

Abstract
Infection with one of the dengue viruses 1–4 (DENV1–4) induces protective antibodies to homotypic infection. However, an exceptional feature of dengue viruses is that they can use preexisting heterotypic antibodies to infect Fcγ receptor-bearing immune cells, leading to higher viral load and immunopathological events that augment disease. We tracked the antigenic dynamics of each DENV serotype using 1,944 sequenced isolates from Bangkok, Thailand between 1994–2014 (n=348) in comparison to regional and global DENV antigenic diversity (n=64 strains). Over the course of 20 years, the Thailand DENV serotypes gradually evolved away from one another. However, for brief periods, the serotypes became more similar, with corresponding changes in epidemic magnitude. Antigenic evolution within a genotype involved a tradeoff in within versus between serotype antigenic change, whereas genotype replacement resulted in antigenic change away from all serotypes. These findings provide insights into theorized dynamics in antigenic evolution.
One sentence summary:
For dengue viruses both individual antibody level and antigenic differences between specific infecting strains may be important for future disease risk.
Antigenic evolution occurs in many viruses. Viral proteins recognized by the immune system change, enabling evasion of host immunity induced by prior infection with similar viruses (1). Dengue viruses 1–4 (DENV1–4) provide an example of how preexisting heterotypic antibodies may not only be evaded but also be exploited by the virus to aid infection by facilitating entry into and replication in immune cells, and inducing higher viral load and immunopathological events that augment disease severity (2–6). DENV1–4 cause ~100 million infections, 50 million febrile dengue cases, ~500,000 hospitalizations, and 10,000–25,000 deaths annually (7). High levels of cross-serotype reactive antibodies can protect against secondary DENV infection with a different serotype, while low to intermediate levels increase risk of Dengue Hemorrhagic Fever/Dengue Shock Syndrome as a result of antibody-dependent enhancement (3–6).
DENV1–4 vary antigenically within serotype but strong evidence for antigenic escape has not been found. Each DENV serotype consists of four to seven genotypes that differ from one another by ≤10% at the amino acid level across the envelope protein (8). Antibodies from naturally infected and vaccinated individuals differentially neutralize distinct genotypes and even distinct clinical isolates, also known as strains, of each serotype (9–12). Homotypic immunity is generally protective, although protection against clinical disease is not always complete against viruses of a different genotype, and this could potentially reduce vaccine efficacy (13, 14). Further, there are large differences in how similar a given DENV strain is antigenically to strains of different DENV serotypes (9, 15). Phylogenetic analyses indicate that genotype replacement events, defined as when a previously common viral lineage vanishes in a given location and a related but distinct lineage becomes dominant, may be driven by natural selection and immune pressure, although population bottlenecks are an alternative explanation (16–18). However, only a few studies have linked genetic or antigenic differences between strains to epidemic magnitude and severity (17, 19, 20). Mechanistic transmission models and empirical observations suggest temporary cross-protection and ‘antibody-dependent enhancement’– including enhanced probability of disease, infectiousness, or susceptibility to second infections – along with other spatial, temporal, and vector-associated parameters are drivers behind DENV epidemic dynamics (18, 21–27). Yet, whether antigenic variation observed among DENV1–4 is biologically relevant and associated with epidemic dynamics of co-circulating strains remains controversial. If DENV1–4 are evolving antigenically, changes are expected to be most evident in a single highly endemic geographic location where strains interact directly with immunity derived from other currently or previously circulating strains.
Here, we tested whether DENV1–4 circulating in Bangkok, Thailand changed antigenically over two decades in relation to each other and a selection of globally representative DENV1–4 strains. We conducted full-genome sequencing of 1,944 clinical DENV isolates systematically sampled between 1994 and 2014 at the Queen Sirikit National Institute of Child Health (QSNICH). QSNICH is a tertiary children’s hospital in central Bangkok, Thailand that serves as the city’s main referral center for hospitalized dengue (28, 29). DENV1–4 have circulated in Bangkok since at least 1962, with transmission observed each year, at all times of year, and larger epidemics occurring periodically (29–31). Between 1994 and 2014, each serotype was dominated by a single genotype: genotype I for DENV1, Asian I genotype for DENV2, genotype II for DENV3, and genotype I for DENV4 (32, 33) (Fig. S1). Another DENV4 genotype circulated at lower levels, and single representatives of distinct DENV2 genotypes were isolated. DENV3 was the only serotype to undergo a genotype replacement event during this period, with genotype III displacing genotype II.
We systematically selected 348 of the sequenced Thailand DENV1–4 strains for antigenic characterization. Sampling was balanced across years and included representation of amino acid variation in the envelope (E) and pre-membrane (prM) proteins to increase the likelihood of capturing any antigenic change (Fig. 1A–E, Fig. S1, Table S1). To place the Thailand DENV1–4 in a temporal, regional, and global context, we also antigenically characterized strains from 20 different countries isolated between 1944 and 2012 (n=64). All strains (n=412 total) were characterized using a plaque (immunofocus) reduction neutralization test (PRNT), the gold-standard method for measuring serological immunity to DENV1–4. High PRNT titers are correlated with vaccine efficacy and reduced risk of dengue, while lower PRNT titers are instead associated with increased risk of severe dengue (4, 34). All viruses were titrated by PRNT on mosquito cells (Aedes albopictus C6/36) against a panel of antisera from 20 African green monkeys (Chlorocebus sabaeus) each inoculated with a distinct DENV strain and used previously to characterize DENV antigenic diversity (9, 35) (n=7,957 titrations, Fig. 1F, Table S2, Methods). Sera collected 90 days post-inoculation had high PRNT titers and closely approximated the long-term antibody response (day 150) (9). PRNT titers were adjusted by virus to control for experimental conditions that systematically modified PRNT titers, regardless of sampling year or serotype: 1) duration of virus-serum incubation and 2) re-aliquoting of old virus stocks versus re-amplification immediately prior to titration. We estimated the antigenic relationships among strains using antigenic cartography, a method that converts serum PRNT50 titers into units of antigenic distance by expressing titer data in maps of reduced dimensions compared to the full data (1, 9). We used cross-validation (100 maps each with a random 75% of titers) to identify the coordinates and map dimensions (exploring 2–10 dimensions) for which map antigenic distances most closely predicted excluded titers (Fig. S2). All antigenic analyses were performed on 3D maps, which were found to optimally represent the titer data (File S1). 2D maps were close in performance and are shown here as well.
