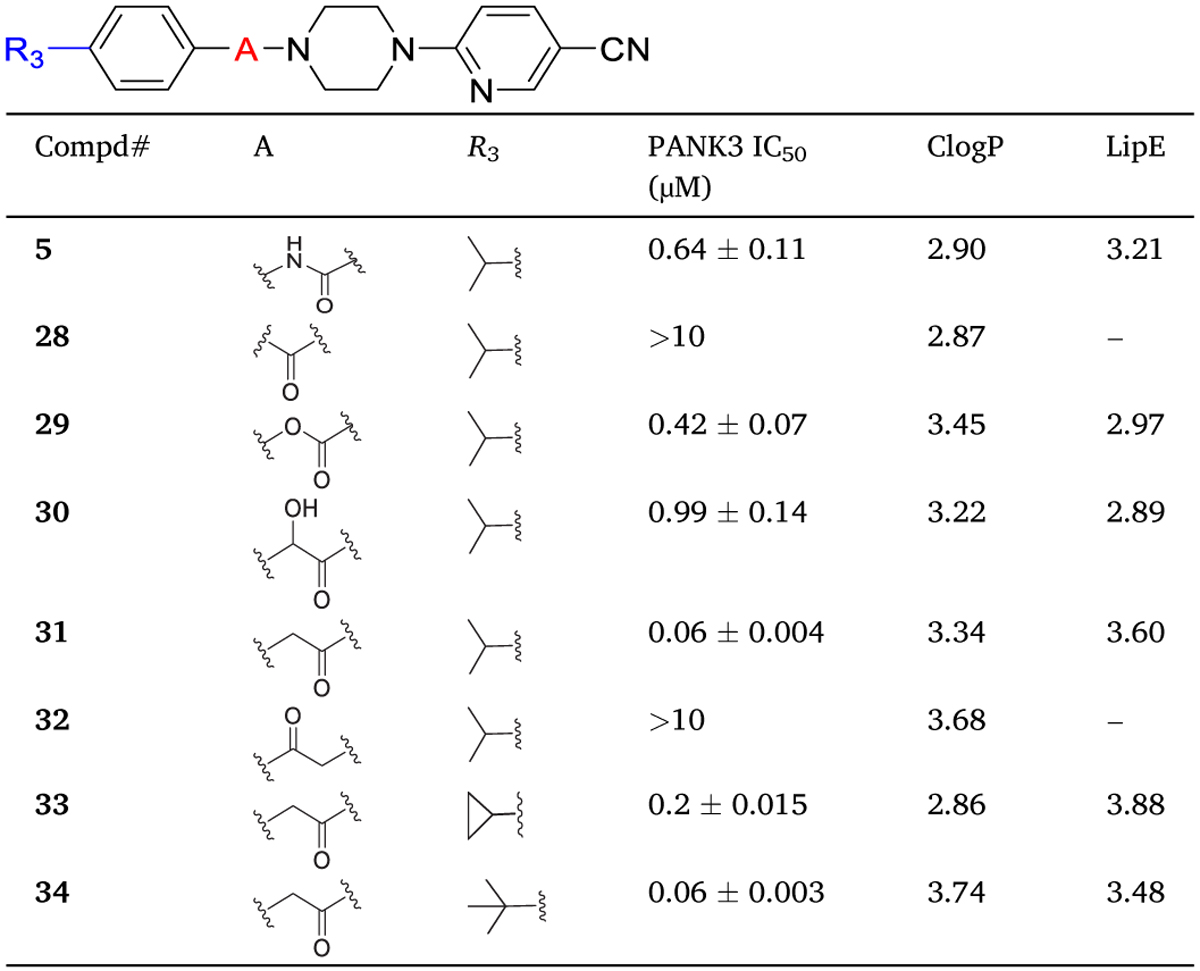
To optimize the potency of 1, systematic structural modifications around the t-butyl phenyl aniline (ring A) (analogs 1–27), urea linker (analogs 28–34), nicotinonitrile (ring C) (analogs 35–61) were performed (Fig. 1B). The optimization was guided by lipophilic ligand efficiency (LipE = pIC50 − CLogP) to balance potency and lipophilicity contributions of the modifications, thus allowing for clear comparison between molecules and prioritizing new enthalpic contributions.14–15 In this study, optimization of piperazine (ring B) is not explored because it serves as a spacer element as described below. All the synthesized analogs of compound 1 were ranked for PANK3 affinity using a pantothenate phosphorylation assay with radiometric detection, as previously described.14 These compounds are orthosteric inhibitors in the PANK3 assay but act as allosteric activators that elevate CoA levels in cells in the presence of pantothenate, in this study the SAR is followed only in the context of orthosteric inhibition.8
2.1. Synthesis of target molecules
The general method for the synthesis of analogs of the A-ring (1–24) is depicted in Scheme 1. First, 1-Boc piperazine was coupled with 6-chloronicotinonitrile (63) utilizing a microwave assisted nucleophilic aromatic substitution. The resulting product was then treated with tri-fluoroacetic acid in dichloromethane to remove the boc-protecting group. Intermediate 64 was then reacted with commercially available isocyanates (or synthesized from corresponding amines) producing the required piperazine ureas derivatives (1–27) (Table 1 and Fig. 2).
Scheme 1.
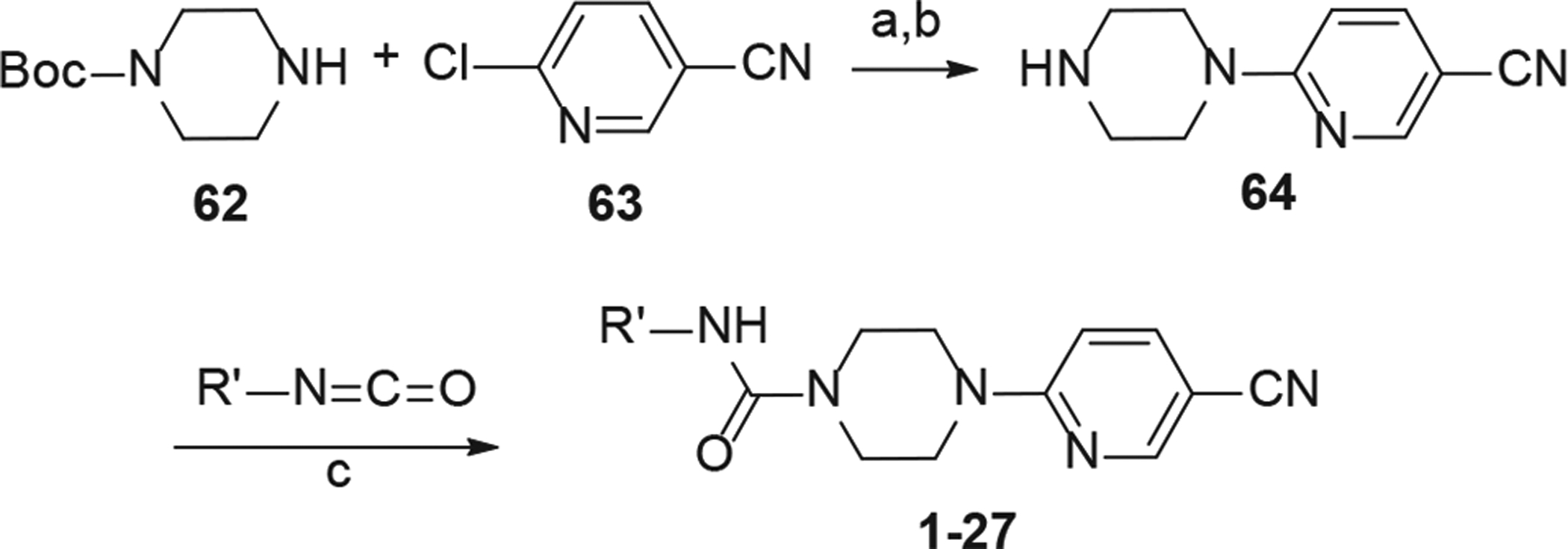
Synthesis of analogs 1–27. Reaction Conditions: (a) Et3N, CH3CN, 160 °C, 30 min, microwave; (b) TFA-CH2Cl2 (1:1), rt, 1 h; (c) R’NCO (1.1 eq), Et2O or CH2Cl2, Et3N, rt, 3 h.
Table 1.
Substitutions on the aniline ring.
|
||||
|---|---|---|---|---|
| Compd | R1 | PANK3 IC50 (μm) | ClogPa | LipEb |
| 1 | 4-t-Butyl | 0.51 ± 0.05 | 3.30 | 2.80 |
| 2 | H | >10 | 1.48 | – |
| 3 | 4-Ethyl | >10 | 2.50 | – |
| 4 | 4-n-Pentyl | >10 | 4.10 | – |
| 5 | 4-i-Propyl | 0.64 ± 0.11 | 2.90 | 3.37 |
| 6 | 2-i-Pr, 6-Me | >10 | 2.03 | – |
| 7 | 4-i-Butyl | 0.49 ± 0.09 | 3.43 | 2.80 |
| 8 | 4-n-Butyl | >10 | 3.56 | – |
| 9 | 2-Me, 4-n-Butyl | 1.2 ± 0.22 | 3.50 | 2.59 |
| 10 | 4-OCH3 | >10 | 1.58 | – |
| 11 | 4-OCF3 | >10 | 2.69 | – |
| 12 | 4-(CH3)2N | >10 | 1.64 | – |
| 13 | 4-COCH3 | >10 | 1.47 | – |
| 14 | 4-Br | >10 | 2.64 | – |
| 15 | 4-Cl | >10 | 2.49 | – |
| 16 | 3-Cl, 4-Cl | >10 | 3.19 | – |
| 17 | 4-CN | >10 | 1.61 | – |
| 18 | 4-CF3 | >10 | 2.89 | – |
LogP calculated by Chemdraw Professional 17.1
IC50 values are reported as the mean. bLipE = pIC50 − ClogP.
Figure 2.
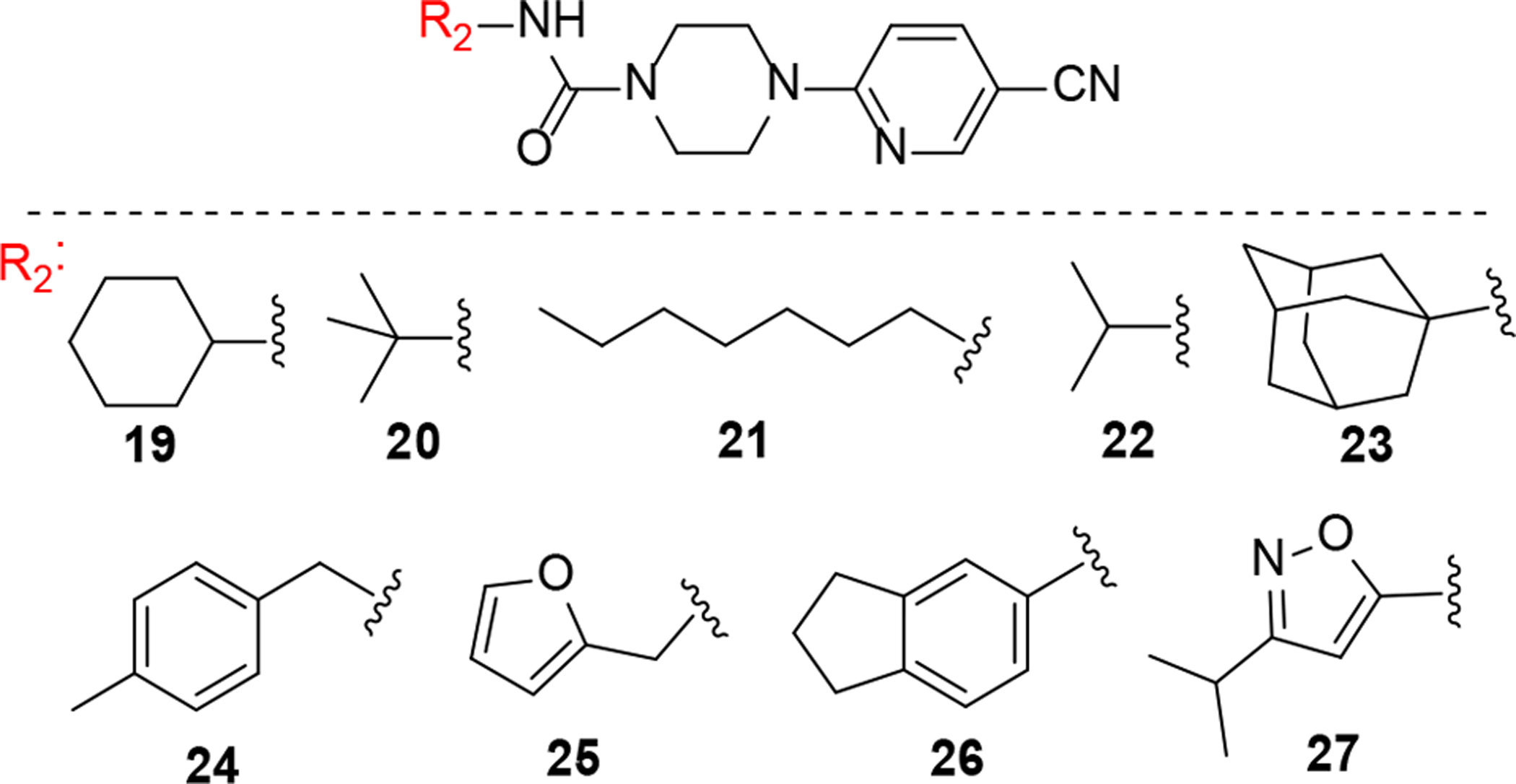
Replacement of aniline ring with aliphatic and aromatic substituents. All these compounds (19–26) showed no activity (IC50 > 10 μM).
The analogs (28–32) with different linkers are synthesized as shown in schemes 2, 3 and 4. HATU assisted coupling of 6-(piperazin-1-yl) nicotinonitrile (64) with acids (65–69) in the presence of diisopropylethylamine (DIPEA) as the base gave analogs 28, 30, 31, 33, and 34 (Scheme 2). The carbamate derivatives 29 and 61 were prepared from tert-butyl piperazine-1-carboxylate (Scheme 3). The treatment of 62 with 1,1′-carbonyldiimidazole, followed by reaction with 4-isopropylphenol led to carbamate 71. The boc-deprotection and reaction with 6-chloronicotinonitrile or 6-chloropyridazine-3-carbonitrile afforded 29 and 61 respectively in good yields.
Scheme 2.