Fig. 1. Genetic and antigenic characteristics of DENV1–4 strains isolated in Bangkok, Thailand in relation to global DENV strains.
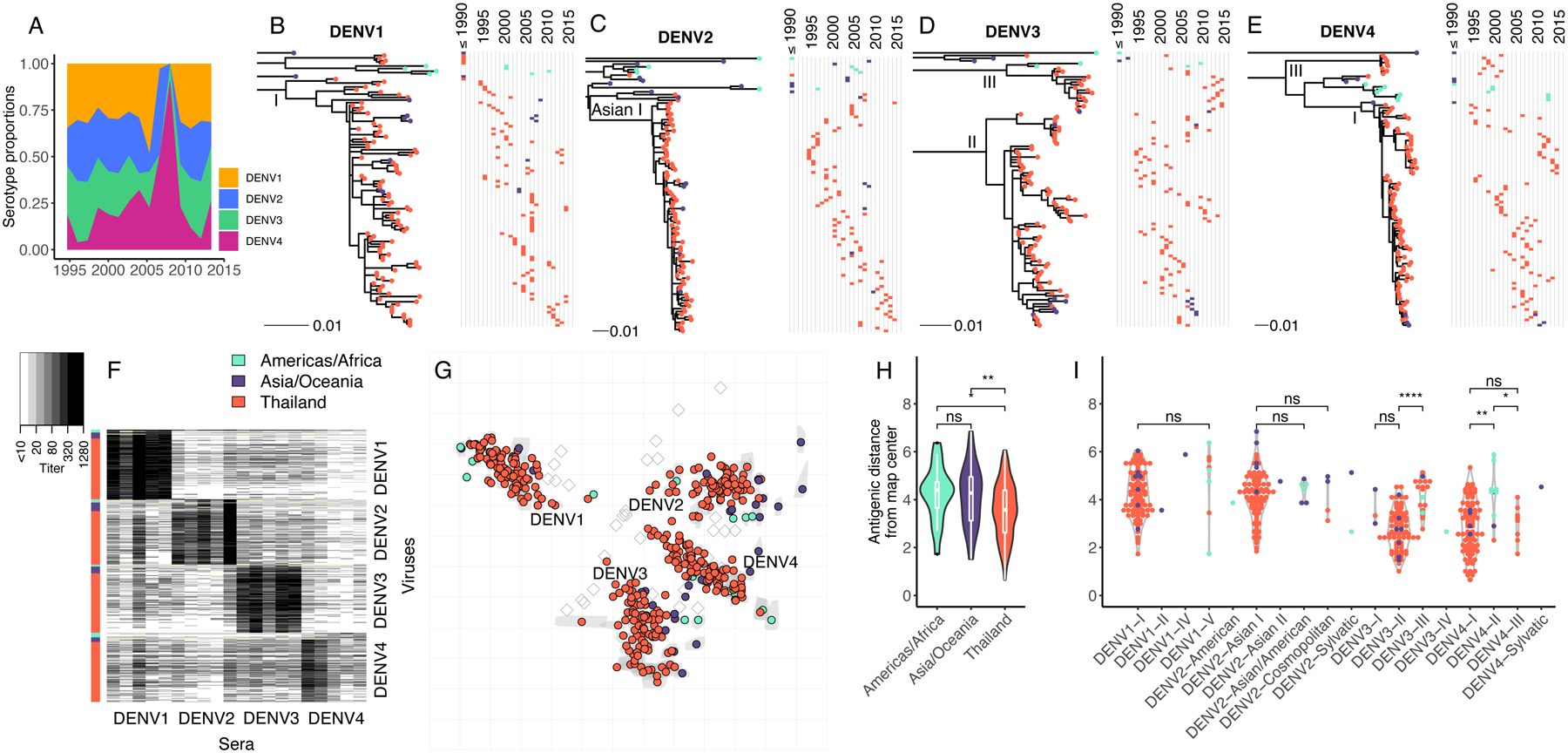
(A) Proportion by serotype of 1,944 clinical DENV strains isolated between 1994 to 2014 at QSNICH. Strains for antigenic characterization were selected from this full set. (B-E) Evolutionary relatedness among E protein sequences of DENV1–4 (n=348) from Thailand (1994–2014) compared to strains from other countries or periods in time (n=64). Maximum likelihood phylogenetic trees were built using generalized time reversible nucleotide substitution models with gamma-distributed rate variation and allowing for invariant sites (GTR+G4+I). Strains are colored to indicate the geographic area where the strain was isolated (Americas/Africa, Asia/Oceania, Thailand). Corresponding time series show the years of strain isolation. (F) Heat map of PRNT50 titers (n=7,957) for all DENV1 (n=105), DENV2 (n=99), DENV3 (n=103) and DENV4 (n=105) strains titrated against antisera from non-human primates (n=20, 5 per serotype) each inoculated with a genetically distinct global DENV strain. Rows correspond to DENV strains (row colors indicate region), while columns correspond to antisera. (G) Antigenic map made in two dimensions of all DENV1–4 strains. Grey shapes indicate interquartile range of coordinates for each virus based on cross-validation maps. Colored circles correspond to median coordinates for each virus. Each grid-square side corresponds to a two-fold dilution in the PRNT50 assay, and distance is interpretable in any direction. Sera are represented as open squares. Violin plots of antigenic distances of each virus from the center of the 3D antigenic map by location of virus isolation (H) or genotype (I). Global significance tests were conducted with a Kruskal-Wallis Rank Sum Test, followed by pairwise comparisons using the Wilcoxon Rank Sum test.