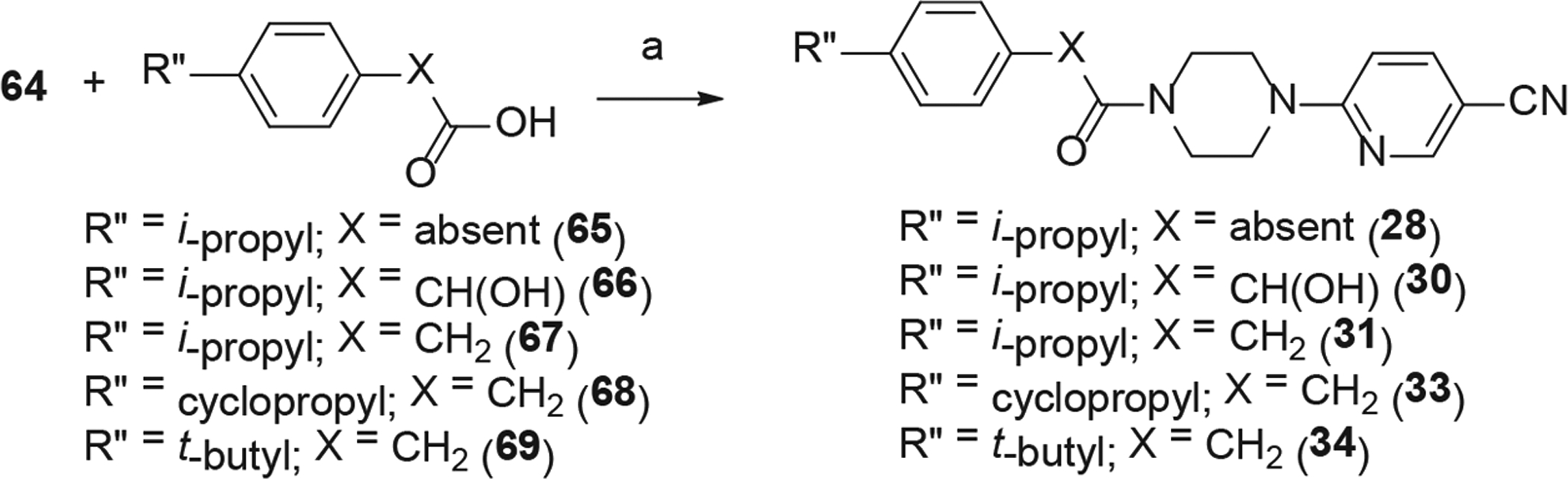
Synthesis of analogs 28, 30, 31, 33 and 34. Reaction Conditions: (a) 6-(piperazin-1-yl)nicotinonitrile (64) (1 eq), acid (1.1 eq), HATU (1.1 eq), DIPEA (5 eq), CH2Cl2, rt, overnight.
Scheme 3.
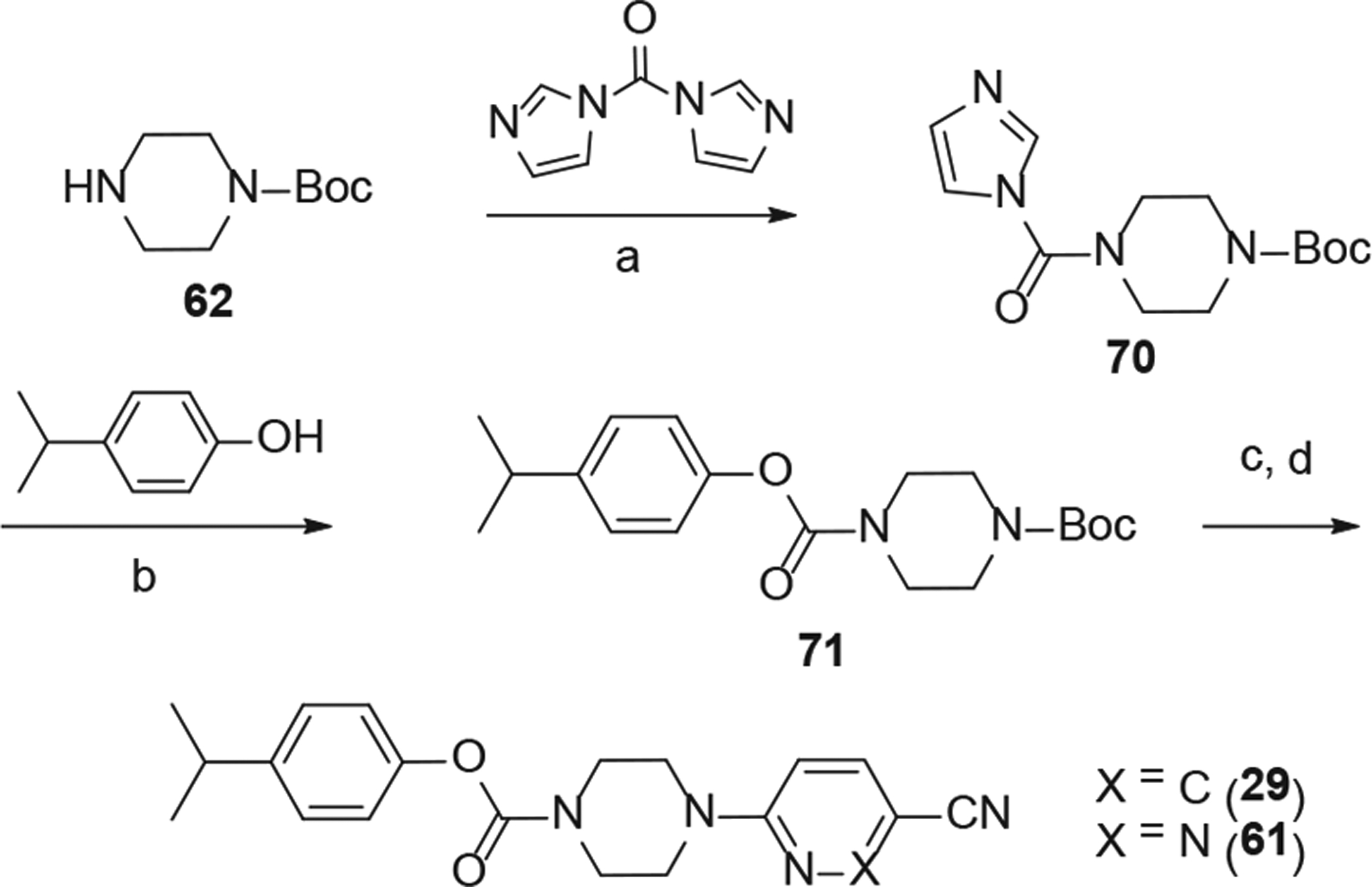
Synthesis of analogs 29 and 61. Reaction Conditions: (a) CDI (2 eq), CH2Cl2, rt, overnight; (b) isopropylphenol (2 eq), Et3N (5 eq), Cs2CO3 (2 eq), acetonitrile, 70 °C, 3–4 h; (c) TFA-CH2Cl2 (1:1), rt, 2 h; (d) 6-chloronicotinonitrile or 6-chloropyridazine-3-carbonitrile (1.1 eq), Et3N (2 eq), acetonitrile, 160 °C, 30 min.
Scheme 4.
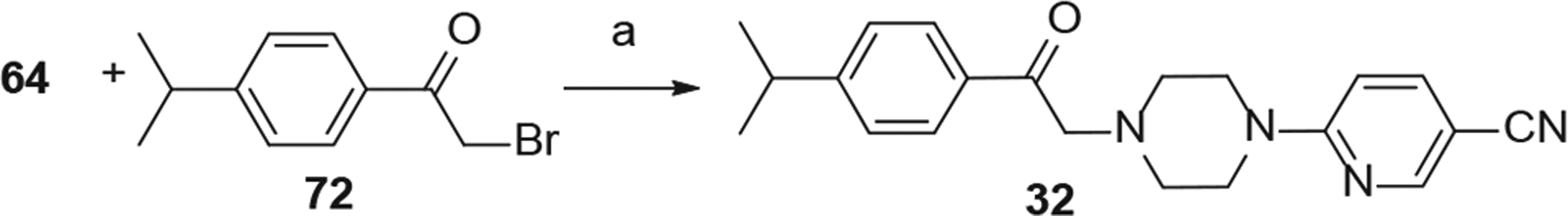
Synthesis of analogs 32. Reaction Conditions: (a) 6-(piperazin-1-yl) nicotinonitrile (1 eq), 2-bromo-1-(4-isopropylphenyl)ethanone (1 eq), DIPEA (1 eq), CH2Cl2, rt, 3 h.
6-(4-(2-(4-isopropylphenyl)-2-oxoethyl)piperazin-1-yl)nico-tinonitrile (32) was synthesized by reaction of 64 with 2-bromo-1-(4-isopropylphenyl)ethan-1-one (72) in the presence of diisopropylethylamine in CH2Cl2 (Scheme 4).
The synthesis of nicotinonitrile (ring C) analogs (49–59) is depicted in Schemes 5 and 6. To obtain urea derivatives, we first reacted 1-isocyanato-4-isopropylbenzene (73) and 62 which provided a urea intermediated 74. Next, a reaction between 74 and heteroaryl halides resulted in required products 49, 50, 52, 54, 56 and 58 (Scheme 5). The intermediate 76 was prepared by coupling of 62 with 2-(4-isopropylphenyl)acetic acid (75). Finally, 51, 53, 55, 57, and 59 were synthesized by reaction of corresponding heteroaryl halides with intermediate 76. 2-oxoacetyl analog 60 was synthesized according to the route of 57 in Scheme 6, using 2-(4-isopropylphenyl)-2-oxoacetic acid in place of 75.
Scheme 5.
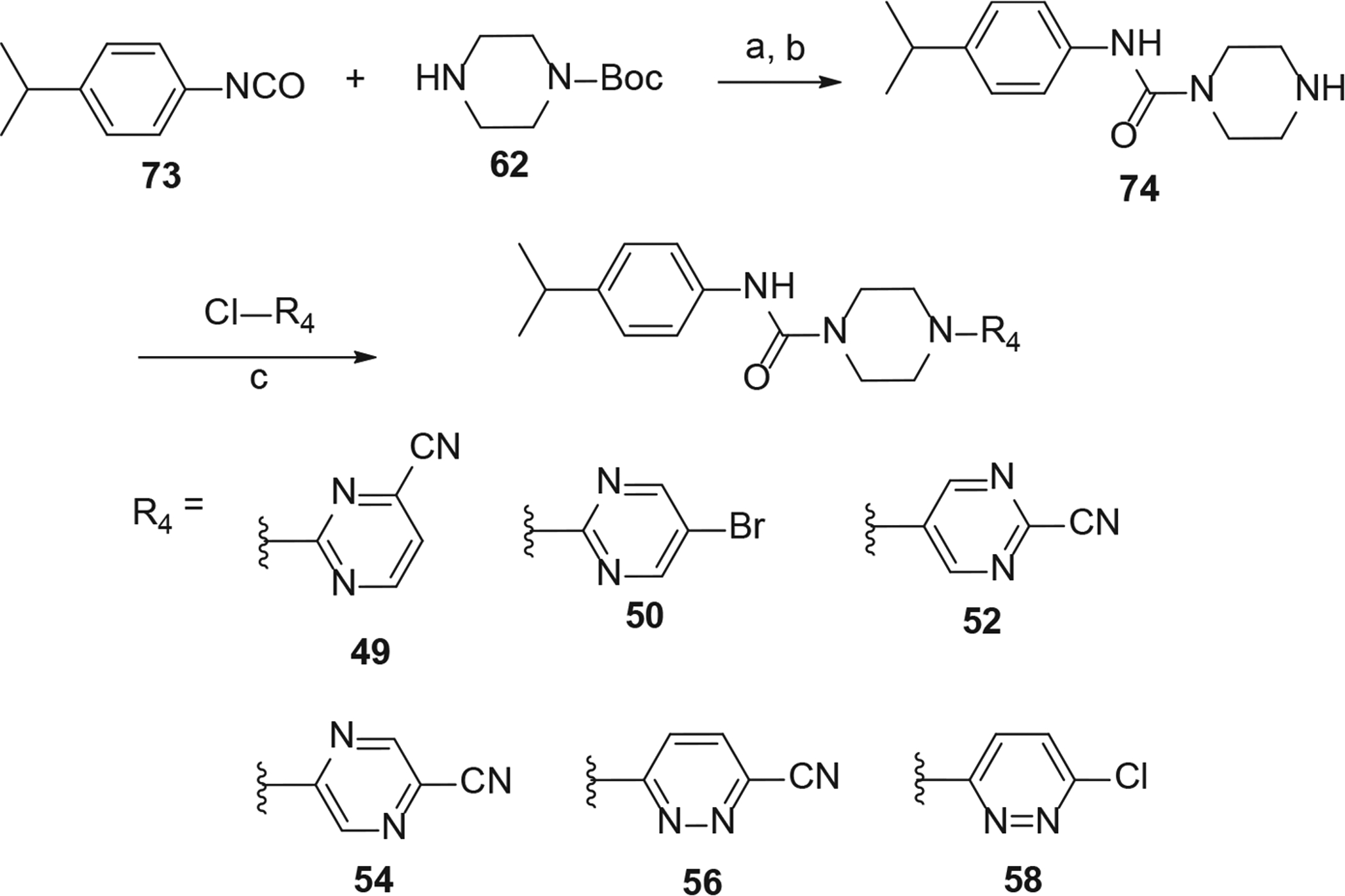
Synthesis of analogs 49, 50, 52, 54, 56 and 58. Reaction Conditions: (a) 1-isocyanato-4-isopropylbenzene (1.1 eq), Et2O, rt, 3 h; (b) TFA-CH2Cl2 (1:1), rt, 1 h; (c) 6-chloronicotinonitrile (1.1 eq), Et3N (2 eq), acetonitrile, MW, 160 °C, 30 min.
Scheme 6.