On the 2D and 3D antigenic maps, extensive antigenic diversity was observed within serotype (Fig. 1G; 95th percentile of within-serotype distances in 3D map, 8.9–19.2-fold, File S1). As we have observed previously, some strains were as close antigenically to strains of other serotypes as to strains of the same serotype (9). Unexpectedly, DENV1–4 strains from Thailand were closer antigenically to each other (measured as distance from the map center) than strains from other countries in Asia/Oceania or the Americas/Africa (Fig. 1H). This effect remained after accounting for the greater number of Thailand strains in the dataset (Fig. S3). These differences were explained in part by the genotypes circulating in Thailand. The dominant genotypes of DENV4 circulating in Thailand, genotypes I and III, were significantly more antigenically central on the maps than DENV4 genotype II strains, which circulate in the Americas/Africa (Fig. 1I). DENV3 genotype II strains were also significantly more similar antigenically to other serotypes than DENV3 genotype III strains (Fig. 1I), which became the dominant genotype in Thailand at the end of the observational period (Fig. 1D).
Thailand DENV1–4 strains appeared to be evolving antigenically over time (Fig. 2, Movie S1). To measure these antigenic dynamics quantitatively, we fitted antigenic distances from the 3D map as a function of time using linear regression and generalized additive models (GAMs) with bootstrap resampling (n=100) to construct confidence intervals (Table S3). Between 1994–2014, the serotypes moved away from the center of the antigenic map, dropping in neutralization by 40% overall (95% confidence interval [CI]: 20 to 53%) (Fig. 3A). DENV1, DENV3, and DENV4 each moved away from other serotypes, with neutralization dropping by 52% for DENV1, 74% for DENV3, and 39% for DENV4 over 20 years (Fig. 3A, vertical plots). Both the raw data (means and standard deviations of annual antigenic data) and GAM fits reveal non-linear antigenic dynamics (estimated degrees of freedom [EDF]: 4.1, p<0.001, Fig. 3A), with antigenic distance fluctuating around an overall increase in time; this effect was observed individually for DENV1, DENV3, and DENV4 (Fig. 3A, vertical plots, detrended antigenic dynamics, Fig. S4). The serotypes also moved away from one another, with an average decrease in neutralization between pairwise serotypes of 65% over the 20-year period (Fig. 3B). Again, the distance between serotypes fluctuated (EDF: 8.5, p<0.001) from as little as 4.9 antigenic units (29-fold difference in PRNT50 titers) up to 6.9 antigenic units (120-fold difference) (Fig. 3B).
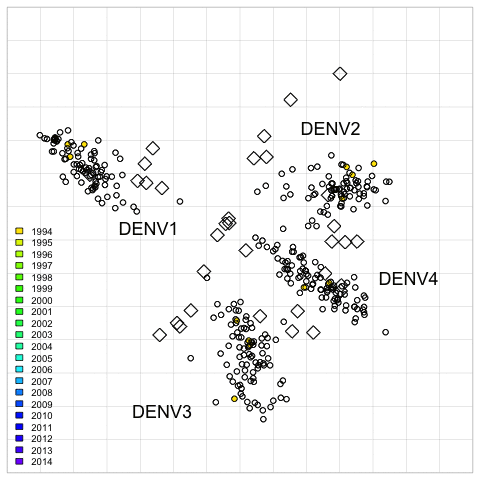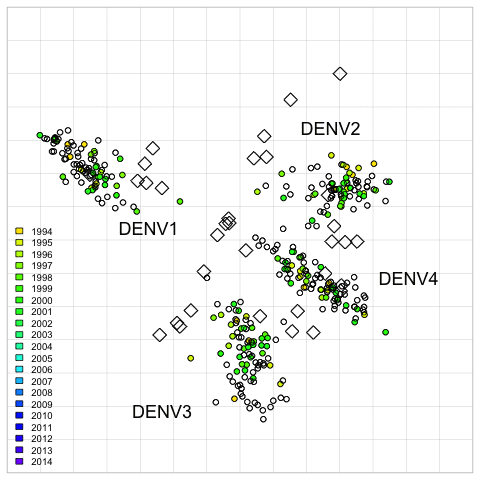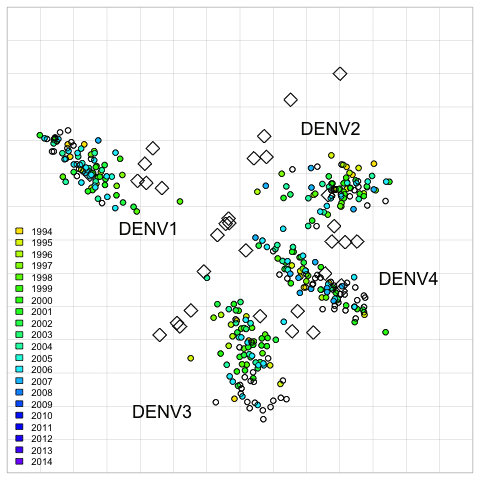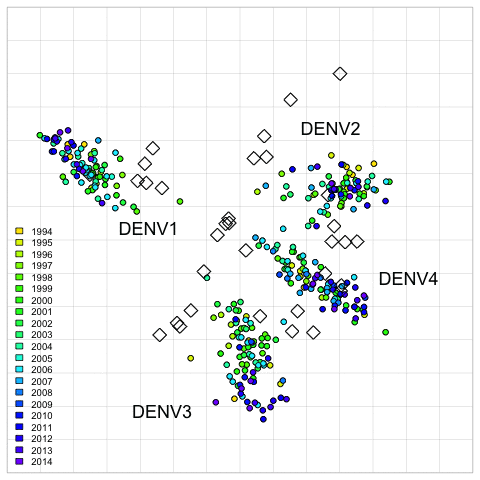
Fig. 2. 2D antigenic map of Thailand DENV1–4 colored by year of isolation.