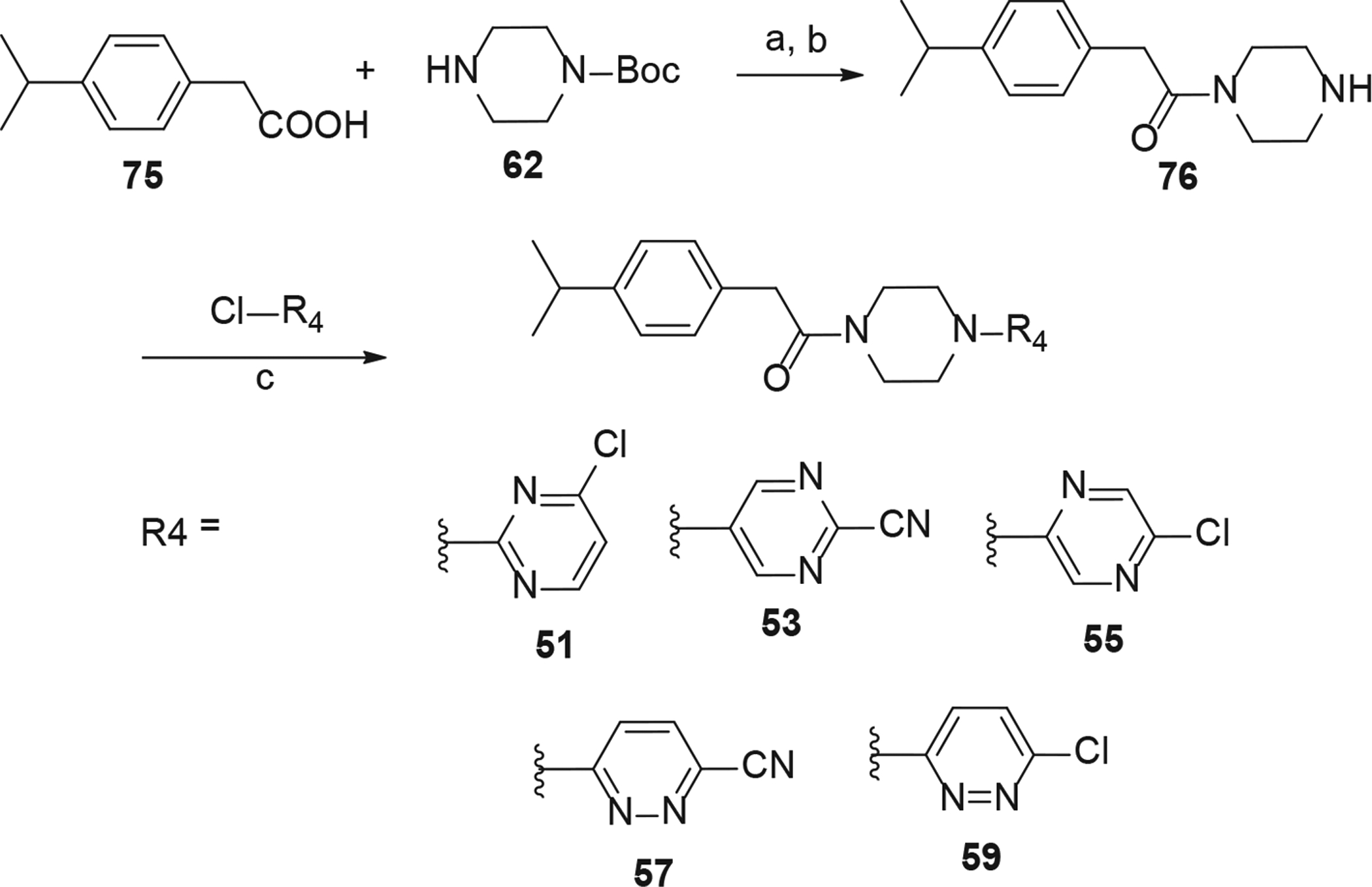
Synthesis of analogs 51, 53, 55, 57 and 59. Reaction Conditions: (a) HATU (1.1 eq), DIPEA (5 eq), CH2Cl2, rt, overnight; (b) TFA-CH2Cl2 (1:1), rt, 1 h; (c) 6-chloronicotinonitrile (1.1 eq), Et3N (2 eq), acetonitrile, MW, 160 °C, 30 min.
2.2. Structure-Activity relationship
2.2.1. Optimization of t-butyl aniline motif
We first explored the effect of aliphatic substitution at the 4-position of the aniline moiety as shown in Table 1. We observed a clear pattern of structural requirements in our homologous series (compounds 1–9). Removal of the aliphatic substituent at the 4-position to obtain compound 2 resulted in a significant loss of PANK3 inhibitory activity (IC50 > 10 μM). This clearly showed the importance of an alkyl side chain at the 4-position of the aniline moiety. The most optimal groups at this position were branched chains such as i-propyl (5), t-butyl (1), and i-butyl (7) at the 4-position. Interestingly, ethyl (3) and n-butyl (8) did not show activity (up to 10 μM), which indicated that a small branched chain aliphatic group is preferred at the 4-position. Consistent with this observation, the bioisosteric replacement of i-propyl with Br (14) and CF3 (18) produced inactive compounds. Also, replacement of the aliphatic side chain at the 4 position with more polar electron donating and electron withdrawing groups resulted in a significant reduction in potency (10–13, 17).
Replacing the aromatic aniline ring with hydrophobic aliphatic substituents (Fig. 2) led to a complete loss of activity (19–23). Similarly, replacing the aniline ring with other aromatic groups also proved to be detrimental to activity (24–27). Overall, our initial SAR exploration on the aniline side of the molecule demonstrated that a branched aliphatic group at the 4-position of the aniline ring is critical for interaction with the PANK3. Even though i-butyl and t-butyl (7 and 1 respectively) at 4-position had similar potency compared to i–propyl (5), the increase in lipophilicity was not desirable, and i-propyl was therefore prioritized for further development based on its LipE score.
2.2.2. Optimization of urea linker
We next investigated the effect of replacing the urea linker. First, replacement of the urea bond with a truncated amide bond (28) resulted in complete loss of activity. The carbamate linker in compound 29 was as efficient as urea (5), which suggested that a hydrogen bond donor group (NH) at this position is not required for interaction with PANK3. This result was supported by the improved activity of 31 which contains acetamide linker. Our interpretation for the improved activity of 31 compared to 5 is that the urea bond has restricted bond rotation that is removed in the acetamide linker in compound 31, thus allowing the molecule to adopt the appropriate orientation for maximum interaction with the PANK3. Importantly, when the position of the carbonyl group in the linker was changed (compound 32), a loss of activity was observed. These data suggested the importance of a hydrogen bond acceptor adjacent to piperazine for maintaining the interactions with the PANK3.
The acetamide linker in compound 31 was clearly superior to other linkers (compare compounds 5, 28–32). However, we also recognized that the increase in potency in 31 relative to 5 came at the cost of an increase in lipophilicity. So, for further structural modification, we investigated both urea and acetamide linkers. We also synthesized compounds 33 and 34 that had cyclopropane and t-butyl at position R3, respectively, and had comparable activity to 31, consistent with the SAR discussed in the prior section. The use of the LipE metric again high-lighted that i-propyl at the R3–position provides an excellent balance between lipophilicity and potency (compare 31, 33 and 34).
2.2.3. Optimization of nicotinonitrile motif
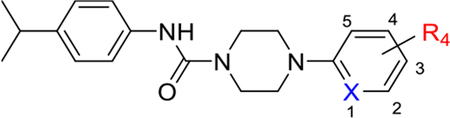
In parallel to our examination of the urea linker, a survey of various substitutions on the nicotinonitrile side chain of the molecule was conducted (Table 3). Analysis of the fourteen synthesized compounds reveals a number of clear trends. Replacement of the nitrile with hydrogen (compound 35), methyl (compound 36), and electron-donating group such as amine (compound 37) result in substantial loss of activity against PANK3. However, electron withdrawing substituents at the 3-position proved to be significant for the activity of compounds against PANK3. Although the order of potency was found to be nitro > bromo = chloro > nitrile > trifluoromethyl (compare 5 and 40–43), we advanced the nitrile at the 3-positon because of its low molecular weight and low lipophilicity. Indeed, this decision was supported by the LipE metric because 5 was more “efficient”. The nitro group was discarded because of the known toxicity associated with this functionality. Interestingly, compound 48 in which the 1-position pyridyl nitrogen atom has been removed, retains the activity illustrating that the nitrogen atom at the 1-position is dispensable. However, removing nitrogen had a detrimental effect on LipE, so this substitution was not advanced. Changing the position of nitrile in the ring was also tolerated with a slight preference to 4-position (39) relative to 5-position (38).
Table 3.
Optimization of nicotinonitrile side chain.
|
|||||
|---|---|---|---|---|---|
| Compd # | X | R4 | PANK3 IC50 (μM) | ClogP | LipE |
| 35 | N | H | 5.3 ± 0.76 | 3.28 | 2.18 |
| 36 | N | 3-CH3 | 2.3 ± 0.27 | 3.78 | 2.04 |
| 37 | N | 3-NH2 | >10 | 2.95 | – |
| 38 | N | 5-CN | 1.1 ± 0.11 | 2.91 | 3.17 |
| 39 | N | 4-CN | 0.23 ± 0.03 | 2.91 | 3.46 |
| 40 | N | 3-NO2 | 0.02 ± 0.004 | 3.20 | 4.49 |
| 41 | N | 3-CF3 | 0.77 ± 0.14 | 4.31 | 1.88 |
| 42 | N | 3-Br | 0.22 ± 0.04 | 4.23 | 2.44 |
| 43 | N | 3-Cl | 0.22 ± 0.03 | 4.07 | 2.60 |
| 44 | C | 3-COCH3 | 3.3 ± 0.36 | 3.97 | 1.46 |
| 45 | C | 3-F | 1.8 ± 0.22 | 4.54 | 1.39 |
| 46 | C | 2-Cl | 1.7 ± 0.21 | 5.11 | 0.65 |
| 47 | C | 3,4-di-Cl | 1.4 ± 0.4 | 5.76 | – |
| 48 | C | 3-CN | 0.58 ± 0.08 | 4.06 | 2.25 |
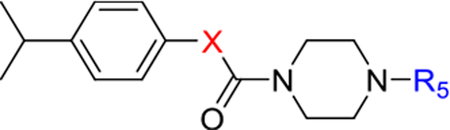
To increase the molecule’s polarity and explore additional hydrogen-bonding interactions with PANK3, we surveyed the effects of incorporating an additional nitrogen atom into the nicotinonitrile ring (Table 4). We observed a slight increase in potency in the pyrimidine-containing analogs. The 2, 6 pyrimidines 49, 50, 51 were similarly active than 3,5 pyrimidines 52, 53, although direct 4-cyano-2,6-pyrimidine analogs analogs were not generated, with 53 demonstrating the best activity. Pyrazines (54 and 55) further increased inhibitory activity. These results suggest that the introduction of nitrogen adjacent to the nitrile is desirable. A major increase in potency was observed in compounds containing a pyridazine ring (e.g., 56–61), subsequently described as pantazines. Most importantly, this 60-fold improvement in potency came with the improvement in lipophilicity (~1 log unit reduction in ClogP), and thus we also observed significantly improved LipE. Such a gain in LipE potency is indicative of additional hydrogen bonding interactions with the protein. The SAR for the X linker motif (Table 2) is further reinforced in this series, with acetamide 53 demonstrating its superior activity compared to direct urea isostere 52. This pattern is repeated for the 4-cyano pyridazine analogs 56, 57, 60, 61 with acetamide > urea > carbamate > alpha-keto linker substitutions.
Table 4.
Exploration of the heterocyclic aromatic ring.
|
|||||
|---|---|---|---|---|---|
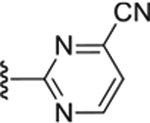
| Compd # | X | R5 | PANK IC50 (μm) | ClogP | LipE |
| 49 | NH |
|
0.14 ± 0.015 | 2.22 | 4.66 |
| 50 | NH |
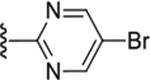
|
0.16 ± 0.012 | 3.40 | 3.42 |
| 51 | CH2 |
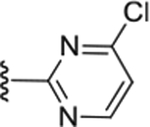
|
0.27 ± 0.054 | 3.69 | 3.07 |
| 52 | NH |
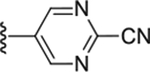
|
0.17 ± 0.017 | 2.22 | 4.54 |
| 53 | CH2 |
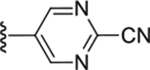
|
0.01 ± 0.001 | 2.66 | 5.34 |
| 54 | NH |
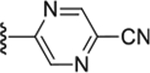
|
0.029 ± 0.004 | 2.22 | 5.33 |
| 55 | CH2 |
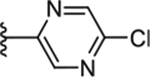
|
0.0018 ± 0.0002 | 3.69 | 5.05 |
| 56 | NH |
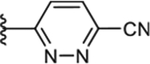
|
0.004 ± 0.0006 | 1.91 | 6.48 |
| 57 | CH2 |
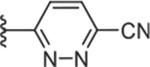
|
0.001 ± 0.0001 | 2.35 | 6.65 |
| 58 | NH |
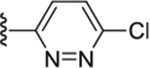
|
0.0007 ± 0.0001 | 3.10 | 6.05 |
| 59 | CH2 |
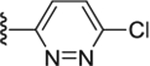
|
0.0004 ± 0.0002 | 3.54 | 5.85 |
| 60 | CO |
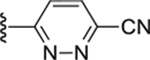
|
>10 | 1.75 | – |
| 61 | O |
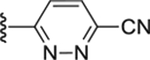
|
0.01 ± 0.001 | 2.45 | 5.55 |
Table 2.
Effect of Variation of the Urea Linker.
|
2.2.4. Structure analysis of PanK3-inhibitor interactions
To understand the SAR that led to the tight binding of 57, we selected three key closely related molecules and determined the crystal structures of their PANK3·AMPNP·Mg2+ complexes (Fig 3, Supplementary Fig S1, Supplementary Table S1). In increasing order of affinity, compounds were pyridine 31 (IC50 60 nM), urea 56 (IC50 4 nM), and chloropyrazine 55 (IC50 1.8 nM). We recently reported the structure of the PANK3•AMPPNP•Mg2+• PZ-2891 (57) complex (PDB ID 6B3V),8 and all three compounds bind in the same location (Fig. 3A, B, C and D). The compounds occupy the pantothenate binding site extending into the PANK3 dimer interface and engage both PANK3 protomers in the dimer, with the two equivalent sites in the dimer both occupied. The heteroaryl C ring and a portion of the piperazine linker ring engage the dimer interface at Trp341′ (prime indicates residues from the opposite monomer) by pi-pi stacking interaction. The acetamide or urea linker and the piperazine ring together generate the required compound length to optimally engage the docking sites on the two monomers. The three compounds were selected to investigate the tight binding of 57; the role of the linker, the positioning and number of nitrogen atoms in the heteroaryl C ring, and the 3-substituent to the heteroaryl ring.
Figure 3.
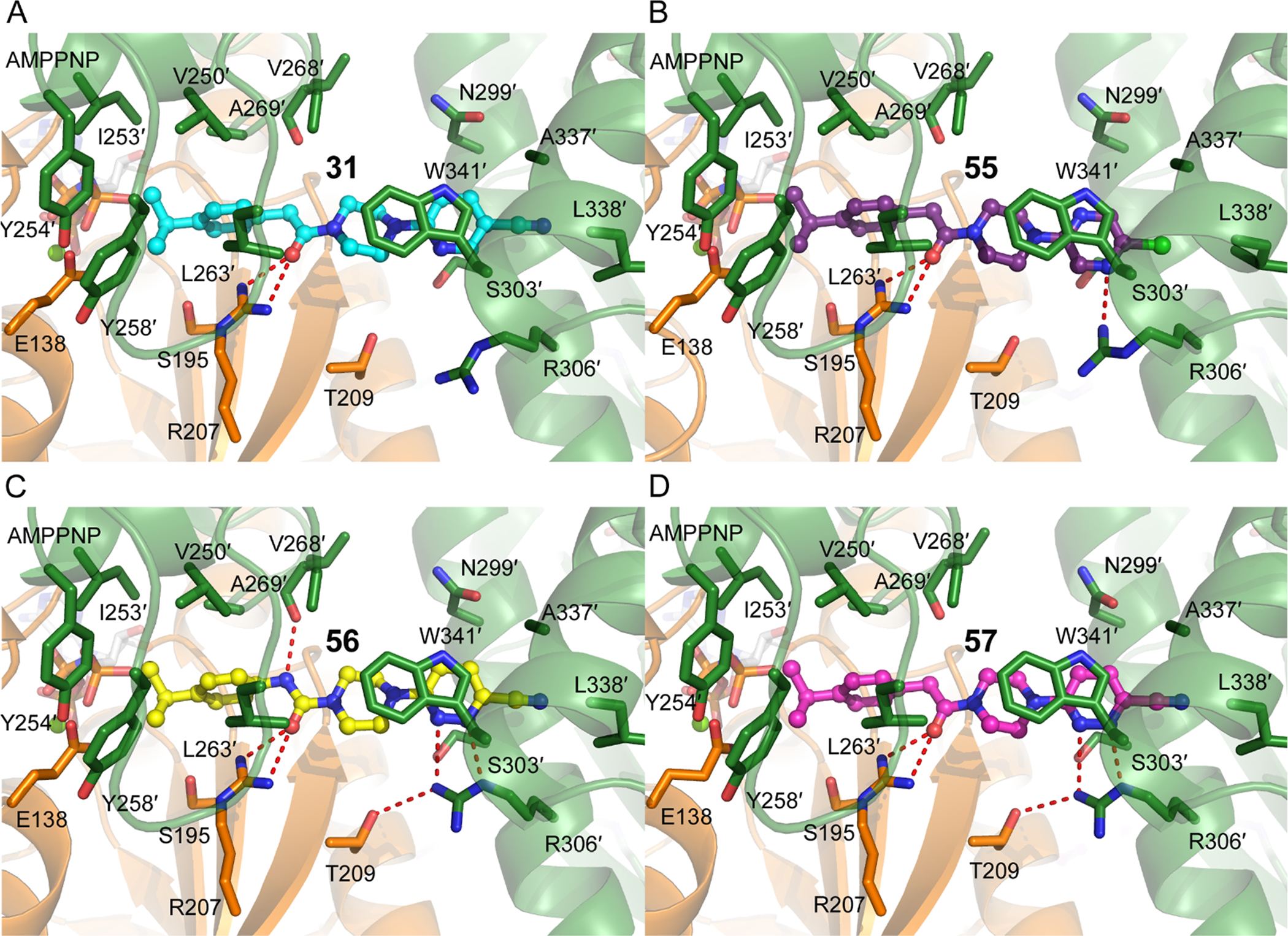
Crystal structures of PANK3 in complex with 4 inhibitors. A. Compound 31 (cyan carbons) (PDB 6X4J); B. Compound 55 (purple carbons) (PDB 6X4L); C. Compound 56 (yellow carbons) (PDB 6X4K); D. Compound 57 (PZ-2891, magenta carbons) (PDB 6B3V). The views are identical in each figure. Monomer A (that contains the bound AMPPNP) is colored orange and monomer B (residues indicated with a prime) is colored green.
Ring A largely occupies the pantothenate binding site adjacent to the catalytic Glu138 and the bound AMPPNP•Mg2+, and packs into a hydrophobic cavity created by V250′, I253′, Y254′, Y258′, L263′, and A269′. These residues reside on a flexible flap that reaches across from the opposite monomer, and the isopropyl substituent optimally fits into this pocket (Fig. 4A). At this site, a branched alkyl 4- phenyl ring is favored in our analog series (see Table 1), with isopropyl proving to be the most desirable. This motif overlays the pantothenate gem-dimethyl group’s binding position found in pantothenate bound structures, [16] explaining the specificity at this position (Fig 4A).
Figure 4.
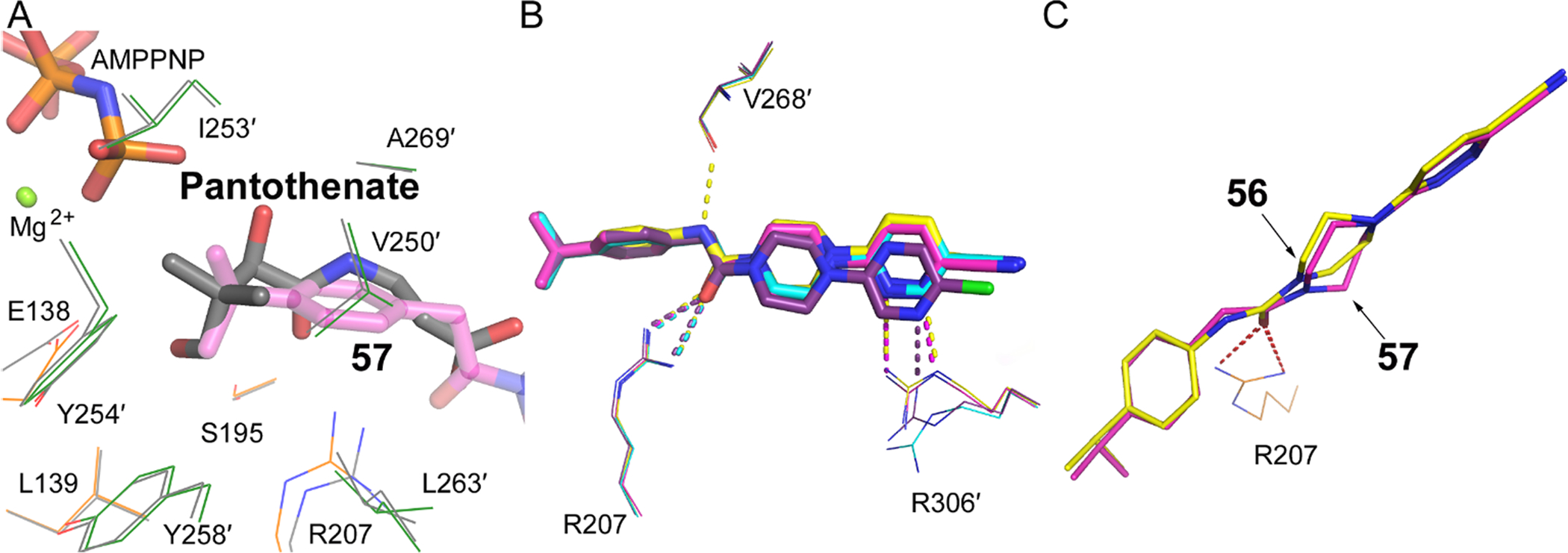
Details of the key compound interactions. A. comparison of the Ring A isopropyl motif with pantothenate gem-dimethyl group’s binding position within the active site; 57 (pink) (PDB 6B3V) overlayed on the ATP:Mg2+:pantothenate (grey) structure (PDB 5KPR). [16] B. Superposition of the 4 compounds showing the displacements of rings B and C, and the interactions with Val268′, Arg207 and Arg306′. Compound 31 (cyan carbons); compound 55 (purple carbons); compound 56 (yellow carbons); compound 57 (PZ-2891, magenta carbons). C. Distortion of ring B in 56 compared to 57.
The preferred linker is an acetamide rather than urea, and this is most evident by comparing the activities 56 (urea) with 57 (acetamide) where this is the only difference. In both linkers, the carbonyl group forms hydrogen bond interactions with Arg207 (Fig. 3A, 3B, 3C, 3D), and the low activity of 32 where the carbonyl group is immediately adjacent to the phenyl ring and unable to engage Arg207 confirms the importance of this interaction. However, in the weaker binding 56, the urea NH group surprisingly forms an additional hydrogen bond interaction with the main chain carbonyl oxygen of Val268′ (Fig. 3C, 4B). This would suggest a tighter binding compound, but closer inspection reveals that the pyridazine and piperazine rings do not perfectly superimpose (Fig. 4B) and piperazine ring of 56 is distorted from its preferred puckered conformation (Fig. 4C). We suggest that the positions and conformations of the pyridazine and piperazine rings are optimal in 57 and that the rigidity of the planar urea linker in 56 has lower affinity because it prevents the two rings from adopting an ideal conformation.
An aromatic stacking interaction with W341 governs the binding affinity of the heteroaryl ring C and a key hydrogen bonding interaction between the heteroatoms and R306′. The 2-pyridyl nitrogen 31 does not interact with R306′ and is the least active (Fig 3A, 4B). In pyrazine 55, the nitrogen at the 3 position forms a hydrogen bonding interaction with a terminal guanidine in R306′ (Fig 3B). In pyridazines 56 and 57, the paired nitrogen atoms at the 2 and 3 positions form strong bidentate hydrogen bonding interaction with the guanidine group (Fig. 3C, 3D, 4B). This aspect of the SAR is evident and explains why electron-withdrawing groups (CN, Cl, NO2) are favored at the 4 position, as they help enhance this hydrogen bonding pattern. The 4-substitution chlorine 55 and nitrile 57 appear to generate equally potent molecules, and both occupy a cavity afforded by Gly302′ and bounded by Ala337′ and Leu338′
2.2.5. Evaluation of physical properties
Based on their single-digit nM potencies and high lipophilicity ligand efficiency (LipE > 6), we evaluated our top candidates (Pantazines) (56, 57) for their physiochemical properties (Table 5). The characterization of the in vitro ADME properties showed that 57 has a modest solubility in PBS buffer (pH 7.4) of 23 μM, approximately 16 times higher than the recorded solubility for our hit compound 1 (1.4 μM).8 56 showed lower solubility (5.5 μM) relative to 57. However, 56 displayed higher stability to mouse, rat, and human microsomes compared to 57. Passive permeability was assessed using the PAMPA assay, and good permeability was observed (316 × 10−6 cm/s for 56 and 413 × 10−6 cm/s for 57). Both the compounds (56 and 57) showed no sign of degradation in mouse, rat, and human plasma (T1/2 = >48 h). The microsomal stability in all animal species was superior for 56 over 57, with 57 being rapidly degraded.
Table 5.
In Vitro ADME Profile of 1, 56 and 57.
| 1 | 56 | 57* | |
|---|---|---|---|
| Solubility at pH = 7.4 (μM) | 1.4 | 5.5 | 23 |
| PAMPA (1 × 10−6 cm/s) | 1232 | 316 | 413 |
| Caco2 Permeability (nm/s) | |||
| A/B | 161 | 243 | 302 |
| Plasma Protein Binding (%) | |||
| mouse | 99 | 92 | 92 |
| rat | 98 | 89 | 91 |
| human | 99 | 92 | 93 |
| Metabolic Stability (T1/2, min) | |||
| mouse | 16 | 113 | 8 |
| rat | 31 | 294 | 47 |
| human | 8 | 64 | 7 |
| Plasma Stability (T1/2, h) | >48 | >48 | >48 |
previously determined.8
3. Conclusion
In summary, we report the rapid LipE-guided optimization of the initial hit 1 to lead 57, as illustrated in Fig. 5. This approach produced potent PANK3 binders with significant improvements in LipE and solubility, overcoming problems encountered with prior attempts to follow-up the PANK3 HTS data set, which focused on simply optimizing the most potent HTS hits. The increased solubility enabled the X-ray structures of the inhibitors bound to the PANK3•AMPPNP•Mg2+complex to be determined. The structures revealed the binding position and the crucial interactions between key molecules in the series and PANK3, rationalizing the emerging SAR and facilitating further design efforts. The LipE metric favors the prioritization of enthalpic-driven binding. The LipE guidance identified the pyridazine-R306′ bidentate hydrogen bonding interaction as a strong and highly favorable interaction. A crucial advantage of the LipE approach is that the Pantazine leads 56 and 57 are naturally selected as highly efficient ligands with desirable in vitro ADME properties. Based in part on this successful study, the clinical development of PANK modulators is ongoing. Our studies suggest that other dormant HTS screening data sets could be re-evaluated using this approach to identify new starting points more amenable to optimization.
Figure 5.