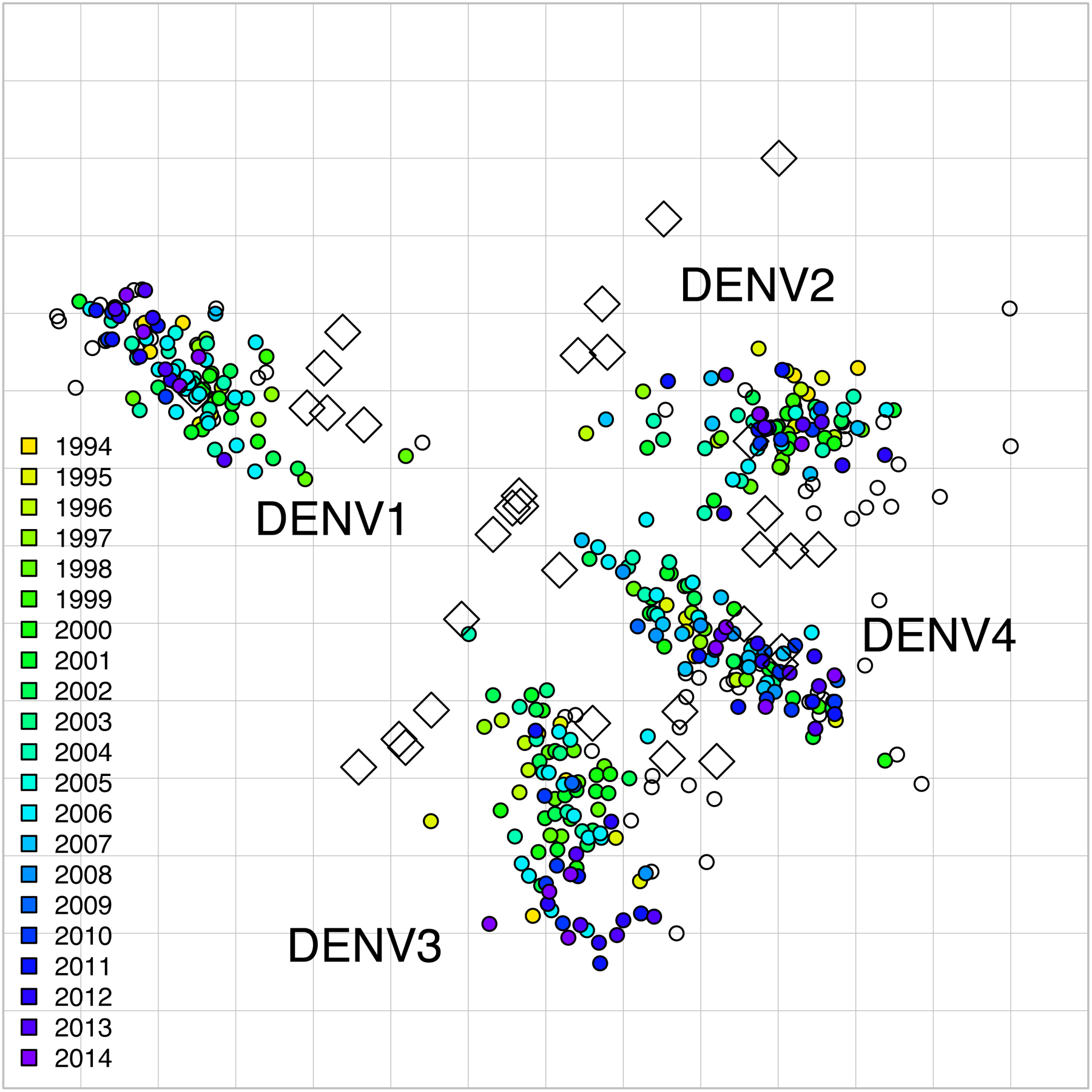
Open circles show global viruses, open squares show the serum positions. Serotype clusters are labeled. Each grid-square side in both dimensions is equivalent to a two-fold dilution in the PRNT50 assay.
Fig. 3. Antigenic dynamics of Thailand DENV1–4 strains isolated between 1994 and 2014.
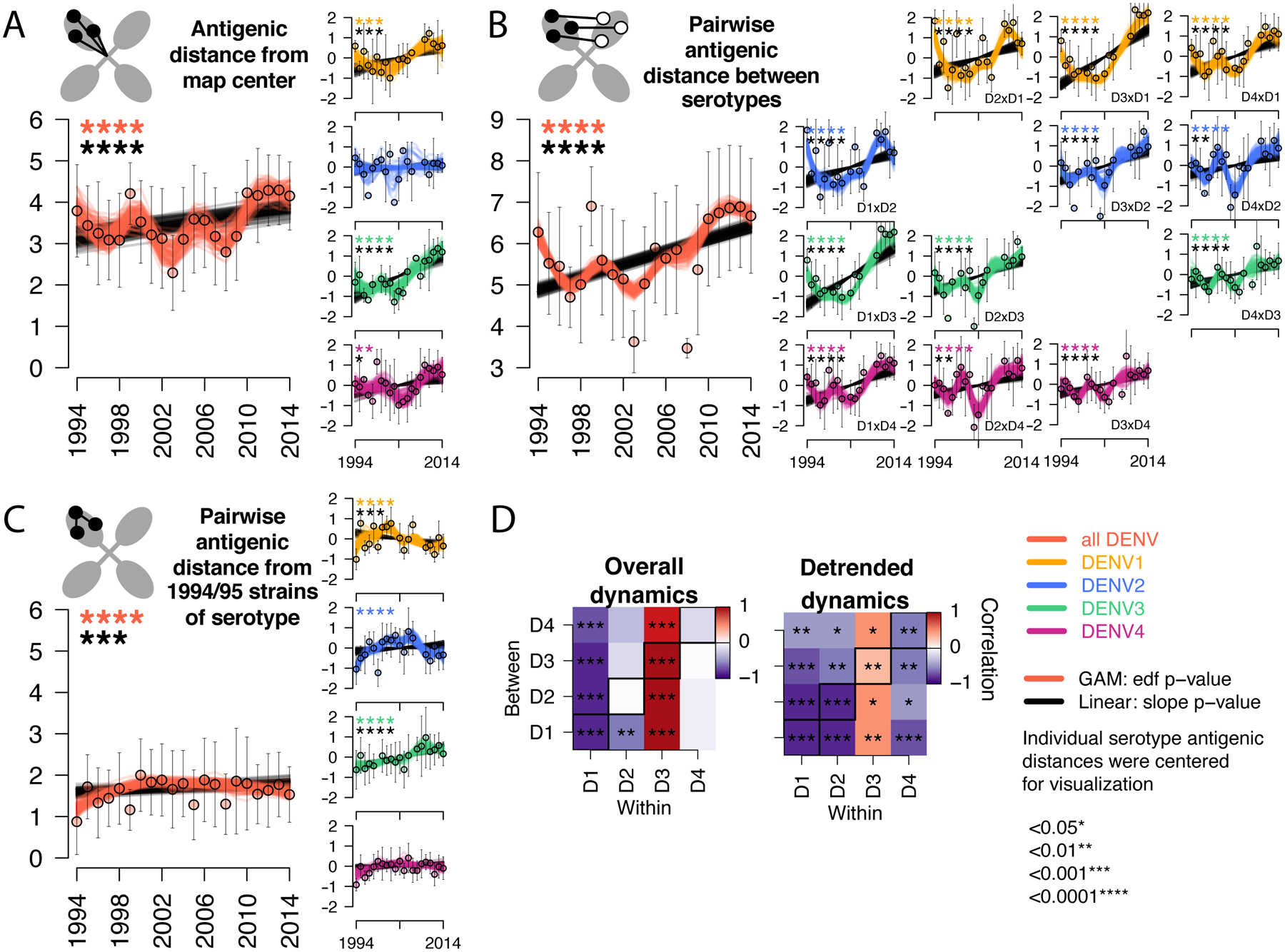
Antigenic change over time for all serotypes, measured as (A) distance from the map center, (B) pairwise distance between serotypes each year, (C) pairwise distance from 1994–1995 strains of the same serotype. Cartoons depict each antigenic distance metric. For each antigenic time series, antigenic distances were bootstrap sampled (n=100) and used to construct 100 linear (black lines) and non-linear generalized additive (color lines) models. Mean and standard deviation of antigenic distances are shown as colored circles with black bars. Black stars indicate significant linear change (slope) while colored stars indicate statistically significant non-linearity (effective degrees of freedom). Models were run for all serotypes combined (with a variable to adjusted for serotype, large plots, y-axis shows measured distances) and for each serotype separately (vertical plots, y-axis shows distances centered at zero to facilitate comparison of relative change across plots). (D) Pearson correlation coefficients and corresponding p-values of bootstrapped (n=1000) within serotype (columns) versus between serotype (rows) overall antigenic dynamics (no adjustment) and detrended dynamics (linear model subtracted from the GAM prior to analysis). Diagonal black boxes correspond to distance from the map center, off diagonal indicates pairwise distance between serotypes. Color indicates correlation (range −1 to 1), while significance is indicated by stars.
The observed increase in antigenic distance between the DENV serotypes is consistent with the hypothesis that mounting immunity to previously circulating strains selects for viruses that are antigenically different (1). Homotypic DENV immunity is potently neutralizing and long-lasting, and thus major epidemics may select for antigenic difference relative to previously circulating strains of the same serotype. Further, high cross-serotype immunity induced in the first months after primary DENV infection or for years after secondary DENV infection may select for antigenic evasion of heterotypic immunity after large epidemics of other serotypes. However, we also observed that periodically, the serotypes evolved to be more antigenically similar. One hypothesis is that cross-serotype antibodies wane after primary infection to titers that mediate antibody-dependent enhancement of infection, viral load, and severity, ‘pulling’ a serotype antigenically toward other serotypes (2). Alternatively, structural constraints on protein function imposed by the need for DENV to efficiently replicate both in the human host and mosquito vector may limit the mode of antigenic change possible for DENV at a given time. For instance, to evade of homotypic immunity, strains may change to resemble other serotypes if heterologous neutralization is a weak selective pressure. If true, we would expect to observe an inverse relationship between homotypic and heterotypic antigenic dynamics and a link between homotypic immune evasion and larger epidemics. It is also possible that within and between serotype antigenic evolution proceeds independently if the epitopes targeted are distinct (36).
We tested the hypothesis that within and between serotype antigenic change is correlated. Within-serotype antigenic change was measured as the pairwise antigenic distance from 1994 and 1995 strains of each serotype (Fig. 3C). On average, the serotypes became more antigenically distinct from earlier strains of the same serotype before gradually switching back, with oscillations in antigenic distance throughout the period (Fig. 3C). These dynamics were significant for DENV1 and DENV2 (Fig. 3C, vertical plots). We then measured the Pearson’s correlation coefficient for within versus between serotype antigenic change using the overall (unadjusted) and detrended bootstrapped (n=1000) antigenic time series. For DENV1, DENV2, and DENV4, change away from homotypic strains correlated with increased similarity to heterotypic strains, with the strongest effects for DENV1 and DENV2, and a weaker effect for DENV4 (Fig. 3D). The kinetics for DENV3 were distinct. DENV3 became more antigenically distant from early homotypic and heterotypic strains linearly and simultaneously (Fig. 3C and D). The largest antigenic change occurred during the replacement of DENV3 genotype II by genotype III between 2010 and 2014 (Fig. 1D). Interestingly, both genotypes were evolving antigenically relative to homotypic and heterotypic strains, but genotype III achieved greater antigenic distance from other serotypes, especially DENV1 (fig. S5).