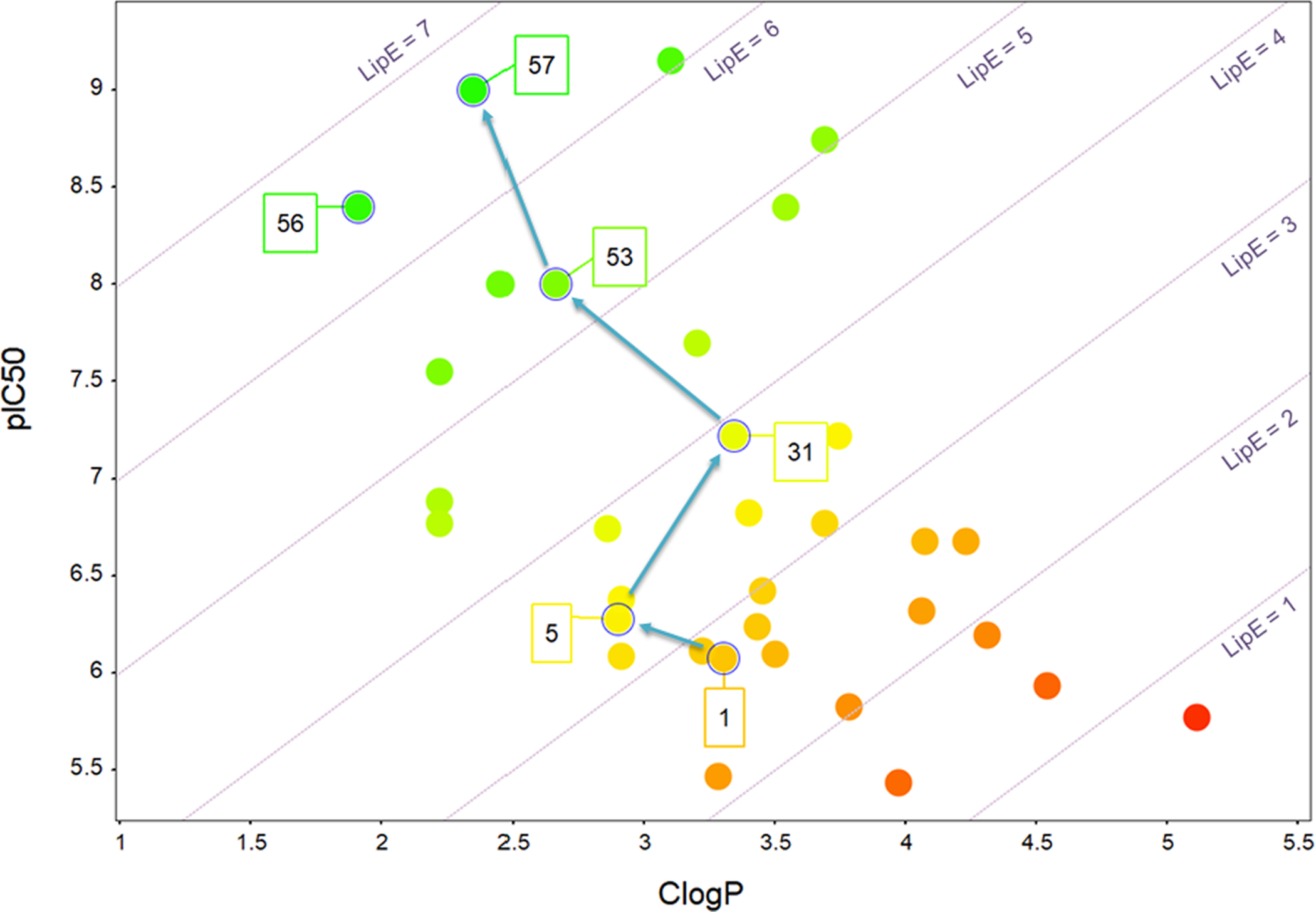
Plot of clogP vs pIC50 showing optimization of HTS-hit (1) guided by LipE and eventually leading to lead 57.
4. Methods
4.1. General methods
Unless otherwise noted, all reactions were carried out in flame-dried glassware under a static nitrogen atmosphere with anhydrous solvent. Chemical reactions were monitored by the thin-layer chromatography (TLC) and the Waters ACQUITY-UPLC-MS-UV system. Microwave-assisted chemical reactions were carried out in the Biotage Initiator+. All reagents were obtained from commercially available sources and used without purification. Purification was handled by reverse phase HPLC or Biotage Flash column chromatography system, with silica cartridges acquired from Biotage Inc. All final compounds were purified to > 95% purity indicated as averages of the total wave count (TWC) and the ELSD readings in LC/MS chromatogram (column: Acquity BEH C18). 1H and 13C spectra were recorded using 400 MHz, using CDCl3 or DMSO as a solvent. The chemical shifts are reported in parts per million (ppm) relative to DMSO (δ 2.50 ppm for proton NMR and δ 39.50 ppm for carbon NMR). Coupling constants are reported in hertz (Hz). The following abbreviations are used to designate the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet.
Compound 1. N-(4-(tert-butyl)phenyl)-4-(5-cyanopyridin-2-yl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.46 (dd, J = 2.4, 0.8 Hz, 1H), 7.69 (dd, J = 9.0, 2.3 Hz, 1H), 7.38−7.32 (m, 2H), 7.30 (d, J = 2.1 Hz, 2H), 6.63 (dd, J = 9.0, 0.9 Hz, 1H), 6.31 (s, 1H), 3.83 (m, 4H), 3.74−3.65 (m, 4H), 1.32 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 155.21, 152.77, 146.72, 140.23, 135.95, 125.99, 120.16, 105.84, 97.27, 43.93, 43.28, 34.45, 31.54. LC-MS (m/z): C21H25N5O Exact Mass 363.21; 364.01 (M++1 observed).
Compound 2. 4-(5-cyanopyridin-2-yl)-N-phenylpiperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.3, 0.7 Hz, 1H), 7.67 (dd, J = 9.0, 2.3 Hz, 1H), 7.41−7.34 (m, 2H), 7.31 (dd, J = 8.7, 7.1 Hz, 2H), 7.11−7.03 (m, 1H), 6.61 (d, J = 9.0 Hz, 1H), 6.33 (s, 1H), 3.82 (dd, J = 6.7, 4.1 Hz, 4H), 3.69 (dd, J = 6.7, 4.1 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 159.04, 154.83, 152.62, 140.10, 138.52, 129.02, 123.55, 120.05, 118.37, 105.70, 43.77, 43.12. LC-MS (m/z): C17H17N5O Exact Mass 307.1; 308.1 (M++1 observed).
Compound 3. 4-(5-cyanopyridin-2-yl)-N-(4-ethylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.4, 0.9 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.27 (m, 2H), 7.14 (d, J = 8.3 Hz, 2H), 6.60 (d, J = 9.0 Hz, 1H), 6.28 (s, 1H), 3.81 (dd, J = 6.6, 4.1 Hz, 4H), 3.72−3.63 (m, 4H), 2.61 (q, J = 7.5 Hz, 2H), 1.27−1.15 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 159.30, 155.32, 152.87, 140.33, 139.94, 136.30, 128.59, 120.67, 118.64, 105.94, 97.37, 44.03, 43.37, 31.19, 28.47, 15.93. LC-MS (m/z): C19H21N5O Exact Mass 335.2; 336.0 (M++1 observed).
Compound 4. 4-(5-cyanopyridin-2-yl)-N-(4-pentylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.4, 0.8 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.23 (d, J = 12.6 Hz, 2H), 7.12 (dd, J = 8.1, 6.1 Hz, 2H), 6.60 (dd, J = 9.1, 0.9 Hz, 1H), 6.30 (s, 1H), 3.80 (dd, J = 6.5, 4.2 Hz, 4H), 3.70−3.63 (m, 4H), 2.56 (m, 2H), 1.57 (m, 2H), 1.31 (m, 4H), 0.88 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 159.19, 155.22, 152.77, 140.23, 138.52, 136.16, 129.39, 129.04, 121.87, 120.47, 118.54, 105.84, 97.27, 43.93, 43.27, 35.40, 31.58, 31.35, 22.69, 14.19. LC-MS (m/z): C22H27N5O Exact Mass 377.2; 378.1 (M++1 observed).
Compound 5. 4-(5-cyanopyridin-2-yl)-N-(4-isopropylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.42 (dd, J = 2.4, 0.8 Hz, 1H), 7.64 (dd, J = 9.0, 2.3 Hz, 1H), 7.28 (m, 2H), 7.20−7.09 (m, 2H), 6.58 (dd, J = 9.0, 0.8 Hz, 1H), 6.46 (s, 1H), 3.83−3.72 (m, 4H), 3.69−3.61 (m, 4H), 2.86 (hept, J = 7.0 Hz, 1H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.17, 155.32, 152.73, 144.42, 140.16, 136.31, 126.99, 120.67, 118.57, 105.82, 97.12, 43.91, 43.25, 33.63, 24.18. LC-MS (m/z): C20H23N5O Exact Mass 349.2; 349.4 (M++1 observed).
Compound 6. 4-(5-cyanopyridin-2-yl)-N-(2-isopropyl-6-methylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.48−8.40 (m, 1H), 7.66 (m, 1H), 7.22−7.13 (m, 2H), 7.08 (m, 1H), 6.66−6.57 (m, 1H), 5.83 (s, 1H), 3.80 (dd, J = 6.7, 4.0 Hz, 4H), 3.68 (dd, J = 6.8, 3.9 Hz, 4H), 3.12 (m, 1H), 2.25 (s, 3H), 1.21 (d, J = 6.9, 0.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.25, 156.14, 152.77, 145.95, 140.21, 136.24, 133.49, 128.28, 127.58, 123.70, 118.56, 105.88, 97.21, 44.09, 43.61, 28.69, 23.68, 18.83. LC-MS (m/z): C21H25N5O Exact Mass 363.2; 363.9 (M++1 observed).
Compound 7. N-(4-(sec-butyl)phenyl)-4-(5-cyanopyridin-2-yl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 2.3 Hz, 1H), 7.66 (dd, J = 8.9, 2.3 Hz, 1H), 7.27 (d, J = 1.9 Hz, 2H), 7.12 (d, J = 8.5 Hz, 2H), 6.60 (d, J = 9.0 Hz, 1H), 6.28 (s, 1H), 3.86−3.74 (m, 4H), 3.71−3.60 (m, 4H), 2.56 (m, 1H), 1.57 (m, 3H), 1.21 (d, J = 6.9 Hz, 2H), 0.81 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.19, 155.23, 152.77, 143.26, 140.23, 136.26, 127.69, 120.47, 118.54, 105.84, 97.26, 43.93, 43.27, 41.22, 31.33, 31.09, 22.05, 12.36. LC-MS (m/z): C21H25N5O Exact Mass 363.2; 364.1 (M++1 observed).
Compound 8. N-(4-butylphenyl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.4, 0.8 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.24 (m, 2H), 7.11 (d, J = 8.2 Hz, 2H), 6.60 (d, J = 9.0 Hz, 1H), 6.27 (s, 1H), 3.81 (m, 4H), 3.75−3.62 (m, 4H), 2.56 (t, J = 7.7 Hz, 2H), 1.59 (m, 2H), 1.34 (h, J = 7.4 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.20, 155.21, 152.77, 140.23, 138.48, 136.17, 129.05, 120.46, 118.54, 105.84, 100.13, 97.27, 43.93, 43.27, 36.52, 35.12, 33.83, 31.09, 22.43, 14.10. LC-MS (m/z): C21H25N5O Exact Mass 363.2; 364.2 (M++1 observed).
Compound 9. N-(4-butyl-2-methylphenyl)-4-(5-cyanopyridin-2-yl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.4, 0.8 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.43 (d, J = 8.1 Hz, 1H), 7.00 (m, 2H), 6.65−6.56 (m, 1H), 6.05 (s, 1H), 3.81 (dd, J = 6.6, 4.2 Hz, 4H), 3.72−3.61 (m, 4H), 2.60−2.49 (m, 2H), 2.23 (s, 3H), 1.58 (m, 2H), 1.43−1.27 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.21, 155.67, 152.77, 140.22, 139.63, 134.12, 130.69, 129.66, 126.90, 123.60, 118.54, 105.84, 97.24, 43.93, 43.37, 35.19, 33.83, 22.50, 18.03, 14.11. LC-MS (m/z): C22H27N5O Exact Mass 377.2; 378.1 (M++1 observed).
Compound 10. 4-(5-cyanopyridin-2-yl)-N-(4-methoxyphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.3, 0.8 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.27 (s, 4H), 7.25 (d, J = 2.3 Hz, 1H), 6.91−6.80 (m, 2H), 6.60 (dd, J = 9.0, 0.9 Hz, 1H), 6.24 (s, 1H), 3.80 (d, J = 6.6 Hz, 8H), 3.71−3.62 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 159.20, 156.37, 155.53, 152.76, 140.22, 131.57, 122.75, 118.54, 114.37, 105.84, 97.25, 55.66, 43.93, 43.24; LC-MS (m/z): C18H19N5O2 Exact Mass 337.2; 338.2 (M++1 observed).
Compound 11. 4-(5-cyanopyridin-2-yl)-N-(4-(trifluoromethoxy) phenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 2.4, 0.8 Hz, 1H), 7.67 (dd, J = 8.9, 2.3 Hz, 1H), 7.43−7.35 (m, 2H), 7.22−7.12 (m, 2H), 6.61 (dd, J = 9.0, 0.8 Hz, 1H), 6.38 (s, 1H), 3.88−3.77 (m, 4H), 3.75−3.63 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 159.22, 154.80, 152.83, 140.36, 137.45, 122.01, 121.33, 118.54, 105.93, 97.51, 43.94, 43.31. LC-MS (m/z): C18H16F3N5O2 Exact Mass 391.1; 391.9 (M++1 observed).
Compound 12. 4-(5-cyanopyridin-2-yl)-N-(4-(dimethylamino) phenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.3, 0.8 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.23−7.14 (m, 2H), 6.76−6.63 (m, 2H), 6.59 (dd, J = 9.1, 0.8 Hz, 1H), 6.16 (s, 1H), 3.87−3.71 (m, 4H), 3.71−3.58 (m, 4H), 2.91 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.23, 155.89, 152.77, 148.10, 140.19, 128.23, 123.08, 118.58, 113.45, 105.84, 97.16, 43.96, 43.27, 41.18. LC-MS (m/z): C19H22N6O Exact Mass 350.2; 351.1 (M++1 observed).
Compound 13. N-(4-acetylphenyl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 2.3, 0.8 Hz, 1H), 7.96−7.89 (m, 2H), 7.68 (dd, J = 9.0, 2.3 Hz, 1H), 7.54−7.44 (m, 2H), 6.61 (dd, J = 9.0, 0.8 Hz, 1H), 6.58 (s, 1H), 3.84 (dd, J = 6.7, 4.0 Hz, 4H), 3.72 (dd, J = 6.7, 4.0 Hz, 4H), 2.57 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 197.10, 159.12, 154.21, 152.76, 143.29, 140.32, 132.30, 130.08, 129.97, 118.80, 118.59, 105.87, 97.52, 43.86, 43.31, 26.57. LC-MS (m/z): C19H19N5O2 Exact Mass 349.1; 350.00 (M++1 observed).
Compound 14. N-(4-bromophenyl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.4, 0.7 Hz, 1H), 7.67 (dd, J = 9.0, 2.3 Hz, 1H), 7.45−7.37 (m, 2H), 7.31−7.26 (m, 2H), 6.60 (d, J = 9.0 Hz, 1H), 6.33 (s, 1H), 3.85−3.77 (m, 4H), 3.74−3.64 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 159.15, 154.63, 152.76, 140.28, 137.82, 132.07, 121.71, 118.47, 116.20, 105.86, 97.42, 43.86, 43.23. LC-MS (m/z): C17H16BrN5O Exact Mass 385.1; 388.1 (M++1 observed).