We hypothesized that these antigenic changes might be associated with epidemic magnitude over the same period. We estimated the Pearson’s correlation coefficient for each antigenic time series with the annual serotype-specific incidence of dengue cases treated at QSNICH in Bangkok (Fig. 4, Fig. S6). On average and independently for DENV1, DENV2, and DENV4, large epidemics occurred when strains were more similar antigenically to other serotypes (Fig. 4A and B) but less similar to earlier strains of the same serotype (Fig. 4C). In contrast, the lowest DENV3 incidence occurred when DENV3 was most antigenically similar to other DENV3 strains and other serotypes. The genotype replacement event followed this period of low incidence, with both DENV3 genotypes evolving antigenically away from earlier DENV3 and heterotypic serotypes as DENV3 incidence rebounded (Fig. 4A to C, vertical plots). Across serotypes, antigenic change correlated over the entire period and on a year-to-year basis with incidence, suggesting a close link between annual epidemiologic change and corresponding shifts in antigenic phenotype at the population level (Fig. 4D, Fig. S4, detrended series).
Fig. 4. Correlation of the antigenic and epidemic time series for DENV1–4 in Bangkok, Thailand from 1994 to 2014.
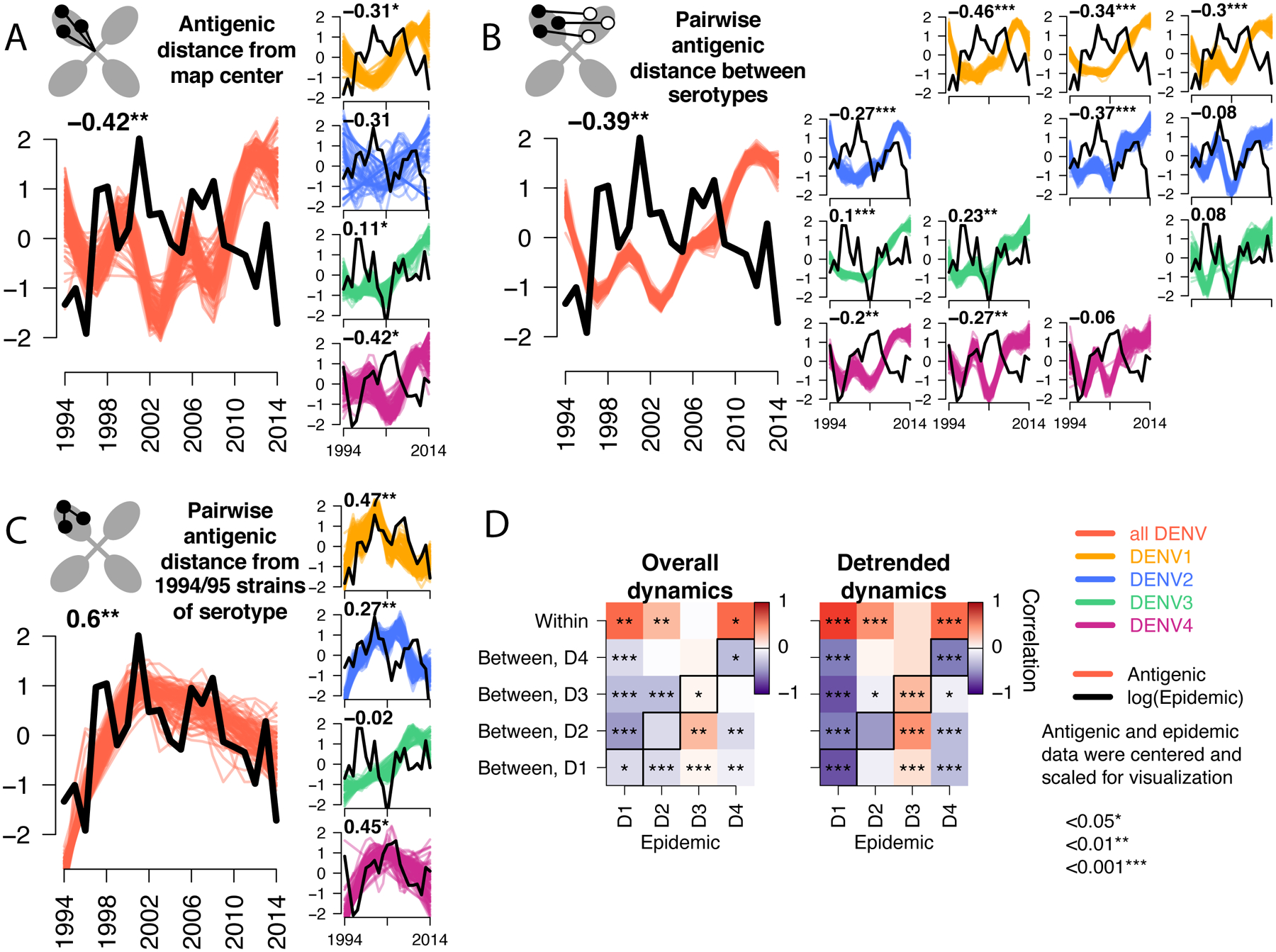
Epidemic (black lines) versus antigenic time series (colored lines) for all serotypes (large plots) or each serotype separately (vertical plots) measured as (A) distance from the map center, (B) pairwise distance between serotypes each year, (C) pairwise distance from 1994 and 1995 strains of the same serotype. All antigenic and epidemic time series are scaled by the standard deviation and centered at zero for visualization. Pearson’s correlation coefficients and corresponding p-values for epidemic versus bootstrapped (n=1000) antigenic time series are shown (top left), with significance indicated by stars. (D) Summary of Pearson correlation coefficients of the epidemic and antigenic time series for overall antigenic dynamics (no adjustment) and detrended dynamics (linear model subtracted from the GAM). Shown for each serotype (columns) and metric of antigenic distance (rows). Diagonal black boxes correspond to distance from the map center, off diagonal indicates pairwise distance between serotypes. Color indicates correlation (range −1 to 1), while significance is indicated by stars.