Compound 15. N-(4-chlorophenyl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.3, 0.8 Hz, 1H), 7.67 (dd, J = 9.0, 2.4 Hz, 1H), 7.36−7.30 (m, 2H), 7.27 (d, J = 6.0 Hz, 2H), 6.60 (d, J = 9.0 Hz, 1H), 6.35 (s, 1H), 3.82 (dd, J = 6.7, 4.1 Hz, 4H), 3.68 (dd, J = 6.7, 4.0 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 159.15, 154.72, 152.76, 140.28, 137.30, 129.13, 128.70, 121.43, 118.49, 105.86, 97.41, 43.87, 43.23. LC-MS (m/z): C17H16ClN5O Exact Mass 341.1; 342.2 (M++1 observed).
Compound 16. 4-(5-cyanopyridin-2-yl)-N-(3,4-dichlorophenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 2.3 Hz, 1H), 7.67 (dd, J = 9.0, 2.4 Hz, 1H), 7.61 (d, J = 2.5 Hz, 1H), 7.35 (d, J = 8.7 Hz, 1H), 7.21 (dd, J = 8.7, 2.6 Hz, 1H), 6.61 (d, J = 9.0 Hz, 1H), 6.39 (s, 1H), 3.88−3.78 (m, 4H), 3.73−3.64 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 159.11, 154.33, 152.75, 140.31, 138.29, 132.86, 130.56, 126.77, 121.73, 119.28, 118.46, 105.87, 97.48, 43.82, 43.22. LC-MS (m/z): C17H15Cl2N5O Exact Mass 375.1; 376.1 (M++1 observed).
Compound 17. N-(4-cyanophenyl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 2.3 Hz, 1H), 7.68 (dd, J = 9.0, 2.3 Hz, 1H), 7.63−7.56 (m, 2H), 7.51 (d, J = 8.8 Hz, 2H), 6.69−6.51 (m, 2H), 3.83 (dd, J = 6.7, 4.0 Hz, 4H), 3.71 (dd, J = 6.8, 4.1 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 159.10, 153.90, 152.75, 143.02, 140.35, 133.41, 119.43, 119.12, 118.41, 106.28, 105.88, 97.60, 43.82, 43.30. LC-MS (m/z): C18H16N6O Exact Mass 332.1; 333.0 (M++1 observed).
Compound 18. 4-(5-cyanopyridin-2-yl)-N-(4-(trifluoromethyl) phenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 2.3, 0.8 Hz, 1H), 7.68 (dd, J = 9.0, 2.3 Hz, 1H), 7.56 (d, J = 8.6 Hz, 2H), 7.50 (d, J = 8.6 Hz, 2H), 6.61 (dd, J = 9.0, 0.9 Hz, 1H), 6.52 (s, 1H), 3.83 (dd, J = 6.5, 4.2 Hz, 4H), 3.77−3.62 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 159.13, 154.36, 152.76, 141.92, 140.31, 126.43, 126.39, 119.36, 119.16, 118.45, 105.87, 97.49, 43.85, 43.27. LC-MS (m/z): C18H16F3N5O Exact Mass 375.1; 376.2 (M++1 observed).
Compound 19. 4-(5-cyanopyridin-2-yl)-N-cyclohexylpiperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 2.3 Hz, 1H), 7.64 (dd, J = 9.0, 2.3 Hz, 1H), 6.57 (d, J = 9.0 Hz, 1H), 4.27 (d, J = 7.7 Hz, 1H), 3.79−3.71 (m, 4H), 3.66 (m, 1H), 3.57−3.48 (m, 4H), 2.06−1.89 (m, 2H), 1.71 (m, 2H), 1.62 (m, 2H), 1.46−1.30 (m, 2H), 1.12 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 159.23, 156.91, 152.75, 140.13, 118.61, 105.80, 97.01, 49.71, 43.90, 42.89, 34.13, 25.77, 25.19. LC-MS (m/z): C17H23N5O Exact Mass 313.2; 314.1(M++1 observed)
Compound 20. N-(tert-butyl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 2.3 Hz, 1H), 7.64 (dd, J = 9.0, 2.4 Hz, 1H), 6.57 (d, J = 9.0 Hz, 1H), 4.30 (s, 1H), 3.80−3.69 (m, 4H), 3.55−3.44 (m, 4H), 1.37 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 159.24, 156.82, 152.76, 140.13, 118.63, 105.80, 96.98, 51.19, 43.94, 42.92, 29.58. LC-MS (m/z): C15H21N5O Exact Mass 287.2; 288.1 (M++1 observed).
Compound 21. 4-(5-cyanopyridin-2-yl)-N-octylpiperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.44 (m, 1H), 7.64 (dd, J = 9.0, 2.3 Hz, 1H), 6.58 (dd, J = 9.0, 0.8 Hz, 1H), 4.42 (s, 1H), 3.80−3.71 (m, 4H), 3.58−3.51 (m, 4H), 3.25 (m, 2H), 1.28 (m, 12H), 0.92−0.83 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 159.24, 157.65, 152.76, 140.15, 118.59, 105.81, 97.07, 43.94, 42.96, 41.22, 31.96, 31.09, 30.41, 29.47, 29.38, 27.11, 22.80, 14.25. LC-MS (m/z): C19H29N5O Exact Mass 343.2; 344.2 (M++1 observed).
Compound 22. 4-(5-cyanopyridin-2-yl)-N-isopropylpiperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.41 (m, 1H), 7.64 (m, 1H), 6.57 (dd, J = 9.1, 1.8 Hz, 1H), 4.21 (d, J = 7.5 Hz, 1H), 4.09−3.92 (m, 1H), 3.74 (m, 4H), 3.52 (m, 4H), 1.18 (d, J = 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.24, 156.95, 152.76, 140.14, 118.60, 105.81, 97.05, 43.92, 42.89, 23.63. LC-MS (m/z): C14H19N5O Exact Mass 273.2; 274.1 (M++1 observed).
Compound 23. N-((3S,5S,7S)-adamantan-1-yl)-4-(5-cyanopyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 2.3 Hz, 1H), 7.64 (dd, J = 9.0, 2.3 Hz, 1H), 6.57 (d, J = 9.0 Hz, 1H), 4.18 (s, 1H), 3.74 (m, 4H), 3.57−3.45 (m, 4H), 2.09 (m, 3H), 1.99 (m, 6H), 1.68 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 159.24, 156.45, 152.76, 140.11, 118.63, 105.78, 96.96, 51.68, 43.93, 42.92, 42.53, 36.58, 29.74. LC-MS (m/z): : C21H27N5O Exact Mass 365.2; 366.4 (M++1 observed).
Compound 24. 4-(5-cyanopyridin-2-yl)-N-(4-methylbenzyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.41 (dd, J = 2.4, 0.7 Hz, 1H), 7.64 (dd, J = 9.0, 2.4 Hz, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 7.9 Hz, 2H), 6.58 (d, J = 9.0 Hz, 1H), 4.66 (m, 1H), 4.41 (d, J = 5.4 Hz, 2H), 3.79−3.70 (m, 4H), 3.60−3.51 (m, 4H), 2.34 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 157.52, 152.85, 140.27, 137.51, 136.25, 129.63, 128.16, 118.67, 105.92, 45.14, 44.03, 43.12, 31.19, 21.36. LC-MS (m/z): C19H21N5O Exact Mass 335.2; 336.1 (M++1 observed).
Compound 25. 4-(5-cyanopyridin-2-yl)-N-(furan-2-ylmethyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.41 (dd, J = 2.4, 0.8 Hz, 1H), 7.64 (dd, J = 9.0, 2.3 Hz, 1H), 7.36 (dd, J = 1.9, 0.9 Hz, 1H), 6.58 (dd, J = 9.1, 0.8 Hz, 1H), 6.33 (dd, J = 3.2, 1.9 Hz, 1H), 6.27−6.21 (m, 1H), 4.77 (m, 1H), 4.45 (d, J = 5.4 Hz, 2H), 3.79−3.71 (m, 4H), 3.60−3.53 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 159.30, 157.23, 152.85, 152.32, 142.40, 140.27, 118.66, 110.72, 107.63, 105.91, 97.24, 43.99, 43.07, 38.17, 31.19. LC-MS (m/z): C16H17N5O2 Exact Mass 311.1; 312.1 (M++1 observed).
Compound 26. Preparation of 4-(5-cyanopyridin-2-yl)-N-(2,3-dihy-dro-1H-inden-5-yl)piperazine-1-carboxamide 1H NMR (400 MHz, CDCl3) δ 8.43 (dd, J = 2.1, 1.2 Hz, 1H), 7.66 (ddd, J = 9.0, 2.3, 0.8 Hz, 1H), 7.29 (d, J = 1.8 Hz, 1H), 7.13 (d, J = 8.0 Hz, 1H), 7.05−6.99 (m, 1H), 6.60 (dd, J = 9.1, 0.9 Hz, 1H), 6.28 (s, 1H), 3.80 (m, 4H), 3.72−3.63 (m, 4H), 2.87 (m, 4H), 2.06 (p, J = 7.3 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 159.30, 155.48, 152.87, 145.50, 140.32, 139.94, 136.79, 124.70, 118.81, 118.65, 117.28, 105.94, 97.34, 44.04, 43.38, 33.26, 32.53, 25.90. LC-MS (m/z): C20H21N5O Exact Mass 347.2; 348.1 (M++1 observed).
Compound 27. 4-(5-cyanopyridin-2-yl)-N-(3-isopropylisoxazol-5-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 2.3, 0.8 Hz, 1H), 7.68 (dd, J = 9.0, 2.3 Hz, 1H), 7.37 (s, 1H), 6.61 (dd, J = 8.9, 0.8 Hz, 1H), 6.10 (s, 1H), 3.91−3.76 (m, 4H), 3.76−3.60 (m, 4H), 2.98 (h, J = 6.9 Hz, 1H), 1.27 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.32, 161.11, 159.06, 152.75, 151.31, 140.37, 118.37, 105.88, 97.69, 85.55, 43.74, 43.30, 27.10, 21.68. LC-MS (m/z): C17H20N6O2 Exact Mass 340.2; 341.2 (M++1 observed).
Compound 28. 6-(4-(4-isopropylbenzoyl)piperazin-1-yl) nicotinonitrile
1H NMR (400 MHz, CDCl3) δ 8.42 (dd, J = 2.3, 0.8 Hz, 1H), 7.66 (dd, J = 9.0, 2.3 Hz, 1H), 7.45−7.32 (m, 2H), 7.31−7.27 (m, 2H), 6.62 (dd, J = 9.0, 0.8 Hz, 1H), 3.73 (m, 8H), 2.94 (h, J = 6.9 Hz, 1H), 1.27 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.03, 159.34, 152.78, 151.43, 140.25, 132.67, 127.48, 126.84, 118.46, 106.01, 97.42, 44.67, 34.24, 23.97. LC-MS (m/z): C20H22N4 Exact Mass 334.2; 335.1 (M++1 observed).
Compound 29. 4-isopropylphenyl 4-(5-cyanopyridin-2-yl)piperazine-1-carboxylate
1H NMR (400 MHz, CDCl3) δ 8.44 (dd, J = 2.3, 0.8 Hz, 1H), 7.67 (dd, J = 9.0, 2.3 Hz, 1H), 7.25−7.18 (m, 2H), 7.08−6.99 (m, 2H), 6.64 (dd, J = 9.0, 0.9 Hz, 1H), 3.75 (m, 8H), 2.91 (hept, J = 7.0 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.31, 154.06, 152.79, 149.16, 146.25, 140.27, 127.44, 121.43, 118.48, 106.00, 97.38, 44.23, 33.76, 24.21. LC-MS (m/z): C20H22N4O2 Exact Mass 350.2; 352.0 (M++1 observed).
Compound 30. 6-(4-(2-hydroxy-2-(4-isopropylphenyl)acetyl)piperazin-1-yl)nicotinonitrile
1H NMR (400 MHz, CDCl3) δ 8.37 (dd, J = 2.3, 0.8 Hz, 1H), 7.62 (dd, J = 9.0, 2.3 Hz, 1H), 7.23 (m, 4H), 6.63−6.51 (m, 1H), 5.23 (d, J = 6.3 Hz, 1H), 4.62 (d, J = 6.3 Hz, 1H), 3.94 (m, 1H), 3.79 (m, 1H), 3.74−3.57 (m, 2H), 3.51 (m, 1H), 3.46−3.27 (m, 2H), 3.12−2.97 (m, 1H), 2.88 (h, J = 6.9 Hz, 1H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.61, 159.05, 152.70, 149.84, 140.27, 136.49, 127.51, 127.49, 118.32, 105.89, 97.59, 71.64, 44.19, 43.55, 42.38, 38.77, 34.01, 31.09, 24.02. LC-MS (m/z): C21H24N4O2 Exact Mass 364.2; 365.3 (M++1 observed).
Compound 31. 6-(4-(2-(4-isopropylphenyl)acetyl)piperazin-1-yl) nicotinonitrile
1H NMR (400 MHz, CDCl3) δ 8.42 (d, J = 2.3 Hz, 1H), 7.65 (dd, J = 9.0, 2.4 Hz, 1H), 7.21 (s, 4H), 6.58 (d, J = 9.0 Hz, 1H), 3.78 (m, 4H), 3.66 (m, 2H), 3.59 (m, 4H), 2.97−2.86 (m, 1H), 1.25 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 170.20, 159.19, 152.73, 147.81, 140.19, 131.95, 128.59, 127.09, 118.46, 105.89, 97.28, 45.58, 44.42, 44.05, 41.26, 40.85, 33.86, 24.11. LC-MS (m/z): C21H24N4O Exact Mass 348.2; 350.3 (M++1 observed).
Compound 32. 6-(4-(2-(4-isopropylphenyl)-2-oxoethyl)piperazin-1-yl)nicotinonitrile
1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 2.3 Hz, 1H), 7.98−7.89 (m, 2H), 7.61 (dt, J = 9.1, 1.8 Hz, 1H), 7.32 (d, J = 7.9 Hz, 2H), 6.60 (d, J = 9.0 Hz, 1H), 3.88 (s, 2H), 3.77 (t, J = 5.0 Hz, 4H), 2.97 (hept, J = 7.0 Hz, 1H), 2.72 (t, J = 5.0 Hz, 4H), 1.27 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 195.54, 159.43, 155.29, 152.83, 139.98, 133.82, 128.46, 126.91, 118.83, 105.85, 96.53, 63.98, 53.11, 44.46, 34.46, 23.79. LC-MS (m/z): C21H24N4O Exact Mass 348.2; 349.9 (M++1 observed).
Compound 33. 6-(4-(2-(4-cyclopropylphenyl)acetyl)piperazin-1-yl) nicotinonitrile
1H NMR (400 MHz, CDCl3) δ 8.39 (dd, J = 2.3, 0.8 Hz, 1H), 7.62 (dd, J = 9.0, 2.3 Hz, 1H), 7.13 (d, J = 8.1 Hz, 2H), 7.02 (d, J = 8.1 Hz, 2H), 6.56 (dd, J = 9.0, 0.9 Hz, 1H), 3.75 (m, 4H), 3.62 (m, 2H), 3.55 (m, 4H), 1.86 (m, 1H), 1.02−0.87 (m, 2H), 0.72−0.58 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 170.15, 159.19, 152.74, 143.00, 140.19, 131.59, 128.52, 126.32, 118.46, 105.90, 97.31, 45.57, 44.42, 44.06, 41.26, 40.91, 15.20, 9.39. LC-MS (m/z): C21H22N4O Exact Mass 346.2; 347.2 (M++1 observed).
Compound 34. 6-(4-(2-(4-(tert-butyl)phenyl)acetyl)piperazin-1-yl) nicotinonitrile
1H NMR (400 MHz, CDCl3) δ 8.45−8.34 (m, 1H), 7.63 (dd, J = 9.0, 2.3 Hz, 1H), 7.38−7.31 (m, 2H), 7.18 (d, J = 8.3 Hz, 2H), 6.56 (d, J = 9.0 Hz, 1H), 3.76 (m, 4H), 3.63 (m, 2H), 3.57 (m, 4H), 1.30 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 170.17, 159.20, 152.75, 140.20, 131.62, 128.35, 125.95, 118.46, 105.89, 100.13, 97.31, 44.45, 44.07, 41.26, 40.72, 34.62, 31.48, 31.09. LC-MS (m/z): C22H26N4O Exact Mass 362.2; 363.1 (M++1 observed).
Compound 35. N-(4-isopropylphenyl)-4-(pyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.21 (ddd, J = 4.9, 2.0, 0.9 Hz, 1H), 7.52 (ddd, J = 8.5, 7.2, 2.0 Hz, 1H), 7.31−7.27 (m, 2H), 7.19−7.13 (m, 2H), 6.71−6.62 (m, 2H), 6.30 (s, 1H), 3.65 (s, 8H), 2.87 (hept, J = 6.7 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.14, 155.37, 148.14, 144.19, 137.82, 136.51, 127.00, 120.45, 113.91, 107.25, 44.88, 43.75, 33.66, 24.22. LC-MS (m/z): C19H24N4O Exact Mass 324.2; 325.0 (M++1 observed).
Compound 36. N-(4-isopropylphenyl)-4-(5-methylpyridin-2-yl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 2.3 Hz, 1H), 7.36 (d, J = 8.9 Hz, 1H), 7.30−7.26 (m, 2H), 7.21−7.13 (m, 2H), 6.61 (d, J = 8.6 Hz, 1H), 6.32 (s, 1H), 3.64 (m, 4H), 3.59 (m, 4H), 2.87 (h, J = 7.0 Hz, 1H), 2.21 (s, 3H), 1.27−1.18 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 155.38, 144.17, 136.52, 127.40, 126.99, 123.08, 120.45, 77.48, 77.36, 77.16, 76.84, 45.50, 43.80, 33.66, 24.22, 17.48. LC-MS (m/z): C20H26N4O Exact Mass 338.2; 339.1 (M++1 observed).
Compound 37. 4-(5-aminopyridin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 7.62 (d, J = 2.8 Hz, 1H), 7.40−7.30 (m, 2H), 7.15−7.05 (m, 2H), 6.94 (dd, J = 8.8, 2.9 Hz, 1H), 6.68 (d, J = 8.8 Hz, 1H), 4.61 (s, 2H), 3.58−3.47 (m, 4H), 3.31−3.19 (m, 4H), 2.80 (hept, J = 6.9 Hz, 1H), 1.16 (d, J = 6.9 Hz, 6H 13C NMR (101 MHz, CDCl3) δ 155.35, 152.18, 141.91, 138.18, 137.60, 133.41, 126.09, 124.63, 120.00, 108.90, 46.55, 43.62, 32.84, 24.13. LC-MS (m/z): C19H25N5O Exact Mass 339.2; 340.3 (M++1 observed).
Compound 38. 4-(3-cyanopyridin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.37 (dd, J = 4.8, 2.0 Hz, 1H), 7.81 (dd, J = 7.6, 2.0 Hz, 1H), 7.28 (m, 2H), 7.20−7.13 (m, 2H), 6.82 (dd, J = 7.7, 4.8 Hz, 1H), 6.29 (s, 1H), 3.85−3.75 (m, 4H), 3.72−3.62 (m, 4H), 2.88 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 160.70, 155.32, 152.03, 144.32, 143.98, 136.40, 127.03, 120.50, 114.83, 95.46, 47.83, 43.79, 33.67, 24.21. LC-MS (m/z): C20H23N5O Exact Mass 349.2; 350.1 (M++1 observed).
Compound 39. 4-(4-cyanopyridin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.34−8.24 (m, 1H), 7.30−7.26 (m, 2H), 7.22−7.12 (m, 2H), 6.81 (m, 2H), 6.28 (s, 1H), 3.77−3.69 (m, 4H), 3.69−3.61 (m, 4H), 2.87 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.59, 155.26, 149.53, 144.41, 136.32, 127.05, 121.83, 120.52, 117.45, 114.00, 108.85, 44.31, 43.45, 33.67, 24.21. LC-MS (m/z): C20H23N5O Exact Mass 349.2; 350.1 (M++1 observed).
Compound 40. N-(4-isopropylphenyl)-4-(5-nitropyridin-2-yl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 9.06 (d, J = 2.8 Hz, 1H), 8.26 (dd, J = 9.5, 2.7 Hz, 1H), 7.31−7.26 (m, 2H), 7.21−7.12 (m, 2H), 6.57 (d, J = 9.5 Hz, 1H), 6.30 (s, 1H), 3.90 (m, 4H), 3.75−3.66 (m, 4H), 2.86 (h, J = 7.0 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 160.32, 155.19, 146.45, 144.55, 136.19, 135.71, 133.39, 127.08, 120.58, 104.71, 44.28, 43.23, 33.67, 24.20. LC-MS (m/z): C19H23N5O3 Exact Mass 369.2; 370.1 (M++1 observed).
Compound 41. N-(4-isopropylphenyl)-4-(5-(trifluoromethyl)pyridin-2-yl)piperazine-1- carboxamide 1H NMR (400 MHz, CDCl3) δ 8.45−8.39 (m, 1H), 7.67 (dd, J = 9.0, 2.5 Hz, 1H), 7.31−7.26 (m, 2H), 7.21−7.13 (m, 2H), 6.64 (d, J = 9.0 Hz, 1H), 6.30 (s, 1H), 3.82−3.73 (m, 4H), 3.71−3.60 (m, 4H), 2.87 (hept, J = 7.0 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 160.10, 155.29, 145.89, 144.38, 136.34, 134.89, 127.04, 120.53, 105.72, 44.23, 43.43, 33.66, 24.20. LC-MS (m/z): C20H23F3N4O Exact Mass 392.2; 394.2 (M++1 observed).
Compound 42. 4-(5-bromopyridin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 2.5 Hz, 1H), 7.57 (dd, J = 9.0, 2.5 Hz, 1H), 7.28 (d, J = 1.9 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 6.55 (d, J = 9.0 Hz, 1H), 6.27 (s, 1H), 3.63 (m, 8H), 2.87 (m, 1H), 1.23 (d, J = 6.9, 6H). 13C NMR (101 MHz, CDCl3) δ 157.60, 155.31, 148.70, 144.28, 140.08, 136.42, 127.02, 120.47, 108.51, 108.31, 44.82, 43.57, 33.66, 24.21. LC-MS (m/z): C19H23BrN4O Exact Mass: 404.11; 405.1 (M++1 observed).
Compound 43. 4-(5-chloropyridin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 2.6 Hz, 1H), 7.46 (dd, J = 9.0, 2.7 Hz, 1H), 7.28 (m, 2H), 7.21−7.12 (m, 2H), 6.59 (d, J = 9.0 Hz, 1H), 6.31 (s, 1H), 3.63 (m, 8H), 2.87 (h, J = 6.9 Hz, 1H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 157.36, 155.33, 146.40, 144.28, 137.53, 136.42, 127.01, 120.84, 120.49, 107.93, 44.96, 43.59, 33.66, 24.21. LC-MS (m/z): C19H23ClN4O Exact Mass 358.1; 359.1 (M++1 observed).
Compound 44. 4-(4-acetylphenyl)-N-(4-isopropylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 7.95−7.86 (m, 2H), 7.31−7.26 (m, 2H), 7.20−7.13 (m, 2H), 6.86 (m, 2H), 6.36 (s, 1H), 3.74−3.64 (m, 4H), 3.49−3.41 (m, 4H), 2.87 (m, 1H), 2.54 (s, 3H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 196.69, 155.29, 153.71, 144.40, 136.35, 130.60, 128.24, 127.31, 127.04, 120.56, 113.61, 47.12, 43.63, 33.66, 26.32, 24.20, 24.18. LC-MS (m/z): C22H27N3O2 Exact Mass 365.2; 366.3 (M++1 observed).
Compound 45. 4-(4-fluorophenyl)-N-(4-isopropylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 7.28 (m, 2H), 7.20−7.13 (m, 2H), 7.02−6.95 (m, 2H), 6.93−6.86 (m, 2H), 6.33 (s, 1H), 3.68−3.60 (m, 4H), 3.18−3.09 (m, 4H), 2.87 (hept, J = 6.8 Hz, 1H), 1.22 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.94, 156.56, 155.30, 147.80, 147.78, 144.23, 136.49, 127.01, 120.44, 118.70, 118.62, 115.97, 115.75, 50.46, 44.33, 33.66, 24.21. LC-MS (m/z): C20H24FN3O Exact Mass 341.2; 342.3 (M++1 observed).
Compound 46. 4-(3-chlorophenyl)-N-(4-isopropylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 7.27 (m, 2H), 7.23−7.12 (m, 3H), 6.92−6.83 (m, 2H), 6.79 (m, 1H), 6.35 (s, 1H), 3.73−3.56 (m, 4H), 3.31−3.16 (m, 4H), 2.87 (m, 1H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.31, 152.04, 144.30, 136.43, 135.21, 130.32, 127.28, 127.01, 120.52, 120.13, 116.32, 114.39, 48.78, 44.01, 33.65, 24.20. LC-MS (m/z): C20H24ClN3O Exact Mass 357.2; 358.2 (M++1 observed).
Compound 47. 4-(3,4-dichlorophenyl)-N-(4-isopropylphenyl) piperazine-1-carboxamide 1H NMR (400 MHz, CDCl3) δ 7.37−7.26 (m, 2H), 7.17 (m, 2H), 7.08 (m, 1H), 6.85 (m, 1H), 6.70 (m 2H), 3.58 (m, 4H), 3.18−3.03 (m, 4H), 2.86 (m, 1H), 1.26−1.17 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 155.58, 152.18, 144.28, 136.56, 135.55, 126.81, 126.78, 121.08, 119.27, 113.93, 47.94, 43.58, 33.53, 24.12. LC-MS (m/z): C20H23Cl2N3O Exact Mass 391.1; 392.2 (M++1 observed).
Compound 48. 4-(4-cyanophenyl)-N-(4-isopropylphenyl)piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 7.56−7.50 (m, 2H), 7.28 (s, 2H), 7.17 (d, J = 8.5 Hz, 2H), 6.89−6.82 (m, 2H), 6.30 (s, 1H), 3.74−3.64 (m, 4H), 3.48−3.39 (m, 4H), 2.87 (hept, J = 7.0 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 155.20, 152.96, 144.46, 136.29, 133.77, 127.06, 120.53, 119.97, 114.34, 101.07, 46.90, 43.53, 33.67, 24.20; LC-MS (m/z): C21H24N4O Exact Mass 348.2; 348.2 (M++1 observed).
Compound 49. 4-(4-cyanopyrimidin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.48 (d, J = 4.7 Hz, 1H), 7.27 (d, J = 6.4 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 6.81 (d, J = 4.7 Hz, 1H), 6.30 (s, 1H), 3.98−3.90 (m, 4H), 3.67−3.56 (m, 4H), 2.87 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 161.28, 159.93, 155.31, 144.39, 141.87, 136.35, 127.05, 120.52, 116.15, 112.71, 43.79, 43.49, 33.67, 24.21. LC-MS (m/z): C19H22N6O Exact Mass 350.2; 351.1 (M++1 observed).
Compound 50. 4-(5-bromopyrimidin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 0.7 Hz, 2H), 7.28 (d, J = 1.9 Hz, 2H), 7.16 (d, J = 8.5 Hz, 2H), 6.29 (s, 1H), 3.97−3.79 (m, 4H), 3.63−3.54 (m, 4H), 2.88 (m, 1H), 1.22 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.88, 158.14, 155.35, 144.29, 136.43, 127.02, 120.49, 106.57, 43.80, 43.71, 33.66, 24.21. LC-MS (m/z): C18H22BrN5O Exact Mass 403.1; 404.3 (M++1 observed).
Compound 51. 1-(4-(4-chloropyrimidin-2-yl)piperazin-1-yl)-2-(4-isopropylphenyl)ethan-1-one 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 5.4 Hz, 1H), 7.18 (s, 4H), 6.53 (d, J = 5.2 Hz, 1H), 3.80 (dd, J = 6.6, 4.1 Hz, 2H), 3.75 (s, 2H), 3.71 (dd, J = 6.7, 4.0 Hz, 2H), 3.63 (dd, J = 6.5, 3.9 Hz, 2H), 3.50 (t, J = 5.2 Hz, 2H), 2.88 (m, 1H), 1.23 (d, J = 7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 170.11, 161.44, 161.41, 158.98, 147.70, 132.16, 128.58, 127.06, 109.86, 45.95, 43.82, 43.72, 41.67, 40.96, 33.86, 24.12. LC-MS (m/z): C19H23ClN4O Exact Mass: 360.2; 361.2 (M++1 observed).