Here, we found that antigenic evolution differed within a genotype versus during a major genotype replacement event. Within genotype for DENV1, DENV2, and DENV4, major outbreaks correlated with evasion of homotypic protection. A large outbreak of a given genotype could lead to a selective sweep that would be followed by reduced antigenic diversity and thus decreased antigenic distance between serotypes. Alternatively, the pressure to evade homotypic immunity may be so strong as to drive strains in the direction of other serotypes if there are structural limits on the amino acids permitted within a given genotype. Strains may also tolerate weak cross-neutralization if such changes improved replication or fitness in mosquitoes or human hosts. For example, laboratory adapted DENV strains have acquired amino acid changes that render them more susceptible to homotypic and heterotypic neutralization but may be advantageous in cell culture (9, 37). It is also possible that antigenic change toward other serotypes facilitates antibody-dependent enhancement (2). Our assay does not directly measure enhancement, but neutralizing antibody titers are correlated both with peak enhancement titers and with increased severity of illness (4, 38). Due to sampling hospitalized dengue cases, our study overrepresents secondary severe infections, which may be under stronger immunologic pressure from enhancement and have higher viral loads with greater within-host viral diversity (29). Our isolates were collected from a pediatric hospital setting and so may not reflect the full diversity of DENV1–4 circulating in Bangkok during this period. Other studies have linked specific infection histories and viral lineages to increased severity. A study in Nicaragua showed that prior infection with DENV1 versus DENV3 differentially modified disease severity during subsequent DENV2 infection with distinct clades (17). In another study, an evolutionarily successful DENV2 lineage had an amino acid change that increased sensitivity to heterotypic neutralization but induced higher viremia during secondary infection (39). Increased severity has been theorized to reduced transmission, and if true, excessive optimization of cross-serotype enhancement may be an evolutionary dead end due to hospitalization reducing transmission opportunities (40). However, the large role that asymptomatic and pre-symptomatic individuals play in transmission may minimize the selection due to this mechanism (41). In the case of DENV3, antigenic similarity to other serotypes as well as to earlier DENV3 strains was associated with the lowest DENV3 incidence, potentially selecting for DENV3 strains that escaped both homotypic and heterotypic immunity. A previous phylogenetic study of DENV in Thailand showed that clade replacements are associated with declining incidence (16). Our findings further support this observation and suggest immunological pressure imposed by co-circulating serotypes, rather than population bottlenecks, help govern replacement events. Additional data from genotype replacement events for DENV and other viruses is needed to further evaluate this hypothesis.
The intense cocirculation of multiple DENV serotypes and genotypes in a single location with high population density is relatively new in most parts of the world, and likely only occurred in Thailand since the beginning of the 20th century. Even in Thailand, DENV1–4 may still be transitioning to an endemic equilibrium, adjusting antigenic distances relative to one another in an ongoing process. Due to its endemicity, the serotypes in Thailand may interact more intensively, explaining why the serotypes were closer together than serotypes in other regions. The antigenic distance between Thailand serotypes increased over time but by a non-linear path, with fluctuations in antigenic distance within and among serotypes that closely correlated with epidemic magnitude. Given that DENV epidemic dynamics are governed by population immunity, demography, host behavior, vector abundance, and environmental factors, it is possible that the observed antigenic fluctuations track with longer-term epidemiological patterns that cannot be disentangled with only 20 years of data (42). However, our study of co-circulating strains, genotypes, and serotypes suggests that multiple selective mechanisms may affect antigenic evolutionary processes simultaneously, including immune evasion, antibody-mediated enhancement, constraints on viral protein structure, introduction of new genotypes, and local dengue incidence. Specifically, the balance between cross-protection and antibody-dependent enhancement has been posited to explain the phylogenetic distance between DENV1–4 and may help explain the more bounded nature of DENV antigenic evolution compared to other well-studied antigenically variable viruses (2). Influenza A and B viruses ‘zig-zag’ through antigenic space but fundamentally evolve linearly away from previously circulating strains (1, 43). While other antigenically variable viruses such as enterovirus 71 and GII.4 noroviruses have complex, non-linear patterns of variation across antigenic space, it is not clear based on available data whether they oscillate over evolutionary time (44, 45). This work constitutes the most comprehensive dataset to date to explore hypothesized evolutionary tradeoffs for DENV and more broadly among antigenically interacting serotypes, with potentially important insights for identifying the determinants of viral antigenic evolution and informing virus surveillance and vaccine evaluation.
Supplementary Material
{kind=link}
Acknowledgements:
Funding:
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (SW, LCK), National Institute of Allergy and Infectious Diseases and National Institutes of Health Grant R01AI114703-01 (DATC, LCK, ACE, AH, BGC, NC, IMB, CC, LM, DS, GG, RJ, HS), the Military Infectious Disease Research Program (AH, IMB, LM, GG, RJ), and a European Research Council Grant 804744 (HS). Sequencing for infectious disease surveillance was additionally supported by the Global Emerging Infections Surveillance (GEIS) Branch (RJ).
Footnotes
Competing interests: P.B. is with GlaxoSmithKline vaccines in Singapore and has stock options with GSK. L.M. now works at Merck. Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The View(s) expressed are those of the authors and do not necessarily reflect the official views of the Uniformed Services University of the Health Sciences, Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., Department of Health and Human Services, the National Institutes of Health, the Departments of the Army, Navy or Air Force, the Department of Defense, or the U.S. Government.