Compound 52. 4-(2-cyanopyrimidin-5-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.34 (s, 2H), 7.25 (m, 2H), 7.21−7.13 (m, 2H), 6.30 (s, 1H), 3.80−3.68 (m, 4H), 3.58−3.48 (m, 4H), 2.88 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.09, 144.77, 144.00, 142.06, 136.01, 133.95, 127.13, 120.66, 116.50, 111.36, 45.58, 43.17, 33.68, 24.19. LC-MS (m/z): C19H22N6O Exact Mass 350.2; 351.2 (M++1 observed).
Compound 53. 5-(4-(2-(4-isopropylphenyl)acetyl)piperazin-1-yl) pyrimidine-2-carbonitrile 1H NMR (500 MHz, CDCl3) δ 8.21 (s, 2H), 7.16 − 7.07 (m, 4H), 3.78 (t, J = 5.4 Hz, 2H), 3.69 (s, 2H), 3.65 − 3.55 (m, 2H), 3.33 (t, J = 5.4 Hz, 2H), 3.13 (t, J = 5.3 Hz, 2H), 2.82 (p, J = 6.9 Hz, 1H), 1.16 (d, J = 6.9 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.94, 147.89, 143.94, 142.14, 133.88, 131.58, 128.41, 127.07, 116.33, 45.64, 44.92, 40.74, 40.71, 33.73, 23.98. LC-MS (m/z): C20H23N5O Exact Mass 349.2; 350.5 (M++1 observed).
Compound 54. 4-(5-cyanopyrazin-2-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 1.5 Hz, 1H), 8.14 (d, J = 1.5 Hz, 1H), 7.27 (m, 2H), 7.17 (m, 2H), 6.28 (s, 1H), 3.87 (m, 4H), 3.70 (m, 4H), 2.88 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 155.25, 153.98, 147.35, 144.77, 136.21, 131.11, 127.21, 120.72, 117.37, 117.30, 43.76, 43.31, 33.78, 24.30. LC-MS (m/z): C19H22N6O Exact Mass 350.2; 351.3 (M++1 observed).
Compound 55 1-(4-(5-chloropyrazin-2-yl)piperazin-1-yl)-2-(4-isopropylphenyl)ethan-1-one
1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 1.5 Hz, 1H), 7.82 (d, J = 1.4 Hz, 1H), 7.18 (s, 4H), 3.76 (m, 4H), 3.56 (m, 4H), 3.39 (m, 2H), 2.88 (hept, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H);). 13C NMR (101 MHz, CDCl3) δ 170.15, 153.56, 147.86, 141.28, 137.08, 132.09, 129.37, 128.65, 127.16, 45.60, 44.80, 44.66, 41.28, 40.92, 33.93, 24.19; LC-MS (m/z): C19H23ClN4O Exact Mass 358.2; 359.3 (M++1 observed)
Compound 56. 4-(6-cyanopyridazin-3-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 9.6 Hz, 1H), 7.27 (m, 2H), 7.22−7.12 (m, 2H), 6.85 (d, J = 9.6 Hz, 1H), 6.35 (s, 1H), 3.98−3.87 (m, 4H), 3.81−3.65 (m, 4H), 2.87 (hept, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.56, 155.18, 144.63, 130.97, 127.09, 120.63, 109.99, 43.96, 43.13, 33.67, 29.01, 24.20. LC-MS (m/z): C19H22N6O Exact Mass 350.2; 351.1 (M++1 observed).
Compound 57. 6-(4-(2-(4-isopropylphenyl)acetyl)piperazin-1-yl) pyridazine-3-carbonitrile
1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 9.6 Hz, 1H), 7.19 (m, 4H), 6.81 (d, J = 9.6 Hz, 1H), 3.82 (m, 2H), 3.76 (s, 2H), 3.75−3.66 (m, 4H), 3.63 (m, 2H), 2.88 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 170.27, 158.54, 147.93, 131.79, 130.91, 130.02, 128.59, 127.16, 116.68, 110.05, 45.42, 44.54, 44.01, 41.12, 40.85, 33.87, 24.11. LC-MS (m/z): C20H23N5O Exact Mass 349.2; 350.2 (M++1 observed).
Compound 58. 4-(6-chloropyridazin-3-yl)-N-(4-isopropylphenyl) piperazine-1-carboxamide
1H NMR (400 MHz, CDCl3) δ 7.27 (m, 2H), 7.20−7.11 (m, 2H), 6.91 (m, 2H), 6.31 (s, 1H), 3.76 (dd, J = 6.7, 3.8 Hz, 4H), 3.68 (dd, J = 6.7, 3.8 Hz, 4H), 2.87 (m, 1H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.89, 155.26, 147.55, 144.42, 136.29, 129.18, 127.05, 120.53, 115.35, 44.73, 43.38, 33.67, 31.10, 24.21. LC-MS (m/z): C18H22ClN5O Exact Mass 359.2; 360.0 (M++1 observed).
Compound 59. 1-(4-(6-chloropyridazin-3-yl)piperazin-1-yl)-2-(4-isopropylphenyl)ethan-1-one
1H NMR (400 MHz, CDCl3) δ 7.31 (m, 3H), 7.26 (m, 2H), 6.95 (d, J = 9.5 Hz, 1H), 3.87 (m, 2H), 3.83 (s, 2H), 3.68 (m, 2H), 3.63 (m, 4H), 2.96 (m, 1H), 1.31 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 170.17, 158.92, 147.79, 147.63, 132.00, 129.12, 128.60, 127.09, 115.48, 45.52, 45.39, 44.78, 41.26, 40.80, 33.86, 24.12. LC-MS (m/z): C19H23ClN4O Exact Mass 358.2; 359.1 (M++1 observed).
Compound 60. 6-(4-(2-(4-isopropylphenyl)-2-oxoacetyl)piperazin-1-yl)pyridazine-3-carbonitrile
1H NMR (400 MHz, CDCl3) δ 8.00−7.81 (m, 2H), 7.51 (d, J = 9.6 Hz, 1H), 7.43−7.34 (m, 2H), 6.89 (d, J = 9.6 Hz, 1H), 4.02−3.80 (m, 6H), 3.61−3.50 (m, 2H), 3.00 (hept, J = 7.0 Hz, 1H), 1.28 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 190.70, 165.97, 158.58, 157.39, 131.06, 130.93, 130.39, 130.23, 127.51, 116.56, 110.32, 45.33, 44.75, 44.46, 40.94, 34.68, 23.69. LC-MS (m/z): C20H21N5O2 Exact Mass 363.2; 363.7 (M++1 observed).
Compound 61. 4-isopropylphenyl 4-(6-cyanopyridazin-3-yl)piperazine-1-carboxylate
1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 9.6 Hz, 1H), 7.25−7.19 (m, 2H), 7.07−7.01 (m, 2H), 6.88 (d, J = 9.6 Hz, 1H), 3.86 (m, 8H), 2.91 (m, 1H), 1.24 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.65, 154.01, 149.09, 146.35, 130.96, 130.07, 127.47, 121.39, 116.72, 110.12, 44.01, 33.76, 24.20. LC-MS (m/z): C19H21N5O2 Exact Mass 351.2; 351.8 (M++1 observed).
4.2. Biochemical methods
4.2.1. Materials
Sources of supplies were: D-[1-14C]pantothenate (specific activity, 55 mCi/mmol), American Radiolabeled Chemicals; Ni-NTA resin, Qiagen corporation; Whatman DE81 filter paper, Sigma; All other materials were reagent grade or better.
4.2.2. PANK activity assays
PANK activity assays were performed as described earlier using purified recombinant human PANK3 proteins.17 Briefly, the reaction mixture contained 100 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 2.5 mM ATP, 45 mM D-[1-14C]pantothenate (specific activity, 22.5 mCi/mmol) and 25 ng of PANK protein ± 0–0.1 μM pantazine or 0–10 μM pantazine. The appropriate range for the IC50 determination was selected based on an initial activity determination for each compound at 10 μM. Each assay was incubated at 37 °C and the reaction was stopped after 10 min by the addition of 4 ml 10% (v/v) acetic acid. The mixture was spotted onto a DE81 ion-exchange filter disk and analyzed by scintillation counting. Curves (6 point) were performed in duplicate, data were combined and fit to Morrison’s quadratic equation18 (GraphPad software, version 9.2.0) that accounts for the impact of enzyme inhibitor binding on the free concentration of inhibitor.
4.3. Structural biology methods
4.3.1. Crystallization and structure determination
PanK3 with two amino acids (DD) added to the carboxy-terminus was expressed, purified and crystallized as previously described.16 Compounds 31, 55 and 56 were dissolved in DMSO at 25 mM concentration. To obtain the three complex cocrystal structures, the crystals of the PanK3•AMPPNP•Mg2+•pantothenate complex were soaked in a solution containing 0.2 M ammonium acetate, 0.1 M citrate, pH 5.6, 50 mM MgCl2, 32% polyethylene glycol 4000, 10 mM AMPPNP (adenosine 5′-(β,γ-imido)triphosphate) and 1 mM of compound (final 4% DMSO) for one to two days. Soaked crystals were cryoprotected with 29% ethylene glycol and diffraction data were collected at the SER-CAT beam line 22-ID and 22-BM at the Advanced Photon Source, and processed using HKL2000.19 The structures were solved by molecular replacement using the PANK3 structure (PDB ID: 5KPR) and the program PHASER.20 The structures were refined and optimized using PHENIX and COOT, respectively.21,22 The refined structures were validated using MolProbity.23 The atomic coordinates and structure factors have been deposited in the Protein Data Bank. The PDB codes, and the data collection and refinement statistics are presented in Supplementary Table S1. All structures were rendered with PyMOL (version 2.3.0, Schrödinger, LLC).
4.4. In vitro ADME profiling
The in vitro ADME values (Solubility, PAMPA and Caco 2 permeability, Plasma Protein binding, metabolic and plasma stability) was deterimined as previously reported.24,25
5. Notes
The authors retain patent rights pertaining to compounds in this series.
Supplementary Material
Acknowledgment
This work was supported by National Institutes of Health, United States grant GM034496 (CR) Cancer Center Support Grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health or other funding agencies. We thank the support of the St Jude Chemical Biology and Therapeutics department Analytical Technologies Center for the generation of the in vitro ADME testing data, and Katie Creed for the performance of the enzyme assays.
Abbreviations:
- PANK
Pantothenate kinase
- CoA
coenzyme A
- PKAN
PanK-associated neurodegeneration
- SAR
structure-activity relationship
- LipE
lipophilic ligand efficiency
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supporting information (SI) includes Fig. S1 - Electron densities of compounds 31, 55, 56 within their respective PANK3 ternary complexes, and Table S1 - Data collection and Refinement Statistics. Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2021.116504.
References
- [1].Leonardi R, Zhang Y, Rock C, Jackowski S. Coenzyme A: back in action. Prog Lipid Res. 2005;44(2–3):125–153. [DOI] [PubMed] [Google Scholar]
- [2].Jackowski S, Rock CO. Regulation of coenzyme A biosynthesis. J Bacteriol. 1981; 148(3):926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28(4):345–349. [DOI] [PubMed] [Google Scholar]
- [4].Zhang Y-M, Rock CO, Jackowski S. Feedback regulation of murine pantothenate kinase 3 by coenzyme A and coenzyme A thioesters. J Biol Chem. 2005;280(38): 32594–32601. [DOI] [PubMed] [Google Scholar]
- [5].Rock CO, Karim MA, Zhang Y-M, Jackowski S. The murine pantothenate kinase (Pank1) gene encodes two differentially regulated pantothenate kinase isozymes. Gene. 2002;291(1–2):35–43. [DOI] [PubMed] [Google Scholar]
- [6].Johnson MA, Kuo YM, Westaway SK, et al. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann N Y Acad Sci. 2004;1012:282–298. [DOI] [PubMed] [Google Scholar]
- [7].Kotzbauer PT, Truax AC, Trojanowski JQ, Lee VM. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci: The Official J Soc Neurosci. 2005;25(3):689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sharma LK, Subramanian C, Yun M-K, et al. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat Commun. 2018;9(1). 10.1038/s41467-018-06703-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Subramanian C, Frank MW, Tangallapally R, et al. Pantothenate kinase activation relieves coenzyme A sequestration and improves mitochondrial function in mice with propionic acidemia. Sci Transl Med. 2021;13(611). 10.1126/scitranslmed.abf5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Garcia M, Leonardi R, Zhang YM, Rehg JE, Jackowski S. Germline deletion of pantothenate kinases 1 and 2 reveals the key roles for CoA in postnatal metabolism. PLoS One. 2012;7(7):e40871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barnard L, Mostert KJ, van Otterlo WAL, Strauss E. Developing Pantetheinase-Resistant Pantothenamide Antibacterials: Structural Modification Impacts on PanK Interaction and Mode of Action. Acs Infect Dis. 2018;4(5):736–743. [DOI] [PubMed] [Google Scholar]
- [12].Spry C, Macuamule C, Lin Z, Virga KG, Lee RE, Strauss E, Saliba KJ. Pantothenamides are potent, on-target inhibitors of Plasmodium falciparum growth when serum pantetheinase is inactivated. PLoS One. 2013;8(2):e54974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sharma LK, Leonardi R, Lin W, et al. A high-throughput screen reveals new small-molecule activators and inhibitors of pantothenate kinases. J Med Chem. 2015;58 (3):1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shultz MD. The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorg Med Chem Lett. 2013;23(21):5992–6000. [DOI] [PubMed] [Google Scholar]
- [15].Johnson TW, Gallego RA, Edwards MP. Lipophilic Efficiency as an Important Metric in Drug Design. J Med Chem. 2018;61(15):6401–6420. [DOI] [PubMed] [Google Scholar]
- [16].Subramanian C, Yun M-K, Yao J, et al. Allosteric Regulation of Mammalian Pantothenate Kinase. J Biol Chem. 2016;291(42):22302–22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hong BS, Senisterra G, Rabeh WM, et al. Crystal structures of human pantothenate kinases. Insights into allosteric regulation and mutations linked to a neurodegeneration disorder. J Biol Chem. 2007;282(38):27984–27993. [DOI] [PubMed] [Google Scholar]
- [18].Morrison JF. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim Biophys Acta. 1969;185(2):269–286. [DOI] [PubMed] [Google Scholar]
- [19].Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. [DOI] [PubMed] [Google Scholar]
- [20].McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63(1):32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Afonine PV, Grosse-Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(4):352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen VB, Arendall WB, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(1): 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Griffith EC, Zhao Y, Singh AP, et al. Ureadepsipeptides as ClpP Activators. Acs Infect Dis. 2019;5(11):1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].North EJ, Scherman MS, Bruhn DF, et al. Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties. Bioorg Med Chem. 2013;21(9):2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.