Data and materials availability:
The viruses and antisera used in this study are covered by material transfer agreements between the institutions in this research (WRAIR, NIH, UF). Requests for sharing of sera or viruses can be directed to the corresponding authors and accommodated subject to institutional and regulatory approvals: Derek Cummings (datc@ufl.edu), Henrik Salje (hs743@cam.ac.uk), and Steve Whitehead (swhitehead@niaid.nih.gov). R code and raw antigenic and epidemic data used in the figures in this manuscript are available on Zenodo (33). All sequence data is publicly available on GenBank (Accession #s KY586306 to KY586946, MW881266, MW945425 to MW945427, MW945430, MW945433 to MW945437, MW945454 to MW945763, MW945772 to MW946604, MW946607 to MW946985).
References
- 1.Smith et al. , Science. 305, 371–376 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Grenfell et al. , Science. 303, 327–32 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Katzelnick et al. , Science. 358, 929–932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salje et al. , Nature. 557, 719–723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halstead, O’Rourke, Nature. 265, 739–41 (1977). [DOI] [PubMed] [Google Scholar]
- 6.Halstead, J. Infect. Dis 140, 527–533 (1979). [DOI] [PubMed] [Google Scholar]
- 7.Cattarinov et al. , Sci. Transl. Med 12, eaax4144 (2020).31996463 [Google Scholar]
- 8.Twiddy, Holmes, Rambaut, Mol. Biol. Evol 20, 122–129 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Katzelnick et al. , Science. 349, 1338–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Messer et al. , PLoS Negl. Trop. Dis 6, e1486 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallichotte et al. , Cell Rep. 25, 1214–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez et al. , Cell Rep. 33, 108226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waggoner et al. , J. Infect. Dis 214, 986–993 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juraska et al. , Proc. Natl. Acad. Sci. U. S. A 115, E8378–E8387 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell, Katzelnick, Bedford, Elife. 8, 1–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang et al. , J. Virol 79, 15123–15130 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.OhAinle et al. , Sci. Transl. Med 3, 114ra128–114ra128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams et al. , Proc. Natl. Acad. Sci 103, 14234–14239 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochel et al. , Lancet. 360, 310–312 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Forshey, Reiner, Olkowski, Morrison, 1–32 (2015).
- 21.Nagao, Koelle, Proc. Natl. Acad. Sci 105, 2238–2243 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wearing, Rohani, Proc. Natl. Acad. Sci 103, 11802–7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reich et al. , J. R. Soc. Interface 10, 20130414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lourenco, Recker, PLoS Comput. Biol 9, e1003308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferguson, Anderson, Gupta, Proc. Natl. Acad. Sci. U. S. A 96, 790–794 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cummings et al. , Proc. Natl. Acad. Sci 102, 15259–15264 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams, Boots, J. Theor. Biol 242, 337–346 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Salje et al. , Science. 355, 1302–1306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nisalak et al. , Am. J. Trop. Med. Hyg 94, 1342–1347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cummings et al. , Nature. 427, 344–347 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Halstead, Scanlon, Umpaivit, Udomsakdi, Am. J. Trop. Med. Hyg 18, 997–1021 (1969). [DOI] [PubMed] [Google Scholar]
- 32.Holmes, Twiddy, Infect. Genet. Evol 3, 19–28 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Katzelnick, Zenodo. V1 (2021), doi: 10.5281/zenodo.5365818. [DOI] [Google Scholar]
- 34.Moodie et al. , J. Infect. Dis 217, 742–753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katzelnick et al. , PLoS Negl. Trop. Dis 12, e0006862 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Alwis et al. , Proc. Natl. Acad. Sci 109, 7439–7444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dowd, DeMaso, Pierson, MBio. 6, e01559–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kliks et al. , Am. J. Trop. Med. Hyg 40, 444–451 (1989). [DOI] [PubMed] [Google Scholar]
- 39.Wang et al. , J. Infect. Dis 213, jiv536 (2015). [Google Scholar]
- 40.Fenner, Marshall, J. Hyg. (Lond) 55, 149–191 (1957). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.ten Bosch et al. , PLoS Pathog. 14, 82–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cummings et al. , PLoS Med. 6, e1000139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bedford et al. , Elife. 3, e01914 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang et al. , J. Clin. Microbiol 47, 3653–3662 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kendra et al. , Proc. Natl. Acad. Sci. U. S. A 118 (2021). [Google Scholar]
- 46.Lefort, Longueville, Gascuel, Mol. Biol. Evol 34, 2422–2424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guindon et al. , Syst. Biol 59, 307–321 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Duong et al. , Infect. Genet. Evol 15, 59–68 (2013). [DOI] [PubMed] [Google Scholar]
- 49.R Core Team, R Foundation for Statistical Computing, Vienna, Austria: (2020), https://www.R-project.org/. [Google Scholar]
- 50.Wood. J. R. Stat. Soc. Ser. B. Statistical Methodol 73, 3–36 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The viruses and antisera used in this study are covered by material transfer agreements between the institutions in this research (WRAIR, NIH, UF). Requests for sharing of sera or viruses can be directed to the corresponding authors and accommodated subject to institutional and regulatory approvals: Derek Cummings (datc@ufl.edu), Henrik Salje (hs743@cam.ac.uk), and Steve Whitehead (swhitehead@niaid.nih.gov). R code and raw antigenic and epidemic data used in the figures in this manuscript are available on Zenodo (33). All sequence data is publicly available on GenBank (Accession #s KY586306 to KY586946, MW881266, MW945425 to MW945427, MW945430, MW945433 to MW945437, MW945454 to MW945763, MW945772 to MW946604, MW946607 to MW946985).